 Tiffany C. Blair
Tiffany C. Blair Alejandro F. Alice
Alejandro F. Alice Lauren Zebertavage
Lauren Zebertavage Marka R. Crittenden2,3
Marka R. Crittenden2,3 Michael J. Gough
Michael J. Gough- 1Molecular Microbiology and Immunology, Oregon Health and Sciences University (OHSU), Portland, OR, United States
- 2Earle A. Chiles Research Institute, Providence Cancer Institute, Providence Portland Medical Center, Portland, OR, United States
- 3The Oregon Clinic, Portland, OR, United States
Analysis of tumor infiltration using conventional methods reveals a snapshot view of lymphocyte interactions with the tumor environment. However, lymphocytes have the unique capacity for continued recirculation, exploring varied tissues for the presence of cognate antigens according to inflammatory triggers and chemokine gradients. We discuss the role of the inflammatory and cellular makeup of the tumor environment, as well as antigen expressed by cancer cells or cross-presented by stromal antigen presenting cells, on recirculation kinetics of T cells. We aim to discuss how current cancer therapies may manipulate lymphocyte recirculation versus retention to impact lymphocyte exclusion in the tumor.
Introduction
The evolutionary development of an adaptive immune system requires different systems to localize responses. An adaptive immune response against a specific virus can result in viral antigen-specific B cells and T cells that remain throughout life (1). The progressive accumulation of such responses over the lifetime of an individual would make it impractical to have all of the different types of antigen-specific cells located at all possible sites of infection. Rather than distribute all immune cells uniformly, the mammalian immune system uses a system of sensors and rapid responses to mobilize responses to a location. Compared to a fully distributed immune system this will result in a delay, but allows a more flexible system.
To achieve this the mammalian immune system employs selective sensors, which provide an initial indication of the type of immune response required, to mobilize suitable cells. For example, intracellular sensors for nucleic acids that might detect a viral infection, such as TLR3, RIG-I-like receptors and cGAS/STING can generate a different pattern of cytokine responses to extracellular sensors for bacterial components such as TLR2 and TLR4 (2, 3). Moreover, the various sensors are not uniformly shared across innate immune cell types, so that specific immune cells can specialize in detecting certain infections (3, 4). In this way the immune response is pre-warned of the nature of the infection, and the appropriate adaptive response that is needed.
Among the cells that participate in the adaptive immune response, T cells have a particular limitation in that they must be in physical proximity to a target cell expressing antigens in order to exert their effect. B cells can retreat to stable niches in the bone marrow and secrete antibody for the duration of the animal’s lifespan (1). This isn’t possible for T cells. To overcome this limitation, the immune system exploits a mechanism to move antigens to T cells, and T cells to antigen (5). Naïve T cells do not travel to tissue sites and so cannot scan for their cognate antigen at the site of infection. Instead, dendritic cells are able to carry antigen from tissues to draining lymph nodes where they are able to present the antigen to naïve T cells and initiate antigen-specific immune responses. Mathematical modeling suggests direct lymph node entry of T cells is low in the absence of inflammation (6). For naïve T cells, entry is classically dictated by CCR7, which also directs T cells to the vicinity of dendritic cells within the T cell zone of the lymph node (7). However, inflammation in the upstream site can lead to remodeling of the lymph node to increase infiltration of naïve T cells and recruitment of all T cells to the node (7, 8). Once in the lymph node, T cells that continue to receive chemokine signals, or are held in place via cognate interaction and retention signals such as CD69, will remain in the lymph node. However, there is an ongoing pull via S1PR1 on the T cells and S1P in the lymphatics that results in exit of T cells that fail to meet their cognate ligand or have disengaged from antigen presenting cells (9). This ongoing pressure to leave ensures continued recirculation of T cells in search of cognate ligands.
Once they are antigen experienced, T cells are subsequently able to travel through the blood to tissues due to a range of changes including altered selectin expression (7) and explore local MHC for their cognate antigens. Importantly, a recirculation system exists to return these T cells through the draining lymphatics and back into blood circulation (5, 7). Without such a system of recirculation, antigen-experienced cells would be ‘lost’ to the tissues resulting in a progressive loss of antigen-experienced cells from the circulation. This is the critical feature that provides our circulating, distributed form of adaptive immune system. While the principle of recirculation is well known and a fundamental of basic immunology, what is often unappreciated is the rate of recirculation. In studies performed over 50 years ago it was demonstrated that the total blood pool of lymphocytes can be refreshed 11 times per day based purely on the output from the thoracic duct (10). The drug FTY720, which prevents lymphocyte egress from lymph nodes by blocking Sphingosine-1-phosphate receptor 1 (S1PR1), can result in complete loss of thoracic duct lymphocytes within 4 hours (11) and 90% loss of peripheral blood lymphocytes in 3-24 hours (12), demonstrating an extremely high recirculation rate. While FTY720 treatment rapidly removes lymphocytes from the blood it has a lesser effect on the tissues, and these data have allowed investigators to calculate transit times of approximately 18 hours through tissue parenchyma (13). Thus, T cells spend their least amount of time in the peripheral blood, with estimates ranging from 1-2 circulations through the heart, which corresponds to timescales of minutes in the bloodstream of mice (6, 14). By contrast lymph node transit even in the absence of cognate antigen can take approximately 10 hours (6, 15). In models of long-term memory to viral infection, it was found that less than 4% of the virus-specific T cell memory population was present in the peripheral blood at any one time (16). Thus, T cells spend the majority of their time in tissues or secondary lymphoid organs scanning for cognate antigens.
As with all rules, there are exceptions. Recently, resident memory T cells (Trm) have been described that remain in peripheral sites long term and provide rapid local antigen-specific responses (7, 16, 17). The majority of experiments that identified these cells were performed in very clean laboratory settings where the mice had an extremely limited history of infection. This makes it difficult to assess how diverse the Trm pool is in peripheral tissues, since there will clearly be a ‘space’ constraint in supporting a fully diverse T cell population at all peripheral sites. Human neonates have increased populations of naïve T cells compared to adult humans, and antigen-experienced populations are less frequent in neonatal tissue where there is likely a limited experience of antigen (18, 19). Therefore, with antigenic experience, peripheral niches are populated with antigen-experienced cells. By analyzing wild and pet-shop mice, Beura et al. demonstrated a dramatically higher population of memory T cells populating peripheral tissues of wild mice compared to laboratory mice (20). In normal human pancreas tissue, Trm were found to express similar defining markers such as CD69 and CD103, but were phenotypically distinct from jejunal Trm (21). This included decreased expression of a range of inflammatory markers (21), which may relate to the lower ongoing exposure to infectious agents in the pancreas. As would be expected given the potential for differing local antigen exposure, Trm clones in the pancreas and jejunum also had limited overlap (21). In agreement with this, Trm in normal lung but not other sites demonstrated reactivity to influenza antigens (22); however, influenza-specific Trm in the lung share clonotypes with non-Trm memory subtypes in the lung (23). These data demonstrate that while Trm provide local recognition, their function is reinforced by recirculating populations. Difficulties in comparing clonotypes is highlighted by Schoettler et al, who demonstrated using lung samples that only approximately 5% of more than 100,000 TCR clones were found in more than one tissue or patient sample and only TCRs expressed by CD4 T cells were identified as shared across multiple memory populations in both the lung and lung-draining lymph nodes (24). It remains unclear quite how comprehensively protective the Trm cell response can be given the limited size of the Trm niche in any one place, compared to the diversity of the combined repertoire that is recirculating or resident elsewhere at any moment. It is possible that the Trm niche reflects recent antigen exposures, and pre-existing cells are displaced to new recruits. In this way the peripheral resident population will proportionally represent frequent infectious agents, including non-pathogenic colonizing organisms and this resident response will therefore be of most use for rapid responses to these most frequently exposed agents. This is supported by data in lung infection, where repeated antigen exposure ensures a durable lung Trm population (25). In this, way, earlier exposures to a common organism may come to dominate the local response. By monitoring the response to viral infections containing model antigens, Muschaweckh et al. demonstrated a selective preference for T cells specific for the model antigen Ovalbumin to form Trm as compared to T cells specific for the viral antigen B8R20 (26). However, if the tissue was first allowed to form Trm specific for the viral antigen B8R20 through prior infection, then B8R20 -specific T cells dominated the Trm niche even after challenge with a B8R20 and Ova-expressing virus (26). These data suggest that Trm formation is affected by immunodominance (discussed more later), and that existing Trm populations can outcompete simultaneous incoming new responses to retain their place in the tissue niche. Importantly, T cells specific for the same antigen can be found as Trm and as classical circulating memory populations (27), so even if Trm that recognize a specific infection are lost from the tissue niche through progressive rounds of infection with other agents, the circulating memory can remain.
The principle of recirculation is also essential to overcome the fact that T cell recruitment to tissue sites is not antigen specific. Recruitment to peripheral sites is dependent on inflammatory patterns. Inflammation in the tissue site generates cytokines such as TNFa, which activates endothelial cells to express adhesion molecules such as ICAM1 [reviewed in (28, 29)]. This is critical to initiate the process of tissue entry by lymphocytes by allowing rolling along the endothelial surface of the blood vessel lumen. In addition, local inflammation results in chemokine secretion, and chemokine binding to receptors on rolling cells permits changes in adhesion to tight binding, and eventually diapedesis through the endothelia and into the tissue (28). This means that an infection that results in a local inflammatory response triggered via infection sensors will non-specifically recruit any T cells expressing selectins and appropriate chemokine receptors, regardless of TCR specificity. Since the recruited T cells need both the correct chemokine receptors and activation-regulated adhesion molecules to permit diapedesis through the vasculature into the tissue, there will be selection for activated T cells (30). In animal models, there is likely only one major ongoing infection at any one time, so the majority of the emerging activated cells are likely specific for the infection (20). However, in a human there are likely multiple ongoing infections simultaneously occurring at different sites, therefore T cells specific for an ongoing flu infection may also be recruited to the site of an infected splinter, and vice versa. Recirculation permits non-specific cells to leave the tissue and be available for recruitment again. This can also result in dominance of a highly inflamed tissue. For example, a lung infection can recruit T cells specific for other pathogens to the lung as part of the local inflammatory response (31). Similarly, in tumors, T cell recruitment is not antigen-directed but instead attracts all T cells with appropriate activation markers (32). To return these cells to the general circulation, and ensure they are available to respond to their cognate targets should they return, requires efficient recirculation.
Using tools such as Kaede mice, where cells can be photoconverted at a specific site then followed for their movement (33), it is clear that not all T cells are equivalent in their recirculation dynamics. By labeling the tumor then analyzing the tumor draining lymph node, within 1 day of tumor labeling the majority of emigrated cells are dendritic cells and T cells (34). Lymph nodes draining tumors exhibited a much higher overall number of recirculating cells than normal skin (34), suggesting a high rate of immunosurveillance in tumors, despite tumor progression. Though the number of T cells in the lymph node that had been in the tumor at conversion peaked at 1 day following conversion, tumor-originating cells were still in the lymph node at day 3 (34). It is not clear whether this is continued emigration or retention of these cells in the lymph node, but the tumor was still observed to hold a large proportion of the converted T cells at day 3 after conversion (34). As would be expected based on the requirements for initial tumor infiltration, the majority of cells recirculating to the draining lymph node are enriched for effector and central memory phenotypes. Using the Kaede system to convert skin resident T cells in infectious models, Park et al. demonstrated that Trm in the skin remain in place following viral rechallenge, and do not recirculate via the draining lymph node (35). However, circulating virus-specific T cells are recruited to the skin site, and themselves become Trm following infection (35). In tumor models, by day 3 following conversion of Kaede cells in the tumor, some of the converted T cells are detectable in the lymph nodes draining an identical tumor at a distant site but the converted cells are poorly detectable in distant lymph nodes that do not drain tumors (34). These data suggest that by this time point dissemination through the peripheral blood has occurred and antigen-mediated retention has allowed accumulation of tumor-specific populations in distant lymph nodes. Thus, even following exit from tumors, recirculation and accumulation at sites of distant antigen are rapid in vivo.
In this review we will discuss how the principle of T cell recirculation impacts lymphocyte exclusion in the tumor environment. In addition, we will explore the effect of therapy on lymphocyte numbers in the tumor, with a focus on the differing effects on recruitment versus retention. In response to cognate antigens T cells can also proliferate locally, which will also result in T cell accumulation at the tumor. In our review we will not discriminate the mechanisms that result in lymphocyte arrest versus proliferation in response to cognate antigen. Together these are grouped as retention mechanisms, rather than recruitment mechanisms. Different treatments may differently affect recruitment versus retention of lymphocytes, and this may play a role in their successes and failures.
Lymphocyte Recirculation Kinetics
Most assessments of tumor infiltration view only a single snapshot in time within the tumor. Although such assessments may show a high degree of tumor infiltration by T cells, it may also represent a tumor with a high throughput of T cells entering and leaving the tumor, so actually representing a high rate of surveillance (Figure 1A). If we consider the lymphocytes in a tumor over time, we can anticipate that some continue to recirculate, while others remain (Figure 1B). Assuming constant inflammatory conditions there is little reason for the overall number of cells to change over time, but when comparing the specificity of the T cells present at any two timepoints we could expect completely different T cells are infiltrating the tumor. With inflammatory flux, the numbers may go up or down as recruitment changes, but their overall time spent in the tumor may be unaffected if their retention is unchanged. Tools such as TCRSeq allow us to sequence the TCR of T cells and examine their diversity (36, 37). Expansions of specific T cells as clonal populations are detectable as repeated TCR sequences with increased frequency and using this technique to examine tumors shows that there are measurable clonal expansions in tumors. If we were to use TCRSeq to compare TCR clones in the tumor over time, we would anticipate an overall change in TCR clonotypes according to the degree of recirculation. Among the population that is retained over time, we would anticipate enrichment for properties of resident cells, such as Trm, or ongoing antigen engagement that results in prolonged adhesive interactions with their targets that impacts retention. This is supported by data from patient tumors, demonstrating that tumor reactive CD8 T cells in tumors express markers associated with tissue residency (38, 39). Multiple additional markers that may define the Trm phenotype have been described (40); however, experimental limitations means that residency has only been proven in murine systems. Nevertheless, ongoing studies in human tissues have identified shared features of cells expressing the canonical CD103/CD69 signature in humans (40). Bystander viral-specific CD8 T cells in tumors can also express these Trm markers (39), therefore CD39, a marker of chronic T cell activation is useful in distinguishing between the bystander and tumor-reactive T cell populations (38, 39). Duhen et al. used the combination of CD39 and the Trm marker CD103 to enrich for tumor-reactive CD8 T cells, and compared these cells in the tumor to those from the blood and lymph nodes (38). The CD103+CD39+ (double positive – DP) CD8 T cells in the tumor were shown to have clear enrichment for specific clonotypes. Duhen et al. demonstrated that the greatest TCR diversity was found in CD8 T cells in the blood, and the lowest TCR diversity was found in the DP CD8 T cells in the tumor (38). DP CD8 T cells did not significantly share TCR sequences with CD8 T cells in the tumor draining lymph node or the peripheral blood, while CD103-CD39- (double negative – DN) CD8 T cells did share TCR sequences with circulating cells (38), suggesting that the DP CD8 T cells are selectively retained within the tumor and the DN CD8 T cells are recirculating. This also means that examining any two different time-points might show a similar proportion of clonally expanded cells, but the non-specific clones would change over time and the cells that are present at both time points would be expected to be enriched for tumor antigen-specific cells. This proposition appears to be supported by current data. Using a combination of scRNASeq and TCRSeq Yost et al. demonstrated clonal expansions of CD8 T cells in tumors were enriched for exhaustion markers (41), and that these same cells also exhibited evidence of tumor reactivity based on expression of CD39 and CD103. Importantly, they found little overlap in TCR clonotypes between the exhausted population and CD8 T cells with effector phenotypes, suggesting that these cells have distinct specificities (41).
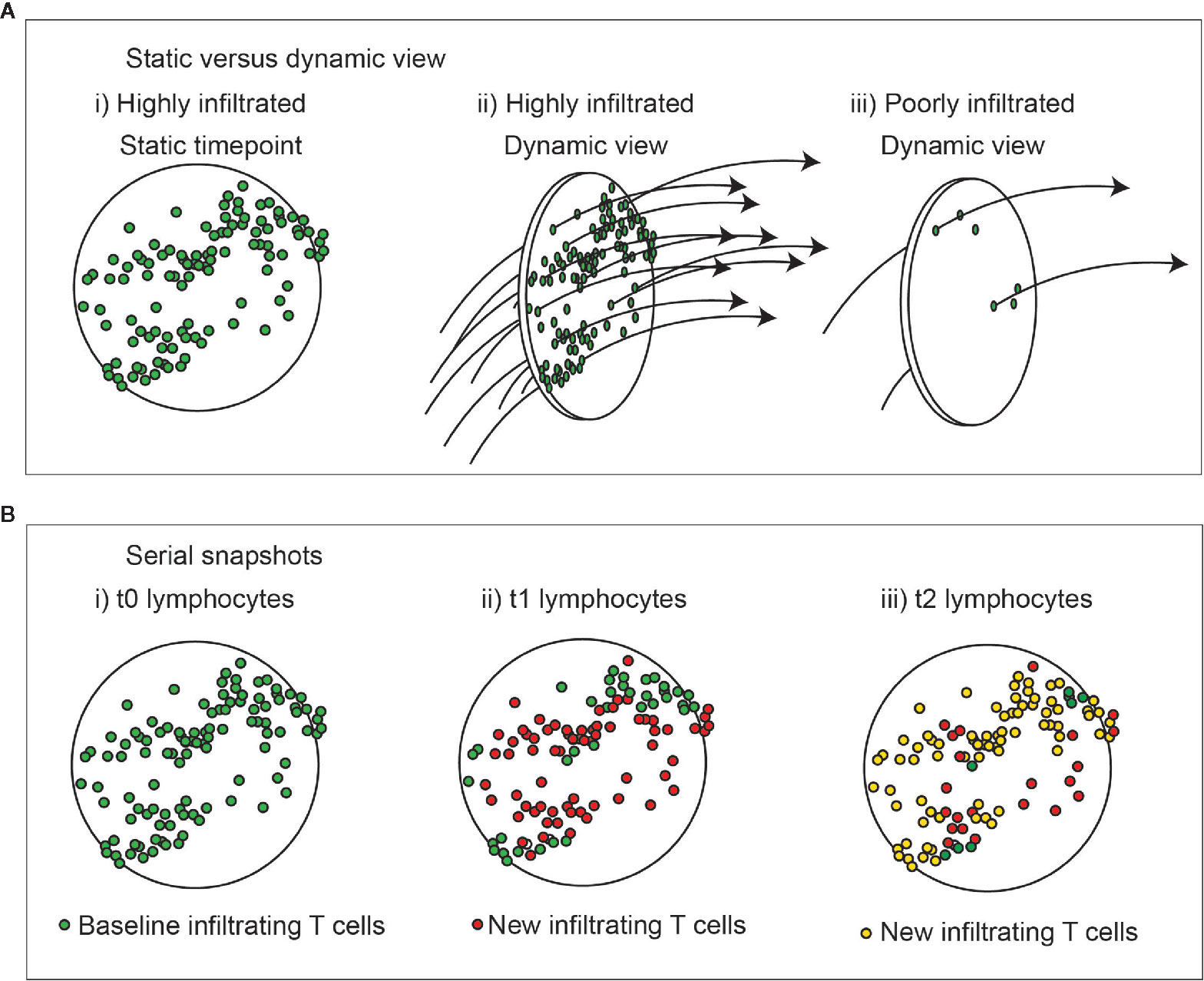
Figure 1 Dynamic view of cell infiltration in tumors. (A) A highly infiltrated tumor may also represent a high rate of throughput of immune cells. i) a static view shows large numbers of T cells in the tumor. ii) a dynamic view shows a high rate of surveillance and recirculation. Iii) a dynamic view of a poorly infiltrated tumor shows a low rate of surveillance and recirculation. Understanding the kinetic will help understand the rate of accrual versus accumulation in the environment. (B) Change in lymphocytes in tumors over time with unchanged overall infiltration. i) Lymphocytes present at baseline are green. ii) Those newly present at the second timepoint are red, and iii) newly present at the third timepoint are yellow. At each timepoint not all cells are replaced, and those exhibiting prolonged interactions in the tumor are more likely to have engaged their cognate antigen.
While we assume that recirculation will result in selective replacement of only non-specific cells, and that while the overall diversity of the infiltrate might remain consistent, there will be some notable caveats. We all have clonally expanded T cells specific for common viruses meaning that our circulating T cell pool is not uniformly distributed among possible TCR sequences. These clonally expanded T cells would also be present in the tumor through non-specific recruitment, but these would not be expected to be selectively retained. However, any chronically active T cell populations, such as those for CMV or EBV might be enriched in the activated T cell pool that is recruited to tumors. Thus, not all clonally expanded cells in tumors can be predicted to be tumor specific. Scheper et al. cloned the TCR from tumor infiltrating T cells and evaluated their specificity for autologous cancer cells. They found that only a small proportion – from 1-10% of T cells in the tumor were specific for cancer cells, and that in some examples T cells specific for EBV were three times more frequent in the tumor (42). As many as 3% of CD8 T cells infiltrating tumors have been shown to be specific for a CMV epitope (39). This number likely varies significantly between tumors, according to the degree of non-specific recruitment and the antigen-specific retention. For example, in the above paper, the authors analyzed a melanoma specimen where 90% of the tumor-infiltrating T cells expressed PD1, and 50-80% of all T cells were estimated to be tumor specific (42). Shitaoka et al. demonstrated that CD8+CD137+ cells represented 10-70% of CD8 T cells infiltrating human tumors, and a large proportion of these were clonally expanded (43). In murine tumors CD8+CD137+ cells represented up to 5% of tumor-infiltrating CD8 T cells, and TCRs from clonally expanded CD8+CD137+ cells were mostly tumor-reactive (43), suggesting that this population also enriches for tumor-reactive cells. Tumor-specific T cells generated ex vivo and adoptively transferred into an animal circulate widely, with no particular selectivity for a tumor expressing the cognate antigen (44). However, functional activation was limited to the site of antigen. These data suggest that non-tumor specific clonally expanded populations would be expected in the tumor, and tumor-specific clonally-expanded populations would be expected elsewhere. In this way clonal expansion may be an insufficient measure of specificity for antigens at that site. This is supported by current data. Penter et al. explored clonal expansions in colorectal tumors as well as uninvolved sites, and found similar rates of clonal expansions inside and outside the tumor (45), indicating that clonal enrichment is not unique to tumors. However, while on aggregate uninvolved and tumor regions had similar expansions, the data suggest that individual patients may exhibit enrichment for clonal expansions in the tumor. The tumor-associated clones exhibited increased expression of activation and exhaustion markers such as PD1, which are not seen in non-tumor clones (45), suggesting they are chronically recognizing antigen while in the tumor. Clonally expanded populations in the peripheral blood were stable over time (45), likely representing circulating memory populations specific for common targets such as EBV and CMV, and importantly a dominant CMV-specific clone was shown to be expanded in the blood, tumor, and uninvolved tissue site, demonstrating that these cells recirculate widely. In addition, clones that were highly expanded in the tumor were also detectable in the uninvolved tissue site (45), suggesting that tumor-specific cells may also be recirculating or can take up residency elsewhere. Analysis of lung tumors versus distant normal lung tissue demonstrated that highly expanded clones were more frequent in normal tissue than the tumor and the T cell in the tumor had greater TCR diversity (46), suggesting that the tumor recruits more non-specific T cells compared to normal tissue. Interestingly, in this and other studies there are data suggesting that a lower clonality and an increased T cell diversity in tumors is associated with worse outcome to conventional therapies and immunotherapies (46, 47). This would fit with clonal populations representing accumulated tumor-specific T cells among a background of diverse non-specific T cells. In this way, overall infiltrate is less informative than infiltrates of specific T cells.
For these reasons, a critical measure of tumor specificity or selectivity may be serial assessment. However, such analyses are rare. In part this is due to the clinical scenarios, since most analyses are performed on single biopsies or a tumor resection specimen. Yost et al. used bulk TCRSeq to compare T cell clonality in untreated tumors at two timepoints and found no significant changes in the TCR sequences present in clonally expanded populations over time (41). This was in contrast to tumors sampled before and after PD1 blockade, which resulted in an influx of new expanded clones (41). Later we will explore the effects of therapy on recirculation kinetics, but it is reasonable that at baseline the T cell infiltration of any tumor will be directly related to recruitment and retention, and be generally split into rapidly recirculating non-specific cells and selective retention of antigen-specific cells. Thus, increasing recruitment via inflammation and chemokines has the potential to increase the diversity of T cells in the tumor, but this occurs without any selectivity for tumor-specific cells (Figure 2A). As we will discuss later, recruitment is not selective for tumor-specific T cells, and will attract any cells expressing the appropriate chemokine receptors, whether tumor-specific or CMV-specific. By contrast increasing antigen presentation and altering recognition thresholds using costimulatory agonists or coinhibitory blockade has the potential to increase clonality by increasing retention of tumor-specific cells (Figure 2B).
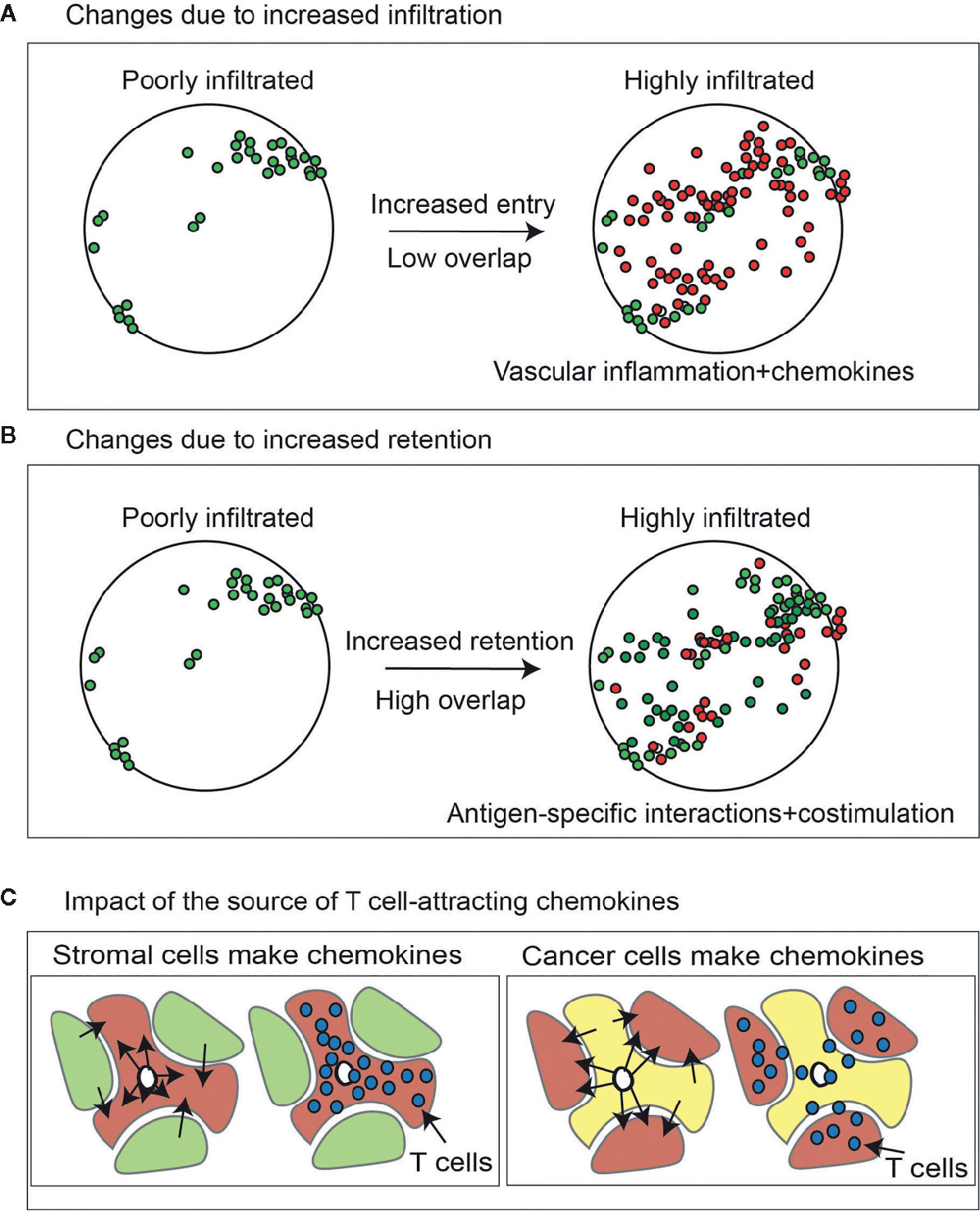
Figure 2 Change in lymphocytes in tumors due to increase infiltration versus increased retention. (A) Effect of increasing lymphocyte infiltration via chemokines and inflammatory changes in the tumor vasculature on T cell diversity in the tumor. Lymphocytes present at baseline are green. Those newly present at the second timepoint are red. (B) As with (A), but the effect of increasing T cell retention via increased antigen-specific interactions via altered antigen presentation or costimulation. (C) Higher expression of chemokines that can attract activated T cells in cancer cells will cause proportional enrichment of immune cells in the vicinity of cancer cell nests. By contrast higher expression of the same chemokines in the tumor stroma may cause their enrichment in the stroma but exclusion from the cancer cell nests, limiting their cytotoxic potential.
Intravital 2 photon microscopy has helped understand the dynamics of T cells in tumors. Using cancer cells expressing ovalbumin as a model antigen to view tumor-specific responses of OT1 TCR transgenic T cells, Breart et al. demonstrated that in a highly responsive model, transferred T cells were first visualized in the vicinity of vascular entry points and rapidly spread throughout the tumor resulting in cure (48). Salmon et al. demonstrated higher motility of T cells in the tumor stroma, and their comparative exclusion from tumor nests (49). Motility was highest in the immediate perivascular location, with fibers of the extracellular matrix serving to prevent direct T cell interactions with cancer cells (49). Initial estimates suggested that tumor-specific T cells and cancer cells required a 6hr duration of interaction to result in cancer cell death (48). This timing is interesting since only a proportion of T cells exhibit a longer duration interaction with cancer cells in tumors, with the majority exhibiting short-term interactions with multiple cells despite having specificity for tumor-associated antigens (50). Random migration of the T cells occurs prior to stable interaction with the target, and the kinetics of T cell movement differs depending on the presence of the cognate antigen (50). While T cells pause when meeting cancer cells that they can recognize, in models where the T cells are known to be responsible for curing tumors, they resume their motility once they successfully kill their targets (51). Interestingly in these models, T cells could be seen actively moving along the exterior of blood vessels in the periphery of the tumor, which would represent a stromal location, and they adopted a non-motile appearance once detached from the blood vessel (51). T cells only entered deeper regions of the tumor when cognate antigen was present, and these changes took more time and followed the kinetic of successful tumor elimination at the periphery. This potentially relates to inflammatory changes in the vicinity of T cells actively engaging antigen that are propagated to neighboring regions that were formerly poorly inflamed and thus poorly infiltrated, or due to the T cells being restrained in the periphery until cancer cells were eliminated and they could resume their motility.
Studies using pertussis toxin demonstrate that chemokine signals are necessary for T cell motility within the tumor stroma (49), and cancer cells engineered to express chemokines can increase recruitment of T cells into the stroma (52), and on into cancer nests (49). In such a scenario, the ability of newly recruited T cells to meet their cognate antigen presented on cancer cells will be highly dependent on local chemokine signals that recruit T cells out of the stroma and towards cancer cell nests (Figure 2C). At the same time, inflamed lymphatic endothelial cells may secrete their own chemokines (53) and direct T cells out of the stroma to recirculation. The pre-existing Trm populations resident within cancer cell nests have both a phenotype that encourages adhesion to the cancer cells (38, 54) and lack SIP1R resulting in limited capacity for lymphatic traffic (55). Thus, once past the tumor stroma circumstances favor tumor residency versus recirculation of tumor antigen specific T cells.
Chemokine Modification of Tumors to Increase Recruitment
To understand recruitment and retention it is useful to take the example of two different tumors, one highly infiltrated and another poorly infiltrated with T cells. At baseline, we have no information as to whether the tumors are highly infiltrated due to an increased recruitment of T cells to the tumor, or increased retention of T cells within the tumor (Figure 2). If recruitment is the key criteria, then manipulating inflammatory signals within the tumor will influence infiltration. Over the last few decades, we and others have explored modification of cancer cells with cytokines and chemokines to increase immune cell infiltrates into tumors. Engineering tumors to express chemokines that attract T cells results in increased T cell infiltration and increased tumor immunogenicity (56–58). These data suggest that T cell recruitment to tumors is limited by suboptimal chemokine expression. Analysis of tumors with high versus low T cell infiltrates demonstrated that expression of a panel of 12 chemokine genes could predict increased T cell infiltration into tumors (59, 60). However, in view of the recirculation behavior of T cells, how will increasing chemokine levels change anti-tumor immunity? As discussed above, increased inflammation in the tumor or increases in chemokine expression will lead to a non-specific influx of T cells into these inflammatory areas, where the majority of cells are not tumor-specific. Among these recruited T cells there may also be populations of unconventional T cells, for example gamma delta T cells. Such cells can share recruitment mechanisms, and while they may not recognize tumor antigens, they may respond to the altered metabolic status in the tumor environment (61).
The fact that tumors may progress despite the presence of targetable neoantigens can in part be explained by immunological ignorance (62). T cells specific for cancer cells can recirculate through the tumor without impacting the tumor growth, unless given some external stimulus. This can be due to limited ongoing cross presentation (63, 64), or limited direct presentation due to low expression of the cognate antigen or low levels of antigen processing and presentation in the cancer cells (65, 66). In some circumstances these limitations can be overcome through increased antigen release from the cancer cells (67, 68). However, T cell ignorance of tumors can also be impacted by a poor rate of recirculation through tumors as well as their limited exposure to tumor-associated antigens. In addition, whether T cells are ignorant of the tumor or are actively recirculating through the tumor, these T cells will still face the broad range of immune suppression mechanisms that operate in the tumor environment. These issues are well reviewed (69–71), and such immune suppression may be the dominant pathway regulating adaptive immune control of growing tumors.
If chemokine levels in a tumor alter over time, the proportion of newly recruited cells that are tumor-specific before and after chemokine expression would be anticipated to remain identical, though their absolute numbers would be expected to change. However, increasing chemokine levels may increase the rate of recirculation, and therefore increase the likelihood that a tumor-specific T cell can meet it’s cognate ligand. In this way, increased T cell recruitment may result in an increased proportion of tumor-specific T cells in the tumor through a more efficient screening of the recirculating T cell repertoire. For this to impact tumor control, entry to the tumor must have been the limiting factor preventing tumor-specific T cells from exerting their function. This is plausible, since as described above, low chemokine expressing tumors and low T cell infiltrated tumors have worse outcome than their matched counterparts (72–76). CXCR6 has been shown to play a role in the recruitment of Trm to tissue sites and their retention in tissues via its ligand CXCL16 (77). However, this chemokine receptor and ligand are not tissue specific. Up to 20% of all peripheral blood CD8+ T cells express CXCR6 in cancer patients (78) and healthy donors, and CXCR6-mediated recruitment occurs in multiple healthy tissues including the lung and liver (77, 79). The percentage of CXCR6+ cells in the peripheral blood is much lower in mice with no history of infection, but is upregulated following infection (80). Therefore, CXCR6-expressing T cells may represent the diverse array of immune responses occurring in humans. Consistent with this, from 20-60% of EBV-specific T cells circulating in patients express CXCR6, and CXCR6 can be rapidly upregulated on antigen rechallenge (78). Importantly, CXCL16, the ligand for CXCR6, is upregulated in normal tissues following infection (79, 81) so all CXCR6-expressing T cells may be recruited to the site along with specific T cells. CXCR6 is enriched on cells that infiltrate tumors, as are other chemokines associated with activated T cells, such as CCR5 (78). In particular, CXCR6 is particularly associated with a Trm population (77, 82, 83). Given the low recirculation of Trm phenotype cells and the high proportion of circulating CD8 T cells that express CXCR6, it is unlikely that CXCR6 is a specific means to recruit Trm. Alternatively, since CXCR6 is an activation marker on T cells, it is potentially a marker of T cells that have received additional cognate stimulation in the tumor environment, and may serve to retain antigen specific cells where both CXCL16 and the cognate antigen co-exist. In this way chemokine receptors that are induced by activation are likely to be enriched on the antigen-specific populations in the tumor environment, akin to the activation markers CD69 and CD39. It may also be important that CXCL16 is an unusual chemokine in that it is membrane bound, until cleaved by proteases that are regulated under inflammatory conditions and during cancer treatment (84–87). Therefore, CXCR6 may generate a retentive niche for antigen-reactive cells in close contact with epithelial cells, or recruitment from systemic circulation under inflammatory conditions.
Where chemokine expression is already high, or T cell infiltration is high, it would seem that recruitment is not a limiting factor in tumor control. It is logical that a tumor that has abundant T cells yet continues to grow may be more impacted by other issues (88). It is possible that these infiltrating T cells are in an unsuitable location, are suppressed, are unable to engage with antigen presented by cancer cells, or are simply not specific for the cancer cells. In addition, as will be discussed below, chemokine expression by the cancer cells might additionally impact the distribution of T cells within the tumor environment, encouraging T cell migration through the stroma and to cancer cells nests. For this reason, experiments that evaluate the role of chemokine expression on immune responses artificially alter the biology of the system. When cancer cells are engineered to express chemokines, as we have used in the past (52, 56), the chemotactic gradient will peak around the cancer cells, so recruitment of immune cells will be to the cancer cell nests. By contrast increased chemokine expression by cells of the tumor stroma may lead to a non-productive accumulation of T cells in the stroma without impacting their contact with cancer cells. Non-cancer cells of the tumor stroma are critical sources of chemokines in tumors, and altering the recruitment of T cells into versus out of the stroma can have therapeutic consequences that do not relate to the overall chemokine production in the broader tumor environment (Figure 2C). A heightened inflammatory environment that is restricted to the stroma may therefore negatively impact functional tumor control. Moreover, the infiltrating non-specific T cells may limit the ability of the specific cells to establish a niche and engage cognate antigen. This phenomenon is evident in a model of diabetes, where increased infiltration of non-specific T cells into the islet actually reduced the ability of islet-specific T cells to cause autoimmune diabetes (89). In this model, there is a threshold number of islet specific T cells that are necessary to bring about diabetes (90); however these islet-specific T cells represent a small proportion of the T cells infiltrating the islet (90). By providing large numbers of T cells of irrelevant specificity through a vaccination approach, the islet-specific population in the islet were less activated and less effective (89). This data is likely very impactful to cancer in patients, where ongoing irrelevant immune responses are likely present to a much greater degree than are present in murine models in clean animal facilities (91), and any attempt to increase recruitment to the tumor will occur regardless of specificity. In this scenario, the mechanisms impacting retention and activation may be more critical than recruitment to improve anti-tumor immunity.
Role of Antigen in Tissue Retention
Advances in genomic analysis of tumors and bioinformatic models to identify tumor mutations has resulted in a dramatic increase in the discovery of patient-specific neoantigens (92). The number of these antigens per patient vary considerably, but thus far most patients tested have been found to have targetable neoantigens and T cells that can recognize them (93–95). However, when considering the additional restriction based on MHC-binding of any neoantigens, it is theoretically possible that a poorly mutated tumor could have no targetable tumor neoantigens. This may be a stronger possibility in pediatric malignancies, where highly penetrant driver translocations/mutations can result in tumorigenesis with few additional passenger mutations (96–98). In these cases, recruited T cells would have no interaction with cognate targets, and would freely recirculate with no possible retention of specific cells. However, since recruitment is not antigen specific, it is possible that such a tumor could still have T cell infiltrates. Spranger et al. examined the neoantigen profile of tumors that were highly or poorly infiltrated with T cells, and found no correlation between the number of antigenic targets and the numbers of T cells infiltrating the tumor (99). These data suggest that T cell infiltration is unrelated to antigen density. However, in addition to neoantigens, there are an array of tumor-associated antigens (100) and in some cases viral antigens that can be effective targets for T cells. For example, in a recent clinical study in head and neck cancer, CD8+ T cells in the tumor did not make measurable responses to any of the mutated neoantigens that were present in the cancer cells (101). However, the T cells made strong responses to the E6 and E7 proteins from human papilloma virus. Notably, these responses were exclusively found in the CD103+CD39+ population of resident CD8 T cells (101), demonstrating that these cells are not restricted to mutated neoantigen reactivity. In B16 tumors in murine models, approximately half of the clonally expanded T cells in the tumor were reactive to the unmodified gp70 epitope that is shared in many murine tumors (43). Thus, mutated neoantigens may not be essential for adaptive immune control of tumors.
In interpreting antigen density we must be cautious not to assume that we will make T cells specific for all potential targets. In infectious disease models, despite a wide range of potential antigenic targets the immune response generally focuses on a small number of antigens. This is known as immunodominance and occurs in antibody and T cell responses (102–104). This suggests that once a tumor passes some antigenic threshold, immunodominant antigens may focus the immune response around a restricted set of T cells, and additional antigens may not impact the T cell response. Immunodominance is a potential problem in the immune response to infectious agents and to cancer, since the high antigen specificity can generate selective pressure that can permit outgrowth of variants. This has been observed as antigenic drift in infection (102) and immunoediting in tumors (105). Thus, despite large numbers of potential neoantigen targets in patient tumors, commonly only a very small number of tumor-specific clones can be cultured, often as low as 1-2 clones per tumor (92, 95). While this may be a technical issue relating to T cell expansion from tumor tissue given their suppressed status, if immunodominance limits the number of responses per patient it may provide an alternative explanation for the disconnect between the number of neoantigens and the degree of T cell infiltrate. However, this is also an opportunity for therapy, since we have the potential to introduce additional T cell responses capable of contributing to tumor control. Linnette et al. identified neoantigens present in tumors using genomic sequencing, and identified T cell specific for these tumor neoantigens using peptide stimulation and a DC vaccination approach in patients (106). Prior to vaccination, T cells specific for tumor antigens were below detection limits in the peripheral blood, but T cells specific for neoantigens could be expanded from blood and tumor ex vivo, with more of them found in the tumor (106). Vaccination resulted in expansions in neoantigen-specific T cells in the peripheral blood, but importantly, these experiments allowed the investigators to identify neoantigen-specific TCR sequences. TCR sequencing of the tumors demonstrated that the majority of these tumor-specific TCR sequences were absent from the tumor prior to vaccination, and even where present only a proportion of the potential specificities were detectable (106). Thus, patient tumors exhibit only a small proportion of the potential reactivity to unique neoantigens, supporting both some degree of immune ignorance and some degree of immunodominance in tumors. Kalaora et al. comprehensively characterized potential neoantigen targets in melanoma patients and corresponding TCR sequences in T cells expanded in vitro from tumors (107). Importantly, while there was significant variability between patients, distinct metastases within an individual patient overlapped in both neoantigen targets and TCR sequences present. In one example, these experiments demonstrated that 11 TCR sequences accounted for 90-99% of the tumor specificity (107). Zhang et al. demonstrated that the degree of clonality in tumors was positively correlated to the overall mutational burden (108). Interestingly, this paper also demonstrated a negative correlation between clonality and the percent tumor in the specimen (108), suggesting that an increased stromal component results in a decreased clonality likely due to increased infiltration of non-specific T cells in the stroma. Recent studies have demonstrated that a ‘mutator phenotype’ associated with loss of mismatch repair pathways is a stronger predictor of outcome than quantity of mutations (109, 110). Such tumors have increased T cell infiltrates counterbalanced by local immune suppression, including increased PDL1 expression (109, 111–113). However, it is unclear whether the mutations in these tumors dictates this infiltration phenotype. Tumors with the mutator phenotype have a more rapid tumorigenesis (114) and these tumors are highly inflamed before they have a high mutational burden (115). It is plausible that the mutator phenotype regulates immune activation through multiple mechanisms of which increased mutational frequency is only one (116).
One significant feature of tumors that is different from acute models of infectious disease is that the antigen target is chronically present in tumors. In most acute infectious disease models, antigen is present for only a few days to weeks in the infection site, and cross-presented antigen has a similarly short half-life. Following elimination of the pathogen, antigen is lost from the environment and as discussed above, the antigen-specific T cells remain as both tissue infiltrating resident memory T cells and circulating conventional memory T cells (17). Broadly, once established these are thought to be non-overlapping populations in the absence of further antigen exposure. Trm show little propensity to reenter circulation, and if they do, they have no directed pressure to establish themselves in other sites. This has best been shown in experiments where two mice – one antigen experienced and the other naïve – are surgically connected so that they share blood circulation. In this setting, only the antigen-experienced animal retains local Trm-mediated responses to rechallenge with the infectious agent (25, 117, 118). In the absence of further stimulation, Trm may traffic as far as the tissue draining lymph node, in a slower event over the course of weeks to months following their initial local antigen exposure (119). These cells retain residency features in the lymph node, and this mechanism can ensure locoregional memory within both the tissue site and the draining lymphatics to provide rapid response to infection (119). The literature is divided on whether these cells can re-enter circulation on rechallenge (27, 35), with some studies showing antigen challenge causes only local proliferation (35), other studies showing that rechallenge with a local infectious agent can cause the Trm to proliferate locally, enter the draining lymphatics and form conventional circulating memory populations (27). These data are impactful for understanding the Trm population in tumors. While cells with a Trm phenotype in tumors do not share TCR sequences with T cells in the draining lymphatics or the peripheral blood (38), it is possible that appropriate activation of these cells can cause recirculation. Similarly, it has been shown that circulating memory cells can become Trm following a subsequent local antigen challenge (120). Thus, in tumors where antigen is chronic, there may be a greater potential for turnover between resident and circulating tumor-specific T cells even though it is difficult to measure this with only steady state data. Yet, as discussed earlier, cells with Trm phenotypes in the tumor have unique clonotypes that are not readily detectable in the tumor-draining lymph node or peripheral circulation (38). These data suggest that despite chronic antigen presence, tumor-associated Trm are not measurably recirculating. As will be discussed later, Trm in the tumor express a range of exhaustion markers (38, 121) that are not typically observed on Trm in post-infection normal tissue. It is possible that by the time cancers become clinically evident, the reactivation potential of the tumor-infiltrating Trm is greatly reduced, and the cells that remain in the tumor have achieved a degree of balance between the antigen-presenting capacity of the cancer cells and their activation state.
Stromal Versus Cancer Distribution of T Cells
The tumor is not a homogenous structure, and there are microenvironments within the broader tumor environment. One of the more critical distinctions is between the nests of cancer cells and the tumor stroma (Figure 3). The extent of tumor stroma varies considerably between individuals and between tumor pathologies. In addition, tumors can incorporate tertiary lymphoid structures, which are lymphocyte aggregates with varying levels of organization that can be found in cancer, and are similarly found in other scenarios where chronic inflammation disrupts the tissue architecture (122). In pancreatic cancer, the presence of these structures does not correlate with the overall tumor mutational burden (123), though the lymphoid structures are more likely where strong MHC-binding neoantigens are present. Tumors with tertiary lymphoid structures are likely to have more T cells infiltrating the tumor, but these T cells are less enriched for CD103+ cells (123), suggesting that the formation of tertiary lymphoid aggregates versus cancer-associated Trm occur through distinct mechanisms. Notably, the 12 chemokine gene signature used to predict T cell infiltration in tumors also predicts the presence of tertiary lymphoid structures in tumors (59, 60). However, it remains unclear whether there is direct movement between the lymphoid structures and the vicinity of the cancer cells. B cells in these structures can recognize tumor-associated antigens (124), so it is reasonable to infer that there are CD4 T cells with similar specificities in the lymphoid structures. However, thus far there is no direct evidence the tertiary lymphoid structures of tumors are significant sources of the tumor-specific effector CD8 T cells that participate in tumor control. Pancreatic cancer is a commonly mentioned example where it is often possible to detect extensive desmoplastic stroma punctuated with relatively small nests of cancer cells (Figure 3). The stroma can incorporate a diverse set of non-cancer cells and plays an important role in tumor growth and immune regulation. T cells are not uniformly distributed between the stroma and cancer cell nests – they are generally enriched in the stroma (49, 125, 126). Importantly, T cell subtypes are differentially distributed between stroma and cancer cell nests. For example, in breast cancer it has been demonstrated that cells with the Trm phenotype are enriched in cancer cell nests rather than the tumor stroma (126). By contrast, stem-like CD8 T cells expressing the Tcf1 marker were shown to be enriched in the immediate perivascular region (127). These stem-like cells have been shown to co-localize with APC in their stromal niche (128), suggesting that their interactions are more impacted by cross-presentation than by direct presentation by cancer cells. Considering features regulating recirculation, it is important to know that lymphatic density is highest in the tumor stroma surrounding cancer cell nests, driven by both cancer cell and stromal cell factors that guide lymphangiogenesis [reviewed in (129)]. Similarly, vascular endothelia are a defining feature of the tumor stroma, and the proximity of these entry and exit vessels and their separation from cancer cells means that recirculation can occur without T cells having an opportunity to directly contact the cancer cells.
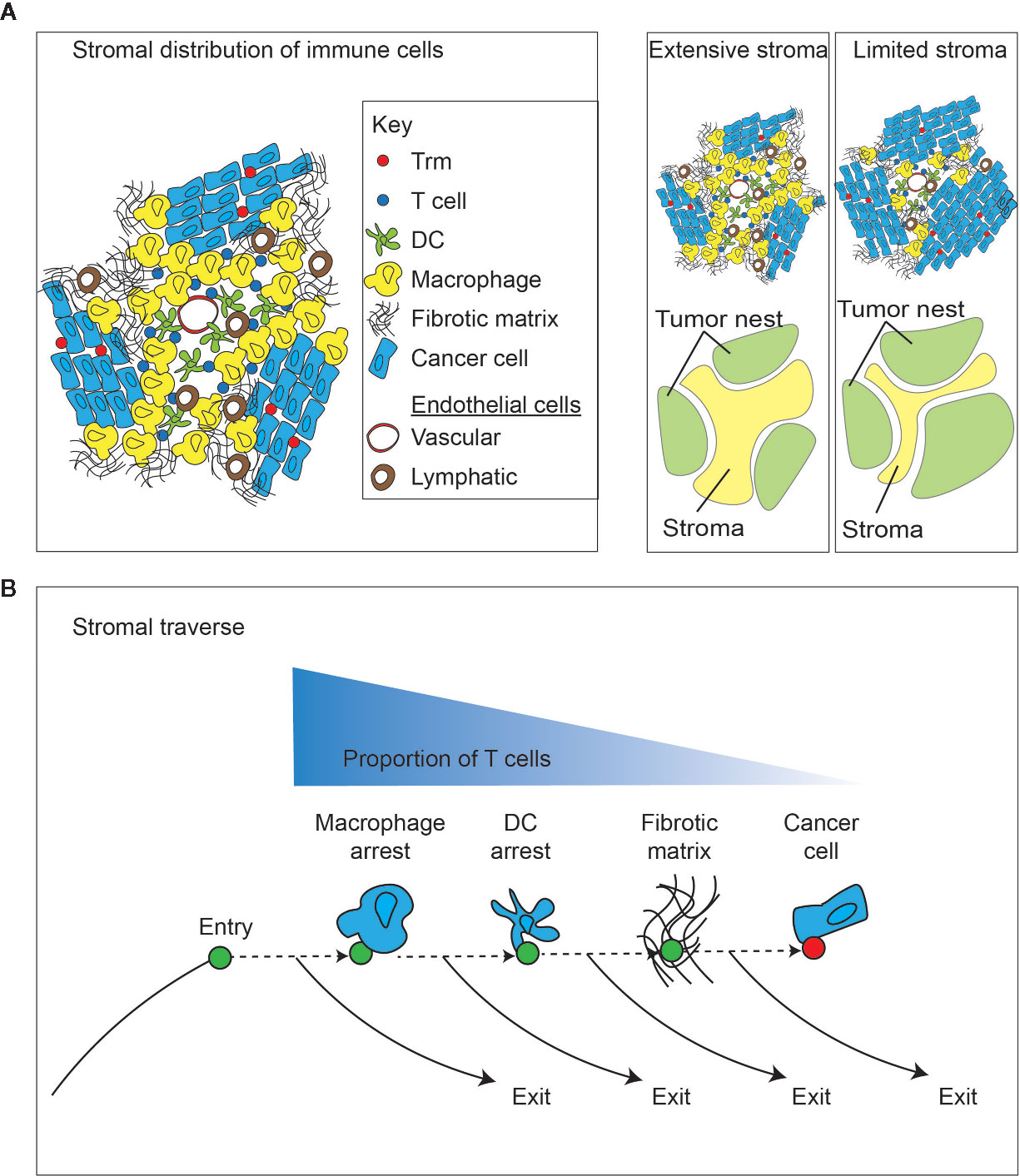
Figure 3 Impact of the tumor stroma and the stromal traverse on T cell dynamics. (A) The tumor is a non-homogenous structure that can be generally split into tumor and stroma. A diverse population of immune cells enrich in stroma away from direct cancer cell contact, and closer to vascular points of entry and lymphovascular points of exit. Different tumors can vary widely in the extent of tumor stroma. capacity for direct cancer cell cytotoxicity. (B) Following entry of lymphocytes into the tumor stroma via the vasculature, there are multiple stromal barriers that can cause T cell arrest and provide opportunities for a dominance of exit signals for continued recirculation through efferent lymphovasculature, rather than continuing through the stroma to meet cancer cells. More extensive stroma may increase the duration of traverse and decrease the likelihood of T cells meeting cancer cells.
The role of lymphatic endothelial cells in tumor immunity is multifaceted (129), but includes direct negative regulation of T cell activation as a result of inflammatory feedback (130). Since lymphatic endothelial cells mediate T cell exit from tissues, loss of lymphatic endothelial cells would be expected to decrease the ability of T cells to leave the tissue site. If ingress into the tumor is sustained and exit decreased, this should result in T cell accumulation. However, since as discussed above, rapidly recirculating T cells are more likely to be non-specific, this may not impact outcomes. Alternatively, since lymphatic endothelial cells can suppress T cells (130), more effective local immune responses might also be expected if there are fewer lymphatic endothelial cells in the tumor. Interestingly, contrary to these expectations tumors implanted into mice that lack functional lymphatic endothelial structures have fewer T cells infiltrating the tumor and reduced overall inflammation in the tumor (131). One possible explanation for this data is that impaired recirculation also results in impaired initial anti-tumor immunity, which will skew these results. As discussed earlier, to initiate new immune response dendritic cells must travel from the antigen site to draining lymph nodes via lymphatics to meet and stimulate naïve T cells. Loss of lymphatics might also mean loss of this initial anti-tumor immunity. Consistent with this, in viral models a lack of lymphatic endothelial structures results in impaired local control because of impaired initial immune activation in the draining lymph nodes (132). This initial immunity to tumor implantation is dependent on cross-presenting cDC1 and requires CD40 to generate both CD4 and CD8 T cell responses to tumor-associated antigens in the draining lymph node (133, 134). Using adoptive transfer to overcome this initial limitation in T cell responses, transferred tumor antigen-specific T cells have been shown to improve tumor control where tumors lack lymphatic endothelial cells (131), and pre-existing virus-specific T cells were equivalently capable of controlling a viral infection in the absence of lymphatic endothelial structures (132). These data suggest that lymphatic endothelial cells are required to prime new immune responses, but do not impair or suppress tumor immunity by existing tumor-specific T cells. Interestingly, increasing lymphangiogenesis in tumors increased their responsiveness to immunotherapies, and is associated with changes in the T cell populations that were recruited into the tumor immune environment (135). It is difficult to isolate the exact contribution of lymphatic structures due to the myriad of mechanisms by which they can interact with immune cells and the tumor stroma. However, given their role as an immunoregulatory component of the tumor stroma, the lymphovascular cells have a significant capacity to regulate the tumor immune environment and T cell recirculation (28).
The extracellular matrix represents an additional important limiting factor in T cell motility within the tumor. As discussed above, Salmon et al. demonstrated high T cell motility in the tumor stroma, and their comparative exclusion from tumor nests (49). Fibers of the extracellular matrix were shown to prevent direct T cell interactions with cancer cells (49). In such a setting the newly infiltrated T cells find it difficult to physically interact with the cognate antigen, but have an easy path to draining lymphatics which may comparatively promote their exit. For these newly-entered T cells the antigen presenting cells within the tumor stroma may play a significant role. Macrophages are prevalent in tumor stroma and are important in driving neoangiogenesis, lymphangiogenesis, and matrix remodeling for cancer cells growth (71, 129). Therefore, these cells are located amidst the key features regulating recirculation. Using live cell imaging, Peranzoni et al. demonstrated that tumor macrophages formed stable interactions with infiltrating CD8 T cells in the tumor stroma which reduced T cell motility (125). Depletion of macrophages using CSF1R inhibition restored T cell mobility and increased direct T cell interaction with cancer cells (125). Thus, macrophages in the tumor stroma may limit T cell mobility and thus limit functional interactions with cancer cells. Importantly, while macrophages can take up tumor antigens and present them to CD4 T cells via MHCII, they cannot cross-present antigen to CD8 T cells via MHCI. Dendritic cells are present in dramatically reduced proportions compared to other myeloid populations in the tumor stroma (136, 137), but they have the unique capacity to cross-present cell-associated antigens to infiltrating CD8 T cells (137, 138). Using cancer cells expressing fluorescent proteins, Englehardt et al. identified that dendritic cells closest to the cancer cells had the most phagocytosed cancer-cell material (139). They demonstrated that T cells closest to cancer cells exhibited reduced motility in vivo, and also that the T cells exhibited a more prolonged interaction with the dendritic cells that were close to the cancer cells compared to those dendritic cells that were further away and that had less phagocytosed cancer cell material (139). These data support the role for stromal dendritic cells in cross-presenting tumor associated antigens and this may increase T cell retention even where the T cells do not directly contact cancer cells. Interestingly, these dendritic cells were poorly able to support T cell proliferation unless first treated with innate adjuvants (139), suggesting that they maintain an immature phenotype and are unable to provide adequate co-stimulatory signals and cytokines to support T cell proliferation or effector function. These data suggest that those APC closest to the cancer cells have more abundant antigen for cross-presentation, but in progressively growing tumors these cells cannot sufficiently activate anti-tumor immunity for tumor control. Thus, they increase retention of tumor-specific clones, but do not necessarily result in tumor elimination. Tumor antigen-specific T cells have been described as being trapped in a dendritic cell network, restricting their access to cancer cells (140). This closely matches the observed state in snapshot views of tumors, where tumor-specific T cell clones are enriched but in poorly functional states permitting progressive tumor growth. Consistent with this, under steady state conditions tumor dendritic cells often have impaired functionality (136, 141). Dendritic cells in tumors can become poorly functional early in tumorigenesis (142, 143), and tumors with poorly functional dendritic cells are also poorly responsive to conventional therapies (136). These data indicate that tumor-infiltrating T cells must pass both physical barriers and intercepting APC that may drive local tolerance before they can even directly access cancer cells (Figure 3B). The role of intratumoral dendritic cells in regulating T cell control of tumors remains controversial, in part due to difficulties in distinguishing myeloid subtypes. While cross-presenting dendritic cells are required to initiate immune responses to tumor-associated antigens (133), they can be dispensable for tumor control by adoptively transferred T cells (144). This continued exit of matured and maturing dendritic cells makes it difficult to interpret the biology of dendritic cells in tumors at steady state, since by definition, the tumor resident dendritic cells should be immature since the mature cells have exited. However, features of the tumor environment that keep dendritic cells in the environment without permitting their maturation have the potential to generate a tolerogenic APC barrier. A wide range of immune interventions aim to provide signals that can drive dendritic cells maturation (145, 146), and many of these have shown efficacy in preclinical settings.
Impact of Cancer Cell Antigen Presentation on Recirculation
Antigen presentation is a regulated process and can be dynamically upregulated in response to stimuli. For both mice and humans, non-MHC genes that are integral to antigen presentation on MHC-I are also contained within the Mhc region, including the genes for tapasin, TAP1, TAP2, and LMP7; however, in both humans and mice the gene for β2m is located on a separate chromosome. Expression of classical MHC-I elements is mediated by three major regulatory elements: enhancer A, IFN-stimulated response element (ISRE), and the SXY module. Within MHC-I promoters, enhancer A elements are bound by NF-κB/rel family members and ISRE elements are bound by interferon regulatory factors (IRFs) including IRF1; these transcription factors notably mediate transcription of MHC-I proteins downstream of IFNγ and TNFα stimulation (147–149). The SXY module, comprised of S/W, X1, X2 and Y boxes, binds a number of nuclear factors including RFX (comprised of RFX5, RFXAP, and RFXANK/B), CREB/ATF, and NF-Y, which require a transcriptional regulator, either the Class I Transactivator (CITA, or NLRC5) or the Class II Transactivator (CIITA), to coordinate enhanceosome assembly (147, 150, 151). NLRC5 is a dominant regulator of MHC-I in most cells (152), and expression of NLRC5 can be induced by IFNγ (153).
Downregulation of antigen presentation on MHC-I is a common immune evasion mechanism employed by tumors (154). While decreased antigen presentation is a common feature of cancer, total loss of MHC-I expression (for example via biallelic loss of B2M) is less common (155), likely due to selective pressure from natural killer cells whose cytotoxic function is inhibited by the presence of MHC-I. Antigen presentation on MHC molecules can be reduced without total ablation by epigenetic suppression or genetic loss of factors regulating MHC-I expression (e.g. NLRC5) (156–158), downregulation of molecules involved in peptide loading onto MHC-I (159–161), loss of specific HLA alleles (162), or suppression of cytokine-activated pathways for augmenting MHC-I expression (e.g. loss of IFNγR/IFNAR or downstream JAK/STAT molecules) (163–165).
Conventional cancer therapies have the potential to regulate MHC expression by cancer cells. Reits et al. demonstrated that MHC-I expression is increased after radiation due to increased availability of intracellular peptides available for loading (166). Cancer cell irradiation can also activate the cGAS/STING pathway, triggering extracellular release of type I IFN (167). Ligation of IFNAR with type I IFN triggers downstream signaling via STAT1, STAT2 and IRF9 and leads to transcription of interferon-stimulated genes including MHC-I related proteins (168, 169); resulting in IFN-dependent upregulation of MHC-I by cancer cells (170). Similar mechanisms have been proposed for chemotherapy-induced activation of the STING-IFN pathway (171, 172). In addition, radiation therapy can upregulate NLRC5 independently of STING and IFN activation (173), potentially via distinct DNA-damage detection mechanisms. This suggests that cancer cell MHC expression can be regulated via an array of conventional approaches to alter lymphocyte dynamics in the tumor environment.
Significantly, alteration of cognate MHC-peptide expression on target cell surfaces can affect the magnitude and efficacy of CD8+ T cell responses (174). Differential responses of CD8+ T cells to varying MHC-peptide concentration have been observed, where increased epitope density corresponds with greater responsiveness to IL-2, enhanced proliferation and increased cytotoxic function including cytokine production (174–176). This phenomenon is better understood in naïve T cells, where high levels of antigen presentation in combination with costimulation and integrin stabilization are required to generate a stable immunological synapse and to cross an activation threshold of TCR signaling (174, 177). In activated T cells, MHC-peptide:TCR interactions at the synapse are much shorter: a single MHC-peptide complex can serially engage with rapidly internalizing TCRs and a CD8+ T cell can exert cytotoxic functions after engaging with as few as 1-3 MHC-peptide complexes per target cell (178, 179). It is clear that higher concentrations of MHC-peptide can engage more TCRs and it has been proposed that serial engagement of the TCR allows increased stability and enhanced signaling within the TCR/MHC-peptide/CD8 molecular complex (176). Functionally, downregulation of MHC-I induced by viral infection can significantly attenuate the ability of CD8+ T cells to kill infected targets (180). Interestingly, expression of the early activation marker CD69 appears to be independent of epitope density (181), and similarly CD69 and PD1 can be induced in the tumor environment independent of cognate antigen (182). Different densities of MHC-peptide can activate different thresholds in T cells for expression of early activation markers, cytolytic degranulation, versus cytotoxic cytokine release (183) (Figure 4). As discussed earlier, we have not explicitly examined the effect of local proliferation on T cell accumulation in tumors, instead grouping that as a retention mechanism. However, the degree of antigen presentation directly impacts the threshold for T cell proliferation in addition to cytotoxic activity. Since antigen presentation below a threshold can eliminate T cell responses, total loss of MHC-I is not necessary for resistance to T cells, and can result in various stages of activation without functional cancer cell cytotoxicity. This can explain why baseline tumor downregulation of antigen presentation via MHC-I results in checkpoint blockade resistance in human patients (164, 184), and presents a role for conventional therapies to increase T cell recognition and killing of cancer cells. In addition, antigen transfer that occurs as a result of cancer cell death driven by chemotherapy been shown to increase T cell infiltration into tumors, and increase T cell interactions with dendritic cells in the tumor (140). Similar mechanisms of antigen transfer to antigen presenting cells occur following radiation therapy (66), and have the potential to further manipulate the kinetics of lymphocyte movement through tumors by altering thresholds for cross-presentation in addition to direct presentation. Finally, T cell interactions that result in successful signalling and cytokine production can result in IFNg secretion and consequently increased antigen presentation in the vicinity of the T cell. While this may be tempered by simultaneous upregulation of negative regulation via PDL1, an initial cognate interaction by T cells can start a positive feedback loop in the microenvironment that can permit the T cell to pass critical activation thresholds that can result in cytotoxicity. For these reasons, manipulating the threshold for T cell activation in the tumor environment has the potential to dramatically alter tumor control.
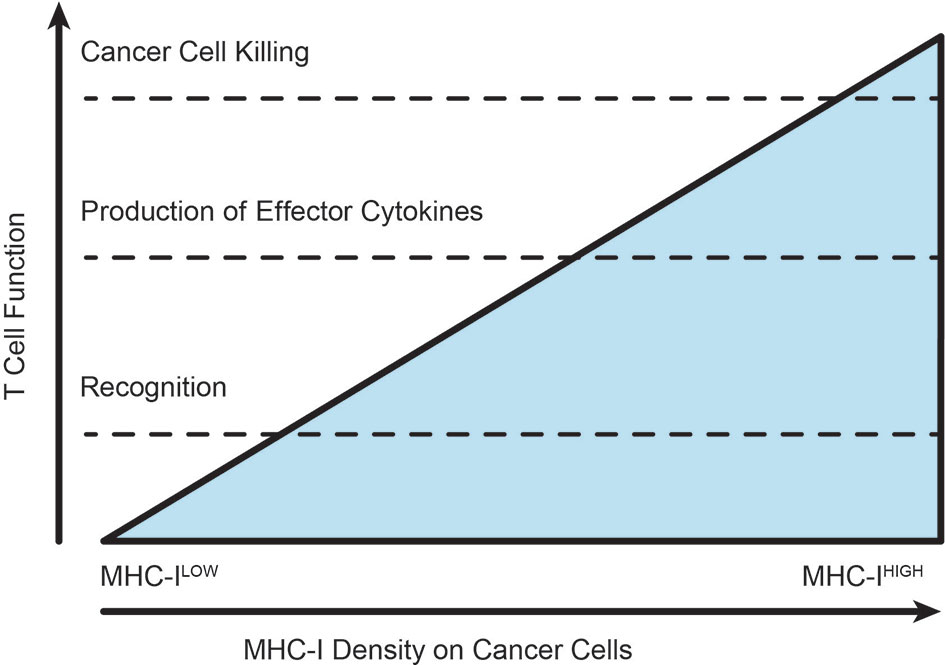
Figure 4 Varying thresholds for T cell function according to extent of TCR-MHC interactions. As the extent of cognate antigen increases on a target, whether by improved antigen expression or increased antigen processing and presentation, the intensity of T cell recognition allows the T cell to pass through various thresholds of response. The position of these thresholds can be altered by checkpoint blockade or costimulatory agonists. T cells in established tumors are typified by phenotypic modification without successful cytotoxic elimination of the cancer cells.
Effect of Checkpoint Regulators on Retention
As described above, while T cell entry into tumor is antigen-independent, retention in the tumor results from antigen-specific interactions with antigen-presenting cells or the cancer cells themselves. Immunotherapy has the potential to increase T cell surveillance of tumors through either mechanism – by increasing the recruitment or increasing their retention. However, to focus our efforts on antigen-specific cells it may be more useful to understand the mechanisms that increase the retention of this subpopulation in the tumor. As discussed earlier, we consider local proliferation to be a feature of retention, resulting in increased numbers of antigen-specific T cells in the site without changes in recruitment. In addition to the direct TCR interaction with cognate MHC-peptide on target cells, there are an array of costimulatory and coinhibitory signals that regulate that interaction (185). These are the targets of most of the immunotherapies currently being tested in clinical studies, and may function by regulating antigen-specific T cell retention and access to cancer cells, as well their function in the tumor, including local proliferation. The effect of checkpoint inhibition on TCR diversity and clonality has recently been reviewed (186), and this is an area of rapid research advancement. For example, Zhang et al. demonstrated that patients exhibiting a major pathological response to PD1 inhibition showed increased sharing of highly expanded clones between the tumor, peripheral blood, and non-tumor tissue (108). These data suggest that PD1 signals ordinarily limit T cell recirculation, or that the poorly responsive tumors in particular have limited active recirculation. Using live cell imaging it was shown that PD1 blockade increased the duration of T cell interactions with cancer cells (125), slowing their overall motility in the tumor environment. However, recent evidence suggests that PD1 blockade may increase the infiltration and function of a new clonally expanded population in the tumor, rather than restoring function of the existing exhausted cells (41). Single cell RNASeq plus TCR clonotype analysis of basal cell carcinoma before and after anti-PD1 therapy identified that T cells with shared TCR clones also tended to share a phenotype and that this phenotype was preserved following treatment (41), with exhausted clones remaining exhausted. Two thirds of the clones that expanded on treatment were new to the tumor, and included newly exhausted clones that made up the majority of exhausted cells in the treated tumor (41). Interestingly, this same precursor-like population is responsible for the proliferative expansion in T cells following PD1 blockade in viral models (187), suggesting that in both cases the response is dependent on expansion, recruitment and accumulation of new cells independent of the pre-existing clonally enriched, but exhausted T cells. Thus, in both viral infection models and tumor models, it has become clear that PD1 blockade is unable to restore the function of exhausted T cells and drive their conversion into memory and effector populations (188, 189). Rather, a distinct population current described as ‘progenitor exhausted cells’ that do not express the antigen-recognition and exhaustion marker CD39 expand following PD1 blockade and are more capable of generating effector function and tumor control (189). Siddiqui et al. demonstrated that these Tcf1+ progenitor-like cells were responsible for the majority of the proliferation following anti-PD1 therapy, and this expansion and tumor control could occur while new recruitment from the periphery was blocked using FTY720 (127). These data suggest that the progenitor-like clones may already be present in the tumor, and the apparent recruitment may be due to the frequency of these cells passing detection thresholds rather than recruitment from elsewhere. Since patients that have a pre-existing clonally expanded population of T cells expressing exhaustion markers is associated with improved outcome following PD1 blockade (47, 190), this suggests two major possibilities. Firstly, that the pre-existing population impacts the subsequent responses even if they are not the proliferative cells. Secondly, that the clonally expanded population in the tumor is evidence of a permissive environment in the cancer, which as discussed above might include effective antigen presentation and recruitment of cells into cancer cell nests to increase accumulation of tumor-specific clones. While it is currently difficult to break down these features, it is notable that clonal expansions of T cells following PD1 blockade occur in neighboring tissues as well as in the tumor (191). This suggests that the biology is not unique to the tumor but to applies to recirculating cells through other tissue sites.
Anti-CTLA4 therapy has been shown to increase T cell diversity in the peripheral blood of cancer patients (192, 193), though this was not necessarily reflected in positive phenotypic changes in the tumor (192). This data is consistent with expansion in functional specificities in the peripheral blood following anti-CTLA4 therapy (194), but little evidence of correlation between clonotypic changes in the peripheral blood versus the tumor following anti-CTLA4 therapy (195). Interestingly, in 4T1 mammary cancer models, anti-CTLA4 treatment increased motility of T cells in the tumor (196). Since this experimental design did not discriminate antigen-specific T cells it is possible that this increased motility reflects an increase in diverse T cell populations that are not tumor specific and so have increased overall motility in the tumor environment. Notably, adding radiation therapy to anti-CTLA4 therapy resulted in decreased overall motility in this model, and the change could be blocked with anti-MHCI antibodies, suggesting that cognate interactions are increased by the combination (196). TCRSeq in the same model shows that the combination of radiation and anti-CTLA4 increases the clonality of T cells in the tumor, and the proportion of antigen-specific T cells in the tumor, but no change in the distribution of TCR clones within the antigen specific population (197). This suggests that the tumor likely experiences increased infiltrates of clonal non-specific cells following anti-CTLA4 therapy. In patients, both anti-CTLA4 therapy and anti-PD1 therapy similarly increased T cell clonality in the tumor (190), but the extent of clonal expansion in the tumor pre-treatment was only predictive of outcome following anti-PD1 therapy (190). These data suggest the two agents have very different effects on T cell dynamics. By correlating patient responses to therapy with T cell populations in the blood, Wei et al. demonstrated that different T cell subpopulations in the peripheral blood correlated with response to single versus dual agent therapy (198). However, the frequency of these populations in the blood did not correlate well with their proportions in the tumor. Lau et al. demonstrated that combined PDL1 and CTLA4 blockade resulted in increased overall numbers of antigen-experienced T cells in murine tumors, and that these cells were more heterogeneously distributed than in untreated tumors (199). As observed in other models and discussed above, in these untreated murine tumor models T cells exhibited greater motility in the tumor periphery than in the tumor core (199). Following PDL1 blockade alone or combined PDL1 and CTLA4 blockade T cell motility was decreased in all regions of the tumor and overall infiltration was increased (199), consistent with increased cognate interactions in the tumor environment.
In contrast to checkpoint inhibitors like anti-PD1, costimulatory agonists like anti-OX40 (CD134) and anti-41BB (CD137) provide additional signals to T cells to overcome limited TCR stimulation, and can dramatically expand new populations of antigen-specific cells (200–202). This may occur because their antigen-specific interaction was below the necessary activation threshold as a lower affinity/avidity interaction, or because limited adjuvant signals have generated APC that are cross-presenting antigen but are not adequately providing costimulatory signals (203). In both cases, costimulation can generate a broader pool of antigen specific cells and also improve the quality of T cells as measured by memory formation and effector function (200–203). In the tumor, administration of anti-OX40 has been shown to increase the clonality of T cells in both the tumor and the spleen, suggesting that only some populations are being expanded by the therapy (204). Consistent with tolerogenic hypotheses limiting T cell responses in the tumor, T cells required anti-OX40 agonism to generate functional TCR signals in the tumor environment (182). In addition, higher affinity T cells in the tumor are more likely to express the costimulatory target OX40 (182), and the effects of anti-OX40 therapy was more pronounced on this tumor-infiltrating T cell population than those in the draining lymph nodes (182). These data suggest that OX40 costimulation specifically targets the existing antigen-specific T cell population that infiltrate but fail to cure tumors. Interestingly, the combination of anti-OX40 therapy and anti-PD1 therapy also enriches for T cells that receive high affinity TCR signals (204), as measured by activation of a Nur77-GFP reporter system (205). Thus, anti-OX40 therapy has been shown to remodel the tumor immune environment via activation of existing CD8 T cells that were previously functionally limited by the tumor immune environment (206, 207). To understand the response to agonist antibodies to 4-1BB, Weigelin et al. performed intravital imaging of tumors expressing ovalbumin as a model antigen to view tumor-specific responses of OT1 TCR transgenic T cells (208). As discussed above in other tumor models, these experiments demonstrated that OT1 T cells moved at slower speeds when tumors expressed their cognate antigen (208). The addition of agonist antibodies to 4-1BB (CD137) slowed the transit of the tumor-specific T cells, and increased the dwell time of T cells with target cells (208). These data demonstrate that as with PD1 blockade, costimulation can increase retention of T cells in the tumor resulting in T cell accumulation.
The consequence of manipulating T cell interactions with their cognate targets via checkpoint blockade and costimulatory agents can therefore be viewed through overlapping mechanisms. Firstly, new cells that previously were able to stably interact with their target can be incorporated into the anti-tumor immune response by decreasing the activation threshold of the T cells. This may occur via removal of negative regulation in T cell activation (209), or through provision of costimulatory support (210). Again, these data are consistent with ongoing immune surveillance of tumors by T cells as part of baseline recirculation. Those clones that are newly able to interact with cancer cells, can arrest and accumulate when checkpoints or costimulation are regulated. Secondly, there is clonal expansion. While the data is limited at present, costimulatory agents appear to expand existing populations that had been limited in their function in the tumor environment (204), while checkpoint inhibition through anti-PD1 appears to permit a new population of T cells to participate in the anti-tumor immune response (41). As the data improves, we will obtain a better picture of how these therapies impact recirculation kinetics versus local function, and how these features explain their effects in tumors.
Conclusions
As we have discussed, lymphocyte exclusion from the tumor environment predominantly revolves around recruitment and recirculation kinetics, but within those contexts the retention of antigen specific T cells and their ability to meet cancer cells are key. A tumor with a high throughput of T cells through high recruitment and high recirculation is very dynamic. Such a tumor has a great deal of potential for T cell-mediated control of cancer cells should cells of the appropriate specificity exist. When faced with a tumor that is not very dynamic, meaning that infiltration is poor, it will likely also lack the inflammatory signals that recruit and mature dendritic cells, so such a tumor may fail to generate cells specific for tumor antigens and also not recruit any that are expanded or provided through immunotherapy.
However, there is little reason to assume that a high recruitment tumor has a higher proportion of tumor antigen specific cells. A random assortment of T cells being recruited to the tumor stroma would not be expected to impact tumor growth and progression, so a high degree of entropy would have no advantage even in a highly infiltrated tumor. Moreover, cells that recirculate through an inflamed tumor stroma may minimally pass out of the stroma and meet the cancer cells to permit antigen-specific destruction. If T cells can be recruited to cancer cell nests, it may not be necessary to have a high recirculation rate to eventually result in an accumulation of cancer-specific cells amongst cancer cells. A wide array of data discussed above suggests that this is the most important feature – high pre-existing clonality, high Trm infiltrates, T cells infiltrated into tumor nests. That is, a low entropy tumor.
Currently, therapies that assist the tumor specific T cells complete their tasks, such as anti-PD1 are the most effective immunotherapy agents in the clinic. As discussed above, recent data suggests that PD1 blockade functions to recruit a new population to participate in tumor control rather than convert the function of terminally exhausted cells. In turn, this suggests that an ability to direct these new T cells to the tumor is essential for responses. Therapies that will help expand the existing population of tumor-specific T cells, such as anti-OX40 and anti-41BB have not yet shown sufficient promise for clinical approval despite their preclinical power. Understanding the critical issues of recruitment and retention of tumor-specific T cells to the tumor, as well as mechanisms that allow us to initiate new anti-tumor immune responses where they are currently lacking, will be key to success. To do this we will need to look carefully so that we can discriminate these responses from the constant recirculation of irrelevant T cells, and understand how these might interact to regulate site specific immune responses, to control the dynamic entropy of tumor lymphocytes.
Author Contributions
TB, writing, editing. AA, writing, editing. LZ, writing, editing. MC, writing, editing. MG, writing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NCI R01CA182311, R01CA244142, and R01CA208644.
Conflict of Interest
MG and MC receive research funding from Bristol Myers-Squibb, Jounce, and Mavupharma that is unrelated to the content of this manuscript.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The decision to frame the title and conclusions in terms of dynamic entropy is courtesy of xkcd (https://xkcd.com/2318/).
References
1. Amanna IJ, Carlson NE, Slifka MK. Duration of Humoral Immunity to Common Viral and Vaccine Antigens. N Engl J Med (2007) 357(19):1903–15. doi: 10.1056/NEJMoa066092
2. Blasius AL, Beutler B. Intracellular Toll-Like Receptors. Immunity (2010) 32(3):305–15. doi: 10.1016/j.immuni.2010.03.012
3. Iwasaki A, Medzhitov R. Toll-Like Receptor Control of the Adaptive Immune Responses. Nat Immunol (2004) 5(10):987–95. doi: 10.1038/ni1112
4. Baird JR, Monjazeb AM, Shah O, McGee H, Murphy WJ, Crittenden MR, et al. Stimulating Innate Immunity to Enhance Radiation Therapy-Induced Tumor Control. Int J Radiat Oncol Biol Phys (2017) 99(2):362–73. doi: 10.1016/j.ijrobp.2017.04.014
5. von Andrian UH. Mackay CR. T-Cell Function and Migration — Two Sides of the Same Coin. New Engl J Med (2000) 343(14):1020–34. doi: 10.1056/NEJM200010053431407
6. Ganusov VV, Auerbach J. Mathematical Modeling Reveals Kinetics of Lymphocyte Recirculation in the Whole Organism. PloS Comput Biol (2014) 10(5):e1003586. doi: 10.1371/journal.pcbi.1003586
7. Masopust D, Schenkel JM. The Integration of T Cell Migration, Differentiation and Function. Nat Rev Immunol (2013) 13(5):309–20. doi: 10.1038/nri3442
8. Soderberg KA, Payne GW, Sato A, Medzhitov R, Segal SS, Iwasaki A. Innate Control of Adaptive Immunity Via Remodeling of Lymph Node Feed Arteriole. Proc Natl Acad Sci U S A (2005) 102(45):16315–20. doi: 10.1073/pnas.0506190102
9. Cyster JG, Schwab SR. Sphingosine-1-phosphate and Lymphocyte Egress From Lymphoid Organs. Annu Rev Immunol (2012) 30:69–94. doi: 10.1146/annurev-immunol-020711-075011
10. Gowans JL. The Effect of the Continuous Re-Infusion of Lymph and Lymphocytes on the Output of Lymphocytes From the Thoracic Duct of Unanaesthetized Rats. Br J Exp Pathol (1957) 38(1):67–78.
11. Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, et al. Alteration of Lymphocyte Trafficking by Sphingosine-1-Phosphate Receptor Agonists. Science (2002) 296(5566):346–9. doi: 10.1126/science.1070238
12. Chiba K, Yanagawa Y, Masubuchi Y, Kataoka H, Kawaguchi T, Ohtsuki M, et al. FTY720, a Novel Immunosuppressant, Induces Sequestration of Circulating Mature Lymphocytes by Acceleration of Lymphocyte Homing in Rats. I. FTY720 Selectively Decreases the Number of Circulating Mature Lymphocytes by Acceleration of Lymphocyte Homing. J Immunol (1998) 160(10):5037–44. doi: 10.1046/j.1365-2567.1998.00639.x
13. Caucheteux SM, Torabi-Parizi P, Paul WE. Analysis of Naive Lung CD4 T Cells Provides Evidence of Functional Lung to Lymph Node Migration. Proc Natl Acad Sci U S A (2013) 110(5):1821–6. doi: 10.1073/pnas.1221306110
14. Smith ME, Ford WL. The Recirculating Lymphocyte Pool of the Rat: A Systematic Description of the Migratory Behaviour of Recirculating Lymphocytes. Immunology (1983) 49(1):83–94.
15. von Andrian UH, Mempel TR. Homing and Cellular Traffic in Lymph Nodes. Nat Rev Immunol (2003) 3(11):867–78. doi: 10.1038/nri1222
16. Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, et al. Quantifying Memory Cd8 T Cells Reveals Regionalization of Immunosurveillance. Cell (2015) 161(4):737–49. doi: 10.1016/j.cell.2015.03.031
17. Masopust D, Soerens AG. Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol (2019) 37:521–46. doi: 10.1146/annurev-immunol-042617-053214
18. Thome JJ, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, et al. Early-Life Compartmentalization of Human T Cell Differentiation and Regulatory Function in Mucosal and Lymphoid Tissues. Nat Med (2016) 22(1):72–7. doi: 10.1038/nm.4008
19. Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial Map of Human T Cell Compartmentalization and Maintenance Over Decades of Life. Cell (2014) 159(4):814–28. doi: 10.1016/j.cell.2014.10.026
20. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the Environment Recapitulates Adult Human Immune Traits in Laboratory Mice. Nature (2016) 532(7600):512–6. doi: 10.1038/nature17655
21. Weisberg SP, Carpenter DJ, Chait M, Dogra P, Gartrell-Corrado RD, Chen AX, et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas Through the PD-1/PD-L1 Pathway. Cell Rep (2019) 29(12):3916–32.e5. doi: 10.1016/j.celrep.2019.11.056
22. Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident Memory T Cells (T(RM)) are Abundant in Human Lung: Diversity, Function, and Antigen Specificity. PloS One (2011) 6(1):e16245. doi: 10.1371/journal.pone.0016245
23. Pizzolla A, Nguyen TH, Sant S, Jaffar J, Loudovaris T, Mannering SI, et al. Influenza-Specific Lung-Resident Memory T Cells are Proliferative and Polyfunctional and Maintain Diverse TCR Profiles. J Clin Invest (2018) 128(2):721–33. doi: 10.1172/JCI96957
24. Schoettler N, Hrusch CL, Blaine KM, Sperling AI, Ober C. Transcriptional Programming and T Cell Receptor Repertoires Distinguish Human Lung and Lymph Node Memory T Cells. Commun Biol (2019) 2(1):411. doi: 10.1038/s42003-019-0657-2
25. Van Braeckel-Budimir N, Varga SM, Badovinac VP, Harty JT. Repeated Antigen Exposure Extends the Durability of Influenza-Specific Lung-Resident Memory Cd8(+) T Cells and Heterosubtypic Immunity. Cell Rep (2018) 24(13):3374–82.e3. doi: 10.1016/j.celrep.2018.08.073
26. Muschaweckh A, Buchholz VR, Fellenzer A, Hessel C, Konig PA, Tao S, et al. Antigen-Dependent Competition Shapes the Local Repertoire of Tissue-Resident Memory CD8+ T Cells. J Exp Med (2016) 213(13):3075–86. doi: 10.1084/jem.20160888
27. Behr FM, Parga-Vidal L, Kragten NAM, van Dam TJP, Wesselink TH, Sheridan BS, et al. Tissue-Resident Memory CD8(+) T Cells Shape Local and Systemic Secondary T Cell Responses. Nat Immunol (2020) 21(9):1070–81. doi: 10.1038/s41590-020-0723-4
28. Lane RS, Lund AW. Non-Hematopoietic Control of Peripheral Tissue T Cell Responses: Implications for Solid Tumors. Front Immunol (2018) 9:2662. doi: 10.3389/fimmu.2018.02662
29. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
30. D’Agostino G, Garcia-Cuesta EM, Gomariz RP, Rodriguez-Frade JM, Mellado M. The Multilayered Complexity of the Chemokine Receptor System. Biochem Biophys Res Commun (2020) 528(2):347–58. doi: 10.1016/j.bbrc.2020.02.120
31. Ely KH, Cauley LS, Roberts AD, Brennan JW, Cookenham T, Woodland DL. Nonspecific Recruitment of Memory CD8+ T Cells to the Lung Airways During Respiratory Virus Infections. J Immunol (2003) 170(3):1423–9. doi: 10.4049/jimmunol.170.3.1423
32. Nolz JC, Starbeck-Miller GR, Harty JT. Naive, Effector and Memory CD8 T-Cell Trafficking: Parallels and Distinctions. Immunotherapy (2011) 3(10):1223–33. doi: 10.2217/imt.11.100
33. Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, et al. Monitoring Cellular Movement In Vivo With Photoconvertible Fluorescence Protein “Kaede” Transgenic Mice. Proc Natl Acad Sci USA (2008) 105(31):10871–6. doi: 10.1073/pnas.0802278105
34. Torcellan T, Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. In Vivo Photolabeling of Tumor-Infiltrating Cells Reveals Highly Regulated Egress of T-cell Subsets From Tumors. Proc Natl Acad Sci U S A (2017) 114(22):5677–82. doi: 10.1073/pnas.1618446114
35. Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, et al. Local Proliferation Maintains a Stable Pool of Tissue-Resident Memory T Cells After Antiviral Recall Responses. Nat Immunol (2018) 19(2):183–91. doi: 10.1038/s41590-017-0027-5
36. Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive Assessment of T-cell Receptor Beta-Chain Diversity in Alphabeta T Cells. Blood (2009) 114(19):4099–107. doi: 10.1182/blood-2009-04-217604
37. Barennes P, Quiniou V, Shugay M, Egorov ES, Davydov AN, Chudakov DM, et al. Benchmarking of T Cell Receptor Repertoire Profiling Methods Reveals Large Systematic Biases. Nat Biotechnol (2021) 39(2):236–45. doi: 10.1038/s41587-020-0656-3
38. Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, et al. Co-Expression of CD39 and CD103 Identifies Tumor-Reactive CD8 T Cells in Human Solid Tumors. Nat Commun (2018) 9(1):2724. doi: 10.1038/s41467-018-05072-0
39. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T Cells are Abundant and Phenotypically Distinct in Human Tumour Infiltrates. Nature (2018) 557(7706):575–9. doi: 10.1038/s41586-018-0130-2
40. Szabo PA, Miron M, Farber DL. Location, Location, Location: Tissue Resident Memory T Cells in Mice and Humans. Sci Immunol (2019) 4(34):eaas9673. doi: 10.1126/sciimmunol.aas9673
41. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal Replacement of Tumor-Specific T Cells Following PD-1 Blockade. Nat Med (2019) 25(8):1251–9. doi: 10.1038/s41591-019-0522-3
42. Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, et al. Low and Variable Tumor Reactivity of the Intratumoral TCR Repertoire in Human Cancers. Nat Med (2019) 25(1):89–94. doi: 10.1038/s41591-018-0266-5
43. Shitaoka K, Hamana H, Kishi H, Hayakawa Y, Kobayashi E, Sukegawa K, et al. Identification of Tumoricidal Tcrs From Tumor-Infiltrating Lymphocytes by Single-Cell Analysis. Cancer Immunol Res (2018) 6(4):378–88. doi: 10.1158/2326-6066.CIR-17-0489
44. Palmer DC, Balasubramaniam S, Hanada K, Wrzesinski C, Yu Z, Farid S, et al. Vaccine-Stimulated, Adoptively Transferred CD8+ T Cells Traffic Indiscriminately and Ubiquitously While Mediating Specific Tumor Destruction. J Immunol (2004) 173(12):7209–16. doi: 10.4049/jimmunol.173.12.7209
45. Penter L, Dietze K, Ritter J, Lammoglia Cobo MF, Garmshausen J, Aigner F, et al. Localization-Associated Immune Phenotypes of Clonally Expanded Tumor-Infiltrating T Cells and Distribution of Their Target Antigens in Rectal Cancer. Oncoimmunology (2019) 8(6):e1586409. doi: 10.1080/2162402X.2019.1586409
46. Wang X, Zhang B, Yang Y, Zhu J, Cheng S, Mao Y, et al. Characterization of Distinct T Cell Receptor Repertoires in Tumor and Distant non-Tumor Tissues From Lung Cancer Patients. Genomics Proteomics Bioinf (2019) 17(3):287–96. doi: 10.1016/j.gpb.2018.10.005
47. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature (2014) 515(7528):568–71. doi: 10.1038/nature13954
48. Breart B, Lemaitre F, Celli S, Bousso P. Two-Photon Imaging of Intratumoral CD8+ T Cell Cytotoxic Activity During Adoptive T Cell Therapy in Mice. J Clin Invest (2008) 118(4):1390–7. doi: 10.1172/JCI34388
49. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix Architecture Defines the Preferential Localization and Migration of T Cells Into the Stroma of Human Lung Tumors. J Clin Invest (2012) 122(3):899–910. doi: 10.1172/JCI45817
50. Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, et al. Random Migration Precedes Stable Target Cell Interactions of Tumor-Infiltrating T Cells. J Exp Med (2006) 203(12):2749–61. doi: 10.1084/jem.20060710
51. Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In Vivo Imaging of Cytotoxic T Cell Infiltration and Elimination of a Solid Tumor. J Exp Med (2007) 204(2):345–56. doi: 10.1084/jem.20061890
52. Crittenden M, Gough M, Harrington K, Olivier K, Thompson J, Vile RG. Expression of Inflammatory Chemokines Combined With Local Tumor Destruction Enhances Tumor Regression and Long-Term Immunity. Cancer Res (2003) 63(17):5505–12.
53. Mancardi S, Vecile E, Dusetti N, Calvo E, Stanta G, Burrone OR, et al. Evidence of CXC, CC and C Chemokine Production by Lymphatic Endothelial Cells. Immunology (2003) 108(4):523–30. doi: 10.1046/j.1365-2567.2003.01613.x
54. Cepek KL, Shaw SK, Parker CM, Russell GJ, Morrow JS, Rimm DL, et al. Adhesion Between Epithelial Cells and T Lymphocytes Mediated by E-cadherin and the Alpha E Beta 7 Integrin. Nature (1994) 372(6502):190–3. doi: 10.1038/372190a0
55. Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte Egress From Thymus and Peripheral Lymphoid Organs is Dependent on S1P Receptor 1. Nature (2004) 427(6972):355–60. doi: 10.1038/nature02284
56. Gough M, Crittenden M, Thanarajasingam U, Sanchez-Perez L, Thompson J, Jevremovic D, et al. Gene Therapy to Manipulate Effector T Cell Trafficking to Tumors for Immunotherapy. J Immunol (2005) 174(9):5766–73. doi: 10.4049/jimmunol.174.9.5766
57. Guo J, Wang B, Zhang M, Chen T, Yu Y, Regulier E, et al. Macrophage-Derived Chemokine Gene Transfer Results in Tumor Regression in Murine Lung Carcinoma Model Through Efficient Induction of Antitumor Immunity. Gene Ther (2002) 9(12):793–803. doi: 10.1038/sj.gt.3301688
58. Bonnotte B, Crittenden M, Larmonier N, Gough M, Vile RG. Mip-3alpha Transfection Into a Rodent Tumor Cell Line Increases Intratumoral Dendritic Cell Infiltration But Enhances (Facilitates) Tumor Growth and Decreases Immunogenicity. J Immunol (2004) 173(8):4929–35. doi: 10.4049/jimmunol.173.8.4929
59. Messina JL, Fenstermacher DA, Eschrich S, Qu X, Berglund AE, Lloyd MC, et al. 12-Chemokine Gene Signature Identifies Lymph Node-Like Structures in Melanoma: Potential for Patient Selection for Immunotherapy? Sci Rep (2012) 2:765. doi: 10.1038/srep00765
60. Coppola D, Nebozhyn M, Khalil F, Dai H, Yeatman T, Loboda A, et al. Unique Ectopic Lymph Node-Like Structures Present in Human Primary Colorectal Carcinoma are Identified by Immune Gene Array Profiling. Am J Pathol (2011) 179(1):37–45. doi: 10.1016/j.ajpath.2011.03.007
61. Hoeres T, Smetak M, Pretscher D, Wilhelm M. Improving the Efficiency of Vgamma9Vdelta2 T-Cell Immunotherapy in Cancer. Front Immunol (2018) 9:800. doi: 10.3389/fimmu.2018.00800
62. Ochsenbein AF. Immunological Ignorance of Solid Tumors. Springer Semin Immunopathol (2005) 27(1):19–35. doi: 10.1007/s00281-004-0192-0
63. Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, et al. Roles of Tumour Localization, Second Signals and Cross Priming in Cytotoxic T-cell Induction. Nature (2001) 411(6841):1058–64. doi: 10.1038/35082583
64. Ochsenbein AF, Klenerman P, Karrer U, Ludewig B, Pericin M, Hengartner H, et al. Immune Surveillance Against a Solid Tumor Fails Because of Immunological Ignorance. Proc Natl Acad Sci (1999) 96(5):2233–8. doi: 10.1073/pnas.96.5.2233
65. Kurts C, Sutherland RM, Davey G, Li M, Lew AM, Blanas E, et al. Cd8 T Cell Ignorance or Tolerance to Islet Antigens Depends on Antigen Dose. Proc Natl Acad Sci USA (1999) 96(22):12703–7. doi: 10.1073/pnas.96.22.12703
66. Spiotto MT, Yu P, Rowley DA, Nishimura MI, Meredith SC, Gajewski TF, et al. Increasing Tumor Antigen Expression Overcomes “Ignorance” to Solid Tumors Via Crosspresentation by Bone Marrow-Derived Stromal Cells. Immunity (2002) 17(6):737–47. doi: 10.1016/S1074-7613(02)00480-6
67. Kurts C, Miller JF, Subramaniam RM, Carbone FR, Heath WR. Major Histocompatibility Complex Class I-restricted Cross-Presentation is Biased Towards High Dose Antigens and Those Released During Cellular Destruction. J Exp Med (1998) 188(2):409–14. doi: 10.1084/jem.188.2.409
68. Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, et al. Induced Sensitization of Tumor Stroma Leads to Eradication of Established Cancer by T Cells. J Exp Med (2007) 204(1):49–55. doi: 10.1084/jem.20062056
69. Tormoen GW, Crittenden MR, Gough MJ. Role of the Immunosuppressive Microenvironment in Immunotherapy. Adv Radiat Oncol (2018) 3(4):520–6. doi: 10.1016/j.adro.2018.08.018
70. Emerson DA, Redmond WL. Overcoming Tumor-Induced Immune Suppression: From Relieving Inhibition to Providing Costimulation With T Cell Agonists. BioDrugs (2018) 32(3):221–31. doi: 10.1007/s40259-018-0277-2
71. Ruffell B, DeNardo DG, Affara NI, Coussens LM. Lymphocytes in Cancer Development: Polarization Towards Pro-Tumor Immunity. Cytokine Growth Factor Rev (2010) 21(1):3–10. doi: 10.1016/j.cytogfr.2009.11.002
72. Bernard B, Rajamanickam V, Dubay C, Piening B, Alonso E, Jutric Z, et al. Transcriptional and Immunohistological Assessment of Immune Infiltration in Pancreatic Cancer. PloS One (2020) 15(8):e0238380. doi: 10.1371/journal.pone.0238380
73. Tang ES, Newell PH, Wolf RF, Hansen PD, Cottam B, Ballesteros-Merino C, et al. Association of Immunologic Markers With Survival in Upfront Resectable Pancreatic Cancer. JAMA Surg (2018) 153(11):1055–7. doi: 10.1001/jamasurg.2018.1757
74. Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International Validation of the Consensus Immunoscore for the Classification of Colon Cancer: A Prognostic and Accuracy Study. Lancet (2018) 391(10135):2128–39. doi: 10.1016/S0140-6736(18)30789-X
75. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science (2006) 313(5795):1960–4. doi: 10.1126/science.1129139
76. Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity (2018) 49(1):178–93.e7. doi: 10.1016/j.immuni.2018.06.006
77. Wein AN, McMaster SR, Takamura S, Dunbar PR, Cartwright EK, Hayward SL, et al. CXCR6 Regulates Localization of Tissue-Resident Memory CD8 T Cells to the Airways. J Exp Med (2019) 216(12):2748–62. doi: 10.1084/jem.20181308
78. Parsonage G, Machado LR, Hui JW-Y, McLarnon A, Schmaler T, Balasothy M, et al. CXCR6 and CCR5 Localize T Lymphocyte Subsets in Nasopharyngeal Carcinoma. Am J Pathol (2012) 180(3):1215–22. doi: 10.1016/j.ajpath.2011.11.032
79. Sato T, Thorlacius H, Johnston B, Staton TL, Xiang W, Littman DR, et al. Role for CXCR6 in Recruitment of Activated CD8+ Lymphocytes to Inflamed Liver. J Immunol (2005) 174(1):277–83. doi: 10.4049/jimmunol.174.1.277
80. Latta M, Mohan K, Issekutz TB. CXCR6 is Expressed on T Cells in Both T Helper Type 1 (Th1) Inflammation and Allergen-Induced Th2 Lung Inflammation But is Only a Weak Mediator of Chemotaxis. Immunology (2007) 121(4):555–64. doi: 10.1111/j.1365-2567.2007.02603.x
81. Lee LN, Ronan EO, de Lara C, Franken KL, Ottenhoff TH, Tchilian EZ, et al. CXCR6 is a Marker for Protective Antigen-Specific Cells in the Lungs After Intranasal Immunization Against Mycobacterium Tuberculosis. Infect Immun (2011) 79(8):3328–37. doi: 10.1128/IAI.01133-10
82. Karaki S, Blanc C, Tran T, Galy-Fauroux I, Mougel A, Dransart E, et al. CXCR6 Deficiency Impairs Cancer Vaccine Efficacy and CD8(+) Resident Memory T-cell Recruitment in Head and Neck and Lung Tumors. J Immunother Cancer (2021) 9(3):e001948. doi: 10.1136/jitc-2020-001948
83. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the Persistence, Vigilance and Control of Human CD8+ Lung-Resident Memory T Cells. Nat Immunol (2016) 17(12):1467–78. doi: 10.1038/ni.3589
84. Shimaoka T, Nakayama T, Fukumoto N, Kume N, Takahashi S, Yamaguchi J, et al. Cell Surface-Anchored SR-PSOX/CXC Chemokine Ligand 16 Mediates Firm Adhesion of CXC Chemokine Receptor 6-Expressing Cells. J Leukoc Biol (2004) 75(2):267–74. doi: 10.1189/jlb.1003465
85. Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, et al. The Transmembrane Cxc-Chemokine Ligand 16 Is Induced by IFN-γ and TNF-α and Shed by the Activity of the Disintegrin-Like Metalloproteinase Adam10. J Immunol (2004) 172(10):6362. doi: 10.4049/jimmunol.172.10.6362
86. Mueller AC, Piper M, Goodspeed A, Bhuvane S, Williams JS, Bhatia S, et al. Induction of ADAM10 by RT Drives Fibrosis, Resistance, and EMT in Pancreatic Cancer. Cancer Res (2021). doi: 10.1158/0008-5472.CAN-20-3892
87. Smith TM, Tharakan A, Martin RK. Targeting ADAM10 in Cancer and Autoimmunity. Front Immunol (2020) 11:499. doi: 10.3389/fimmu.2020.00499
88. Medler TR, Blair TC, Crittenden MR, Gough MJ. Defining Immunogenic and Radioimmunogenic Tumors. Front Oncol (2021) 11:667075. doi: 10.3389/fonc.2021.667075
89. Christoffersson G, Chodaczek G, Ratliff SS, Coppieters K, von Herrath MG. Suppression of Diabetes by Accumulation of non-Islet-Specific CD8(+) Effector T Cells in Pancreatic Islets. Sci Immunol (2018) 3(21):eaam6533. doi: 10.1126/sciimmunol.aam6533
90. Oldstone MB, Edelmann KH, McGavern DB, Cruite JT, Welch MJ. Molecular Anatomy and Number of Antigen Specific CD8 T Cells Required to Cause Type 1 Diabetes. PloS Pathog (2012) 8(11):e1003044. doi: 10.1371/journal.ppat.1003044
91. Masopust D, Sivula CP, Jameson SC. Of Mice, Dirty Mice, and Men: Using Mice to Understand Human Immunology. J Immunol (2017) 199(2):383–8. doi: 10.4049/jimmunol.1700453
92. Tran E, Robbins PF, Rosenberg SA. ‘Final Common Pathway’ of Human Cancer Immunotherapy: Targeting Random Somatic Mutations. Nat Immunol (2017) 18(3):255–62. doi: 10.1038/ni.3682
93. Yossef R, Tran E, Deniger DC, Gros A, Pasetto A, Parkhurst MR, et al. Enhanced Detection of Neoantigen-Reactive T Cells Targeting Unique and Shared Oncogenes for Personalized Cancer Immunotherapy. JCI Insight (2018) 3(19):e122467. doi: 10.1172/jci.insight.122467
94. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med (2016) 375(23):2255–62. doi: 10.1056/NEJMoa1609279
95. Tran E, Ahmadzadeh M, Lu YC, Gros A, Turcotte S, Robbins PF, et al. Immunogenicity of Somatic Mutations in Human Gastrointestinal Cancers. Science (2015) 350(6266):1387–90. doi: 10.1126/science.aad1253
96. Pon JR, Marra MA. Driver and Passenger Mutations in Cancer. Annu Rev Pathol: Mech Dis (2015) 10(1):25–50. doi: 10.1146/annurev-pathol-012414-040312
97. Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, et al. The Genetic Landscape of the Childhood Cancer Medulloblastoma. Science (2011) 331(6016):435–9. doi: 10.1126/science.331.6016.375-e
98. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The Landscape of Genomic Alterations Across Childhood Cancers. Nature (2018) 555(7696):321–7. doi: 10.1038/nature25480
99. Spranger S, Luke JJ, Bao R, Zha Y, Hernandez KM, Li Y, et al. Density of Immunogenic Antigens Does Not Explain the Presence or Absence of the T-cell-inflamed Tumor Microenvironment in Melanoma. Proc Natl Acad Sci U.S.A. (2016) 113(48):E7759–E68. doi: 10.1073/pnas.1609376113
100. Haen SP, Loffler MW, Rammensee HG, Brossart P. Towards New Horizons: Characterization, Classification and Implications of the Tumour Antigenic Repertoire. Nat Rev Clin Oncol (2020) 17(10):595–610. doi: 10.1038/s41571-020-0387-x
101. Duhen R, Ballesteros-Merino C, Frye AK, Tran E, Rajamanickam V, Chang SC, et al. Neoadjuvant anti-OX40 (MEDI6469) Therapy in Patients With Head and Neck Squamous Cell Carcinoma Activates and Expands Antigen-Specific Tumor-Infiltrating T Cells. Nat Commun (2021) 12(1):1047. doi: 10.1038/s41467-021-21383-1
102. Altman MO, Angeletti D, Yewdell JW. Antibody Immunodominance: The Key to Understanding Influenza Virus Antigenic Drift. Viral Immunol (2018) 31(2):142–9. doi: 10.1089/vim.2017.0129
103. Akram A, Inman RD. Immunodominance: A Pivotal Principle in Host Response to Viral Infections. Clin Immunol (2012) 143(2):99–115. doi: 10.1016/j.clim.2012.01.015
104. Cheuk E, D’Souza C, Hu N, Liu Y, Lang H, Chamberlain JW. Human MHC Class I Transgenic Mice Deficient for H2 Class I Expression Facilitate Identification and Characterization of New HLA Class I-restricted Viral T Cell Epitopes. J Immunol (2002) 169(10):5571–80. doi: 10.4049/jimmunol.169.10.5571
105. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and Genetic Properties of Tumors Associated With Local Immune Cytolytic Activity. Cell (2015) 160(1-2):48–61. doi: 10.1016/j.cell.2014.12.033
106. Linette GP, Becker-Hapak M, Skidmore ZL, Baroja ML, Xu C, Hundal J, et al. Immunological Ignorance is an Enabling Feature of the Oligo-Clonal T Cell Response to Melanoma Neoantigens. Proc Natl Acad Sci USA (2019) 116(47):23662–70. doi: 10.1073/pnas.1906026116
107. Kalaora S, Wolf Y, Feferman T, Barnea E, Greenstein E, Reshef D, et al. Combined Analysis of Antigen Presentation and T-cell Recognition Reveals Restricted Immune Responses in Melanoma. Cancer Discovery (2018) 8(11):1366–75. doi: 10.1158/2159-8290.CD-17-1418
108. Zhang J, Ji Z, Caushi JX, El Asmar M, Anagnostou V, Cottrell TR, et al. Compartmental Analysis of T-cell Clonal Dynamics as a Function of Pathologic Response to Neoadjuvant Pd-1 Blockade in Resectable non-Small Cell Lung Cancer. Clin Cancer Res (2020) 26(6):1327–37. doi: 10.1158/1078-0432.CCR-19-2931
109. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer Immunology. Mutational Landscape Determines Sensitivity to PD-1 Blockade in non-Small Cell Lung Cancer. Science (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
110. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. Pd-1 Blockade in Tumors With Mismatch-Repair Deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
111. Gatalica Z, Snyder C, Maney T, Ghazalpour A, Holterman DA, Xiao N, et al. Programmed Cell Death 1 (PD-1) and its Ligand (PD-L1) in Common Cancers and Their Correlation With Molecular Cancer Type. Cancer Epidemiol Biomarkers Prev (2014) 23(12):2965–70. doi: 10.1158/1055-9965.EPI-14-0654
112. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The Vigorous Immune Microenvironment of Microsatellite Instable Colon Cancer is Balanced by Multiple Counter-Inhibitory Checkpoints. Cancer Discovery (2015) 5(1):43–51. doi: 10.1158/2159-8290.CD-14-0863
113. Lin A, Zhang J, Luo P. Crosstalk Between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front Immunol (2020) 11:2039. doi: 10.3389/fimmu.2020.02039
114. Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch Syndrome: 1895-2015. Nat Rev Cancer (2015) 15(3):181–94. doi: 10.1038/nrc3878
115. Chang K, Taggart MW, Reyes-Uribe L, Borras E, Riquelme E, Barnett RM, et al. Immune Profiling of Premalignant Lesions in Patients With Lynch Syndrome. JAMA Oncol (2018) 4(8):1085–92. doi: 10.1001/jamaoncol.2018.1482
116. Willis JA, Reyes-Uribe L, Chang K, Lipkin SM, Vilar E. Immune Activation in Mismatch Repair-Deficient Carcinogenesis: More Than Just Mutational Rate. Clin Cancer Res (2020) 26(1):11–7. doi: 10.1158/1078-0432.CCR-18-0856
117. Schenkel JM, Masopust D. Tissue-Resident Memory T Cells. Immunity (2014) 41(6):886–97. doi: 10.1016/j.immuni.2014.12.007
118. Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and Alarm Function of Resident Memory CD8(+) T Cells. Nat Immunol (2013) 14(5):509–13. doi: 10.1038/ni.2568
119. Stolley JM, Johnston TS, Soerens AG, Beura LK, Rosato PC, Joag V, et al. Retrograde Migration Supplies Resident Memory T Cells to Lung-Draining LN After Influenza Infection. J Exp Med (2020) 217(8):e20192197. doi: 10.1084/jem.20192197
120. Osborn JF, Hobbs SJ, Mooster JL, Khan TN, Kilgore AM, Harbour JC, et al. Central Memory CD8+ T Cells Become CD69+ Tissue-Residents During Viral Skin Infection Independent of CD62L-mediated Lymph Node Surveillance. PloS Pathog (2019) 15(3):e1007633. doi: 10.1371/journal.ppat.1007633
121. Canale FP, Ramello MC, Nunez N, Araujo Furlan CL, Bossio SN, Gorosito Serran M, et al. Cd39 Expression Defines Cell Exhaustion in Tumor-Infiltrating Cd8(+) T Cells. Cancer Res (2018) 78(1):115–28. doi: 10.1158/0008-5472.CAN-16-2684
122. Sautes-Fridman C, Lawand M, Giraldo NA, Kaplon H, Germain C, Fridman WH, et al. Tertiary Lymphoid Structures in Cancers: Prognostic Value, Regulation, and Manipulation for Therapeutic Intervention. Front Immunol (2016) 7(407):407. doi: 10.3389/fimmu.2016.00407
123. Gunderson A, Rajamanickam V, Bui C, Bernard B, Pucilowska J, Ballesteros-Merino C, et al. Germinal Center Reactions in Tertiary Lymphoid Structures Associate With Neoantigen Burden, Humoral Immunity and Long-Term Survivorship in Pancreatic Cancer. OncoImmunology (2021) 10(1). doi: 10.1080/2162402X.2021.1900635
124. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B Cells in Tertiary Lymphoid Structures is Associated With a Protective Immunity in Patients With Lung Cancer. Am J Respir Crit Care Med (2014) 189(7):832–44. doi: 10.1164/rccm.201309-1611OC
125. Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, et al. Macrophages Impede CD8 T Cells From Reaching Tumor Cells and Limit the Efficacy of anti-PD-1 Treatment. Proc Natl Acad Sci USA (2018) 115(17):E4041–E50. doi: 10.1073/pnas.1720948115
126. Egelston CA, Avalos C, Tu TY, Rosario A, Wang R, Solomon S, et al. Resident Memory CD8+ T Cells Within Cancer Islands Mediate Survival in Breast Cancer Patients. JCI Insight (2019) 4(19):e130000. doi: 10.1172/jci.insight.130000
127. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)Pd-1(+)Cd8(+) T Cells With Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity (2019) 50(1):195–211.e10. doi: 10.1016/j.immuni.2018.12.021
128. Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An Intra-Tumoral Niche Maintains and Differentiates Stem-Like CD8 T Cells. Nature (2019) 576(7787):465–70. doi: 10.1038/s41586-019-1836-5
129. Lund AW, Medler TR, Leachman SA, Coussens LM. Lymphatic Vessels, Inflammation, and Immunity in Skin Cancer. Cancer Discov (2016) 6(1):22–35. doi: 10.1158/2159-8290.CD-15-0023
130. Lane RS, Femel J, Breazeale AP, Loo CP, Thibault G, Kaempf A, et al. Ifngamma-Activated Dermal Lymphatic Vessels Inhibit Cytotoxic T Cells in Melanoma and Inflamed Skin. J Exp Med (2018) 215(12):3057–74. doi: 10.1084/jem.20180654
131. Lund AW, Wagner M, Fankhauser M, Steinskog ES, Broggi MA, Spranger S, et al. Lymphatic Vessels Regulate Immune Microenvironments in Human and Murine Melanoma. J Clin Invest (2016) 126(9):3389–402. doi: 10.1172/JCI79434
132. Loo CP, Nelson NA, Lane RS, Booth JL, Loprinzi Hardin SC, Thomas A, et al. Lymphatic Vessels Balance Viral Dissemination and Immune Activation Following Cutaneous Viral Infection. Cell Rep (2017) 20(13):3176–87. doi: 10.1016/j.celrep.2017.09.006
133. Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, et al. cDC1 Prime and are Licensed by CD4(+) T Cells to Induce Anti-Tumour Immunity. Nature (2020) 584(7822):624–9. doi: 10.1038/s41586-020-2611-3
134. Crittenden MR, Zebertavage L, Kramer G, Bambina S, Friedman D, Troesch V, et al. Tumor Cure by Radiation Therapy and Checkpoint Inhibitors Depends on Pre-Existing Immunity. Sci Rep (2018) 8(1):7012. doi: 10.1038/s41598-018-25482-w
135. Fankhauser M, Broggi MAS, Potin L, Bordry N, Jeanbart L, Lund AW, et al. Tumor Lymphangiogenesis Promotes T Cell Infiltration and Potentiates Immunotherapy in Melanoma. Sci Transl Med (2017) 9(407):eaal4712. doi: 10.1126/scitranslmed.aal4712
136. Blair TC, Bambina S, Alice AF, Kramer GF, Medler TR, Baird JR, et al. Dendritic Cell Maturation Defines Immunological Responsiveness of Tumors to Radiation Therapy. J Immunol (2020) 204(12):3416–24. doi: 10.4049/jimmunol.2000194
137. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell (2014) 26(5):638–52. doi: 10.1016/j.ccell.2014.09.007
138. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell (2016) 30(2):324–36. doi: 10.1016/j.ccell.2016.06.003
139. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating Dendritic Cells of the Tumor Microenvironment Cross-Present Tumor Antigens and Stably Engage Tumor-Specific T Cells. Cancer Cell (2012) 21(3):402–17. doi: 10.1016/j.ccr.2012.01.008
140. Boissonnas A, Licata F, Poupel L, Jacquelin S, Fetler L, Krumeich S, et al. CD8+ Tumor-Infiltrating T Cells are Trapped in the Tumor-Dendritic Cell Network. Neoplasia (2013) 15(1):85–94. doi: 10.1593/neo.121572
141. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors At the Tumor Site Enhances Tumor Responses to Therapeutic Pd-L1 and BRAF Inhibition. Immunity (2016) 44(4):924–38. doi: 10.1016/j.immuni.2016.03.012
142. Lin JH, Huffman AP, Wattenberg MM, Walter DM, Carpenter EL, Feldser DM, et al. Type 1 Conventional Dendritic Cells are Systemically Dysregulated Early in Pancreatic Carcinogenesis. J Exp Med (2020) 217(8):e20190673. doi: 10.1084/jem.20190673
143. Meyer MA, Baer JM, Knolhoff BL, Nywening TM, Panni RZ, Su X, et al. Breast and Pancreatic Cancer Interrupt IRF8-dependent Dendritic Cell Development to Overcome Immune Surveillance. Nat Commun (2018) 9(1):1250. doi: 10.1038/s41467-018-03600-6
144. Diao J, Gu H, Tang M, Zhao J, Cattral MS. Tumor Dendritic Cells (Dcs) Derived From Precursors of Conventional Dcs Are Dispensable for Intratumor Ctl Responses. J Immunol (2018) 201(4):1306–14. doi: 10.4049/jimmunol.1701514
145. Medler T, Patel JM, Alice A, Baird JR, Hu HM, Gough MJ. Activating the Nucleic Acid-Sensing Machinery for Anticancer Immunity. Int Rev Cell Mol Biol (2019) 344:173–214. doi: 10.1016/bs.ircmb.2018.08.006
146. Takeda Y, Kataoka K, Yamagishi J, Ogawa S, Seya T. Matsumoto M. A Tlr3-Specific Adjuvant Relieves Innate Resistance to PD-L1 Blockade Without Cytokine Toxicity in Tumor Vaccine Immunotherapy. Cell Rep (2017) 19(9):1874–87. doi: 10.1016/j.celrep.2017.05.015
147. van den Elsen PJ. Expression Regulation of Major Histocompatibility Complex Class I and Class II Encoding Genes. Front Immunol (2011) 2:48. doi: 10.3389/fimmu.2011.00048
148. Logeat F, Israel N, Ten R, Blank V, Le Bail O, Kourilsky P, et al. Inhibition of Transcription Factors Belonging to the Rel/NF-Kappa B Family by a Transdominant Negative Mutant. EMBO J (1991) 10(7):1827–32. doi: 10.1002/j.1460-2075.1991.tb07708.x
149. Gobin SJ, Keijsers V, van Zutphen M, van den Elsen PJ. The Role of Enhancer A in the Locus-Specific Transactivation of Classical and Nonclassical Hla Class I Genes by Nuclear Factor κb. J Immunol (1998) 161(5):2276–83.
150. Ludigs K, Seguin-Estevez Q, Lemeille S, Ferrero I, Rota G, Chelbi S, et al. NLRC5 Exclusively Transactivates MHC Class I and Related Genes Through a Distinctive SXY Module. PloS Genet (2015) 11(3):e1005088. doi: 10.1371/journal.pgen.1005088
151. Meissner TB, Liu YJ, Lee KH, Li A, Biswas A, van Eggermond MC, et al. NLRC5 Cooperates With the RFX Transcription Factor Complex to Induce MHC Class I Gene Expression. J Immunol (2012) 188(10):4951–8. doi: 10.4049/jimmunol.1103160
152. Meissner TB, Li A, Biswas A, Lee KH, Liu YJ, Bayir E, et al. NLR Family Member NLRC5 is a Transcriptional Regulator of MHC Class I Genes. Proc Natl Acad Sci USA (2010) 107(31):13794–9. doi: 10.1073/pnas.1008684107
153. Neerincx A, Castro W, Guarda G, Kufer TA. NLRC5, At the Heart of Antigen Presentation. Front Immunol (2013) 4:397. doi: 10.3389/fimmu.2013.00397
154. Garrido F, Algarra I. MHC Antigens and Tumor Escape From Immune Surveillance. Adv Cancer Res (2001) 83:117–58. doi: 10.1016/S0065-230X(01)83005-0
155. Garrido F, Cabrera T, Concha A, Glew S, Ruiz-Cabello F, Stern PL. Natural History of HLA Expression During Tumour Development. Immunol Today (1993) 14(10):491–9. doi: 10.1016/0167-5699(93)90264-L
156. Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell (2019) 36(4):385–401.e8. doi: 10.1016/j.ccell.2019.08.008
157. Yoshihama S, Vijayan S, Sidiq T, Kobayashi KS. Nlrc5/Cita: A Key Player in Cancer Immune Surveillance. Trends Cancer (2017) 3(1):28–38. doi: 10.1016/j.trecan.2016.12.003
158. Yoshihama S, Roszik J, Downs I, Meissner TB, Vijayan S, Chapuy B, et al. NLRC5/MHC Class I Transactivator is a Target for Immune Evasion in Cancer. Proc Natl Acad Sci USA (2016) 113(21):5999–6004. doi: 10.1073/pnas.1602069113
159. Ling A, Lofgren-Burstrom A, Larsson P, Li X, Wikberg ML, Oberg A, et al. TAP1 Down-Regulation Elicits Immune Escape and Poor Prognosis in Colorectal Cancer. Oncoimmunology (2017) 6(11):e1356143. doi: 10.1080/2162402X.2017.1356143
160. Henle AM, Nassar A, Puglisi-Knutson D, Youssef B, Knutson KL. Downregulation of TAP1 and TAP2 in Early Stage Breast Cancer. PloS One (2017) 12(11):e0187323. doi: 10.1371/journal.pone.0187323
161. Yang T, McNally BA, Ferrone S, Liu Y, Zheng P. A Single-Nucleotide Deletion Leads to Rapid Degradation of TAP-1 mRNA in a Melanoma Cell Line. J Biol Chem (2003) 278(17):15291–6. doi: 10.1074/jbc.M300954200
162. Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discovery (2018) 8(6):730–49. doi: 10.1158/2326-6074.TUMIMM17-PR03
163. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br J Cancer (2018) 118(1):9–16. doi: 10.1038/bjc.2017.434
164. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated With Acquired Resistance to PD-1 Blockade in Melanoma. New Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
165. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discovery (2017) 7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223
166. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation Modulates the Peptide Repertoire, Enhances MHC Class I Expression, and Induces Successful Antitumor Immunotherapy. J Exp Med (2006) 203(5):1259–71. doi: 10.1084/jem.20052494
167. Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic Progression Following DNA Damage Enables Pattern Recognition Within Micronuclei. Nature (2017) 548(7668):466–70. doi: 10.1038/nature23470
168. Kuchtey J, Chefalo PJ, Gray RC, Ramachandra L, Harding CV. Enhancement of Dendritic Cell Antigen Cross-Presentation by CpG DNA Involves Type I IFN and Stabilization of Class I MHC Mrna. J Immunol (2005) 175(4):2244–51. doi: 10.4049/jimmunol.175.4.2244
169. Wan S, Pestka S, Jubin RG, Lyu YL, Tsai YC, Liu LF. Chemotherapeutics and Radiation Stimulate MHC Class I Expression Through Elevated Interferon-Beta Signaling in Breast Cancer Cells. PloS One (2012) 7(3):e32542. doi: 10.1371/journal.pone.0032542
170. Zhang Q, Green MD, Lang X, Lazarus J, Parsels JD, Wei S, et al. Inhibition of ATM Increases Interferon Signaling and Sensitizes Pancreatic Cancer to Immune Checkpoint Blockade Therapy. Cancer Res (2019) 79(15):3940–51. doi: 10.1158/0008-5472.CAN-19-0761
171. Wang Z, Chen J, Hu J, Zhang H, Xu F, He W, et al. cGAS/STING Axis Mediates a Topoisomerase II Inhibitor-Induced Tumor Immunogenicity. J Clin Invest (2019) 129(11):4850–62. doi: 10.1172/JCI127471
172. Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, et al. Parp Inhibitor Efficacy Depends on CD8(+) T-Cell Recruitment Via Intratumoral Sting Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discovery (2019) 9(6):722–37. doi: 10.1158/2159-8290.CD-18-1218
173. Zebertavage LK, Alice A, Crittenden MR, Gough MJ. Transcriptional Upregulation of NLRC5 by Radiation Drives STING- and Interferon-Independent Mhc-I Expression on Cancer Cells and T Cell Cytotoxicity. Sci Rep (2020) 10(1):7376. doi: 10.1038/s41598-020-64408-3
174. Cosma GL, Eisenlohr LC. Impact of Epitope Density on CD8(+) T Cell Development and Function. Mol Immunol (2019) 113:120–5. doi: 10.1016/j.molimm.2019.03.010
175. Valitutti S, Muller S, Dessing M, Lanzavecchia A. Different Responses are Elicited in Cytotoxic T Lymphocytes by Different Levels of T Cell Receptor Occupancy. J Exp Med (1996) 183(4):1917–21. doi: 10.1084/jem.183.4.1917
176. Anikeeva N, Gakamsky D, Scholler J, Sykulev Y. Evidence That the Density of Self peptide-MHC Ligands Regulates T-cell Receptor Signaling. PloS One (2012) 7(8):e41466. doi: 10.1371/journal.pone.0041466
177. Curtsinger JM, Lins DC, Mescher MF. CD8+ Memory T Cells (CD44high, Ly-6C+) are More Sensitive Than Naive Cells to (CD44low, Ly-6C-) to TCR/CD8 Signaling in Response to Antigen. J Immunol (1998) 160(7):3236–43.
178. Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN. Evidence That a Single Peptide–Mhc Complex on a Target Cell Can Elicit a Cytolytic T Cell Response. Immunity (1996) 4(6):565–71. doi: 10.1016/S1074-7613(00)80483-5
179. Dustin ML. T-Cell Activation Through Immunological Synapses and Kinapses. Immunol Rev (2008) 221:77–89. doi: 10.1111/j.1600-065X.2008.00589.x
180. Halle S, Keyser KA, Stahl FR, Busche A, Marquardt A, Zheng X, et al. In Vivo Killing Capacity of Cytotoxic T Cells Is Limited and Involves Dynamic Interactions and T Cell Cooperativity. Immunity (2016) 44(2):233–45. doi: 10.1016/j.immuni.2016.01.010
181. Balyan R, Gund R, Ebenezer C, Khalsa JK, Verghese DA, Krishnamurthy T, et al. Modulation of Naive Cd8 T Cell Response Features by Ligand Density, Affinity, and Continued Signaling Via Internalized Tcrs. J Immunol (2017) 198(5):1823–37. doi: 10.4049/jimmunol.1600083
182. Moran AE, Polesso F, Weinberg AD. Immunotherapy Expands and Maintains the Function of High-Affinity Tumor-Infiltrating Cd8 T Cells In Situ. J Immunol (2016) 197(6):2509–21. doi: 10.4049/jimmunol.1502659
183. Betts MR, Price DA, Brenchley JM, Lore K, Guenaga FJ, Smed-Sorensen A, et al. The Functional Profile of Primary Human Antiviral CD8+ T Cell Effector Activity is Dictated by Cognate Peptide Concentration. J Immunol (2004) 172(10):6407–17. doi: 10.4049/jimmunol.172.10.6407
184. Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC Proteins Confer Differential Sensitivity to CTLA-4 and PD-1 Blockade in Untreated Metastatic Melanoma. Sci Transl Med (2018) 10(450):eaar3342. doi: 10.1126/scitranslmed.aar3342
185. Korman AJ, Peggs KS, Allison JP. Checkpoint Blockade in Cancer Immunotherapy. Adv Immunol (2006) 90:297–339. doi: 10.1016/S0065-2776(06)90008-X
186. Kidman J, Principe N, Watson M, Lassmann T, Holt RA, Nowak AK, et al. Characteristics of TCR Repertoire Associated With Successful Immune Checkpoint Therapy Responses. Front Immunol (2020) 11:587014. doi: 10.3389/fimmu.2020.587014
187. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T Cells That Provide the Proliferative Burst After PD-1 Therapy. Nature (2016) 537(7620):417–21. doi: 10.1038/nature19330
188. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic Stability of Exhausted T Cells Limits Durability of Reinvigoration by PD-1 Blockade. Science (2016) 354(6316):1160–5. doi: 10.1126/science.aaf2807
189. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of Exhausted CD8(+) T Cells Differentially Mediate Tumor Control and Respond to Checkpoint Blockade. Nat Immunol (2019) 20(3):326–36. doi: 10.1038/s41590-019-0312-6
190. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated Molecular Analysis of Tumor Biopsies on Sequential CTLA-4 and PD-1 Blockade Reveals Markers of Response and Resistance. Sci Transl Med (2017) 9(379):eaah3560. doi: 10.1126/scitranslmed.aah3560
191. Wu TD, Madireddi S, de Almeida PE, Banchereau R, Chen YJ, Chitre AS, et al. Peripheral T Cell Expansion Predicts Tumour Infiltration and Clinical Response. Nature (2020) 579(7798):274–8. doi: 10.1038/s41586-020-2056-8
192. Robert L, Tsoi J, Wang X, Emerson R, Homet B, Chodon T, et al. CTLA4 Blockade Broadens the Peripheral T-cell Receptor Repertoire. Clin Cancer Res (2014) 20(9):2424–32. doi: 10.1158/1078-0432.CCR-13-2648
193. Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, et al. Improved Survival With T Cell Clonotype Stability After anti-CTLA-4 Treatment in Cancer Patients. Sci Transl Med (2014) 6(238):238ra70. doi: 10.1126/scitranslmed.3008211
194. Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti-CTLA-4 Therapy Broadens the Melanoma-Reactive CD8+ T Cell Response. Sci Transl Med (2014) 6(254):254ra128. doi: 10.1126/scitranslmed.3008918
195. Page DB, Yuan J, Redmond D, Wen YH, Durack JC, Emerson R, et al. Deep Sequencing of T-cell Receptor DNA as a Biomarker of Clonally Expanded TILs in Breast Cancer After Immunotherapy. Cancer Immunol Res (2016) 4(10):835–44. doi: 10.1158/2326-6066.CIR-16-0013
196. Ruocco MG, Pilones KA, Kawashima N, Cammer M, Huang J, Babb JS, et al. Suppressing T Cell Motility Induced by anti-CTLA-4 Monotherapy Improves Antitumor Effects. J Clin Invest (2012) 122(10):3718–30. doi: 10.1172/JCI61931
197. Rudqvist NP, Pilones KA, Lhuillier C, Wennerberg E, Sidhom JW, Emerson RO, et al. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol Res (2018) 6(2):139–50. doi: 10.1158/2326-6066.CIR-17-0134
198. Wei SC, Anang NAS, Sharma R, Andrews MC, Reuben A, Levine JH, et al. Combination anti-CTLA-4 Plus anti-PD-1 Checkpoint Blockade Utilizes Cellular Mechanisms Partially Distinct From Monotherapies. Proc Natl Acad Sci USA (2019) 116(45):22699–709. doi: 10.1073/pnas.1821218116
199. Lau D, Garcon F, Chandra A, Lechermann LM, Aloj L, Chilvers ER, et al. Intravital Imaging of Adoptive T-Cell Morphology, Mobility and Trafficking Following Immune Checkpoint Inhibition in a Mouse Melanoma Model. Front Immunol (2020) 11:1514. doi: 10.3389/fimmu.2020.01514
200. Lathrop SK, Huddleston CA, Dullforce PA, Montfort MJ, Weinberg AD, Parker DC. A Signal Through OX40 (CD134) Allows Anergic, Autoreactive T Cells to Acquire Effector Cell Functions. J Immunol (2004) 172(11):6735–43. doi: 10.4049/jimmunol.172.11.6735
201. Weatherill AR, Maxwell JR, Takahashi C, Weinberg AD, Vella AT. OX40 Ligation Enhances Cell Cycle Turnover of Ag-activated Cd4 T Cells In Vivo. Cell Immunol (2001) 209(1):63–75. doi: 10.1006/cimm.2001.1783
202. Maxwell JR, Weinberg A, Prell RA, Vella AT. Danger and OX40 Receptor Signaling Synergize to Enhance Memory T Cell Survival by Inhibiting Peripheral Deletion. J Immunol (2000) 164(1):107–12. doi: 10.4049/jimmunol.164.1.107
203. Redmond WL, Gough MJ, Charbonneau B, Ratliff TL, Weinberg AD. Defects in the Acquisition of CD8 T Cell Effector Function After Priming With Tumor or Soluble Antigen can be Overcome by the Addition of an OX40 Agonist. J Immunol (2007) 179(11):7244–53. doi: 10.4049/jimmunol.179.11.7244
204. Polesso F, Weinberg AD, Moran AE. Late-Stage Tumor Regression After PD-L1 Blockade Plus a Concurrent Ox40 Agonist. Cancer Immunol Res (2019) 7(2):269–81. doi: 10.1158/2326-6066.CIR-18-0222
205. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T Cell Receptor Signal Strength in Treg and iNKT Cell Development Demonstrated by a Novel Fluorescent Reporter Mouse. J Exp Med (2011) 208(6):1279–89. doi: 10.1084/jem.20110308
206. Redmond WL, Gough MJ, Weinberg AD. Ligation of the OX40 Co-Stimulatory Receptor Reverses self-Ag and Tumor-Induced CD8 T-Cell Anergy In Vivo. Eur J Immunol (2009) 39(8):2184–94. doi: 10.1002/eji.200939348
207. Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 Agonist Therapy Enhances CD8 Infiltration and Decreases Immune Suppression in the Tumor. Cancer Res (2008) 68(13):5206–15. doi: 10.1158/0008-5472.CAN-07-6484
208. Weigelin B, Bolanos E, Teijeira A, Martinez-Forero I, Labiano S, Azpilikueta A, et al. Focusing and Sustaining the Antitumor CTL Effector Killer Response by Agonist anti-CD137 Mab. Proc Natl Acad Sci USA (2015) 112(24):7551–6. doi: 10.1073/pnas.1506357112
209. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T Cell Costimulatory Receptor CD28 is a Primary Target for PD-1-mediated Inhibition. Science (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
Keywords: T cell, recirculation, tumor, clonality analysis, retention, recruitment
Citation: Blair TC, Alice AF, Zebertavage L, Crittenden MR and Gough MJ (2021) The Dynamic Entropy of Tumor Immune Infiltrates: The Impact of Recirculation, Antigen-Specific Interactions, and Retention on T Cells in Tumors. Front. Oncol. 11:653625. doi: 10.3389/fonc.2021.653625
Received: 14 January 2021; Accepted: 06 April 2021;
Published: 22 April 2021.
Edited by:
Sarah E. Church, NanoString Technologies, United StatesReviewed by:
Rodney Macedo Gonzales, Albert Einstein College of Medicine, United StatesTodd Triplett, Dell Medical School, The University of Texas at Austin, United States
Copyright © 2021 Blair, Alice, Zebertavage, Crittenden and Gough. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Gough, bWljaGFlbC5nb3VnaEBwcm92aWRlbmNlLm9yZw==