 Ping Zhang1,2
Ping Zhang1,2 Mingzhi Zhang1*
Mingzhi Zhang1*- 1Department of Oncology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou City, China
- 2Department of Oncology, Academy of Medical Sciences of Zhengzhou University, Zhengzhou City, China
Cutaneous T-cell lymphomas (CTCLs) comprise a group of heterogeneous diseases involving malignant T cells. The pathogenesis and etiology of CTCL are still unclear, although a large number of genetic and epidemiological studies on CTCL have been conducted. Most CTCLs have an indolent course, making early diagnosis difficult. Once large-cell transformation occurs, CTCL progresses to more aggressive types, resulting in an overall survival of less than five years. Epigenetic drugs, which have shown certain curative effects, have been selected as third-line drugs in patients with relapsing and refractory CTCL. Many studies have also identified epigenetic biomarkers from tissues and peripheral blood of patients with CTCL and suggested that epigenetic changes play a role in malignant transformation and histone deacetylase inhibitor (HDACi) resistance in CTCL. Single-cell sequencing has been applied in CTCL studies, revealing heterogeneity in CTCL malignant T cells. The mechanisms of HDACi resistance have also been described, further facilitating the discovery of novel HDACi targets. Despite the heterogeneity of CTCL disease and its obscure pathogenesis, more epigenetic abnormalities have been gradually discovered recently, which not only enables us to understand CTCL disease further but also improves our understanding of the specific role of epigenetics in the pathogenesis and treatment. In this review, we discuss the recent discoveries concerning the pathological roles of epigenetics and epigenetic therapy in CTCL.
Introduction
Cutaneous T-cell lymphomas (CTCLs) comprise a heterogeneous group of non-Hodgkin lymphomas derived from skin-homing T cells. Therefore, the classification of CTCL is ill-defined and continuously updated. The incidence of CTCL has been increasing for the past decade and is currently 6.4 per million persons worldwide. The incidence of CTCL increases significantly with age and is thus highest in elderly individuals, especially in patients over 70 years of age (1, 2) Mycosis fungoides (MF) and Sézary syndrome (SS) are the most common types of CTCL, accounting for 50% of CTCLs (3).
Most early stage MF cases have an indolent clinical course. On the other hand, cases of advanced-stage MF (stages IIB–IV) and SS present with an aggressive clinical course, with a median survival of one to five years (4). SS is an aggressive form of CTCL involving mature T cells. It typically has a poor prognosis and limited therapeutic options.
The pathogenesis of CTCL remains elusive. The genetic aberrations and epigenetic modifications associated with the expansion of CTCL T cells have not been elucidated. Recently, next-generation sequencing (NGS) has been used in CTCL studies to offer new insights into the pathogenesis of this condition at the genetic level. Moreover, a large number of studies have shown that the occurrence of CTCL is closely related to epigenetics (5). Epigenetic changes can alter gene expression and function without changing the DNA sequence by regulating gene transcription. These changes mainly involve modification of histones, methylation of DNA, and methylation of microRNA (miRNA) host genes. With the successful clinical application of epigenetic drugs, many investigations have demonstrated the role of epigenetics in CTCL pathogenesis and progression. Emerging data also suggest that treatment strategies aimed at regulating multiple epigenetic targets may be achieved through combination regimens. This review discusses the latest advancements in CTCL, including data on epigenetic markers and their role in CTCL diagnosis, epigenetic therapies for CTCL, and histone deacetylase inhibitor (HDACi) resistance in CTCL.
Histone Modification in CTCL
Gene transcription is regulated by a number of complex enzymes that modify chromatin accessibility. It is controlled by several factors, including the balance between the activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs) that acetylate and deacetylate histones, respectively. HATs facilitate the activation of gene transcription, primarily occurring near the proximal enhancers and promoters. In contrast, HDACs inhibit the expression of specific tumor suppressor genes. Many studies have reported that HDACs consist of 18 subtypes, which can be subdivided into four classes according to their homology with yeast HDACs. Nevertheless, the most frequently studied HDACs in CTCL are HDAC1, HDAC2, and HDAC6 (6). HDAC2 and HDAC6 levels in MF have been reported to be significantly higher than those in healthy individuals. In a study that investigated the differential expression and prognostic significance of HDACs in CTCL, HDAC2 was found to be more highly expressed in the aggressive types of CTCL than in the indolent ones. Meanwhile, HDAC6 overexpression has been associated with favorable outcomes in all CTCL types (7). The overexpression of HDAC1 and HDAC6 may be mediated by IL-15 overexpression in CTCL, making these HDACs attractive molecular targets of IL-15 downstream signaling in CTCL (8). IL-15 is a significant component in the pathogenesis of CTCLs, as the overexpression of IL-15 induces spontaneous CTCL and MF progression (9–11). The inhibition of HDAC6 and the presence of PI3K inhibitors were observed to synergistically inhibit cell proliferation in CTCL cell lines (12) (Figure 1). Moreover, HDAC6 inhibition was found to diminish the activation of Akt, a downstream kinase involved in the PI3K pathway (14), and to affect the development of T cells (15). The inhibition of HDAC3 was also reported to be useful in the treatment of CTCL (16). Since HDACs are involved in many signaling pathways that affect cellular death and apoptosis, an increasing number of studies have focused on developing specific HDACis.
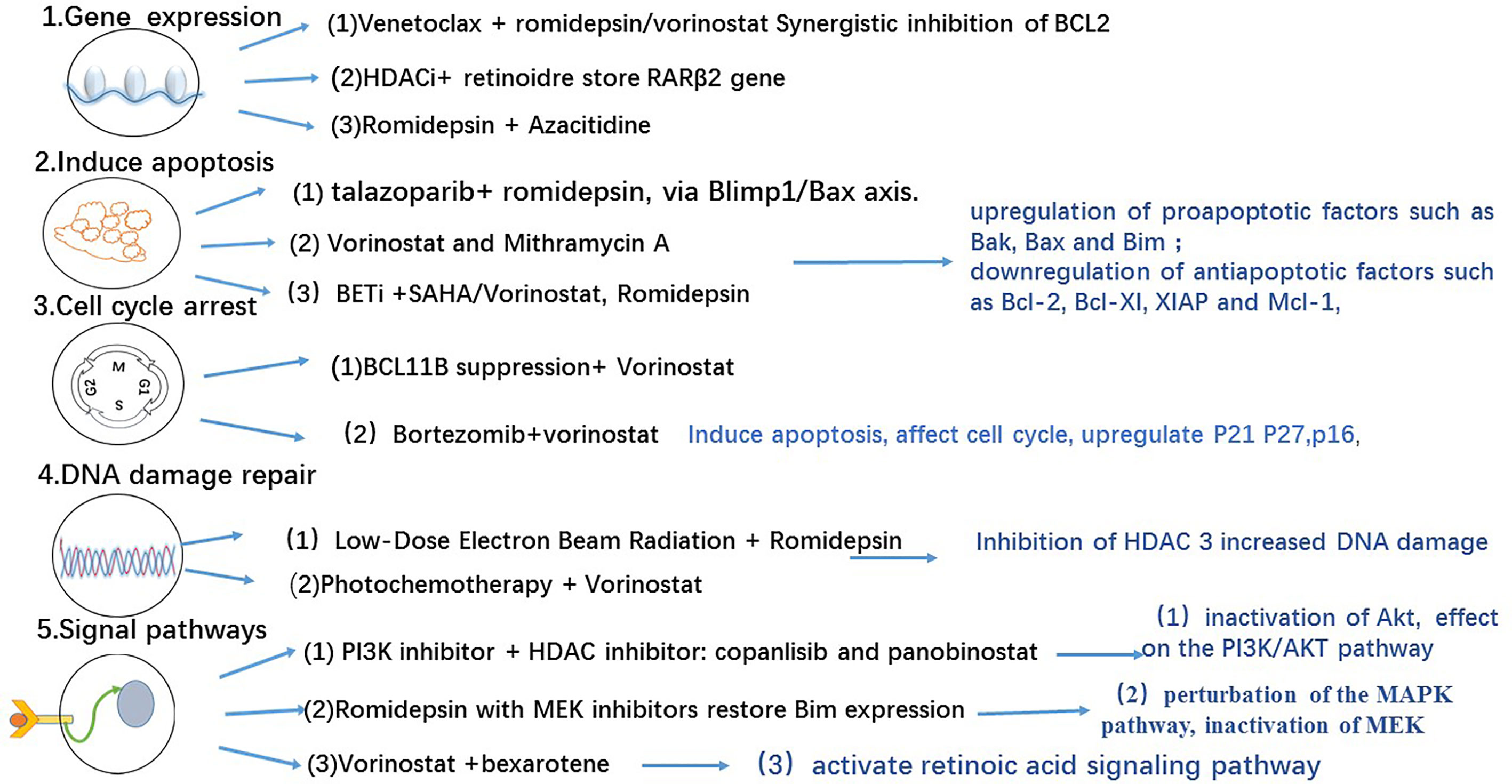
Figure 1 HDACI combination therapy and effects (13).
Romidepsin
Romidepsin (depsipeptide) was demonstrated to be effective in patients with CTCL in a study conducted by the National Cancer Institute in 2009 (17). Moreover, two phase II multi-institutional trials showed that romidepsin had a significant single-agent clinical activity against CTCL and yielded robust responses(ORR 34%) in patients with CTCL (18). The NCI1312 clinical trial provided a more comprehensive and detailed description of the responses and tolerated toxicities associated with romidepsin use (Table 1). Clinical and correlative data have shown that HDAC inhibitors cannot modify gene expression but affect NFκB target genes. DNA damage may be the dominant mechanism of romidepsin to induce cell death (23). Many retrospective studies have suggested that romidepsin is safe and effective in patients with CTCL who received prior systemic chemotherapy (24, 25). A retrospective comparative analysis of 198 MF/SS patients showed that the median time to next treatment (TTNT) for romidepsin monotherapy (4.5 months) was comparable to that for multi-agent chemotherapy (3.9 months) (26). Long-term use of a dose-reducing romidepsin regimen was also determined to be an alternative treatment strategy in patients with CTCL (27).
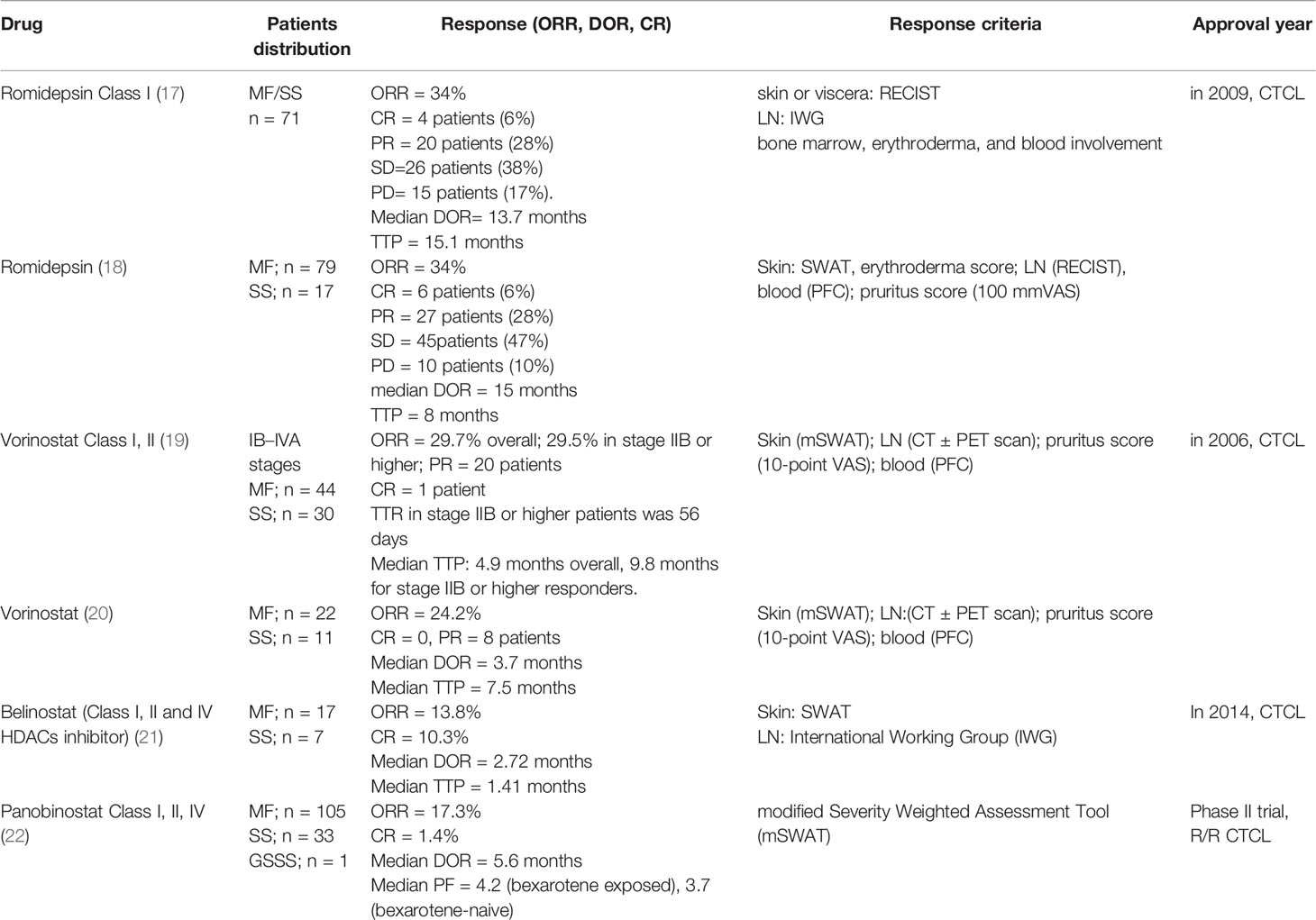
Table 1 Clinical effects of HDAC Inhibitors in Cutaneous T−cell Lymphoma [refer to Adriana T. Lopez et al. (13)].
Vorinostat
Vorinostat, an oral HDACi, was approved by the Food and Drug Administration (FDA) in 2006 for use in the treatment of MF and SS. Many small clinical trials involving the use of vorinostat in patients with CTCL have been performed consecutively (28, 29). The overall response rate of vorinostat in patients with advanced refractory CTCL was 24–30%. According to these trials, vorinostat, which was found to cause reversible side effects, is a safe and well-tolerated third-line drug for patients with CTCL (30, 31). Therefore, many trials that investigated combination therapies with vorinostat have been conducted (32). Some of these combinations have been shown to induce genetic changes, causing malignant cell death, but their effectiveness in the clinical setting is still uncertain (33–35).
Belinostat
Patients with CTCL who had received one or more prior systemic therapies were enrolled in a belinostat phase II trial (21). The effects of belinostat are listed in Table 1.
HDACi Combination Therapy
The aforementioned epigenetic drugs and their efficacies have been well studied in advanced CTCL. Other effective epigenetic drugs are currently being investigated (36) in patients with advanced MF/SS with variable courses and poor outcomes. Domatinostat, panobinostat, remetinostat, and andresminostat have also been studied in patients with CTCL. Remetinostat gel is the only HDACi that has been investigated for the treatment of topical cutaneous disease in MF. In phase Ib and II trials of remetinostat, patients with limited-stage (IA–IIA) CTCL benefited from remetinostat gel based on the improvements in the composite assessment of index lesions (CAILs) score and modified severity-weighted assessment tool (mSWAT) score, and the clinically significant reductions in pruritus (37). Synergistic HDACi combination therapies have also been investigated to determine their benefits in terms of objective response and durable response time (37). Results from preclinical findings (38) and a phase I clinical trial show that combining vorinostat and bexarotene can provide clinical relief of pruritus (39). Other HDACi combinations have been investigated in preclinical and clinical studies (40) involving the use of interferon-gamma (IFNγ) (41, 42), retinoids (43), ultraviolet A (UV-A) phototherapy (44), extracorporeal photopheresis (ECP) (45), PI3K inhibitors (12, 46) the proteasome inhibitor bortezomib (47), and hypomethylated agents such as azacytidine (48) (Figure 1). However, these combinations do not show remarkable outcomes. Nevertheless, other HDACi combination chemotherapies and topical skin treatments have shown promising therapeutic potential in patients with CTCL (44, 49). A number of HDACi combination therapies are currently undergoing clinical trials (Table 2).

Table 2 HDACi combination therapies under investigation are recruiting for relapsed/refractory cutaneous T-cell lymphomas.
HDACi Therapy Limitations and Strengths
Few studies have directly compared the efficacy and safety profiles of HDACis in patients with MF/SS. A retrospective study compared the TTNT for romidepsin, vorinostat, and panobinostat in patients with MF/SS and reported that there were no significant differences between HDACi therapies, as the overall median TTNT was 5.5 months (50).
Based on the literature, HDACis exhibit similar toxicity profiles. Adverse events include gastrointestinal disturbance, myelosuppression, transient prolongation of QTc interval, nausea, asthenia/fatigue, histone acetylation in peripheral blood mononuclear cells, and infections. Among these events, the most remarkable are the cardiac events, particularly ST-T segment abnormalities and QTc prolongation (51). Differences in the chemical structures of the inhibitors may contribute to the development of these adverse effects.
The National Comprehensive Cancer Network (NCCN) recommends a wide range of therapies for CTCL; however, curative options for CTCL are limited to autologous stem cell transplantations. Among the recommended therapies are those that use vorinostat and romidepsin for systemic therapy. Studies have shown that vorinostat and romidepsin therapies result in unremarkable outcomes compared with other therapies (52). However, data from the outcomes of these therapies were still able to support the use of HDACis as a third-line therapeutic option in advanced CTCL, without increasing morbidity due to toxicity (53). In the phase III MAVORIC trial (n = 372, with 186 patients treated with vorinostat), mogamulizumab was reported to be more effective than vorinostat. For mogamulizumab and vorinostat, the median progression-free survival (PFS) values were 7.7 and 3.1 months, respectively; objective response rates (ORRs) in the MF cohort were 21 and 7.1%, respectively; and ORRs in the SS cohort were 37and 4.1%, respectively (54). A subsequent study compared mogamulizumab and vorinostat in terms of quality of life (QOL) measurements and showed that mogamulizumab was superior to vorinostat. This study also demonstrated that mogamulizumab exhibited a frequency of adverse events that was almost twice as high as that of vorinostat and showed inferior tolerability compared to vorinostat in patients with MF/SS (55). These findings of poor tolerance and adverse effects, such as frequent granulomatous drug eruption, may influence the preference for mogamulizumab (56). The effects of HDACi are non-specific compared to antibody-targeting drugs such as mogamulizumab. These effects on the pathogenesis of CTCL have been reported in many preclinical studies (57). In addition, the mechanisms of HDACi resistance in terms of the heterogeneity of advanced MF/SS have been investigated (58).
Predictive Biomarkers for Epigenetic Therapy Responses
Previous studies have demonstrated that the apoptotic effects of HDACis have a significant role in the treatment of patients with MF/SS. HDACis have been reported to activate intrinsic and extrinsic apoptosis in malignant T cells (59) by increasing the transcription of tumor suppression genes (60), dysregulating cell cycle progression (16), and inhibiting cell proliferation (61). Specifically, a study reported that HDACis induced apoptosis by regulating the expression of pro- and anti-apoptotic genes [p21 (WAF1) and bax] or inducing the transcription of multiple immediate–early (IE) genes (ATF3) (62). Vorinostat affects a wide range of signal pathways (46), including the STAT signaling pathway, and the acetylation of tumor suppressors, including P53 (Figure 1) (63). The low overall response rate (approximately 30–40%) of HDACis in CTCL is probably related to HDACi resistance in malignant T cells. Cytogenetic and genomic studies have recently provided data on the molecular mechanism for apoptosis resistance in CTCL malignant T cells and data on the molecular heterogeneity of CTCL cell populations (Figure 2) (58). In one study, STAT3 and RAD23B genotypes were reported to influence primary HDACi sensitivity in Sézary cells (64). In another study, persistent activation of STAT1 and pSTAT3 was shown to correlate with resistance to vorinostat in patients with CTCL (65). Other studies showed that increased tyrosine phosphorylation of STAT3 (pYSTAT3) expression reduced the response to suberoylanilide hydroxamic acid (SAHA) in Sézary cells, while increased HR23B expression was identified as a determinant of sensitivity to SAHA (66, 67). HR23B expression evaluation in a unique collection of CTCL biopsies taken from a phase II trial of SAHA suggested that HR23B could be an informative biomarker for predicting clinical responses to HDAC inhibitors. These studies also reported that anti-apoptotic cell adhesion/migration genes were highly expressed in MF/SS, suggesting the occurrence of a possible mechanism of HDACi resistance involving the overexpression of such genes in MF/SS. The HDACi resistance genes CCR6, CXCR4, BCL2, BIRC5, CDK1, and LAIR2 were also highly acetylated, possibly inducing the high expression of these genes and promoting disease progression and HDACi resistance. Some of the upregulated genes, STAT4, TNFRSF17, TNFAIP3, GSTM1, GSTM3, and TXNDC5, are known drivers of HDACi resistance (Figure 2) (68). In a previous study, LAIR2 was identified as a potential predictor of HDACi resistance in CTCL, as LAIR2 was significantly higher in skin biopsies and blood from patients with HDACi-resistant MF/SS. Mitogen-activated protein kinase (MAPK) pathway activation, which can be reversed by MEK (MAPK kinase) inhibitors, has been speculated to be involved in HDACi resistance (69, 70). Therefore, another probable reason for HDACi resistance is the non-specific off-target effects of HDACis on malignant T cells. Chromatin accessibility has been investigated in CTCL, as changes in chromatin accessibility may predict clinical response to HDACi therapy (69). A study found an association between the clinical response to HDACi treatment and the dynamic increase in DNA accessibility and identified HDACi-responsive (FOXP3) and non-responsive (IFIT3) genes. Both genes were accessible in clinical responders but not in non-responders (shown as Figure 2) (71). HDACi resistance may be related to subpopulation heterogeneity within malignant T cells in SS. A high degree of single-cell heterogeneity was identified in SS using single-cell RNA sequencing (72). Terkild Brink et al. demonstrated that distinct subpopulations showed selective sensitivity toward HDACi, causing HDACi resistance (73) that may lead to relapsing or aggressive diseases (73). Although HDACis did not achieve satisfactory responses and had considerable side effects in patients with CTCL (74), the preliminary investigations of HDACi use in CTCL paved the way for novel HDACi drug development for CTCL treatment and provided an increased understanding of the distinct effects and deficiencies of HDACi. In addition, studies on histone modification in CTCL have not only promoted drug discovery and therapy optimization involving HDACis, but also provided a thorough investigation of CTCL epigenomes (75).
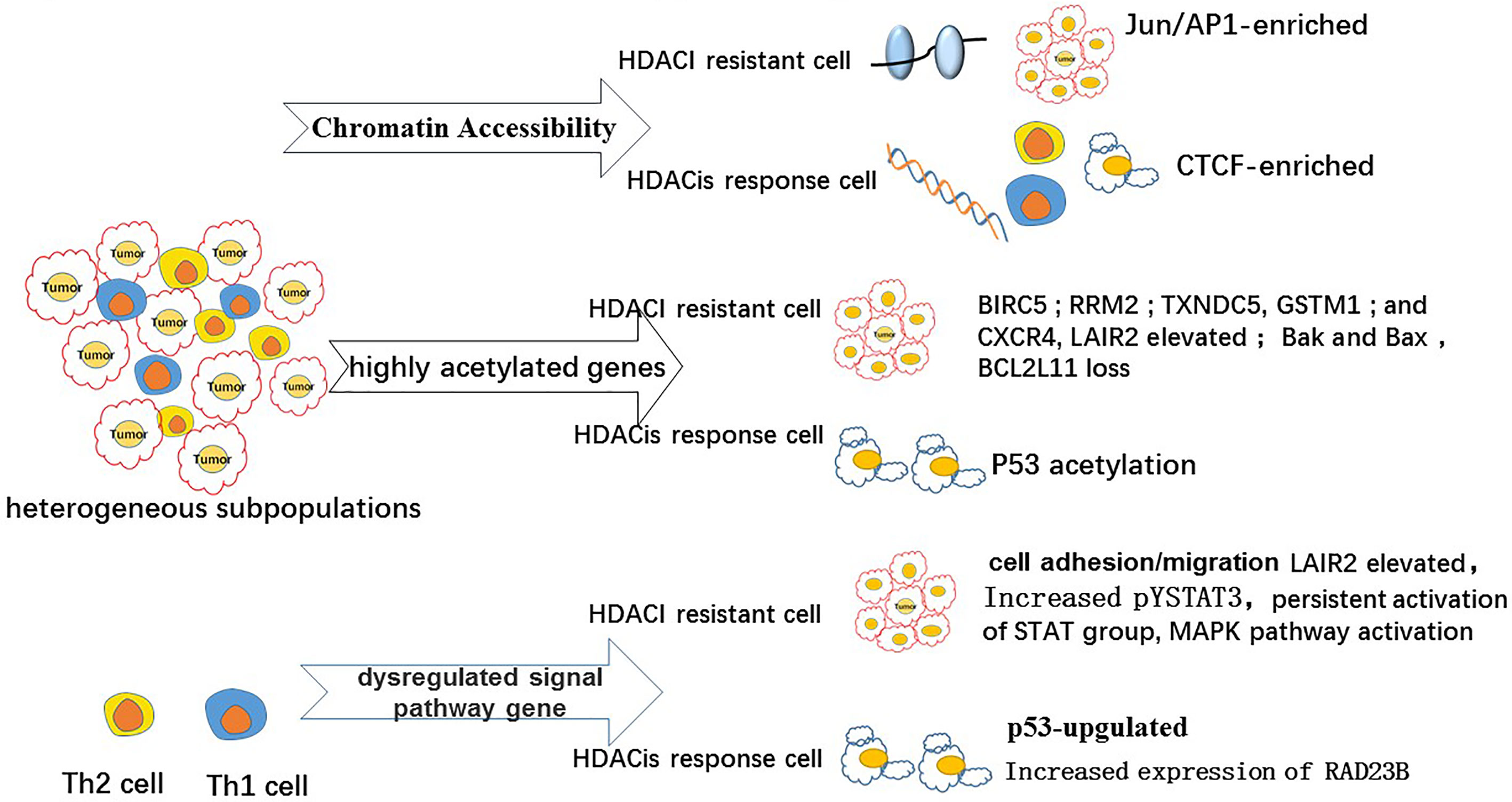
Figure 2 Mechanism of HDACI therapy for drug resistance.
DNA Methylation in CTCL
DNA methylation is one of the most studied epigenetic events in the development and progression of CTCL (76). Methylation abnormalities not only serve as diagnostic biomarkers but also contribute to the development of treatment options for CTCL. DNA methylation is the process catalyzed by DNA methyltransferases (DNMTs), specifically DNMT1, DNMT3A, and DNMT3B, which transfer methyl groups from S-adenosyl methionine (SAM) to the 5-carbon (C5) position of cytosine bases. On the other hand, “epigenetic erasers” from the TET family of proteins are involved in DNA demethylation. Hypermethylation is often associated with gene inactivation and silencing, inhibiting the expression of genes, such as SAMHD1 (77). In CTCL, silenced genes mainly include tumor suppressor genes, including CDKN2A, BCL7A, and MLH1, which regulate cell proliferation and apoptosis resistance during malignant cell transformation (Table 3). Defective Fas signaling has been reported as a possible causative agent in MF pathogenesis due to defects in apoptosis signaling in skin-homing T cells. In SS, a reduced Fas expression due to the hypermethylation of the Fas gene promoter region was found to cause apoptosis resistance (89, 94). In contrast, hypomethylation is often associated with genomic instability, leading to gene activation. In a previous study, the demethylation of the SATB1 promoter was found to cause SATB1 overexpression, which promoted malignant T cell proliferation by directly reducing the expression of the inhibitor p21, contributing to the progression of cutaneous CD30+ lymphoproliferative disease (95). Many studies have reported that SATB1 dysregulation is involved in CTCL pathogenesis (96–98). SATB1 is considered a pivotal epigenetic biomarker for CTCL (99), and its dysregulation can be reversed by methyltransferase inhibitors (100). The expression of SATB1 is heterogeneous in CTCL and is associated with the clinical prognosis of patients with MF (101). A reduced SATB1 expression is associated with disease progression and poor prognosis in SS and MF (102). The dysregulation of DNA methylation has been extensively studied in CTCL. Because CTCL presents with a highly variable disease course, CTCL usually cannot be diagnosed before extensive disease progression. To find specific epigenetic markers that can stage and diagnose CTCL, many studies have analyzed the global methylation patterns and epigenetic abnormalities in samples from patients with CTCL, particularly SS (103). The genomic methylation patterns in SS, which are consistent with those of other cancers, are characterized by global genome hypomethylation and specific gene hypermethylation. Hypermethylated promoter CpG loci, such as those in the CMTM2 gene, were found to be sufficient diagnostic biomarkers for SS (92). Specific gene promoter methylation is also a significant predictor of disease diagnosis prognosis, and progression (85) (Table 3). Highly expressed PLS3 and TWIST (104, 105) have been well investigated as specific markers for Sézary cells and differentiators between SS and erythrodermic inflammatory dermatoses (EIDs) (106, 107), and these overexpressions have been demonstrated to result from DNA promoter hypomethylation (108). Increased PLS3 was reported to be able to differentiate SS from MF and inflammatory skin diseases and monitor disease progression (107). A broad spectrum of genes, including DNMT3A (109), TET2 (110), CREBBP, MLL, SETDA/B, KDM6B (111, 112), BRD9, SMARCA4, and NuRD (CHD3) (113), involved in epigenetic regulation has been consistently associated with loss-of-function mutations or deletions in CTCL, according to several investigations of whole-genome sequencing (Table 4). DNMT3A and TET, which have been frequently reported to be deleted in CTCL, are tumor suppressor genes that play a crucial role in the malignant transformation of mature T cells (114). These genes involved in chromatin modifications were also reported to be highly mutated. ARID1A, which has also been repeatedly shown to be deleted together with ARID5B or SMARC in SS, is a component of the SWI/SNF chromatin remodeling complex and functions as an epigenetic tumor suppressor in CTCL (111, 112). KDM6A, CREBBP, and SETDB2, which are histone-modifying genes, were also reported to be highly mutated in CTCL, based on genomic analysis of 220 cases of CTCLs (115). Both genomic instability and transcriptional dysregulation lead to malignant T-cell transformation and CTCL progression, mostly due to alterations in methylation. Specifically, hypermethylation of the IL-15 promoter region was shown to prevent the binding of the transcriptional repressor Zeb1, increasing the transcription of IL-15 and subsequently initiating CTCL pathogenesis (8).
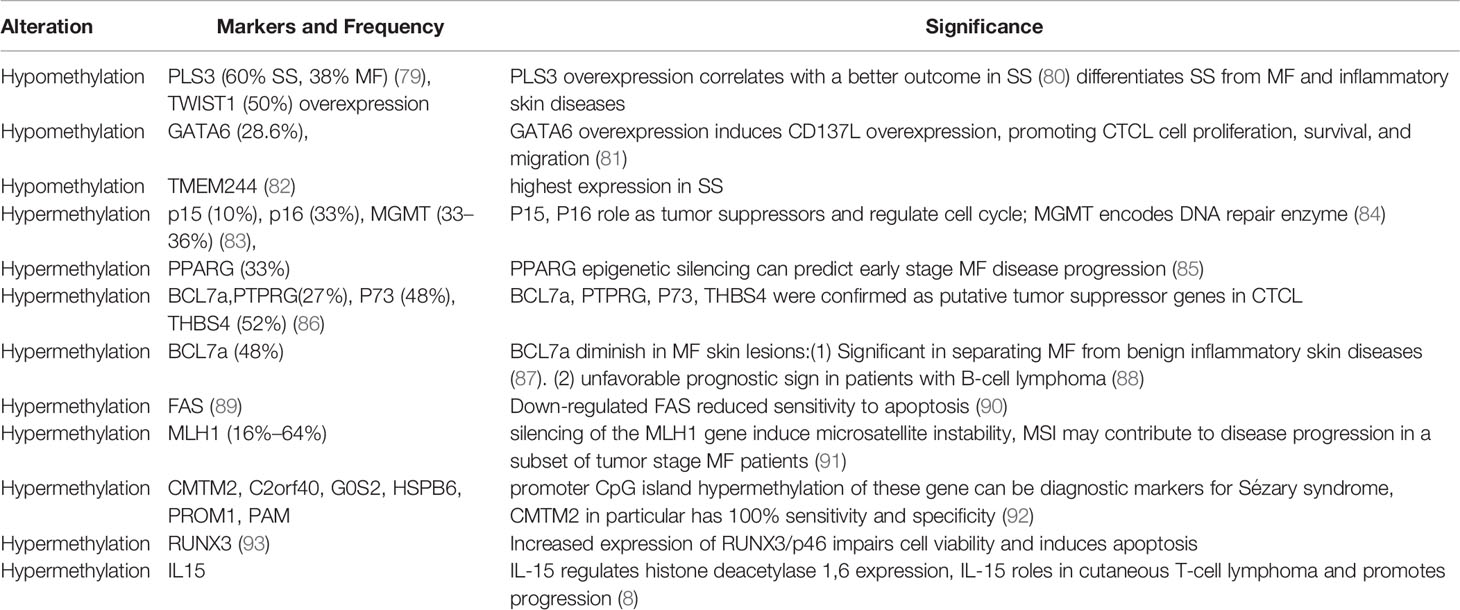
Table 3 Distinct methylation gene of cutaneous T-cell lymphomas and significance [partly refer to Iżykowska K et al. (78)].
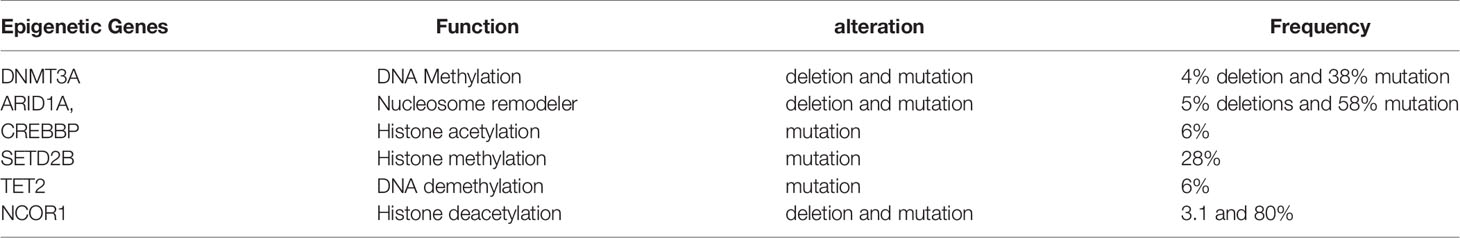
Table 4 The mutational frequency of epigenetic related gene in CTCL (according to Park J et al. genomic analysis of 220 CTCLs).
DNA Methylation for Dysregulated miRNAs in CTCL
MiRNAs are a set of small (18–25 bp), single-stranded RNA molecules that regulate gene expression at the post-transcriptional level. With the use of quantitative real-time PCR (RT-qPCR) and comparative genomic hybridization techniques, a differentially expressed miRNA profile has been defined in CTCL (116). Aberrant miRNA levels are common in CTCL. Many investigations have validated miRNA classifiers that can discriminate CTCL from benign inflammation (117) and predict disease progression (118, 119) and prognosis in patients with CTCL (120, 121). Many studies have shown that miRNAs are involved in many signaling pathways that regulate the cell cycle (122) and apoptosis resistance, particularly the Notch, STAT, and (123) NFκB pathogenic pathways. The promoter regions of miR-200c and miR-124-2/3 were reported to be hypermethylated in MF tumor stage (MFt). The repression of miR-200c elevated the expression of Jagged1, contributing to Notch activation in MFt (124). STAT3 is also frequently dysregulated in CTCL (112). STAT3 activation plays a role in CTCL pathogenesis (125) and progression (126) and large-cell transformations (127) MiRNAs, such as miR-337 (123) and miR-124 (128), mediate the expression of STAT3. In particular, miR-124 silencing caused by the hypermethylation of the miR-124 promoter region was shown to increase STAT3 levels in CTCL. Therefore, dysregulated miRNAs are thought to be involved in the pathogenesis of CTCL (129), as miRNA dysregulation is related to miRNA promoter methylation, which has been shown to induce the downregulation of miRNA expression (130). Specifically, DNA methylation in miR-10b, miR-193b, and miR-141 promoter regions was reported to downregulate miRNA expression in CTCL.
Discussion
Accumulated evidence suggests that intrinsically epigenetic events participate in CTCL malignant transformation and disease progression. Previous reports have identified histone modifications, miRNA regulation, chromatin accessibility aberrations, and abnormal DNA methylation signatures in advanced CTCL, including MF and SS, suggesting that epigenetic dysregulation contributes to the pathogenesis and progression of MF/SS. The expression of epigenetic markers by malignant T cells in CTCL has been extensively exploited to evaluate the role of these markers in diagnosing and managing MF/SS. Stage progression and HDACi resistance have also been discovered to be associated with intratumoral heterogeneity and divergent subclonal evolution. Furthermore, whole‐genome sequencing and single-cell sequencing have been employed to characterize the intratumoral transcriptional heterogeneity of malignant CD4+ T cells, revealing the mechanism of HDACi resistance at the molecular level. Currently, the mechanisms of HDACis are not clear; however, there has been increasing evidence of possible resistance mechanisms. HDACi therapy has already been shown to have positive effects on aggressive CTCL types. HDACis in combination with other therapy, such as chemotherapeutic drugs, immunomodulatory drugs, monoclonal antibody, may provide a novel treatment option that can improve clinical outcomes in patients with CTCL (34, 35, 57, 70, 131–133) (Table 2). Currently, more and more HDACI combination therapy regimens are undergoing clinical trials, indicating the important role of HDACi drug in the treatment of CTCL. Indeed, studies on the epigenetic changes in CTCL contribute not only to a comprehensive understanding of CTCL but also to drug development. Moreover, efforts to further elucidate and validate the diagnostic, prognostic, and predictive epigenetic biomarkers for CTCL can facilitate early diagnosis, risk assessment of disease progression, and prediction of treatment outcomes in CTCL. Furthermore, the heterogeneity and diversity of CTCLs allow us to better understand the limitations of HDACi therapy and to optimize HDACi combination therapy.
Author Contributions
PZ searched and collected papers on Pubmed and wrote the paper. MZ was responsible for reviewing and providing guidance. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Science and Technology Major Project of China (Grant No. 2020ZX09201- 009). This study was supported by National natural Science Foundation of China (81970184, U1904139) and Department of Science & Technology of Henan province (182102310114).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was supported by the Oncology Department of the First Affiliated Hospital of Zhengzhou University and the Medical Science Academy of Zhengzhou University, and I would like to show great gratitude to them all. We would like to thank Editage (www.editage.com) for English language editing.
References
1. Hristov AC, Tejasvi T, Wilcox RA. Mycosis Fungoides and Sézary Syndrome: 2019 Update on Diagnosis, Risk-Stratification, and Management. Am J Hematol (2019) 94:1027–41. doi: 10.1002/ajh.25577
2. Phyo ZH, Shanbhag S, Rozati S. Update on Biology of Cutaneous T-Cell Lymphoma. Front Oncol (2020) 10:765. doi: 10.3389/fonc.2020.00765
3. Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, Swerdlow SH, et al. The 2018 Update of the WHO-EORTC Classification for Primary Cutaneous Lymphomas. Blood (2019) 133:1703–14. doi: 10.1182/blood-2018-11-881268
4. Scarisbrick JJ, Prince HM, Vermeer MH, Quaglino P, Horwitz S, Porcu P, et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J Clin Oncol (2015) 33:3766–73. doi: 10.1200/JCO.2015.61.7142
5. Walia R, Yeung C. An Update on Molecular Biology of Cutaneous T Cell Lymphoma. Front Oncol (2019) 9:1558. doi: 10.3389/fonc.2019.01558
6. El Tawdy A, Amin I, Abdel Hay R, Rashed L, Gad Z. Assessment of Tissue Level of Histone Deactylase-2 (Hdac-2) in Patients With Mycosis Fungoides. J Cutan Med Surg (2016) 20:40–3. doi: 10.1177/1203475415589055
7. Marquard L, Gjerdrum LM, Christensen IJ, Jensen PB, Sehested M, Ralfkiaer E. Prognostic Significance of the Therapeutic Targets Histone Deacetylase 1, 2, 6 and Acetylated Histone H4 in Cutaneous T-Cell Lymphoma. Histopathology (2008) 53:267–77. doi: 10.1111/j.0309-0167.2008.03109.x
8. Mishra A, La Perle K, Kwiatkowski S, Sullivan LA, Sams GH, Johns J, et al. Mechanism, Consequences, and Therapeutic Targeting of Abnormal Il15 Signaling in Cutaneous T-Cell Lymphoma. Cancer Discovery (2016) 6:986–1005. doi: 10.1158/2159-8290.CD-15-1297
9. Fehniger TA, Suzuki K, Ponnappan A, VanDeusen JB, Cooper MA, Florea SM, et al. Fatal Leukemia in Interleukin 15 Transgenic Mice Follows Early Expansions in Natural Killer and Memory Phenotype CD8+ T Cells. J Exp Med (2001) 193:219–31. doi: 10.1084/jem.193.2.219
10. Leroy S, Dubois S, Tenaud I, Chebassier N, Godard A, Jacques Y, et al. Interleukin-15 Expression in Cutaneous T-Cell Lymphoma (Mycosis Fungoides and Sézary Syndrome). Br J Dermatol (2001) 144:1016–23. doi: 10.1046/j.1365-2133.2001.04192.x
11. Asadullah K, Haeussler-Quade A, Gellrich S, Hanneken S, Hansen-Hagge TE, Döcke WD, et al. Il-15 and IL-16 Overexpression in Cutaneous T-Cell Lymphomas: Stage-Dependent Increase in Mycosis Fungoides Progression. Exp Dermatol (2000) 9:248–51. doi: 10.1034/j.1600-0625.2000.009004248.x
12. Bobrowicz M, Slusarczyk A, Domagala J, Dwojak M, Ignatova D, Chang YT, et al. Selective Inhibition of HDAC6 Sensitizes Cutaneous T-Cell Lymphoma to PI3K Inhibitors. Oncol Lett (2020) 20:533–40. doi: 10.3892/ol.2020.11587
13. Lopez AT, Bates S, Geskin L. Current Status of HDAC Inhibitors in Cutaneous T-Cell Lymphoma. Am J Clin Dermatol (2018) 19:805–19. doi: 10.1007/s40257-018-0380-7
14. Iaconelli J, Lalonde J, Watmuff B, Liu B, Mazitschek R, Haggarty SJ, et al. Lysine Deacetylation by HDAC6 Regulates the Kinase Activity of AKT in Human Neural Progenitor Cells. ACS Chem Biol (2017) 12:2139–48. doi: 10.1021/acschembio.6b01014
15. Stengel KR, Zhao Y, Klus NJ, Kaiser JF, Gordy LE, Joyce S, et al. Histone Deacetylase 3 Is Required for Efficient T Cell Development. Mol Cell Biol (2015) 35:3854–65. doi: 10.1128/MCB.00706-15
16. Wells CE, Bhaskara S, Stengel KR, Zhao Y, Sirbu B, Chagot B, et al. Inhibition of Histone Deacetylase 3 Causes Replication Stress in Cutaneous T Cell Lymphoma. PloS One (2013) 8:e68915. doi: 10.1371/journal.pone.0068915
17. Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II Multi-Institutional Trial of the Histone Deacetylase Inhibitor Romidepsin as Monotherapy for Patients With Cutaneous T-Cell Lymphoma. J Clin Oncol (2009) 27:5410–7. doi: 10.1200/JCO.2008.21.6150
18. Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final Results From a Multicenter, International, Pivotal Study of Romidepsin in Refractory Cutaneous T-Cell Lymphoma. J Clin Oncol (2010) 28:4485–91. doi: 10.1200/JCO.2010.28.9066
19. Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase IIb Multicenter Trial of Vorinostat in Patients With Persistent, Progressive, or Treatment Refractory Cutaneous T-Cell Lymphoma. J Clin Oncol (2007) 25:3109–15. doi: 10.1200/JCO.2006.10.2434
20. Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, et al. Phase 2 Trial of Oral Vorinostat (Suberoylanilide Hydroxamic Acid, SAHA) for Refractory Cutaneous T-Cell Lymphoma (CTCL). Blood (2007) 109:31–9. doi: 10.1182/blood-2006-06-025999
21. Foss F, Advani R, Duvic M, Hymes KB, Intragumtornchai T, Lekhakula A, et al. A Phase II Trial of Belinostat (PXD101) in Patients With Relapsed or Refractory Peripheral or Cutaneous T-Cell Lymphoma. Br J Haematol (2015) 168:811–9. doi: 10.1111/bjh.13222
22. Duvic M, Dummer R, Becker JC, Poulalhon N, Ortiz Romero P, Grazia Bernengo M, et al. Panobinostat Activity in Both Bexarotene-Exposed and -Naïve Patients With Refractory Cutaneous T-Cell Lymphoma: Results of a Phase II Trial. Eur J Cancer (2013) 49:386–94. doi: 10.1016/j.ejca.2012.08.017
23. Bates SE, Eisch R, Ling A, Rosing D, Turner M, Pittaluga S, et al. Romidepsin in Peripheral and Cutaneous T-Cell Lymphoma: Mechanistic Implications From Clinical and Correlative Data. Br J Haematol (2015) 170:96–109. doi: 10.1111/bjh.13400
24. Duvic M, Bates SE, Piekarz R, Eisch R, Kim YH, Lerner A, et al. Responses to Romidepsin in Patients With Cutaneous T-Cell Lymphoma and Prior Treatment With Systemic Chemotherapy. Leuk Lymphoma (2018) 59:880–7. doi: 10.1080/10428194.2017.1361022
25. Shimony S, Horowitz N, Ribakovsky E, Rozovski U, Avigdor A, Zloto K, et al. Romidepsin Treatment for Relapsed or Refractory Peripheral and Cutaneous T-Cell Lymphoma: Real-Life Data From a National Multicenter Observational Study. Hematol Oncol (2019) 37:569–77. doi: 10.1002/hon.2691
26. Hughes CF, Khot A, McCormack C, Lade S, Westerman DA, Twigger R, et al. Lack of Durable Disease Control With Chemotherapy for Mycosis Fungoides and Sézary Syndrome: A Comparative Study of Systemic Therapy. Blood (2015) 125:71–81. doi: 10.1182/blood-2014-07-588236
27. Martinez-Escala ME, Kuzel TM, Kaplan JB, Petrich A, Nardone B, Rosen ST, et al. Durable Responses With Maintenance Dose-Sparing Regimens of Romidepsin in Cutaneous T-Cell Lymphoma. JAMA Oncol (2016) 2:790–3. doi: 10.1001/jamaoncol.2016.0004
28. Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, et al. Vorinostat for Treatment of Cutaneous Manifestations of Advanced Primary Cutaneous T-Cell Lymphoma. Clin Cancer Res (2007) 13:2318–22. doi: 10.1158/1078-0432.CCR-06-2672
29. Wada H, Tsuboi R, Kato Y, Sugaya M, Tobinai K, Hamada T, et al. Phase I and Pharmacokinetic Study of the Oral Histone Deacetylase Inhibitor Vorinostat in Japanese Patients With Relapsed or Refractory Cutaneous T-Cell Lymphoma. J Dermatol (2012) 39:823–8. doi: 10.1111/j.1346-8138.2012.01554.x
30. Duvic M, Vu J. Vorinostat: A New Oral Histone Deacetylase Inhibitor Approved for Cutaneous T-Cell Lymphoma. Expert Opin Investig Drugs (2007) 16:1111–20. doi: 10.1517/13543784.16.7.1111
31. Kogge A, Volteau C, Saint-Jean M, Peuvrel L, Brocard A, Knol AC, et al. Vorinostat for Refractory or Relapsing Epidermotropic T-Cell Lymphoma: A Retrospective Cohort Study of 15 Patients. Acta Derm Venereol (2015) 95:72–7. doi: 10.2340/00015555-1886
32. Fu W, Yi S, Qiu L, Sun J, Tu P, Wang Y. Bcl11b-Mediated Epigenetic Repression Is a Crucial Target for Histone Deacetylase Inhibitors in Cutaneous T-Cell Lymphoma. J Invest Dermatol (2017) 137:1523–32. doi: 10.1016/j.jid.2017.02.980
33. Nihal M, Ahmad N, Wood GS. SIRT1 is Upregulated in Cutaneous T-Cell Lymphoma, and its Inhibition Induces Growth Arrest and Apoptosis. Cell Cycle (2014) 13:632–40. doi: 10.4161/cc.27523
34. Ragheb R, Venton G, Chelbi R, Bonnet N, Le Treut T, Ivanov V, et al. Vorinostat and Mithramycin A in Combination Therapy as an Interesting Strategy for the Treatment of Sézary T Lymphoma: A Transcriptomic Approach. Arch Dermatol Res (2017) 309:611–23. doi: 10.1007/s00403-017-1761-0
35. Cyrenne BM, Lewis JM, Weed JG, Carlson KR, Mirza FN, Foss FM, et al. Synergy of BCL2 and Histone Deacetylase Inhibition Against Leukemic Cells From Cutaneous T-Cell Lymphoma Patients. Blood (2017) 130:2073–83. doi: 10.1182/blood-2017-06-792150
36. Wobser M, Weber A, Glunz A, Tauch S, Seitz K, Butelmann T, et al. Elucidating the Mechanism of Action of Domatinostat (4SC-202) in Cutaneous T Cell Lymphoma Cells. J Hematol Oncol (2019) 12:30. doi: 10.1186/s13045-019-0719-4
37. Trager MH, Geskin LJ. Current Status of Histone Deacetylase Inhibitors in Cutaneous T-Cell Lymphoma. G Ital Dermatol Venereol (2019) 154:681–95. doi: 10.23736/S0392-0488.19.06503-9
38. Epping MT, Wang L, Plumb JA, Lieb M, Gronemeyer H, Brown R, et al. A Functional Genetic Screen Identifies Retinoic Acid Signaling as a Target of Histone Deacetylase Inhibitors. Proc Natl Acad Sci USA (2007) 104:17777–82. doi: 10.1073/pnas.0702518104
39. Dummer R, Beyer M, Hymes K, Epping MT, Bernards R, Steinhoff M, et al. Vorinostat Combined With Bexarotene for Treatment of Cutaneous T-Cell Lymphoma: In Vitro and Phase I Clinical Evidence Supporting Augmentation of Retinoic Acid Receptor/Retinoid X Receptor Activation by Histone Deacetylase Inhibition. Leuk Lymphoma (2012) 53:1501–8. doi: 10.3109/10428194.2012.656625
40. Rangwala S, Zhang C, Duvic M. HDAC Inhibitors for the Treatment of Cutaneous T-Cell Lymphomas. Future Med Chem (2012) 4:471–86. doi: 10.4155/fmc.12.6
41. Gardner JM, Introcaso CE, Nasta SD, Kim EJ, Vittorio CC, Rook AH. A Novel Regimen of Vorinostat With Interferon Gamma for Refractory Sézary Syndrome. J Am Acad Dermatol (2009) 61:112–6. doi: 10.1016/j.jaad.2008.11.889
42. Samimi S, Morrissey K, Anshelevich S, Evans K, Gardner J, Musiek A, et al. Romidepsin and Interferon Gamma: A Novel Combination for Refractory Cutaneous T-Cell Lymphoma. J Am Acad Dermatol (2013) 68:e5–6. doi: 10.1016/j.jaad.2011.06.043
43. Kato Y, Egusa C, Maeda T, Tsuboi R. Combination of Retinoid and Histone Deacetylase Inhibitor Produced an Anti-Tumor Effect in Cutaneous T-Cell Lymphoma by Restoring Tumor Suppressor Gene, Retinoic Acid Receptorβ2, Via Histone Acetylation. J Dermatol Sci (2016) 81:17–25. doi: 10.1016/j.jdermsci.2015.10.016
44. Sung JJ, Ververis K, Karagiannis TC. Histone Deacetylase Inhibitors Potentiate Photochemotherapy in Cutaneous T-Cell Lymphoma MyLa Cells. J Photochem Photobiol B (2014) 131:104–12. doi: 10.1016/j.jphotobiol.2014.01.009
45. Sanli H, Akay BN, Anadolu R, Ozcan M, Saral S, Akyol A. The Efficacy of Vorinostat in Combination With Interferon Alpha and Extracorporeal Photopheresis in Late Stage Mycosis Fungoides and Sezary Syndrome. J Drugs Dermatol (2011) 10:403–8.
46. Wozniak MB, Villuendas R, Bischoff JR, Aparicio CB, Martínez Leal JF, de La Cueva P, et al. Vorinostat Interferes With the Signaling Transduction Pathway of T-Cell Receptor and Synergizes With Phosphoinositide-3 Kinase Inhibitors in Cutaneous T-Cell Lymphoma. Haematologica (2010) 95(4):613–21. doi: 10.3324/haematol.2009.013870
47. Heider U, Rademacher J, Lamottke B, Mieth M, Moebs M, von Metzler I, et al. Synergistic Interaction of the Histone Deacetylase Inhibitor SAHA With the Proteasome Inhibitor Bortezomib in Cutaneous T Cell Lymphoma. Eur J Haematol (2009) 82:440–9. doi: 10.1111/j.1600-0609.2009.01239.x
48. Rozati S, Cheng PF, Widmer DS, Fujii K, Levesque MP, Dummer R. Romidepsin and Azacitidine Synergize in Their Epigenetic Modulatory Effects to Induce Apoptosis in CTCL. Clin Cancer Res (2016) 22:2020–31. doi: 10.1158/1078-0432.CCR-15-1435
49. Akilov OE, Grant C, Frye R, Bates S, Piekarz R, Geskin LJ. Low-Dose Electron Beam Radiation and Romidepsin Therapy for Symptomatic Cutaneous T-Cell Lymphoma Lesions. Br J Dermatol (2012) 167:194–7. doi: 10.1111/j.1365-2133.2012.10905.x
50. Papps T, McCormack C, Buelens O, Van der Weyden C, Twigger R, Campbell BA, et al. A Comparative Analysis of Histone Deacetylase Inhibitors for the Treatment of Mycosis Fungoides and Sézary Syndrome. Br J Dermatol (2020) 182:497–8. doi: 10.1111/bjd.18522
51. Noonan AM, Eisch RA, Liewehr DJ, Sissung TM, Venzon DJ, Flagg TP, et al. Electrocardiographic Studies of Romidepsin Demonstrate Its Safety and Identify a Potential Role for K(ATP) Channel. Clin Cancer Res (2013) 19:3095–104. doi: 10.1158/1078-0432.CCR-13-0109
52. Zinzani PL, Bonthapally V, Huebner D, Lutes R, Chi A, Pileri S. Panoptic Clinical Review of the Current and Future Treatment of Relapsed/Refractory T-Cell Lymphomas: Cutaneous T-Cell Lymphomas. Crit Rev Oncol Hematol (2016) 99:228–40. doi: 10.1016/j.critrevonc.2015.12.018
53. Kim EJ, Kim YH, Rook AH, Lerner A, Duvic M, Reddy S, et al. Clinically Significant Responses Achieved With Romidepsin Across Disease Compartments in Patients With Cutaneous T-Cell Lymphoma. Leuk Lymphoma (2015) 56:2847–54. doi: 10.3109/10428194.2015.1014360
54. Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, et al. Mogamulizumab Versus Vorinostat in Previously Treated Cutaneous T-Cell Lymphoma (MAVORIC): An International, Open-Label, Randomised, Controlled Phase 3 Trial. Lancet Oncol (2018) 19:1192–204. doi: 10.1016/S1470-2045(18)30379-6
55. Porcu P, Hudgens S, Horwitz S, Quaglino P, Cowan R, Geskin L, et al. Quality of Life Effect of the Anti-CCR4 Monoclonal Antibody Mogamulizumab Versus Vorinostat in Patients With Cutaneous T-Cell Lymphoma. Clin Lymphoma Myeloma Leuk (2021) 21(2):97–105. doi: 10.1016/j.clml.2020.09.003
56. Chen L, Carson KR, Staser KW, Mehta-Shah N, Schaffer A, Rosman IS, et al. Mogamulizumab-Associated Cutaneous Granulomatous Drug Eruption Mimicking Mycosis Fungoides But Possibly Indicating Durable Clinical Response. JAMA Dermatol (2019) 155(8):968–71. doi: 10.1001/jamadermatol.2019.0369
57. Zhao L, Okhovat JP, Hong EK, Kim YH, Wood GS. Preclinical Studies Support Combined Inhibition of BET Family Proteins and Histone Deacetylases as Epigenetic Therapy for Cutaneous T-Cell Lymphoma. Neoplasia (2019) 21:82–92. doi: 10.1016/j.neo.2018.11.006
58. Rassek K, Iżykowska K. Single-Cell Heterogeneity of Cutaneous T-Cell Lymphomas Revealed Using Rna-Seq Technologies. Cancers (Basel) (2020) 12(8):2129. doi: 10.3390/cancers12082129
59. Zhang C, Richon V, Ni X, Talpur R, Duvic M. Selective Induction of Apoptosis by Histone Deacetylase Inhibitor SAHA in Cutaneous T-Cell Lymphoma Cells: Relevance to Mechanism of Therapeutic Action. J Invest Dermatol (2005) 125:1045–52. doi: 10.1111/j.0022-202X.2005.23925.x
60. Smolewski P, Robak T. The Discovery and Development of Romidepsin for the Treatment of T-Cell Lymphoma. Expert Opin Drug Discovery (2017) 12:859–73. doi: 10.1080/17460441.2017.1341487
61. Jain S, Zain J, O’Connor O. Novel Therapeutic Agents for Cutaneous T-Cell Lymphoma. J Hematol Oncol (2012) 5:24. doi: 10.1186/1756-8722-5-24
62. Chüeh AC, Tse J, Dickinson M, Ioannidis P, Jenkins L, Togel L, et al. Atf3 Repression of BCL-X(L) Determines Apoptotic Sensitivity to HDAC Inhibitors Across Tumor Types. Clin Cancer Res (2017) 23:5573–84. doi: 10.1158/1078-0432.CCR-17-0466
63. Yu X, Li H, Zhu M, Hu P, Liu X, Qing Y, et al. Involvement of P53 Acetylation in Growth Suppression of Cutaneous T-Cell Lymphomas Induced by HDAC Inhibition. J Invest Dermatol (2020) 140:2009–22.e4. doi: 10.1016/j.jid.2019.12.041
64. Butler RM, McKenzie RC, Jones CL, Flanagan CE, Woollard WJ, Demontis M, et al. Contribution of STAT3 and RAD23B in Primary Sézary Cells to Histone Deacetylase Inhibitor Fk228 Resistance. J Invest Dermatol (2019) 139:1975–84.e2. doi: 10.1016/j.jid.2019.03.1130
65. Fantin VR, Loboda A, Paweletz CP, Hendrickson RC, Pierce JW, Roth JA, et al. Constitutive Activation of Signal Transducers and Activators of Transcription Predicts Vorinostat Resistance in Cutaneous T-Cell Lymphoma. Cancer Res (2008) 68:3785–94. doi: 10.1158/0008-5472.CAN-07-6091
66. Fotheringham S, Epping MT, Stimson L, Khan O, Wood V, Pezzella F, et al. Genome-Wide Loss-of-Function Screen Reveals an Important Role for the Proteasome in HDAC Inhibitor-Induced Apoptosis. Cancer Cell (2009) 15:57–66. doi: 10.1016/j.ccr.2008.12.001
67. Khan O, Fotheringham S, Wood V, Stimson L, Zhang C, Pezzella F, et al. HR23B is a Biomarker for Tumor Sensitivity to HDAC Inhibitor-Based Therapy. Proc Natl Acad Sci USA (2010) 107:6532–7. doi: 10.1073/pnas.0913912107
68. Andrews JM, Schmidt JA, Carson KR, Musiek AC, Mehta-Shah N, Payton JE. Novel Cell Adhesion/Migration Pathways are Predictive Markers of HDAC Inhibitor Resistance in Cutaneous T Cell Lymphoma. EBioMedicine (2019) 46:170–83. doi: 10.1016/j.ebiom.2019.07.053
69. Ott CJ, Wu CJ. Hdac Inhibitors Finally Open Up: Chromatin Accessibility Signatures of CTCL. Cancer Cell (2017) 32:1–3. doi: 10.1016/j.ccell.2017.06.008
70. Chakraborty AR, Robey RW, Luchenko VL, Zhan Z, Piekarz RL, Gillet JP, et al. MAPK Pathway Activation Leads to Bim Loss and Histone Deacetylase Inhibitor Resistance: Rationale to Combine Romidepsin With an MEK Inhibitor. Blood (2013) 121:4115–25. doi: 10.1182/blood-2012-08-449140
71. Qu K, Zaba LC, Satpathy AT, Giresi PG, Li R, Jin Y, et al. Chromatin Accessibility Landscape of Cutaneous T Cell Lymphoma and Dynamic Response to HDAC Inhibitors. Cancer Cell (2017) 32:27–41.e4. doi: 10.1016/j.ccell.2017.05.008
72. Borcherding N, Voigt AP, Liu V, Link BK, Zhang W, Jabbari A. Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated With Disease Progression. Clin Cancer Res (2019) 25:2996–3005. doi: 10.1158/1078-0432.CCR-18-3309
73. Buus TB, Willerslev-Olsen A, Fredholm S, Blümel E, Nastasi C, Gluud M, et al. Single-Cell Heterogeneity in Sézary Syndrome. Blood Adv (2018) 2:2115–26. doi: 10.1182/bloodadvances.2018022608
74. Hanel W, Briski R, Ross CW, Anderson TF, Kaminski MS, Hristov AC, et al. A Retrospective Comparative Outcome Analysis Following Systemic Therapy in Mycosis Fungoides and Sezary Syndrome. Am J Hematol (2016) 91:E491–491E495. doi: 10.1002/ajh.24564
75. Méhul B, Perrin A, Grisendi K, Galindo AN, Dayon L, Ménigot C, et al. Mass Spectrometry and DigiWest Technology Emphasize Protein Acetylation Profile From Quisinostat-Treated Hut78 CTCL Cell Line. J Proteomics (2018) 187:126–43. doi: 10.1016/j.jprot.2018.07.003
76. Whittaker S. Global Patterns of Methylation In Sézary Syndrome Provide Insight Into The Role of Epigenetics In Cutaneous T-Cell Lymphoma. J Invest Dermatol (2016) 136:1753–4. doi: 10.1016/j.jid.2016.05.114
77. de Silva S, Wang F, Hake TS, Porcu P, Wong HK, Wu L. Downregulation of SAMHD1 Expression Correlates With Promoter DNA Methylation in Sézary Syndrome Patients. J Invest Dermatol (2014) 134:562–5. doi: 10.1038/jid.2013.311
78. Iżykowska K. Methylation Patterns of Cutaneous T-Cell Lymphomas. Exp Dermatol (2020). doi: 10.1111/exd.14108
79. Wong HK, Gibson H, Hake T, Geyer S, Frederickson J, Marcucci G, et al. Promoter-Specific Hypomethylation is Associated With Overexpression of PLS3, GATA6, and TWIST1 in the Sezary Syndrome. J Invest Dermatol (2015) 135:2084–92. doi: 10.1038/jid.2015.116
80. Boonk SE, Zoutman WH, Putter H, Ram-Wolff C, Felcht M, Klemke CD, et al. Increased Expression of PLS3 Correlates With Better Outcome in Sézary Syndrome. J Invest Dermatol (2017) 137:754–7. doi: 10.1016/j.jid.2016.10.025
81. Kamijo H, Miyagaki T, Shishido-Takahashi N, Nakajima R, Oka T, Suga H, et al. Aberrant CD137 Ligand Expression Induced by GATA6 Overexpression Promotes Tumor Progression in Cutaneous T-Cell Lymphoma. Blood (2018) 132:1922–35. doi: 10.1182/blood-2018-04-845834
82. Iżykowska K, Rassek K, Żurawek M, Nowicka K, Paczkowska J, Ziółkowska-Suchanek I, et al. Hypomethylation of the Promoter Region Drives Ectopic Expression of TMEM244 in Sézary Cells. J Cell Mol Med (2020) 24:10970–7. doi: 10.1111/jcmm.15729
83. Scarisbrick JJ, Woolford AJ, Calonje E, Photiou A, Ferreira S, Orchard G, et al. Frequent Abnormalities of the p15 and p16 Genes in Mycosis Fungoides and Sezary Syndrome. J Invest Dermatol (2002) 118:493–9. doi: 10.1046/j.0022-202x.2001.01682.x
84. Gallardo F, Esteller M, Pujol RM, Costa C, Estrach T, Servitje O. Methylation Status of the p15, p16 and MGMT Promoter Genes in Primary Cutaneous T-Cell Lymphomas. Haematologica (2004) 89:1401–3.
85. Ferrara G, Pancione M, Votino C, Quaglino P, Tomasini C, Santucci M, et al. A Specific DNA Methylation Profile Correlates With a High Risk of Disease Progression in Stage I Classical (Alibert-Bazin Type) Mycosis Fungoides. Br J Dermatol (2014) 170:1266–75. doi: 10.1111/bjd.12717
86. van Doorn R, Zoutman WH, Dijkman R, de Menezes RX, Commandeur S, Mulder AA, et al. Epigenetic Profiling of Cutaneous T-Cell Lymphoma: Promoter Hypermethylation of Multiple Tumor Suppressor Genes Including BCL7a, PTPRG, and P73. J Clin Oncol (2005) 23:3886–96. doi: 10.1200/JCO.2005.11.353
87. Tracey L, Villuendas R, Dotor AM, Spiteri I, Ortiz P, Garcia JF, et al. Mycosis Fungoides Shows Concurrent Deregulation of Multiple Genes Involved in the TNF Signaling Pathway: An Expression Profile Study. Blood (2003) 102:1042–50. doi: 10.1182/blood-2002-11-3574
88. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature (2000) 403:503–11. doi: 10.1038/35000501
89. Jones CL, Wain EM, Chu CC, Tosi I, Foster R, McKenzie RC, et al. Downregulation of Fas Gene Expression in Sézary Syndrome Is Associated With Promoter Hypermethylation. J Invest Dermatol (2010) 130:1116–25. doi: 10.1038/jid.2009.301
90. Wu J, Nihal M, Siddiqui J, Vonderheid EC, Wood GS. Low FAS/CD95 Expression by CTCL Correlates With Reduced Sensitivity to Apoptosis That can be Restored by FAS Upregulation. J Invest Dermatol (2009) 129:1165–73. doi: 10.1038/jid.2008.309
91. Scarisbrick JJ, Mitchell TJ, Calonje E, Orchard G, Russell-Jones R, Whittaker SJ. Microsatellite Instability is Associated With Hypermethylation of the hMLH1 Gene and Reduced Gene Expression in Mycosis Fungoides. J Invest Dermatol (2003) 121:894–901. doi: 10.1046/j.1523-1747.2003.12496.x
92. van Doorn R, Slieker RC, Boonk SE, Zoutman WH, Goeman JJ, Bagot M, et al. Epigenomic Analysis of Sézary Syndrome Defines Patterns of Aberrant DNA Methylation and Identifies Diagnostic Markers. J Invest Dermatol (2016) 136:1876–84. doi: 10.1016/j.jid.2016.03.042
93. Haider A, Steininger A, Ullmann R, Hummel M, Dimitrova L, Beyer M, et al. Inactivation of RUNX3/p46 Promotes Cutaneous T-Cell Lymphoma. J Invest Dermatol (2016) 136:2287–96. doi: 10.1016/j.jid.2016.05.126
94. Contassot E, French LE. Epigenetic Causes of Apoptosis Resistance in Cutaneous T-Cell Lymphomas. J Invest Dermatol (2010) 130:922–4. doi: 10.1038/jid.2009.427
95. Wang Y, Gu X, Zhang G, Wang L, Wang T, Zhao Y, et al. SATB1 Overexpression Promotes Malignant T-Cell Proliferation in Cutaneous CD30+ Lymphoproliferative Disease by Repressing P21. Blood (2014) 123:3452–61. doi: 10.1182/blood-2013-10-534693
96. Fredholm S, Willerslev-Olsen A, Met Ö, Kubat L, Gluud M, Mathiasen SL, et al. SATB1 in Malignant T Cells. J Invest Dermatol (2018) 138:1805–15. doi: 10.1016/j.jid.2018.03.1526
97. Wang Y, Su M, Zhou LL, Tu P, Zhang X, Jiang X, et al. Deficiency of SATB1 Expression in Sezary Cells Causes Apoptosis Resistance by Regulating FasL/CD95L Transcription. Blood (2011) 117:3826–35. doi: 10.1182/blood-2010-07-294819
98. Herrera A, Fredholm S, Cheng A, Mimitou EP, Seffens A, Bar-Natan M, et al. Low SATB1 Expression Promotes IL-5 and IL-9 Expression in Sézary Syndrome. J Invest Dermatol (2020) 140:713–6. doi: 10.1016/j.jid.2019.07.714
99. Poglio S, Merlio JP. Satb1 Is a Pivotal Epigenetic Biomarker in Cutaneous T-Cell Lymphomas. J Invest Dermatol (2018) 138:1694–6. doi: 10.1016/j.jid.2018.04.018
100. Harro CM, Perez-Sanz J, Costich TL, Payne KK, Anadon CM, Chaurio RA, et al. Methyltransferase Inhibitors Restore SATB1 Protective Activity Against Cutaneous T Cell Lymphoma in Mice. J Clin Invest (2021) 131(3):e135711. doi: 10.1172/JCI135711
101. Gao Y, Liu F, Sun J, Wen Y, Tu P, Kadin ME, et al. Differential SATB1 Expression Reveals Heterogeneity of Cutaneous T-Cell Lymphoma. J Invest Dermatol (2020) 141(3):607–18.e6. doi: 10.1016/j.jid.2020.05.120
102. Grzanka D, Gagat M, Izdebska M, Marszałek A. Expression of Special AT-rich Sequence-Binding Protein 1 Is an Independent Prognostic Factor in Cutaneous T-Cell Lymphoma. Oncol Rep (2015) 33:250–66. doi: 10.3892/or.2014.3597
103. van Doorn R. Mycosis Fungoides: Promoter Hypermethylation Predicts Disease Progression. Br J Dermatol (2014) 170:1216. doi: 10.1111/bjd.12870
104. Michel L, Jean-Louis F, Begue E, Bensussan A, Bagot M. Use of PLS3, Twist, CD158k/KIR3DL2, and NKp46 Gene Expression Combination for Reliable Sézary Syndrome Diagnosis. Blood (2013) 121:1477–8. doi: 10.1182/blood-2012-10-460535
105. Boonk SE, Zoutman WH, Marie-Cardine A, van der Fits L, Out-Luiting JJ, Mitchell TJ, et al. Evaluation of Immunophenotypic and Molecular Biomarkers for Sézary Syndrome Using Standard Operating Procedures: A Multicenter Study of 59 Patients. J Invest Dermatol (2016) 136:1364–72. doi: 10.1016/j.jid.2016.01.038
106. Nebozhyn M, Loboda A, Kari L, Rook AH, Vonderheid EC, Lessin S, et al. Quantitative PCR on 5 Genes Reliably Identifies CTCL Patients With 5% to 99% Circulating Tumor Cells With 90% Accuracy. Blood (2006) 107:3189–96. doi: 10.1182/blood-2005-07-2813
107. Tang N, Gibson H, Germeroth T, Porcu P, Lim HW, Wong HK. T-Plastin (PLS3) Gene Expression Differentiates Sézary Syndrome From Mycosis Fungoides and Inflammatory Skin Diseases and can Serve as a Biomarker to Monitor Disease Progression. Br J Dermatol (2010) 162:463–6. doi: 10.1111/j.1365-2133.2009.09587.x
108. Jones CL, Ferreira S, McKenzie RC, Tosi I, Caesar JA, Bagot M, et al. Regulation of T-Plastin Expression by Promoter Hypomethylation in Primary Cutaneous T-Cell Lymphoma. J Invest Dermatol (2012) 132:2042–9. doi: 10.1038/jid.2012.106
109. Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic Landscape of Cutaneous T Cell Lymphoma. Nat Genet (2015) 47:1011–9. doi: 10.1038/ng.3356
110. Beltzung F, Ortonne N, Pelletier L, Beylot-Barry M, Ingen-Housz-Oro S, Franck F, et al. Primary Cutaneous Cd4+ Small/Medium T-Cell Lymphoproliferative Disorders: A Clinical, Pathologic, and Molecular Study of 60 Cases Presenting With a Single Lesion: A Multicenter Study of the French Cutaneous Lymphoma Study Group. Am J Surg Pathol (2020) 44:862–72. doi: 10.1097/PAS.0000000000001470
111. Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, et al. Genomic Profiling of Sézary Syndrome Identifies Alterations of Key T Cell Signaling and Differentiation Genes. Nat Genet (2015) 47:1426–34. doi: 10.1038/ng.3444
112. Kiel MJ, Sahasrabuddhe AA, Rolland D, Velusamy T, Chung F, Schaller M, et al. Genomic Analyses Reveal Recurrent Mutations in Epigenetic Modifiers and the JAK-STAT Pathway in Sézary Syndrome. Nat Commun (2015) 6:8470. doi: 10.1038/ncomms9470
113. da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The Mutational Landscape of Cutaneous T Cell Lymphoma and Sézary Syndrome. Nat Genet (2015) 47:1465–70. doi: 10.1038/ng.3442
114. Woollard WJ, Pullabhatla V, Lorenc A, Patel VM, Butler RM, Bayega A, et al. Candidate Driver Genes Involved in Genome Maintenance and DNA Repair in Sézary Syndrome. Blood (2016) 127:3387–97. doi: 10.1182/blood-2016-02-699843
115. Park J, Yang J, Wenzel AT, Ramachandran A, Lee WJ, Daniels JC, et al. Genomic Analysis of 220 CTCLs Identifies a Novel Recurrent Gain-of-Function Alteration in RLTPR (P.Q575E). Blood (2017) 130:1430–40. doi: 10.1182/blood-2017-02-768234
116. Ballabio E, Mitchell T, van Kester MS, Taylor S, Dunlop HM, Chi J, et al. MicroRNA Expression in Sezary Syndrome: Identification, Function, and Diagnostic Potential. Blood (2010) 116:1105–13. doi: 10.1182/blood-2009-12-256719
117. Shen X, Wang B, Li K, Wang L, Zhao X, Xue F, et al. Microrna Signatures in Diagnosis and Prognosis of Cutaneous T-Cell Lymphoma. J Invest Dermatol (2018) 138:2024–32. doi: 10.1016/j.jid.2018.03.1500
118. Talaat IM, Abdelmaksoud RE, Guimei M, Agamia NF, Nugud A, El-Serafi AT. Potential Role for microRNA-16 (miR-16) and microRNA-93 (miR-93) in Diagnosis and Prediction of Disease Progression in Mycosis Fungoides in Egyptian Patients. PloS One (2019) 14:e0224305. doi: 10.1371/journal.pone.0224305
119. Dusílková N, Bašová P, Polívka J, Kodet O, Kulvait V, Pešta M, et al. Plasma miR-155, miR-203, and miR-205 are Biomarkers for Monitoring of Primary Cutaneous T-Cell Lymphomas. Int J Mol Sci (2017) 18(10):2136. doi: 10.3390/ijms18102136
120. Lindahl LM, Besenbacher S, Rittig AH, Celis P, Willerslev-Olsen A, Gjerdrum L, et al. Prognostic miRNA Classifier in Early-Stage Mycosis Fungoides: Development and Validation in a Danish Nationwide Study. Blood (2018) 131:759–70. doi: 10.1182/blood-2017-06-788950
121. Gluud M, Willerslev-Olsen A, Gjerdrum L, Lindahl LM, Buus TB, Andersen MH, et al. MicroRNAs in the Pathogenesis, Diagnosis, Prognosis and Targeted Treatment of Cutaneous T-Cell Lymphomas. Cancers (Basel) (2020) 12(5):1229. doi: 10.3390/cancers12051229
122. Rittig AH, Johansen C, Celis P, Odum N, Litman T, Woetmann A, et al. Suppressed microRNA-195-5p Expression in Mycosis Fungoides Promotes Tumor Cell Proliferation. Exp Dermatol (2020). doi: 10.1111/exd.14124
123. Xia L, Wu L, Xia H, Bao J, Li Q, Chen X, et al. miR-337 Suppresses Cutaneous T-Cell Lymphoma Via the STAT3 Pathway. Cell Cycle (2019) 18:1635–45. doi: 10.1080/15384101.2019.1629789
124. Gallardo F, Sandoval J, Díaz-Lagares A, Garcia R, D’Altri T, González J, et al. Notch1 Pathway Activation Results From the Epigenetic Abrogation of Notch-Related MicroRNAs in Mycosis Fungoides. J Invest Dermatol (2015) 135:3144–52. doi: 10.1038/jid.2015.328
125. Olszewska B, Żawrocki A, Lakomy J, Karczewska J, Gleń J, Zabłotna M, et al. Mapping Signal Transducer and Activator of Transcription (STAT) Activity in Different Stages of Mycosis Fungoides and Sezary Syndrome. Int J Dermatol (2020) 59:1106–12. doi: 10.1111/ijd.15036
126. Olszewska B, Gleń J, Zabłotna M, Nowicki RJ, Sokołowska-Wojdyło M. The Polymorphisms of IL-6/STAT3 Signaling Pathway may Contribute to Cutaneous T-Cell Lymphomas Susceptibility. Arch Dermatol Res (2021) 313:25–31. doi: 10.1007/s00403-020-02062-5
127. Pérez C, Mondéjar R, García-Díaz N, Cereceda L, León A, Montes S, et al. Advanced-Stage Mycosis Fungoides: Role of the Signal Transducer and Activator of Transcription 3, Nuclear Factor-κb and Nuclear Factor of Activated T Cells Pathways. Br J Dermatol (2020) 182:147–55. doi: 10.1111/bjd.18098
128. García-Colmenero L, González J, Sandoval J, Guillén Y, Diaz-Lagares A, Andrades E, et al. Epigenetic Silencing of Tumor Suppressor Mir-124 Directly Supports STAT3 Activation in Cutaneous T-Cell Lymphoma. Cells (2020) 9(12):2692. doi: 10.3390/cells9122692
129. Lindahl LM, Gluud M, Emmanuel T, Thomsen EA, Hu T, Rittig AH, et al. Microrna-106b Regulates Expression of the Tumour Suppressors p21 and TXNIP and Promotes Tumour Cell Proliferation in Mycosis Fungoides. Acta Derm Venereol (2020) 100:adv00270. doi: 10.2340/00015555-3574
130. Sandoval J, Díaz-Lagares A, Salgado R, Servitje O, Climent F, Ortiz-Romero PL, et al. MicroRNA Expression Profiling and DNA Methylation Signature for Deregulated microRNA in Cutaneous T-Cell Lymphoma. J Invest Dermatol (2015) 135:1128–37. doi: 10.1038/jid.2014.487
131. Kruglov O, Wu X, Hwang ST, Akilov OE. The Synergistic Proapoptotic Effect of PARP-1 and HDAC Inhibition in Cutaneous T-Cell Lymphoma is Mediated Via Blimp-1. Blood Adv (2020) 4:4788–97. doi: 10.1182/bloodadvances.2020002049
132. Wu CH, Yang CY, Wang L, Gao HX, Rakhshandehroo T, Afghani S, et al. Cutaneous T-Cell Lymphoma PDX Drug Screening Platform Identifies Cooperation Between Inhibitions of PI3Kα/δ and HDAC. J Invest Dermatol (2021) 141(2):364–73. doi: 10.1016/j.jid.2020.05.110
Keywords: epigenetics, cutaneous T-cell lymphoma, epigenetic biomarkers, HDACi resistance, epigenetic therapy, histone modification, histone deacetylase inhibitor
Citation: Zhang P and Zhang M (2021) Epigenetics in the Pathogenesis and Treatment of Cutaneous T-Cell Lymphoma. Front. Oncol. 11:663961. doi: 10.3389/fonc.2021.663961
Received: 26 February 2021; Accepted: 12 May 2021;
Published: 24 June 2021.
Edited by:
Anjali Mishra, Sidney Kimmel Cancer Center, United StatesReviewed by:
Albrecht Reichle, University Medical Center Regensburg, GermanyPier Paolo Piccaluga, University of Bologna, Italy
Copyright © 2021 Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingzhi Zhang, bWluZ3poaV96aGFuZzFAMTYzLmNvbQ==