 Stefan Weiler1,2
Stefan Weiler1,2 Manfred Nairz3*
Manfred Nairz3*- 1National Poisons Information Centre, Tox Info Suisse, Associated Institute of the University of Zurich, Zurich, Switzerland
- 2Department of Chemistry and Applied Biosciences, Institute of Pharmaceutical Sciences, Eidgenossische Technische Hochschule Zurich, Zurich, Switzerland
- 3Department of Internal Medicine II, Infectious Diseases, Immunology, Rheumatology, Pneumology, Medical University of Innsbruck, Innsbruck, Austria
Cancer-induced anemia (CIA) is a common consequence of neoplasia and has a multifactorial pathophysiology. The immune response and tumor treatment, both intended to primarily target malignant cells, also affect erythropoiesis in the bone marrow. In parallel, immune activation inevitably induces the iron-regulatory hormone hepcidin to direct iron fluxes away from erythroid progenitors and into compartments of the mononuclear phagocyte system. Moreover, many inflammatory mediators inhibit the synthesis of erythropoietin, which is essential for stimulation and differentiation of erythroid progenitor cells to mature cells ready for release into the blood stream. These pathophysiological hallmarks of CIA imply that the bone marrow is not only deprived of iron as nutrient but also of erythropoietin as central growth factor for erythropoiesis. Tumor-associated macrophages (TAM) are present in the tumor microenvironment and display altered immune and iron phenotypes. On the one hand, their functions are altered by adjacent tumor cells so that they promote rather than inhibit the growth of malignant cells. As consequences, TAM may deliver iron to tumor cells and produce reduced amounts of cytotoxic mediators. Furthermore, their ability to stimulate adaptive anti-tumor immune responses is severely compromised. On the other hand, TAM are potential off-targets of therapeutic interventions against CIA. Red blood cell transfusions, intravenous iron preparations, erythropoiesis-stimulating agents and novel treatment options for CIA may interfere with TAM function and thus exhibit secondary effects on the underlying malignancy. In this Hypothesis and Theory, we summarize the pathophysiological hallmarks, clinical implications and treatment strategies for CIA. Focusing on TAM, we speculate on the potential intended and unintended effects that therapeutic options for CIA may have on the innate immune response and, consequently, on the course of the underlying malignancy.
Cancer-Induced Anemia Is a Frequent Consequence of Malignancy
Cancer-induced anemia (CIA) occurs in roughly one to two thirds of patients with solid tumors and complicates the course of malignancy (1–5). Its incidence is highly dependent on patient-related factors, on the entity and stage of the underlying disease and on therapeutic interventions. Specifically, the frequency and degree of anemia is higher in metastatic cancers, in aggressive hematologic malignancies and following treatment with high-dose chemotherapy, multi-targeted tyrosine kinase inhibitors and chimeric antigen receptor T cells (6–9). Therefore, CIA forms a spectrum which can broadly be categorized into three principal etiologies: First, CIA present before the initiation of anti-tumor therapy is typical of advanced disease stages with infiltration and replacement of the bone marrow or when the primary neoplasia results in substantial bleeding such as in colorectal or genitourinary malignancies. Second, indirect effects of products of neoplasms can lead to hemophagocytosis, autoantibody induced hemolysis or cytokine inhibition of erythropoiesis. Third, CIA with initial presentation only after the onset of anti-neoplastic treatment is one of the most common side effects of chemotherapy, yet also occurs as sign of progressive disease (10). According to the common terminology criteria for adverse events by the World Health Organization and National Cancer Institute, anemia is categorized into 5 grades from mild (Hemoglobin (Hb) 10 g/dL – lower limit of normal), to moderate (Hb 8.0 – 9.9 g/dL), severe (Hb <8 g/dL) and life-threatening with urgent interventions indicated (grade 4) or even death (grade 5) (11).
Cancer-Induced Anemia Has Distinct Pathophysiological Hallmarks
The pathophysiology of CIA is complex and involves several contributing mechanisms. First, the immune response against malignant cells inevitably induces the iron-regulatory hormone hepcidin, which then directs iron fluxes away from the erythron and into compartments of the mononuclear phagocyte system (MPS) (Figure 1). In addition, certain inflammatory mediators, many of them cytokines such as tumor necrosis factor (TNF), interleukin (IL)-1, IL-6, transforming growth factor (TGF)-ß and IL-10, stimulate iron uptake into the MPS, induce iron storage in the form of ferritin (FT) and/or block iron recycling (12–15). Together, these effects of inflammatory mediators result in a functional iron deficiency, and erythropoietic cells are cut off their iron supply by macrophages. Presumably, this iron-storing macrophage phenotype deprives infectious agents as well as malignant cells from circulating iron sources. However, in the tumor microenvironment (TME), tumor-associated macrophages (TAM) may lose their ability to store iron because they are re-programmed by neoplastic cells to resume iron export. The mechanisms that result in metabolic reprogramming of TAM are incompletely understood but may involve transcriptional regulations and epigenetic changes (16).
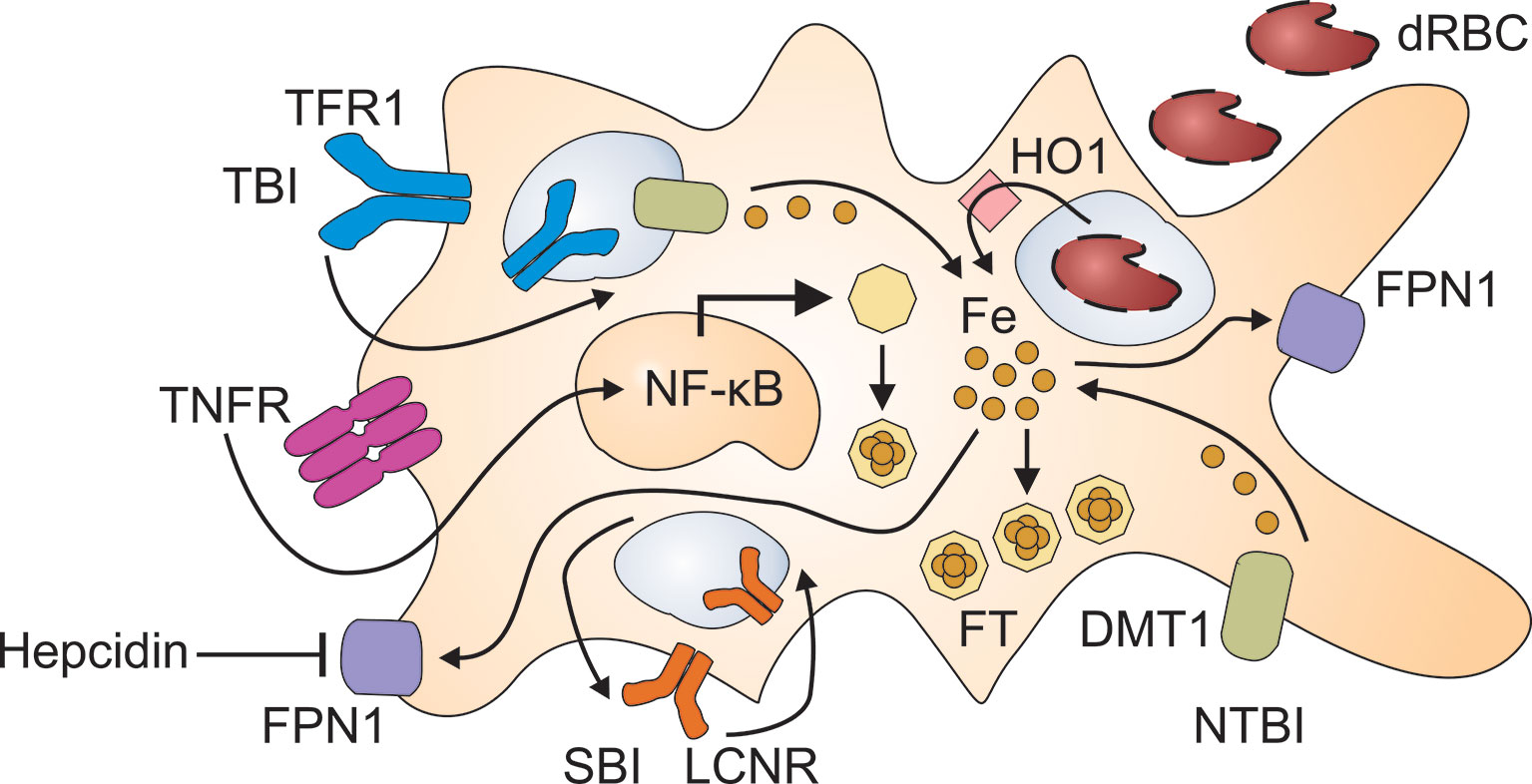
Figure 1 Pathways for the retention of iron in macrophages. Immune activation results in the production of inflammatory cytokines such as tumor necrosis factor (TNF) which damage red blood cells (dRBC) and activate nuclear factor (NF)-κB. The uptake of dRBC delivers large amounts of heme to macrophages which recycle heme-derived iron after its release by heme oxygenase (HO)-1. Moreover, inflammatory cytokines stimulate the uptake of transferrin-bound iron (TBI) via transferrin receptor (TFR)-1 and of non-transferrin-bound iron (NTBI) via divalent metal transporter (DMT)-1. TBI incorporated via TFR1 undergoes reduction to its ferrous form in the endosome and subsequent transfer to the cytoplasm through DMT1. The lipocalin-2 receptor (LCNR) can mediate both the uptake and the release of siderophore-bound iron (SBI). Inflammatory cytokines and hepcidin reduce ferroportin (FPN)-1 mediated iron export, which further contributes to iron retention in macrophages. To avoid elevated iron levels in the cytoplasm, labile iron is incorporated into ferritin (FT) which is upregulated both by iron and NF-κB.
Second, inflammatory mediators such as TNF and hydrogen peroxide inhibit the production of erythropoietin (EPO) in renal peritubular fibroblasts (Figure 2) (17, 18). Again, this mechanism aims at reducing the oxygen supply to tumor cells. However, these cells may switch to anaerobic glycolysis and induce tumor neovascularization by starting to generate angiogenetic factors. However, macrophages, endothelial cells or other cell types in the TME are also able to secrete angiogenetic mediators (19–21). Therefore, hypoxia in the TME is also a potential driving force for disease progression and metastasis (22). Although EPO levels are elevated in patients with CIA, this elevation remains insufficient for the degree of anemia (23–25).
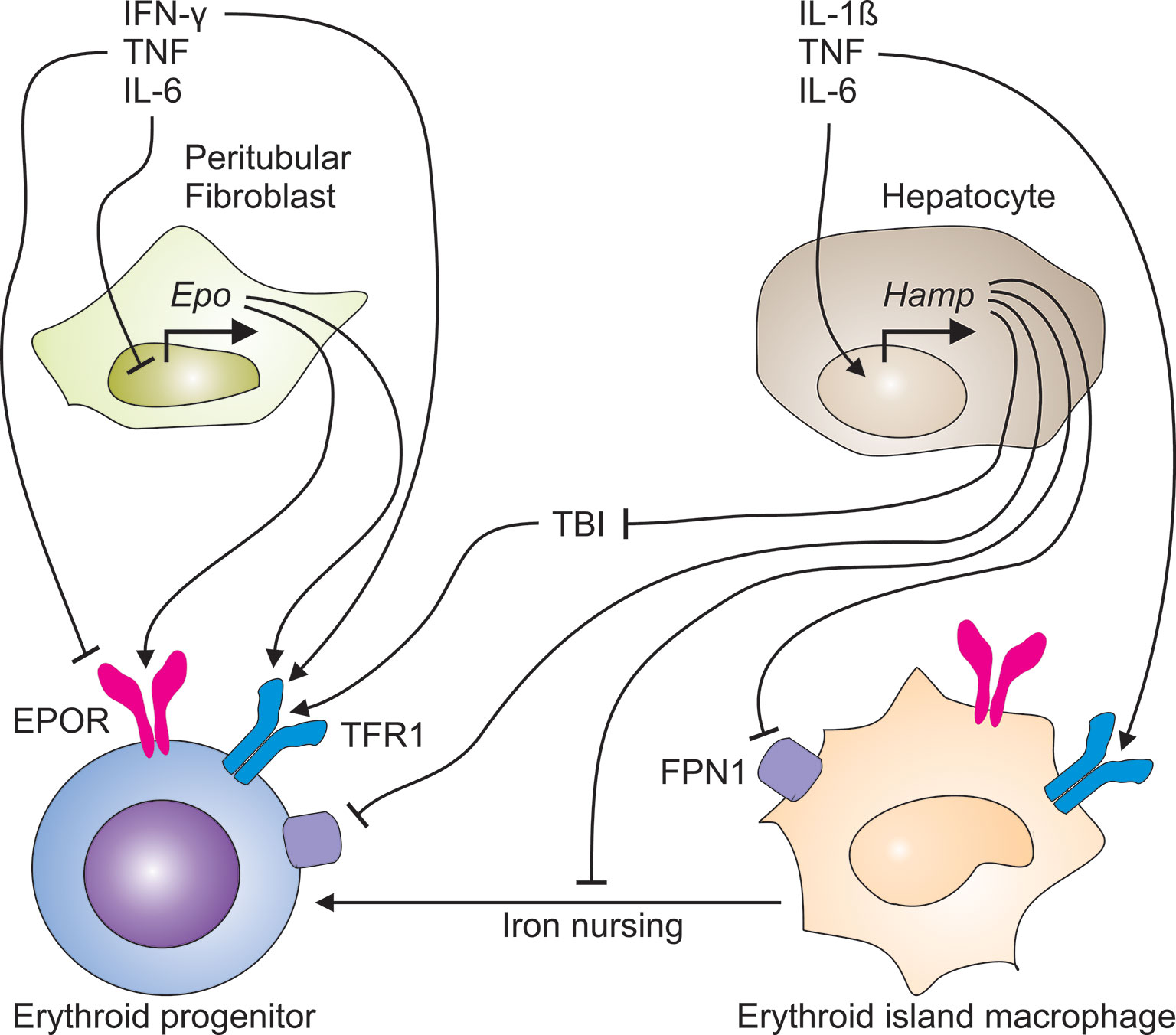
Figure 2 Immune-mediated pathways contribute to the development of cancer-induced anemia. The immune response to cancer cells generates a plethora of pro-inflammatory cytokines. TNF and other TH1 cytokines inhibit both, the production of erythropoietin (EPO) in peritubular fibroblasts in the kidney and the expression of the EPO receptor (EPOR) on erythroid progenitor cells in the bone marrow. In parallel, TNF, IL-1ß and IL-6 induce the transcriptional expression of the hepcidin antimicrobial peptide (Hamp) gene in hepatocytes. Liver-derived hepcidin acts on ferroportin (FPN)-1 and blocks its iron export function, thus reducing the transfer of iron to the circulation and the amount of transferrin-bound iron (TBI), consequently. In addition, hepcidin may undermine the transport of iron from erythroid island macrophages to erythroid progenitors, thus impairing their proliferation and hemoglobin synthesis.
Third, the immune response, intended to primarily target malignant cells, also impairs erythropoiesis in the bone marrow (Figure 2) due to inhibition of and damage to erythroid progenitors (EP) and hematopoietic stem cells (26–28).
Fourth, in advanced solid tumors, proliferating cancer cells may deprive EP of cobalamin and folate, infiltrate the bone marrow, displace normal hematopoiesis and destroy its niches similar to what is typical of hematologic malignancies. Without cancer treatment, all these mechanisms interact in a vicious circle to aggravate the severity of CIA. However, when cancer treatment is initiated, the toxic effects of chemotherapeutics or radiation therapy on the bone marrow and on other organs such as the kidney may contribute to anemia, too. Moreover, cellular iron retention counteracts the ability of hypoxia inducible factors (HIF) to stimulate EPO production, a mechanisms which is relevant to peritubular fibroblasts (29).
In summary, many of the major pathways that contribute to anemia in cancer patients interact with each other, and specific treatment for CIA needs to be taken into consideration to break this circle of mechanisms causing and aggravating conditions of anemia (30).
Inflammation and Cancer Dysregulate Iron Metabolism
Iron metabolism and the immune response are interlinked (31). Iron is an essential nutrient to almost all microbes (32, 33). One important role of the acute phase response (APR) is the reduction of iron levels in extracellular compartments (i.e. the serum) and iron storage in the intracellular compartment (i.e. in FT). As for many other immune functions, this important role is fulfilled by monocytes, macrophages and other cellular players of the mononuclear phagocyte system (MPS). Macrophages possess a broad spectrum of pattern recognition and scavenger receptors for the sensing and uptake of potentially harmful macromolecules and the recognition of malignant cells. These receptors are linked to intracellular signaling cascades which converge at the level of key inflammatory transcription factors including the nuclear factor (NF)-κB (Figure 1). Once activated, NF-κB orchestrates the transcriptional responses of macrophages and other immune-competent cell types to trans-activate inflammatory gene products such as TNF, IL-6 and IL-22 which also play an important role in the cancer development and aggravation of CIA (34). These pro-inflammatory cytokines, especially the APR-initiator IL-6, target the liver and stimulate hepatocytes to increase hepcidin output (35–37). In inflammatory conditions such as in patients with malignancies, hepcidin expression is induced by the concerted interaction of two pathways involving IL-6, Janus kinases (JAK) and signal transducers and activators of transcription (STAT) or activin B and activation of the SMAD (for sisters of mothers against decapentaplegic)-1/5/8 signaling pathway (38–42). On the contrary, patients treated with an anti-TNF antibody or an anti-IL-6 antibodies exhibit reduced levels of inflammatory markers such as IL-6, hepcidin, and/or C-reactive protein, correlating with improvement in anemia related to autoimmune inflammatory conditions (43).
The Acute Phase Response Drives Iron Retention in Macrophages
The immune response to pathogen associated molecular patterns (PAMP) such as lipopolysaccharide and to danger associated molecular patterns (DAMP), present in the TME, such as free heme, adenosine, IL-1α, high-mobility group box-1 (HMGB1) and S100 proteins are similar. The recognition of either PAMP or DAMP results in the activation of NF-κB, p53, mitogen-activated (MAP) kinases and inflammasomes (44). As a consequence, the hypoferremia of the APR also limits the availability of iron for malignant cells. Yet, prolonged hypoferremia may be regarded as a maladaptation to persistent immune stimulation in the setting of chronic infections, autoimmune or neoplastic diseases.
T cells are major mediators of the immune response against malignant cells. Specifically, major histo-compatibility (MHC) class I molecules present on tumor cells present neoantigens on their surfaces. These neoantigens are detected by cytotoxic T cells and elicit the secretion of granzyme B, perforins and of cytokines such as TNF and IL-6. Both cytokines induce hepcidin, a hormone with a unique mode of action: hepcidin binds to its receptor ferroportin (FPN)-1, an iron export channel, and blocks its transport function. Hepcidin thus mediates a negative feedback loop because it reduces iron recycling by macrophages and iron absorption by enterocytes when serum and tissue iron levels are elevated. Therefore, excess hepcidin deactivates iron transport to the blood stream resulting in a reduction of serum iron concentrations during ongoing iron consumption by transferrin receptor (TFR)-1 expressing cells in the face of reduced resupply (Figure 2).
To further support the iron withdrawal from malignant cells, IL-6, IL-10 and other pathways induce the iron storage protein FT (45, 46). FT is composed of 24 subunits of heavy and light chains which assemble in variable proportions to form a shell-shaped heteromultimer. Serum FT is primarily produced and secreted by macrophages (47). Therefore, the levels of serum FT reflect body iron stores and the state of immune activation. In other words, in the presence of cancer cells or other immune stimuli, serum FT does not accurately predict the amount of iron stored in macrophages and other cell types such as hepatocytes. Consequently, in these scenarios, normal serum FT does not rule out the depletion of iron stores that characterizes absolute iron deficiency. Vice versa, increased serum iron (hyperferritinemia) can indicate either parenchymal iron overload or immune activation with subsequent uptake and storage of iron in macrophages. In clinical settings, it is fundamental to distinguish the different etiologies, as treatment is directed toward the underlying morbidity and does not automatically result in iron reduction approaches.
Iron Has Pleiotropic Effects on the Immune Response
The adaptation of iron metabolism during the APR may have evolved to deal with acute stressors such as bacterial infections. However, when inflammatory stimuli persist, as is the case in neoplasms that cannot be fully resected, this immune-mediated storage of iron in the MPS may be of disadvantage for the affected individual for at least two reasons. First, iron storage in the MPS is the basis for a functional iron deficiency in the erythron. Second, while iron promotes the non-enzymatic generation of reactive oxygen species (ROS), it has negative effects on many other immune effector pathways. Macrophage iron overload, commonly resulting from chronic hemolysis and/or repetitive red blood cell (RBC) transfusions, impairs their effector functions which are promoted by TNF and interferon (IFN)-γ. These key cytokines are produced by cytotoxic T cells, T helper type 1 (TH1) cells and natural killer (NK) cells. Specifically, an increased macrophage iron content results in impaired production of nitric oxide (NO). This is a consequence of iron inhibiting the abilities of HIF-1 and of NF-IL6 to trans-activate the NOS2 (for NO synthase-2) gene (Figure 3) (48, 49). Similarly, iron impairs MHC class II expression in macrophages (50). The transcriptional mechanisms are unknown yet. Therefore, iron may inhibit most TH cell responses. In a similar fashion, surplus iron can also be directly toxic to T cells and inhibit their proliferation or induce ferroptosis (Tymoszuk et al., 2020). The latter is a specific form of cell death that is dependent on iron, ROS and lipid peroxides. Ferroptosis is mediated by inactivation of the lipid repair enzyme glutathione peroxidase 4 (GPX4) (51). In macrophages, the induction of ferroptosis results in degradation of FT. This process is known as ferritinophagy. Another effect of ferroptosis is the release of iron by nuclear receptor coactivator (NCOA)-4-mediated autophagy. As ferroptosis is an increasingly recognized mechanism of action of chemotherapeutics, the pathway provides another possible mechanism of interaction between iron metabolism, anti-tumor immunity and cancer biology (52–54).
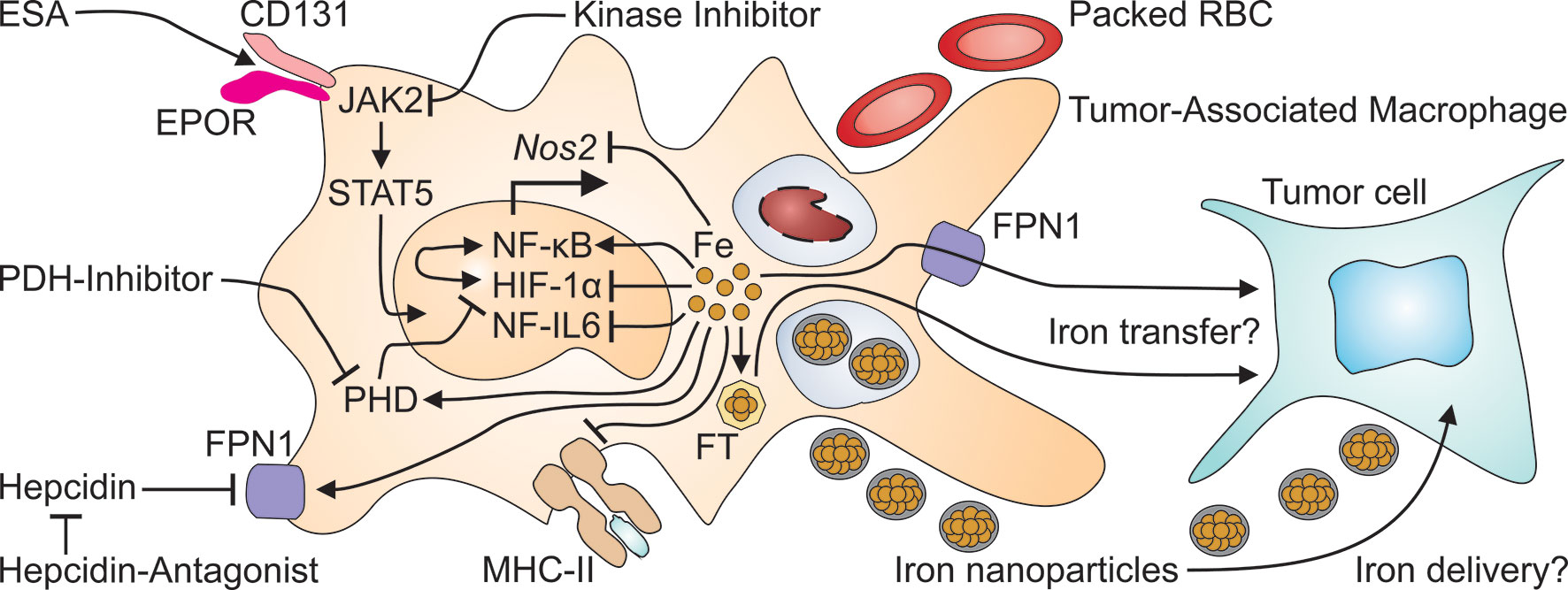
Figure 3 Many therapeutics for cancer-induced anemia may exert off-target effects on tumor-associated macrophages. Erythropoiesis-stimulating agents (ESA) activate the EPOR/CD131 heterodimer to initiate signaling via Janus kinase (JAK)-2 and signal transducer and activator of transcription (STAT)-5. On the other hand, kinase inhibitors, prescribed to treat cancer or to reduce hepcidin production, may interfere with JAK2 signaling. Packed red blood cells (RBC) are taken up by macrophages including TAM at the end of their life span and increase intramacrophage iron levels. Intravenous iron preparations are nanoparticles which are taken up by TAM by phagocytosis and increase the cellular iron content. Surplus intracellular iron in turn, can inhibit the transcriptional expression of the nitric oxide synthase (Nos)-2 and of major histocompatibility (MHC) class II unless it is exported through FPN1 or stored in ferritin (FT). Presumably, either form of iron may be transferred from TAM to adjacent tumor cells. This may also be relevant for the application of hepcidin antagonists which prevent the action of hepcidin on FPN1, thus restoring macrophage iron export. Intracellular iron also has profound effects on TAM themselves because iron stimulates the binding activity of nuclear factor (NF)-κB, while reducing the activation of NF-IL6 and of hypoxia-inducible factor (HIF)-1α. The latter effect is also relevant to treatment with prolyl hydroxylase domain (PHD) inhibitors which impair the degradation of HIF-1α, much like iron deficiency which stabilizes HIF-1α's active form.
In conclusion, iron has multiple, predominately negative effects on the immune response.
Some Macrophage Populations Professionally Handle Iron
Macrophages are dispersed throughout the human body to assume organ- and tissue-specific forms and functions. Several macrophage populations exist for example in the spleen. Of major importance for the maintenance of body iron homeostasis are SpiC- and FPN1-expressing iron-recycling red pulp macrophages (RPM) (55, 56). Under steady-state conditions, RBC have a normal life span of 120 days. Finally, they display altered molecular surface patterns and begin to lose ‘don’t eat me’ signals. Moreover, RBC start to flip phosphatidylserine to their outer surface (57–59). RPM respond to these alterations and take up these RBC marked as ‘aged’ by a process known as erythrophagocytosis. In brief, engulfed RBC are degraded and their heme-iron is recycled to the systemic circulation by FPN1-mediated iron export (Figure 3). Alternatively, a novel study suggests that RBC may undergo physiologic hemolysis in the spleen and that their remnants are taken up by RPM for rapid turn-over (60). In situations when this capacity of RPM to take up damaged RBC (dRBC) and to recycle iron is overwhelmed, such as in massive hemolysis, Kupffer cells (KC) in the liver take over RBC uptake and degradation as a back-up mechanism. In addition, the liver-derived chemokines CCL2 and CCL5 stimulate the bone marrow to release new monocytes (61). These will then also participate in RBC uptake and iron recycling as a support to RPM and KC. Apart of these iron homeostatic functions, KC may play a role in tumor progression, too. Per se the liver is one of the organs most commonly affected by metastasis. Therefore, resident KC may be considered as TAM analogues for metastatic cells that have reached the liver. In this setting, iron-recycling by KC may facilitate the growth of liver metastasis. Similarly, in hepatocellular carcinoma, KC may be re-programmed to supply cancer cells with iron, resulting in adverse outcome (62, 63).
In summary, KC are a paradigm for macrophage populations that recycle iron and deliver this nutrient to cancer cells.
TAM Are Macrophages in the TME
TAM are located in the TME and thus have a strategic position in immunity against malignant cells. However, tumor cells enter a cross-talk with TAM by producing soluble mediators and metabolites with which they can manipulate anti-tumor immune responses. Iron is a decisive factor in this interaction: The immune-mediated uptake of Hb by and the sequestration of iron in TAM both aim at withholding this nutrient from malignant cells to counteract disease progression (64, 65). Vice versa, tumor cells seek at undermining these mechanisms and may promote the release of iron from TAM (66). FPN1-mediated export of ionic iron and the secretion of iron-laden FT or lipocalin-2 may be the most relevant pathways by which re-programmed TAM supply neoplastic cells with iron (67, 68). In addition, malignant cells themselves produce lipocalin-2 and its receptor for the uptake of siderophore-bound iron (SBI) which supports cancer cell growth (69, 70). Moreover, tumor cells can express ionic iron importers such as divalent metal transporter (DMT)-1 and solute carrier family 39 member 14 (SLC39A14; also known as ZIP14) as well as receptors for FT and transferrin-bound iron (TBI) (71–73).
By producing the vascular endothelial growth factor (VEGF), TAM may also be implicated in tumor neovascularization which is triggered by tissue hypoxia, influenced by cardiovascular function and CIA, respectively (74). Hypoxia is sensed by oxygen sensitive prolyl hydroxylases (PHD) which stabilize HIF, a heterodimeric transcription factor composed of α and ß subunits. Under normoxic conditions, PHD continuously hydroxylate HIF-1α at two specific proline residues. This enzymatic process is modified by concentrations of ferrous iron and 2-oxoglutarate and tags HIF-1α for proteosomal degradation following binding by the von Hippel Lindau (VHL) E3 ubiquitin ligase complex. During cellular hypoxia, which commonly occurs in the TME, HIF-1α is stabilized thus enabling trans-activation of HIF target genes such as VEGF. VEGF also constitutes a relevant target for monoclonal antibodies, which are used as add-ons in combination with chemotherapeutics to counteract disease progression. The cross-talk between TAM and cancer cells is bidirectional, though. On the one hand, cells release lactate in breast cancer in order to stimulate TAM to transfer a HIF-1α stabilizing RNA into malignant cells. This impairs the action of chemotherapy (75).
On the other hand, PDH inhibitors, which are of potential use to treat CIA, may exert effects on TAM: PDH3 and HIF-1α play central roles in the activation of macrophages and in their interaction with T cells (76, 77). In addition, the central transcription factors HIF-1α and NF-κB are linked and put the PDH-HIF pathway right at the interface between the sensing of reduced oxygen and iron levels and immune effector functions (78, 79). Despite the key role of HIF-1α in TAM however (Figure 3), it is hard to predict whether these immunologic effects or potential pharmacologic intervention will favor or impair the progression of the underlying malignancy. In line, we currently lack a comprehensive understanding, how compounds for the treatment of CIA may affect the pleiotropic functions of TAM. In addition, many studies on the cross-regulation of iron metabolism and macrophage function address the functions of inflammatory cytokines and infectious agents but still need to be carefully translated into cancer models and, subsequently, cancer patients.
TAM as Potential Targets of CIA-Directed Therapies
Macrophages form the central executive part of the MPS and are equipped with a plethora of pattern recognition and scavenger receptors. In order to fulfill their immune and homeostatic functions in the human body, macrophages need to respond to endocrine, paracrine and metabolic signals. The corresponding receptors present on macrophages include – but are not limited to – bone morphogenic protein receptor (BMPR)-I and -RII, hemojuvelin (HJV), transferrin receptor (TFR)-2, FPN1, IL6R, TNFRI, EPOR and CD (for cluster of differentiation)-131. Therefore, many therapeutic options for CIA are predicted to affect macrophage functions in general and TAM functions, specifically (Figure 3).
Intravenous Iron Preparations Target Myeloid Cells
Intravenous iron preparations can be administered to cancer patients for two indications. First, patients with CIA and relevant absolute iron deficiency may benefit from intravenous iron supplementation as long as the underlying malignancy is under therapeutic control as well. Second, superparamagnetic iron oxide nanoparticles (SPION) are used for diagnostic purposes in imaging studies. Both classes of intravenous iron preparations are nanoparticles which primarily target classical monocytes and macrophages. Concretely, classical monocytes and macrophages take up iron-containing nanoparticles via their set of scavenger receptors and degrade them in phagolysosomes (80, 81). As a consequence, these cells accumulate their total and cytoplasmatic iron content before they transfer iron to the circulation (82). However, it is increasingly recognized that iron-containing nanoparticles exert immune-modulation on macrophages (83). For example, the clinically used intravenous iron preparations iron sucrose and sodium ferric gluconate impair the adhesion and phagocytosis of monocytes, while ferric carboxymaltose and iron isomaltoside lack these effects (84). However, also the latter two compounds affect the MPS. This is also evident from the fact that monocytes and macrophages take up iron carboxymaltose and deliver these iron-nanoparticles to tumors. There, they inhibit monocyte chemoattractant protein (MCP)-1 and to a lesser degree NO production (85). In contrast, the uptake of iron sucrose by circulating monocytes results in activation of NF-κB and in enhanced production of TNF, IL-6 and IL-8 (86, 87).
The uptake of iron nanoparticles by monocytes and macrophages is also relevant for the use of SPION as contrast enhancers for magnetic resonance imaging. SPION coated with dextran are phagocytozed by human monocytes, in which they activate mitogen-activated protein (MAP) kinases and stimulate TNF and IL-1ß production (88). Other SPION preparations however, counteract toll-like receptor (TLR)-4 signaling and NF-κB activation, thus reducing TNF, IL-1ß and IL-6 production (89). Further results obtained with monocyte-derived dendritic cells suggest that SPION impair antigen processing and TH cell stimulation which may have important implications for anti-tumor immunity (90). Furthermore, efforts have been made to selectively label distinct macrophage phenotypes with SPION, which may be relevant for cancer patients because TAM represent distinct macrophage phenotypes (91).
In the face of these heterogenous data, it is currently difficult or even impossible to predict the clinical implications of the use of iron nanoparticles from their specific effects on TAM function. Therefore, more preclinical studies and following clinical data are desirable to better understand the effects of intravenous iron preparations on CIA and cancer.
Blocking IL-6 or BMP-6 to Inhibit Hepcidin in CIA
The inhibition of hepcidin is an attractive treatment strategy for CIA patients, too. Hepcidin antagonism can be achieved via multiple approaches such as inhibitors of hepcidin expression, HJV inhibitors, hepcidin-binding oligoribonucleotides, neutralizing antibodies to hepcidin or antibodies which block its interaction with FPN1 (92, 93).
Tocilizumab, a monoclonal antibody approved for several indications such as rheumatoid arthritis, giant cell vasculitis or cytokine release syndrome, targets the IL-6R and thus one of the major pathways to induce the transcription of the HAMP (for hepcidin-antimicrobial peptide) gene (37, 94, 95). Therefore, tocolizumab ameliorates disease activity and lowers circulating hepcidin levels in rheumatologic diseases (96, 97). The latter effect improves ACD, presumably by a direct effect on hepatocytes (98). Nevertheless, one might speculate that blockade of IL-6R may also modulated the function of macrophages including TAM (99). For example, in macrophage activation syndromes, tocilizumab results in lower serum FT levels (100). The ligand IL-6 is also involved in the cross-talk between TAM and tumor cells and promotes the survival of tumor cells in hypoxic conditions and the differentiation of cancer stem cells (101–104). Whether or not this is relevant for the clinical application of tocilizumab in cancer patients remains to be addressed in further clinical trials. However, a plethora of studies has implicated chronic inflammation in general and IL-6, specifically, in tumor initiation and progression (105, 106). Therefore, IL-6R blockade may impair the progression and metastasis of some forms of cancer (107). Yet, these data do not exclude the possibility that tocilizumab facilitates malignant transformation in other malignant diseases (108). Thus, only clinical trials in cancer patients will give relevant answers as to whether or not tocilizumab is a safe and efficient therapy for CIA.
Other potential targets for the treatment of CIA are bone morphogenetic proteins (BMP) and their receptors. As members of the TGF-ß superfamily of cytokines, BMP-2 and BMP-6, are specifically involved in the maintenance of iron homeostasis, yet also immune regulation and cancer progression (41, 109). BMP-6 is secreted by endothelial cells in the liver to inform adjacent hepatocytes about elevated serum iron levels and replenished iron stores (110). Accordingly, BMP-6 is a major inducer of HAMP transcription and a potential mediator of CIA (111–113). Given the key role of BMP-6 for the induction of hepcidin, it is not surprising that neutralizing antibodies have been used to block hepcidin induction in preclinical and clinical models of ACD (114–116). As many cell types including normal and neoplastic epithelial cells as well as macrophages possess BMPR-I and -II, BMP-6 can affect both tumor cells and TAM.
Several studies have implicated BMP-6 in the linkage of immune regulation and cancer progression. In a mouse model of malignant melanoma for example, the absence of BMP-6 resulted in a substantial delay in tumor onset and progression by a mechanism depending on mast cells (117). In non-small cell lung cancer however, reduced BMP-6 expression was associated with reduced overall survival and BMP-6 inhibited the proliferation of lung cancer cells (118). Similarly, BMP-6 inhibited the growth of breast cancer cells induced by estrogens (119). A high expression of BMP-6 was associated with higher immune cell infiltration and better survival in estrogen receptor-positive breast cancer in a cohort study (120). In prostate cancer, BMP-6 produced by neoplastic cells acts on adjacent macrophages and activates NF-κB and SMAD1 signaling to increase IL-1α and IL-6 secretion. IL-1α, in turn, stimulates endothelial cells and promotes tumor angiogenesis (121). In parallel, IL-6 acts on BMP-6 secreting malignant cells and enhances the expression of the androgen receptor, a major determinant of tumor growth and treatment response in prostate cancer (122). In renal cell carcinoma (RCC), BMP-6 mediates a crosstalk between tumor cells and TAM, too. BMP-6 instructs TAM to assume an anti-inflammatory phenotype with increased IL-10 production. The expression of BMP-6 in RCC cell lines compared to that in a nonmalignant renal cell line correlated with RCC cell line proliferation Increased IL-10 levels predicted poor prognosis of RCC in human subjects (123, 124).
Given the pleiotropic effects of BMP-6 in the crosstalk between immune and neoplastic cells in the TME, it will be important to assess the effects of BMP-6 neutralizing antibodies on the clinical course of the underlying malignancies. In light of the available data, it is tempting to speculate that the gene signatures in biopsies from primary lesions and the role played by BMP-6 in a given tumor entity may enable us to predict whether BMP-6 blockade exerts stimulatory or inhibitory effects on tumor growth in an individual patient.
The Hepcidin-FPN1 Axis Itself Is a Pharmacologic Target
Located downstream of IL-6 and BMP-6, the hepcidin-FPN1 axis itself is an attractive target for the therapy of CIA and other forms of ACD (125, 126). However, both FPN1 and its ligand hepcidin are potential regulators of tumor growth and the immune response directed against it. Loss-of-function mutations in SLC40A1, the gene encoding for FPN1, are in discussion to produce a molecule that does not traffic appropriately to the cell surface or that has limited ability to export iron (127–129). Excess accumulation of iron in macrophages is the result with accompanying high serum FT levels. In gain-of-function mutations of SLC40A1, the binding site of hepcidin is altered, resulting in a hepcidin-resistant protein and in iron overload (130–132). In TAM in RCC, FPN1 expression is elevated, especially in high grade lesions. Importantly, high FPN1 levels predict poor overall survival because iron export by TAM supports the proliferation and migration of RCC cells (133). Similarly, in breast cancer biopsies taken from the primary lesion and axillary lymph node metastases, TAM exhibit high FPN1 expression and cancer cells display high TFR1 levels, suggesting that TAM supply tumor cells with iron (134). Importantly, the influence that FPN1 levels on TAM exert on disease outcome, extend to other relevant malignancies including hepatocellular carcinoma (135). This may be a general observation for cancers with predominant infiltration by monocytes and macrophages. However, this effect may also partly be attributable to the role of FPN1 on macrophages in the control of their cellular iron status and immune response. Specifically, macrophages lacking FPN1 are impaired in their function and secrete higher amounts of TNF and IL-6, possibly because intracellular iron can stimulate the translation of these cytokines (136–138). On the other hand, over-expression of FPN1 in macrophages enhances their NO output because low intramacrophage iron levels promote the transcription of the NOS2 gene (139, 140). FPN1 inhibitors are being evaluated in clinical trials for the treatment of thalassemia (141). However, to date, no data exist on the use of allosteric FPN1 modulators for ACD or CIA.
In contrast, humanized monoclonal antibodies which display a high affinity toward hepcidin and lead to its premature degradation have been developed. One of these, LY2787106, was shown to be tolerated well during its phase one clinical trial, demonstrating a significant increase in serum iron levels (142). Short hairpin RNA (shRNA) which target hepcidin were demonstrated to cause a reduction in hepcidin production and alleviate anemia when used in conjunction with erythropoiesis stimulating agents (ESA) because they may inhibit hepcidin more robustly than anti-hepcidin antibodies (143). Similarly, aptamers, an emerging class of synthetic, structured oligonucleotide therapeutics, can inhibit HAMP expression with high affinity and specificity, thus increasing iron availability for erythropoiesis in a preclinical ACD model (144, 145).
In conclusion, several approaches targeting the hepcidin-FPN1 axis may be effective in CIA because they have the potential to improve anemia in preclinical models of ACD (93, 146, 147). Not only does hepcidin target macrophages to limit iron recycling, it is also produced by macrophages themselves, possibly to autoregulate their iron homeostasis (148–150). Therefore, it is feasible to assume that hepcidin-targeting therapies will affect the immune functions of macrophage populations including TAM, raising safety concerns.
The Hepcidin-FPN1 Interaction in Malignancies
The functional importance of the hepcidin-FPN1 interaction is not limited to TAM, though. In cancer cells, up-regulation of iron uptake pathways such as DMT1 or TFR1 and down-regulation of FPN1 keep cellular iron levels high for metabolism and proliferation (151–153). Also, low FPN1 expression in malignant cells has been linked to the proliferation of malignant myeloma (154, 155). It therefore comes as a surprise that low FPN1 levels are associated with improved prognosis in acute myeloid leukemia (156). However, this observation has been linked to increased sensitivity to chemotherapy, and it is currently being investigated whether iron-induced cytotoxicity or ferroptosis are contributing mechanisms (157).
As for solid tumors, the most conclusive data are available for breast cancer (134). In this tumor entity, several mechanisms cooperate to reduce FPN1 expression on tumor cells including epigenetic modifications in the FPN1 promoter region and down-regulation of FPN1 protein by hepcidin which is secreted by cancer cells and adjacent fibroblasts (158–160). Of relevance, expression levels of hepcidin and FPN1 govern disease outcome in breast cancer and decreased SCL40A1 gene expression is an independent predictor of reduced metastasis-free and disease-specific survival (161).
In conclusion, there is overwhelming evidence that the hepcidin-FPN1 axis regulates both, the function of TAM and the growth of cancer cells. While antagonizing the hepcidin-FPN1 interaction may ameliorate CIA because one of the key molecular mechanisms of functional iron deficiency is undermined, there may be relevant further consequences for the underlying disease (Figure 4). Blocking the hepcidin-FPN1 interaction on TAM will reduce their iron content and may promote NO production, MHC class II expression and alter cytokine production, consecutively increasing their anti-tumor activities. In contrast, FPN1 mediated iron export from TAM may increase the availability of iron not only for EP, as intended, but also for cancer cells. On the other hand, the neutralization of hepcidin’s effects on FPN1 present on cancer cells may inhibit their proliferation yet impair ROS-mediated effects of chemotherapeutics. Therefore, any compound that targets the hepcidin-FPN1 axis exerts both systemic and local effects and needs to be thoroughly tested in preclinical cancer models and clinical cancer trials to carefully balance the benefit-risk ratio of such products before approval for clinical use.
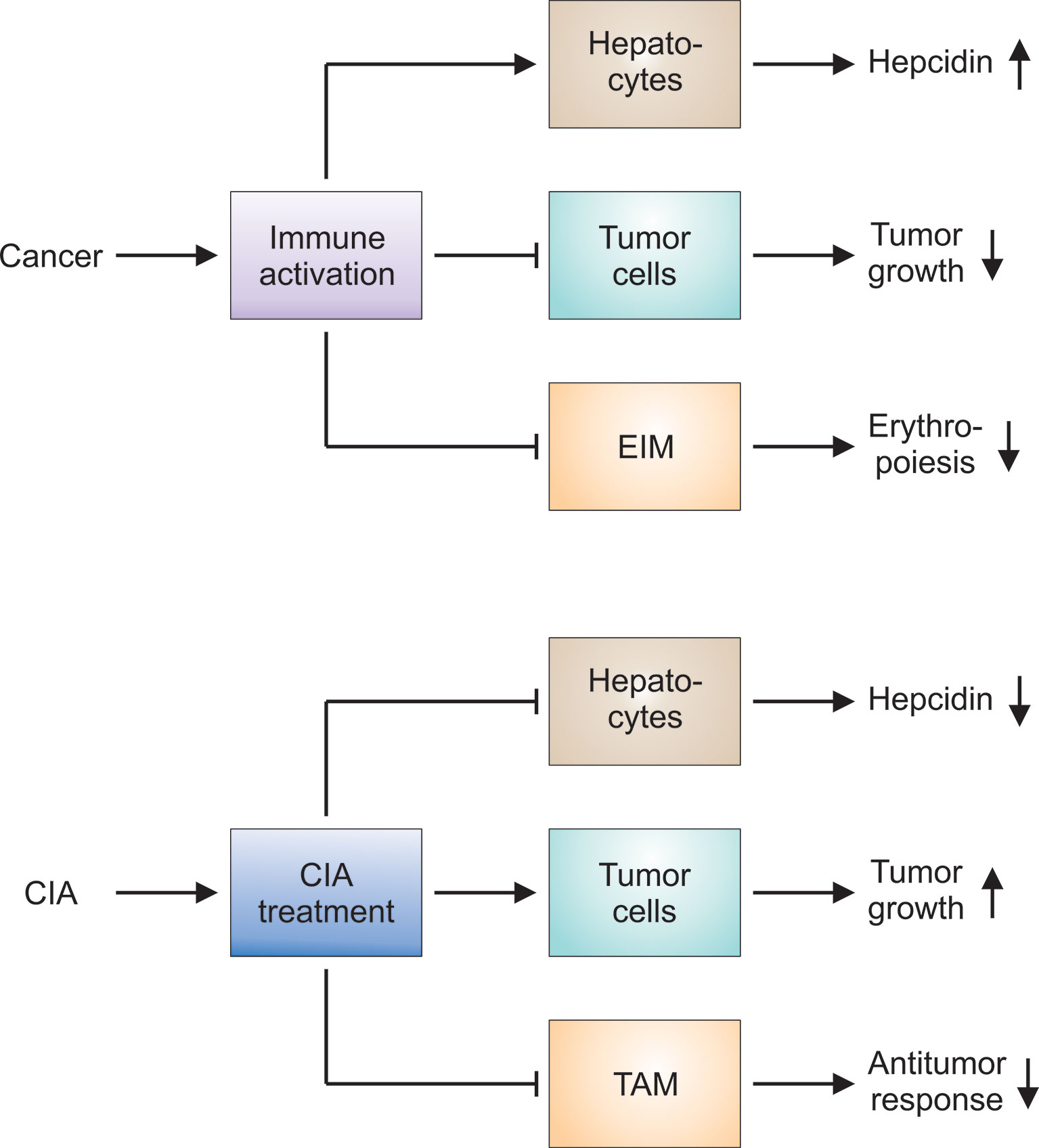
Figure 4 Hypotheses on the pathophysiology of CIA and the effects of CIA treatment on the underlying tumor. Upper panel: Cancer results in prolonged immune activation, especially when the disease is advanced or progressive. The immune response directed against malignant cells impairs tumor growth. At the same time, proinflammatory mediators stimulate hepatocytes to produce hepcidin: Hepcidin in turn counteracts iron export by erythroid island macrophages (EIM). Other immune mechanisms exert similar effects on EIM and aggravate the functional iron deficiency observed in cancer patients. Thus, several mechanisms contribute to the development of cancer-induced anemia (CIA). Lower panel: CIA and the underlying functional iron deficiency can be considered maladaptations to prolonged immune activation. CIA treatment is intended to counteract these immune-mediated pathways and to promote erythropoiesis. Many of these therapeutic approaches reduce hepcidin production or block its negative action on ferroportin-mediated iron export. However, CIA treatment may exert unintended side effects on tumor cells and on tumor-associated macrophages (TAM). The latter may be attributable to the fact that EIM cannot be selectively targeted and other macrophage populations such as TAM are inhibited by CIA treatment, too. Therefore, all therapeutic interventions for CIA have to be thoroughly tested in preclinical models and clinical trials to check for unintended effects on tumor growth and the antitumor response.
Diagnosis of CIA
Individual Approach for Cancer Patients
CIA is a subtype of ACD and may be viewed as spectrum by itself that ranges from mild and asymptomatic forms in individuals with well-controlled residual malignancy to severe forms that, if left uncorrected, will limit patients’ life expectancy just as does the underlying neoplasia (162). Anemia in cancer patients can be classified as CIA when the anemia has hyporegenerative features as indicated by a reduced reticulocyte count or reticulocyte production index (163). Often, the underlying cancer is advanced or not in remission or patients are under chemotherapy (164). In many cases of CIA however, substantial hemolysis, evident from haptoglobin consumption, contributes to anemia as do other contributing factors such as EPO and vitamin deficiencies. Given this multifactorial etiology of CIA, cancer patients often require an individual approach in the diagnosis and treatment.
CIA and Laboratory Markers
CIA is often normocytic or microcytic but occasionally macrocytic. Because of the fact that serum FT is elevated as part of the APR, a higher cut-off of 100 rather than 30 ng/ml for the assessment of body iron stores in cancer patients may be necessary (165). In cancer patients, serum FT < 100 ng/ml and TSAT < 20% suggest absolute iron deficiency but the Hb content of reticulocytes may be a more reliable parameter (166). Similarly, the soluble TFR is less affected by inflammation and may thus be a reliable indicator of absolute iron deficiency, even in the setting of cancer (167). This may be attributable to the idea that the Hb content of reticulocytes is a marker of the functional ID whereas the FT index (soluble TFR/log FT) primarily reflects the iron availability for erythropoiesis (168, 169). Therefore, the FT index facilitates the differential diagnosis between iron deficiency and ACD (170). An sTFR-FT index <1 suggests ACD, whereas, an sTFR-FT index >2 suggests ACD accompanied by absolute iron deficiency anemia. The use of the FT index for daily clinical routine is limited by the lack of internationally standardized assays, though (171).
BMP-6 and Hepcidin Elevation in CIA
BMP-6 and hepcidin levels are elevated in CIA which may aid in the diagnosis (112, 113). Yet it remains unclear whether serum concentrations of these mediators predict the response to currently available treatments such as ESA and to therapies specifically targeting them (172). Also, hepcidin levels may be regulated by cancer-induced inflammation through IL-6 dependent and independent pathways (167, 172, 173).
In conclusion, both patient’s history as well as classical and novel laboratory parameters enable the accurate classification of CIA. Given the increasing number of parameters available, we expect that improved algorithms for the differential diagnosis of CIA and the prediction of treatment responses will become available in the near future.
Treatment Indications and Options for CIA
Aim of CIA Treatment
All inner organs depend on a sufficient supply with oxygen from its iron-containing carrier Hb. CIA thus impairs organ functions and activities of daily living in patients with neoplastic disorders. In addition, CIA may negatively affect the survival of cancer patients (174, 175). Therefore, a correction of the reduced Hb levels is warranted although the desired target levels remain incompletely studied and thus still under debate.
Different Treatment Options in CIA
The therapeutic options to correct the CIA have increased in recent years (Table 1). Apart of RBC transfusions and intravenous iron preparations, ESA have long been used to treat CIA. Recently, hepcidin antagonists such as monoclonal antibodies, short interfering RNA (siRNA), shRNA, aptamers, TFR2 inhibitors as well as pharmacologic inhibitors of HIF prolyl hydroxylases, e.g. vadadustat, roxadustat and daproustat, have emerged as novel therapeutic concepts for CIA. RBC transfusions and ESA are described more in detail as cornerstones of CIA treatment in the further sections.

Table 1 Selected approved and experimental treatment options for CIA.
Red Blood Cell Transfusions
RBC transfusions have long been used to correct anemia, including CIA. The direct replacement of RBC and rise in Hb levels promptly ameliorates oxygen supply to vital organs including the central nervous system and myocardium and thus promotes quality of life and exercise capacity. Yet, this strategy can also worsen the underlying malignancy because oxygen delivery to neoplastic cells will increase, too. Furthermore, RBC transfusions may also modulate the immune response in recipients because packed RBC contain up to 0.8% of hemolyzed cells (176). Therefore, during transfusion, substantial amounts of free Hb, heme and iron as well as microvesicles and membrane fragments can enter the circulation. To avoid tissue damage and inflammation, strategically located macrophage populations such as RPM in the spleen and KC in the liver will neutralize these compounds by CD163, CD91 and other scavenger receptors. CD163 recognizes both free and haptoglobin-bound Hb and eliminates it by receptor-mediated endocytosis. CD91, on the other hand, binds heme-hemopexin complexes. Subsequently, intracellular heme is degraded by heme oxygenase-1 (HO1) and detoxified to bilirubin. When HO1’s enzymatic capacity is overwhelmed, and heme starts to accumulate within cells, free heme activates the NLRP3 inflammasome (177). Similarly, in the extracellular space, macrophages sense free heme as DAMP and initiate the APR (178). Therefore, by capturing free Hb and free heme, the CD163 and CD91 pathways protect from pro-oxidative tissue damage at the systemic level. In the TME, however, their functions may be different because CD163 is a marker of TAM and may enhance the delivery of iron to the tumor (179).
In clinical practice, the major advantage of RBC transfusions is the rapid improvement of Hb levels, which must be balanced against the risks for immune-mediated adverse reactions and transmissible infections.
Erythropoiesis-Stimulating Agents for CIA Treatment
EPO Functions and Signaling in CIA
In general, ESA are derivatives of the endogenous hormone EPO. EPO itself has a dual function in human biology: On the one hand, it is the key growth factor for EP in the bone marrow. There, EPO activates its homodimeric receptor to promote the differentiation of multipotent hematopoietic progenitors along the erythroid lineage and inhibit the apoptotic elimination of surplus cells (Figure 2) (180). On the other hand, EPO exerts functions of an anti-inflammatory and tissue-protective cytokine throughout the body. These latter functions are mediated by a distinct molecular form of its receptor, a heteroreceptor of EPOR and CD131 (181). This extraerythropoietic receptor is also expressed by immune cells including T cells and macrophages (Figure 3). EPO’s effects on the immune system may be either pro- or anti-inflammatory dependent on the type of EPO-responsive cells, the context of the tissue microenvironment or the entity of underlying disease. In CIA, ESA raise Hb levels and reduce the frequency of RBC transfusion in patients.
Cell-, Organ- and Malignancy-Specificities of EPO
In the liver, EPO enhances the phagocytotic capacity of KC and the production of CCL2. This chemokine, in turn, promotes the recruitment of monocytes from the bone marrow to the liver, but it is also implicated in tumor metastasis (182–184). Also, in lung epithelial cells, EPOR expression is higher in malignant cells than in normal cell types (185). This suggests that unintended side effects of EPO are more likely to occur in CIA than in other forms of ACD. In malignant myeloma for instance, EPO stimulates IFN-γ production and counteracts disease progression. In bone marrow derived macrophages in contrast, EPO promotes the secretion of angiogenetic factors which may drive myeloma progression. This function may be especially relevant in the setting of multiple myeloma because in these patients, bone marrow macrophages overexpress EPOR (186). On the other hand, EPO may induce apoptosis in myeloma cells (187). This may be relevant for erythroid island macrophages, on which EPOR is highly expressed and may be important for their nursing function and for the delivery of iron to adjacent EP (188).
As EPO and ESA can act on both, TAM and cancer cells, these compounds may impact on the malignant disease underlying CIA. ESA in the presence of EPOR may promote angiogenesis, tumor growth, tumor cell survival, or resistance to treatment. However, it is impossible to predict the net effect of ESA therapy in a given tumor entity. In gastroesophageal cancer for example, ESA therapy initiated at a similar Hb cut-off of 11 g/dl, tended to have improved clinical outcome, implying that in this context, ESA is a valuable adjunct therapy (189). In breast cancer patients with CIA however, treatment with ESA may not affect overall survival yet increase the risk of venous thromboembolic events (190, 191).
Benefit/Risk Considerations of ESA in CIA
Previous meta-analysis have suggested that ESA may be efficient and safe for the treatment of CIA (192). However, more recent work has led to opposite conclusions, questioning the safety of ESA and raising concerns about the increased risk for thromboembolic events and deaths in CIA patients receiving these compounds: A systematic review and meta-analysis of 6,769 cancer patients in 35 clinical trials exhibited an increased risk of thromboembolic events with recombinant human erythropoietin compared with controls (relative risk (RR), 1.67; 95% confidence interval (CI), 1.35 to 2.06) (193). A meta-analysis of patient-level data from 53 controlled trials in cancer patients who received chemotherapy, radiation therapy, chemoradiotherapy, or no therapy with epoetin therapy (n=13,933) reported a consistently significantly increased risk of thromboembolic events (194). The absolute event rates ranged from 0 to 30.8% (pooled 5.8%) in the treatment arms and from 0 to 14.5% (pooled 3.2%) in the control arms. Besides, other off-target effects of ESA might contribute to mechanisms of tumor regulation, such as cell activation and neovascularization. Based on the results on thromboembolic safety and mortality, ESA are not recommended for the treatment of anemia that is unrelated to chemotherapy in patients with malignancy. Potential exceptions of use of ESA are in patients with lower risk myelodysplastic syndromes to avoid RBC transfusions and the use in patients with concomitant renal failure. The main advantage of ESA is the decreased need for RBC transfusions.
To date, it remains unknown whether and how ESA affect tumor growth or their control by the immune system. From a clinical standpoint, in the absence of conclusive evidence, a personalized approach is mandatory in order to balance the potential benefits of ESA treatment, taking into account each patient’s individual circumstances and preferences, against the increased risk of thromboembolic events and death.
In summary, we need further preclinical and clinical research to characterize the cellular mechanisms and molecular pathways by which ESA affect thrombogenesis and tumor growth in patients with CIA (195).
Conclusions
As for other forms of ACD, treating the underlying disease is the preferred therapeutic approach to patients with CIA. In patients with progressive disease however, it may be required to treat CIA per se in order to positively influence the cancer patient’s quality of life, physical performance and life expectancy.
Nowadays, physicians have an increasing armamentarium at hand to treat CIA. In patients, in whom the underlying malignancy is in full remission and in regular follow-up, treatment of CIA may be relatively safe and the erythropoietic bone marrow is likely to benefit from ESA, iron supplementation or hepcidin antagonism. In progressive cancers however, there may be an increased risk that CIA-directed interventions in fact provide malignant cells with growth promoting nutrients and/or signals. Therefore, a combination therapy that stimulates erythropoiesis with ESA on the one hand and provides EP with iron by medications targeting the IL-6R, BMP-6, hepcidin or FPN1 may be the preferred approach because in combination, these compounds may preferentially supply EP with signals and nutrients for proliferation.
In the future, we expect to have improved mathematical models and IT supported algorithms to diagnose and subclassify CIA, select multimodal therapies and predict treatment responses. We need to conduct appropriately powered randomized controlled trials in order to evaluate the benefits and risks of therapeutic interventions for CIA. Further objectives are trials with end points that focus on overall survival, disease free survival, progression free survival, exercise capacity, infection rate and quality of life. Before complementing these clinical studies, ongoing work in preclinical cancer models aims to gain further mechanistic insight in the effects that CIA-directed therapies exert both locally, on TAM and other tumor infiltrating leukocyte populations, as well as systemically (Figure 4).
Therefore, both preclinical and clinical investigations are inevitable to ameliorate – or TAM-e – CIA with acceptable risks of medicine.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
SW has written the manuscript. MN has written the manuscript and drawn the figures. All authors contributed to the article and approved the submitted version.
Funding
Work in MN’s laboratory is supported by grants from the Austrian Research Fund (FWF stand-alone project P 33062) and the Tyrolean Research Fund (TWF).
Disclaimer
SW is a member of the Pharmacovigilance Risk Assessment Committee (PRAC) of the EMA and the Human Medicines Expert Committee (HMEC) of Swissmedic. The views expressed in this article are the personal views of the authors and may not be understood or quoted as being made on behalf of or reflecting the position of an agency or one of the committees or working parties.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Gianni L, Cole BF, Panzini I, Snyder R, Holmberg SB, Byrne M, et al. Anemia during adjuvant non-taxane chemotherapy for early breast cancer: Incidence and risk factors from two trials of the International Breast Cancer Study Group. Support Care Cancer (2008) 16(1):67–74. doi: 10.1007/s00520-007-0295-y
2. Modi S, Saura C, Yamashita T, Park YH, Kim SB, Tamura K, et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N Engl J Med (2020) 382(7):610–21. doi: 10.1056/NEJMoa1914510
3. Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol (2017) 18(7):946–57. doi: 10.1016/S1470-2045(17)30355-8
4. Tong H, Fan Z, Liu B, Lu T. The benefits of modified FOLFIRINOX for advanced pancreatic cancer and its induced adverse events: a systematic review and meta-analysis. Sci Rep (2018) 8(1):8666. doi: 10.1038/s41598-018-26811-9
5. Ludwig H, Van Belle S, Barrett-Lee P, Birgegard G, Bokemeyer C, Gascon P, et al. The European Cancer Anaemia Survey (ECAS): a large, multinational, prospective survey defining the prevalence, incidence, and treatment of anaemia in cancer patients. Eur J Cancer (2004) 40(15):2293–306. doi: 10.1016/j.ejca.2004.06.019
6. Funakoshi T, Latif A, Galsky MD. Risk of hematologic toxicities in cancer patients treated with sunitinib: a systematic review and meta-analysis. Cancer Treat Rev (2013) 39(7):818–30. doi: 10.1016/j.ctrv.2013.01.004
7. Colita A, Bumbea H, Croitoru A, Orban C, Lipan LE, Craciun OG, et al. LEAM vs. BEAM vs. CLV Conditioning Regimen for Autologous Stem Cell Transplantation in Malignant Lymphomas. Retrospective Comparison of Toxicity and Efficacy on 222 Patients in the First 100 Days After Transplant, On Behalf of the Romanian Society for Bone Marrow Transplantation. Front Oncol (2019) 9:892. doi: 10.3389/fonc.2019.00892
8. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med (2019) 380(18):1726–37. doi: 10.1056/NEJMoa1817226
9. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
10. Madeddu C, Gramignano G, Astara G, Demontis R, Sanna E, Atzeni V, et al. Pathogenesis and Treatment Options of Cancer Related Anemia: Perspective for a Targeted Mechanism-Based Approach. Front Physiol (2018) 9:1294. doi: 10.3389/fphys.2018.01294
11. WHO. Common Terminology Criteria for Adverse Events. (2017). Available at: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5x7.pdf.
12. Schaefer B, Haschka D, Finkenstedt A, Petersen BS, Theurl I, Henninger B, et al. Impaired hepcidin expression in alpha-1-antitrypsin deficiency associated with iron overload and progressive liver disease. Hum Mol Genet (2015) 24(21):6254–63. doi: 10.1093/hmg/ddv348
13. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest (2004) 113(9):1271–6. doi: 10.1172/JCI20945
14. Weiss G, Bogdan C, Hentze MW. Pathways for the regulation of macrophage iron metabolism by the anti-inflammatory cytokines IL-4 and IL-13. J Immunol (1997) 158(1):420–5.
15. Alvarez-Hernandez X, Liceaga J, McKay IC, Brock JH. Induction of hypoferremia and modulation of macrophage iron metabolism by tumor necrosis factor. Lab Invest (1989) 61(3):319–22.
16. Puthenveetil A, Dubey S. Metabolic reprograming of tumor-associated macrophages. Ann Transl Med (2020) 8(16):1030. doi: 10.21037/atm-20-2037
17. Imagawa S, Yamamoto M, Ueda M, Miura Y. Erythropoietin gene expression by hydrogen peroxide. Int J Hematol (1996) 64(3-4):189–95. doi: 10.1016/0925-5710(96)00475-6
18. Jelkmann W. Proinflammatory cytokines lowering erythropoietin production. J Interferon Cytokine Res (1998) 18(8):555–9. doi: 10.1089/jir.1998.18.555
19. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
20. Rohlenova K, Goveia J, Garcia-Caballero M, Subramanian A, Kalucka J, Treps L, et al. Single-Cell RNA Sequencing Maps Endothelial Metabolic Plasticity in Pathological Angiogenesis. Cell Metab (2020) 31(4):862–877 e814. doi: 10.1016/j.cmet.2020.03.009
21. Sormendi S, Wielockx B. Hypoxia Pathway Proteins As Central Mediators of Metabolism in the Tumor Cells and Their Microenvironment. Front Immunol (2018) 9:40. doi: 10.3389/fimmu.2018.00040
22. Hockel M, Vaupel P. Biological consequences of tumor hypoxia. Semin Oncol (2001) 28(2 Suppl 8):36–41. doi: 10.1016/S0093-7754(01)90211-8
23. Miller CB, Jones RJ, Piantadosi S, Abeloff MD, Spivak JL. Decreased erythropoietin response in patients with the anemia of cancer. N Engl J Med (1990) 322(24):1689–92. doi: 10.1056/NEJM199006143222401
24. Ozguroglu M, Arun B, Demir G, Demirelli F, Mandel NM, Buyukunal E, et al. Serum erythropoietin level in anemic cancer patients. Med Oncol (2000) 17(1):29–34. doi: 10.1007/BF02826213
25. Adamson JW. The anemia of inflammation/malignancy: mechanisms and management. Hematol Am Soc Hematol Educ Program (2008) 1:159–65. doi: 10.1182/asheducation-2008.1.159
26. Swann JW, Koneva LA, Regan-Komito D, Sansom SN, Powrie F, Griseri T. IL-33 promotes anemia during chronic inflammation by inhibiting differentiation of erythroid progenitors. J Exp Med (2020) 217(9):e20200164. doi: 10.1084/jem.20200164
27. Rusten LS, Jacobsen SE. Tumor necrosis factor (TNF)-alpha directly inhibits human erythropoiesis in vitro: role of p55 and p75 TNF receptors. Blood (1995) 85(4):989–96. doi: 10.1182/blood.V85.4.989.bloodjournal854989
28. de Bruin AM, Voermans C, Nolte MA. Impact of interferon-gamma on hematopoiesis. Blood (2014) 124(16):2479–86. doi: 10.1182/blood-2014-04-568451
29. Suzuki N, Matsuo-Tezuka Y, Sasaki Y, Sato K, Miyauchi K, Kato K, et al. Iron attenuates erythropoietin production by decreasing hypoxia-inducible transcription factor 2alpha concentrations in renal interstitial fibroblasts. Kidney Int (2018) 94(5):900–11. doi: 10.1016/j.kint.2018.06.028
30. Gilreath JA, Stenehjem DD, Rodgers GM. Diagnosis and treatment of cancer-related anemia. Am J Hematol (2014) 89(2):203–12. doi: 10.1002/ajh.23628
31. Recalcati S, Locati M, Cairo G. Systemic and cellular consequences of macrophage control of iron metabolism. Semin Immunol (2013) 24(6):393–8. doi: 10.1016/j.smim.2013.01.001
32. Schaible UE, Kaufmann SH. Iron and microbial infection. Nat Rev Microbiol (2004) 2(12):946–53. doi: 10.1038/nrmicro1046
33. Nairz M, Schroll A, Sonnweber T, Weiss G. The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol (2010) 12(12):1691–702. doi: 10.1111/j.1462-5822.2010.01529.x
34. Parks OB, Pociask DA, Hodzic Z, Kolls JK, Good M. Interleukin-22 Signaling in the Regulation of Intestinal Health and Disease. Front Cell Dev Biol (2015) 3:85. doi: 10.3389/fcell.2015.00085
35. Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswoode N, Selvakumar TA, et al. Hepcidin regulation by innate immune and infectious stimuli. Blood (2011) 118(15):4129–39. doi: 10.1182/blood-2011-04-351957
36. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood (2006) 108(9):3204–9. doi: 10.1182/blood-2006-06-027631
37. Pietrangelo A, Dierssen U, Valli L, Garuti C, Rump A, Corradini E, et al. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology (2007) 132(1):294–300. doi: 10.1053/j.gastro.2006.10.018
38. Fillebeen C, Wilkinson N, Charlebois E, Katsarou A, Wagner J, Pantopoulos K. Hepcidin-mediated hypoferremic response to acute inflammation requires a threshold of Bmp6/Hjv/Smad signaling. Blood (2018) 132(17):1829–41. doi: 10.1182/blood-2018-03-841197
39. Canali S, Core AB, Zumbrennen-Bullough KB, Merkulova M, Wang CY, Schneyer AL, et al. Activin B Induces Noncanonical SMAD1/5/8 Signaling via BMP Type I Receptors in Hepatocytes: Evidence for a Role in Hepcidin Induction by Inflammation in Male Mice. Endocrinology (2015) 157(3):1146–62. doi: 10.1210/en.2015-1747
40. Besson-Fournier C, Latour C, Kautz L, Bertrand J, Ganz T, Roth MP, et al. Induction of activin B by inflammatory stimuli up-regulates expression of the iron-regulatory peptide hepcidin through Smad1/5/8 signaling. Blood (2012) 120(2):431–9. doi: 10.1182/blood-2012-02-411470
41. Maes K, Nemeth E, Roodman GD, Huston A, Esteve F, Freytes C, et al. In anemia of multiple myeloma, hepcidin is induced by increased bone morphogenetic protein 2. Blood (2010) 116(18):3635–44. doi: 10.1182/blood-2010-03-274571
42. Sharma S, Nemeth E, Chen YH, Goodnough J, Huston A, Roodman GD, et al. Involvement of hepcidin in the anemia of multiple myeloma. Clin Cancer Res (2008) 14(11):3262–7. doi: 10.1158/1078-0432.CCR-07-4153
43. De Benedetti F, Brunner HI, Ruperto N, Kenwright A, Wright S, Calvo I, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med (2012) 367(25):2385–95. doi: 10.1056/NEJMoa1112802
44. Eller K, Schroll A, Banas M, Kirsch AH, Huber JM, Nairz M, et al. Lipocalin-2 expressed in innate immune cells is an endogenous inhibitor of inflammation in murine nephrotoxic serum nephritis. PloS One (2013) 8(7):e67693. doi: 10.1371/journal.pone.0067693
45. Rogers J, Lacroix L, Durmowitz G, Kasschau K, Andriotakis J, Bridges KR. The role of cytokines in the regulation of ferritin expression. Adv Exp Med Biol (1994) 356:127–32. doi: 10.1007/978-1-4615-2554-7_14
46. Nairz M, Ferring-Appel D, Casarrubea D, Sonnweber T, Viatte L, Schroll A, et al. Iron Regulatory Proteins Mediate Host Resistance to Salmonella Infection. Cell Host Microbe (2015) 18(2):254–61. doi: 10.1016/j.chom.2015.06.017
47. Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Zhang DL, Crooks DR, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood (2009) 116(9):1574–84. doi: 10.1182/blood-2009-11-253815
48. Dlaska M, Weiss G. Central role of transcription factor NF-IL6 for cytokine and iron-mediated regulation of murine inducible nitric oxide synthase expression. J Immunol (1999) 162(10):6171–7.
49. Melillo G, Taylor LS, Brooks A, Musso T, Cox GW, Varesio L. Functional requirement of the hypoxia-responsive element in the activation of the inducible nitric oxide synthase promoter by the iron chelator desferrioxamine. J Biol Chem (1997) 272(18):12236–43. doi: 10.1074/jbc.272.18.12236
50. Oexle H, Kaser A, Most J, Bellmann-Weiler R, Werner ER, Werner-Felmayer G, et al. Pathways for the regulation of interferon-gamma-inducible genes by iron in human monocytic cells. J Leukoc Biol (2003) 74(2):287–94. doi: 10.1189/jlb.0802420
51. Conrad M, Friedmann Angeli JP. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol Cell Oncol (2014) 2(3):e995047. doi: 10.4161/23723556.2014.995047
52. Lu B, Chen XB, Ying MD, He QJ, Cao J, Yang B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front Pharmacol (2017) 8:992. doi: 10.3389/fphar.2017.00992
53. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520(7545):57–62. doi: 10.1038/nature14344
54. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2013) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
55. Haldar M, Kohyama M, So AY, Kc W, Wu X, Briseno CG, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell (2014) 156(6):1223–34. doi: 10.1016/j.cell.2014.01.069
56. Kohyama M, Ise W, Edelson BT, Wilker PR, Hildner K, Mejia C, et al. Role for Spi-C in the development of red pulp macrophages and splenic iron homeostasis. Nature (2009) 457(7227):318–21. doi: 10.1038/nature07472
57. Korolnek T, Hamza I. Macrophages and iron trafficking at the birth and death of red cells. Blood (2014) 125(19):2893–7. doi: 10.1182/blood-2014-12-567776
58. Bernhardt I, Nguyen DB, Wesseling MC, Kaestner L. Intracellular Ca(2+) Concentration and Phosphatidylserine Exposure in Healthy Human Erythrocytes in Dependence on in vivo Cell Age. Front Physiol (2019) 10:1629. doi: 10.3389/fphys.2019.01629
59. Burger P, Hilarius-Stokman P, de Korte D, van den Berg TK, van Bruggen R. CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood (2011) 119(23):5512–21. doi: 10.1182/blood-2011-10-386805
60. Klei TRL, Dalimot J, Nota B, Veldthuis M, Mul FPJ, Rademakers T, et al. Hemolysis in the spleen drives erythrocyte turnover. Blood (2020) 136(14):1579–89. doi: 10.1182/blood.2020005351
61. Theurl I, Hilgendorf I, Nairz M, Tymoszuk P, Haschka D, Asshoff M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med (2016) 22(8):945–51. doi: 10.1038/nm.4146
62. Chung JW, Shin E, Kim H, Han HS, Cho JY, Choi YR, et al. Hepatic iron overload in the portal tract predicts poor survival in hepatocellular carcinoma after curative resection. Liver Int (2018) 38(5):903–14. doi: 10.1111/liv.13619
63. Dou L, Shi X, He X, Gao Y. Macrophage Phenotype and Function in Liver Disorder. Front Immunol (2019) 10:3112. doi: 10.3389/fimmu.2019.03112
64. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron Induces Anti-tumor Activity in Tumor-Associated Macrophages. Front Immunol (2017) 8:1479. doi: 10.3389/fimmu.2017.01479
65. Thielmann CM, Costa da Silva M, Muley T, Meister M, Herpel E, Muckenthaler MU. Iron accumulation in tumor-associated macrophages marks an improved overall survival in patients with lung adenocarcinoma. Sci Rep (2019) 9(1):11326. doi: 10.1038/s41598-019-47833-x
66. Mertens C, Akam EA, Rehwald C, Brune B, Tomat E, Jung M. Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype with Consequences on Tumor Progression. PloS One (2016) 11(11):e0166164. doi: 10.1371/journal.pone.0166164
67. Mertens C, Mora J, Oren B, Grein S, Winslow S, Scholich K, et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology (2017) 7(3):e1408751. doi: 10.1080/2162402X.2017.1408751
68. Alkhateeb AA, Han B, Connor JR. Ferritin stimulates breast cancer cells through an iron-independent mechanism and is localized within tumor-associated macrophages. Breast Cancer Res Treat (2013) 137(3):733–44. doi: 10.1007/s10549-012-2405-x
69. Chi Y, Remsik J, Kiseliovas V, Derderian C, Sener U, Alghader M, et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science (2020) 369(6501):276–82. doi: 10.1126/science.aaz2193
70. Rehwald C, Schnetz M, Urbschat A, Mertens C, Meier JK, Bauer R, et al. The iron load of lipocalin-2 (LCN-2) defines its pro-tumour function in clear-cell renal cell carcinoma. Br J Cancer (2020) 122(3):421–33. doi: 10.1038/s41416-019-0655-7
71. Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab (2016) 24(3):447–61. doi: 10.1016/j.cmet.2016.07.015
72. Zhao N, Zhang AS, Wortham AM, Jue S, Knutson MD, Enns CA. The Tumor Suppressor, P53, Decreases the Metal Transporter, ZIP14. Nutrients (2017) 9(12):1–16. doi: 10.3390/nu9121335
73. Chen TT, Li L, Chung DH, Allen CD, Torti SV, Torti FM, et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J Exp Med (2005) 202(7):955–65. doi: 10.1084/jem.20042433
74. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
75. Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y, et al. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol (2019) 21(4):498–510. doi: 10.1038/s41556-019-0299-0
76. Escribese MM, Sierra-Filardi E, Nieto C, Samaniego R, Sanchez-Torres C, Matsuyama T, et al. The prolyl hydroxylase PHD3 identifies proinflammatory macrophages and its expression is regulated by activin A. J Immunol (2012) 189(4):1946–54. doi: 10.4049/jimmunol.1201064
77. Doedens AL, Stockmann C, Rubinstein MP, Liao D, Zhang N, DeNardo DG, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res (2010) 70(19):7465–75. doi: 10.1158/0008-5472.CAN-10-1439
78. Siegert I, Schodel J, Nairz M, Schatz V, Dettmer K, Dick C, et al. Ferritin-Mediated Iron Sequestration Stabilizes Hypoxia-Inducible Factor-1alpha upon LPS Activation in the Presence of Ample Oxygen. Cell Rep (2015) 13(10):2048–55. doi: 10.1016/j.celrep.2015.11.005
79. Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature (2008) 453(7196):807–11. doi: 10.1038/nature06905
80. Chao Y, Karmali PP, Simberg D. Role of carbohydrate receptors in the macrophage uptake of dextran-coated iron oxide nanoparticles. Adv Exp Med Biol (2011) 733:115–23. doi: 10.1007/978-94-007-2555-3_11
81. Settles M, Etzrodt M, Kosanke K, Schiemann M, Zimmermann A, Meier R, et al. Different capacity of monocyte subsets to phagocytose iron-oxide nanoparticles. PloS One (2011) 6(10):e25197. doi: 10.1371/journal.pone.0025197
82. Garbowski MW, Bansal S, Porter JB, Mori C, Burckhardt S, Hider RC. Intravenous iron preparations transiently generate non-transferrin-bound iron from two proposed pathways. Haematologica (2020). doi: 10.3324/haematol.2020.250803. Online ahead of print.
83. Camiolo G, Barbato A, Giallongo C, Vicario N, Romano A, Parrinello NL, et al. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol (2020) 36:101611. doi: 10.1016/j.redox.2020.101611
84. Fell LH, Seiler-Mussler S, Sellier AB, Rotter B, Winter P, Sester M, et al. Impact of individual intravenous iron preparations on the differentiation of monocytes towards macrophages and dendritic cells. Nephrol Dial Transplant (2016) 31(11):1835–45. doi: 10.1093/ndt/gfw045
85. Dalzon B, Guidetti M, Testemale D, Reymond S, Proux O, Vollaire J, et al. Utility of macrophages in an antitumor strategy based on the vectorization of iron oxide nanoparticles. Nanoscale (2019) 11(19):9341–52. doi: 10.1039/c8nr03364a
86. Sonnweber T, Theurl I, Seifert M, Schroll A, Eder S, Mayer G, et al. Impact of iron treatment on immune effector function and cellular iron status of circulating monocytes in dialysis patients. Nephrol Dial Transplant (2010) 26(3):977–87. doi: 10.1093/ndt/gfq483
87. Garcia-Fernandez N, Echeverria A, Sanchez-Ibarrola A, Paramo JA, Coma-Canella I. Randomized clinical trial on acute effects of i.v. iron sucrose during haemodialysis. Nephrol (Carlton) (2010) 15(2):178–83. doi: 10.1111/j.1440-1797.2009.01174.x
88. Wu Q, Miao T, Feng T, Yang C, Guo Y, Li H. Dextrancoated superparamagnetic iron oxide nanoparticles activate the MAPK pathway in human primary monocyte cells. Mol Med Rep (2018) 18(1):564–70. doi: 10.3892/mmr.2018.8972
89. Grosse S, Stenvik J, Nilsen AM. Iron oxide nanoparticles modulate lipopolysaccharide-induced inflammatory responses in primary human monocytes. Int J Nanomed (2016) 11:4625–42. doi: 10.2147/IJN.S113425
90. Blank F, Gerber P, Rothen-Rutishauser B, Sakulkhu U, Salaklang J, De Peyer K, et al. Biomedical nanoparticles modulate specific CD4+ T cell stimulation by inhibition of antigen processing in dendritic cells. Nanotoxicology (2010) 5(4):606–21. doi: 10.3109/17435390.2010.541293
91. Zini C, Venneri MA, Miglietta S, Caruso D, Porta N, Isidori AM, et al. USPIO-labeling in M1 and M2-polarized macrophages: An in vitro study using a clinical magnetic resonance scanner. J Cell Physiol (2018) 233(8):5823–8. doi: 10.1002/jcp.26360
92. Torti SV, Lemler E, Mueller BK, Popp A, Torti FM. Effects of Anti-repulsive Guidance Molecule C (RGMc/Hemojuvelin) Antibody on Hepcidin and Iron in Mouse Liver and Tumor Xenografts. Clin Exp Pharmacol (2017) 6(6):1–18. doi: 10.4172/2161-1459.1000223
93. Theurl I, Schroll A, Sonnweber T, Nairz M, Theurl M, Willenbacher W, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood (2011) 118(18):4977–84. doi: 10.1182/blood-2011-03-345066
94. Stone JH, Klearman M, Collinson N. Trial of Tocilizumab in Giant-Cell Arteritis. N Engl J Med (2017) 377(15):1494–5. doi: 10.1056/NEJMc1711031
95. Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, et al. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist (2018) 23(8):943–7. doi: 10.1634/theoncologist.2018-0028
96. Isaacs JD, Harari O, Kobold U, Lee JS, Bernasconi C. Effect of tocilizumab on haematological markers implicates interleukin-6 signalling in the anaemia of rheumatoid arthritis. Arthritis Res Ther (2013) 15(6):R204. doi: 10.1186/ar4397
97. Song SN, Iwahashi M, Tomosugi N, Uno K, Yamana J, Yamana S, et al. Comparative evaluation of the effects of treatment with tocilizumab and TNF-alpha inhibitors on serum hepcidin, anemia response and disease activity in rheumatoid arthritis patients. Arthritis Res Ther (2013) 15(5):R141. doi: 10.1186/ar4323
98. Hashizume M, Uchiyama Y, Horai N, Tomosugi N, Mihara M. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, improved anemia in monkey arthritis by suppressing IL-6-induced hepcidin production. Rheumatol Int (2010) 30(7):917–23. doi: 10.1007/s00296-009-1075-4
99. Strippoli R, Carvello F, Scianaro R, De Pasquale L, Vivarelli M, Petrini S, et al. Amplification of the response to Toll-like receptor ligands by prolonged exposure to interleukin-6 in mice: implication for the pathogenesis of macrophage activation syndrome. Arthritis Rheum (2012) 64(5):1680–8. doi: 10.1002/art.33496
100. Schulert GS, Minoia F, Bohnsack J, Cron RQ, Hashad S, Kon EPI, et al. Effect of Biologic Therapy on Clinical and Laboratory Features of Macrophage Activation Syndrome Associated With Systemic Juvenile Idiopathic Arthritis. Arthritis Care Res (Hoboken) (2018) 70(3):409–19. doi: 10.1002/acr.23277
101. Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, et al. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer (2019) 18(1):64. doi: 10.1186/s12943-019-0976-4
102. Yin Y, Yao S, Hu Y, Feng Y, Li M, Bian Z, et al. The Immune-microenvironment Confers Chemoresistance of Colorectal Cancer through Macrophage-Derived IL6. Clin Cancer Res (2017) 23(23):7375–87. doi: 10.1158/1078-0432.CCR-17-1283
103. Jeong SK, Kim JS, Lee CG, Park YS, Kim SD, Yoon SO, et al. Tumor associated macrophages provide the survival resistance of tumor cells to hypoxic microenvironmental condition through IL-6 receptor-mediated signals. Immunobiology (2015) 222(1):55–65. doi: 10.1016/j.imbio.2015.11.010
104. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology (2014) 147(6):1393–404. doi: 10.1053/j.gastro.2014.08.039
105. He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell (2013) 155(2):384–96. doi: 10.1016/j.cell.2013.09.031
106. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell (2009) 140(2):197–208. doi: 10.1016/j.cell.2009.12.052
107. Jayatilaka H, Tyle P, Chen JJ, Kwak M, Ju J, Kim HJ, et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat Commun (2017) 8:15584. doi: 10.1038/ncomms15584
108. Kleinegger F, Hofer E, Wodlej C, Golob-Schwarzl N, Birkl-Toeglhofer AM, Stallinger A, et al. Pharmacologic IL-6Ralpha inhibition in cholangiocarcinoma promotes cancer cell growth and survival. Biochim Biophys Acta Mol Basis Dis (2018) 1865(2):308–21. doi: 10.1016/j.bbadis.2018.11.006
109. Eddowes LA, Al-Hourani K, Ramamurthy N, Frankish J, Baddock HT, Sandor C, et al. Antiviral activity of bone morphogenetic proteins and activins. Nat Microbiol (2019) 4(2):339–51. doi: 10.1038/s41564-018-0301-9
110. Canali S, Zumbrennen-Bullough KB, Core AB, Wang CY, Nairz M, Bouley R, et al. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood (2016) 129(4):405–14. doi: 10.1182/blood-2016-06-721571
111. Andriopoulos B Jr., Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet (2009) 41(4):482–7. doi: 10.1038/ng.335
112. Shi YJ, Pan XT. BMP6 and BMP4 expression in patients with cancer-related anemia and its relationship with hepcidin and s-HJV. Genet Mol Res (2016) 15(1):1–5. doi: 10.4238/gmr.15017130
113. Cheng Z, Yan M, Lu Y, Pan XT. Expression of serum BMP6 and hepcidin in cancer-related anemia. Hematology (2020) 25(1):134–8. doi: 10.1080/16078454.2020.1738098
114. Sheetz M, Barrington P, Callies S, Berg PH, McColm J, Marbury T, et al. Targeting the hepcidin-ferroportin pathway in anaemia of chronic kidney disease. Br J Clin Pharmacol (2019) 85(5):935–48. doi: 10.1111/bcp.13877
115. Petzer V, Tymoszuk P, Asshoff M, Carvalho J, Papworth J, Deantonio C, et al. A fully human anti-BMP6 antibody reduces the need for erythropoietin in rodent models of the anemia of chronic disease. Blood (2019) 136(9):1080–90. doi: 10.1182/blood.2019004653
116. Wang CY, Babitt JL. Liver iron sensing and body iron homeostasis. Blood (2018) 133(1):18–29. doi: 10.1182/blood-2018-06-815894
117. Stieglitz D, Lamm S, Braig S, Feuerer L, Kuphal S, Dietrich P, et al. BMP6-induced modulation of the tumor micro-milieu. Oncogene (2019) 38(5):609–21. doi: 10.1038/s41388-018-0475-x
118. Xiong W, Wang L, Yu F. Expression of bone morphogenetic protein 6 in non-small cell lung cancer and its significance. Oncol Lett (2018) 17(2):1946–52. doi: 10.3892/ol.2018.9781
119. Takahashi M, Otsuka F, Miyoshi T, Otani H, Goto J, Yamashita M, et al. Bone morphogenetic protein 6 (BMP6) and BMP7 inhibit estrogen-induced proliferation of breast cancer cells by suppressing p38 mitogen-activated protein kinase activation. J Endocrinol (2008) 199(3):445–55. doi: 10.1677/JOE-08-0226
120. Katsuta E, Maawy AA, Yan L, Takabe K. High expression of bone morphogenetic protein (BMP) 6 and BMP7 are associated with higher immune cell infiltration and better survival in estrogen receptorpositive breast cancer. Oncol Rep (2019) 42(4):1413–21. doi: 10.3892/or.2019.7275
121. Kwon SJ, Lee GT, Lee JH, Iwakura Y, Kim WJ, Kim IY. Mechanism of pro-tumorigenic effect of BMP-6: neovascularization involving tumor-associated macrophages and IL-1a. Prostate (2014) 74(2):121–33. doi: 10.1002/pros.22734
122. Lee GT, Jung YS, Ha YS, Kim JH, Kim WJ, Kim IY. Bone morphogenetic protein-6 induces castration resistance in prostate cancer cells through tumor infiltrating macrophages. Cancer Sci (2013) 104(8):1027–32. doi: 10.1111/cas.12206
123. Gelbrich N, Ahrend H, Kaul A, Brandenburg LO, Zimmermann U, Mustea A, et al. Different Cytokine and Chemokine Expression Patterns in Malignant Compared to Those in Nonmalignant Renal Cells. Anal Cell Pathol (Amst) (2017) 7190546. doi: 10.1155/2017/7190546
124. Lee JH, Lee GT, Woo SH, Ha YS, Kwon SJ, Kim WJ, et al. BMP-6 in renal cell carcinoma promotes tumor proliferation through IL-10-dependent M2 polarization of tumor-associated macrophages. Cancer Res (2013) 73(12):3604–14. doi: 10.1158/0008-5472.CAN-12-4563
125. Petzer V, Theurl I, Weiss G. Established and Emerging Concepts to Treat Imbalances of Iron Homeostasis in Inflammatory Diseases. Pharm (Basel) (2018) 11(4):1–24. doi: 10.3390/ph11040135
126. Sebastiani G, Wilkinson N, Pantopoulos K. Pharmacological Targeting of the Hepcidin/Ferroportin Axis. Front Pharmacol (2016) 7:160. doi: 10.3389/fphar.2016.00160
127. Mayr R, Griffiths WJ, Hermann M, McFarlane I, Halsall DJ, Finkenstedt A, et al. Identification of mutations in SLC40A1 that affect ferroportin function and phenotype of human ferroportin iron overload. Gastroenterology (2011) 140(7):2056–63.E1. doi: 10.1053/j.gastro.2011.02.064
128. Viveiros A, Panzer M, Baumgartner N, Schaefer B, Finkenstedt A, Henninger B, et al. Reduced iron export associated with hepcidin resistance can explain the iron overload spectrum in ferroportin disease. Liver Int (2020) 40(8):1941–51. doi: 10.1111/liv.14539
129. Griffiths WJ, Mayr R, McFarlane I, Hermann M, Halsall DJ, Zoller H, et al. Clinical presentation and molecular pathophysiology of autosomal dominant hemochromatosis caused by a novel ferroportin mutation. Hepatology (2010) 51(3):788–95. doi: 10.1002/hep.23377
130. Fernandes A, Preza GC, Phung Y, De Domenico I, Kaplan J, Ganz T, et al. The molecular basis of hepcidin-resistant hereditary hemochromatosis. Blood (2009) 114(2):437–43. doi: 10.1182/blood-2008-03-146134
131. Letocart E, Le Gac G, Majore S, Ka C, Radio FC, Gourlaouen I, et al. A novel missense mutation in SLC40A1 results in resistance to hepcidin and confirms the existence of two ferroportin-associated iron overload diseases. Br J Haematol (2009) 147(3):379–85. doi: 10.1111/j.1365-2141.2009.07834.x
132. Aschemeyer S, Qiao B, Stefanova D, Valore EV, Sek AC, Ruwe TA, et al. Structure-function analysis of ferroportin defines the binding site and an alternative mechanism of action of hepcidin. Blood (2017) 131(8):899–910. doi: 10.1182/blood-2017-05-786590
133. Schnetz M, Meier JK, Rehwald C, Mertens C, Urbschat A, Tomat E, et al. The Disturbed Iron Phenotype of Tumor Cells and Macrophages in Renal Cell Carcinoma Influences Tumor Growth. Cancers (Basel) (2020) 12(3):1–20. doi: 10.3390/cancers12030530
134. Marques O, Porto G, Rema A, Faria F, Cruz Paula A, Gomez-Lazaro M, et al. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer (2016) 16:187. doi: 10.1186/s12885-016-2228-y
135. Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell (2019) 179(4):829–45.e820. doi: 10.1016/j.cell.2019.10.003
136. Zhang Z, Zhang F, An P, Guo X, Shen Y, Tao Y, et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood (2011) 118(7):1912–22. doi: 10.1182/blood-2011-01-330324
137. Wang L, Johnson EE, Shi HN, Walker WA, Wessling-Resnick M, Cherayil BJ. Attenuated inflammatory responses in hemochromatosis reveal a role for iron in the regulation of macrophage cytokine translation. J Immunol (2008) 181(4):2723–31. doi: 10.4049/jimmunol.181.4.2723
138. Recalcati S, Gammella E, Buratti P, Doni A, Anselmo A, Locati M, et al. Macrophage ferroportin is essential for stromal cell proliferation in wound healing. Haematologica (2018) 104(1):47–58. doi: 10.3324/haematol.2018.197517
139. Weiss G, Werner-Felmayer G, Werner ER, Grunewald K, Wachter H, Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med (1994) 180(3):969–76. doi: 10.1084/jem.180.3.969
140. Nairz M, Schleicher U, Schroll A, Sonnweber T, Theurl I, Ludwiczek S, et al. Nitric oxide-mediated regulation of ferroportin-1 controls macrophage iron homeostasis and immune function in Salmonella infection. J Exp Med (2012) 210(5):855–73. doi: 10.1084/jem.20121946
141. Richard F, van Lier JJ, Roubert B, Haboubi T, Gohring UM, Durrenberger F. Oral ferroportin inhibitor VIT-2763: First-in-human, phase 1 study in healthy volunteers. Am J Hematol (2020) 95(1):68–77. doi: 10.1002/ajh.25670
142. Vadhan-Raj S, Abonour R, Goldman JW, Smith DA, Slapak CA, Ilaria RL Jr., et al. A first-in-human phase 1 study of a hepcidin monoclonal antibody, LY2787106, in cancer-associated anemia. J Hematol Oncol (2017) 10(1):73. doi: 10.1186/s13045-017-0427-x
143. Sasu BJ, Cooke KS, Arvedson TL, Plewa C, Ellison AR, Sheng J, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood (2009) 115(17):3616–24. doi: 10.1182/blood-2009-09-245977
144. Schwoebel F, van Eijk LT, Zboralski D, Sell S, Buchner K, Maasch C, et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood (2012) 121(12):2311–5. doi: 10.1182/blood-2012-09-456756
145. Boyce M, Warrington S, Cortezi B, Zollner S, Vauleon S, Swinkels DW, et al. Safety, pharmacokinetics and pharmacodynamics of the anti-hepcidin Spiegelmer lexaptepid pegol in healthy subjects. Br J Pharmacol (2016) 173(10):1580–8. doi: 10.1111/bph.13433
146. Sun CC, Vaja V, Chen S, Theurl I, Stepanek A, Brown DE, et al. A hepcidin lowering agent mobilizes iron for incorporation into red blood cells in an adenine-induced kidney disease model of anemia in rats. Nephrol Dial Transplant (2013) 28(7):1733–43. doi: 10.1093/ndt/gfs584
147. Asshoff M, Petzer V, Warr MR, Haschka D, Tymoszuk P, Demetz E, et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood (2016) 129(13):1823–30. doi: 10.1182/blood-2016-09-740092
148. Theurl I, Theurl M, Seifert M, Mair S, Nairz M, Rumpold H, et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood (2008) 111(4):2392–9. doi: 10.1182/blood-2007-05-090019
149. Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood (2006) 107(9):3727–32. doi: 10.1182/blood-2005-06-2259
150. Andriopoulos B, Pantopoulos K. Hepcidin generated by hepatoma cells inhibits iron export from co-cultured THP1 monocytes. J Hepatol (2006) 44(6):1125–31. doi: 10.1016/j.jhep.2005.10.025
151. Jamnongkan W, Thanan R, Techasen A, Namwat N, Loilome W, Intarawichian P, et al. Upregulation of transferrin receptor-1 induces cholangiocarcinoma progression via induction of labile iron pool. Tumour Biol (2017) 39(7):1010428317717655. doi: 10.1177/1010428317717655
152. Jiang XP, Elliott RL, Head JF. Manipulation of iron transporter genes results in the suppression of human and mouse mammary adenocarcinomas. Anticancer Res (2010) 30(3):759–65.
153. Brookes MJ, Hughes S, Turner FE, Reynolds G, Sharma N, Ismail T, et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut (2006) 55(10):1449–60. doi: 10.1136/gut.2006.094060
154. Kong Y, Hu L, Lu K, Wang Y, Xie Y, Gao L, et al. Ferroportin downregulation promotes cell proliferation by modulating the Nrf2-miR-17-5p axis in multiple myeloma. Cell Death Dis (2019) 10(9):624. doi: 10.1038/s41419-019-1854-0
155. Gu Z, Wang H, Xia J, Yang Y, Jin Z, Xu H, et al. Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res (2015) 75(11):2211–21. doi: 10.1158/0008-5472.CAN-14-3804
156. Gasparetto M, Pei S, Minhajuddin M, Stevens B, Smith CA, Seligman P. Low ferroportin expression in AML is correlated with good risk cytogenetics, improved outcomes and increased sensitivity to chemotherapy. Leuk Res (2019) 80:1–10. doi: 10.1016/j.leukres.2019.02.011
157. Trujillo-Alonso V, Pratt EC, Zong H, Lara-Martinez A, Kaittanis C, Rabie MO, et al. FDA-approved ferumoxytol displays anti-leukaemia efficacy against cells with low ferroportin levels. Nat Nanotechnol (2019) 14(6):616–22. doi: 10.1038/s41565-019-0406-1
158. Chen Y, Zhang S, Wang X, Guo W, Wang L, Zhang D, et al. Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell Signal (2014) 27(1):168–76. doi: 10.1016/j.cellsig.2014.11.002
159. Blanchette-Farra N, Kita D, Konstorum A, Tesfay L, Lemler D, Hegde P, et al. Contribution of three-dimensional architecture and tumor-associated fibroblasts to hepcidin regulation in breast cancer. Oncogene (2018) 37(29):4013–32. doi: 10.1038/s41388-018-0243-y
160. Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y, et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal (2014) 26(11):2539–50. doi: 10.1016/j.cellsig.2014.07.029
161. Pinnix ZK, Miller LD, Wang W, D’Agostino R Jr., Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med (2010) 2(43):43ra56. doi: 10.1126/scisignal.3001127
162. Caro JJ, Salas M, Ward A, Goss G. Anemia as an independent prognostic factor for survival in patients with cancer: a systemic, quantitative review. Cancer (2001) 91(12):2214–21. doi: 10.1002/1097-0142(20010615)91
163. Weiss G, Ganz T, Goodnough LT. Anemia of inflammation. Blood (2018) 133(1):40–50. doi: 10.1182/blood-2018-06-856500
164. Maccio A, Madeddu C, Gramignano G, Mulas C, Tanca L, Cherchi MC, et al. The role of inflammation, iron, and nutritional status in cancer-related anemia: results of a large, prospective, observational study. Haematologica (2014) 100(1):124–32. doi: 10.3324/haematol.2014.112813
165. Ludwig H, Muldur E, Endler G, Hubl W. Prevalence of iron deficiency across different tumors and its association with poor performance status, disease status and anemia. Ann Oncol (2013) 24(7):1886–92. doi: 10.1093/annonc/mdt118
166. Peerschke EI, Pessin MS, Maslak P. Using the hemoglobin content of reticulocytes (RET-He) to evaluate anemia in patients with cancer. Am J Clin Pathol (2014) 142(4):506–12. doi: 10.1309/AJCPCVZ5B0BOYJGN
167. Durigova A, Lamy PJ, Thezenas S, Pouderoux S, Montels F, Romieu G, et al. Anemia and iron biomarkers in patients with early breast cancer. Diagnostic value of hepcidin and soluble transferrin receptor quantification. Clin Chem Lab Med (2013) 51(9):1833–41. doi: 10.1515/cclm-2013-0031
168. Thomas C, Kirschbaum A, Boehm D, Thomas L. The diagnostic plot: a concept for identifying different states of iron deficiency and monitoring the response to epoetin therapy. Med Oncol (2006) 23(1):23–36. doi: 10.1385/MO:23:1:23
169. Steinmetz HT, Tsamaloukas A, Schmitz S, Wiegand J, Rohrberg R, Eggert J, et al. A new concept for the differential diagnosis and therapy of anaemia in cancer patients. Support Care Cancer (2010) 19(2):261–9. doi: 10.1007/s00520-010-0812-2
170. Skikne BS, Punnonen K, Caldron PH, Bennett MT, Rehu M, Gasior GH, et al. Improved differential diagnosis of anemia of chronic disease and iron deficiency anemia: a prospective multicenter evaluation of soluble transferrin receptor and the sTfR/log ferritin index. Am J Hematol (2011) 86(11):923–7. doi: 10.1002/ajh.22108
171. Nairz M, Theurl I, Wolf D, Weiss G. Iron deficiency or anemia of inflammation? : Differential diagnosis and mechanisms of anemia of inflammation. Wien Med Wochenschr (2016) 166(13-14):411–23. doi: 10.1007/s10354-016-0505-7
172. Hohaus S, Massini G, Giachelia M, Vannata B, Bozzoli V, Cuccaro A, et al. Anemia in Hodgkin’s lymphoma: the role of interleukin-6 and hepcidin. J Clin Oncol (2009) 28(15):2538–43. doi: 10.1200/JCO.2009.27.6873
173. Shu T, Jing C, Lv Z, Xie Y, Xu J, Wu J. Hepcidin in tumor-related iron deficiency anemia and tumor-related anemia of chronic disease: pathogenic mechanisms and diagnosis. Eur J Haematol (2015) 94(1):67–73. doi: 10.1111/ejh.12402
174. Wilson MJ, van Haaren M, Harlaar JJ, Park HC, Bonjer HJ, Jeekel J, et al. Long-term prognostic value of preoperative anemia in patients with colorectal cancer: A systematic review and meta-analysis. Surg Oncol (2017) 26(1):96–104. doi: 10.1016/j.suronc.2017.01.005
175. An MS, Yoo JH, Kim KH, Bae KB, Choi CS, Hwang JW, et al. T4 stage and preoperative anemia as prognostic factors for the patients with colon cancer treated with adjuvant FOLFOX chemotherapy. World J Surg Oncol (2015) 13:64. doi: 10.1186/s12957-015-0488-7
176. Grabmer C, Holmberg J, Popovsky M, Amann E, Schonitzer D, Falaize S, et al. Up to 21-day banked red blood cells collected by apheresis and stored for 14 days after automated wash at different times of storage. Vox Sang (2006) 90(1):40–4. doi: 10.1111/j.1423-0410.2005.00719.x
177. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci USA (2014) 111(39):E4110–4118. doi: 10.1073/pnas.1405023111
178. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem (2007) 282(28):20221–9. doi: 10.1074/jbc.M610737200
179. Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, Puig-Kroger A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology (2010) 215(9-10):788–95. doi: 10.1016/j.imbio.2010.05.020
180. Grover A, Mancini E, Moore S, Mead AJ, Atkinson D, Rasmussen KD, et al. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J Exp Med (2013) 211(2):181–8. doi: 10.1084/jem.20131189
181. Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci USA (2004) 101(41):14907–12. doi: 10.1073/pnas.0406491101
182. Gilboa D, Haim-Ohana Y, Deshet-Unger N, Ben-Califa N, Hiram-Bab S, Reuveni D, et al. Erythropoietin enhances Kupffer cell number and activity in the challenged liver. Sci Rep (2017) 7(1):10379. doi: 10.1038/s41598-017-11082-7
183. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut (2015) 66(1):157–67. doi: 10.1136/gutjnl-2015-310514
184. Zhao L, Lim SY, Gordon-Weeks AN, Tapmeier TT, Im JH, Cao Y, et al. Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology (2013) 57(2):829–39. doi: 10.1002/hep.26094
185. Yasuda Y, Hara S, Hirohata T, Koike E, Yamasaki H, Okumoto K, et al. Erythropoietin-responsive sites in normal and malignant human lung tissues. Anat Sci Int (2010) 85(4):204–13. doi: 10.1007/s12565-010-0081-7
186. De Luisi A, Binetti L, Ria R, Ruggieri S, Berardi S, Catacchio I, et al. Erythropoietin is involved in the angiogenic potential of bone marrow macrophages in multiple myeloma. Angiogenesis (2013) 16(4):963–73. doi: 10.1007/s10456-013-9369-2
187. Vatsveen TK, Sponaas AM, Tian E, Zhang Q, Misund K, Sundan A, et al. Erythropoietin (EPO)-receptor signaling induces cell death of primary myeloma cells in vitro. J Hematol Oncol (2016) 9(1):75. doi: 10.1186/s13045-016-0306-x
188. Li W, Wang Y, Zhao H, Zhang H, Xu Y, Wang S, et al. Identification and transcriptome analysis of erythroblastic island macrophages. Blood (2019) 134(5):480–91. doi: 10.1182/blood.2019000430
189. Thomaidis T, Weinmann A, Sprinzl M, Kanzler S, Raedle J, Ebert M, et al. Erythropoietin treatment in chemotherapy-induced anemia in previously untreated advanced esophagogastric cancer patients. Int J Clin Oncol (2014) 19(2):288–96. doi: 10.1007/s10147-013-0544-7
190. Aapro M, Leonard RC, Barnadas A, Marangolo M, Untch M, Malamos N, et al. Effect of once-weekly epoetin beta on survival in patients with metastatic breast cancer receiving anthracycline- and/or taxane-based chemotherapy: results of the Breast Cancer-Anemia and the Value of Erythropoietin (BRAVE) study. J Clin Oncol (2008) 26(4):592–8. doi: 10.1200/JCO.2007.11.5378
191. Nitz U, Gluz O, Zuna I, Oberhoff C, Reimer T, Schumacher C, et al. Final results from the prospective phase III WSG-ARA trial: impact of adjuvant darbepoetin alfa on event-free survival in early breast cancer. Ann Oncol (2014) 25(1):75–80. doi: 10.1093/annonc/mdt505
192. Ross SD, Allen IE, Henry DH, Seaman C, Sercus B, Goodnough LT. Clinical benefits and risks associated with epoetin and darbepoetin in patients with chemotherapy-induced anemia: a systematic review of the literature. Clin Ther (2006) 28(6):801–31. doi: 10.1016/j.clinthera.2006.06.003
193. Bohlius J, Wilson J, Seidenfeld J, Piper M, Schwarzer G, Sandercock J, et al. Recombinant human erythropoietins and cancer patients: updated meta-analysis of 57 studies including 9353 patients. J Natl Cancer Inst (2006) 98(10):708–14. doi: 10.1093/jnci/djj189
194. Grant MD, Piper M, Bohlius J, Tonia T, Robert N, Vats V, et al. Epoetin and Darbopoetin for managing anemia in patients undergoing cancer treatment: Comparative effectiveness update. In: AHRQ Comparative Effectiveness Reviews. Rockville, Maryland, USA: Agency for Healthcare Research and Quality (2013).
Keywords: cancer-induced anemia (CIA), tumor-associated macrophage (TAM), iron, hepcidin, ferroportin, BMP - Smad signaling pathway, IL-6 (interleukin 6)
Citation: Weiler S and Nairz M (2021) TAM-ing the CIA—Tumor-Associated Macrophages and Their Potential Role in Unintended Side Effects of Therapeutics for Cancer-Induced Anemia. Front. Oncol. 11:627223. doi: 10.3389/fonc.2021.627223
Received: 08 November 2020; Accepted: 01 March 2021;
Published: 25 March 2021.
Edited by:
Stefania Recalcati, University of Milan, ItalyReviewed by:
Robert F. Paulson, Pennsylvania State University (PSU), United StatesPaola Allavena, University of Milan, Italy
Fernando Torres Andón, University of Santiago de Compostela, Spain
Copyright © 2021 Weiler and Nairz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manfred Nairz, bWFuZnJlZC5uYWlyekBpLW1lZC5hYy5hdA==