 Zainab A. Bazzi1,2
Zainab A. Bazzi1,2 Isabella T. Tai1,2*
Isabella T. Tai1,2*- 1Division of Gastroenterology, Department of Medicine, University of British Columbia, Vancouver, BC, Canada
- 2Canada’s Michael Smith Genome Sciences Centre, British Columbia (BC) Cancer, Vancouver, BC, Canada
Cyclin-dependent kinase 10 (CDK10) is a CDC2-related serine/threonine kinase involved in cellular processes including cell proliferation, transcription regulation and cell cycle regulation. CDK10 has been identified as both a candidate tumor suppressor in hepatocellular carcinoma, biliary tract cancers and gastric cancer, and a candidate oncogene in colorectal cancer (CRC). CDK10 has been shown to be specifically involved in modulating cancer cell proliferation, motility and chemosensitivity. Specifically, in CRC, it may represent a viable biomarker and target for chemoresistance. The development of therapeutics targeting CDK10 has been hindered by lack a specific small molecule inhibitor for CDK10 kinase activity, due to a lack of a high throughput screening assay. Recently, a novel CDK10 kinase activity assay has been developed, which will aid in the development of small molecule inhibitors targeting CDK10 activity. Discovery of a small molecular inhibitor for CDK10 would facilitate further exploration of its biological functions and affirm its candidacy as a therapeutic target, specifically for CRC.
Introduction
Cyclin-dependent kinases (CDKs) are a family of serine/threonine protein kinases that play a critical role in regulating cellular processes, including cell division and cell death (1). Currently, more than 20 members of the CDK family have been identified by their characteristic ATP-binding pocket, PSTAIRE-like cyclin-binding domain and activating T-loop motif (1, 2). CDKs become active when non-covalently bound to their cyclin partner, via association with the PSTAIRE-like cyclin binding domain. The interaction of a CDK to its cyclin partner forms a heterodimer, in which the CDK acts as the catalytic subunit and the cyclin functions as the regulatory subunit. Cyclins are responsible for regulation of a CDK’s kinase activity and substrate specificity. CDKs, their cyclin interacting partners, and functions are summarized in Table 1.
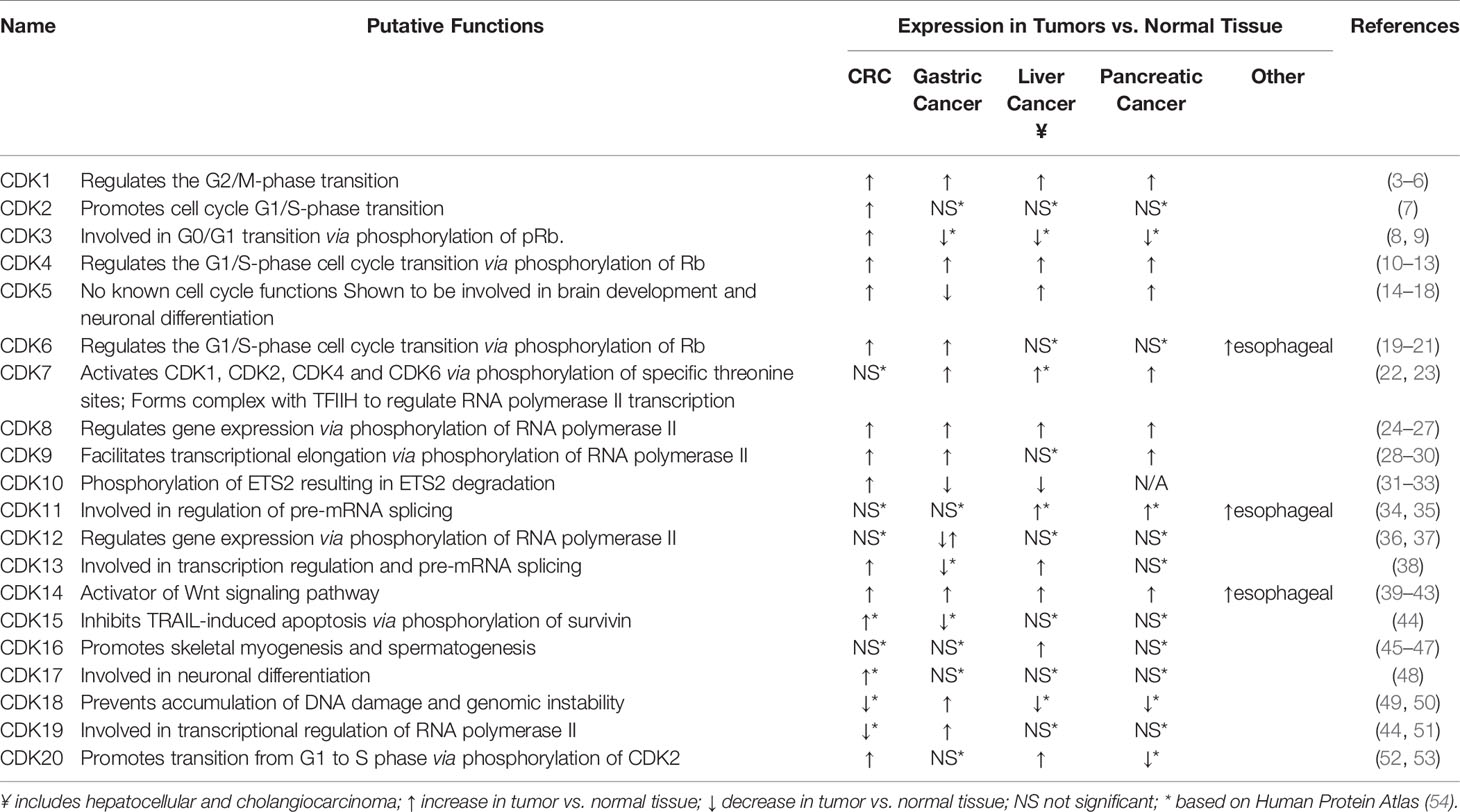
Table 1 CDKs in gastrointestinal cancers.
CDKs are generally categorized into two groups, based on their functions (1): cell cycle regulators; and (2) transcription regulators. CDKs involved in cell cycle regulation include CDK1, CDK2, CDK4 and CDK6. These CDKs are regulated through oscillation of expression throughout the cell cycle (55). CDK1 triggers the G2/M phase transition, while CDK2, CDK4 and CDK6 are regulators of the G1/S phase transition (56). CDKs involved in transcription regulation include CDK7, CDK8, CDK9, CDK10 and CDK11. Expression of CDKs involved in transcription regulation do not oscillate and are instead regulated by protein-protein interactions (56). They regulate transcription through phosphorylation of RNA polymerase II and through pre-mRNA splicing regulation.
CDKs often are dysregulated in malignancies, as shown in Table 1, causing dysregulation to cell cycle and transcription, leading to abnormal cell proliferation and inhibition of cell death (56). Genetic aberrations of CDKs and cyclins in tumor cells result in continuous cell proliferation or unscheduled cell cycle progression (56). Given their dysregulation in cancer, and their roles in mediating cell cycle progression, CDKs have been considered viable therapeutic targets for cancers, including gastrointestinal cancers.
Cyclin-dependent kinase 10 (CDK10) is a Cdc2-related kinase that was discovered based on its homology to the Cdc2 PSTA1RE amino acid domain (57). CDK10 plays a pivotal role in the regulation of fundamental cellular processes, including cell proliferation, transcription regulation and cell cycle regulation. Initial reports have indicated that CDK10 may act as a tumor suppressor in breast cancer. CDK10 is significantly downregulated in breast cancer compared to normal breast tissue (58). Additionally, CDK10 expression was inversely correlated with tumor stage and lymph node metastasis (58). Importantly, CDK10 expression was associated with better overall survival and may be a predictor of prognosis in breast cancer (58). Additional studies have demonstrated tumor suppressive and oncogenic roles for CDK10 in other malignancies. Specifically, CDK10 has been identified as a candidate tumor suppressor in hepatobiliary cancers, gastric cancer, glioma and nasopharyngeal carcinoma (31, 32, 59–61). Additionally, CDK10 has been shown to promote tumorigenesis in colorectal cancer (CRC) (62). Herein, we present a review of CDK10: its interacting partners, its role in gastrointestinal malignancies and its viability as a therapeutic target.
Molecular Genetics of CDK10
CDK10 was discovered in 1994 based on its homology to the Cdc2 PSTA1RE amino acid domain. PCR-based cloning was used to amplify cDNA encoding a novel human Cdc2-related kinase, which was called PISSLRE, and later termed CDK10 (57). Amino acid analysis revealed 38-45% identity with other CDKs (57). The chromosomal location of the gene encoding CDK10 was determined to be 16q24.3. The gene CDK10 (as designed by HUGO) consists of thirteen exons, distributed over approximately 15,000 kilobases of genomic DNA.
A putative method of regulation of CDK10 is through alternative splicing of pre-mRNA transcripts. Several CDK10 alternatively spliced isoforms have been identified. These alternatively spliced transcripts differ in exon 11 and in their 5’ and 3’ untranslated regions (UTRs). Of the differentially spliced transcripts identified, two produce functional proteins: the full-length transcript, which encodes for a 360 amino acid protein and a second transcript that encodes a truncated 272 amino acid variant, as shown in Figure 1. The latter protein is missing the ATP-binding domain and is therefore enzymatically inactive. Additionally, the shorter isoform does not interact with ETS2 and only weakly interacts with Cyclin M (33). It is therefore thought that alternative splicing is an important method of regulating CDK10 kinase activity.
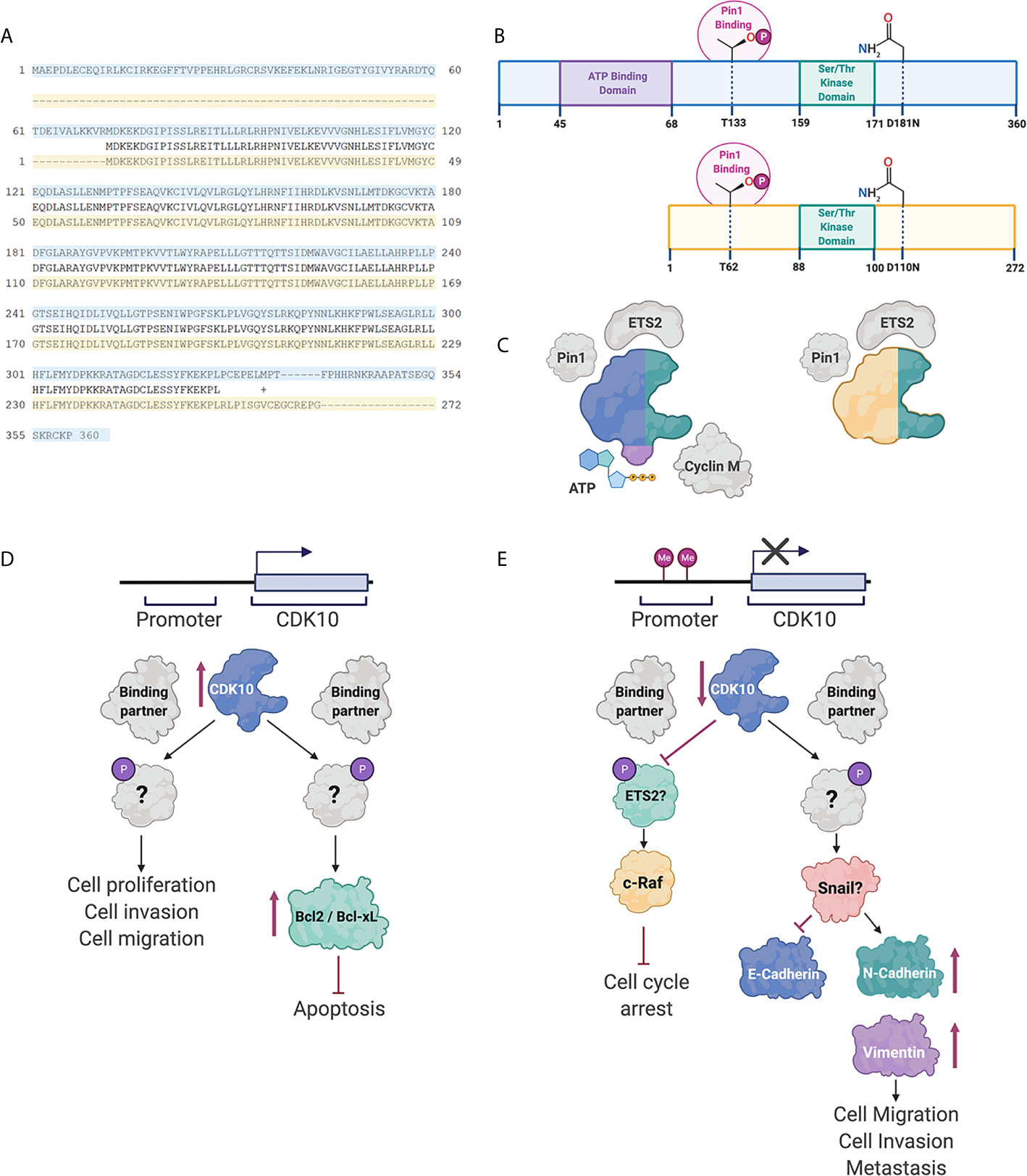
Figure 1 CDK10 isoforms and interacting partners. (A) Protein sequence alignment of the full length CDK10 isoform (blue) and the splice variant (yellow). (B) Schematics of the full length CDK10 isoform and splice isoform, showing the ATP binding domain, the Ser/Thr kinase domain and Thr133, which is involved in Pin1 binding. (C) Models of CDK10 full-length protein and splice isoform with putative binding partners. Figures were created using BioRender. (D) Proposed CDK10 oncogenic signaling pathways. (E) Proposed CDK10 tumor suppressive signaling pathways. “?” denotes not yet shown in gastrointestinal and hepatobiliary cancers (60, 62–64).
Interacting Partners of CDK10
Binding of a CDK to its partner cyclin is generally required for its activation and subsequent kinase function. CDK10 stood alone as the last CDK lacking identification of a cyclin partner. Recently, cyclin M was identified as a CDK10 binding partner by yeast two-hybrid screening and immunoprecipitation (33). The binding of CDK10 to cyclin M is independent of the kinase domain, however, the interaction of CDK10 with cyclin M was shown to regulate the kinase activity of CDK10, in STAR syndrome, (Figure 1) (33). This interaction, however, has not been shown in cancers involving the GI tract or hepatobiliary system.
Using a yeast interaction trap, CDK10 was shown to bind to transcription factor ETS2, both in vitro and in vivo, using human embryonic kidney 293 cells (Figure 1) (65). This interaction occurs via the N-terminus of ETS2, and mutation of the kinase domain of CDK10 (resulting in CDK10DN variant) did not impact the binding of ETS2 to CDK10. CDK10 has been shown to regulate transactivation of ETS2. Mass spectrometric analysis revealed ETS2 as a substrate for CDK10/cyclin M phosphorylation (33) and it has been reported that phosphorylation of ETS2 by CDK10/Cyclin M results in inhibition of ETS2 transactivation.
Subcellular localization of CDK10 and Cyclin M at the base of the primary cilia, and specific co-localization with centrosomal proteins, suggests a role for CDK10/Cyclin M in ciliogenesis (33). Knockdown of CDK10/Cyclin M significantly decreases stress fiber formation and ciliogenesis in human telomerase reverse transcriptase retinal pigmented epithelial (hTERT RPE-1) cells (33). Using an in vitro kinase assay, known core centrosomal proteins and regulators of ciliogenesis and actin dynamics were screened as candidate substrates for CDK10/Cyclin M. This method was used to identify protein kinase C-like 2 (PKN2) as an interacting partner and substrate for CDK10/Cyclin M, both in vitro and in vivo (33). CDK10/Cyclin M was shown to phosphorylate residues T121 and T124 of PKN2 and furthermore, to repress ciliogenesis in a RhoA-dependent manner. Cyclin M, however, has yet to be shown to be a CDK10 binding partner in gastrointestinal and hepatobiliary cancers.
Pin1 interaction with CDK10 has been documented in ER-positive breast cancer cells (Figure 1) (63). Pin1 is known to interact with Ser/Thr-Pro motifs, where the serine or threonine preceding the proline is phosphorylated. CDK10 does not contain any Ser-Pro motifs, however, does contain three Thr-Pro motifs. Mutagenesis of the three Thr-Pro motifs in CDK10 revealed Thr133 as an important residue for Pin1/CDK10 binding. Furthermore, treatment of CDK10 with a phosphatase demonstrated that Pin1 interacts with CDK10 in a phosphorylation-dependent manner. Consequently, interaction of Pin1 with CDK10 results in ubiquitination of CDK10 and subsequent degradation.
There is also evidence of CDK10 interaction with additional binding partners in non-human cell types. Specifically, CDK10 was shown to interact with Hsc70, Hsp90 and EcRB1 in Helicoverpa armigera (66). The interaction between CDK10 and Hsc70 and Hsp90 was augmented upon CDK10 phosphorylation. CDK10 forms a complex with Hsc70 and Hsp90, which sequentially binds with EcRB1 to facilitate the interaction between EcRB1 and EcRE to regulate 20E-mediated gene expression (66). Further studies are warranted to assess these as candidate binding partners for CDK10 in humans, and specifically, cancers involving the gastrointestinal tract and hepatobiliary system.
CDK10 as a Tumor Suppressor in Gastrointestinal and Hepatobiliary Cancers
Hepatobiliary Cancer
CDK10 has been identified as a candidate tumor suppressor in hepatobiliary cancers, including hepatocellular carcinoma (HCC) and biliary tract cancers (BTC) (31, 32, 64, 67). Examination of HCC tumor tissue revealed decreased expression of CDK10 mRNA and CDK10 protein compared to adjacent normal liver tissue (31). Immunohistological staining of CDK10 showed weak or no staining in HCC tissue samples. Significantly, CDK10 abundance was found to be inversely correlated to tumor size and tumor stage in HCC. In BTC, downregulation of CDK10 gene expression and CDK10 protein was observed in cancer tissue and cell lines, and was adversely associated with tumor stage, and lymph node invasion (64). Specifically, CDK10 was significantly downregulated in intrahepatic cholangiocarcinoma and gallbladder cancer, compared to normal tissue.
Several studies have characterized the effects of CDK10 on proliferation of hepatobiliary cancers in vitro (31, 64). Zhong et al. found that ectopic expression of CDK10 in the HCC cell line SMMC-7721 resulted in inhibition of cell proliferation (31). Similarly, in BTC, ectopic expression of CDK10 decreased cell proliferation, and downregulation of CDK10 expression significantly increased cell proliferation (64). Furthermore, cell cycle analysis of HCC cells following CDK10 overexpression revealed a significant increase in the G0-G1 phase population of cells, and a decrease in the S phase population (31). Consistently, BTC cells overexpressing CDK10 had a significant increase in the population of cells in G1 phase, and a significant decrease in the population of cells in G2/M phase (64).
Studies have also assessed the impact of CDK10 expression on cell invasion and migration (31, 64). In HCC, ectopic expression of CDK10 significantly delayed wound healing (31). Yu et al. also reported a significant decrease in BTC cell migration upon CDK10 overexpression (64). Consistently, they also reported a significant increase in BTC cell migration following downregulation of CDK10 (64). These studies, however, did not examine the mechanism by which CDK10 inhibits cancer cell invasion and migration. In glioma, CDK10 was shown to regulate cell motility through inhibition of epithelial to mesenchymal transition (EMT) (60). CDK10 knockdown decreased E-cadherin and increased vimentin and N-cadherin expression. Conversely, overexpression of CDK10 increased E-cadherin and decreased vimentin and N-cadherin expression. CDK10 was shown to regulate expression of the EMT transcription factor, Snail, and the effects of CDK10 on EMT in glioma were partially reversed by manipulation of Snail expression (60). Importantly, Snail is a known regulator of EMT in hepatobiliary cancersCDK10 may regulate cell motility of hepatobiliary cancer cells through inhibition of EMT via Snail downregulation.
Therapeutic resistance is a major hindrance on treatment of gastrointestinal cancers. There is significant evident identifying CDK10 as an important modulator of tamoxifen sensitivity in breast cancer, suggesting a potential role for CDK10 in chemosensitivity in other cancers (63, 68). In patients with ER-positive breast cancer, low CDK10 expression was associated with shorter overall survival and clinical resistance to tamoxifen (68). This study used gene silencing to identify CDK10 as a modulator of tamoxifen resistance in breast cancer through regulation of p42/p44 MAPK pathway (68). In BTC, Yu et al. found that knockdown of CDK10 significantly decreased sensitivity to 5-fluorouracil (64). Furthermore, overexpression of CDK10 increased BTC cell sensitivity to 5-fluorouracil, adriamycin/epirubicin, cisplatin and hydroxylcamptothecin in vitro (64). Similar to endocrine resistance in breast cancer, cell cycle arrest at G1 phase was observed in 5-fluorouracil-treated cells overexpressing CDK10 (64, 68). In HCC, overexpression of CDK10 increased chemosensitivity to cisplatin and epirubicin in SMMC-7721 cells but not HepG2 cells (31). Furthermore, overexpression of CDK10 increased gall bladder cancer sensitivity to gemcitabine (69). Anticipating a mechanism similar to endocrine therapy resistance in breast cancer, CDK10 expression was shown to downregulate c-Raf levels in BTC (64, 69). Furthermore, in gall bladder cancer, knockdown of c-Raf resulted in a significant increase in gemcitabine sensitivity in cells overexpressing CDK10 (69). Downregulation of CDK10 increased ETS2-mediated transcription of c-Raf, resulting in activation of the MAPK pathway. Additionally, Khanal et al. investigated the association between CDK10 and Pin1 expression in tamoxifen-resistant breast cancer cells (63). This study observed a significant inverse correlation between Pin1 and CDK10 expression in tamoxifen-resistant breast cancer. Khanal et al. also found that overexpression of CDK10 increased breast cancer cell sensitivity to tamoxifen treatment, and decreased Pin1-mediated c-Raf phosphorylation (63). However, further research is warranted to delineate the mechanism by which CDK10 regulates c-Raf levels in gastrointestinal cancers and how that results in cell cycle arrest.
Gastric Cancer
In addition to hepatobiliary cancers, CDK10 has been identified as a candidate tumor suppressor in gastric cancer. Independent studies found decreased expression of CDK10 in gastric cancer compared to normal gastric tissue (32, 67). Consistently, these studies found a significant correlation between loss of CDK10 expression and advanced tumor stage, lymph node invasion and distant metastasis, in patients with gastric cancer. Furthermore, these studies also identified loss of CDK10 expression as an unfavorable prognostic marker in gastric cancer. However, Fukui et al. found that CDK10 was upregulated in peritoneal and liver metastases in human gastric cancer cell lines established following injection into nude mice (70).
Studies have also assessed the effect of CDK10 on cell proliferation and cell motility in gastric cancers. Ectopic expression of CDK10 decreased cell proliferation, while downregulation of CDK10 expression significantly increased cell proliferation (32). Treatment of gastric cancer cells with quercetin, a flavonol shown to induce apoptosis, significantly decreased expression of CDK10 (71). Additionally, in gastric cancer, overexpression of CDK10 decreased cell invasion, while knockdown of CDK10 promoted cell invasion (32).
Mechanisms of CDK10 Downregulation in Gastrointestinal Cancers
As previously mentioned, CDK10 expression is downregulated in hepatobiliary cancers and gastric cancers (31, 32, 64). Mechanisms of CDK10 downregulation have not been examined in gastrointestinal cancers. Chromosomal deletions at the q24 region of chromosome 16 are associated with human cancers, including gastric cancer and HCC (72, 73). Furthermore, aberrant methylation of chromosome 16 is a mechanism of gene expression dysregulation in chronic hepatitis, liver cirrhosis and HCC (74, 75). Promoter hypermethylation was found to be a mechanism of CDK10 suppression in breast cancer and nasopharyngeal carcinoma (61, 68). Therefore, suppression of CDK10 may result from loss of heterozygosity and hypermethylation at the q24 region of chromosome 16. Further research is warranted to assess loss of heterozygosity and promoter hypermethylation as potential mechanisms of CDK10 downregulation in gastrointestinal cancers.
CDK10 in Colorectal Cancer
A meta-analysis of CRC gene expression profiling studies identified CDK10 as a gene consistently upregulated in CRC (76). Consistent with this meta-analysis, Weiswald et al. found overexpression of CDK10 in CRC tissue and CRC cell lines, compared to matched normal tissue and normal colon cells, respectively (62). This observation is similar to other studies that found upregulation of CDK10 in prostate cancer and seminomas (77, 78). Furthermore, increased expression of CDK10 was found to be correlated with lymph node positive tumors in CRC (79).
Weiswald et al. also found that CRC growth and survival were significantly affected by manipulation of CDK10 gene expression. Specifically, CDK10 knockdown decreased cell survival and promoted apoptosis in CRC cell lines. Similarly, overexpression of CDK10 increased cell proliferation and inhibited apoptosis in vitro, in a Bcl-2/Bcl-XL-dependent manner. Consistently, suppression of CDK10 in patient-derived xenograft CRC tumors inhibited tumor growth and decreased expression of Bcl-2 in vivo. Interestingly, CDK10 expression did not affect cell cycle progression in CRC, indicating that the impact of CDK10 on cell proliferation and apoptosis is independent of cell cycle regulation.
Furthermore, Weiswald et al. evaluated the effects of CDK10 on chemotherapy resistance. CRC cell lines overexpressing CDK10 were significantly less responsive to 5-fluorouracil compared to controls (62). Additionally, compared to CDK10WT, cell lines expressing CDK10DN were more sensitive to 5-fluorouracil, indicating kinase involvement in CDK10-mediated chemoresistance in CRC. Indeed, CDK10 expression may be a viable biomarker for chemotherapy resistance and should be assessed as a potential biomarker for CRC recurrence. Importantly, while this study suggests a kinase-dependent role for CDK10, an interacting partner to CDK10 has yet to be identified and implicated in CRC.
The role of CDK10 in CRC cell invasion and migration is yet to be determined. However, Zehra et al. demonstrated upregulation of CDK10 and ETS2 in a corneal epithelial wound healing model (80). This study infers a potential role for CDK10 in cancer metastasis, as inhibition of CDK10 resulted in a significant delay in corneal epithelial cell migration.
CDK10 as a Therapeutic Target for Gastrointestinal Cancers
Research has been dedicated to the development of candidate CDK small molecule inhibitors, however, the majority of CDK inhibitors lack specificity and clinical trials have had disappointing outcomes. Several strategies have been employed in drug discovery to develop effective CDK inhibitors, including reversible ATP-competitive and non-competitive inhibition, reversible and irreversible allosteric inhibition, antibodies, and CDK-targeted degradation. Non-specific pan-CDK inhibitors exhibit low anti-cancer activity and high toxicity. Due to their non-specificity, these inhibitors block several cell processes, including cell proliferation, transcription, and translation. Given the tumor suppressive nature of CDK10 in breast cancer, HCC, BTC and gastric cancer, targeting CDK10 via pan-CDK inhibitors may have limited the therapeutic response.
The development of therapeutics targeting CDK10 has been hindered by lack of a CDK10 activity assay, and lack of a specific small molecule inhibitor to identify novel therapies. Discovery of a small molecule inhibitor for CDK10 would facilitate further exploration of its biological functions and affirm its candidacy as a therapeutic target, specifically for CRC. Recently, Robert et al. described a novel CDK10/Cyclin M in vitro activity assay (81). This luminescence-based assay uses a synthetic peptide phosphorylation substrate for the CDK10/Cyclin M complex.
Flavopiridol is the most extensively studied pan-CDK inhibitor. It has been shown to inhibit CDK1, CDK2, CDK4, CDK6, CDK9 and CDK10. While flavopiridol can inhibit CDK10, the IC50 for flavopiridol inhibition against CDK10/Cyclin M is less potent than flavopiridol inhibition of other CDKs (81). In vitro studies in gastrointestinal cells demonstrated that flavopiridol was effective in inducing apoptosis through blockage of cell cycle progression at G1 (82, 83). However, clinical trials in patients with gastrointestinal cancers did not result in favorable outcomes (84, 85) and reported significant toxicity among patients.
Additional CDK inhibitors such as dinaciclib, SDS-032, AZD4573, AT7519 and riviciclib were all tested on CDK10/Cyclin M, however none of these potently inhibited CDK10 kinase activity (81). Ibrahim et al. synthesized novel flavopiridol analogs and assessed their inhibitory activity on CDK2, CDK5 and CDK9 (86). This series of inhibitors was more potent towards CDK9 than the other CDKs examined in the study. Given the close relation between CDK9 and CDK10, these inhibitors may exhibit inhibitory activity towards CDK10 (2).
The development of therapeutics targeting CDK10 should account for the tissue-specific biological activity of CDK10. Given that CDK10 acts as a tumor suppressor in some gastrointestinal and hepatobiliary cancers, future drug development should focus on inhibiting other CDKs, while maintaining activity of CDK10. In these cancers, CDK10 expression levels may be indicative of chemoresistance. The promotion of tumorigenesis by CDK10 in CRC suggests its inhibition is a promising therapeutic strategy. Given that the kinase domain has been implicated CDK10-mediated inhibition of apoptosis in CRC, inhibition of CDK10 kinase activity may be an effective therapeutic approach. The development of a CDK10-specific inhibitor may be a viable therapeutic target for the treatment of CRC.
Conclusion
CDK10 has been implicated as both a tumor suppressor and an oncogene in gastrointestinal and hepatobiliary cancers. CDK10 is involved in cell proliferation, cell motility, and plays an important role in chemosensitivity and chemoresistance. Further studies are warranted to understand the tissue-specific functions of CDK10 and the mechanisms that influence its oncogenic and tumor suppressive potential in gastrointestinal cancer. Implications of CDK10 as an oncogene in CRC make inhibition of CDK10 a viable therapeutic strategy. The development of therapeutics targeting CDK10 has been hindered by lack of a high throughput CDK10 activity screening assay. Detection of CDK10 kinase activity will allow for identification of small molecule inhibitors of CDK10. This will aid in further understanding the role of CDK10 in disease progression, and the development of therapeutics for the treatment of gastrointestinal cancers.
Author Contributions
ZB: drafting and editing the manuscript. IT: editing and critically reading manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Canadian Institutes of Health Research (CIHR) (Grant #MOP-82881) and CIHR New Investigator salary award to IT (MSH-95344).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figures were created using biorender.com.
References
1. Lim S, Kaldis P. Cdks, Cyclins and CKIs: Roles Beyond Cell Cycle Regulation. Development (2013) 140:3079–93. doi: 10.1242/dev.091744
2. Cao L, Chen F, Yang X, Xu W, Xie J, Yu L. Phylogenetic Analysis of CDK and Cyclin Proteins in Premetazoan Lineages. BMC Evol Biol (2014) 14:10. doi: 10.1186/1471-2148-14-10
3. Piao J, Zhu L, Sun J, Li N, Dong B, Yang Y, et al. High Expression of CDK1 and BUB1 Predicts Poor Prognosis of Pancreatic Ductal Adenocarcinoma. Gene (2019) 701:15–22. doi: 10.1016/j.gene.2019.02.081
4. Gan W, Zhao H, Li T, Liu K, Huang J. CDK1 Interacts With iASPP to Regulate Colorectal Cancer Cell Proliferation Through p53 Pathway. Oncotarget (2017) 8:71618–29. doi: 10.18632/oncotarget.17794
5. Shi Q, Ni X, Lei M, Xia Q, Dong Y, Zhang Q, et al. Phosphorylation of Islet-1 Serine 269 by CDK1 Increases its Transcriptional Activity and Promotes Cell Proliferation in Gastric Cancer. Mol Med (2021) 27:47. doi: 10.1186/s10020-021-00302-6
6. Yang W-X, Pan Y-Y, You C-G. Cdk1, CCNB1, Cdc20, BUB1, Mad2l1, MCM3, Bub1b, MCM2, and RFC4 may Be Potential Therapeutic Targets for Hepatocellular Carcinoma Using Integrated Bioinformatic Analysis. BioMed Res Int (2019) 2019:1245072. doi: 10.1155/2019/1245072
7. Li J-Q, Miki H, Ohmori M, Wu F, Funamoto Y. Expression of Cyclin E and Cyclin-Dependent Kinase 2 Correlates With Metastasis and Prognosis in Colorectal Carcinoma. Hum Pathol (2001) 32:945–53. doi: 10.1053/hupa.2001.27116
8. Ren S, Rollins BJ. Cyclin C/Cdk3 Promotes Rb-Dependent G0 Exit. Cell (2004) 117:239–51. doi: 10.1016/S0092-8674(04)00300-9
9. Lu J, Zhang ZL, Huang D, Tang N, Li Y, Peng Z, et al. Cdk3-promoted Epithelial-Mesenchymal Transition Through Activating AP-1 is Involved in Colorectal Cancer Metastasis. Oncotarget (2016) 7:7012–28. doi: 10.18632/oncotarget.6875
10. Lu JW, Lin YM, Chang JG, Yeh KT, Chen RM, Tsai JJ, et al. Clinical Implications of Deregulated CDK4 and Cyclin D1 Expression in Patients With Human Hepatocellular Carcinoma. Med Oncol (2013) 30:379. doi: 10.1007/s12032-012-0379-5
11. Lindberg D, Hessman O, Åkerström G, Westin G. Cyclin-Dependent Kinase 4 (Cdk4) Expression in Pancreatic Endocrine Tumors. Neuroendocrinology (2007) 86:112–8. doi: 10.1159/000106762
12. Wang S, Wang X, Gao Y, Peng Y, Dong N, Xie Q, et al. RN181 is a Tumor Suppressor in Gastric Cancer by Regulation of the ERK/MAPK-cyclin D1/CDK4 Pathway. J Pathol (2019) 248:204–16. doi: 10.1002/path.5246
13. Ikeda K, Monden T, Tsujie M, Izawa H, Yamamoto H, Ohnishi T, et al. [Cyclin D, CDK4 and p16 Expression in Colorectal Cancer]. Nihon Rinsho (1996) 54:1054–9.
14. Zhuang K, Zhang J, Xiong M, Wang X, Luo X, Han L, et al. CDK5 Functions as a Tumor Promoter in Human Colorectal Cancer Via Modulating the ERK5–AP-1 Axis. Cell Death Dis (2016) 7:e2415–5. doi: 10.1038/cddis.2016.333
15. Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, et al. Targeted Disruption of the Cyclin-Dependent Kinase 5 Gene Results in Abnormal Corticogenesis, Neuronal Pathology and Perinatal Death. Proc Natl Acad Sci (1996) 93:11173–8. doi: 10.1073/pnas.93.20.11173
16. Zhang R, Lin P, Yang H, He Y, Dang Y-W, Feng Z-B, et al. Clinical Role and Biological Function of CDK5 in Hepatocellular Carcinoma: A Study Based on Immunohistochemistry, RNA-seq and In Vitro Investigation. Oncotarget (2017) 8:108333–54. doi: 10.18632/oncotarget.22659
17. Eggers JP, Grandgenett PM, Collisson EC, Lewallen ME, Tremayne J, Singh PK, et al. Cyclin-Dependent Kinase 5 Is Amplified and Overexpressed in Pancreatic Cancer and Activated by Mutant K-Ras. Clin Cancer Res (2011) 17:6140–50. doi: 10.1158/1078-0432.CCR-10-2288
18. Cao L, Zhou J, Zhang J, Wu S, Yang X, Zhao X, et al. Cyclin-Dependent Kinase 5 Decreases in Gastric Cancer and Its Nuclear Accumulation Suppresses Gastric Tumorigenesis. Clin Cancer Res (2015) 21:1419–28. doi: 10.1158/1078-0432.CCR-14-1950
19. Feng L, Xie Y, Zhang H, Wu Y. miR-107 Targets Cyclin-Dependent Kinase 6 Expression, Induces Cell Cycle G1 Arrest and Inhibits Invasion in Gastric Cancer Cells. Med Oncol (2012) 29:856–63. doi: 10.1007/s12032-011-9823-1
20. Tadano T, Kakuta Y, Hamada S, Shimodaira Y, Kuroha M, Kawakami Y, et al. MicroRNA-320 Family is Downregulated in Colorectal Adenoma and Affects Tumor Proliferation by Targeting CDK6. World J gastrointestinal Oncol (2016) 8:532–42. doi: 10.4251/wjgo.v8.i7.532
21. Baba Y, Watanabe M, Murata A, Shigaki H, Miyake K, Ishimoto T, et al. : LINE-1 Hypomethylation, DNA Copy Number Alterations, and CDK6 Amplification in Esophageal Squamous Cell Carcinoma. Clin Cancer Res (2014) 20:1114–24. doi: 10.1158/1078-0432.CCR-13-1645
22. Lu P, Geng J, Zhang L, Wang Y, Niu N, Fang Y, et al. THZ1 Reveals CDK7-dependent Transcriptional Addictions in Pancreatic Cancer. Oncogene (2019) 38:3932–45. doi: 10.1038/s41388-019-0701-1
23. Wang Q, Li M, Zhang X, Huang H, Huang J, Ke J, et al. Upregulation of CDK7 in Gastric Cancer Cell Promotes Tumor Cell Proliferation and Predicts Poor Prognosis. Exp Mol Pathol (2016) 100:514–21. doi: 10.1016/j.yexmp.2016.05.001
24. Xu W, Wang Z, Zhang W, Qian K, Li H, Kong D, et al. Mutated K-ras Activates CDK8 to Stimulate the Epithelial-to-Mesenchymal Transition in Pancreatic Cancer in Part Via the Wnt/β-Catenin Signaling Pathway. Cancer Lett (2015) 356:613–27. doi: 10.1016/j.canlet.2014.10.008
25. Seo JO, Han SI, Lim SC. Role of CDK8 and Beta-Catenin in Colorectal Adenocarcinoma. Oncol Rep (2010) 24:285–91. doi: 10.3892/or_00000858
26. Kim M-Y, Han SI, Lim S-C. Roles of Cyclin-Dependent Kinase 8 and β-Catenin in the Oncogenesis and Progression of Gastric Adenocarcinoma. Int J Oncol (2011) 38:1375–83. doi: 10.3892/ijo.2011.948
27. Han SI, Lim S-C. Expression and Prognostic Significance of CDK8 and β-Catenin in Hepatocellular Carcinoma. In Vivo (Athens Greece) (2020) 34:1387–94. doi: 10.21873/invivo.11918
28. Kretz AL, Schaum M, Richter J, Kitzig EF, Engler CC, Leithäuser F, et al. CDK9 is a Prognostic Marker and Therapeutic Target in Pancreatic Cancer. Tumor Biol (2017) 39:1010428317694304. doi: 10.1177/1010428317694304
29. Lu Y, Tang L, Zhang Q, Zhang Z, Wei W. MicroRNA-613 Inhibits the Progression of Gastric Cancer by Targeting CDK9. Artif Cells Nanomed Biotechnol (2018) 46:980–4. doi: 10.1080/21691401.2017.1351983
30. Wang J, Liu J, Tian F, Zhan Y, Kong D. Cyclin-Dependent Kinase 9 Expression and its Association With CD8(+) T Cell Infiltration in Microsatellite-Stable Colorectal Cancer. Oncol Lett (2019) 18:6046–56. doi: 10.3892/ol.2019.10970
31. Zhong XY, Xu XX, Yu JH, Jiang GX, Yu Y, Tai S, et al. Clinical and Biological Significance of Cdk10 in Hepatocellular Carcinoma. Gene (2012) 498:68–74. doi: 10.1016/j.gene.2012.01.022
32. You Y, Bai F, Ye Z, Zhang N, Yao L, Tang Y, et al. Downregulated CDK10 Expression in Gastric Cancer: Association With Tumor Progression and Poor Prognosis. Mol Med Rep (2018) 17:6812–8. doi: 10.3892/mmr.2018.8662
33. Guen VJ, Gamble C, Flajolet M, Unger S, Thollet A, Ferandin Y, et al. CDK10/Cyclin M is a Protein Kinase That Controls ETS2 Degradation and is Deficient in STAR Syndrome. Proc Natl Acad Sci USA (2013) 110:19525–30. doi: 10.1073/pnas.1306814110
34. Hu D, Mayeda A, Trembley JH, Lahti JM, Kidd VJ. CDK11 Complexes Promote pre-mRNA Splicing. J Biol Chem (2003) 278:8623–9. doi: 10.1074/jbc.M210057200
35. Du Y, Yan D, Yuan Y, Xu J, Wang S, Yang Z, et al. Cdk11(p110) Plays a Critical Role in the Tumorigenicity of Esophageal Squamous Cell Carcinoma Cells and is a Potential Drug Target. Cell Cycle (Georgetown Tex) (2019) 18:452–66. doi: 10.1080/15384101.2019.1577665
36. Liu M, Fan H, Li T, Sihong L, Qiao S, Bi J. Low Expression of CDK12 in Gastric Cancer is Correlated With Advanced Stage and Poor Outcome. Pathol - Res Pract (2020) 216:152962. doi: 10.1016/j.prp.2020.152962
37. Ji J, Zhou C, Wu J, Cai Q, Shi M, Zhang H, et al. Expression Pattern of CDK12 Protein in Gastric Cancer and its Positive Correlation With CD8(+) Cell Density and CCL12 Expression. Int J Med Sci (2019) 16:1142–8. doi: 10.7150/ijms.34541
38. Kim H-E, Kim D-G, Lee KJ, Son JG, Song M-Y, Park Y-M, et al. Frequent Amplification of CENPF, GMNN and CDK13 Genes in Hepatocellular Carcinomas. PloS One (2012) 7:e43223. doi: 10.1371/journal.pone.0043223
39. Chen L, Wang Y, Jiang W, Ni R, Wang Y, Ni S. CDK14 Involvement in Proliferation Migration and Invasion of Esophageal Cancer. Ann Trans Med (2019) 7:681–1. doi: 10.21037/atm.2019.11.105
40. Sun Y, Zhu Q, Yang W, Shan Y, Yu Z, Zhang Q, et al. Lncrna H19/miR-194/PFTK1 Axis Modulates the Cell Proliferation and Migration of Pancreatic Cancer. J Cell Biochem (2019) 120:3874–86. doi: 10.1002/jcb.27669
41. Yang L, Zhu J, Huang H, Yang Q, Cai J, Wang Q, et al. Pftk1 Promotes Gastric Cancer Progression by Regulating Proliferation, Migration and Invasion. PloS One (2015) 10:e0140451. doi: 10.1371/journal.pone.0140451
42. Pang EY, Bai AH, To KF, Sy SM, Wong NL, Lai PB, et al. Identification of PFTAIRE Protein Kinase 1, a Novel Cell Division Cycle-2 Related Gene, in the Motile Phenotype of Hepatocellular Carcinoma Cells. Hepatology (2007) 46:436–45. doi: 10.1002/hep.21691
43. Mao Y, Jia Y, Zhu H, Wang W, Jin Q, Huang F, et al. High Expression of PFTK1 in Cancer Cells Predicts Poor Prognosis in Colorectal Cancer. Mol Med Rep (2017) 16:224–30. doi: 10.3892/mmr.2017.6560
44. Park MH, Kim SY, Kim YJ, Chung Y-H. ALS2CR7 (CDK15) Attenuates TRAIL Induced Apoptosis by Inducing Phosphorylation of Survivin Thr34. Biochem Biophys Res Commun (2014) 450:129–34. doi: 10.1016/j.bbrc.2014.05.070
45. Mikolcevic P, Sigl R, Rauch V, Hess MW, Pfaller K, Barisic M, et al. Cyclin-Dependent Kinase 16/PCTAIRE Kinase 1 is Activated by Cyclin Y and is Essential for Spermatogenesis. Mol Cell Biol (2012) 32:868–79. doi: 10.1128/MCB.06261-11
46. Shimizu K, Uematsu A, Imai Y, Sawasaki T. Pctaire1/Cdk16 Promotes Skeletal Myogenesis by Inducing Myoblast Migration and Fusion. FEBS Lett (2014) 588:3030–7. doi: 10.1016/j.febslet.2014.05.060
47. Liu Q, Wang C, Jiang Z, Li S, Li F, Tan H-B, et al. circRNA 001306 Enhances Hepatocellular Carcinoma Growth by Up-Regulating CDK16 Expression Via Sponging Mir-584-5p. J Cell Mol Med (2020) 24:14306–15. doi: 10.1111/jcmm.16047
48. Kerr N, Pintzas A, Holmes F, Hobson SA, Pope R, Wallace M, et al. The Expression of ELK Transcription Factors in Adult DRG: Novel Isoforms, Antisense Transcripts and Upregulation by Nerve Damage. Mol Cell Neurosci (2010) 44:165–77. doi: 10.1016/j.mcn.2010.03.005
49. Barone G, Staples CJ, Ganesh A, Patterson KW, Bryne DP, Myers KN, et al. Human CDK18 Promotes Replication Stress Signaling and Genome Stability. Nucleic Acids Res (2016) 44:8772–85. doi: 10.1093/nar/gkw615
50. Li P, Ge D, Li P, Hu F, Chu J, Chen X, et al. CXXC Finger Protein 4 Inhibits the CDK18-ERK1/2 Axis to Suppress the Immune Escape of Gastric Cancer Cells With Involvement of ELK1/MIR100HG Pathway. J Cell Mol Med (2020) 24:10151–65. doi: 10.1111/jcmm.15625
51. Zhao JQ, Li XN, Fu LP, Zhang N, Cai JH. ISOC1 Promotes the Proliferation of Gastric Cancer Cells by Positively Regulating CDK19. Eur Rev Med Pharmacol Sci (2020) 24:11602–9. doi: 10.26355/eurrev_202011_23803
52. Feng H, Yu Z, Tian Y, Lee YY, Li MS, Go MY, et al. A CCRK-EZH2 Epigenetic Circuitry Drives Hepatocarcinogenesis and Associates With Tumor Recurrence and Poor Survival of Patients. J Hepatol (2015) 62:1100–11. doi: 10.1016/j.jhep.2014.11.040
53. An X, Ng SS, Xie D, Zeng YX, Sze J, Wang J, et al. Functional Characterisation of Cell Cycle-Related Kinase (CCRK) in Colorectal Cancer Carcinogenesis. Eur J Cancer (2010) 46:1752–61. doi: 10.1016/j.ejca.2010.04.007
54. Uhlén M, Björling E, Agaton C, Szigyarto CA-K, Amini B, Andersen E, et al. : A Human Protein Atlas for Normal and Cancer Tissues Based on Antibody Proteomics. Mol Cell Proteomics (2005) 4:1920–32. doi: 10.1074/mcp.M500279-MCP200
55. Simmons Kovacs LA, Mayhew MB, Orlando DA, Jin Y, Li Q, Huang C, et al. Cyclin-Dependent Kinases are Regulators and Effectors of Oscillations Driven by a Transcription Factor Network. Mol Cell (2012) 45:669–79. doi: 10.1016/j.molcel.2011.12.033
56. Malumbres M, Barbacid M. Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat Rev Cancer (2009) 9:153–66. doi: 10.1038/nrc2602
57. Graña X, Claudio PP, De Luca A, Sang N, Giordano A. PISSLRE, a Human Novel CDC2-related Protein Kinase. Oncogene (1994) 9:2097–103.
58. You Y, Li H, Qin X, Zhang Y, Song W, Ran Y, et al. Decreased CDK10 Expression Correlates With Lymph Node Metastasis and Predicts Poor Outcome in Breast Cancer Patients - A Short Report. Cell Oncol (Dordr) (2015) 38:485–91. doi: 10.1007/s13402-015-0246-4
59. Crawford J, Ianzano L, Savino M, Whitmore S, Cleton-Jansen AM, Settasatian C, et al. The PISSLRE Gene: Structure, Exon Skipping, and Exclusion as Tumor Suppressor in Breast Cancer. Genomics (1999) 56:90–7. doi: 10.1006/geno.1998.5676
60. Li H, You Y, Liu J. Cyclin−Dependent Kinase 10 Prevents Glioma Metastasis Via Modulation of Snail Expression. Mol Med Rep (2018) 18:1165–70. doi: 10.3892/mmr.2018.9059
61. You Y, Yang W, Wang Z, Zhu H, Li H, Lin C, et al. Promoter Hypermethylation Contributes to the Frequent Suppression of the CDK10 Gene in Human Nasopharyngeal Carcinomas. Cell Oncol (Dordr) (2013) 36:323–31. doi: 10.1007/s13402-013-0137-5
62. Weiswald L-B, Hasan MR, Wong JCT, Pasiliao CC, Rahman M, Ren J, et al. Inactivation of the Kinase Domain of CDK10 Prevents Tumor Growth in a Preclinical Model of Colorectal Cancer, and is Accompanied by Downregulation of Bcl-2. Mol Cancer Ther (2017) 16(10):2292–303. doi: 10.1158/1535-7163.MCT-16-0666
63. Khanal P, Yun HJ, Lim SC, Ahn SG, Yoon HE, Kang KW, et al. Proyl Isomerase Pin1 Facilitates Ubiquitin-Mediated Degradation of Cyclin-Dependent Kinase 10 to Induce Tamoxifen Resistance in Breast Cancer Cells. Oncogene (2012) 31:3845–56. doi: 10.1038/onc.2011.548
64. Yu JH, Zhong XY, Zhang WG, Wang ZD, Dong Q, Tai S, et al. CDK10 Functions as a Tumor Suppressor Gene and Regulates Survivability of Biliary Tract Cancer Cells. Oncol Rep (2012) 27:1266–76. doi: 10.3892/or.2011.1617
65. Kasten M, Giordano A. Cdk10, a Cdc2-related Kinase, Associates With the Ets2 Transcription Factor and Modulates its Transactivation Activity. Oncogene (2001) 20:1832–8. doi: 10.1038/sj.onc.1204295
66. Liu W, Cai M-J, Wang J-X, Zhao X-F. In a Nongenomic Action, Steroid Hormone 20-Hydroxyecdysone Induces Phosphorylation of Cyclin-Dependent Kinase 10 to Promote Gene Transcription. Endocrinology (2014) 155:1738–50. doi: 10.1210/en.2013-2020
67. Zhao BW, Chen S, Li YF, Xiang J, Zhou ZW, Peng JS, et al. Low Expression of CDK10 Correlates With Adverse Prognosis in Gastric Carcinoma. J Cancer (2017) 8:2907–14. doi: 10.7150/jca.20142
68. Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, et al. Identification of CDK10 as an Important Determinant of Resistance to Endocrine Therapy for Breast Cancer. Cancer Cell (2008) 13:91–104. doi: 10.1016/j.ccr.2008.01.001
69. Yu J, Zhang W, Lu B, Qian H, Tang H, Zhu Z, et al. miR-433 Accelerates Acquired Chemoresistance of Gallbladder Cancer Cells by Targeting Cyclin M. Oncol Lett (2018) 15:3305–12. doi: 10.3892/ol.2017.7708
70. Fukui R, Nishimori H, Hata F, Yasoshima T, Ohno K, Nomura H, et al. Et Al: Metastases-related Genes in the Classification of Liver and Peritoneal Metastasis in Human Gastric Cancer. J Surg Res (2005) 129:94–100. doi: 10.1016/j.jss.2005.04.030
71. Shang HS, Lu HF, Lee CH, Chiang HS, Chu YL, Chen A, et al. Quercetin Induced Cell Apoptosis and Altered Gene Expression in AGS Human Gastric Cancer Cells. Environ Toxicol (2018) 33:1168–81. doi: 10.1002/tox.22623
72. Mori Y, Matsunaga M, Abe T, Fukushige S, Miura K, Sunamura M, et al. Chromosome Band 16q24 is Frequently Deleted in Human Gastric Cancer. Br J Cancer (1999) 80:556–62. doi: 10.1038/sj.bjc.6690391
73. Riou P, Saffroy R, Comoy J, Gross-Goupil M, Thiery JP, Emile JF, et al. Investigation in Liver Tissues and Cell Lines of the Transcription of 13 Genes Mapping to the 16q24 Region That are Frequently Deleted in Hepatocellular Carcinoma. Clin Cancer Res (2002) 8:3178–86.
74. Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Sugimura T, Hirohashi S. Aberrant DNA Methylation on Chromosome 16 is an Early Event in Hepatocarcinogenesis. Japanese J Cancer Res Gann (1996) 87:1210–7. doi: 10.1111/j.1349-7006.1996.tb03135.x
75. Kanai Y, Ushijima S, Tsuda H, Sakamoto M, Hirohashi S. Aberrant DNA Methylation Precedes Loss of Heterozygosity on Chromosome 16 in Chronic Hepatitis and Liver Cirrhosis. Cancer Lett (2000) 148:73–80. doi: 10.1016/S0304-3835(99)00316-X
76. Chan SK, Griffith OL, Tai IT, Jones SJ. Meta-Analysis of Colorectal Cancer Gene Expression Profiling Studies Identifies Consistently Reported Candidate Biomarkers. Cancer Epidemiol Biomarkers Prev (2008) 17:543–52. doi: 10.1158/1055-9965.EPI-07-2615
77. Magee JA, Araki T, Patil S, Ehrig T, True L, Humphrey PA, et al. Expression Profiling Reveals Hepsin Overexpression in Prostate Cancer. Cancer Res (2001) 61:5692–6.
78. Leman ES, Magheli A, Yong KM, Netto G, Hinz S, Getzenberg RH. Identification of Nuclear Structural Protein Alterations Associated With Seminomas. J Cell Biochem (2009) 108:1274–9. doi: 10.1002/jcb.22357
79. Han SW, Ahn JY, Lee S, Noh YS, Jung HC, Lee MH, et al. Gene Expression Network Analysis of Lymph Node Involvement in Colon Cancer Identifies AHSA2, CDK10, and CWC22 as Possible Prognostic Markers. Sci Rep (2020) 10:7170. doi: 10.1038/s41598-020-63806-x
80. Zehra M, Mushtaq S, Ghulam Musharraf S, Ghani R, Ahmed N. Association of Cyclin Dependent Kinase 10 and Transcription Factor 2 During Human Corneal Epithelial Wound Healing In Vitro Model. Sci Rep (2019) 9:11802. doi: 10.1038/s41598-019-48092-6
81. Robert T, Johnson JL, Guichaoua R, Yaron TM, Bach S, Cantley LC, et al. Development of a CDK10/CycM In Vitro Kinase Screening Assay and Identification of First Small-Molecule Inhibitors. Front Chem (2020) 8:147. doi: 10.3389/fchem.2020.00147
82. Jung C, Motwani M, Kortmansky J, Sirotnak FM, She Y, Gonen M, et al. The Cyclin-Dependent Kinase Inhibitor Flavopiridol Potentiates Gamma-Irradiation-Induced Apoptosis in Colon and Gastric Cancer Cells. Clin Cancer Res (2003) 9:6052–61.
83. Miyashita K, Shiraki K, Fuke H, Inoue T, Yamanaka Y, Yamaguchi Y, et al. The Cyclin-Dependent Kinase Inhibitor Flavopiridol Sensitizes Human Hepatocellular Carcinoma Cells to TRAIL-induced Apoptosis. Int J Mol Med (2006) 18:249–56. doi: 10.3892/ijmm.18.2.249
84. Ang C, O’Reilly EM, Carvajal RD, Capanu M, Gonen M, Doyle L, et al. A Nonrandomized, Phase II Study of Sequential Irinotecan and Flavopiridol in Patients With Advanced Hepatocellular Carcinoma. Gastrointestinal Cancer Res GCR (2012) 5:185–9.
85. Aklilu M, Kindler HL, Donehower RC, Mani S, Vokes EE. Phase II Study of Flavopiridol in Patients With Advanced Colorectal Cancer. Ann Oncol (2003) 14:1270–3. doi: 10.1093/annonc/mdg343
Keywords: colorectal cancer, cyclin-dependent kinases, gastrointestinal cancers, hepatocellular carcinoma, gastric cancer, biliary tract cancer, CDK10
Citation: Bazzi ZA and Tai IT (2021) CDK10 in Gastrointestinal Cancers: Dual Roles as a Tumor Suppressor and Oncogene. Front. Oncol. 11:655479. doi: 10.3389/fonc.2021.655479
Received: 18 January 2021; Accepted: 16 June 2021;
Published: 30 June 2021.
Edited by:
Jaw-Yuan Wang, Kaohsiung Medical University Hospital, TaiwanReviewed by:
Pei-Jung Lu, National Cheng Kung University, TaiwanPeng-Chan Lin, National Cheng Kung University, Taiwan
Long-sen Chang, National Sun Yat-sen University, Taiwan
Copyright © 2021 Bazzi and Tai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabella T. Tai, aXRhaUBiY2dzYy5jYQ==