 Anjali Rohatgi
Anjali Rohatgi John M. Kirkwood
John M. Kirkwood- Hillman Cancer Center, University of Pittsburgh Medical Center, Pittsburgh, PA, United States
The advent of first and second-generation immune checkpoint blockade (ICI) has resulted in improved survival of patients with metastatic melanoma over the past decade. However, the majority of patients ultimately progress despite these treatments, which has served as an impetus to consider a range of subsequent therapies. Many of the next generation of immunotherapeutic agents focus on modifying the immune system to overcome resistance to checkpoint blockade. ICI resistance can be understood as primary, or acquired—where the latter is the most common scenario. While there are several postulated mechanisms by which resistance, particularly acquired resistance, occurs, the predominant escape mechanisms include T cell exhaustion, upregulation of alternative inhibitory checkpoint receptors, and alteration of the tumor microenvironment (TME) into a more suppressive, anti-inflammatory state. Therapeutic agents in development are designed to work by combating one or more of these resistance mechanisms. These strategies face the added challenge of minimizing immune-related toxicities, while improving antitumor efficacy. This review focuses upon the following categories of novel therapeutics: 1) alternative inhibitory receptor pathways; 2) damage- or pathogen-associated molecular patterns (DAMPs/PAMPs); and 3) immune cell signaling mediators. We present the current state of these therapies, including preclinical and clinical data available for these targets under development.
Introduction
The use of checkpoint inhibitors in melanoma has dramatically changed treatment options for patients with melanoma. Prior to 2011 and the FDA approval of ipilimumab, standard of care options included chemotherapy and high dose IL-2, and in the adjuvant setting, high-dose interferon alpha-2, all of which were associated with limited efficacy and significant toxicity. Targeted therapy with BRAF and MEK inhibition has also been approved for the 50% of melanoma patients with activating BRAF mutations. Despite a promising overall response rate and evidence of durable responses for some, the majority of patients with advanced melanoma have ultimately exhibited progression of disease on or after checkpoint blockade (1). Further, while the majority of patients tolerate therapy, there is risk of significant and sometimes fatal toxicity.
Significant effort has been put into finding ways to re-sensitize tumors after immunotherapy resistance has developed as well as alternative strategies for checkpoint blockade. In fact, the number of trials of combination inhibitors has increased significantly each year, though success of this approach remains to be seen (2). In this review, we will profile some of the most promising strategies in broad categories, including 1) alternative checkpoint receptors 2) DAMPs/PAMPs and 3) immune cell signaling modulators of the TME.
In order to understand the rationale for many of these novel therapies, mechanisms of anti-PD-1/PDL-1 resistance need to be discussed. Resistance is characterized as primary or secondary. The Society for Immunotherapy of Cancer taskforce recently published consensus guidelines to define these terms (3). Primary resistance is defined as progression of disease or at best stable disease for less than 6 months for patients who received a minimum of 6 weeks of therapy. Secondary resistance is defined as nonresponse with progression of disease after initial response to therapy with at least complete response (CR), partial response (PR) or stable disease (SD) of greater than 6 months duration. The mechanisms of resistance to anti-PD-1 therapy are postulated to be diverse, but this remains an area of exploration. In particular, identifying which mechanism or mechanisms are responsible for disease progression in individual patients is an area of ongoing interest.
A full review of immunotherapy escape mechanisms is outside the scope of this review; this topic has been reviewed extensively elsewhere and is summarized briefly here (4–7). Mechanisms attributed to resistance include a lack of target tumor neoantigens, or impaired antigen presentation in the tumor. Further, a lack of tumor immune cell infiltration described as a “cold” tumor, within which the non-inflamed tumor lacks the effector T cell populations that are the basis of benefit from ICI has been reported. Further, impairment of IFNγ secretion or signaling or other inflammatory cytokine responses can lead to resistance (8). This is often accompanied by presence of other types of suppressive immune cells, including M2 macrophages, T regulatory cells (Tregs), and myeloid derived suppressor cells (MDSCs). Alternative checkpoints that may govern the antitumor function of T cells, such as LAG3, TIM3 and other inhibitory receptors are also discussed here, as they can lead to reduction of antitumor cytotoxicity of T cells and are observed in exhausted T cells after chronic antigen stimulation. We will focus on strategies for which clinical data from ongoing trials are anticipated.
A combinatorial approach with ICI has been favored for many of these therapies, both because of limited efficacy seen thus far with many of the single agents being explored, and also because of postulated mechanisms of resistance to immunotherapy. For example, an alternative inhibitory receptor may reduce T-cell exhaustion, but may not be sufficient to promote T effector cytotoxicity without the concurrent administration of anti-PD-1. An example of successful combinatorial therapy is seen with dual-checkpoint inhibition using nivolumab and ipilimumab, as seen in several disease types including melanoma, lung cancer, and renal cell carcinoma. However, increases in response rates compared to ipilimumab monotherapy have come at the expense of increased toxicity (9). Where available, efficacy and toxicity with combination therapy are reported in this review.
Alternative Checkpoint Receptor Pathways
Additional inhibitory checkpoint receptors are being explored as a potential avenue for single agent or combined therapy with anti-PD-1. Many of these receptors were identified in the setting of chronic viral infection, which leads to T cell exhaustion and unresponsiveness to stimuli. Therapies targeting inhibitory receptors are postulated to augment anti-tumor response, perhaps by reversing T cell exhaustion. Most of these are monoclonal antibodies that act via inhibitory receptors to relieve inhibition of T cell activity. This review will focus on effector T cell receptors, however it is worth noting that additional strategies for targeting other suppressive immune cell actors including MDSCs and Tregs are under development.
Lymphocyte Activation Gene-3
Lymphocyte activation gene-3 (LAG3) (CD223) is a type I membrane protein found on the surface of activated T cells, T regulatory cells, NK cells, and plasmacytoid dendritic cells (10). LAG3 demonstrates homology to the costimulatory membrane protein CD4 and binds major histocompatibility complex II (MHCII) (11). LAG3 expression on T cells is upregulated after continued antigenic stimulation, and often co-expressed with additional inhibitory receptors such as PD-1 and TIGIT (12). LAG3 can also be proteolytically cleaved, releasing the external portion to become soluble LAG3, the role of which remains unclear (13). LAG3 has multiple functions in suppressing the immune response, as elucidated by murine knockout models. First, LAG3 decreases CD4+ T cell proliferation and secretion of inflammatory cytokines such as IL-2, IFNγ, and TNFα (14). LAG3 also affects the development of memory CD4+ T cells (15). LAG3 promotes the suppressive activity of Tregs, and inhibition of LAG3 can reduce Treg formation (16, 17). Similar immunosuppressive properties of LAG3 have been observed in human tumor samples (18). These data support LAG3 as an additional promising clinical immunotherapy target.
LAG3-targetting agents are currently under clinical investigation from several companies. Soluble LAG3 peptide (IMP321; eftilagimod alpha) has been tested in several early phase clinical trials for patients with solid tumors, both as monotherapy and as an adjuvant to vaccine development. In a phase I/IIa trial in resected melanoma, IMP321 was used as an adjuvant with a peptide vaccine in which the primary objective was to evaluate the T cell response and toxicity of the therapy (19). Indeed, CD8+ and CD4+ T cell responses were induced in the majority of patients. Additional studies have been conducted in combination with gemcitabine in patients with pancreatic cancer with a best response of stable disease (20), and in combination with paclitaxel for breast cancer with an objective tumor response rate of 50% (partial responses in 15/30 patients) (21). Partial responses have also been seen in combination with pembrolizumab in head and neck carcinoma in a phase II trial (22). Ongoing combinatorial strategies for IMP321 are under investigation.
Further, a number of anti-LAG3 monoclonal antibodies is under development, including BMS-986016 (relatlimab), LAG525, TSR-033, REGN3767, and MK-4280. These antibodies are under therapeutic evaluation in patients with melanoma, both as monotherapy and in combination with anti-PD-1. Further, they are being explored in the neoadjuvant and metastatic settings. Recruitment is ongoing for the majority of these studies. Preliminary data from NCT01968109, reporting the results of relatlimab plus nivolumab in melanoma patients who have received prior immunotherapy was reported at ESMO 2019 with an ORR of 11.5% (23). In a Phase I study of advanced solid tumors, LAG525 was given with or without spartalizumab resulting in the majority of patients discontinuing treatment for progressive disease (79% and 67% respectively) (24). Efficacy was reported as 11 PRs and 1 CR in the combination arm. Together, these data demonstrate less success with second line LAG3 inhibition than had been anticipated, but larger phase II studies in the treatment refractory and treatment naïve setting are needed to assess the potential role of this agent and are forthcoming.
T Cell Immunoglobulin and Mucin-Domain Containing-3
T cell immunoglobulin and mucin-domain containing-3 (Tim-3) is a type I transmembrane protein found on the surface of T cells, NK cells, dendritic cells and macrophages (25). Tim-3 has several ligands including galactin-9, engagement of which results in cell death of Th1 cells (26). Other ligands include ceacam1, which may stabilize Tim-3 on the cell surface, and HMGB1 and phosphatidylserine (27). Tim-3 is also a marker of exhausted T cells, and is often co-expressed on CD8+ T cells with PD-1 (28). Tim-3 is associated with decreased inflammatory cytokine production of IFNγ, and can also enhance the immunosuppressive activity of Foxp3 Tregs, (29) (30). Tim-3 can also contribute to the suppressive TME by promoting the generation of MDSCs (31). In humans, Tim-3 is implicated in autoimmunity as well as chronic viral infections (29, 32). Patients whose tumors exhibit high levels of Tim-3 expression are more likely to have worse prognosis in several tumor types (27).
Similar to LAG3, Tim-3 is an attractive clinical target and several monoclonal antibodies targeting Tim-3 are under investigation including MGB453, TSR-022, Sym023, BGBA425, RO7121661, ICAGN02390, LY3321367, and BMS-986258. Ongoing trials with these antibodies were recently summarized in the review by Acharya, et al (33). Clinical data is forthcoming. LY3321367 alone, or with an anti-PD-L1 therapy, did not produce any dose-limiting toxicities, and was associated with >20% tumor regression (1 PR) in the monotherapy arm (NCT03099109) (34). For patients with NSCLC and melanoma treated with prior anti-PD-1/PD-L1, MBG453 was given with spartalizumab in a phase II study. Of the 33 patients in that study, 15.2% were being treated at the time of abstract presentation, with the remainder discontinuing study due to progressive disease. Grade 3/4 adverse effects including pruritis, amylase and lipase elevation, increased ALT were noted (35). As with the studies of LAG3, mature data and larger studies are in development.
T Cell Immunoreceptor With Immunoglobulin and ITIM Domain
T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT) is another inhibitory receptor on T cells, as its name implies. TIGIT is a found on activated CD8+ and CD4+ T cells, NK cells, Tregs and T follicular cells (36). TIGIT binds to multiple ligands including CD155 and CD112, for which binding competes with the co-stimulatory receptor CD226/DNAM-1 (37). TIGIT is highly expressed in tumor samples and T cells that also express PD-1, suggesting a role in T cell exhaustion (38). In murine models of CT26 colorectal carcinoma, monotherapy with anti-TIGIT therapy did not effect tumor growth; however, when introduced with anti-PD-1 it resulted in a reduction of tumor growth (38). Further, this combination increased percent of tumor-infiltrating IFNγ+ CD8+ T cells (38). The anti-tumor effect of TIGIT may also be mediated by NK cells and enhanced by IL-15 (39).
A number of anti-TIGIT monoclonal antibodies are under development as listed in Table 1. A recent review by Chauvin, et al. lists ongoing Phase I/II clinical trials involving TIGIT, which are primarily being conducted with anti-PD-1/PD-L1 therapies (36). Results from the CITYSCAPE Phase II trial of anti-TIGIT tiragolumab and atezolizumab in patients with PD-L1+ advanced NSCLC demonstrated grade >=3 TRAE in 15% of patients. ORR was higher in patients receiving tiragolumab and atezolizumab (37.3% (CI 25–49.6) compared to those receiving placebo and atezoliumab (20.6% (CI 10.2–30.9), with an odds ratio of 2.57 (CI 1.07–6.14) (40). Tiragolumab with or without atezolizumab was also tested in patients with advanced solid tumors, in a Phase Ia/Ib dose escalation trial with TRAE of ≥ grade 3 in 4% of patients in each phase (41). There were three responses greater than stable disease in the Phase 1b portion, all of which occurred in PD-L1 positive patients. In an additional NSCLC expansion cohort ORR was 50%. Results are not yet available for additional clinical trials.
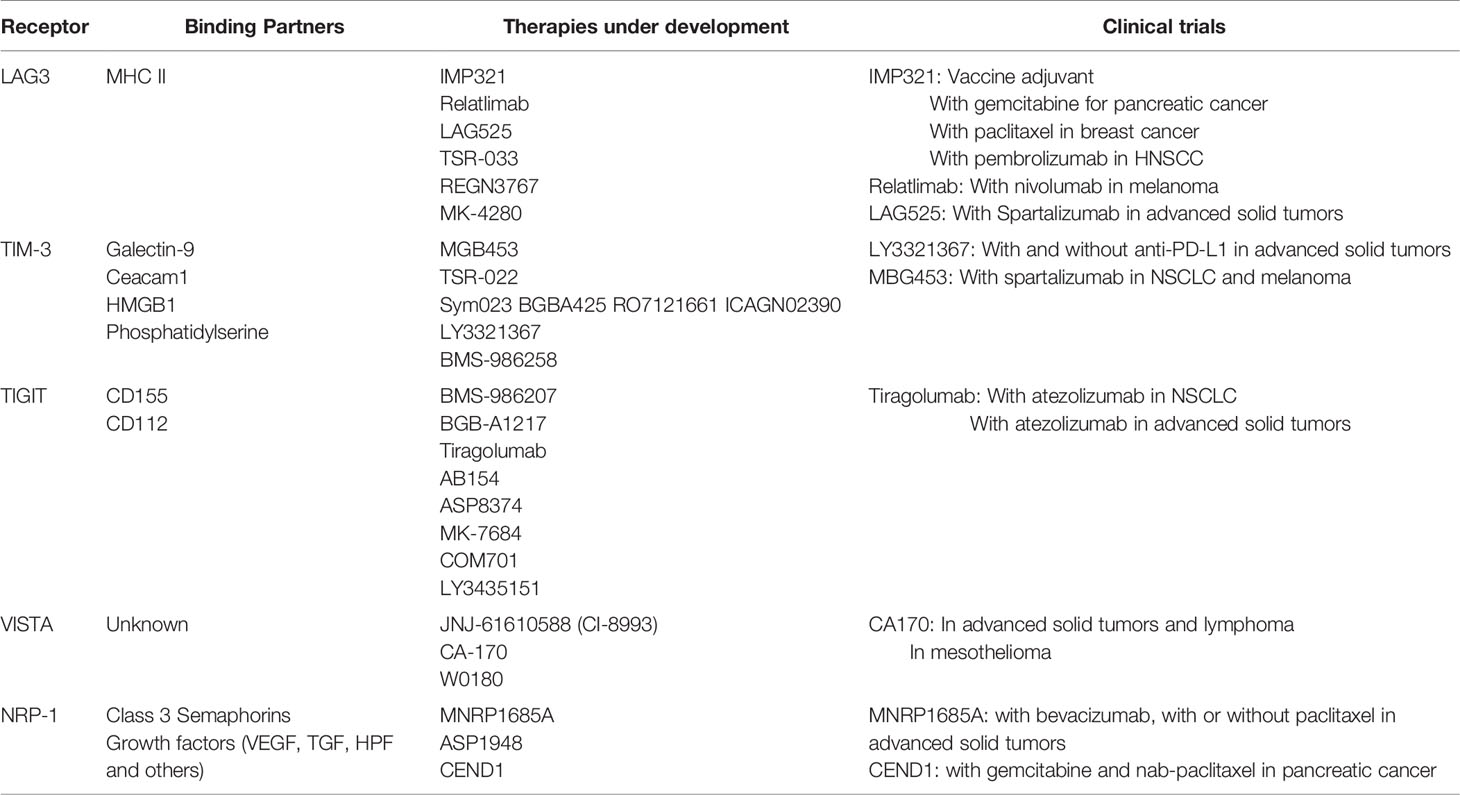
Table 1 Summary of inhibitory receptors in clinical trials.
Additional Inhibitory Receptors
Additional inhibitory receptors under current clinical investigation include V-domain Ig-containing suppressor of T cell Activation (VISTA) (42). VISTA has homology to CD28 family members including PD-1 (43). Data is primarily available in the preclinical setting, but suggests VISTA blockade may reduce tumor growth in melanoma models, and alter the TME by reducing MDSCs and Tregs (44). The small molecule CA-170 which binds both VISTA and PD-1 has been evaluated in phase I trials with patients with advanced solid tumors, lymphomas and mesotheliomas (45, 46). Phase I trials with anti-VISTA monoclonal antibody is also currently underway.
Neuropilin-1 (NRP1) is another inhibitory receptor under investigation for its clinical potential (47). NRP1 may play a role in T cell dysfunction and is highly expressed on PD1+ intratumoral CD8+ T cells. Murine melanoma models exhibited decreased tumor growth with treatment of combination anti-PD-1 and anti-Nrp-1 (48). Early phase clinical trials with two anti-NRP1 agents were published, but do not appear to have been pursued further, in part due to toxicity (49, 50). An anti-NRP1 monoclonal antibody is currently under clinical development in a Phase 1b trial.
Co-Stimulatory Receptors
Another strategy has been to target co-stimulatory receptors with monoclonal antibodies that behave as receptor agonists, with or without anti-PD-1 blockade. These receptors include OX40, CD27, 4-1BB, and GITR. Co-stimulatory receptors are present on T cells and counter-act the negative regulation of inhibitory receptors such as PD-1 and CTLA-4 (51). Phase I studies of anti-OX40 agonists have yielded disappointing results, with a single partial response noted in the trial of MEDI0562 in advanced solid tumors, and best response of stable disease with GSK998 (52, 53). When combined with pembrolizumab in a trial enrolling 96 patients, anti-OX40, gave 2 CRs and 7PRs (53). 4-1BB targeted therapy was complicated by hepatic toxicity that was mitigated at lower doses (54). However, efficacy of monotherapy and combination treatment with anti-PD-1 was not particularly impressive (55). Several companies have dropped their pursuit of co-stimulatory monoclonal antibodies, such as OX40 from their pipelines.
Damage- or Pathogen-Associated Molecular Patterns
Pattern recognition receptors (PRRs) were identified as part of the innate immune system as a first line defense to pathogens. These receptors can recognize pathogen-associated molecular patterns (PAMPs) and include a family of receptors called toll-like receptors (TLRs). Additional receptors have been identified to recognize damage-associated molecular patterns (DAMPs). TLRs are present on both immune and non-immune cell types. Presence of a PAMP/DAMP leads to TLR activation, and downstream activation of transcription factors that result in the production of interferons, and interferon-stimulated responses. In addition to triggering the production of inflammatory cytokines and chemokines, the IFN response is also important for antigen presentation and the priming of an adaptive immune response. Interferon triggers maturation of antigen-presenting cells, leading to presentation of proteins in the context of MHC. This ultimately leads to the induction of specific T and B cell responses. Preclinical work with tumor cell lines and murine models however, have also shown data suggesting TLR stimulation can lead to tumor proliferation (56, 57). Whether the anti-tumor effects of TLR-stimulation can be specifically harnessed is a work in progress.
Although the mechanisms were not known at the time, TLR-agonists were used in early cancer therapy by William Coley (58). In his historic experiments, patients with cancer were injected with lipopolysaccharide (LPS)-containing bacterial concoctions, and tumor regression was occasionally noted. Unsurprisingly, these patients also developed high fevers and other intolerable side effects. LPS was ultimately identified in these bacterial cocktails as the active agent. Since that time many strategies to utilize PRRs in cancer therapy have been exploited as described below and summarized in Table 2.
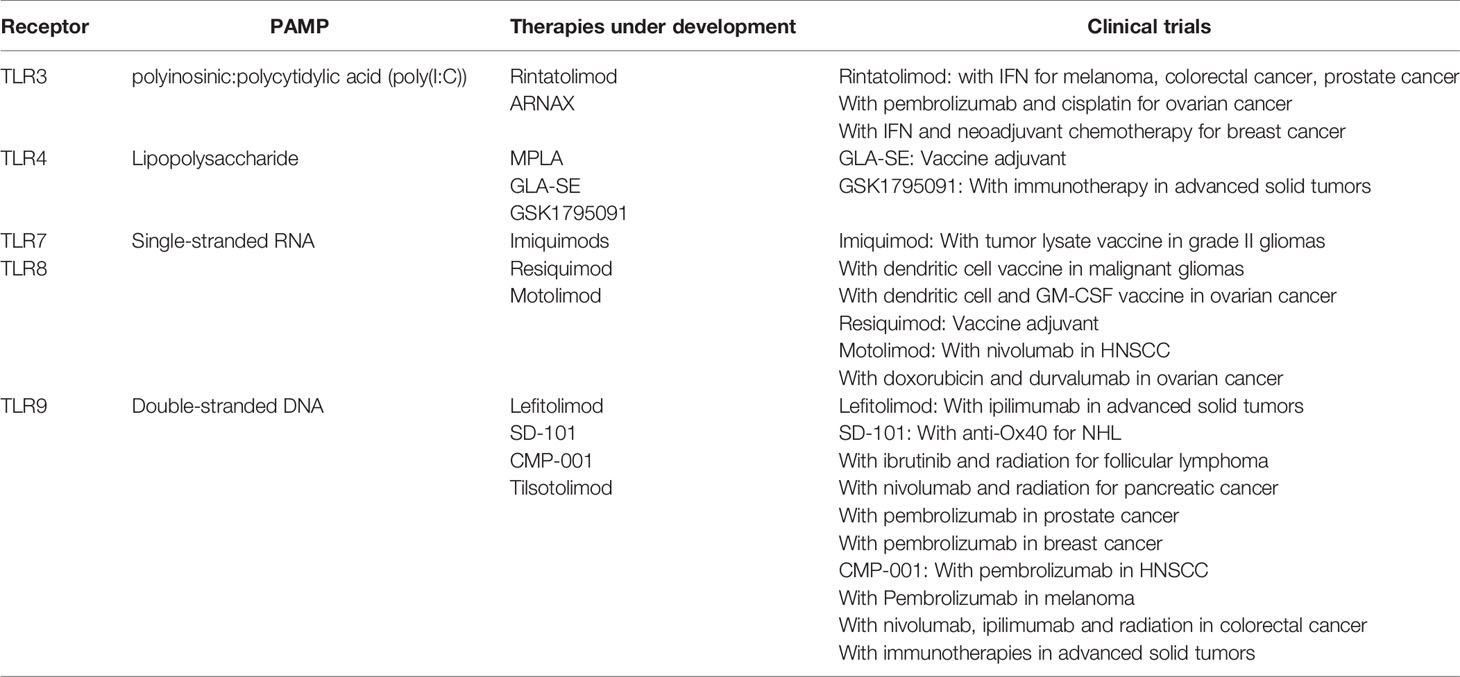
Table 2 Toll-like receptor (TLR) agonists under investigation.
Toll-Like Receptor Agonists
Toll-Like Receptors That Bind Nucleic Acids
TLRs recognizing nucleic acids have been under investigation and promising for some time as therapeutic agents for cancer. These include TLR3, TLR7, TLR8, and TLR9. TLR3 recognizes dsRNA, and a common synthetic nucleic acid used for stimulation is polyinosinic:polycytidylic acid (poly(I:C)). Early studies had issues with toxicity and stability and were discontinued (59). Further, phase I studies with poly(I:C) monotherapy appeared to have little clinical efficacy (60). Since that time, several compounds modified to enhance stability have been produced and are being studied in various malignancies in combination with immunotherapy and as adjuvants in vaccine-based strategies (61). Early phase clinical trials are ongoing with the formulations rintatolimod and ARNAX.
TLR7 and TLR8 recognize single-stranded RNA. Stimulation results in activation of MyD88, and secretion of cytokines, including type I interferons. The synthetic imidazoquinolones have been tested as antiviral treatments, and now derivatives are being explored as cancer therapeutics. Imiquimod is compound used topically to treat several skin conditions, including basal cell carcinomas and is being explored in pre-cancerous lesions, such as cervical intraepithelial neoplasia. Imiquimod has also been used in early phase clinical trials for treatment of cutaneous metastases for breast cancer and melanoma, however has not been pursued further (62, 63). Imiquimods have been explored as an adjuvant cancer vaccines and is being used in a variety of these trials (NCT01678352, NCT00799110, NCT01792505) (64). Resiquimod binds both TLR7 and TLR8 and is similarly being explored as an adjuvant in cancer vaccines (65). Motolimod (VTX-2337) is another imidazoquinolone with preclinical evidence supporting activation of NK cells and priming of CD8+ T cells when given with cetuximab (66, 67). A phase 1b clinical trial of motolimod and cetuximab in patients with head and neck squamous cell carcinoma resulted in demonstration of maximum tolerated dose with 2/13 patients achieving partial responses (68). Studies are ongoing with motolimod in combination with anti-PD1 for patients with head and neck cancer (NCT03906526), as well as with doxorubicin and durvalumab in patients with ovarian cancer (NCT02431559).
TLR9 recognizes double-stranded DNA, typically in the form of unmethylated cytidine phosphate guanosine (CpG) oligonucleotides (ODN) present in pathogens. Several synthetic CpG-ODNs have been investigated for cancer therapy, including in combination with chemotherapy, immunotherapy or as vaccine adjuvants. Lefitolimod (MGN1703) is a DNA molecule tested in a phase I dose escalation with ipilimumab, with planned dose escalation (NCT02668770) (69). SD-101 is another CpG-ODN that has been tested intratumorally in combination with pembrolizumab in a phase 1b clinical trial with an ORR of 15% (70). A number of trials are ongoing evaluating SD-101 in hematologic malignancies, as well as in pancreatic cancer (NCT04050085) and prostate cancer (NCT03007732). CMP-001 is a CpG-A virus-like particle that stimulates TLR9. Intratumoral CMP-001 with pembrolizumab has been shown in a Phase 1b trial demonstrating 24% ORR in melanoma patients who have previously progressed on anti-PD-1 therapy (71). Additional trials are evaluating CMP-001 by subcutaneous administration (NCT03084640), in the neoadjuvant setting (NCT04401995), and in other solid tumors and hematologic malignancies. Tilsotolimod (IMO-2125) is another TLR9 agonist being investigated in combination with nivolumab and ipilimumab for patients with solid tumors (NCT03865082), with preliminary data from a phase 1/2 trial only available by press release thus far.
Toll-Like Receptors That Recognize Other Bacterial Products
Additional TLRs target bacterial components. TLR1,2,6, and 4 bind bacterial cell wall products and TLR5 binds flagellin. TLR2 is found on the cell surface and forms a heterodimer with either TLR1 or TLR6 that recognized lipoproteins (72). Although there are several synthetic lipoproteins targeting these receptors being explored in autoimmunity or as adjuvants in therapy of viral infection, none are actively explored in malignancies. BCG is used to treat non-muscle-invasive bladder cancers, and may work in part by inducing inflammation through TLR2 and TLR4 (73). TLR4 agonists have been explored extensively since Coley’s initial observations. An LPS derivative of Salmonella, monophosphoryl lipid A (MPLA) is a TLR4 agonist that is currently used as an adjuvant in the Cervarix HPV vaccine (74). A dose escalation studies of lipid A formulations as monotherapy for were tolerated at lower doses, but did not demonstrate anti-tumor responses as monotherapy (75, 76). TLR4 agonists such as GLA-SE have been used as adjuvants in vaccines directed at malignancies or infectious agents, but are not actively being developed in oncologic trials (77). Currently, GSK1795091 synthetic agonist was tested in healthy volunteers, and a planned trial in combination with immunotherapy in advanced solid tumors is planned (NCT03447314) (78). TLR5 binds flagella, and has been targeted by Mobilan, an adenoviral vector expressing flagellin. Mobilan has been tested in a phase I trial with intratumoral injection for prostate cancer, though has not been pursued further (79).
Additional Pathogen Recognition Receptors
Additional PRRs have been identified since the initial discovery of TLRs and include RIG-I like receptors (RLRs), NOD-like receptors (NLRs) and C-type lectin receptors (CLRs). The work on these receptors has not been as well-developed as for the TLRs, but are now being explored in cancer immunity. RLRs include two receptors, retinoic acid-inducible gene I protein (RIG-I) and melanoma differentiation associated protein 5 (MDA5) that bind viral RNA molecules. Downstream signaling results in interferon secretion. The RIG-I agonist, MK4621, was tested in a phase I/II trial for advanced solid tumors without dose-limiting toxicities observed (80) and is planned for study in combination with pembrolizumab (NCT03739138). NLRs recognize bacterial products and lead to activation of the inflammasome and IL-1β production. NLR activation may contribute to carcinogenesis, and is being explored for potential therapeutic targets (81, 82). CLRs are a large group of PRRs that bind a wide variety of ligands and can result in pro-inflammatory and anti-inflammatory responses, and are trying to be understood (83). STING is an additional nucleic acid PRR located in the endoplasmic reticulum (84). Many early phase clinical trials are underway with STING agonists as monotherapy, in combination with immunotherapy, chemotherapy, or radiation (85).
Immune Signaling Mediators
Cytokines
Cytokines in the TME are produced by infiltrating tumor and stromal cells, and can contribute to either a pro-inflammatory or anti-inflammatory milieu (86). Cytokine therapy has FDA approved indications with IL-2 for renal cell carcinoma and melanoma, and type I interferon for adjuvant therapy of melanoma, and in CML and MPNs. While these therapies are used infrequently in melanoma in favor of current ICI immunotherapy, a small number of patients do well, and may have been cured with IL-2 in metastatic RCC (87). GM-CSF has also been investigated for its potential benefits in cancer therapy. The GM-CSF modality is currently an underlying basis of the benefits of talimogene laherparepvec, an FDA-approved oncolytic immunotherapy for melanoma (88). The body of literature discussing the many functions of cytokines in cancer therapy is vast, and this review will focus on those therapies currently under development.
Toxicity has been a significant issue with cytokine administration in clinical trials and standard therapies, as evidenced by the experience with IL-12. Preclinical data demonstrated the anti-tumor effects of IL-12 by multiple mechanisms and in several murine tumor models (89). However, although a dose was selected in phase I trials to minimize toxicity, the phase II study was halted after significant toxicity and 2 patient deaths (90, 91). The additional toxicities seen in the phase II trial were thought to be a result of a change in the dosing schedule, when a priming initial dose was no longer given. Interest in IL-12 persists, with formulations such as NHS-IL12, an IL12 heterodimer fused to an antibody, tested in a phase I trial (92). A number of ongoing trials with this compound are ongoing, as monotherapy and in combination with immunotherapy. Additional studies use IL-12 expressing viral or CART constructs.
IL-15 shares part of the IL-2 receptor and signaling pathway, and similarly results in NK and T cell proliferation (93). Unlike IL-2, however, IL-15 is not thought to stimulate T regulatory cells, making it an attractive target. Recombinant IL-15 (rhIL15) has been studied in patients with advanced solid tumors in a phase I study, notable for increased NK and CD8+ T cell proliferation seen in the peripheral blood (94). Subcutaneous rhIL15 along with haploidentical NK cell infusion was used to treat patients with acute myeloid leukemia, demonstrating NK cell proliferation and 40% remission rates, though cytokine release syndrome was noted in 56% of these patients (95). ALT-803 is a IL-15/IL-15Ra Fc fusion complex, referred to a superagonist, that has been tested in PD-1 refractory NSCLC patients evaluated in phase Ib study, with additional studies ongoing (96). Other formulations of IL-15 have been developed and are undergoing clinical trials, including recombinant proteins BJ-001, PF-07209960, NIZ985, and N-803. Additional combinations of IL-15 products are being conducted with immunotherapy, and as part of adoptive cell therapy products.
Interferon-γ is a type III interferon potently induced by IL-12, whose expression is associated with immunotherapy response in melanoma (8). However, IFNγ has been associated with both anti-tumor and pro-tumor effects (97). Clinical trials with IFNγ as monotherapy have not been fruitful, perhaps due to its seemingly contradictory role in the TME. Other cytokines initially pursued and since abandoned for toxicity and lack of efficacy include IL-21 and IL-7 (98).
Small Molecule Inhibitors
In addition to traditional cytokines and chemokines, the TME also contains a number of small molecules that effect the inflammatory state of the tumor. Indoleamine 2,3-dioxygenase 1 (IDO1) helps convert tryptophan to kynurenine, which has an immunosuppressive effect on the TME. Kynurenine promotes development of Tregs and MDSCs (99). Epacadostat is an IDO1 inhibitor that has been studied in combination with pembrolizumab. Although the initial phase I/II trial in advanced solid tumors showed promise, the phase III trial in melanoma did not show a difference in progression-free survival or overall survival versus placebo with pembrolizumab (100, 101). Many of the ongoing trials of IDO1 inhibitors have since been terminated, though a few trials with IDO1 inhibitor BMS-986205 are still recruiting.
The phosphoinositide 3-kinase (PI3K) signaling pathway functions at many stages of cancer biology including cell division, differentiation, motility and metabolism (102). Inhibitors downstream of the PI3K pathway are active in some solid tumors, including everolimus in neuroendocrine tumors (103) and everolimus with exemestane in breast cancer (104). Targeting the PI3K isoforms γ and δ, that are specifically expressed in hematopoietic cells, is an area of investigation supported by preclinical work showing alterations in the TME to a pro-inflammatory phenotype (105). PI3Kγ and γ/δ inhibition are being studied in clinical trials.
Concluding Remarks
The strategies discussed in this review highlight a number of promising approaches for overcoming immunotherapy resistance, a significant treatment dilemma for patients with advanced melanoma. These therapies all aim to increase local inflammation in the TME but by drastically different mechanisms, with varying routes of administration and toxicities. Of these, use of DAMPs/PAMPs are of particular promise, and early phase trials have shown intratumoral and administration with anti-PD-1 to be tolerable with early signs of efficacy. Development of intravenous formulations or formulations compounded with anti-PD-1 monoclonal antibodies could be an interesting avenue to explore. We look forward to more data in this field and with other tumor types.
One major challenge in the development and testing of novel immunotherapeutics is the heterogeneity of mechanisms of resistance. Patients have varying expression of inhibitory receptors after immunotherapy, differing levels and types of immune infiltrates, and differences in mutational profiles and epigenetic changes that can all alter response to immunotherapy (106). This heterogeneity may also contribute to the limited efficacy observed in some trials. However, without definitive biomarkers to reliably sort patients by mechanism, this truly personalized approach remains currently out of reach. While some biomarkers are certainly helpful, such as TMB and PD-L1 expression, even these do not always correlate with response (107).
Ongoing efforts to identify biomarkers are underway, including with gene-expression profiling, such as those signatures associated with IFNγ (8, 108). Until robust biomarkers are identified and then correlated with response to specific therapies, an all-comers approach must be utilized. After validation of a biomarker-based treatment approach, these therapies could also be explored in the front-line setting, perhaps identifying those at risk for primary resistance to immunotherapy, and ultimately leading to greater portion of those with durable responses. Cost-benefit analysis would also be an important factor in the design of a biomarker-driven, personalized medicine approach to avoid contributing to the already egregious cost of oncologic care that may benefit a small portion of patients.
The strategies discussed in this review are only a part of the approach being considered for overcoming immunotherapy resistance, and a number of other promising strategies that are under development. These include vaccine development with tumor-associated antigens, in part with the adjuvants mentioned here. Other strategies that may in the near future gain regulatory approval include adoptive cell transfer, both with the use of TILs and perhaps with CAR-T therapies. Finally, targeting other elements of the TME that are a more fundamental basis of immunotherapy resistance, such as myeloid derived suppressor cells and T regulatory cells are also under development. There is much reason for excitement given the breadth and pace of development of immunonotherapeutics and forthcoming results in the next several years will dictate the future of the field.
Author Contributions
AR contributed to the design, concept, research, writing, and critical review of this review. JK contributed to the design, concept, critical review, and supervision of this review. All authors contributed to the article and approved the submitted version.
Funding
The work of the authors is supported by the NIH National Cancer Institute SPORE in Skin CAncer grant P50 CA-121973.
Conflict of Interest
JK has received grants and personal fees for consultancy from Amgen, Bristol-Myers Squibb, Checkmate and Novartis, and grants from Castle Biosciences, Immunocore, and Iovance.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated Melanoma. N Engl J Med (2015) 373:23–34. doi: 10.1056/NEJMc1509660
2. Tang J, Yu JX, Hubbard-Lucey VM, Neftelinov ST, Hodge JP, Lin Y. The clinical trial landscape for PD1 / PD-L1 immune checkpoint inhibitors. Nat Rev Drug Discov (2018) 17:854–55. doi: 10.1038/nrd.2018.210
3. Kluger HM, Zito CR, Barr ML, Baine MK, Chiang VLS, Sznol M, et al. Characterization of PD-L1 expression and associated T-cell infiltrates in metastatic melanoma samples from variable anatomic sites. Clin Cancer Res (2015) 21:3052–60. doi: 10.1158/1078-0432.CCR-14-3073
4. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer (2018) 118:9–16. doi: 10.1038/bjc.2017.434
5. Kim TK, Herbst RS, Chen L. De fi ning and Understanding Adaptive Resistance in Cancer Immunotherapy. Trends Immunol (2018) 39:624–31. doi: 10.1016/j.it.2018.05.001
6. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B. Tumor-Intrinsic and -Extrinsic Factors. Cancer (2016) 28:1255–69. doi: 10.1016/j.immuni.2016.06.001
7. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer (2019) 19:133–50. doi: 10.1038/s41568-019-0116-x
8. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest (2017) 127:2930–40. doi: 10.1172/JCI91190
9. Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol (2018) 19:1480–92. doi: 10.1016/S1470-2045(18)30700-9
10. Workman CJ, Rice DS, Dugger KJ, Kurschner C, Vignali DAA. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3). Eur J Immunol (2002) 32:2255–63. doi: 10.1002/1521-4141(200208)32:8<2255::AID-IMMU2255>3.0.CO;2-A
11. Huard B, Mastrangeli R, Prigent P, Bruniquel D, Donini S, El-Tayar N, et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc Natl Acad Sci U S A (1997) 94:5744–9. doi: 10.1073/pnas.94.11.5744
12. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
13. Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J (2007) 26:494–504. doi: 10.1038/sj.emboj.7601520
14. Huard B, Tournier M, Hercend T, Triebel F, Faure F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. Eur J Immunol (1994) 24:3216–21. doi: 10.1002/eji.1830241246
15. Workman CJ, Cauley LS, Kim I-J, Blackman MA, Woodland DL, Vignali DAA, et al. Lymphocyte Activation Gene-3 (CD223) Regulates the Size of the Expanding T Cell Population Following Antigen Activation In Vivo. J Immunol (2004) 172:5450–5. doi: 10.4049/jimmunol.172.9.5450
16. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
17. Durham NM, Nirschl CJ, Jackson CM, Elias J, Kochel CM, Anders RA, et al. Lymphocyte activation gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed In Vivo. PloS One (2014) 9:1–13. doi: 10.1371/journal.pone.0109080
18. Andrews LP, Marciscano AE, Drake CG, Vignali DAA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev (2017) 276:80–96. doi: 10.1111/imr.12519
19. Legat A, Maby-El Hajjami H, Baumgaertner P, Cagnon L, Maillard SA, Geldhof C, et al. Vaccination with LAG-3Ig (IMP321) and peptides induces specific CD4 and CD8 T-cell responses in metastatic melanoma patients-report of a phase I/IIa clinical trial. Clin Cancer Res (2016) 22:1330–40. doi: 10.1158/1078-0432.CCR-15-1212
20. Wang-Gillam A. A phase i study of IMP321 and gemcitabine as the front-line therapy in patients with advanced pancreatic adenocarcinoma. Invest New Drugs (2013) 31:707–13. doi: 10.1007/s10637-012-9866-y
21. Brignone C, Gutierrez M, Mefti F, Brain E, Jarcau R, Cvitkovic F, et al. First-line chemoimmunotherapy in metastatic breast carcinoma: Combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J Transl Med (2010) 8:1–11. doi: 10.1186/1479-5876-8-71
22. Felip E, Doger B, Majem M, Carcereny E, Krebs M, Peguero JA, et al. Initial results from a phase II study (TACTI-002) in metastatic non-small cell lung or head and neck carcinoma patients receiving eftilagimod alpha (soluble LAG-3 protein) and pembrolizumab. J Clin Oncol (2020) 38:3100. doi: 10.1200/JCO.2020.38.15_suppl.3100
23. Ascierto PA, Bono P, Bhatia S, Melero I, Nyakas MS, Svane I-M, et al. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti–PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched popu. Ann Oncol (2017) 28:v611–2. doi: 10.1093/annonc/mdx440.011
24. Hong DS, Schoffski P, Calvo A, Sarantopoulos J, Ochoa De Olza M, Carvajal RD, et al. Phase I/II study of LAG525 ± spartalizumab (PDR001) in patients (pts) with advanced malignancies. J Clin Oncol (2018) 36:3012. doi: 10.1200/JCO.2018.36.15_suppl.3012
25. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415:536–41. doi: 10.1038/415536a
26. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6:1245–52. doi: 10.1038/ni1271
27. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev (2017) 276:97–111. doi: 10.1111/imr.12520
28. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207:2175–86. doi: 10.1084/jem.20100637
29. Koguchi K, Anderson DE, Yang L, O’Connor KC, Kuchroo VK, Hafler DA. Dysregulated T cell expression of TIM3 in multiple sclerosis. J Exp Med (2006) 203:1413–8. doi: 10.1084/jem.20060210
30. Sakuishi K, Ngiow SF, Sullivan JM, Teng MWL, Kuchroo VK, Smyth MJ, et al. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology (2013) 2. doi: 10.4161/onci.23849
31. Dardalhon V, Anderson AC, Karman J, Apetoh L, Chandwaskar R, Lee DH, et al. Tim-3/Galectin-9 Pathway: Regulation of Th1 Immunity through Promotion of CD11b + Ly-6G + Myeloid Cells. J Immunol (2010) 185:1383–92. doi: 10.4049/jimmunol.0903275
32. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med (2008) 205:2763–79. doi: 10.1084/jem.20081398
33. Acharya N, Acharya N, Sabatos-Peyton C, Anderson AC, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer (2020) 8:1–11. doi: 10.1136/jitc-2020-000911
34. Harding JJ, Patnaik A, Moreno V, Stein M, Jankowska AM, Velez de Mendizabal N, et al. A phase Ia/Ib study of an anti-TIM-3 antibody (LY3321367) monotherapy or in combination with an anti-PD-L1 antibody (LY3300054): Interim safety, efficacy, and pharmacokinetic findings in advanced cancers. J Clin Oncol (2019) 37:12. doi: 10.1200/JCO.2019.37.8_suppl.12
35. Mach N, Curigliano G, Santoro A, Kim D-W, Tai DWM, Hodi S, et al. Phase (Ph) II study of MBG453 + spartalizumab in patients (pts) with non-small cell lung cancer (NSCLC) and melanoma pretreated with anti–PD-1/L1 therapy. Ann Oncol (2019) 30:v491–2. doi: 10.1093/annonc/mdz253.028
36. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer (2020) 8:1–7. doi: 10.1136/jitc-2020-000957
37. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med (2003) 198:557–67. doi: 10.1084/jem.20030788
38. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
39. Chauvin J-M, Ka M, Pagliano O, Menna C, Ding Q, DeBlasio R, et al. IL15 Stimulation with TIGIT Blockade Reverses CD155-mediated NK-Cell Dysfunction in Melanoma. Clin Cancer Res (2020) 26:5520–33. doi: 10.1158/1078-0432.CCR-20-0575
40. Rodriguez-Abreu D, Johnson ML, Hussein MA, Cobo M, Patel AJ, Secen NM, et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J Clin Oncol (2020) 38:9503. doi: 10.1200/JCO.2020.38.15_suppl.9503
41. Bendell JC, Bedard P, Bang Y-J, LoRusso P, Hodi S, Gordon M, et al. CT302 - Phase Ia / Ib dose-escalation study of the anti-TIGIT antibody tiragolumab as a single agent and in combination with atezolizumab in patients with advanced solid tumors. Cancer Res (2020) 80:CT302–CT302. doi: 10.1158/1538-7445. AM2020-CT302
42. Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory functions of VISTA. Immunol Rev (2017) 276:66–79. doi: 10.1111/imr.12525
43. Wang L, Rubinstein P, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med (2011) 208:577–92. doi: 10.1084/jem.20100619
44. Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res (2014) 74:1933–45. doi: 10.1158/0008-5472.CAN-13-1506
45. Zauderer M, Brody J, Marron T, Pacey S, Martell R, Wang H, et al. First-in-class small molecule CA-170 targeting VISTA: a report on efficacy outcomes from a cohort of 12 malignant pleural mesothelioma (MPM) patients in study CA170-101. J Immunother Cancer (2019) 7:283. doi: 10.1186/s40425-019-0764-0
46. Bang Y-L, Sosman JA, Daud A, Meric-Bernstam F, Garcia-Corbacho J, Patel MR, et al. Phase 1 study of CA-170, a first-in-class, orally available, small molecule immune checkpoint inhibitor (ICI) dually targeting VISTA and PD-L1, in patients with advanced solid tumors or lymphomas. in P341. (2018) 6:175. doi: 10.1186/s40425-018-0422-y
47. Chuckran CA, Liu C, Bruno TC, Workman CJ, Vignali DAA. Neuropilin-1: a checkpoint target with unique implications for cancer immunology and immunotherapy. J Immunother Cancer (2020) 8:1–12. doi: 10.1136/jitc-2020-000967
48. Leclerc M, Voilin E, Gros G, Corgnac S, de Montpréville V, Validire P, et al. Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nat Commun (2019) 10:1–14. doi: 10.1038/s41467-019-11280-z
49. Weekes CD, LoRusso P, Ramakrishnan V, Shih LM, Darbonne WC, Hegde P, et al. A phase Ib study for MNRP1685A (anti-NRP1) administered intravenously with bevacizumab with or without paclitaxel to patients with advanced solid tumors. J Clin Oncol (2011) 29:3050. doi: 10.1200/jco.2011.29.15_suppl.3050
50. Dean A, Gill S, McGregor M, Broadbridge V, Jarvelainen HA, Price TJ. 1528P Phase I trial of the first-in-class agent CEND-1 in combination with gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer. Ann Oncol (2020) 31:S941. doi: 10.1016/j.annonc.2020.08.2011
51. Krummel BMF, Allison JR. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med (1995) 182:459–65. doi: 10.1084/jem.182.2.459
52. Glisson B, Leidner R, Ferris RL, Powderly J, Rizvi NA, Keam B, et al. Safety and clinical activity of MEDI0562, a humanized OX40 agonist monoclonal antibody, in adult patients with advanced solid tumors. Ann Oncol (2018) 29:viii410. doi: 10.1093/annonc/mdy288.025
53. Postel-Vinay S, Lam VK, Ros W, Bauer TM, Hansen AR, Cho DC, et al. Abstract CT150: A first-in-human phase I study of the OX40 agonist GSK3174998 (GSK998) +/- pembrolizumab in patients (Pts) with selected advanced solid tumors (ENGAGE-1). Tumor Biol (2020) 3174998:CT150–0. doi: 10.1158/1538-7445.AM2020-CT150
54. Segal NH, He AR, Doi T, Levy TM, Bhatia S, Pishvaian MJ, et al. Phase i study of single-agent utomilumab (PF-05082566), a 4-1bb/cd137 agonist, in patients with advanced cancer. Clin Cancer Res (2018) 24:1816–23. doi: 10.1158/1078-0432.CCR-17-1922
55. Massarelli E. Clinical safety and efficacy as- sessment of the CD137 agonist urelumab alone and in combination with nivolumab in patients with hematologic and solid tumor malignancies. In: Proceeding of the 31st Annual Meeting and Associated Programs of the Society for Immunotherapy of Cancer (2016) 4:5. Available at: https://jitc.biomedcentral.com/articles/10.1186/s40425-016-0172-7
56. Pinto A, Morello S, Sorrentino R. Lung cancer and toll-like receptors. Cancer Immunol Immunother (2011) 60:1211–20. doi: 10.1007/s00262-011-1057-8
57. Rakoff-nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer (2009) 9:57–63. doi: 10.1038/nrc2541
58. Birbriar A. Tumor Microenvironment. In: Tumor Microenvironment Siemann DW (ed.). Chichester, UK: John Wiley and Sons, Ltd (2010). doi: 10.1002/9780470669891
59. Vanpouille-Box C, Hoffmann JA, Galluzzi L. Pharmacological modulation of nucleic acid sensors — therapeutic potential and persisting obstacles. Nat Rev Drug Discovery (2019) 18:845–67. doi: 10.1038/s41573-019-0043-2
60. Stevenson HC, Abrams PG, Schoenberger CS, Smalley RB, Herberman RB, Foon KA. A phase I evaluation of poly(I,C)-LC in cancer patients. J Biol Response Mod (1985) 4:650–5.
61. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T, Matsumoto M. A TLR3-Specific Adjuvant Relieves Innate Resistance to PD-L1 Blockade without Cytokine Toxicity in Tumor Vaccine Immunotherapy. Cell Rep (2017) 19:1874–87. doi: 10.1016/j.celrep.2017.05.015
62. Green DS, Bodman-Smith MD, Dalgleish AG, Fischer MD. Phase I/II study of topical imiquimod and intralesional interleukin-2 in the treatment of accessible metastases in malignant melanoma. Br J Dermatol (2007) 156:337–45. doi: 10.1111/j.1365-2133.2006.07664.x
63. Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L, et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res (2012) 18:6748–57. doi: 10.1158/1078-0432.CCR-12-1149
64. Adams S, O’Neill DW, Nonaka D, Hardin E, Chiriboga L, Siu K, et al. Immunization of Malignant Melanoma Patients with Full-Length NY-ESO-1 Protein Using TLR7 Agonist Imiquimod as Vaccine Adjuvant. J Immunol (2008) 181:776–84. doi: 10.4049/jimmunol.181.1.776
65. Sabado RL, Pavlick A, Gnjatic S, Cruz CM, Vengco I, Hasan F, et al. Resiquimod as an immunologic adjuvant for NY-ESO-1 protein vaccination in patients with high-risk melanoma. Cancer Immunol Res (2015) 3:278–87. doi: 10.1158/2326-6066.CIR-14-0202
66. Lu H, Dietsch GN, Matthews MAH, Yang Y, Ghanekar S, Inokuma M, et al. VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin Cancer Res (2012) 18:499–509. doi: 10.1158/1078-0432.CCR-11-1625
67. Stephenson RM, Lim CM, Matthews M, Dietsch G, Hershberg R, Ferris RL. TLR8 stimulation enhances cetuximab-mediated natural killer cell lysis of head and neck cancer cells and dendritic cell cross-priming of EGFR-specific CD8+ T cells. Cancer Immunol Immunother (2013) 62:1347–57. doi: 10.1007/s00262-013-1437-3
68. Chow LQM, Morishima C, Eaton KD, Baik CS, Goulart BH, Anderson LN. Phase Ib trial of the toll-like receptor 8 agonist, motolimod (VTX-2337), combined with cetuximab in patients with recurrent or metastatic SCCHN. Clin Cancer Res (2017) 23:2442–50. doi: 10.1158/1078-0432.CCR-16-1934
69. Reilley M, Tsimberidou AM, Piha-Paul SA, Yap TA, Fu S, Naing A. Phase 1 trial of TLR9 agonist lefitolimod in combination with CTLA-4 checkpoint inhibitor ipilimumab in advanced tumors. J Clin Oncol (2019) 37:TPS2669–TPS2669. doi: 10.1200/JCO.2019.37.15_suppl.TPS2669
70. Ribas A, Medina T, Kummar S, Amin A, Kalbasi A, Drabick JJ, et al. Sd-101 in combination with pembrolizumab in advanced melanoma: Results of a phase ib, multicenter study. Cancer Discovery (2018) 8:1250–7. doi: 10.1158/2159-8290.CD-18-0280
71. Milhem M, Zakharia Y, Davar D, Buchbinder E, Medina T, Daud A, et al. Durable responses in anti-PD-1 refractory melanoma following intratumoral injection of a Toll-like receptor 9 (TLR9) agonist, CMP-001, in combination with pembrolizumab. SITC 2019 Annu Meet (2019) O85. doi: 10.1136/LBA2019.4
72. Oliveira-Nascimento L, Massari P, Wetzler LM. The role of TLR2 ininfection and immunity. Front Immunol (2012) 3:1–17. doi: 10.3389/fimmu.2012.00079
73. Méndez-Samperio P, Belmont L, Miranda E. Mycobacterium bovis BCG Toll-Like Receptors 2 and 4 Cooperation Increases the Innate Epithelial Immune Response. Arch Med Res (2008) 39:33–9. doi: 10.1016/j.arcmed.2007.06.019
74. Vacchelli E, Galluzzi L, Eggermont A, Fridman WH, Galon J, Sautès-Fridman C, et al. Trial watch: FDA-approved toll-like receptor agonists for cancer therapy. Oncoimmunology (2012) 1:894–907. doi: 10.4161/onci.20931
75. Vosika GJ, Barr C, Gilbertson D. Phase-I study of intravenous modified lipid A. Cancer Immunol Immunother (1984) 18:107–12. doi: 10.1007/BF00205743
76. Isambert N, Fumoleau P, Paul C, Ferrand C, Zanetta S, Bauer J, et al. Phase I study of OM-174, a lipid A analogue, with assessment of immunological response, in patients with refractory solid tumors. BMC Cancer (2013) 13:1–10. doi: 10.1186/1471-2407-13-172
77. Mahipal A, Ejadi S, Gnjatic S, Kim-Schulze S, Lu H, ter Meulen JH, et al. First-in-human phase 1 dose-escalating trial of G305 in patients with advanced solid tumors expressing NY-ESO-1. Cancer Immunol Immunother (2019) 68:1211–22. doi: 10.1007/s00262-019-02331-x
78. Hug BA, Matheny CJ, Burns O, Struemper H, Wang X, Washburn ML, et al. Safety, Pharmacokinetics, and Pharmacodynamics of the TLR4 Agonist GSK1795091 in Healthy Individuals: Results from a Randomized, Double-blind, Placebo-controlled, Ascending Dose Study. Clin Ther (2020) 42:1519–1534.e33. doi: 10.1016/j.clinthera.2020.05.022
79. Eremina NV, Kazey VI, Mishugin SV, Leonenkov RV, Pushkar DY, Mett VL, et al. First-in-human study of anticancer immunotherapy drug candidate mobilan: Safety, pharmacokinetics and pharmacodynamics in prostate cancer patients. Oncotarget (2020) 11:1273–88. doi: 10.18632/oncotarget.27549
80. Middleton MR, Wermke M, Calvo E, Chartash E, Zhou H, Zhao X, et al. Phase I/II, multicenter, open-label study of intratumoral/intralesional administration of the retinoic acid–inducible gene I (RIG-I) activator MK-4621 in patients with advanced or recurrent tumors. Ann Oncol (2018) 29:viii712. doi: 10.1093/annonc/mdy424.016
81. Saxena M, Yeretssian G. NOD-like receptors: Master regulators of inflammation and cancer. Front Immunol (2014) 5:1–16. doi: 10.3389/fimmu.2014.00327
82. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol Cancer (2018) 17:158. doi: 10.1186/s12943-018-0900-3
83. Brown GD, Willment JA. & Whitehead, L. C-type lectins in immunity and homeostasis. Nat Rev Immunol (2018) 18:374–89. doi: 10.1038/s41577-018-0004-8
84. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
85. Le Naour J, Zitvogel L, Galluzzi L, Vacchelli E, Kroemer G. Trial watch: STING agonists in cancer therapy. Oncoimmunology (2020) 9:1–12. doi: 10.1080/2162402X.2020.1777624
86. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer (2004) 4:11–22. doi: 10.1038/nrc1252
87. Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol (1995) 13:688–96. doi: 10.1200/JCO.1995.13.3.688
88. Andtbacka RHI, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol (2015) 33:2780–8. doi: 10.1200/JCO.2014.58.3377
89. Lasek W, Zagożdżon R, Jakobisiak M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother (2014) 63:419–35. doi: 10.1007/s00262-014-1523-1
90. Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res (1997) 3:409–17.
91. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of Single-Dose Interleukin-12 Exposure on Interleukin-12–Associated Toxicity and Interferon-γ Production. Blood (1997) 90:2541–8.
92. Strauss J, Heery CR, Kim JW, Jochems C, Donahue RN, Montgomery AS, et al. First-in-human phase I trial of a tumor-targeted cytokine (NHS-IL12) in subjects with metastatic solid tumors. Clin Cancer Res (2019) 25:99–109. doi: 10.1158/1078-0432.CCR-18-1512
93. Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol (2006) 24:657–79. doi: 10.1146/annurev.immunol.24.021605.090727
94. Miller JS, Morishima C, McNeel DG, Patel MR, Kohrt HEK, Thompson JA, et al. A first-in-human phase I study of subcutaneous outpatient recombinant human IL15 (rhIL15) in adults with advanced solid tumors. Clin Cancer Res (2018) 24:1525–35. doi: 10.1158/1078-0432.CCR-17-2451
95. Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv (2019) 3:1970–80. doi: 10.1182/bloodadvances.2018028332
96. Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol (2018) 19:694–704. doi: 10.1016/S1470-2045(18)30148-7
97. Burke JD, Young HA. IFN-Γ: A cytokine at the right time, is in the right place. Semin Immunol (2019) 43:101280. doi: 10.1016/j.smim.2019.05.002
98. Conlon KC, Miljkovic MD, Waldmann TA. Cytokines in the Treatment of Cancer. J Interf Cytokine Res (2019) 39:6–21. doi: 10.1089/jir.2018.0019
99. Le Naour J, Galluzzi L, Zitvogel L, Kroemer G, Vacchelli E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology (2020) 9:1–16. doi: 10.1080/2162402X.2020.1777625
100. Mitchell TC, Hamid O, Smith CD, Bauer TM, Wasser JS, Olszanski AJ, et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: Phase I results from a multicenter, open-label phase I/II trial (ECHO-202/KEYNOTE-037). J Clin Oncol (2018) 36:3223–30. doi: 10.1200/JCO.2018.78.9602
101. Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol (2019) 20:1083–97. doi: 10.1016/S1470-2045(19)30274-8
102. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell (2017) 170:605–35. doi: 10.1016/j.cell.2017.07.029
103. Yao JC, Shah MH, Ito T, Bohas CL. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med (2011) 364:514–23. doi: 10.1056/NEJMoa1009290
104. Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med (2012) 366:520–9. doi: 10.1056/NEJMoa1109653
105. Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, et al. Activity of the PI3K-δ,g inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood (2018) 131:888–98. doi: 10.1182/blood-2017-08-802470
106. Huang Q, Lei Y, Li X, Guo F. & Liu, M. A Highlight of the Mechanisms of Immune Checkpoint Blocker Resistance. Front Cell Dev Biol (2020) 8:1–12. doi: 10.3389/fcell.2020.580140
107. Chae YK, Oh MS, Giles FJ. Molecular Biomarkers of Primary and Acquired Resistance to T-Cell-Mediated Immunotherapy in Cancer: Landscape, Clinical Implications, and Future Directions. Oncologist (2018) 23:410–21. doi: 10.1634/theoncologist.2017-0354
Keywords: melanoma, checkpoint inhibition/blockade, pathogen recognition receptor (PRR), cytokines, TLR (Toll-like receptors)
Citation: Rohatgi A and Kirkwood JM (2021) Beyond PD-1: The Next Frontier for Immunotherapy in Melanoma. Front. Oncol. 11:640314. doi: 10.3389/fonc.2021.640314
Received: 11 December 2020; Accepted: 07 January 2021;
Published: 01 March 2021.
Edited by:
Giuseppe Palmieri, National Research Council (CNR), ItalyReviewed by:
Inna Smalley, Moffitt Cancer Center, United StatesKathleen Marie Kokolus, University at Buffalo, United States
Copyright © 2021 Rohatgi and Kirkwood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John M. Kirkwood, a2lya3dvb2RqbUB1cG1jLmVkdQ==