 Lorena Carvajal
Lorena Carvajal Jaime Gutiérrez
Jaime Gutiérrez Eugenia Morselli
Eugenia Morselli Andrea Leiva
Andrea Leiva- 1Department of Physiology, Faculty of Biological Sciences, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2School of Medical Technology, Health Sciences Faculty, Universidad San Sebastian, Santiago, Chile
- 3Autophagy Research Center, Santiago, Chile
Early human placental development begins with blastocyst implantation, then the trophoblast differentiates and originates the cells required for a proper fetal nutrition and placental implantation. Among them, extravillous trophoblast corresponds to a non-proliferating trophoblast highly invasive that allows the vascular remodeling which is essential for appropriate placental perfusion and to maintain the adequate fetal growth. This process involves different placental cell types as well as molecules that allow cell growth, cellular adhesion, tissular remodeling, and immune tolerance. Remarkably, some of the cellular processes required for proper placentation are common between placental and cancer cells to finally support tumor growth. Indeed, as in placentation trophoblasts invade and migrate, cancer cells invade and migrate to promote tumor metastasis. However, while these processes respond to a controlled program in trophoblasts, in cancer cells this regulation is lost. Interestingly, it has been shown that autophagy, a process responsible for the degradation of damaged proteins and organelles to maintain cellular homeostasis, is required for invasion of trophoblast cells and for vascular remodeling during placentation. In cancer cells, autophagy has a dual role, as it has been shown both as tumor promoter and inhibitor, depending on the stage and tumor considered. In this review, we summarized the similarities and differences between trophoblast cell invasion and cancer cell metastasis specifically evaluating the role of autophagy in both processes.
Introduction
The placentation is a complex process that involves different stages, which quickly and efficiently leads to the development of the placenta, a temporary organ. The placenta is developed through regulated and dynamic cellular processes that include embryo pre-implantation and implantation, decidua formation, trophoblast proliferation, trophoblast differentiation into the invasive phenotype, and vascular remodeling (1). Interestingly, during placentation, the ability of trophoblast cells to proliferate, invade, and evade the immune system, resemble those induced by cancer cells during tumor growth (2). Indeed, the processes of proliferation, migration, and invasion in cancer cells and trophoblast derived cells share different molecules such as growth factors, cell adhesion molecules, surface receptors, matrix-digesting enzymes, and enzymes inhibitors, proto-oncogenes, hormones, and peptides, among others (3). These molecules regulate different processes that are highly controlled in trophoblasts, with trophoblast-derived cells following an organized pattern without metastasizing to new tissues, while the same pathways are dysregulated in cancer, driving metastasis (4).
In addition to sharing proliferative and invasive features, trophoblasts and cancer cells, actively modulate the host immune response to develop and sustain nutrient supply (5). Interestingly, it has been described that activation of autophagy occurs in both processes, regulating placental and cancer development (6, 7). However, how autophagy modulation affects trophoblast function is not entirely known (8). Consistently, the role of autophagy in cancer development is still a matter of study due to its dual role in tumor onset and progression (9, 10). Indeed, the role of autophagy in tumor development is controversial and dependent on tumor stage and type. It has been suggested that autophagy could promote aggressive characteristics of cancer cells, such as increased cellular invasion (11, 12), but also be a barrier to cancer proliferation (13–15). Additionally, autophagy also provides the microenvironment for placentation and cancer growth. This review will summarize the parallels between trophoblast-derived cells in placentation and cancer cells in tumor growth and metastasis with a final focus on the role of autophagy in both processes.
Development of the Human Placenta
The placenta is a temporary organ that maintains and protects the fetus during pregnancy controlling the maternal-fetal exchange of nutrients, gases, and metabolic waste. Human pregnancy begins with the physiological preparation of the endometrium modulated by hormones such as progesterone and estrogen, which regulate growth factors, cytokines, and adhesion molecules that allow the blastocyst’s implantation (16).
The placenta develops from the trophectoderm (TE), the outer layer of the blastocyst from which derives the undifferentiated cytotrophoblast (CTB). The CTB originates two main villus structures: the floating villus, where CTBs fuse to form the multinuclear syncyiotrophoblast (STB) and the anchoring villus (17–20) (Figure 1). The STB acts as an exchange barrier with the maternal blood to assure nutrients as well as waste and gases exchange with the fetal blood (21). The floating villus cells proliferate to form primary villi, which show further branching, forming the intervillous space. The branching to secondary and tertiary villi, allows the expansion of the STB surface area, which favors an efficient nutrient exchange with the fetal blood (19).
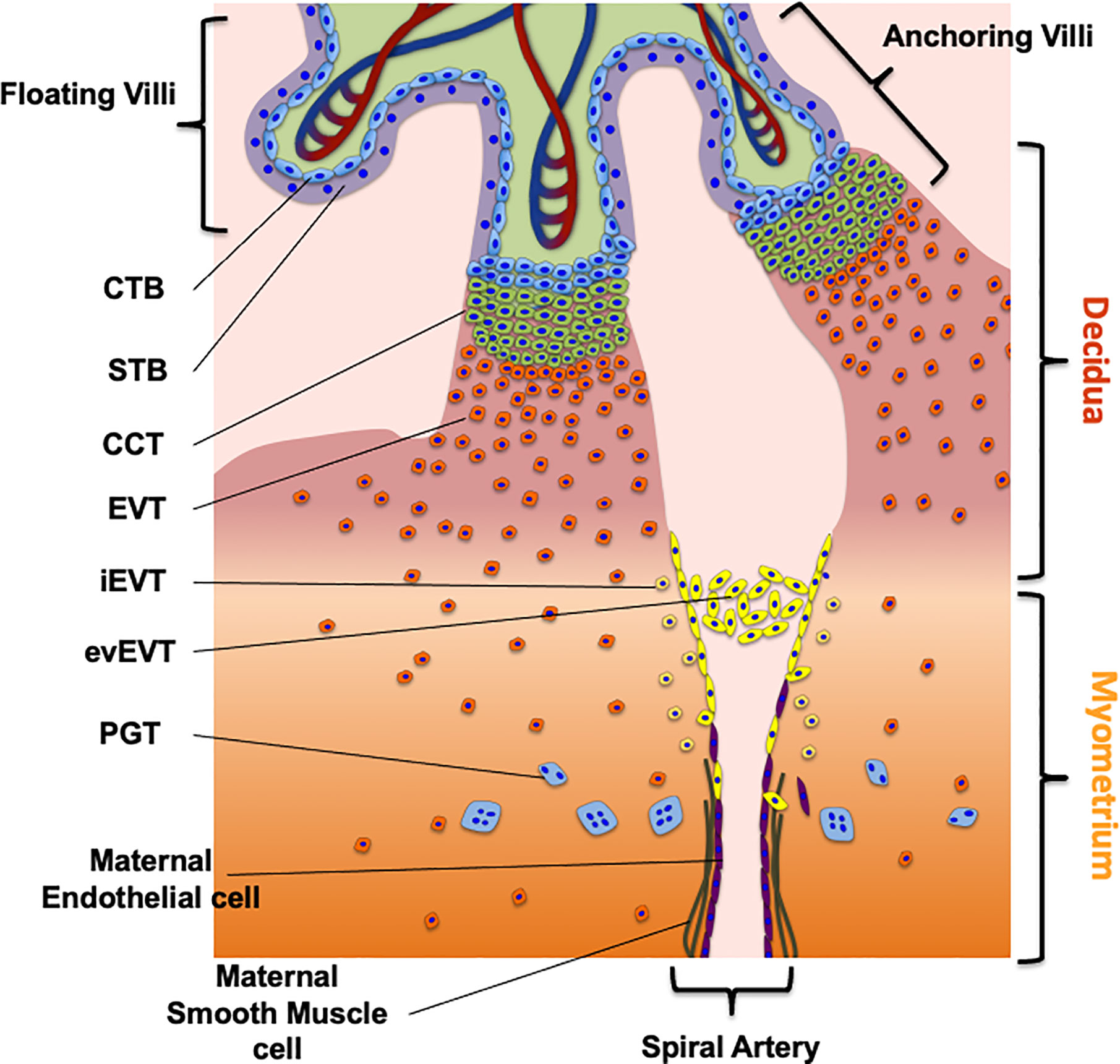
Figure 1 The maternal fetal interface and trophoblast cells subtypes. The figure shows the placental cell types required for the early first trimester human placentation as well as the route to migrate and invade the decidua and myometrium. The different trophoblast subtypes are villous cytotrophoblast (CTB), synctiotrophoblast (STB), cell column trophoblast (CCT), extravillous trophoblast (EVT), endovascular EVT (evEVT), interstitial EVT (iEVT), placental giant trophoblast (PGT). The complete description of the process is in the section “Development of the Human Placenta” of the review.
As mentioned, the second villus structure derived from the CTB is the anchoring villus, whose main function is to mediate the placental attachment to the endometrium in the uterine wall, to sustain fetal growth (17–20) (Figure 1). In the distal tip of the anchoring villus it is possible to find a group of proliferating cells known as cell column trophoblast (CCT). From these cells emerge the placental giant trophoblast (PGT) that mediates the early histotrophic nutrition of the embryo and of extravillous trophoblast cells (EVT). EVTs are a group of non-proliferating trophoblast cells characterized by a highly invasive phenotype. They invade the maternal decidua and the first third of the myometrium, playing a crucial role in the histotrophic nutrition of the fetus, immunomodulation and remodeling of the uterine spiral arteries (Figure 1) (19).
These activities are mediated by specialized subgroups of EVTs, characterized by specific markers: the endovascular EVT (evEVT) and the interstitial EVT (iEVT) (Table 1). The evEVTs migrate through the lumen of the spiral arteries forming a trophoblast plug reducing the maternal blood flow towards the intervillous space during the early stages of placenta development, permitting histiotrophic nutrition (46). Additionally, evEVT maintain the oxygen concentration low, which is required for placental development and successful trophoblast differentiation and may also promote favorable trophoblast migration and endothelial cell replacement, both required for vascular remodeling. Finally, at the end of first trimester the trophoblast plug formed by evEVT is disintegrated (47, 48).
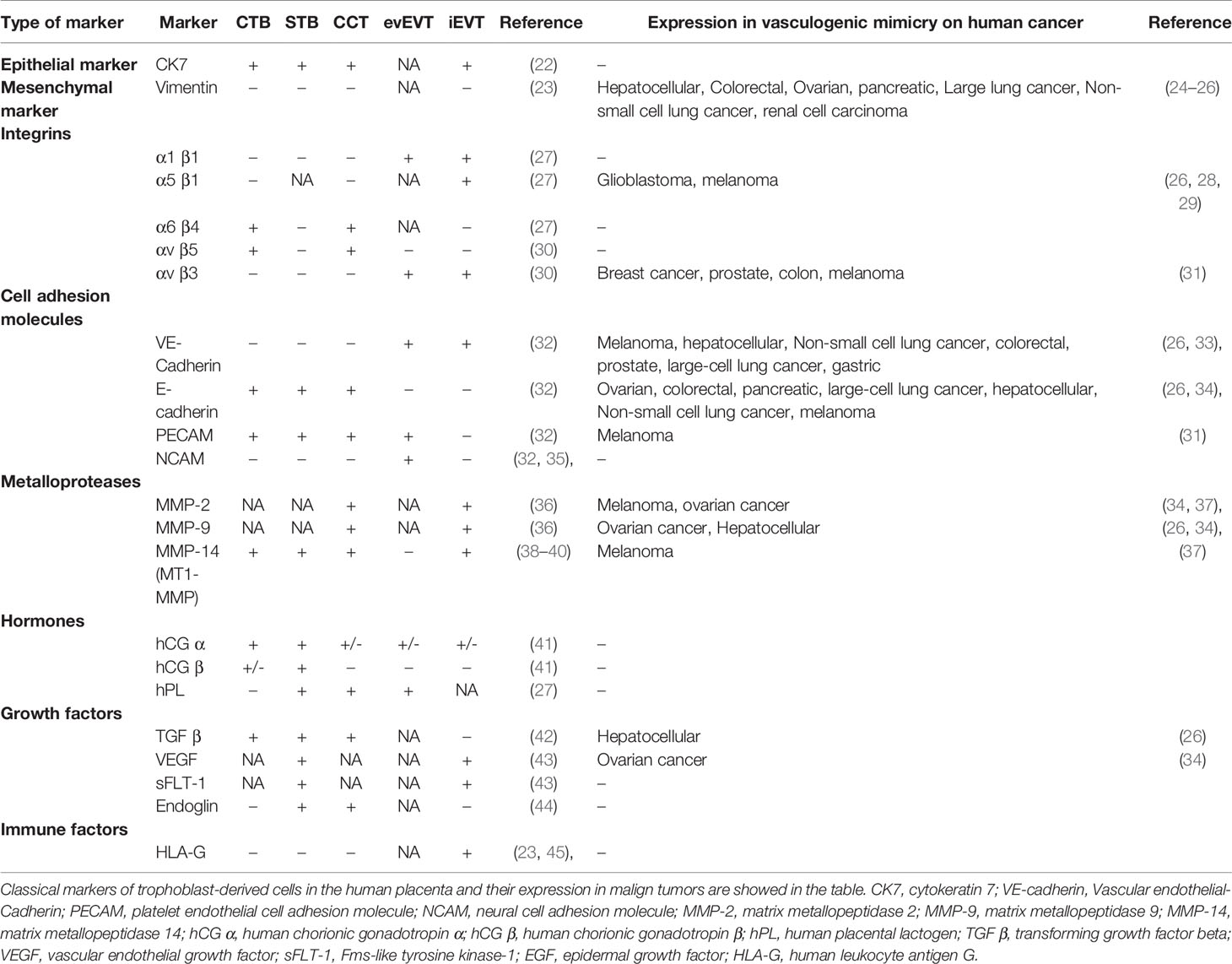
Table 1 Classical markers of trophoblast-derived cells in the human placenta and its expression in vasculogenic mimicry on human cancer.
The main function of iEVT is to participate to the immune tolerance and placental invasion of maternal tissues. iEVTs express Human Leukocyte antigen-G (HLA-G), a nonclassical major histocompatibility complex (MHC) (Table 1), which is essential to modulate the immune tolerance at the maternal-fetal interface by regulating the interaction and communication with the Uterine Natural Killers (uNK) (49).
iEVTs invade trough the interstitium of the decidua and myometrium towards the maternal spiral arteries (46). Once there, the iEVT acquires an endothelial-like phenotype under conditions of low oxygen concentration (approximately 8% oxygen) replacing the maternal endothelial cells of the uterine spiral arteries. It is important to highlight that many studies refer to this low oxygen concentration as an “hypoxic” environment. However, the reduced oxygen level corresponds to the physiological level required for placenta development, which is maintained until the end of the first trimester (below 20 mmHg). Thus, as this occurs in normal placental development, we will refer to this low oxygen condition as normoxic and not hypoxia (50–52).
The process of invasion by EVTs allows the replacement of the endothelial layer of the maternal spiral arteries, which is essential for an appropriate placental perfusion that maintains an adequate fetal growth. This complex process not only involves different placental cell types, but also a wide range of molecules related to cell growth (i.e., hormones and growth factors), cellular adhesion (i.e., Integrins, Cadherins), tissular remodeling (i.e., Metalloproteases) and proteins related to immune tolerance (Table 1) (19, 53). In addition to the classical markers, other molecules including structural and adhesion related proteins, proteins associated to invasion, immunity, embryonic stem cell-associated transcription regulators and oncogenes have been recently described as markers of CTB, STB, iEVT or eEVT (54–58).
CTB and CCT but not EVT cells show proliferative activity and generate cells that stop the proliferation and start to differentiate (20). The process of proliferation and differentiation of CTB into migratory, invasive EVT and endothelial-like trophoblast shows similarities with the process of tumor formation and metastasis of cancer cells. The main similarities are: (i) tissue invasion, (ii) immune system modulation and (iii) vascularization. Despite this, a crucial difference between trophoblast derived and cancer cells is that while in trophoblast cells these processes are regulated, this regulation is lost in cancer cells (4).
The physiological process of placentation responds to a controlled program that results in changes in gene expression and cell cycle. As such, when placentation is not kept under control, malformation of the placenta, pregnancy pathologies and abortions can occur (17, 20, 59). Different abnormal placentation processes have been described, which are characterized by abnormal trophoblast invasion such as abnormally invasive placentas. Abnormal placentation processes are: (i) placenta accreta (abnormal adherence with direct contact to myometrium), (ii) placenta increta (placental villi penetrate into the myometrium) and (iii) percreta (placental villi penetrate trough myometrium to uterine serosa and into the surrounding structures such as the bladder). It has been suggested that trophoblast cells of this abnormally invasive placentas lose their physiological regulation, leading to increased proliferative activity during the invasion, behaving like cancer cells (60, 61).
Similarities and Differences Between Cancer Cells and Trophoblast Derived Cells
Cellular Proliferation, Migration, and Invasion
Both cancer cells and trophoblast derived cells express different molecules such as growth factors, proto-oncogenes, enzymes, cell surface receptors, enzyme receptors, hormones and peptides, whose activation mediates their high proliferative, migratory and invasive capacity. During placentation, growth factors such as Epidermal Growth Factor (EGF), Hepatocyte Growth Factor (HGF), Vascular Endothelial Growth Factor (VEGF), Placental Growth Factor (PLGF), Insulin-like Growth Factor (IGF), Transforming Growth Factor (TGF) and their corresponding receptors are among the main factors that regulate CTB proliferation, acting in a paracrine and autocrine manner (62). These growth factors bind to Tyrosine Kinase Receptors to activate the Mitogen-activated Protein/extracellular Signal-regulated kinase/Extracellular Signal-Regulated Kinase (MEK/ERK) proliferation pathway and the Phosphatidylinositol 3-kinase/Protein Kinase B (PI3K/Akt) anti-apoptosis pathway (3). Moreover, proto-oncogenes play an essential role in the etiology of cancer inducing its growth. As cancer cells, trophoblast derived cells express several proto-oncogenes; for example, CTB and STB exclusively express proto-oncogenes that encode Growth Factor Receptor c-erbB1 (Human Epidermal Growth Factor Receptor 1 (HER1), Epidermal Growth Factor Receptor 1 (ERBB1 or EGF-receptor)) (63). Also, trophoblast cells such as CTB, STB, and EVT encode for a Receptor Tyrosine Kinase (RTK), namely c-erbB2 (HER2/neu, ERBB2), c-fms (CSF1R), c-met (MET) and c-kit (KIT) (64–67), as well as for transcription factors that have been implicated in trophoblast invasion such as c-fos (FOS) and c-jun (JUN), in addition to c-myc (MYC) and c-ets1 (ETS) (68–71). Additionally, in iEVT, c-sis (SIS, Platelet-derived Growth Factor Beta (PDGFB)) is expressed, which encodes for one of the two chains (the B-chains) constituting Platelet-derived Growth Factor (PDGF) (72) and in EVT the c-ras family (Kirsten rat Sarcoma viral oncogene (K-RAS), Neuroblastoma RAS viral oncogene homolog (N-RAS), and Harvey rat sarcoma (H-RAS)) is expressed encoding for Rat sarcoma (RAS) proteins that regulate cellular proliferation and inflammation in the human placenta (73, 74). All the aforementioned proto-oncogenes are crucial in the first step of malignant transformation and its physiological expression occurs during the first week of pregnancy promoting proliferation, migration, and invasion of the trophoblast (2).
The Telomerase is a factor that regulates the proliferative capacity of a cell, as it maintains chromosome stability in actively dividing cells (75). CTB expresses a functional Telomerase, which is downregulated during differentiation, but expressed in term placenta. During human pregnancy, Telomerase activity is the highest during the first trimester, and decreases with the maturation of the placenta (76). Telomerase activity ensures a high rate of proliferation and could be a factor controlling placental growth (77–79). Consistently, in cancer cells, the Telomerase allows uncontrolled cell proliferation, which is essential for tumor progression (80). Additionally, Survivin, a protein overexpressed in many cancers (81), where it promotes proliferation and inhibits apoptosis, is expressed in trophoblast cells, however its role in this location has not been elucidated yet (82, 83). Altogether these studies indicate that the Telomerase and Survivin have an important role in cell proliferation in both trophoblast and cancer cells.
As mentioned, placental development during the first trimester occurs in a stable state of low oxygen concentration (84); by comparison, in tumors, hypoxia is necessary to support tumor growth and metastasis (85). In response to low oxygen levels, cells upregulate Hypoxia-Inducible Factor (HIF), a family of transcription factors that functions as a Heterodimer with a regulatory α subunit (HIF-α) and a constitutive β subunit (HIF- β) (86, 87). The activation of the different HIF isoforms leads to the transcription of genes involved in several processes such as metabolism, angiogenesis, and immunomodulation (86). Thus, this low oxygen concentration environment in trophoblast and cancer cells could be considered as key to stimulate proliferation, invasion, and vasculogenesis in host tissues (88).
During placentation and cancer growth, invasion is required to provide blood and nutrient supply. Different events need to occur for a successful invasion process: (i) changes in the expression of Cell Adhesion molecules (ii) secretion of Proteases, and (iii) availability of Growth Factors (5). One feature shared by both cell types is the process of epithelial to mesenchymal transition (EMT), which leads to the loss of cell-to-cell contact inhibition, and to the increased expression of proteins that degrade the extracellular matrix. During EMT the Integrin expression pattern changes, and the expression of E-cadherin decreases, enhancing cell movement through tissues by reducing cell polarity (89, 90).
EVT and invasive cancer cells also share enzymes required for the degradation of the basal membrane that allow the process of invasion. Among those there are Serine Proteases, Cathepsins and Matrix Metalloproteinases (MMPs), the Heparan Sulfate-degrading Endoglycosidase, the Protease-Activated Receptor (PAR) and the Receptor of Thrombin (91, 92). These enzymes are expressed transiently in the trophoblast, in a very regulated manner, while in cancer cells their expression becomes constitutive (2, 4). As an example, the expression of MMP-2 and MMP-9 is increased during trophoblast invasion, promoting proteolysis and therefore invasion. Importantly, when the invasion is completed, decidual cells inhibit MMP-2 and MMP-9 activity by the release of protease inhibitors (53, 93, 94). When the control of the protease activity is lost abnormally invasive placentas develop, consistently, this regulation disappears in cancer invasive cells (60).
Additionally, Placenta-Specific Protein 8 (PLAC-8) is a placental protein implicated in embryo implantation, which is expressed in iEVT on the feto-maternal interface promoting trophoblast invasion and migration (57, 95) nevertheless PLAC-8 is also expressed in cells from different cancers such as lung adenocarcinoma, pancreatic cancer, colorectal cancer, gastrointestinal cancer, and cervical cancer (96–100), where it is involved in malignant tumor progression by regulating cell differentiation (100), proliferation (101), apoptosis (102) and autophagy by mediating autophagosome/autolysosome fusion (103).
In conclusion, for the physiological invasion of iEVT and for the pathological metastasis of cancer cells similar mechanisms are used. However, despite the similarities between them, they show a key difference: while the trophoblast follows an organized pattern of proliferation, differentiation and invasion without metastasizing to new tissues; cancer cells spread through the host tissue with a high proliferation rate, with the final objective of being able to metastasize to other tissues (3, 5).
Vasculogenic Capacity
The vascularization capacity is also a common feature between trophoblast and cancer cells as an abundant blood supply is necessary both for the growth of the tumor nodule, and for the implanting embryo. To date, three processes of vessel growth have been described: vasculogenesis, angiogenesis and vascular mimicry (104, 105). Vasculogenesis is the process of new blood vessel formation from angioblast precursor cells; angiogenesis is the process of growth and development of new capillary blood vessels from pre-existing vessels like new branches; vascular mimicry corresponds to vessel growth from adult cells into a vascular-like phenotype (105, 106).
During the first trimester of pregnancy, vasculogenesis and angiogenesis are consecutive processes. Mesenchymal stem cells differentiate to become hemangiogenic stem cells, then, in a paracrine manner, the CTB induces the formation of the first vessels via induction of VEGF signaling. After that, the existing vessels become longer, a process mediated by VEGF and PLGF (107). In cancer, angiogenesis is crucial for the newly formed tumor nodule, since it provides blood continuously to initiate progression and tumor growth (108). This process involves molecular and cellular interactions between cancerous cells, endothelial cells, and some components of the Extra-Cellular Matrix (ECM), such as matrix proteins (Fibronectin, Laminin, Collagen), receptors (Integrins) and enzymes that degrade the ECM [MMP and Tissue Inhibitor of Metalloproteinase (TIMP)]. Specific proteins such as VEGF and FGF are secreted by cancer cells to stimulate the proliferation of capillary endothelial cells leading to the sprout and branching of them through the ECM (109). Recent evidence suggests that in tumors resistant to different anti-angiogenic drugs, in addition to angiogenesis, other processes that contribute to tumoral vascularization occur, namely vasculogenesis and vascular mimicry (110, 111).
Interestingly, human EVT and invasive cancer cells have similar patterns of integrins expression (Table 1), which allows the EVT to adopt a vascular phenotype capable of invading maternal spiral arterioles, a process similar to what occurs in endothelial cells when they migrate towards the tumor (5). This turnover of endothelial cells to form new vessels requires different angiogenesis regulators that are similar between EVT and cancer cells (3). Among those, VEGF and PLGF promote angiogenesis and are regulated by hypoxia and Fibroblast Growth Factor (FGF) can initiate angiogenesis in both cell types. Conversely, Angiostatin, Fibronectin, and Tissue inhibitor of Metalloproteinases act as angiogenesis inhibitors (109, 112, 113).
Additionally, both cell types are able to directly contribute to their own blood supply by inducing vascular mimicry (88), enhancing gene expression patterns and signaling pathways shared by the two cell types (5). As an example, the Galactose-binding protein Galectin-3, which is known to provide a vascular phenotype, is highly expressed in EVT (114) and is also a key factor for the development of aggressive melanomas (115).
In summary, the process of angiogenesis is essential both in trophoblast and cancer cells. In cancer it drives tumor growth and metastasis, and in pregnancy it allows proper embryo implantation and placentation. However, while trophoblast cells create new blood vessels inducing a controlled process of vasculogenesis, the angiogenesis in cancer is uncontrolled (3).
Immune Evasion
For proper development, trophoblast and cancer cells evade the immune response of the host. During placentation, for the development of the maternal-fetal interface, the maternal decidua basalis, where the maternal immune cells are located, interacts with the fetal derived placental iEVT. Additionally the placenta produces anti-inflammatory Cytokines, TGF-β2, Interleukin (IL)-4 and IL-10, which reduce the deleterious effects of pro-inflammatory cytokines (4). Fas Ligand (Fas-L) expression on trophoblast promotes apoptosis of Fas-expressing lymphocytes of maternal origin, having a role in placental invasion during implantation (116). The position of trophoblast cells in the placenta encasing the embryo produces a barrier between maternal and fetal cells, finally being the placenta the main separation of fetal and maternal blood and lymphatic systems, preventing the immune system of the mother to perceive fetal antigens. During the first trimester the immune cells located in the decidua basalis are Natural Killer (NK, 70%), Macrophages (20-25%) and T Lymphocytes [3-10%, (117–119)]. It has been suggested that the presence of progesterone and TGF-β1 in the decidua promotes the differentiation of these NK into mature Uterine Natural Killers (uNK) (120, 121). uNK cells are more immunomodulatory than cytotoxic, they secrete Growth factors, Angiogenic factors and Cytokines facilitating immune tolerance which suggests uNK play a role in implantation, invasion and vascular remodeling of spiral artery remodeling, regulating EVT invasion (122) by a mechanism that has not yet been totally clarified (49, 123, 124). Additionally, macrophages have been shown as capable of regulating the process of spiral artery remodeling, metabolic regulation of lipids, tissue regeneration, inflammation and fetal antigen recognition (125). Furthermore, they can influence EVT function as they are more abundant at the invasive front and implantation site (126, 127). Despite these studies, their role in placentation, as support of trophoblast cells, has not been fully elucidated. The role of T lymphocytes is also poorly understood, however, it has been described that they could have a role in controlling infections caused by bacteria located at the maternal-fetal interface (119).
In cancer, NK cells are known to contribute to tumor development via secretion of Cytokines (128, 129). Additionally, cancer cells express tumor-associated Macrophages, which can have an inflammatory and immunosuppressive role, being key in tumor progression and metastasis (130). Fas-mediated apoptosis and the expression of Fas-L allow many cancers to attack the immune system (131, 132). Regulatory T cells are implicated in mediating tolerance in cancer and pregnancy; immunophenotypically expressing Cluster of differentiation (CD); CD4, CD25 and Forkead Box P3 (FOXP3) (133). In pregnancy, regulatory T cells are induced by paternal/fetal alloantigens (134), which is crucial for maternal-fetal tolerance. In cancer, regulatory T cells are implicated in impaired antitumor immunity, suppression of effector T lymphocytes proliferation, and increased tumor blood vessel density, suggesting an essential link between immunity and angiogenesis (5). iEVT express Human leukocyte antigen- G (HLA-G) (19), which suppresses cytolytic killing by NK and cytotoxic T cells inducing apoptosis of immune cells (49). HLA-G regulates cytokine production in blood mononuclear cells, reducing stimulatory capacity and impairing the maturation of dendritic cells (5). In tumors, HLA-G promotes immune evasion by interacting with NK cells via Inhibitory receptors and Killer cell Immunoglobulin-like receptor (KIR) (135). This molecule can directly mediate immune tolerance by inhibiting receptors, predominantly Immunoglobulin-like Transcript (ILT) 2 and 4 expressed on immune effectors (136). Finally, HLA-G has been detected in melanoma and solid tumors including cervical cancer, gastrointestinal cancer and breast cancer (137–139).
In conclusion, both trophoblast and cancer cells actively modulate the host immune response by different mechanisms that are induced by similar cells and molecules, finally promoting cell invasion.
Autophagy
Autophagy is a catabolic process highly conserved among eukaryotic organisms, which allows the lysosomal-mediated degradation of cytoplasmic components, thus contributing to cell homeostasis. Three types of autophagy have been described based on the mechanism by which the cargo is delivered to the lysosome: (i) microautophagy, where the cytosolic material is delivered to the lysosome by a direct invagination or protrusion of the lysosomal membrane (140) (ii) chaperone-mediated autophagy, where unfolded soluble proteins containing a specific consensus motif translocate across the lysosomal membrane (141–143), and macroautophagy, herein referred to as autophagy, where the cargo is sequestered in a special double membrane organelle known as autophagosome and then delivered to the lysosome. Briefly, during autophagy the autophagosome fuses to lysosome, forming the autolysosome, where the cargo is degraded (144) (Figure 2). The new metabolites derived from the degradation return then back to the cytosol and will be used for the synthesis of new macromolecules and/or energy production (145). Different autophagy-related (ATG) proteins are required for autophagy to occur, these are organized in protein complexes that are necessary in the different steps of the autophagic process. These can be divided into five stages (initiation, nucleation, elongation, fusion with the lysosome, and cargo degradation) (Figure 2). During “initiation” the unc-51-like kinase 1 (ULK1)/focal adhesion kinase family interacting protein of 200 kDa (FIP200)/ATG13 complex (ULK1 complex) is activated, in response to the metabolic status of the cell (146). Once active, the ULK1 complex translocates to membranous sites, known as omegasomes, where the autophagosome will form (i.e. endoplasmic reticulum and mitochondria contact sites) (147, 148). Then, during the “nucleation”, the isolation membrane of the new autophagosome is generated. This process is mediated by the kinase complex formed by Vacuolar Sorting Protein (VPS) 34 (VPS34), Beclin-1, and VPS15 and Autophagy related 14-like protein (ATGL14), which generates phosphatidylinositol 3-phosphate (PI3P), necessary for the recruitment of the machinery required for the generation of the new autophagosome (146, 149). ATG9-containing vesicles cycle between the omegasome and the Golgi/endosomes, and they contribute to the recruitment of membranes for the nucleation of the phagophore (147, 150, 151). Then, the phagophore extends during the “elongation stage”, a process that is tightly regulated by two ubiquitin-like systems: the microtubule-associated protein 1A/1B-light chain 3 (MAP1LC3A, also known as LC3-I) system and the ATG5–ATG12 system (152) and by the ATG5–ATG12 complex. The ATG5–ATG12 complex then interacts with ATG16L, forming a new complex that works like an E3 enzyme, assisting the incorporation of LC3-II into the membrane of the phagophore (153). In parallel with the elongation the autophagic cargo is selected. Proteins targeted for autophagy are labeled with the receptor p62/Sequestosome-1 (p62/SQSTM1), which interacts with LC3 through an LC3 interacting region (LIR) (154, 155). Following elongation, the elongated phagophore is finally closed forming the autophagosome. This step is completed by a membrane abscission process mediated by the endosomal-sorting complex required for transport (ESCRT) (156, 157). Upon closure, the nascent autophagosome dissociates from the assembly site and undergoes maturation (158). The mature autophagosome then fuses with the lysosome generating autolysosomes (159), a process mediated by Rab GTPases, membrane-tethering complexes and soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) (160). The inner membrane of the autolysosome breaks down and the process of autophagosomal cargo degradation begins (161). The degradation products are recycled and turn back to the cytosol for being reused (162, 163) (Figure 2).
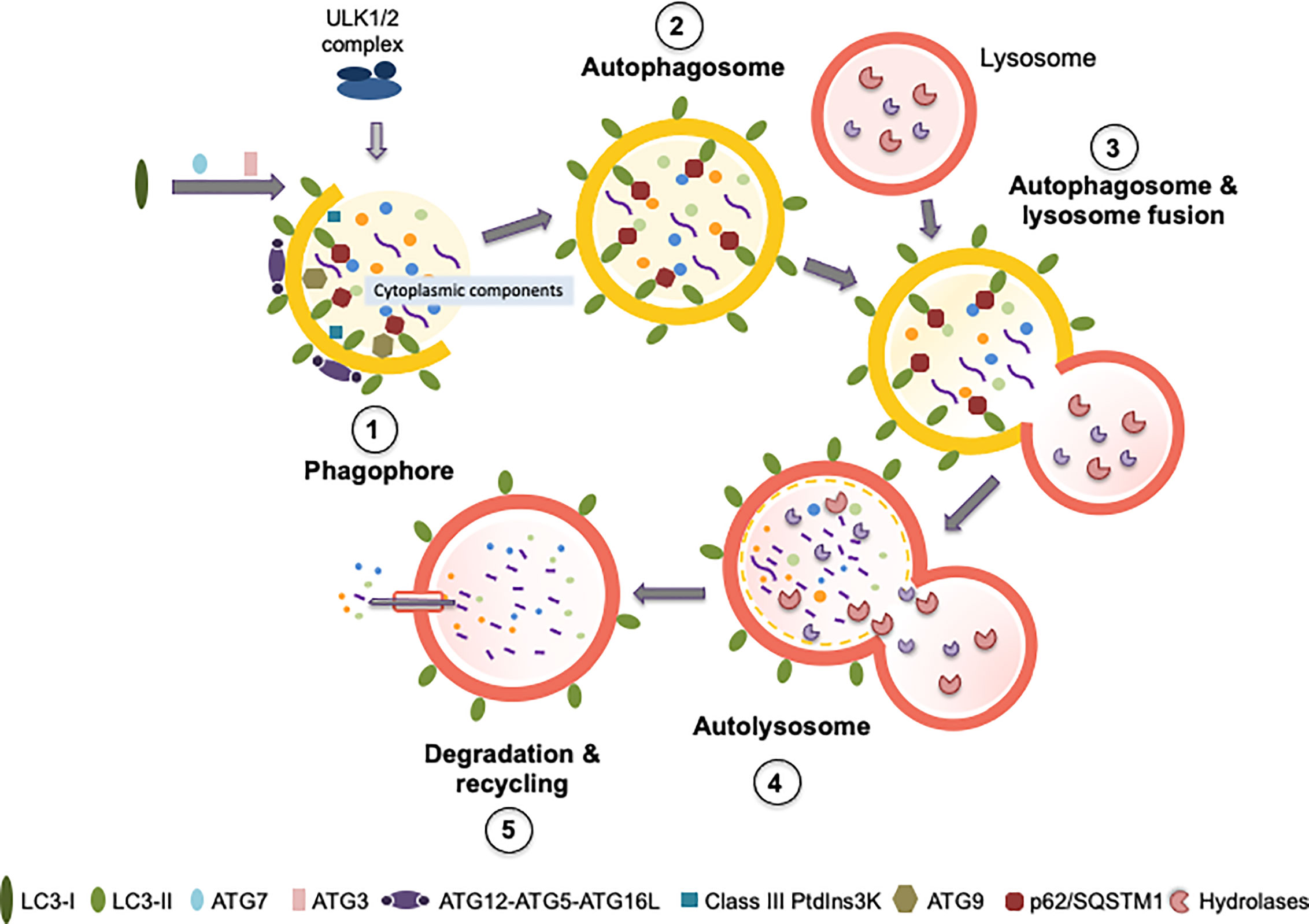
Figure 2 Autophagy process and principal proteins involved in the different steps. The figure shows the principal proteins required for autophagy process, the different steps of the process are described in the section “Autophagy” (initiation, nucleation, elongation, fusion with the lysosome, and cargo degradation and recycling). Figure (1) corresponds to phagophore formation that includes initiation and nucleation. (2) Autophagosome maturation includes the elongation process. (3) Autophagosome and lysosome fusion. (4) Represent the structure of the autolysosome, and (5) corresponds to degradation and recycling. In each step are indicated the main proteins required: ULK1/2 complex, LC3 I, LC3II, ATG7, ATG3, ATG12-ATG5-ATG16L, Class III Ptdlns3K, ATG9 and p62/SQSTM1. The yellow semi-circumferences and circumferences correspond to the phagosome membrane. See the main text for further details.
Autophagy is a complex and highly regulated process that under stress conditions such as hypoxia, low glucose concentration and oxidative stress is triggered to promote cell survival or leads to cell death. Physiologically autophagy maintains cellular and energy homeostasis, cooperates with the immune system to promote adaptation, and represents a quality control system for proteins and organelles (164). Impaired autophagy contributes to the development of neurodegenerative (165), infectious (166) and metabolic diseases (167, 168), due to the accumulation of abnormal and damaged proteins and/or organelles, forming intracellular aggregates that induce cellular stress, finally promoting cell death.
During placentation it has been reported that autophagy could be relevant for different processes required for a proper development of the placenta; however, how this occurs is still under investigation (7). On the other hand, in the context of cancer, autophagy has a dual role, where it could be tumor-suppressive or tumor-promoting depending on the stage of cancer development and the type of cancer considered (10).
Role of Autophagy in Immune Evasion and Vascular Remodeling: Differences Between Placentation and Cancer
As previously described, the processes of placentation and tumor development share similarities and autophagy activation has been described in both (6, 7). The role of autophagy in cancer has been widely explored, however, as previously mentioned, its role has not completely been elucidated (169). On the other hand, how modulation of autophagy affects trophoblast function is still largely unknown.
Role of Autophagy in Placental and Cancer Immune Evasion
The role of autophagy in the placentation process remains unclear, and its contribution to immune evasion is still unknown (170). It has been described that autophagy is highly activated in decidualized endometrium of early pregnancy, which increases NK cell adhesion and retention in the decidua. Also, when autophagy is inhibited, decidual NK (dNK) cell residence is decreased, contributing to spontaneous abortion (171). Tan et al. described that autophagy levels are highly reduced in cases of recurrent miscarriage. Indeed, suppression of autophagy in an in vitro model of trophoblast cells enhances the cytotoxicity activity of dNK, impairing trophoblast invasion, finally causing abortion (172).
On the other hand, autophagy has been describes as an important regulator of cancer immunity in the tumor microenvironment; however, the exact mechanisms involved remain unclear (173, 174). The tumor microenvironment contains different factors that promote autophagy, such as hypoxia or inflammation (166, 175). Remarkably, it has been described that autophagic activation correlates with immune evasion (176, 177). Conversely, inhibition of autophagy associates with NK-dependent immune responses. In breast cancers, in tumors presenting hypoxia, blocking autophagy restores NK-mediated lysis in vitro, facilitating breast tumor elimination by NK cells in mice (178). Inhibition of autophagy also reduces NK cell-mediated cytotoxicity in melanoma (175), non-small cell lung cancer (179) and liver cancer (180). In contrast, the role of autophagy is dual in the response to immune cell recognition, being a suppressor or inductor of tumorigenesis depending on the specific context (181). Altogether these data suggest that autophagy actively participates and regulates the immune evasion of dNK in placental development and NK activity in cancer cells. However, the mechanism involved in both phenomena remains to be elucidated, a crucial aspect that needs to be studied for the development of immunotherapy in each field.
Role of Autophagy in Placental Vascular Remodeling
As indicated, trophoblast invasion and vascular remodeling allows the replacement of the endothelial layer of the maternal spiral arteries, which is essential for proper placental perfusion and adequate fetal growth. It has been described that activation of autophagy occurs in human placentas from normal pregnancies at weeks 8 to 12 of gestation, as indicated by LC3 and Beclin-1 protein in CTB and STB cells (182) (Table 2). Moreover, autophagosomes have been identified in human placentas throughout gestation from early (8 weeks) (189, 194) to term pregnancies (39 weeks) (189, 194, 261).

Table 2 Changes in protein involved in the autophagic process described in human placental tissues and trophoblast cell lines.
The key role of autophagy in implantation was demonstrated by studies in ATG5-deficient mouse oocytes, where pre-implantation cannot occur correctly. Indeed, autophagy increases in the oocytes after fertilization, and it is necessary for pre-implantation development, which is essential to allow the differentiation from zygote to blastocyst in mammals (262). In different mouse models it has been shown that proteins of the LC3 family are expressed in the labyrinth zone and in the decidua basalis, which suggests a possible role in the placentation process (263).
In vitro assays in the trophoblast cell line HTR8/SVneo (i.e., a first-trimester human trophoblast cell line) showed increased LC3 lipidation and LC3 puncta in cells cultured in 2% of oxygen, which mimics the physiological O2 concentration in the early pregnancy period (186, 189, 192). In the same cell line, higher LC3 and Beclin-1 expression was determined in conditions of enhanced oxidative stress (264). Additionally, using a model of autophagy-deficient EVT cells (cells expressing a ATG4B- negative mutant), the relevance of autophagy in the trophoblast in the process of invasion was shown, as the process was impaired in autophagy-deficient cells (189). Consistently, in a mouse model where the ATG7 gene was deleted only in trophoblast (not in fetuses), the placentas were smaller than in wild type, due to reduced trophoblast invasion and low vascular remodeling. Remarkably, this result needs to be compared with those described in cancer cells lacking ATG7, which is described in the next section. Altogether, these studies demonstrate that autophagy plays a key role in trophoblast function, especially in invasion and vascular remodeling during placentation (Table 2). Despite this, how modulation of autophagy affects trophoblast function in pathological conditions has not been elucidated.
Importantly, even if previous research indicates a positive correlation between autophagy and cell invasion and vice-versa (192, 264), the role of the whole autophagic process, intended as autophagic flux, defined as the whole process from autophagosome formation up to its fusion with lysosome and cargo degradation (7), in the development of pregnancy-associated diseases such as preeclampsia (PE), gestational diabetes, or fetal growth (FGR) is still controversial. Indeed, for example, in homogenized tissue from PE placenta and in trophoblast cells obtained from PE placentas has been described that LC3 and Beclin-1 are increased (197, 215), suggesting that increased markers of autophagy correlate with a poor placentation process (187). However, another study in PE placenta and in the cell line JEG-3 showed an increase in LC3 without changes in Beclin-1 (265). Furthermore, additional work showed, in homogenized tissue from PE placentas, a decrease in LC3 and Beclin-1 (206) and an increase in Beclin-1 and p62/SQSTM1 (219). These controversial results could be due to different factors: (i) the placenta is a complex organ, with different cell types that perform different functions, so it is not appropriate to use placenta homogenates and evaluate autophagy in these samples, as the levels of autophagy can be different in the different cell types. (ii) The time at which the analysis is performed is important. Indeed, as reported, it has been described that autophagy plays different roles in embryogenesis and implantation, while its role in the later stages of pregnancy is still unknown (8). (iii) It is key to evaluate a set of autophagic markers to study the autophagic flux to reach a conclusion (at least LC3 and an autophagic receptor such as p62/SQSTM1), unfortunately, some of the studies only evaluate a single autophagic protein, which is not sufficient to clearly indicate what is happening in autophagy but only suggest that the condition reported might affect this cellular process (Table 2). Thus, the available information related with the role of autophagy in placentation in terms of specific cells involved, cellular processes affected beyond migration of invasion (i.e., processes of differentiation to endothelial phenotype, angiogenesis, vasculogenesis or immune control) and the modulation of autophagy according to gestational age, as well as the complete autophagic flux in these different processes still needs to be elucidated.
Role of Autophagy in Vascular Remodeling in Cancer
As mentioned, the role of autophagy in tumor development is controversial and dependent of the tumor characteristics and stage of tumor development (266). Briefly, it has been suggested that autophagy could promote aggressive characteristics of cancer cells such as increased cellular invasion (11, 12), but it also represents a barrier for cancer proliferation (13–15).
In cancer cells, the inhibition of autophagy results in impaired metabolism proliferation, survival, and spontaneous tumor malignancy depending not only on the tumor type but also of its temporal development (267). This has been demonstrated in different types of cancers using genetically engineered mouse models with ablation of ATGs and consequently autophagy. For instance, in pancreatic ductal adenocarcinoma, loss of ATG5 increases tumor initiation but avoids invasive cancer progression (268). Consistently, in prostate cancer, lack of ATG7 delayed tumor cell proliferation (269) and in lung cancer driven by oncogenic Kras, the deletion of ATG7 reduces cell proliferation and tumor weight compared with mice with intact ATG7 (270). Conversely, Rao et al. show that ATG5 deletion accelerates early oncogenesis, increasing the number of tumor foci and the transition from hyperplasia to adenomas; however as cancer develops, lack of ATG5 reduces the progression from adenoma to adenocarcinoma, resulting in a decrease of tumors mass and enhanced lifespan in mice (271). Altogether, these studies demonstrate that autophagy plays a crucial role in cancer cells.
According to the stage, during the early phases of solid tumor formation, autophagy plays an anti-tumorigenic (272) effect because it limits the production of DNA damaging agents [i.e. Reactive Oxygen Species (ROS)], it promotes the elimination of oncogenic proteins, and stimulates the induction of the immune response in response to cellular stress (273). Additionally, it has been shown that autophagy could promote senescence in tumor cells in response to oncogenic stress, which results in decreased tumor growth (274, 275). On the other hand, it has been described that during tumor progression, autophagy increases the tolerance to stressful conditions such as metabolic changes and hypoxia within the tumor microenvironment, leading to enhanced tumor cell survival and playing a pro-tumorigenic role (276, 277). Autophagy can also increase metastasis, supporting tumor growth, interacting with pathways involved in cell motility and invasion (6), including the promotion of Focal Adhesion (FA) turnover, which is a component of the cell migration machinery, being Paxillin the essential FA protein degraded by autophagy (278) and ECM proteins. For example, in pancreatic ductal adenocarcinomas hypoxia induces autophagy resulting in degradation of Lumican, an extracellular matrix protein highly upregulated in different cancers (279). Autophagy is also enhanced upon oncogenic RAS activation (280, 281) and is required for the production of multiple secreted factors, which include IL-6 and MMP2 in tumors bearing RAS mutations, facilitating cancer cell invasion (282). All the described data indicate that cancer cell migration could be molecularly regulated by autophagy and vice versa, providing metabolites and nutrients in stress conditions to the different cell types that form the tumor microenvironment (169).
Thus, autophagy has a dual role in cancer since in tumor initiation limits DNA damage agents such as ROS and increases tumor cell senescence leading to an anti-tumorigenic environment, preventing tumor promotion. However, in established tumors autophagy provides the necessary conditions for tumors to growth, regulating the invasion and migration process enhancing tumor cell survival increasing resistance to stressful conditions (176). Something similar occurs in trophoblast cells, where it has been suggested that autophagy regulates invasion, migration and vascular remodeling of trophoblasts, allowing the optimal development of the placenta (7). One difference between both processes is that autophagy has a role in the promotion of the placentation process from fertilization, whereas, as mentioned above, at the beginning of tumor development autophagy exerts anticarcinogenic functions protecting the host tissue, but as the tumor progresses, autophagy supports tumor metastasis, enhancing tumor cell survival by increasing the resistance to stressful conditions (283). Finally, the role of autophagy in cancer cells and trophoblast derived cells appears quite similar, since it provides the conditions to carry out cellular functions depending on the timing or stage in cancer, promoting or stopping tumor growth, while in the trophoblast autophagy could favor optimal placentation. Nevertheless, the precise role of autophagy in modulating the described cellular processes involved in vascular remodeling in cancer progression or placentation needs to be fully studied.
Conclusion
In conclusion, the physiological placentation process of trophoblast and the pathological metastasis of cancer cells share similar mechanisms to proliferate, migrate, and invade both trophoblast and cancer cells, modulating host immune response. However, the main difference is that trophoblast follows an organized pattern without metastasizing new tissues. On the other hand, another shared process is autophagy, which is required for invasion of trophoblast, and it has been shown in cancer has a dual role being a tumor promoter and inhibitor, depending on the stage and tumor considered. Nevertheless, the precise role of autophagy in cancer progression or placentation needs to be thoroughly studied. These studies could give a new insight in cancer biology by evaluating the similarities with trophoblast cells and the highly regulated behavior they have in placentation.
Author Contributions
LC and JG: review of the literature. AL and EM: preparation of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT 1180935 JG, 1200499 EM, 1190250 AL). LC holds ANID (Chile) fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Turco MY, Moffett A. Development of the human placenta. Development (2019) 146(22):dev163428. doi: 10.1242/dev.163428
2. Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum Reprod Update (2007) 13(2):121–41. doi: 10.1093/humupd/dml048
3. West RC, Bouma GJ, Winger QA. Shifting perspectives from "oncogenic" to oncofetal proteins; how these factors drive placental development. Reprod Biol Endocrinol (2018) 16(1):1–12. doi: 10.1186/s12958-018-0421-3
4. Soundararajan R, Rao AJ. Trophoblast ‘pseudo-tumorigenesis’: Significance and contributory factors. Reprod Biol Endocrinol (2004) 2:15. doi: 10.1186/1477-7827-2-15
5. Holtan SG, Creedon DJ, Haluska P, Markovic SN. Cancer and pregnancy: Parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clin Proc (2009) 84(11):985–1000. doi: 10.4065/84.11.985
6. Mowers EE, Sharifi MN, Macleod KF. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J (2018) 285(10):1751–66. doi: 10.1111/febs.14388
7. Nakashima A, Tsuda S, Kusabiraki T, Aoki A, Ushijima A, Shima T, et al. Current understanding of autophagy in pregnancy. Int J Mol Sci (2019) 20(9):1–13. doi: 10.3390/ijms20092342
8. Nakashima A, Aoki A, Saito S. The Role of Autophagy in Maintaining Pregnancy. Autophagy Curr Trends Cell Physiol Pathol (2016) 329. doi: 10.5772/63475
9. Grandér D, Panaretakis T. Autophagy: cancer therapy’s friend or foe? Future Med Chem (2010) 2(2):285–97. doi: 10.4155/fmc.09.155
10. Saha S, Panigrahi DP, Patil S, Bhutia SK. Autophagy in health and disease: A comprehensive review. Biomed Pharmacother (2018) 104:485–95. doi: 10.1016/j.biopha.2018.05.007
11. Gao L, Wang Q, Ren W, Zheng J, Li S, Dou Z, et al. The RBP1–CKAP4 axis activates oncogenic autophagy and promotes cancer progression in oral squamous cell carcinoma. Cell Death Dis (2020) 11(6):1–16. doi: 10.1038/s41419-020-2693-8
12. Li X, Gao L, Zheng L, Shi J, Ma J. BMP4-mediated autophagy is involved in the metastasis of hepatocellular carcinoma via JNK/Beclin1 signaling. Am J Transl Res (2020) 12(6):3068–77.
13. Sudsaward S, Khunchai S, Thepmalee C, Othman A, Limjindaporn T, Yenchitsomanus PT, et al. Endoplasmic reticulum stress, unfolded protein response and autophagy contribute to resistance to glucocorticoid treatment in human acute lymphoblastic leukaemia cells. Int J Oncol (2020) 57(3):835–44. doi: 10.3892/ijo.2020.5089
14. Wu L, Wang Z, Li X, He X, Han Y, Chen Y, et al. Autophagy mediates bronchial cell malignant transformation induced by chronic arsenic exposure via MEK/ERK1/2 pathway. Toxicol Lett (2020) 332:155–63. doi: 10.1016/j.toxlet.2020.06.006
15. Jiao YJ, Dan ZD, Cui B, Zhang C, Wei TF, Chang S, et al. Disruption of the EGFR-SQSTM1 interaction by a stapled peptide suppresses lung cancer via activating autophagy and inhibiting EGFR signaling. Cancer Lett (2020) 474:23–35. doi: 10.1016/j.canlet.2020.01.004
16. Cross JC, Werb Z, Fisher SJ. Implantation and the placenta: Key pieces of the development puzzle. Science (1994) 266(5190):1508–18. doi: 10.1126/science.7985020
17. Gude NM, Roberts CT, Kalionis B, King RG. Growth and function of the normal human placenta. Thromb Res (2004) 114(5–6):397–407. doi: 10.1016/j.thromres.2004.06.038
18. Wang Y, Zhao S. Vascular biology of the placenta. In Colloquium Series on Integrated Systems Physiology: From Molecule to Function. Morgan & Claypool Life Sciences (2010) 2(1):1–98. doi: 10.4199/C00016ED1V01Y201008ISP009
19. Ji L, Brkić J, Liu M, Fu G, Peng C, Wang YL. Placental trophoblast cell differentiation: Physiological regulation and pathological relevance to preeclampsia. Mol Aspects Med (2013) 34(5):981–1023. doi: 10.1016/j.mam.2012.12.008
20. Huppertz B. Trophoblast Invasion: Remodelling of Spiral Arteries and Beyond. In: S Saito, editor. Preeclampsia. Comprehensive Gynecology and Obstetrics. Springer, Singapore (2018). doi: 10.1007/978-981-10-5891-2_3
21. Frank H-G. Placental Development. In: Fetal and Neonatal Physiology. Elsevier: WB Saunders (2017). p. 101–13. doi: 10.1016/B978-0-323-35214-7.00010-X
22. Blaschitz A, Weiss U, Dohr G, Desoye G. Antibody reaction patterns in first trimester placenta: Implications for trophoblast isolation and purity screening. Placenta (2000) 21(7):733–41. doi: 10.1053/plac.2000.0559
23. Shorter SC, Starkey PM, Ferry BL, Clover LM, Sargent IL, Redman CWG. Antigenic heterogeneity of human cytotrophoblast and evidence for the transient expression of MHC class I antigens distinct from HLA-G. Placenta (1993) 14(5):571–82. doi: 10.1016/S0143-4004(05)80210-3
24. Song H, Ci H, Xu J, Xu Z, Zhang Y, Wang Y, et al. Vasculogenic mimicry and expression of slug and vimentin correlate with metastasis and prognosis in non-small cell lung cancer. Int J Clin Exp Pathol (2018) 11(5):2749–58.
25. Lin H, Hong Y, Huang B, Liu X, Zheng J, Qiu S. Vimentin Overexpressions Induced by Cell Hypoxia Promote Vasculogenic Mimicry by Renal Cell Carcinoma Cells. BioMed Res Int (2019) 2019:7259691. doi: 10.1155/2019/7259691
26. Hernández de la Cruz ON, López-González JS, García-Vázquez R, Salinas-Vera YM, Muñiz-Lino MA, Aguilar-Cazares D, et al. Regulation Networks Driving Vasculogenic Mimicry in Solid Tumors. Front Oncol (2020) 9:1419. doi: 10.3389/fonc.2019.01419
27. Damsky CH, Fitzgerald ML, Fisher SJ. Distribution patterns of extracellular matrix components and adhesion receptors are intricately modulated during first trimester cytotrophoblast differentiation along the invasive pathway, in vivo. J Clin Invest (1992) 89(1):210–22. doi: 10.1172/JCI115565
28. Iyengar B, Singh AV. Embryonic vasculogenesis in nodular melanomas and tumour differentiation. Pathol Oncol Res (2011) 17(3):569–77. doi: 10.1007/s12253-010-9350-y
29. Liu Y, Li F, Yang YT, Xu XD, Chen JS, Chen TL, et al. IGFBP2 promotes vasculogenic mimicry formation via regulating CD144 and MMP2 expression in glioma. Oncogene (2019) 38(11):1815–31. doi: 10.1038/s41388-018-0525-4
30. Zhou Y, Fisher SJ, Janatpour M, Genbacev O, Dejana E, Wheelock M, et al. Human cytotrophoblasts adopt a vascular phenotype as they differentiate: A strategy for successful endovascular invasion? J Clin Invest (1997) 99(9):2139–51. doi: 10.1172/JCI119387
31. Tímár J, Tóvári J, Rásó E, Mészáros L, Bereczky B, Lapis K. Platelet-mimicry of cancer cells: Epiphenomenon with clinical significance. Oncology (2005) 69(3):185–201. doi: 10.1159/000088069
32. Damsky CH, Fisher SJ. Trophoblast pseudo-vasculogenesis: Faking it with endothelial adhesion receptors. Curr Opin Cell Biol (1998) 10(5):660–6. doi: 10.1016/S0955-0674(98)80043-4
33. Hess AR, Margaryan NV, Seftor EA, Hendrix MJC. Deciphering the signaling events that promote melanoma tumor cell vasculogenic mimicry and their link to embryonic vasculogenesis: Role of the Eph receptors. Dev Dyn (2007) 236(12):3283–96. doi: 10.1002/dvdy.21190
34. Wang JY, Sun T, Zhao XL, Zhang SW, Zhang DF, Gu Q, et al. Functional significance of VEGF-a in human ovarian carcinoma: Role in vasculogenic mimicry. Cancer Biol Ther (2008) 7(5):758–66. doi: 10.4161/cbt.7.5.5765
35. Burrows TD, King A, Loke YW. Expression of adhesion molecules by endovascular trophoblast and decidual endothelial cells: Implications for vascular invasion during implantation. Placenta (1994) 15(1):21–33. doi: 10.1016/S0143-4004(05)80233-4
36. Huppertz B, Kertschanska S, Demir AY, Frank HG, Kaufmann P. Immunohistochemistry of matrix metalloproteinases (MMP), their substrates, and their inhibitors (TIMP) during trophoblast invasion in the human placenta. Cell Tissue Res (1997) 291(1):133–48. doi: 10.1007/s004410050987
37. Seftor REB, Seftor EA, Koshikawa N, Meltzer PS, Gardner LMG, Bilban M, et al. Cooperative Interactions of Laminin 5 γ2 Chain, Matrix Metalloproteinase-2, and Membrane Type-1-Matrix/Metalloproteinase Are Required for Mimicry of Embryonic Vasculogenesis by Aggressive Melanoma. Cancer Res (2001) 61(17):6322–7.
38. Hurskainen T, Seiki M, Apte SS, Syrjäkallio-Ylitalo M, Sorsa T, Oikarinen A, et al. Production of membrane-type matrix metalloproteinase-1 (MT-MMP-1) in early human placenta. A possible role in placental implantation? J Histochem Cytochem (1998) 46(2):221–9. doi: 10.1177/002215549804600211
39. Xu P, Wang Y, Zhu S, Luo S, Piao Y, Zhuang L. Expression of Matrix Metalloproteinase-2, -9, and -14, Tissue Inhibitors of Metalloproteinase-1, and Matrix Proteins in Human Placenta During the First Trimester1. Biol Reprod (2000) 62(4):988–94. doi: 10.1095/biolreprod62.4.988
40. Hiden U, Ghaffari-Tabrizi N, Gauster M, Tam-Amersdorfer C, Cetin I, Dieber-Rotheneder M, et al. Membrane-type matrix metalloproteinase 1 regulates trophoblast functions and is reduced in fetal growth restriction. Am J Pathol (2013) 182(5):1563–71. doi: 10.1016/j.ajpath.2013.01.011
41. Handschuh K, Guibourdenche J, Tsatsaris V, Guesnon M, Laurendeau I, Evain-Brion D, et al. Human Chorionic Gonadotropin Expression in Human Trophoblasts from Early Placenta: Comparative Study Between Villous and Extravillous Trophoblastic Cells. Placenta (2007) 28(2–3):175–84. doi: 10.1016/j.placenta.2006.01.019
42. Selick CE, Horowitz GM, Gratch M, Scott RT, Navot D, Hofmann GE. Immunohistochemical localization of transforming growth factor-beta in human implantation sites. J Clin Endocrinol Metab (1994) 78(3):592–6. doi: 10.1210/jcem.78.3.7510303
43. Pietro L, Daher S, Rudge MVC, Calderon IMP, Damasceno DC, Sinzato YK, et al. Vascular endothelial growth factor (VEGF) and VEGF-receptor expression in placenta of hyperglycemic pregnant women. Placenta (2010) 31(9):770–80. doi: 10.1016/j.placenta.2010.07.003
44. St-Jacques S, Forte M, Lye SJ, Letarte M. Localization of endoglin, a transforming growth factor-β binding protein, and of CD44 and integrins in placenta during the first trimester of pregnancy. Biol Reprod (1994) 51(3):405–13. doi: 10.1095/biolreprod51.3.405
45. Kovats S, Main EK, Librach C, Stubblebine M, Fisher SJ, Demars R. A class I antigen, HLA-G, expressed in human trophoblasts. Sci (80- ) (1990) 248(4952):220–3. doi: 10.1126/science.2326636
46. Moser G, Windsperger K, Pollheimer J, Chuva de Sousa S, Huppertz B. Human trophoblast invasion: new and unexpected routes and functions. Histochem Cell Biol (2018) 150:361–70. doi: 10.1007/s00418-018-1699-0
47. Weiss G, Sundl M, Glasner A, Huppertz B, Moser G. The trophoblast plug during early pregnancy: a deeper insight. Histochem Cell Biol (2016) 146(6):749–56. doi: 10.1007/s00418-016-1474-z
48. James JL, Saghian R, Perwick R, Clark AR. Trophoblast plugs: Impact on uteroplacental haemodynamics and spiral artery remodelling. Hum Reprod (2018) 33(8):1430–41. doi: 10.1093/humrep/dey225
49. Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal-fetal interface. Sci Immunol (2019) 4(31):eaat6114. doi: 10.1126/sciimmunol.aat6114
50. Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress: A possible factor in human early pregnancy failure. Am J Pathol (2000) 157(6):2111–22. doi: 10.1016/S0002-9440(10)64849-3
51. Huppertz B, Weiss G, Moser G. Trophoblast invasion and oxygenation of the placenta: Measurements versus presumptions. J Reprod Immunol (2014) 101–102(1):74–9. doi: 10.1016/j.jri.2013.04.003
52. Soares MJ, Iqbal K, Kozai K. Hypoxia and Placental Development. Birth Defects Res (2017) 109(17):1309–29. doi: 10.1002/bdr2.1135
53. Sharma S, Godbole G, Modi D. Decidual Control of Trophoblast Invasion. Am J Reprod Immunol (2016) 75(3):341–50. doi: 10.1111/aji.12466
54. Weber M, Göhner C, San Martin S, Vattai A, Hutter S, Parraga M, et al. Unique trophoblast stem cell- and pluripotency marker staining patterns depending on gestational age and placenta-associated pregnancy complications. Cell Adhes Migr (2016) 10(1–2):56–65. doi: 10.1080/19336918.2016.1142035
55. Mayhew TM. Villous trophoblast of human placenta: A coherent view of its turnover, repair and contributions to villous development and maturation. Histol Histopathol (2001) 16(4):1213–24. doi: 10.14670/HH-16.1213
56. Wong FTM, Lin C, Cox BJ. Cellular systems biology identifies dynamic trophoblast populations in early human placentas. Placenta (2019) 76:10–8. doi: 10.1016/j.placenta.2018.12.012
57. Chang WL, Liu YW, Dang YL, Jiang XX, Xu H, Huang X, et al. Plac8, a new marker for human interstitial extravillous trophoblast cells, promotes their invasion and migration. Dev (2018) 145(2):dev148932. doi: 10.1242/dev.148932
58. Founds SA, Fallert-Junecko B, Reinhart TA, Parks WT. LAIR2-expressing extravillous trophoblasts associate with maternal spiral arterioles undergoing physiologic conversion. Placenta (2013) 34(3):248–55. doi: 10.1016/j.placenta.2012.09.017
59. Huppertz B. The anatomy of the normal placenta. J Clin Pathol (2008) 61(12):1296–302. doi: 10.1136/jcp.2008.055277
60. Jansen CHJR, Kastelein AW, Kleinrouweler CE, Van Leeuwen E, De Jong KH, Pajkrt E, et al. Development of placental abnormalities in location and anatomy. Acta Obstetricia Gynecol Scandinavica (2020) 99(8):983–93. doi: 10.1111/aogs.13834
62. Rizov M, Andreeva P, Dimova I. Molecular regulation and role of angiogenesis in reproduction. Taiwanese J Obstetrics Gynecol (2017) 56(2):127–32. doi: 10.1016/j.tjog.2016.06.019
63. Maruo T, Mochizuki M. Immunohistochemical localization of epidermal growth factor receptor and myc oncogene product in human placenta: Implication for trophoblast proliferation and differentiation. Am J Obstet Gynecol (1987) 156(3):721–7. doi: 10.1016/0002-9378(87)90086-X
64. Fulop V, Mok SC, Genest DR, Szigetvari I, Cseh I, Berkowitz RS. c-myc, c-erbb-2, c-fms and bcl-2 Oncoproteins: Expression in normal placenta, partial and complete mole, and choriocarcinoma. J Reprod Med Obstet Gynecol (1998) 43(2):101–10.
65. Jokhi PP, King A, Loke YW. Reciprocal expression of epidermal growth factor receptor (EGF-R) and c-erbB2 by non-invasive and invasive human trophoblast populations. Cytokine (1994) 6(4):433–42. doi: 10.1016/1043-4666(94)90068-X
66. Tarrade A, Lai Kuen R, Malassiné A, Tricottet V, Blain P, Vidaud M, et al. Characterization of human villous and extravillous trophoblasts isolated from first trimester placenta. Lab Investig (2001) 81(9):1199–211. doi: 10.1038/labinvest.3780334
67. Wang Y, Cao Y, Sun Y. Expression of c-erbB2 in gestational trophoblastic disease and its clinical significance. J Huazhong Univ Sci Technol - Med Sci (2002) 22(2):123–5. doi: 10.1007/bf02857672
68. Rahat B, Hamid A, Najar R, Bagga R, Kaur J. Epigenetic mechanisms regulate placental c-myc and hTERT in normal and pathological pregnancies; c-myc as a novel fetal DNA epigenetic marker for pre-eclampsia. Mol Hum Reprod (2014) 20(10):1026–40. doi: 10.1093/MOLEHR/GAU053
69. Xinyuan C, Jiaqin S, Caixia L, Qin Z, Xuesong Y, Shuai L, et al. Progesterone promotes embryo adhesion by upregulating c-Fos/c-Jun transcription factor-mediated poFUT1 expression†. Biol Reprod (2019) 101(4):675–85. doi: 10.1093/BIOLRE/IOZ110
70. Peng B, Zhu H, Ma L, Wang YL, Klausen C, Leung PCK. AP-1 transcription factors c-FOS and c-JUN mediate GnRH-induced cadherin-11 expression and trophoblast cell invasion. Endocrinology (2015) 156(6):2269–77. doi: 10.1210/en.2014-1871
71. Renaud SJ, Kubota K, Rumi MAK, Soares MJ. The FOS transcription factor family differentially controls trophoblast migration and invasion. J Biol Chem (2014) 289(8):5025–39. doi: 10.1074/jbc.M113.523746
72. Goustin AS, Betsholtz C, Pfeifer-Ohlsson S, Persson H, Rydnert J, Bywater M, et al. Coexpression of the sis and myc proto-oncogenes in developing human placenta suggests autocrine control of trophoblast growth. Cell (1985) 41(1):301–12. doi: 10.1016/0092-8674(85)90083-2
73. Sarkar S, Kacinski BM, Kohorn EI, Merino MJ, Carter D, Blakemore KJ. Demonstration of myc and ras oncogene expression by hybridization in situ in hydatidiform mole and in the BeWo choriocarcinoma cell line. Am J Obstet Gynecol (1986) 154(2):390–3. doi: 10.1016/0002-9378(86)90677-0
74. Liong S, Barker G, Lappas M. Placental ras regulates inflammation associated with maternal obesity. Mediators Inflamm (2018) 2018:3645386. doi: 10.1155/2018/3645386
75. Blagoev KB. Cell proliferation in the presence of telomerase. PloS One (2009) 4(2):e4622. doi: 10.1371/journal.pone.0004622
76. Kyo S, Takakura M, Tanaka M, Kanaya T, Sagawa T, Kohama T, et al. Expression of telomerase activity in human chorion. Biochem Biophys Res Commun (1997) 241(2):498–503. doi: 10.1006/bbrc.1997.7767
77. Chen RJ, Chu CT, Huang SC, Chow SN, Hsieh CY. Telomerase activity in gestational trophoblastic disease and placental tissue from early and late human pregnancies. Hum Reprod (2002) 17(2):463–8. doi: 10.1093/humrep/17.2.463
78. Fragkiadaki P, Tsoukalas D, Fragkiadoulaki I, Psycharakis C, Nikitovic D, Spandidos DA, et al. Telomerase activity in pregnancy complications (Review). Mol Med Rep (2016) 14(1):16–21. doi: 10.3892/mmr.2016.5231
79. Rama S, Suresh Y, Rao AJ. Regulation of telomerase during human placental differentiation: A role for TGFβ1. Mol Cell Endocrinol (2001) 182(2):233–48. doi: 10.1016/S0303-7207(01)00550-0
80. Trybek T, Kowalik A, Góźdź S, Kowalska A. Telomeres and telomerase in oncogenesis (review). Oncol Lett (2020) 20(2):1015–27. doi: 10.3892/ol.2020.11659
81. Warrier NM, Agarwal P, Kumar P. Emerging Importance of Survivin in Stem Cells and Cancer: the Development of New Cancer Therapeutics. Stem Cell Rev Rep (2020) 16(5):828–52. doi: 10.1007/s12015-020-09995-4
82. Lehner R, Bobak J, Kim NW, Shroyer AL, Shroyer KR. Localization of telomerase hTERT protein and survivin in placenta: Relation to placental development and hydatidiform mole. Obstet Gynecol (2001) 97(6):965–70. doi: 10.1016/S0029-7844(01)01131-0
83. Muschol-Steinmetz C, Friemel A, Kreis NN, Reinhard J, Yuan J. and Louwen, F., “Function of Survivin in Trophoblastic Cells of the Placenta. PloS One (2013) 8(9):e73337. doi: 10.1371/journal.pone.0073337
84. Burton GJ, Jauniaux E, Murray AJ. Oxygen and placental development; parallels and differences with tumour biology. Placenta (2017) 56:14–8. doi: 10.1016/j.placenta.2017.01.130
85. Zhuyan J, Chen M, Zhu T, Bao X, Zhen T, Xing K, et al. Critical steps to tumor metastasis: Alterations of tumor microenvironment and extracellular matrix in the formation of pre-metastatic and metastatic niche. Cell Biosci (2020) 10(1):1–9. doi: 10.1186/s13578-020-00453-9
86. Corrado C, Fontana S. Hypoxia and HIF signaling: One axis with divergent effects. Int J Mol Sci (2020) 21(6):1–17. doi: 10.3390/ijms21165611
87. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A (1995) 92(12):5510–4. doi: 10.1073/pnas.92.12.5510
88. Macklin PS, McAuliffe J, Pugh CW. and Yamamoto, A., “Hypoxia and HIF pathway in cancer and the placenta. Placenta (2017) 56:8–13. doi: 10.1016/j.placenta.2017.03.010
89. Yang J, Weinberg RA. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev Cell (2008) 14(6):818–29. doi: 10.1016/j.devcel.2008.05.009
90. Davies E, Pollheimer J, Yong HEJ, Kokkinos MI, Kalionis B, Knöfler M, et al. Epithelial-mesenchymal transition during extravillous trophoblast differentiation. Cell Adhesion Migration (2016) 10(3):310–21. doi: 10.1080/19336918.2016.1170258
91. Pollheimer J, Fock V, Knöfler M. Review: The ADAM metalloproteinases - Novel regulators of trophoblast invasion? Placenta (2014) 35(SUPPL):S57–63. doi: 10.1016/j.placenta.2013.10.012
92. Uekita T, Yamanouchi K, Sato H, Tojo H, Seiki M, Tachi C. Expression and localization of matrix metalloproteinases (MT1-MMP, MMP-2) and tissue inhibitor of metalloproteinase-2 (TIMP-2) during synepitheliochorial placentation of goats (Capra hircus). Placenta (2004) 25(10):810–9. doi: 10.1016/j.placenta.2004.03.007
93. Hamutoğlu R, Bulut HE, Kaloğlu C, Önder O, Dağdeviren T, Aydemir MN, et al. The regulation of trophoblast invasion and decidual reaction by matrix metalloproteinase-2, metalloproteinase-7, and metalloproteinase-9 expressions in the rat endometrium. Reprod Med Biol (2020) 19(4):385–97. doi: 10.1002/rmb2.12342
94. Plaks V, Rinkenberger J, Dai J, Flannery M, Sund M, Kanasaki K, et al. Matrix metalloproteinase-9 deficiency phenocopies features of preeclampsia and intrauterine growth restriction. Proc Natl Acad Sci U S A (2013) 110:11109–14. doi: 10.1073/pnas.1309561110
95. Li M, Liu D, Wang L, Wang W, Wang A, Yao Y. Expression of placenta-specific 8 in human oocytes, embryos, and models of in vitro implantation. Fertil Steril (2016) 106(3):781–9.e2. doi: 10.1016/j.fertnstert.2016.05.018
96. Huang CC, Shen MH, Chen SK, Yang SH, Liu CY, Guo JW, et al. Gut butyrate-producing organisms correlate to Placenta Specific 8 protein: Importance to colorectal cancer progression. J Adv Res (2020) 22:7–20. doi: 10.1016/j.jare.2019.11.005
97. Hung CS, Wang YC, Guo JW, Yang RN, Lee CL, Shen MH, et al. Expression pattern of placenta specific 8 and keratin 20 in different types of gastrointestinal cancer. Mol Med Rep (2020) 21(2):659–66. doi: 10.3892/mmr.2019.10871
98. Zeng X, Liu Q, Yang Y, Jia W, Li S, He D, et al. Placenta-specific protein 8 promotes the proliferation of lung adenocarcinoma PC-9 cells and their tolerance to an epidermal growth factor receptor tyrosine kinase inhibitor by activating the ERK signaling pathway. Oncol Lett (2019) 18(5):5621–7. doi: 10.3892/ol.2019.10911
99. Ma X, Liu J, Wang H, Jiang Y, Wan Y, Xia Y, et al. Identification of crucial aberrantly methylated and differentially expressed genes related to cervical cancer using an integrated bioinformatics analysis. Biosci Rep (2020) 40(5):BSR20194365. doi: 10.1042/BSR20194365
100. Kaistha BP, Lorenz H, Schmidt H, Sipos B, Pawlak M, Gierke B, et al. PLAC8 localizes to the inner plasma membrane of pancreatic cancer cells and regulates cell growth and disease progression through critical cell-cycle regulatory pathways. Cancer Res (2016) 76(1):96–107. doi: 10.1158/0008-5472.CAN-15-0216
101. Tatura M, Schmidt H, Haijat M, Stark M, Rinke A, Diels R, et al. Placenta-Specific 8 Is Overexpressed and Regulates Cell Proliferation in Low-Grade Human Pancreatic Neuroendocrine Tumors. Neuroendocrinology (2020) 110(1–2):23–34. doi: 10.1159/000500541
102. Mao M, Chen Y, Jia Y, Yang J, Wei Q, Li Z, et al. PLCA8 suppresses breast cancer apoptosis by activating the PI3k/AKT/NF-κB pathway. J Cell Mol Med (2019) 23(10):6930–41. doi: 10.1111/jcmm.14578
103. Kinsey C, Balakrishnan V, O’Dell MR, Huang JL, Newman L, Whitney-Miller CL, et al. Plac8 links oncogenic mutations to regulation of autophagy and is critical to pancreatic cancer progression. Cell Rep (2014) 7(4):1143–55. doi: 10.1016/j.celrep.2014.03.061
104. Demir R, Kayisli UA, Cayli S, Huppertz B. Sequential steps during vasculogenesis and angiogenesis in the very early human placenta. Placenta (2006) 27(6–7):535–9. doi: 10.1016/j.placenta.2005.05.011
105. Fernández-Cortés M, Delgado-Bellido D, Oliver FJ. Vasculogenic Mimicry: Become an Endothelial Cell ‘But Not So Much’. Front Oncol (2019) 9(AUG):803. doi: 10.3389/fonc.2019.00803
106. Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res (2001) 49(3):507–21. doi: 10.1016/S0008-6363(00)00281-9
107. Demir R, Seval Y, Huppertz B. Vasculogenesis and angiogenesis in the early human placenta. Acta Histochem (2007) 109(4):257–65. doi: 10.1016/j.acthis.2007.02.008
108. Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manage (2006) 2(3):213–9. doi: 10.2147/vhrm.2006.2.3.213
109. Murray MJ, Lessey BA. Embryo implantation and tumor metastasis: Common pathways of invasion and angiogenesis. Semin Reprod Endocrinol (1999) 17(3):275–90. doi: 10.1055/s-2007-1016235
110. Brown JM. Vasculogenesis: A crucial player in the resistance of solid tumours to radiotherapy. Br J Radiol (2014) 87(1035):20130686. doi: 10.1259/bjr.20130686
111. Testa U, Pelosi E, Castelli G. Endothelial Progenitors in the Tumor Microenvironment. In. Advances in Experimental Medicine and Biology Springer (2020) Vol. 1263, pp. 85–115.
112. Li X, Sun X, Carmeliet P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab (2019) 30(3):414–33. doi: 10.1016/j.cmet.2019.08.011
113. Zecchin A, Kalucka J, Dubois C, Carmeliet P. How endothelial cells adapt their metabolism to form vessels in tumors. Front Immunol (2017) 8:1750(DEC). doi: 10.3389/fimmu.2017.01750
114. Maquoi E, Van den Brule FA, Castronovo V, Foidart JM. Changes in the distribution pattern of galectin-1 and galectin-3 in human placenta correlates with the differentiation pathways of trophoblasts. Placenta (1997) 18(5–6):433–9. doi: 10.1016/S0143-4004(97)80044-6
115. Mourad-Zeidan AA, Melnikova VO, Wang H, Raz A, Bar-Eli M. Expression profiling of galectin-3-depleted melanoma cells reveals its major role in melanoma cell plasticity and vasculogenic mimicry. Am J Pathol (2008) 173(6):1839–52. doi: 10.2353/ajpath.2008.080380
116. Sagrillo-Fagundes L, Bienvenue-Pariseault J, Legembre P, Vaillancourt C. An insight into the role of the death receptor CD95 throughout pregnancy: Guardian, facilitator, or foe. Birth Defects Res (2019) 111(4):197–211. doi: 10.1002/bdr2.1470
117. Liu S, Diao L, Huang C, Li Y, Zeng Y, Kwak-Kim JYH. The role of decidual immune cells on human pregnancy. J Reprod Immunol (2017) 124:44–53. doi: 10.1016/j.jri.2017.10.045
118. Manaster I, Mandelboim O. The Unique Properties of Uterine NK Cells. Am J Reprod Immunol (2010) 63(6):434–44. doi: 10.1111/j.1600-0897.2009.00794.x
119. Nancy P, Erlebacher A. T cell behavior at the maternal-fetal interface. Int J Dev Biol (2014) 58(2–4):189–98. doi: 10.1387/ijdb.140054ae
120. Male V, Sharkey A, Masters L, Kennedy PR, Farrell LE, Moffett A. The effect of pregnancy on the uterine NK cell KIR repertoire. Eur J Immunol (2011) 41(10):3017–27. doi: 10.1002/eji.201141445
121. Sojka DK, Yang L, Yokoyama WM. Uterine natural killer cells. Front Immunol (2019) 10:960. doi: 10.3389/fimmu.2019.00960
122. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med (2006) 12(9):1065–74. doi: 10.1038/nm1452
123. Cartwright JE, James-Allan L, Buckley RJ, Wallace AE. The role of decidual NK cells in pregnancies with impaired vascular remodelling. J Reprod Immunol (2017) 119:81–4. doi: 10.1016/j.jri.2016.09.002
124. Lash GE, Naruse K, Robson A, Innes BA, Searle RF, Robson SC, et al. Interaction between uterine natural killer cells and extravillous trophoblast cells: effect on cytokine and angiogenic growth factor production. Hum Reprod (2011) 26(9):2289–95. doi: 10.1093/humrep/der198
125. Ning F, Liu H, Lash GE. The Role of Decidual Macrophages During Normal and Pathological Pregnancy. Am J Reprod Immunol (2016) 75(3):298–309. doi: 10.1111/aji.12477
126. Helige C, Ahammer H, Moser G, Hammer A, Dohr G, Huppertz B, et al. Distribution of decidual natural killer cells and macrophages in the neighbourhood of the trophoblast invasion front: A quantitative evaluation. Hum Reprod (2014) 29(1):8–17. doi: 10.1093/humrep/det353
127. Lash GE, Pitman H, Morgan HL, Innes BA, Agwu CN, Bulmer JN. Decidual macrophages: key regulators of vascular remodeling in human pregnancy. J Leukoc Biol (2016) 100(2):315–25. doi: 10.1189/jlb.1a0815-351r
128. Morvan MG, Lanier LL. NK cells and cancer: You can teach innate cells new tricks. Nat Rev Cancer (2016) 16(1):7–19. doi: 10.1038/nrc.2015.5
129. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol (2015) 35:S185–98. doi: 10.1016/j.semcancer.2015.03.004
130. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front Immunol (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
131. Modiano JF, Bellgrau D. Fas Ligand Based Immunotherapy: A Potent and Effective Neoadjuvant with Checkpoint Inhibitor Properties, or a Systemically Toxic Promoter of Tumor Growth? - Jaime F Modiano - Discovery Medicine (2016). Available at: http://www.discoverymedicine.com/Jaime-F-Modiano/2016/02/fas-ligand-based-immunotherapy-a-potent-and-effective-neoadjuvant-with-checkpoint-inhibitor-properties-or-a-systemically-toxic-promoter-of-tumor-growth/ (Accessed Jul. 14, 2020).
132. Houston A, O’Connell J. The Fas signalling pathway and its role in the pathogenesis of cancer. Curr Opin Pharmacol (2004) 4(4):321–6. doi: 10.1016/j.coph.2004.03.008
133. Wilczynski JR, Kalinka J, Radwan M. The role of T-regulatory cells in pregnancy and cancer. Front Biosci (2008) 13(6):2275–89. doi: 10.2741/2841
134. Zhao JX, Zeng YY, Liu Y. Fetal alloantigen is responsible for the expansion of the CD4+CD25+ regulatory T cell pool during pregnancy. J Reprod Immunol (2007) 75(2):71–81. doi: 10.1016/j.jri.2007.06.052
135. Loustau M, Anna F, Dréan R, Lecomte M, Langlade-Demoyen P, Caumartin J. HLA-G Neo-Expression on Tumors. Front Immunol (2020) 11:1685. doi: 10.3389/fimmu.2020.01685
136. Lin A, Yan WH. Intercellular transfer of HLA-G: its potential in cancer immunology. Clin Trans Immunol (2019) 8(9):e1077. doi: 10.1002/cti2.1077
137. Gimenes F, Teixeira JJV, de Abreu ALP, Souza RP, Pereira MW, da Silva VRS, et al. Human leukocyte antigen (HLA)-G and cervical cancer immunoediting: A candidate molecule for therapeutic intervention and prognostic biomarker? Biochim Biophys Acta - Rev Cancer (2014) 1846(2):576–89. doi: 10.1016/j.bbcan.2014.10.004
138. Steven A, Seliger B. The Role of Immune Escape and Immune Cell Infiltration in Breast Cancer. Breast Care (2018) 13(1):16–21. doi: 10.1159/000486585
139. Farjadian S, Tabebordbar M, Mokhtari M, Safaei A, Malekzadeh M, Gaderi A. HLA-G expression in tumor tissues and soluble HLA-G plasma levels in patients with gastrointestinal cancer. Asian Pacific J Cancer Prev (2018) 19(10):2731–5. doi: 10.22034/APJCP.2018.19.10.2731
140. Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy (2011) 7(7):673–82. doi: 10.4161/auto.7.7.14733
141. Wu H, Chen S, Ammar AB, Xu J, Wu Q, Pan K, et al. Crosstalk Between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol Neurobiol (2015) 52(3):1284–96. doi: 10.1007/s12035-014-8933-0
142. Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res (2010) 20(7):748–62. doi: 10.1038/cr.2010.82
143. Parzych KR, Klionsky DJ. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signaling (2014) 20(3):460–73. doi: 10.1089/ars.2013.5371
144. Eskelinen EL, Saftig P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta - Mol Cell Res (2009) 1793(4):664–73. doi: 10.1016/j.bbamcr.2008.07.014
145. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol (2015) 16(8):461–72. doi: 10.1038/nrm4024
146. Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol Rev (2010) 90(4):1383–435. doi: 10.1152/physrev.00030.2009
147. Reggiori F, Ungermann C. Autophagosome Maturation and Fusion. J Mol Biol (2017) 429(4):486–96. doi: 10.1016/j.jmb.2017.01.002
148. Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell (2011) 147(4):728–41. doi: 10.1016/j.cell.2011.10.026
149. Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy (2010) 6(6):764–76. doi: 10.4161/auto.6.6.12709
150. Reggiori F, Tucker KA, Stromhaug PE. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell (2004) 6(1):79–90. doi: 10.1016/S1534-5807(03)00402-7
151. Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol (2012) 198(2):219–33. doi: 10.1083/jcb.201202061
152. Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol (2004) 15(2):231–6. doi: 10.1016/j.semcdb.2003.12.004
153. Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, et al. The Atg12-Atg5 Conjugate Has a Novel E3-like Activity for Protein Lipidation in Autophagy. J Biol Chem (2007) 282(52):37298–302. doi: 10.1074/jbc.C700195200
154. Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy (2011) 7(3):279–96. doi: 10.4161/auto.7.3.14487
155. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy*[S]. J Biol Chem (2007) 282(33):24131–45. doi: 10.1074/jbc.M702824200
156. Takahashi Y, He H, Tang Z, Hattori T, Liu Y, Young MM, et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun (2018) 9(1):1–13. doi: 10.1038/s41467-018-05254-w
157. Yu S, Melia TJ. The coordination of membrane fission and fusion at the end of autophagosome maturation. Curr Opin Cell Biol (2017) 47:92–8. doi: 10.1016/j.ceb.2017.03.010
158. Zhao YG, Zhang H. Autophagosome maturation: An epic journey from the ER to lysosomes. J Cell Biol (2019) 218(3):757–70. doi: 10.1083/jcb.201810099
159. Korolchuk VI, Rubinsztein DC. Regulation of autophagy by lysosomal positioning. Autophagy (2011) 7(8):927–8. doi: 10.4161/auto.7.8.15862
160. Nakamura S, Yoshimori T. New insights into autophagosome-lysosome fusion. J Cell Sci (2017) 130(7):1209–16. doi: 10.1242/jcs.196352
161. Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy (2018) 14(2):207–15. doi: 10.1080/15548627.2017.1378838
162. Rong Y, McPhee C, Denga S, Huanga L, Chen L, Liu M, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci U S A (2011) 108(19):7826–31. doi: 10.1073/pnas.1013800108
163. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature (2010) 465(7300):942–6. doi: 10.1038/nature09076
164. Kawabata T, Yoshimori T. Autophagosome biogenesis and human health. Cell Discovery (2020) 6(1):1–14. doi: 10.1038/s41421-020-0166-y
165. Fujikake N, Shin M, Shimizu S. Association between autophagy and neurodegenerative diseases. Front Neurosci (2018) 12(MAY):255. doi: 10.3389/fnins.2018.00255
166. Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy (2016) 12(2):245–60. doi: 10.1080/15548627.2015.1071759
167. Rocchi A, He C. Emerging roles of autophagy in metabolism and metabolic disorders. Front Biol (2015) 10(2):154–64. doi: 10.1007/s11515-015-1354-2
168. Ryter SW, Koo JK, Choi AMK. Molecular regulation of autophagy and its implications for metabolic diseases. Curr Opin Clin Nutr Metab Care (2014) 17(4):329–37. doi: 10.1097/MCO.0000000000000068
169. Yang Y, Klionsky DJ. Autophagy and disease: unanswered questions. Cell Death Differentiation (2020) 27(3):858–71. doi: 10.1038/s41418-019-0480-9
170. Nakashima A, Shima T, Aoki A, Kawaguchi M, Yasuda I, Tsuda S, et al. Molecular and immunological developments in placentas. Hum Immunol (2021). doi: 10.1016/j.humimm.2021.01.012
171. Lu H, Yang HL, Zhou WJ, Lai ZZ, Qiu XM, Fu Q, et al. Rapamycin prevents spontaneous abortion by triggering decidual stromal cell autophagy-mediated NK cell residence. Autophagy (2020) 1:1–17. doi: 10.1080/15548627.2020.1833515
172. Tan HX, Yang SL, Li MQ, Wang HY. Autophagy suppression of trophoblast cells induces pregnancy loss by activating decidual NK cytotoxicity and inhibiting trophoblast invasion. Cell Commun Signal (2020) 18(1):1–16. doi: 10.1186/s12964-020-00579-w
173. Noman MZ, Janji B, Berchem G, Mami-Chouaib F, Chouaib S. Hypoxia-induced autophagy: A new player in cancer immunotherapy? Autophagy (2012) 8(4):704–6. doi: 10.4161/auto.19572
174. Janji B, Berchem G, Chouaib S. Targeting autophagy in the tumor microenvironment: New challenges and opportunities for regulating tumor immunity. Front Immunol (2018) 9:887(APR). doi: 10.3389/fimmu.2018.00887
175. Tittarelli A, Janji B, Van Moer K, Noman MZ, Chouaib S. The selective degradation of synaptic connexin 43 protein by hypoxia-induced autophagy impairs natural killer cell-mediated tumor cell killing. J Biol Chem (2015) 290(39):23670–9. doi: 10.1074/jbc.M115.651547
176. Gerada C, Ryan KM. Autophagy, the innate immune response and cancer. Mol Oncol (2020) 14(9):1913–29. doi: 10.1002/1878-0261.12774
177. Jiang GM, Tan Y, Wang H, Peng L, Chen HT, Meng XJ, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer (2019) 18(1):1–22. doi: 10.1186/s12943-019-0944-z
178. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, Van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A (2013) 110(43):17450–5. doi: 10.1073/pnas.1304790110
179. Yao C, Ni Z, Gong C, Zhu X, Wang L, Xu Z, et al. Rocaglamide enhances NK cell-mediated killing of non-small cell lung cancer cells by inhibiting autophagy. Autophagy (2018) 14(10):1831–44. doi: 10.1080/15548627.2018.1489946
180. Zhao H, Jia H, Han Q, Zhang J. Homeobox containing 1 inhibits liver cancer progression by promoting autophagy as well as inhibiting stemness and immune escape. Oncol Rep (2018) 40(3):1657–65. doi: 10.3892/or.2018.6551
181. Zaarour RF, Azakir B, Hajam EY, Nawafleh H, Zeinelabdin NA, Engelsen AST, et al. Role of hypoxia-mediated autophagy in tumor cell death and survival. Cancers (2021) 13(3):1–20. doi: 10.3390/cancers13030533
182. Chifenti B, Locci MT, Lazzeri G, Guagnozzi M, Dinucci D, Chiellini F, et al. Autophagy-related protein LC3 and Beclin-1 in the first trimester of pregnancy. Clin Exp Reprod Med (2013) 40(1):33–7. doi: 10.5653/cerm.2013.40.1.33
183. Signorelli P, Avagliano L, Virgili E, Gagliostro V, Doi P, Braidotti P, et al. Autophagy in term normal human placentas. Placenta (2011) 32(6):482–5. doi: 10.1016/j.placenta.2011.03.005
184. Chen GQ, Zhang H, Qi H, Yao ZW, Gao L, Qiu CL. [Effects and mechanisms of autophagy of trophoblast cells in severe preeclampsia]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2012) 28(3):294–6.
185. Hung TH, Chen SF, Lo LM, Li MJ, Yeh YL, Hsieh TT. an., “Increased autophagy in placentas of intrauterine growth-restricted pregnancies. PloS One (2012) 7(7):e40957. doi: 10.1371/journal.pone.0040957
186. Choi J-H, Lee H-J, Yang T-H, Kim GJ. Effects of hypoxia inducible factors-1α on autophagy and invasion of trophoblasts. Clin Exp Reprod Med (2012) 39(2):73–80. doi: 10.5653/cerm.2012.39.2.73
187. Chen B, Longtine MS, Nelson DM. Hypoxia induces autophagy in primary human trophoblasts. Endocrinology (2012) 153(10):4946–54. doi: 10.1210/en.2012-1472
188. Chang YL, Wang TH, Chang SD, Chao AS, Hsieh PCC. and Wang, C. N., “Increased autophagy in the placental territory of selective intrauterine growth-restricted monochorionic twins. Prenat Diagn (2013) 33(2):187–90. doi: 10.1002/pd.4040
189. Nakashima A, Yamanaka-Tatematsu M, Fujita N, Koizumi K, Shima T, Yoshida T, et al. Impaired autophagy by soluble endoglin, under physiological hypoxia in early pregnant period, is involved in poor placentation in preeclampsia. Autophagy (2013) 9(3):303–16. doi: 10.4161/auto.22927
190. Avagliano L, Virgili E, Garò C, Quadrelli F, Doi P, Samaja M, et al. Autophagy and human parturition: Evaluation of LC3 expression in placenta from spontaneous or medically induced onset of labor. BioMed Res Int (2013) 2013:1–9. doi: 10.1155/2013/689768
191. Avagliano L, Danti L, Doi P, Felis S, Guala M, Locatelli A, et al. Autophagy in placentas from acidotic newborns: An immunohistochemical study of LC3 expression. Placenta (2013) 34(11):1091–4. doi: 10.1016/j.placenta.2013.09.004
192. Yamanaka-Tatematsu M, Nakashima A, Fujita N, Shima T, Yoshimori T, Saito S. Autophagy induced by HIF1α overexpression supports trophoblast invasion by supplying cellular energy. PloS One (2013) 8(10):e76605. doi: 10.1371/journal.pone.0076605
193. Kalkat M, Garcia J, Ebrahimi J, Melland-Smith M, Todros T, Post M, et al. Placental autophagy regulation by the BOK-MCL1 rheostat. Autophagy (2013) 9(12):2140–53. doi: 10.4161/auto.26452
194. Hung T-H, Hsieh T-T, Chen S-F, Li M-J, Yeh Y-L. Autophagy in the Human Placenta throughout Gestation. PloS One (2013) 8(12):e83475. doi: 10.1371/journal.pone.0083475
195. Cha HH, Hwang JR, Kim HY, Choi SJ, Oh SY, Roh CR. Autophagy induced by tumor necrosis factor mediates intrinsic apoptosis in trophoblastic cells. Reprod Sci (2014) 21(5):612–22. doi: 10.1177/1933719113508816
196. Akaishi R, Yamada T, Nakabayashi K, Nishihara H, Furuta I, Kojima T, et al. Autophagy in the placenta of women with hypertensive disorders in pregnancy. Placenta (2014) 35(12):974–80. doi: 10.1016/j.placenta.2014.10.009
197. Gaoa L, Qi HB, Kc K, Zhang XM, Zhang H, Baker PN. Excessive autophagy induces the failure of trophoblast invasion and vasculature: Possible relevance to the pathogenesis of preeclampsia. J Hypertens (2015) 33(1):106–17. doi: 10.1097/HJH.0000000000000366
198. Avagliano L, Terraneo L, Virgili E, Martinelli C, Doi P, Samaja M, et al. Autophagy in normal and abnormal early human pregnancies. Reprod Sci (2015) 22(7):838–44. doi: 10.1177/1933719114565036
199. Melland-Smith M, Ermini L, Chauvin S, Craig-Barnes H, Tagliaferro A, Todros T, et al. Disruption of sphingolipid metabolism augments ceramide-induced autophagy in preeclampsia. Autophagy (2015) 11(4):653–69. doi: 10.1080/15548627.2015.1034414
200. Brickle A, Tran HT, Lim R, Liong S, Lappas M. Autophagy, which is decreased in labouring fetal membranes, regulates IL-1β production via the inflammasome. Placenta (2015) 36(12):1393–404. doi: 10.1016/j.placenta.2015.10.015
201. He B, Zhang N, Zhao R. Dexamethasone Downregulates SLC7A5 Expression and Promotes Cell Cycle Arrest, Autophagy and Apoptosis in BeWo Cells. J Cell Physiol (2016) 231(1):233–42. doi: 10.1002/jcp.25076
202. Nakabayashi Y, Nakashima A, Yoshino O, Shima T, Shiozaki A, Adachi T, et al. Impairment of the accumulation of decidual T cells, NK cells, and monocytes, and the poor vascular remodeling of spiral arteries, were observed in oocyte donation cases, regardless of the presence or absence of preeclampsia. J Reprod Immunol (2016) 114:65–74. doi: 10.1016/j.jri.2015.07.005
203. Wang Y, Chen B, Longtine MS, Nelson DM. Punicalagin promotes autophagy to protect primary human syncytiotrophoblasts from apoptosis. Reproduction (2016) 151(2):97–104. doi: 10.1530/REP-15-0287
204. Muralimanoharan S, Gao X, Weintraub S, Myatt L, Maloyan A. Sexual dimorphism in activation of placental autophagy in obese women with evidence for fetal programming from a placenta-specific mouse model. Autophagy (2016) 12(5):752–69. doi: 10.1080/15548627.2016.1156822
205. He MY, Wang G, Han SS, Jin Y, Li H, Wu X, et al. Nrf2 signalling and autophagy are involved in diabetes mellitus-induced defects in the development of mouse placenta. Open Biol (2016) 6(7):160064. doi: 10.1098/rsob.160064
206. Zhang Y, Hu X, Gao G, Wang Y, Chen P, Ye Y. Autophagy protects against oxidized low density lipoprotein-mediated inflammation associated with preeclampsia. Placenta (2016) 48:136–43. doi: 10.1016/j.placenta.2016.09.015
207. Cao B, Macones C, Mysorekar IU. ATG16L1 governs placental infection risk and preterm birth in mice and women. JCI Insight (2016) 1(21):e86654. doi: 10.1172/jci.insight.86654
208. Xu Y, Huang X, Xie J, Chen Y, Fu J, Wang L. Let-7i-Induced Atg4B Suppression Is Essential for Autophagy of Placental Trophoblast in Preeclampsia. J Cell Physiol (2017) 232(9):2581–9. doi: 10.1002/jcp.25661
209. Avagliano L, Massa V, Zullino S, Doi P, Marconi AM, Ferrazzi E, et al. Inflammation modulates LC3 expression in human preterm delivery. J Matern Neonatal Med (2017) 30(6):698–704. doi: 10.1080/14767058.2016.1183630
210. Prokesch A, Blaschitz A, Bauer T, Moser G, Hiden U, Zadora J, et al. Placental DAPK1 and autophagy marker LC3B-II are dysregulated by TNF-α in a gestational age-dependent manner. Histochem Cell Biol (2017) 147(6):695–705. doi: 10.1007/s00418-016-1537-1
211. Avagliano L, Massa V, Terraneo L, Samaja M, Doi P, Bulfamante GP, et al. Gestational diabetes affects fetal autophagy. Placenta (2017) 55:90–3. doi: 10.1016/j.placenta.2017.05.002
212. Zhang QX, Na Q, Song W. Altered expression of mTOR and autophagy in term normal human placentas. Rom J Morphol Embryol (2017) 2012(1):61–6.
213. Ji L, Chen Z, Xu Y, Xiong G, Liu R, Wu C, et al. Systematic Characterization of Autophagy in Gestational Diabetes Mellitus. Endocrinology (2017) 158(8):2522–32. doi: 10.1210/en.2016-1922
214. Holland OJ, Hickey AJR, Alvsaker A, Moran S, Hedges C, Chamley LW, et al. Changes in mitochondrial respiration in the human placenta over gestation. Placenta (2017) 57:102–12. doi: 10.1016/j.placenta.2017.06.011
215. Hutabarat M, Wibowo N, Huppertz B. The trophoblast survival capacity in preeclampsia. PloS One (2017) 12(11):e0186909. doi: 10.1371/journal.pone.0186909
216. Dimasuay KG, Gong L, Rosario F, McBryde E, Spelman T, Glazier J, et al. Impaired placental autophagy in placental malaria. PloS One (2017) 12(11):e0187291. doi: 10.1371/journal.pone.0187291
217. Pan T, He G, Chen M, Bao C, Chen Y, Liu G, et al. Abnormal CYP11A1 gene expression induces excessive autophagy, contributing to the pathogenesis of preeclampsia. Oncotarget (2017) 8(52):89824–36. doi: 10.18632/oncotarget.21158
218. Hung TH, Hsieh TT, Wu CP, Li MJ, Yeh YL, Chen SF. Mammalian target of rapamycin signaling is a mechanistic link between increased endoplasmic reticulum stress and autophagy in the placentas of pregnancies complicated by growth restriction. Placenta (2017) 60:9–20. doi: 10.1016/j.placenta.2017.10.001
219. Akcora Yildiz D, Irtegun Kandemir S, Agacayak E, Deveci E. Evaluation of protein levels of autophagy markers (Beclin 1 and SQSTM1/p62) and phosphorylation of cyclin E in the placenta of women with preeclampsia. Cell Mol Biol (2017) 63(12):51. doi: 10.14715/cmb/2017.63.12.12
220. Lee H-M, Choi K-C. Cigarette smoke extract and isoprene resulted in the induction of apoptosis and autophagy in human placenta choriocarcinoma JEG-3 cells. Environ Toxicol (2018) 33(2):178–90. doi: 10.1002/tox.22506
221. Gauster M, Maninger S, Siwetz M, Deutsch A, El-Heliebi A, Kolb-Lenz D, et al. Downregulation of p53 drives autophagy during human trophoblast differentiation. Cell Mol Life Sci (2018) 75(10):1839–55. doi: 10.1007/s00018-017-2695-6
222. Hong Y-J, Ahn H-J, Shin J, Lee JH, Kim J-H, Park H-W, et al. Unsaturated fatty acids protect trophoblast cells from saturated fatty acid-induced autophagy defects. J Reprod Immunol (2018) 125:56–63. doi: 10.1016/j.jri.2017.12.001
223. Hawkins SJ, Crompton LA, Sood A, Saunders M, Boyle NT, Buckley A, et al. Nanoparticle-induced neuronal toxicity across placental barriers is mediated by autophagy and dependent on astrocytes. Nat Nanotechnol (2018) 13(5):427–33. doi: 10.1038/s41565-018-0085-3
224. Ozsoy AZ, Cayli S, Sahin C, Ocakli S, Sanci TO, Ilhan DB. Altered expression of p97/Valosin containing protein and impaired autophagy in preeclamptic human placenta. Placenta (2018) 67:45–53. doi: 10.1016/j.placenta.2018.05.013
225. Pan YJ, He L, Zhou SJ, Zhang LJ, Zhang AH, Zhao YY. Expression of urotensin II is associated with placental autophagy in patients with severe preeclampsia. J Hum Hypertens (2018) 32(11):759–69. doi: 10.1038/s41371-018-0083-9
226. Sagrillo-Fagundes L, Assunção Salustiano EM, Ruano R, Markus RP, Vaillancourt C. Melatonin modulates autophagy and inflammation protecting human placental trophoblast from hypoxia/reoxygenation. J Pineal Res (2018) 65(4):e12520. doi: 10.1111/jpi.12520
227. Hu J, Liu L, Gong Y, Zhang L, Gan X, Luo X, et al. Linc02527 promoted autophagy in Intrahepatic cholestasis of pregnancy. Cell Death Dis (2018) 9(10):1–14. doi: 10.1038/s41419-018-1013-z
228. Zhang Y, Xu B, Yao M, Dong T, Mao Z, Hang B, et al. Titanium dioxide nanoparticles induce proteostasis disruption and autophagy in human trophoblast cells. Chem Biol Interact (2018) 296:124–33. doi: 10.1016/j.cbi.2018.09.015
229. Cai H, Chen L, Zhang M, Xiang W, Su P. Low expression of MFN2 is associated with early unexplained miscarriage by regulating autophagy of trophoblast cells. Placenta (2018) 70:34–40. doi: 10.1016/j.placenta.2018.08.005
230. Mao Z, Yao M, Li Y, Fu Z, Li S, Zhang L, et al. MiR-96-5p and miR-101-3p as potential intervention targets to rescue TiO2 NP-induced autophagy and migration impairment of human trophoblastic cells. Biomater Sci (2018) 6(12):3273–83. doi: 10.1039/c8bm00856f
231. Zhang L, Deng X, Shi X, Dong X. Silencing H19 regulated proliferation, invasion, and autophagy in the placenta by targeting miR-18a-5p. J Cell Biochem (2019) 120(6):9006–15. doi: 10.1002/jcb.28172
232. Sagrillo-Fagundes L, Bienvenue-Pariseault J, Vaillancourt Id C. Melatonin: The smart molecule that differentially modulates autophagy in tumor and normal placental cells. PloS One (2019) 14(1):e0202458. doi: 10.1371/journal.pone.0202458
233. Nakashima A, Higashisaka K, Kusabiraki T, Aoki A, Ushijima A, Ono Y, et al. Autophagy is a new protective mechanism against the cytotoxicity of platinum nanoparticles in human trophoblasts. Sci Rep (2019) 9(1):1–13. doi: 10.1038/s41598-019-41927-2
234. Wang Y, Liu LL, Tian Y, Chen Y, Zha WH, Li Y, et al. Upregulation of DAPK2 ameliorates oxidative damage and apoptosis of placental cells in hypertensive disorder complicating pregnancy by suppressing human placental microvascular endothelial cell autophagy through the mTOR signaling pathway. Int J Biol Macromol (2019) 121:488–97. doi: 10.1016/j.ijbiomac.2018.09.111
235. Chen Y, Wang L, Bao J, Sha X, Cui L, Huang Q, et al. Persistent hypoxia induced autophagy leading to invasiveness of trophoblasts in placenta accreta. J Matern Neonatal Med (2019) 34(8):1291–303. doi: 10.1080/14767058.2019.1635582
236. Nakashima A, Cheng SB, Kusabiraki T, Motomura K, Aoki A, Ushijima A, et al. Endoplasmic reticulum stress disrupts lysosomal homeostasis and induces blockade of autophagic flux in human trophoblasts. Sci Rep (2019) 9(1):1–12. doi: 10.1038/s41598-019-47607-5
237. Zhu HL, Xu XF, Shi XT, Feng YJ, Xiong YW, Nan Y, et al. Activation of autophagy inhibits cadmium-triggered apoptosis in human placental trophoblasts and mouse placenta. Environ Pollut (2019) 254(Pt A). doi: 10.1016/j.envpol.2019.112991
238. Bastida-Ruiz D, Yart L, Wuillemin C, Ribaux P, Morris N, Epiney M, et al. The fine-tuning of endoplasmic reticulum stress response and autophagy activation during trophoblast syncytialization. Cell Death Dis (2019) 10(9):651. doi: 10.1038/s41419-019-1905-6
239. Lima FA, Barateiro A, Dombrowski JG, de Souza RM, de Sousa Costa D, Murillo O, et al. Plasmodium falciparum infection dysregulates placental autophagy. PloS One (2019) 14(12):e0226117. doi: 10.1371/journal.pone.0226117
240. Wang Y, Ji L, Peng Z, Lai R, Zhang X, Xu Y, et al. Silencing DAPK3 blocks the autophagosome-lysosome fusion by mediating SNAP29 in trophoblast cells under high glucose treatment. Mol Cell Endocrinol (2020) 502. doi: 10.1016/j.mce.2019.110674
241. Zhang H, He Y, Wang JX, Chen MH, Xu JJ, Jiang MH, et al. miR-30-5p-mediated ferroptosis of trophoblasts is implicated in the pathogenesis of preeclampsia. Redox Biol (2020) 29:101402. doi: 10.1016/j.redox.2019.101402
242. Wang P, Huang CX, Gao JJ, Shi Y, Li H, Yan H, et al. Resveratrol induces SIRT1-Dependent autophagy to prevent H2O2-Induced oxidative stress and apoptosis in HTR8/SVneo cells. Placenta (2020) 91:11–8. doi: 10.1016/j.placenta.2020.01.002
243. Yang D, Ding J, Wang Y, Yuan M, Xian S, Zhang L, et al. YY1-PVT1 affects trophoblast invasion and adhesion by regulating mTOR pathway-mediated autophagy. J Cell Physiol (2020) 235(10):6637–46. doi: 10.1002/jcp.29560
244. Han Q, Zhang W, Lu C, Wu J, An S, Zhang S. Repression of Kisspeptin1 weakens hydrogen peroxide-caused injury in HTR8 cells via adjusting PI3K/AKT/mTOR pathway. J Biochem Mol Toxicol (2020) 34(5):e22461. doi: 10.1002/jbt.22461
245. Hung TH, Huang SY, Chen SF, Wu CP, Hsieh TT. an., “Decreased placental apoptosis and autophagy in pregnancies complicated by gestational diabetes with large-for-gestational age fetuses. Placenta (2020) 90:27–36. doi: 10.1016/j.placenta.2019.12.003
246. Mao Q, Chu S, Shapiro S, Yao H, De Paepe ME. Discordant placental oxygenation and autophagy in twin anemia-polycythemia sequence (TAPS). Placenta (2020) 90:9–17. doi: 10.1016/j.placenta.2019.11.003
247. Zaugg J, Melhem H, Huang X, Wegner M, Baumann M, Surbek D, et al. Gestational diabetes mellitus affects placental iron homeostasis: Mechanism and clinical implications. FASEB J (2020) 34(6):7311–29. doi: 10.1096/fj.201903054R
248. Yang P, Li Z, Tye KD, Chen Y, Lu T, He Z, et al. Effects of an orally supplemented probiotic on the autophagy protein LC3 and Beclin1 in placentas undergoing spontaneous delivery during normal pregnancy. BMC Pregnancy Childbirth (2020) 20(1):1–8. doi: 10.1186/s12884-020-02905-z
249. Cohen M, Guo E, Pucchio A, de Vrijer B, Shepherd TG, Eastabrook G. Maternal obesity reduces placental autophagy marker expression in uncomplicated pregnancies. J Obstet Gynaecol Res (2020) 46(8):1282–91. doi: 10.1111/jog.14315
250. Dong X, Zhang Y, Chen X, Xue M. Long noncoding RNA LINC00511 regulates the proliferation, apoptosis, invasion and autophagy of trophoblast cells to mediate pre-eclampsia progression through modulating the miR-31-5p/homeobox protein A7 axis. J Obstet Gynaecol Res (2020) 46(8):1298–309. doi: 10.1111/jog.14344
251. Du J, Ji Q, Dong L, Meng Y, Xin G. HDAC4 Knockdown Induces Preeclampsia Cell Autophagy and Apoptosis by miR-29b. Reprod Sci (2020) 28(2):334–42. doi: 10.1007/s43032-020-00286-4
252. Ji Y, Zhang W, Yang J, Li C. MiR-193b inhibits autophagy and apoptosis by targeting IGFBP5 in high glucose-induced trophoblasts. Placenta (2020) 101:185–93. doi: 10.1016/j.placenta.2020.09.015
253. Li YX, Long DL, Liu J, Qiu D, Wang J, Cheng X, et al. Gestational diabetes mellitus in women increased the risk of neonatal infection via inflammation and autophagy in the placenta. Med (Baltimore) (2020) 99(40):e22152. doi: 10.1097/MD.0000000000022152
254. Pan Y, Yan L, Chen Q, Wei C, Dai Y, Tong X, et al. Dysfunction of Shh signaling activates autophagy to inhibit trophoblast motility in recurrent miscarriage. Exp Mol Med (2021) 53(1):52–66. doi: 10.1038/s12276-020-00530-6
255. de la Torre P, Fernández-de la Torre M, Flores AI. Premature senescence of placental decidua cells as a possible cause of miscarriage produced by mycophenolic acid. J Biomed Sci (2021) 28(1):1–14. doi: 10.1186/s12929-020-00704-4
256. Dai Y, Li TH, He X, Yan SB, Gao Y, Chen Y. The Effect and Mechanism of Asymmetric Dimethylarginine Regulating Trophoblastic Autophagy on Fetal Growth Restriction. Reprod Sci (2021) 1–11. doi: 10.1007/s43032-020-00442-w
257. Vangrieken P, Al-Nasiry S, Bast A, Leermakers PA, Tulen CBM, Janssen GMJ, et al. Hypoxia-induced mitochondrial abnormalities in cells of the placenta. PloS One (2021) 16(1):e0245155. doi: 10.1371/journal.pone.0245155
258. Chen Z, Li C, Yuan A, Gu T, Zhang F, Fan X, et al. α-Solanine Causes Cellular Dysfunction of Human Trophoblast Cells via Apoptosis and Autophagy. Toxins (Basel) (2021) 13(1):67. doi: 10.3390/toxins13010067
259. Vangrieken P, Al-Nasiry S, Bast A, Leermakers PA, Tulen CBM, Schiffers PMH, et al. Placental Mitochondrial Abnormalities in Preeclampsia. Reprod Sci (2021) 1–14. doi: 10.1007/s43032-021-00464-y
260. Feng X, Wei Z, Tao X, Du Y, Wu J, Yu Y, et al. PLAC8 promotes the autophagic activity and improves the growth priority of human trophoblast cells. FASEB J (2021) 35(3):e21351. doi: 10.1096/fj.202002075rr
261. Curtis S, Jones CJP, Garrod A, Hulme CH, Heazell AEP. Identification of autophagic vacuoles and regulators of autophagy in villous trophoblast from normal term pregnancies and in fetal growth restriction. J Matern Fetal Neonatal Med (2013) 26(4):339–46. doi: 10.3109/14767058.2012.733764
262. Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Sci (80- ) (2008) 321(5885):117–20. doi: 10.1126/science.1154822
263. Hiyama M, Kusakabe KT, Takeshita A, Sugi S, Kuniyoshi N, Imai H, et al. Nutrient starvation affects expression of LC3 family at the feto-maternal interface during murine placentation. J Vet Med Sci (2015) 77(3):305–11. doi: 10.1292/jvms.14-0490
264. Gao L, Qi H-B, Kamana KC, Zhang X-M, Zhang H, Baker PN. Excessive autophagy induces the failure of trophoblast invasion and vasculature: possible relevance to the pathogenesis of preeclampsia. J Hypertens (2015) 33(1):106–17. doi: 10.1097/HJH.0000000000000366
265. Oh S-Y, Choi S-J, Kyung Hee Kim KH, Cho E, Kim J-H, Roh C-R. Autophagy-Related Proteins, LC3 and Beclin-1, in Placentas From Pregnancies Complicated by Preeclampsia. Reprod Sci (2008) 15(9):912–20. doi: 10.1177/1933719108319159
266. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: Therapeutic implications. Mol Cancer Ther (2011) 10(9):1533–41. doi: 10.1158/1535-7163.MCT-11-0047
267. Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev (2016) 30(17):1913–30. doi: 10.1101/gad.287524.116
268. Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Nature (2014) 504(7479):296–300. doi: 10.1158/2159-8290.CD-14-0362
269. Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, Dipaola RS. Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev (2016) 30(4):399–407. doi: 10.1101/gad.274134.115
270. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev (2013) 27(13):1447–61. doi: 10.1101/gad.219642.113
271. Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun (2014) 5(1):1–15. doi: 10.1038/ncomms4056
272. Mah LY, Ryan KM. Autophagy and cancer. Cold Spring Harbor Perspect Biol (2012) 4(1):a008821. doi: 10.1101/cshperspect.a008821
273. Chung C, Seo W, Silwal P, Jo EK. Crosstalks between inflammasome and autophagy in cancer. J Hematol Oncol (2020) 13(1):100. doi: 10.1186/s13045-020-00936-9
274. Slobodnyuk K, Radic N, Ivanova S, Llado A, Trempolec N, Zorzano A, et al. Autophagy-induced senescence is regulated by p38α signaling. Cell Death Dis (2019) 10(6):1–18. doi: 10.1038/s41419-019-1607-0
275. Lee S, Schmitt CA. The dynamic nature of senescence in cancer. Nat Cell Biol (2019) 21(1):94–101. doi: 10.1038/s41556-018-0249-2
276. Huang T, Song X, Yang Y, Wan X, Alvarez AA, Sastry N, et al. Autophagy and hallmarks of cancer. Crit Rev Oncog (2018) 23(5–6):247–67. doi: 10.1615/CritRevOncog.2018027913
277. Singh SS, Vats S, Chia AYQ, Tan TZ, Deng S, Ong MS, et al. Dual role of autophagy in hallmarks of cancer. Oncogene (2018) 37(9):1142–58. doi: 10.1038/s41388-017-0046-6
278. Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M, et al. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep (2016) 15(8):1660–72. doi: 10.1016/j.celrep.2016.04.065
279. Sarcar B, Li X, Fleming JB. Hypoxia-Induced Autophagy Degrades Stromal Lumican into Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma: A Mini-Review HHS Public Access. J Cancer Treat Diagn (2019) 3(1):22–7. doi: 10.29245/2578-2967/2019/1.1165
280. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev (2011) 25(5):460–70. doi: 10.1101/gad.2016311
281. Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell (2011) 22(2):165–78. doi: 10.1091/mbc.E10-06-0500
282. Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-Driven invasion. Cancer Discovery (2014) 4(4):466–79. doi: 10.1158/2159-8290.CD-13-0841
Keywords: autophagy, placentation, trophoblast and cancer cells, cellular proliferation, migration and invasion, vasculogenic capacity, vascular remodeling, immune evasion
Citation: Carvajal L, Gutiérrez J, Morselli E and Leiva A (2021) Autophagy Process in Trophoblast Cells Invasion and Differentiation: Similitude and Differences With Cancer Cells. Front. Oncol. 11:637594. doi: 10.3389/fonc.2021.637594
Received: 03 December 2020; Accepted: 08 March 2021;
Published: 15 April 2021.
Edited by:
Benjamin Kidder, Wayne State University, United StatesReviewed by:
Xiaoyin Lu, University of Texas MD Anderson Cancer Center, United StatesHua Zhang, First Affiliated Hospital of Chongqing Medical University, China
Copyright © 2021 Carvajal, Gutiérrez, Morselli and Leiva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea Leiva, YW5kcmVhLmxlaXZhQHVzcy5jbA==; Eugenia Morselli, ZW1vcnNlbGxpQGJpby5wdWMuY2w=