 Bin Wang
Bin Wang Rui Li
Rui Li Shuai Wu1
Shuai Wu1 Hongyan Jia
Hongyan Jia- 1Department of Breast Surgery, First Hospital of Shanxi Medical University, Taiyuan, China
- 2Department of Microbiology and Immunology, Shanxi Medical University, Taiyuan, China
Breast cancer is a common malignant tumor in women, with a highest incidence and mortality among all of the female malignant tumors. Notably, targeted therapy has achieved impressive success in the treatment of breast cancer. As one class of the anti-tumor targeted therapeutics, Cyclin-Dependent Kinases 4/6CDK4/6inhibitors have shown good clinical activity in treating breast cancer. Nevertheless, despite the promising clinical outcomes, intrinsic or acquired resistance to CDK4/6 inhibitors has limited the benefits of this novel target therapy. In the present review, we provide an overview of the currently known molecular mechanisms of resistance to CDK4/6 inhibitors, and discuss the potential strategies to overcoming drug resistance improving the outcomes for breast cancer patients treated with CDK4/6 inhibitors.
Introduction
Cyclin-dependent kinases (CDKs) are serine/threonine kinases that play key roles in regulating cell cycle (1). CDK 4 and 6, two critical kinases among CDKs mediate the cellular transition from G0/G1 phase to S phase during cell cycle: dysregulation of CDK 4/6, result in uncontrolled cell division. The main effect of CDK4/6 inhibitor is to bind with cyclin D specifically, block cell cycle transformation, and stop cell cycle in G1 phase, thereby inhibiting tumor cell proliferation (2). Importantly, CDK4/6 inhibitors have showed great efficacy in treatment of breast cancer. Based on the PALOMA-1trail, FDA approved palbociclib, the first CDK 4/6 inhibitor, in combination with letrozole as first-line treatment for patients with ER-positive, HER2-negative advanced breast cancer (ABC) or metastatic breast cancer (MBC) (3). At present, three selective CDK4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) have been approved by FDA (4, 5). These three CDK4/6 inhibitors are used in combination with endocrine therapies or fulvestrant for patients with ER+ Her− metastatic breast cancer. Clinical trials PALOMA-2, MONALEESA-2, and MONARCH-3 have showed that when combined with aromatase inhibitors, CDK4/6 inhibitors could significantly prolong the progression-free survival in postmenopausal women with HR-positive metastatic breast cancer (6–8). Nevertheless, despite promising clinical outcomes, acquired or intrinsic resistance to CDK4/6 inhibitors often occurs, and this constitutes a major hindrance to successful treatment and limits the therapeutic benefits of those targeted therapeutics for patients with this disease. Therefore, understanding the molecular mechanisms and pathways involved in resistance to CDK4/6 inhibitors may help develop effective strategies to circumventing drug resistance and selecting patient populations who can benefit from this targeted therapy. Here, we review and discuss the known molecular mechanisms and pathways that modulate the cellular sensitivity or resistance to CDK4/6 inhibitors, and provide our outlook on this subject (6–8).
Potential Resistance Mechanisms
Breast cancer cells can be intrinsically resistant to CDK4/6 or develop acquired resistance to those agents. CDK4/6 can phosphorylate retinoblastoma protein (Rb1), and the phosphorylation leads to Rb1 functional inactivation, then Rb1 uncoupling from E2Fs transcription factors and release E2Fs. CDK4/6 inhibitors exert their effects through breaking the CDK4/6-Rb-E2F pathway (9, 10). The tumor cells with loss of Rb1 and lack of the major targets, intrinsic resistance to CDK4/6 inhibitors may occur (11, 12). The major obstacle to successful treatment with CDK4/6 inhibitors is the acquired resistance that frequently occurs in the patients who have received this therapy. Tumor cells can acquire the ability to escape CDK4/6 action (13). Understanding potential mechanisms of acquired resistance to CDK4/6 inhibitors may help find effective ways to preventing or overcoming drug resistance to this class of therapeutics (Figure 1).
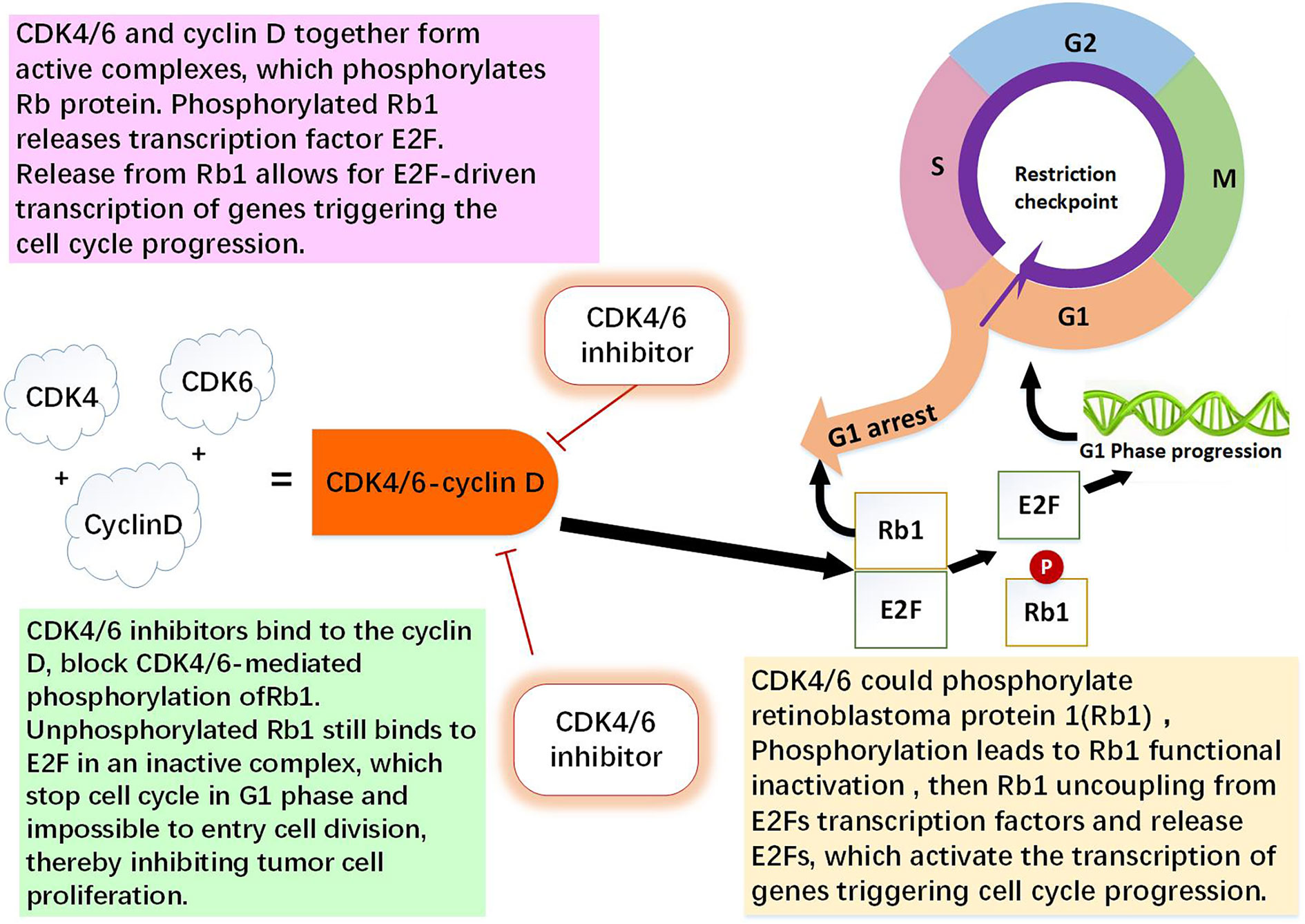
Figure 1 The role of CDK4/6-cyclin D and CDK4/6 inhibitor. CDK4 and CDK6 play key roles in cell proliferation. Cyclin D is regulator of the CDK4 and CDK6 kinases. CDK4/6 and cyclin D together form active complexes, which phosphorylates Rb1 protein. Rb1 is an onco-suppressor which repress the transcription of genes required for the cell cycle, limit the expression of transcription factor E2F target genes which are involved in cell cycle progression. Phosphorylated Rb1 releases E2F. Release from Rb1 allows for E2F-driven genes triggering the cell cycle progression. CDK4/6 inhibitors bind to the cyclin D specifically, thereby block CDK4/6-mediated phosphorylation of Rb1. Non-phosphorylated Rb1 still binds to E2F in an inactive complex, which leading to cell cycle arrest in G0/G1 phase and impossible to entry cell division, thereby inhibiting tumor cell proliferation (1, 9, 10, 13).
Direct Cell Cycle Mechanisms
Loss of Drug Target Genes
RB1
The tumor suppressor Rb1 is a key checkpoint in the cell cycle and a major target of CDK4/6 inhibitors. In both of preclinical and clinical settings, Rb1 mutations were found (14). In the tumor cell line with acquired resistance to palbociclib, it was demonstrated that resistance to CDK4/6 inhibitors was mediated through Rb1 loss, and restoration of Rb1 expression rendered tumor cells sensitivity to the CDK4/6 inhibitor (15). Chronic loss of Rb1 was found to be a cause of resistance to CDK4/6 inhibitors in breast cancer (16, 17). Using the breast cancer cell lines sensitive or resistant to palbociclib, it was showed that the complex change of Rb1 pathway was related to resistance to CDK4/6 inhibitor, Rb1 deficient in function is an important factor that contributes to palbociclib and abemaciclib resistance in breast cancer patients (18, 19). In clinical settings, researchers sequenced the somatic genomic mutations of three HR+ breast cancer samples before and after drug resistance to CDK4/6 inhibitors occurred and found that Rb1 mutation, allele substitution or exon deletion only existed in the blood samples after but not before drug resistance (20). Many researches showed that Rb1 loss could activate bypass of cyclin D1-CDK4/6-dependent pathway, leading to acquired resistance to CDK4/6 inhibition (14). These observations suggest that despite loss of Rb1, progression of the cell cycle continues via the activation of other cell cycle machinery, and inhibition of the bypass axis in combination with the CDK4/6 inhibitors may be effective in overcoming resistance to these targeted therapies. However, in the PALOMA-3 randomized phase III trial, the circulating tumor DNA sequencing from patients showed that Rb1 mutations occurred only in 6 of 127 (4.7%) patients (21). Thus, further clinical evidence is needed to analyze the frequency of Rb1 mutation in breast cancer patients receiving CDK 4/6 treatment (Figure 2, Table 1).
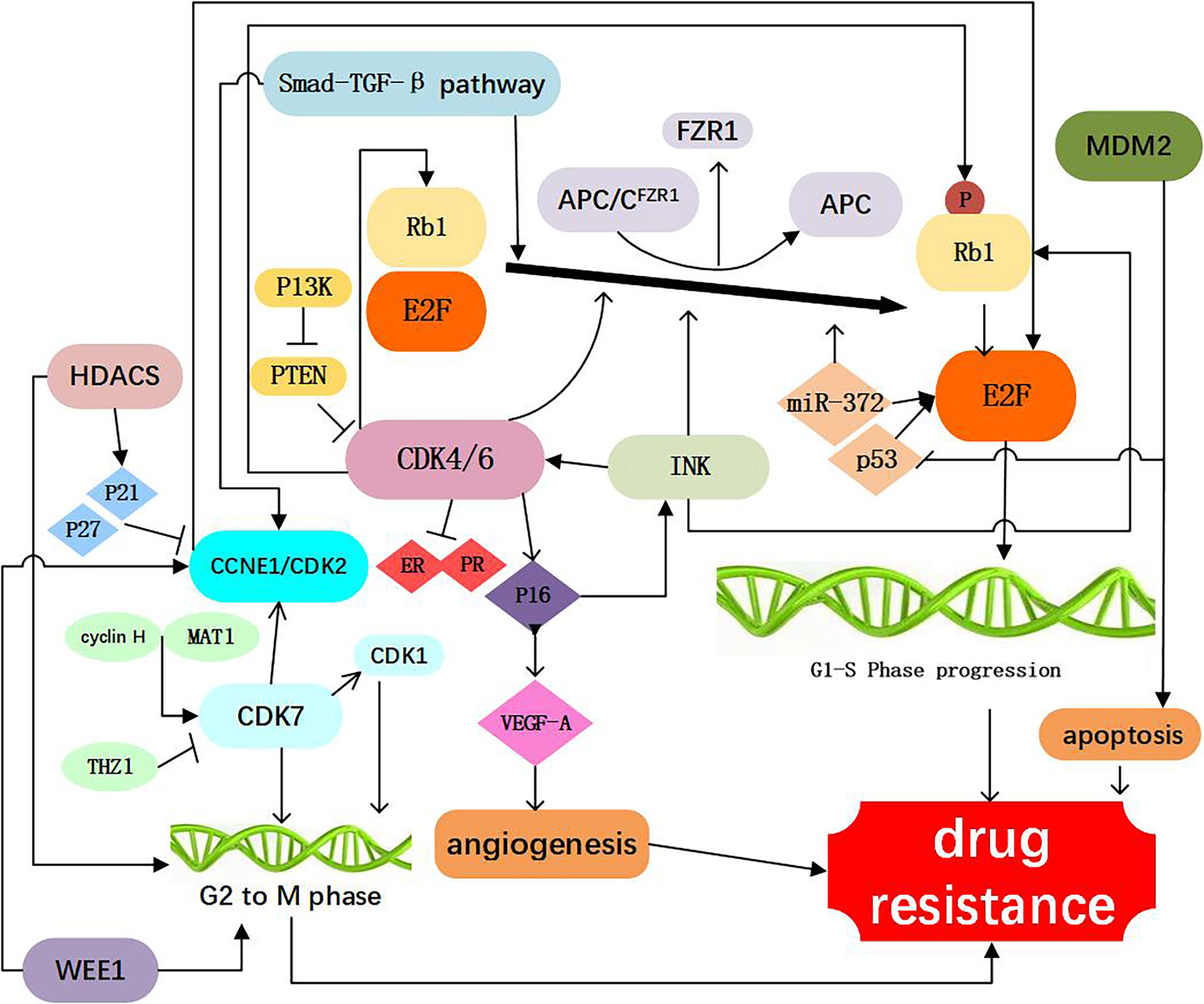
Figure 2 Resistance to CDK4/6 inhibitors: Direct Cell Cycle Mechanism: 1. Loss of drug target genes: APC/CFZR1 promote the phosphorylation of Rb1 and regulate cell transition from G1 to S. knockdown of Rb1 and FZR1 synergistically bypassed cell division arrest induced by the CDK4/6 inhibitor (14–23); 2. Increased activity of the CDK4 and CDK6: amplification of CDK4/6 account for a decreased CDK4/6 targeted phosphorylation of Rb1 and a decreased sensitivity of breast cancer cells to CDK4/6 inhibitor (2, 24–34); 3. Abnormal regulations of upstream and Downstream kinases: CCNE1/CDK2, CDK7, E2F, INK, PTEN, Smad-TGF-β pathway which are involved in the progression of cell cycle, as shown in Figure 1, are responsible for resistance to CDK4/6 inhibitors (12, 15, 17, 26, 35–60); 4. Activation of alternate genes like HDACS, WEE1, MDM2, partly help the cancer cell escape from the drugs work (61–67).

Table 1 Mechanisms of acquired resistance to CDK4/6 inhibitors: Direct cell cycle mechanisms.
APC/CFZR1
Similar to Rb1, the ubiquitin ligase anaphase promoting complex/cyclosome (APC/C) play an important role in cell cycle regulation. APC/C and pRb interact via the co-activator of APC/CFZR1, providing an alternative pathway to regulate transition from G1 to S by pRb through a post-translational mechanism (22). FZR1 is a candidate CDK4/6-cyclin D substrate and as an important determinant in response to CDK4/6 inhibitors. It was found that the loss of FZR1 resulted in uncontrolled cell cycle progression from G1 to S phase. In human breast cancer cell lines, simultaneous knockdown of Rb and FZR1 synergistically bypassed cell division arrest induced by the CDK4/6 inhibitor PD-0332991 (23). The precise mechanism of resistance to CDK4/6 inhibitors associated with the loss of FZR1 remains unclear. It is likely that loss of FZR1 corresponds with the loss of Rb; however, this possibility remains to be further investigated (Figure 2, Table 1).
Increased Activity of the Target Genes
CDK4
CDK4 is an important component of the cyclind-CDK4/6-Rb1 pathway, and was observed in 25% luminal B and 14% Luminal A breast cancers (24). In addition, aberrant expression of CDK4 activates the cyclind-CDK4/6-Rb1 pathway and results in drug resistance (25). It has been demonstrated that CDK4 was elevated in palbociclib resistant cell lines (26). Also, amplification of CDK4 has been reported in melanoma, glioma, rhabdomyosarcoma, and lung cancer and confers resistance to CDK4/6 inhibitors in these malignancies (27–30). The researchers found that increasing phosphorylation of p27 could inhibit CDK4 and regulate the cyclin D/CDK4/p27 complex activity, which could make breast cancer cells more resistant to palbociclib (2, 31), above study suggesting a potential strategy to prevent adaptation to CDK4/6 inhibitors (Figure 2, Table 1).
CDK6
The functions of CDK6 are both kinase-dependent and non-kinase-dependent (32). After a prolonged exposure to CDK4/6 inhibitor LY2835219, a significant amplification of CDK6 was found in several breast cancer cell lines, and this may account for a decreased CDK4/6 targeted phosphorylation of Rb1 and a decreased sensitivity of breast cancer cells to CDK4/6 inhibitor (32). Further experiments confirmed that forced overexpression of CDK6 indeed mediated drug resistance. Overexpression of CDK6 not only mediates resistance to CDK4/6 inhibitors, but also leads to decreased expression of estrogen and progesterone receptors. These studies also suggest that the efficacy of CDK4/6 inhibitors in breast cancer cells is modulated by ER. Therefore, CDK6 amplification can decrease the tumor cell sensitivity to both ER antagonists and CDK4/6 inhibitors. Knockdown of CDK6 can restore sensitivity, while enforced overexpression of CDK6 can confer resistance to CDK4/6 inhibitors
A decrease in ER/PR expression was observed in the tumor specimens from patients receiving treatment of CDK4/6 inhibitor and showing insensitivity to CDK4/6 inhibitors (33). The non-kinase dependent function of CD6 lies in its transcriptional regulation function. In the STAT3 and Cyclin D pathways, CDK6 could up-regulate the transcription of P16 and the expression of VEGF-A that can promote angiogenesis, contributing to the progression and drug resistance of breast cancer (32, 34) (Figure 2, Table 1).
Abnormal Regulations of Upstream and Downstream Kinases
CCNE1/CDK2
The cyclin E (encoded by CCNE1 gene)-CDK2 complexes play a key role in the cell cycle from G1 to S phase. Cyclin E-CDK2 can phosphorylate Rb1, release E2F, and promote entry into the S phase (35, 36). In an analysis of global gene expressions, increased expression of CDK2 was found in the palbociclib-resistant breast cancer cell lines. Also it was suggested that loss of p21 and p27, which has an inhibitory effect function on CDK2, may represent a mechanism leading to bypass of palbociclib (17). It has been reported that when combined CDK2 and CDK4 inhibitors, resistance to palbociclib was no longer obvious, suggesting that cyclin E-CDK2 complexes protein might mediate resistance to CDK4/6 inhibitors (37). Hopefully, next generation CDK inhibitors can target CDK2 to prevent or conquer drug resistance (Figure 2, Table 1).
CDK7
CDK7, one of the major cell cycle regulators, acts as a CDK-activating kinase (CAK) by maintaining CDK1 and CDK2 activity. CDK7 promotes the cell transition from G2 phase to M phase (38). It has been demonstrated that CDK7 overexpression occurred in the estrogen receptor-positive, palbociclib-resistant breast cancer cells (26), suggesting that CDK7 is involved in cellular resistance to CDK4/6 inhibitors. The CDK7 selective inhibitor, THZ1, can significantly inhibit the proliferation of triple negative breast cancer cells at the nmol/L concentration (39, 40). Also, the sensitivity of breast cancer cells to CDK7 inhibitors appears to be associated with the loss of ER and Rb1 CN expression (26). Thus, CDK7 inhibitors may play an important role in both of the targeted therapy and cellular resistance to CDK4/6 inhibitors (Figure 2, Table 1).
E2F
The CDK-Rb-E2F pathway plays a critical role in the control of cell cycle in breast cancer. At the early stage of G1, E2F binds to Rb1 protein and forms a functional complex. Phosphorylation of Rb1 protein by CDK activates E2F. Activation of E2F can promote the transition of cells from G1 phase to S phase. It has been reported that in the CDK4/6 inhibitor-resistant cell lines, the CDK-Rb-E2F pathway reactivate (41). Researchers found that in tumor biopsies resistant to palbociclib, CCND3, CCNE1, and CDKN2D are persistently elevated before palbociclib used, all three genes are known E2F1 transcription targets, suggesting persistent E2F activity in resistant tumors (42). It was also revealed that E2F1 was up-regulated in patients with tumor lymph node metastasis and advanced stage (43) and patients with increased E2F expression was associated with lower overall survival (OS), relapse-free survival (RFS), distant metastasis-free survival (DMFS) (44). Therefore, E2F might be exploited as a therapeutic target both for suppressing drug resistance to CDK4/6 inhibitors and biomarkers and therapeutic targets for breast cancer in breast cancer.
INK
CDK4/6 activity is regulated by the INK4 family proteins (p16INK4A, p15INK4B, p18INK4C, and p19INK4D), can inhibit the expression of CDK4 and lead to cell cycle arrest in the G1/S phase, thus considered as a natural tumor inhibitor (45). The P16 (p16INK4A) protein, encoded by the CDKN2Aink4a gene, play an important role of the INK4 family. It has been reported that CDK4/6 inhibitors can inhibit cancer cell cycle progression because of P16 gene deletion (46). Cancer cells with P16 methylation are more sensitive to palbociclib than those control (47, 48). It has been found that overexpression of p16 and loss of Rb1 often occur simultaneously. When p16 overexpression is accompanied by Rb1 deficiency, CDK4/6 inhibitors are inactive due to the Rb1 deficiency. With the presence of Rb1, overexpression of p16 (be consistent) leads to a decrease of CDK4 and resistance to CDK4/6 inhibitors (12). Further studies are needed to delineate the precise mechanistic association between Rb1 loss and P16 overexpression, which may help design novel therapeutic strategies to overcoming the acquired resistance to CDK4/6 inhibitors (Figure 2, Table 1).
PTEN
PTEN a tumor suppressor gene, is one of the frequently mutated genes in human cancers (49). The increased expression of PTEN leads to the inactivation of CDK, which enables the Rb1 keep dephosphorylating, while binding to transcription factor E2F, which ultimately inhibits cell proliferation. these ways may influence the effect of CDK4/6 inhibitors (49). Researchers analyzed serial biopsies from breast cancer patients treated with the combination of ribociclib and letrozole and found that ablation of PTEN was sufficient to promote resistance to CDK4/6 inhibition (50). The increased AKT expression could reduce PTEN expression and render breast cancer cells resistant to CDK4/6 inhibitors (51). In breast cancer cells, loss of PTEN also conferred resistance to alpelisib. Moreover, loss of PTEN expression can cause dual resistance to CDK4/6 inhibitors and PI3K inhibitors (52) (Figure 2, Table 1).
Smad-TGF-β Pathway
Smad–transforming growth factor β (TGF-β) pathway contributes to G1 arrest in breast cancer cells (53). TGF-β signaling is transduced through Smad2 and Smad3 and forms a complex with Smad4 to regulate target gene expression relevant to cell growth and differentiation (54, 55). Smad3, which has antiproliferative effects, has a key role in TGF-β signaling cascade. Smad3 can regulate cell cycle arrest, and has been shown to be correlated with resistance to CDK4/6 inhibitors (53). Mechanistically, cyclin E-CDK2 and cyclin D1-CDK4/6 complexes can suppress Smad3 through its phosphorylation, and the suppression of Smad3 releases the Rb1-E2F blockade and restore cell cycle arrest in breast cancer cells (53, 56). TGF-β can phosphorylate and activate Smad2 and Smad3 and form a complex with Smad4, and this contributes to the induction and progression of EMT. EMT can promote invasion and metastasis of cancer cells and increase drug resistance (57). Consistently, inhibition of the CDK2-mediated phosphorylation of Smad3 reduces TNBC cell migration and invasion through changes in EMT-related signaling factors (58). According to these findings, resistance of tumor cells to CDK4/6 inhibitors may result from suppression of Smad3 that is associated with the activated cyclin E-CDK2 axis and EMT (15, 36, 59, 60). Thus, the Smad-TGF-β pathway might be considered as a potential therapeutic target for overcome drug resistance to CDK4/6 inhibitors (Figure 2, Table 1).
Activation of Alternate Genes Are Involved in the Progression of Cell Cycle
WEE1
WEE1 is a protein tyrosine kinase that phosphorylates CDK1 and CDK2 and causes their inhibition (61). WEE1 inhibits CDK1 to maintain the cell in an inactive state and prevent mitosis. WEE1 also inhibits CDK2 to delay the replication process and allow time for DNA repair. Both of these events occur in breast cancer cells (61, 62). Inhibiting the expression of WEE1 can sensitize the drug resistant cancer cells to CDK4/6 inhibitors, probably because that inhibiting WEE1 can increase the expression of CD4 (63). In the ribociclib-resistant cancer cells, a down-regulation of the G2/M checkpoint was observed (64). Drug resistant cancer cells exhibited collateral sensitivity to the Wee-1 inhibitor, adavosertib (AZD1775). Combined treatment with ribociclib and adavosertib can elicit significantly stronger antiproliferative effect on drug resistant tumor cells cells than ribociclib alone (64) (Figure 2, Table 1).
MDM2
Mouse double minute 2 homolog (MDM2) is a negative regulatory protein of tumor suppressor p53 and can inhibit cellular senescence. MDM2 binds to p53 protein and inhibits the function of this tumor suppressor (65). Overexpression of MDM2 drives breast oncogenesis and blocks apoptosis of breast cancer cells, resulting in resistance of tumor cells to CDK4/6 inhibitors. Therefore, the use of MDM2 inhibitors may reverse cellular resistance to CDK4/6 inhibitors, and this has been in human liposarcoma (66). Indeed, the MDM2 inhibitor, CGM097, in combination with a CDK4/6 inhibitor palbociclib and fulvestrant has shown promising therapeutic benefits in reversing the tumor resistance to CDK4/6 inhibitors and to endocrine therapy (67) (Figure 2, Table 1).
Indirect Cell Cycle Mechanisms
Bypass Pathways of the Cell Cycle
mTOR Pathway
Abnormal activation of mammalian target of rapamycin (mTOR) pathway is an important target for development of anti-cancer drug, the most common mechanism of mTOR activation in breast cancer is via phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling, PI3K/AKT/mTOR pathway is closely associated with cellular resistance to CDK4/6 inhibitors (15, 68–70). It was reported that mTOR signaling is dysregulated in breast cancer patients following abemaciclib treatment (70) and PI3K/mTOR pathway has been shown to be upregulated in response to chronic exposure to CDK4/6 inhibitors (71). Also, pre-treatment with mTOR inhibitors was shown to prevent or delay the resistance to CDK4/6 inhibitors (72). In a kinome-wide siRNA screen, it was found that the AKT pathway is highly activated in the ribociclib resistant breast cancer cells (73). Combination of PI3K and CDK 4/6 inhibitors could reduce cell viability and overcome intrinsic and adaptive resistance leading to tumor regressions (74). Further studies demonstrated that ribociclib in combination with an AKT inhibitor or PI3K inhibitor has a significantly stronger inhibitory effect on the growth of transplanted tumor in mouse models, as compared with ribociclib alone, supporting the role of PI3K signaling pathway in mediating resistance to the CDK4/6 inhibitor (73). Thus, coinhibition of the PI3K/mTOR and CDK4/6 pathways may prevent induction of drug resistance. Furthermore, it has been showed in a preclinical model that a PI3K inhibitor combined with a CDK4/6 inhibitor has a significant stronger inhibitory effect on proliferation of breast cancer cells than the single drug (41). Taken together, the PI3K/AKT/mTOR inhibitors may represent a class of sensitizers in CDK4/6-targeted therapy (Figure 3, Table 2).
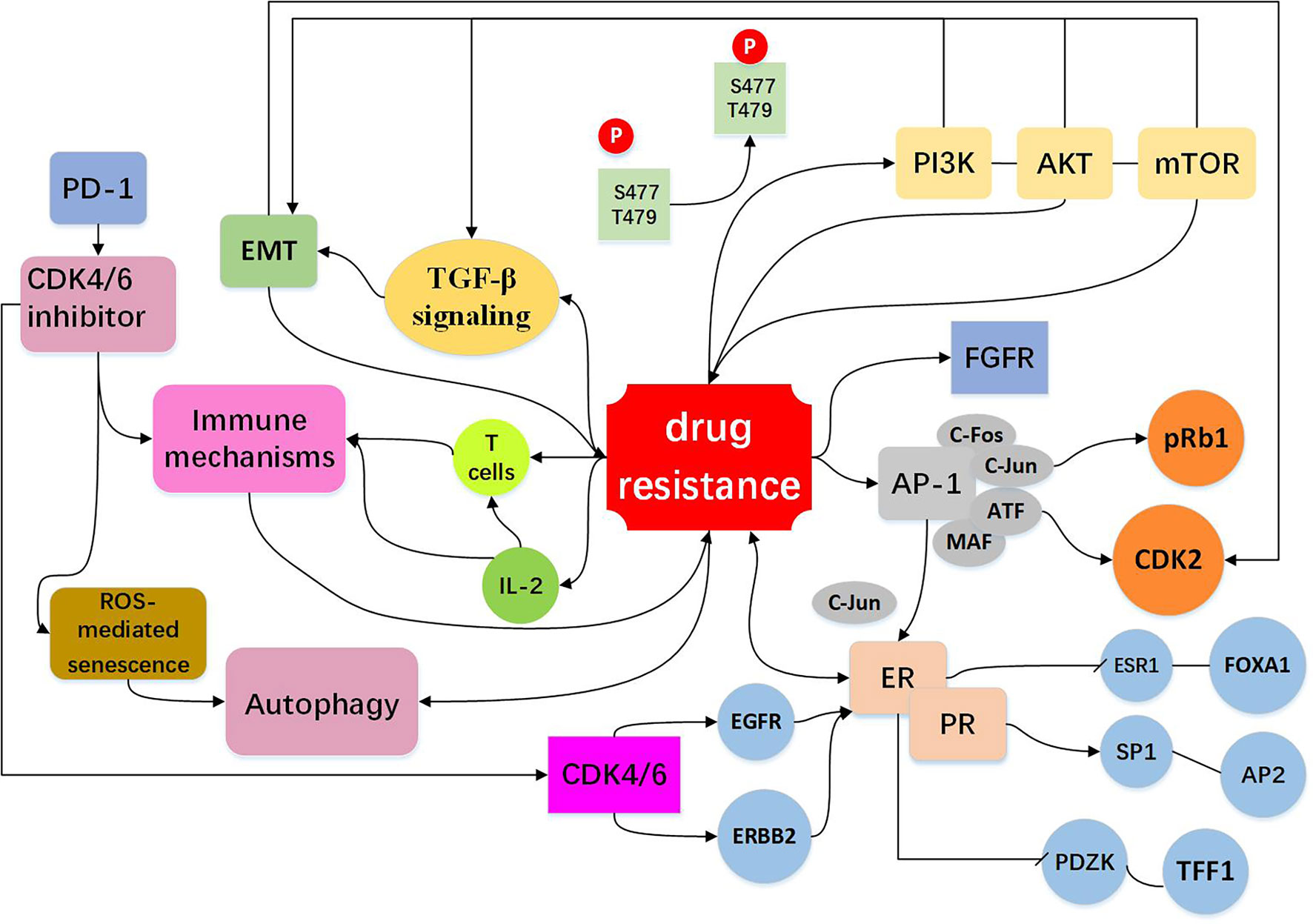
Figure 3 Resistance to CDK4/6 inhibitors: Indirect Cell Cycle Mechanism Bypass pathways of the cell cycle: mTOR activation is via phosphoinositide PI3K/AKT signaling. The PI3K/AKT/mTOR pathway regulate cell signal transduction, have extensive links with other bypasses, for example EMT and TGF-β pathway (15, 41, 68–74). High expression of AP-1 (75–78), FGFR amplification (79–82), loss of ER or PR (13, 16, 26, 72) expression drives cells to escape CDK4/6 inhibition and act as bypass pathways for the progression of the cell. Other mechanisms include EMT (10, 83–87), immune mechanisms (88–91) and autophagy directly or indirectly influence drug resistance shown in the figure (10, 92–96).
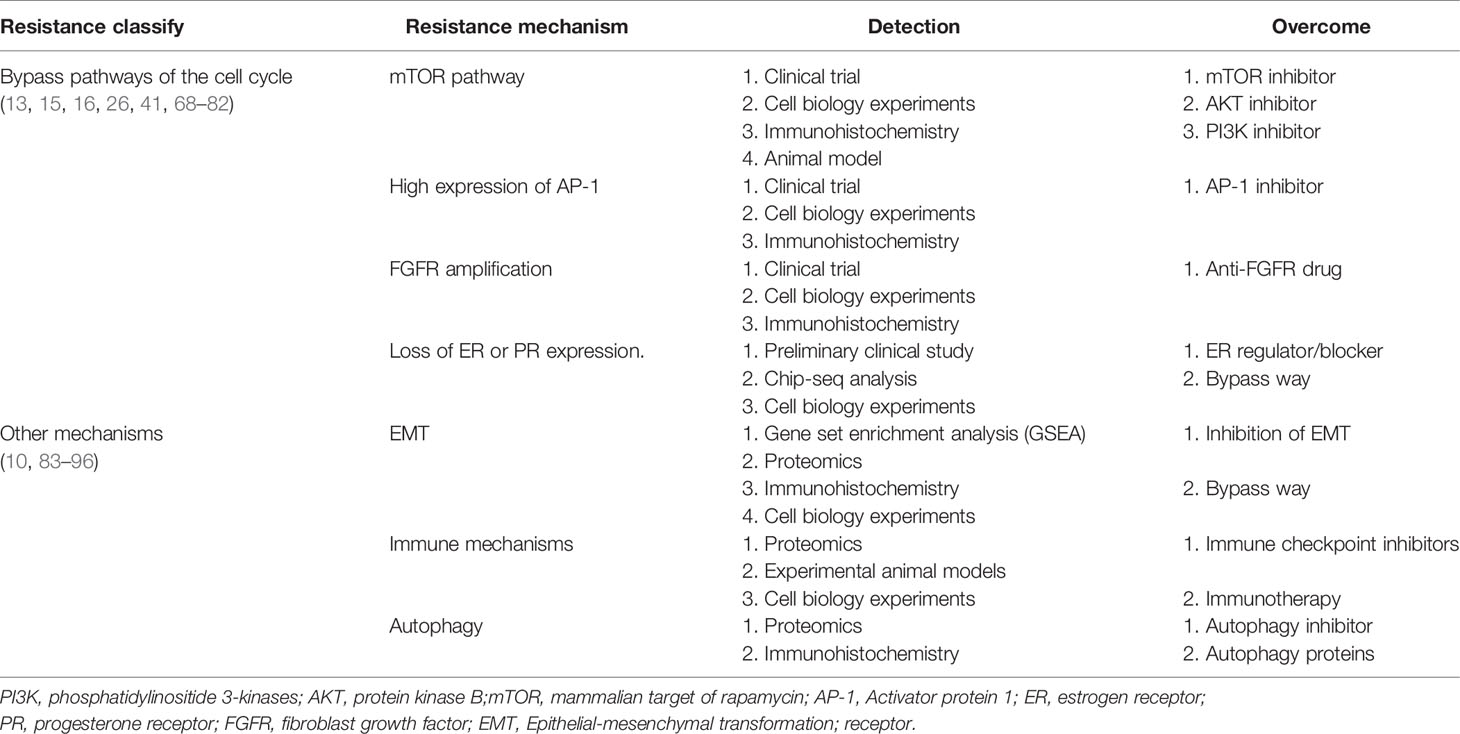
Table 2 Mechanisms of acquired resistance to CDK4/6 inhibitors: Indirect cell cycle mechanisms.
AP-1
High expression of AP-1 can lead to resistance to CDK4/6 inhibitors. AP-1 family consists of C-FOS, C-Jun, ATF, and MAF, and is involved in the regulation of a variety of genes, including cyclinD (75). The high expression of C-Jun is common in breast cancer and affects the expression of ER (76). It was found in breast cancer cells that are resistant to palbociclib which the transcriptions of AP-1 and C-FOS were increased, and AP-1 blockade in combination with palbociclib could effectively inhibit cell proliferation and reduce pRb and CDK2 levels as compared to single agent treatment (77). These observations suggest that co-treatment with Ap-1 specific inhibitors and CDK4/6 inhibitors may elicit anti-tumor synergistic effects. AP-1 and c-FOS inhibitors have entered Phase II clinical trial (T-5224) (78) (Figure 3, Table 2).
FGFR
The fibroblast growth factor receptor (FGFR) is growth factor receptor tyrosine kinases (79). Development of normal mammary gland requires active transcription of FGFR mediated proto-protein kinase and FGFR is closely associated with the development and progression of breast cancer (80, 81). Based on the combination of letrozole with ribociclib, the clinical trial MONALESA-2 observed that FGFR1 amplification was related to a lower PFS (79). It was also demonstrated that FGFR1 expression was increased in breast cancer MCF-7 cells treated with fulvestrant and palbociclib (82), and lucitanib, an anti-FGFR drug, can decrease drug resistance. As FGFR1 can stimulate the proliferation capacity of cancer cells, inhibiting both FGFR/FGF and the CDK4/6 pathways might be an effective approach to preventing or circumventing resistance to a single agent (Figure 3, Table 2).
ER and PR
ER and PR are the major factors that mediate cyclinD-CDK4/6 activity in estrogen receptor-positive (ER+) and progesterone receptor-positive (PR+) breast cancer cells (13). Effect of ER on resistance to CDK4/6 inhibitors involves both cell cycle and non-cell cycle mechanisms. In a preliminary clinical study, it was found that the expressions of ER/PR were lost in the palbociclib resistant tumor samples and down-regulated in the palbociclib resistant breast cancer cells (16, 26). Chip-seq analysis uncovered that ER was deficient in binding to ESR1 and FOXA1, but enriched in binding to SP1 and AP2, and these were accompanied by decreased expression of regulatory genes such as PDZK1 and TFF1.These data indicate that drug-resistant cells are genetically altered by chromosome remodeling. In other pathways discussed above, high expression of AP-1 leads to overexpression of C-Jun, which inhibits ER activity and modulates the efficacy of CDK4/6 inhibitors (76). Similarly, CDK4/6 blockade can lead to up-regulation of EGFR/ERBB and down-regulation of ER signaling pathway, and this negative feedback regulation can impact the efficacy of CDK4/6 inhibitors (26) (Figure 3, Table 2).
Other Mechanisms
EMT
Epithelial-mesenchymal transformation (EMT) is a biological process in which epithelial cells lose their polarity obtain the ability to invade and migrate. EMT has important roles in tumor cell metastasis, tumor stem cell formation, drug resistance, and other malignant phenotypes. A number of EMT-related signaling pathways are involved in drug resistance in cancer cells (83–85). The gene set enrichment analysis (GSEA) revealed enrichment of pathways that regulate EMT and cancer stem cells (IL-6/Stat3, IL-2/STAT-5, Notch, Wnt) in the cells resistant to palbociclib (10). Indeed, anti-CDK4/6 therapy can induce EMT and enhance cell invasion through activating TGF-β signaling (60, 86). It was suggested that EMT is an important determinant of success/failure of targeted therapies by interfering with the compensatory changes such as deregulation of CDK2 activity (87). Low cyclin D1 (CCND1) expression displays increased expression of EMT markers, increased migration of breast cancer cells and drug resistance (86) (Figure 3, Table 2).
Immune Mechanisms
CDK4/6 inhibitors not only induce tumor cell cycle arrest, but also promote anti-tumor immunity (88–90). In murine models of breast carcinoma, it was found that CDK4/6 inhibitors can activate tumor expression of endogenous retroviral elements that enhance tumor antigen presentation. CDK4/6 inhibitors also suppress the proliferation of suppressive regulatory T cells (Tregs) and enhance the cytotoxic T cell-mediated killing of tumor cells. It was also found that CDK4/6 inhibitors could promote anti-tumor immunity by phosphorylating NFAT4, a transcription factor of T cells, thereby increasing IL-2 levels (91). CDK4/6 inhibitors reduced the proliferation of T cells, but increased tumor infiltration and activation of effector T cells. In addition, CDK4/6 inhibition can augment the response to PD-1 blockade in multiple in vivo murine syngeneic tumor models (91). These studies provide a rationale for combining CDK4/6 inhibitors with immunotherapy to more effectively killing tumor cells and preventing drug resistance (Figure 3, Table 2).
Autophagy
Autophagy is a cellular process that eliminates the damaged or aged cells and is the key machinery for bulk degradation of superfluous or aberrant cytoplasmic components. Autophagy is a double-edged sword in drug sensitivity/drug resistance (92–94). Autophagy could elevate the maintenance of cancer stem cells which may enhance drug resistance, while autophagy may help tumor cells to clear the drug-induced damage which decreasing the impact of chemotherapy and enhances therapeutic response (95, 96). It was demonstrated that CDK4/6 inhibition induces ROS mediated senescence and autophagy, blockade of autophagy significantly improves the efficacy of CDK4/6 inhibition (10). It was reported that high expression of autophagy proteins like LC3B can be utilized to combat resistance to cell-cycle-targeted therapies, such as CDK4/6 inhibitors (94). More research is needed to clarify the relationship between the CDK4/6 inhibitor and autophagy, this will provide a better prospect for the clinical application (Figure 3, Table 2).
Summary and Perspectives
CDK4/6 inhibitors are an effective therapeutic option for patients. A number of clinical trials have demonstrated the effectiveness and benefits of CDK4/6 inhibitors in improving the progression-free survival (PFS) of patients with ER-positive, HER2-negative advanced breast cancer (ABC) or metastatic breast cancer (MBC) when combined with endocrine therapy. The approval of palbociclib was based on the results from the PALOMA-1/TRIO-18, PALOMA-2, and PALOMA-3 trials. In the PALOMA-1 trail, combined therapy of letrozole with palbociclib significantly improved PFS as compared with single-agent letrozole. The PALOMA-2 trial confirmed the clinical activity of combination of palbociclib with letrozole. In PALOMA-3 trial, combined treatment of palbociclib with fulvestrant has shown benefits in patients with HR-positive, HER2-negative ABC or MBC. Thus, FDA approved the combined use of palbociclib with fulvestran based on this trial (3, 6, 21, 97). Abemaciclib was approved based on the results of MONARCH 1, MONARCH2, and MONARCH3, and combination of abemaciclib with fulvestrant has been approved for treatment of patients with HR-positive, HER2-negative ABC or MBC. MONARCH 3 trial showed that abemaciclib plus anastrozole or letrozole produced a significantly longer median PFS than the placebo plus anastrozole or letrozole. FDA has approved the combined therapy of abemaciclib in with an aromatase inhibitor as first-line treatment for postmenopausal women with HR-positive, HER2-negative ABC (8, 98, 99). In addition, ribociclib in combination with letrozole was approved as the first-line treatment for postmenopausal women with HR-positive and HER2-negative ABC or MBC, and the combination of ribociclib with fulvestrant was approved for the treatment of postmenopausal women with HR-positive and HER2-negative ABC, based on the outcomes from clinical trials. MONALEESA-7 trial compared patience received ovarian function suppression and endocrine therapy plus ribociclib or not, in the ribociclib group, the PFS and overall survival (OS) was significantly long than placebo group (100–102). The recent study SOLAR-1, indicated that when alpelisib was combined with fulvestrant to treat the patients with PIK3CA-mutated, HR+, HER2- ABC patients, the PFS was increased from 5.7 to 11.2 months, a statistically significant prolongation (103). In China, the CDK4/6 inhibitors have been introduced into the first-line treatment for patients with advanced estrogenic receptor positive breast cancer. While this new targeted therapy has benefited numerous patients with advanced breast cancer, drug resistance to CDK4/6 inhibitors remain to be a major impediment to successful treatment of the disease. Novel approaches to preventing or overcoming the resistance to CDK4/6 inhibitors would certainly increase the value and benefits of these agents to breast cancer patients. However, to reach this goal, we need to have a better understanding of the multiplicity and complexity of the molecular mechanisms involved in resistance to CDK4/6 inhibitors. Also, despite enormous advances in this targeted therapy in treating breast cancer, its clinical efficacy and benefits are limited by the patient populations that do not benefit from this remedy, and this might be associated with a variety of factors such as tumor heterogeneity and target alterations. Identification and development of predictive and reliable biomarkers for the response to CD4/6 inhibitors shall significantly improve the outcome and value of the CD4/6-targeted therapy through better selecting appropriate patients for specific therapeutic regimens, thus are urgently needed. With a better understanding of the molecular mechanism behind resistance to CDK4/6 inhibitors, we could anticipate that patients can better benefit from novel therapeutic strategies that prevent and circumvent drug resistance and reinforce the efficacy of this targeted therapy.
Author Contributions
HJ contributed to the conception of the study. RL finished the first manuscript preparation. BW revised the manuscript. SW, XL, JR, JL, KB, YW helped perform the analysis with constructive discussions. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Balter M, Vogel G. NOBEL PRIZE IN PHYSIOLOGY OR MEDICINE: Cycling Toward Stockholm. Science (2001) 294(5542):502–3. doi: 10.1126/science.294.5542.502
2. Guarducci C, Bonechi M, Boccalini G, Benelli M, Risi E, Di Leo A, et al. Mechanisms of Resistance to CDK4/6 Inhibitors in Breast Cancer and Potential Biomarkers of Response. Breast Care (Basel Switzerland) (2017) 12(5):304–8. doi: 10.1159/000484167
3. Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, et al. The Cyclin-Dependent Kinase 4/6 Inhibitor Palbociclib in Combination with Letrozole Versus Letrozole Alone as First-Line Treatment of Oestrogen Receptor-Positive, HER2-Negative, Advanced Breast Cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol (2015) 16(1):25–35. doi: 10.1016/s1470-2045(14)71159-3
4. Corona SP, Generali D. Abemaciclib: A CDK4/6 Inhibitor for the Treatment of HR+/HER2- Advanced Breast Cancer. Drug Des Devel Ther (2018) 12:321–30. doi: 10.2147/DDDT.S137783
5. Spring LM, Wander SA, Andre F, Moy B, Turner NC, Bardia A. Cyclin-Dependent Kinase 4 and 6 Inhibitors for Hormone Receptor-Positive Breast Cancer: Past, Present, and Future. Lancet (2020) 395(10226):817–27. doi: 10.1016/s0140-6736(20)30165-3
6. Diéras V, Harbeck N, Joy AA, Gelmon K, Ettl J, Verma S, et al. Palbociclib with Letrozole in Postmenopausal Women with ER+/HER2– Advanced Breast Cancer: Hematologic Safety Analysis of the Randomized PALOMA-2 Trial. Oncologist (2019) 24(12):1514–25. doi: 10.1634/theoncologist.2019-0019
7. Duso B, Trapani D, Viale G, Criscitiello C, D’Amico P, Belli C, et al. Clinical Efficacy of Ribociclib as a First-Line Therapy for HR-Positive, Advanced Breast Cancer. Expert Opin Pharmacotherapy (2018) 19(3):299–305. doi: 10.1080/14656566.2018.1429407
8. Johnston S, Martin M, Di Leo A, Im S-A, Awada A, Forrester T, et al. MONARCH 3 Final PFS: A Randomized Study of Abemaciclib as Initial Therapy For Advanced Breast Cancer. NPJ Breast Cancer (2019) 5(1):244–59. doi: 10.1038/s41523-018-0097-z
10. Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M, et al. CDK4/6 and Autophagy Inhibitors Synergistically Induce Senescence in Rb Positive Cytoplasmic Cyclin E Negative Cancers. Nat Commun (2017) 8(27):15916. doi: 10.1038/ncomms15916
11. Jeffry LD. Therapeutic Response to CDK4/6 Inhibition in Breast Cancer Defined by Ex Vivo Analyses of Human Tumors. Cell Cycle (Georgetown Tex) (2012) 14(11):2756–61. doi: 10.4161/cc.21195
12. Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic Response to CDK4/6 Inhibition in Breast Cancer Defined by Ex Vivo Analyses of Human Tumors. Cell Cycle (Georgetown Tex) (2012) 11(14):2756–61. doi: 10.4161/cc.21195
13. Finn RS, Aleshin A, Slamon DJ. Targeting the Cyclin-Dependent Kinases (CDK) 4/6 in Estrogen Receptor-Positive Breast Cancers. Breast Cancer Res (2016) 18(1):17. doi: 10.1186/s13058-015-0661-5
14. Xi J, Ma CX. Sequencing Endocrine Therapy for Metastatic Breast Cancer: What Do We Do After Disease Progression on a CDK4/6 Inhibitor? Curr Oncol Rep (2020) 22(6):57. doi: 10.1007/s11912-020-00917-8
15. Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res (2016) 76(8):2301–13. doi: 10.1158/0008-5472.CAN-15-0728
16. Yang C, Li Z, Bhatt T, Dickler M, Giri D, Scaltriti M, et al. Acquired CDK6 Amplification Promotes Breast Cancer Resistance to CDK4/6 Inhibitors and Loss of ER Signaling and Dependence. Oncogene (2017) 36(16):2255–64. doi: 10.1038/onc.2016.379
17. Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 Inhibition in Breast Cancer: Key Mechanisms of Response and Failure. Oncogene (2010) 29(28):4018–32. doi: 10.1038/onc.2010.154
18. Malorni L, Piazza S, Ciani Y, Guarducci C, Bonechi M, Biagioni C, et al. A Gene Expression Signature of Retinoblastoma Loss-of-Function is a Predictive Biomarker of Resistance to Palbociclib in Breast Cancer Cell Lines and is Prognostic in Patients with ER Positive Early Breast Cancer. Oncotarget (2016) 7(42):68012–22. doi: 10.18632/oncotarget.12010
19. Gong X, Litchfield LM, Webster Y, Chio LC, Wong SS, Stewart TR, et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell (2017) 32(6):761–76.e766. doi: 10.1016/j.ccell.2017.11.006
20. Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1 Mutations and Acquired Resistance to CDK 4/6 Inhibitors in Patients With Metastatic Breast Cancer. Ann Oncol (2018) 29(3):640–5. doi: 10.1093/annonc/mdx784
21. O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov (2018) 8(11):1390–403. doi: 10.1158/2159-8290.Cd-18-0264
22. Ramanujan A, Tiwari S. APC/C and Retinoblastoma Interaction: Cross-Talk of Retinoblastoma Protein With the Ubiquitin Proteasome Pathway. Biosci Rep (2016) 36(5):e00377–7. doi: 10.1042/bsr20160152
23. The I, Ruijtenberg S, Bouchet BP, Cristobal A, Prinsen MBW, van Mourik T, et al. Rb and FZR1/Cdh1 Determine CDK4/6-Cyclin D Requirement in C. elegans and Human Cancer Cells. Nat Commun (2015) 6(1):5906. doi: 10.1038/ncomms6906
24. Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive Molecular Portraits of Human Breast Tumours. Nature (2012) 490(7418):61–70. doi: 10.1038/nature11412
25. Hamilton E, Infante JR. Targeting CDK4/6 in Patients With Cancer. Cancer Treat Rev (2016) 45:129–38. doi: 10.1016/j.ctrv.2016.03.002
26. Pancholi S, Ribas R, Simigdala N, Schuster E, Nikitorowicz-Buniak J, Ressa A, et al. Tumour Kinome Re-Wiring Governs Resistance to Palbociclib in Oestrogen Receptor Positive Breast Cancers, Highlighting New Therapeutic Modalities. Oncogene (2020) 39(25):4781–97. doi: 10.1038/s41388-020-1284-6
27. Eran H. A Landscape of Driver Mutations in Melanoma. Cell (2012) 2(150):251–63. doi: 10.1016/j.cell.2012.06.024
28. Ling C. p16-Cdk4-Rb Axis Controls Sensitivity to a Cyclin-Dependent Kinase Inhibitor PD0332991 in Glioblastoma Xenograft Cells. Neuro-oncology (2012) 7(14):810–81. doi: 10.1093/neuonc/nos114
29. Olanich ME, Sun W, Hewitt SM, Abdullaev Z, Pack SD, Barr FG. CDK4 Amplification Reduces Sensitivity to CDK4/6 Inhibition in Fusion-Positive Rhabdomyosarcoma. Clin Cancer Res (2015) 21(21):4947–59. doi: 10.1158/1078-0432.Ccr-14-2955
30. Jiang H, Luo N, Zhang X, Song C, Zang J. EGFR L861Q and CDK4 Amplification Responding to Afatinib Combined With Palbociclib Treatment in a Patient With Advanced Lung Squamous Cell Carcinoma. Lung Cancer (2020) 145:216–218. doi: 10.1016/j.lungcan.2020.04.001
31. Patel P, Asbach B, Shteyn E, Gomez C, Coltoff A, Bhuyan S, et al. Brk/Protein Tyrosine Kinase 6 Phosphorylates p27KIP1, Regulating the Activity of Cyclin D-Cyclin-Dependent Kinase 4. Mol Cell Biol (2015) 35(9):1506–22. doi: 10.1128/mcb.01206-14
32. Kollmann K, Heller G, Schneckenleithner C, Warsch W, Scheicher R, et al. A Kinase-Independent Function of CDK6 Links the Cell Cycle to Tumor Angiogenesis. Cancer Cell (2016) 30(2):359–360. doi: 10.1016/j.ccell.2016.07.003
33. Finn RS, Aleshin A, Slamon DJ. Targeting the Cyclin-Dependent Kinases (CDK) 4/6 in Estrogen Receptor-Positive Breast Cancers. Breast Cancer Res BCR (2016) 18(1):17. doi: 10.1186/s13058-015-0661-5
34. Gacche RN, Assaraf YG. Redundant Angiogenic Signaling and Tumor Drug Resistance. Drug Resist Update (2018) 36:47–76. doi: 10.1016/j.drup.2018.01.002
35. Pandey K, An HJ, Kim SK, Lee SA, Kim S, Lim SM, et al. Molecular Mechanisms of Resistance to CDK4/6 Inhibitors in Breast Cancer: A Review. Int J Cancer (2019) 145(5):1179–88. doi: 10.1002/ijc.32020
36. GA B, Alan DJ. Cell Cycle Progression Without Cyclin E/CDK2: Breaking Down the Walls of Dogma. Cancer Cell (2003) 4(3):160–2. doi: 10.1016/S1535-6108(03)00217-4
37. Caldon CE, Sergio CM, Kang J, Muthukaruppan A, Boersma MN, Stone A, et al. Cyclin E2 Overexpression is Associated With Endocrine Resistance But Not Insensitivity to CDK2 Inhibition in Human Breast Cancer Cells. Mol Cancer Ther (2012) 11(7):1488–99. doi: 10.1158/1535-7163.Mct-11-0963
38. Schachter MM, Merrick KA, Larochelle S, Hirschi A, Zhang C, Shokat KM, et al. A Cdk7-Cdk4 T-Loop Phosphorylation Cascade Promotes G1 Progression. Mol Cell (2013) 50(2):250–60. doi: 10.1016/j.molcel.2013.04.003
39. Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell (2015) 163(1):174–86. doi: 10.1016/j.cell.2015.08.063
40. Zhenghui Z, Wei L, Liyan. Yangshune Correlation Analysis of Cyclin D1, Aurora Kinase-A, Fibroblast Growth Factor Receptor2 Gene Single Nucleotide Polymorphism With Pathological Indicators and Prognosis of Breast Cancer. Chin J Exp Surgery|Chin J Exp Surg (2020) 7(37):1208–11. doi: 10.3760/cma.j.cn421213-20200709-00496
41. Michaloglou C, Crafter C, Siersbæk R, Delpuech O, Curwen J, Carnevalli L, et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-Positive Breast Cancer. Mol Cancer Ther (2018) 17:molcanther.0537.2017. doi: 10.1158/1535-7163.MCT-17-0537
42. Ma CX, Gao F, Luo J, Northfelt DW, Goetz M, Forero A, et al. NeoPalAna: Neoadjuvant Palbociclib, a Cyclin-Dependent Kinase 4/6 Inhibitor, and Anastrozole for Clinical Stage 2 or 3 Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res (2017) 23(15):4055–65. doi: 10.1158/1078-0432.Ccr-16-3206
43. Yan Y, Xu H, Wang J, Wu X, Wen W, Liang Y, et al. Inhibition of Breast Cancer Cells by Targeting E2F-1 Gene and Expressing IL15 Oncolytic Adenovirus. Biosci Rep (2019) 39(7):BSR20190384. doi: 10.1042/bsr20190384
44. Li Y, Huang J, Yang D, Xiang S, Sun J, Li H, et al. Expression Patterns of E2F Transcription Factors and Their Potential Prognostic Roles in Breast Cancer. Oncol Lett (2018) 15(6):9216–30. doi: 10.3892/ol.2018.8514
45. Sun F, Li N, Tong X, Zeng J, He S, Gai T, et al. Ara-c Induces Cell Cycle G1/S Arrest by Inducing Upregulation of the INK4 Family Gene or Directly Inhibiting the Formation of the Cell Cycle-Dependent Complex CDK4/Cyclin D1. Cell Cycle (2019) 18(18):2293–306. doi: 10.1080/15384101.2019.1644913
46. Kong Y, Sheng X, Wu X, Yan J, Ma M, Yu J, et al. Frequent Genetic Aberrations in the CDK4 Pathway in Acral Melanoma Indicate the Potential for CDK4/6 Inhibitors in Targeted Therapy. Clin Cancer Res (2017) 23(22):6946–57. doi: 10.1158/1078-0432.Ccr-17-0070
47. Li P, Zhang X, Gu L, Zhou J, Deng D. P16 Methylation Increases the Sensitivity of Cancer Cells to the CDK4/6 Inhibitor Palbociclib. PloS One (2019) 14(10):e0223084. doi: 10.1371/journal.pone.0223084
48. Medema RH, Herrera RE, Lam F, Weinberg RA. Growth Suppression by p16ink4 Requires Functional Retinoblastoma Protein. Proc Natl Acad Sci U S A (1995) 92(14):6289–93. doi: 10.1073/pnas.92.14.6289
49. Dosil MA, Mirantes C, Eritja N, Felip I, Navaridas R, Gatius S, et al. Palbociclib has Antitumour Effects on Pten-Deficient Endometrial Neoplasias. J Pathol (2017) 242(2):152–64. doi: 10.1002/path.4896
50. Wang Y, Ly A, Hosono Y, Murchie E, Walmsley CS. PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kα Inhibitors in Breast Cancer. Cancer Discov (2020) 10(1):72–85. doi: 10.1158/2159-8290.CD-18-0830.
51. Juric D, Castel P, Griffith M, Griffith OL, Won HH, Ellis H. Convergent Loss of PTEN Leads to Clinical Resistance to a PI(3)Kα Inhibitor. Nature (2015) 518(7538):240–4. doi: 10.1038/nature13948
52. Razavi P, Dickler MN, Shah PD, Toy W, Brown DN, Won HH, et al. Alterations in PTEN and ESR1 Promote Clinical Resistance to Alpelisib Plus Aromatase Inhibitors. Nat Cancer (2020) 1(4):382–93. doi: 10.1038/s43018-020-0047-1
53. Zelivianski S, Cooley A, Kall R, Jeruss JS. Cyclin-Dependent Kinase 4-Mediated Phosphorylation Inhibits Smad3 Activity in Cyclin D-overexpressing breast cancer cells. Mol Cancer Res (2010) 8(10):1375–87. doi: 10.1158/1541-7786.Mcr-09-0537
54. Kretschmer A, Moepert K, Dames S, Sternberger M, Kaufmann J, Klippel A. Differential Regulation of TGF-Beta Signaling Through Smad2, Smad3 and Smad4. Oncogene (2003) 22(43):6748–63. doi: 10.1038/sj.onc.1206791
55. Meirson T, Gil-Henn H. Targeting Invadopodia for Blocking Breast Cancer Metastasis. Drug Resist Updat (2018) 39:1–17. doi: 10.1016/j.drup.2018.05.002
56. Yang J, Song K, Krebs TL, Jackson MW. Danielpour D Rb/E2F4 and Smad2/3 link Survivin to TGF-Beta-Induced Apoptosis and Tumor Progression. Oncogene (2008) 27(40):5326–38. doi: 10.1038/onc.2008.165
57. Lamouille S, Xu J, Derynck R. Molecular Mechanisms of Epithelial–Mesenchymal Transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi: 10.1038/nrm3758
58. Thomas AL, Lind H, Hong A, Dokic D, Oppat K, Rosenthal E, et al. Inhibition of CDK-Mediated Smad3 Phosphorylation Reduces the Pin1-Smad3 Interaction and Aggressiveness of trIple Negative Breast Cancer Cells. Cell Cycle (2017) 16(15):1453–64. doi: 10.1080/15384101.2017.1338988
59. Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 Inhibitors Have Potent Activity in combination With Pathway Selective Therapeutic Agents in Models of Pancreatic Cancer. Oncotarget (2014) 5(15):6512–25. doi: 10.18632/oncotarget.2270
60. Liu F, Korc M. Cdk4/6 Inhibition Induces Epithelial-Mesenchymal Transition and Enhances Invasiveness in Pancreatic Cancer Cells. Mol Cancer Ther (2012) 10(11):2138–48. doi: 10.1158/1535-7163.MCT-12-0562
61. O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell (2015) 60(4):547–60. doi: 10.1016/j.molcel.2015.10.040
62. Geenen JJJ, Schellens JHM. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin Cancer Res (2017) 23(16):4540–44. doi: 10.1158/1078-0432.CCR-17-0520
63. Martin L-A, Pancholi S, Ribas R, Gao NS Q, Nikitorowicz-Buniak J, Johnston S, et al. Resistance to Palbociclib Depends on Multiple Targetable Mechanisms Highlighting the Potential of Drug Holidays and Drug Switching To Improve Therapeutic Outcome, Abstracts: 2016 San Antonio Breast Cancer Symposium. Cancer Res Cancer Res (2017) 77(4 Suppl):P3-03-09-P3-03-09. doi: 10.1158/1538-7445.
64. Grolmusz VK, Chen J, Emond R, Cosgrove PA, Pflieger L, Nath A, et al. Exploiting collateral sensitivity controls growth of mixed culture of sensitive and resistant cells and decreases selection for resistant cells in a cell line model. Cancer Cell Int (2020) 20:253. doi: 10.1186/s12935-020-01337-1
65. Kundu N, Brekman A, Kim JY, Xiao G, Gao C, Bargonetti J. Estrogen-Activated MDM2 Disrupts Mammary Tissue Architecture Through a p53-Independent Pathway. Oncotarget (2017) 8(29):47916–30. doi: 10.18632/oncotarget.18147
66. Laroche-Clary A, Chaire V, Algeo M-P, Derieppe M-A, Loarer FL, Italiano A. Combined Targeting of MDM2 and CDK4 is Synergistic in Dedifferentiated Liposarcomas. J Hematol Oncol (2017) 10(1):123. doi: 10.1186/s13045-017-0482-3
67. Portman N, Milioli HH, Alexandrou S, Coulson R, Yong A, Fernandez KJ, et al. MDM2 Inhibition in Combination With Endocrine Therapy and CDK4/6 Inhibition For the Treatment of ER-Positive Breast Cancer. Breast Cancer Res (2020) 22(1):87. doi: 10.1186/s13058-020-01318-2
68. Michaloglou C, Crafter C, Siersbaek R, Delpuech O, Curwen JO, Carnevalli LS, et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-positive Breast Cancer. Mol Cancer Ther (2018) 17(5):908–20. doi: 10.1158/1535-7163.Mct-17-0537
69. Dey N, De P, Leyland-Jones B. PI3K-AKT-mTOR Inhibitors in Breast Cancers: From Tumor Cell Signaling to Clinical Trials. Pharmacol Ther (2017) 175:91–106. S0163725817300517. doi: 10.1016/j.pharmthera.2017.02.037
70. Litchfield LM, Boehnke K, Brahmachary M, Mur C, Bi C, Stephens JR, et al. Combined Inhibition of PIM and CDK4/6 Suppresses Both mTOR Signaling and Rb Phosphorylation and Potentiates PI3K Inhibition in Cancer Cells. Oncotarget (2020) 11(17):1478–92. doi: 10.18632/oncotarget.27539
71. McCartney A, Migliaccio I, Bonechi M, Biagioni C, Romagnoli D, De Luca F, et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front Oncol (2019) 9:666. doi: 10.3389/fonc.2019.00666
72. Occhipinti G, Romagnoli E, Santoni M, Cimadamore A, Sorgentoni G, Cecati M, et al. Sequential or Concomitant Inhibition of Cyclin-Dependent Kinase 4/6 Before mTOR Pathway in Hormone-Positive HER2 Negative Breast Cancer: Biological Insights and Clinical Implications. Front Genet (2020) 15(11):349. doi: 10.3389/fgene.2020.00349
73. Jansen VM, Bhola NE, Bauer JA, Formisano L, Lee K-M, Hutchinson KE, et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res (2017) 77(9):2488–99. doi: 10.1158/0008-5472.CAN-16-2653
74. Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, et al. CDK 4/6 Inhibitors Sensitize PIK3CA Mutant Breast Cancer to PI3K Inhibitors. Cancer Cell (2014) 26(1):136–49. doi: 10.1016/j.ccr.2014.05.020
75. Shaulian E, Karin M. AP-1 in Cell Proliferation and Survival. Oncogene (2001) 20(19):2390–400. doi: 10.1038/sj.onc.1204383
76. Shen Q, Uray IP, Li Y, Zhang Y, Hill J, Xu XC, et al. Targeting the Activator Protein 1 Transcription Factor for the Prevention of Estrogen Receptor-Negative Mammary Tumors. Cancer Prev Res (Phila) (2008) 1(1):45–55. doi: 10.1158/1940-6207.Capr-08-0034
77. Angelis C, Nardone A, Cataldo ML, Veeraraghavan J, Fu X, Giuliano M, et al. AP-1 as a Potential Mediator of Resistance to the Cyclin-Dependent Kinase (CDK) 4/6-Inhibitor Palbociclib in ER-Positive Endocrine-Resistant Breast Cancer. Cancer Res (2018) 78:P4–03. doi: 10.1158/1538-7445.SABCS17-P4-03-05
78. Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small Molecule Inhibitors Targeting Activator Protein 1 (AP-1). J Med Chem (2014) 16(57):6930–48. doi: 10.1021/jm5004733
79. Sobhani N, Fan C, Flores-Villanueva PO, Generali D, Li Y. The Fibroblast Growth Factor Receptors in Breast Cancer: from Oncogenesis to Better Treatments. Int J Mol Sci (2020) 21(6):2021. doi: 10.3390/ijms21062011
80. Perez-Garcia J, Muñoz-Couselo E, Soberino J, Racca F, Cortes J. Targeting FGFR Pathway in Breast Cancer. Breast (2018) 37:126–33. doi: 10.1016/j.breast.2017.10.014
81. Sahores A, May M, Sequeira GR, Fuentes C, Jacobsen B, Lanari C, et al. Targeting FGFR with BGJ398 in Breast Cancer: Effect on Tumor Growth and Metastasis. Curr Cancer Drug Targets (2018) 18(10):979–87. doi: 10.2174/1568009618666171214114706
82. Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR Signaling Mediates Resistance to CDK4/6 Inhibitors in ER+ Breast Cancer. Nat Commun (2019) 10(1):1373. doi: 10.1038/s41467-019-09068-2
83. Pradella D, Naro C, Sette C, Ghigna C. EMT And Stemness: Flexible Processes Tuned by Alternative Splicing in Development and Cancer Progression. Mol Cancer (2017) 16(1):8. doi: 10.1186/s12943-016-0579-2
84. Kim DH, Xing T, Yang Z, Dudek R, Lu Q, Chen Y-H. Epithelial Mesenchymal Transition in Embryonic Development, Tissue Repair and Cancer: A Comprehensive Overview. J Clin Med (2017) 1. doi: 10.3390/jcm7010001
85. Du B, Shim JS. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules (2016) 21(7):965. doi: 10.3390/molecules21070965
86. Tobin NP, Sims AH, Lundgren KL, Lehn S, Landberg G. Cyclin D1, Id1 and EMT in Breast Cancer. BMC Cancer (2011) 11:417. doi: 10.1186/1471-2407-11-417
87. Yaswen P. Reinforcing Targeted Therapeutics With Phenotypic Stability Factors. Cell Cycle (2014) 13(24):3818–22. doi: 10.4161/15384101.2014.985071
88. Ernst B, Anderson KS. Immunotherapy for the Treatment of Breast Cancer. Curr Oncol Rep (2015) 2(17):5. doi: 10.1007/s11912-014-0426-9
89. Jia H, Truica CI, Wang B, Wang Y, Ren X, Harvey HA, et al. Immunotherapy For Triple-Negative Breast Cancer: Existing Challenges and Exciting Prospects. Drug Resist Update (2017) 32:1–15. doi: 10.1016/j.drup.2017.07.002
90. Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature (2017) 548(7668):471–5. doi: 10.1038/nature23465
91. Deng J, Wang ES, Jenkins RW, Li S, Dries R, Yates K, et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov (2018) 8(2):216–33. doi: 10.1158/2159-8290.CD-17-0915
92. Sisinni L, Pietrafesa M, Lepore S, Maddalena F, Condelli V, Esposito F, et al. Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance Between Apoptosis and Autophagy and Its Role in Drug Resistance. Int J Mol Sci (2019) 20(4):10.3390/ijms20040857. doi: 10.3390/ijms20040857
93. Chen S, Jiang Y-Z, Huang L, Zhou R-J, Yu K-D, Liu Y, et al. The Residual Tumor Autophagy Marker LC3B Serves as a Prognostic Marker in Local Advanced Breast Cancer after Neoadjuvant Chemotherapy. Clin Cancer Res (2013) 19(24):6853–62. doi: 10.1158/1078-0432.CCR-13-1617
94. Lefort S, Joffre C, Kieffer Y, Givel A-M, Bourachot B, Zago G, et al. Inhibition of Autophagy as a New Means of Improving Chemotherapy Efficiency in High-LC3B Triple-Negative Breast Cancers. Autophagy (2014) 10(12):2122–42. doi: 10.4161/15548627.2014.981788
95. Rubinstein AD. Kimchi A Life in the Balance - A Mechanistic View of the Crosstalk Between Autophagy and Apoptosis. J Cell Sci (2012) 125(Pt 22):5259–68. doi: 10.1242/jcs.115865
96. Gangemi R, Paleari L, Orengo AM, Cesario A, Chessa L, Ferrini S, et al. Cancer Stem Cells: A New Paradigm for Understanding Tumor Growth and Progression and Drug Resistance. Curr Med Chem (2009) 16(14):1688–703. doi: 10.2174/092986709788186147
97. Verma S, Bartlett CH, Schnell P, DeMichele AM, Loi S, Ro J, et al. Palbociclib in Combination With Fulvestrant in Women With Hormone Receptor-Positive/HER2-Negative Advanced Metastatic Breast Cancer: Detailed Safety Analysis From a Multicenter, Randomized, Placebo-Controlled, Phase III Study (PALOMA-3). Oncologist (2016) 21(10):1165–75. doi: 10.1634/theoncologist.2016-0097
98. Goetz MP, Toi M, Campone M, Sohn J, Paluch-Shimon S, Huober J, et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J Clin Oncol (2017) 35(32):3638–46. doi: 10.1200/JCO.2017.75.6155
99. Dickler MN, Tolaney SM, Rugo HS, Cortés J, Diéras V, Patt D, et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(-) Metastatic Breast Cancer. Clin Cancer Res (2017) 23(17):5218–24. doi: 10.1158/1078-0432.Ccr-17-0754
100. Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, et al. Updated Results from MONALEESA-2, a Phase III Trial of First-Line Ribociclib Plus Letrozole Versus Placebo Plus Letrozole in Hormone Receptor-Positive, HER2-Negative Advanced Breast Cancer. Ann Oncol (2018) 29(7):1541–7. doi: 10.1093/annonc/mdy155
101. Tripathy D, Im SA, Colleoni M, Franke F, Bardia A, Harbeck N, et al. Ribociclib Plus Endocrine Therapy for Premenopausal Women With Hormone-Receptor-Positive, Advanced Breast Cancer (MONALEESA-7): A Randomised Phase 3 Trial. Lancet Oncol (2018) 19(7):904–15. doi: 10.1016/s1470-2045(18)30292-4
102. Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III Randomized Study of Ribociclib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J Clin Oncol (2018) 36(24):2465–72. doi: 10.1200/jco.2018.78.9909
103. André F, Ciruelos EM, Juric D, Loibl S, Campone M, Mayer IA, et al. Alpelisib Plus Fulvestrant for PIK3CA-Mutated, Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor-2-Negative Advanced Breast Cancer: Final Overall Survival Results From SOLAR-1. Ann Oncol (2021) 32(2):208–17. doi: 10.1016/j.annonc.2020.11.011
Keywords: breast cancer, CDK4/6 inhibitors, drug resistance, molecular mechanisms, combination administration
Citation: Wang B, Li R, Wu S, Liu X, Ren J, Li J, Bi K, Wang Y and Jia H (2021) Breast Cancer Resistance to Cyclin-Dependent Kinases 4/6 Inhibitors: Intricacy of the Molecular Mechanisms. Front. Oncol. 11:651541. doi: 10.3389/fonc.2021.651541
Received: 10 January 2021; Accepted: 01 February 2021;
Published: 26 May 2021.
Edited by:
Jin-Ming Yang, University of Kentucky, United StatesReviewed by:
Jinghui Liu, University of Kentucky, United StatesZhuangzhuang Zhang, University of Kentucky, United States
Copyright © 2021 Wang, Li, Wu, Liu, Ren, Li, Bi, Wang and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyan Jia, c3dhbGxvd19qaHlAMTYzLmNvbQ==; Yanhong Wang, d2FuZ3lhbmhvbmdtYWlsQDEyNi5jb20=
†These authors have contributed equally to this work