 Anthony Lima
Anthony Lima Danilo Maddalo
Danilo Maddalo- 1Department of Translational Oncology, Genentech, Inc., South San Francisco, CA, United States
- 2Roche Pharmaceuticals, Basel, Switzerland
Most experimental oncology therapies fail during clinical development despite years of preclinical testing rationalizing their use. This begs the question of whether the current preclinical models used for evaluating oncology therapies adequately capture patient heterogeneity and response to therapy. Most of the preclinical work is based on xenograft models where tumor mis-location and the lack of the immune system represent a major limitation for the translatability of many observations from preclinical models to patients. Genetically engineered mouse models (GEMMs) hold great potential to recapitulate more accurately disease models but their cost and complexity have stymied their widespread adoption in discovery, early or late drug screening programs. Recent advancements in genome editing technology made possible by the discovery and development of the CRISPR/Cas9 system has opened the opportunity of generating disease-relevant animal models by direct mutation of somatic cell genomes in an organ or tissue compartment of interest. The advent of CRISPR/Cas9 has not only aided in the production of conventional GEMMs but has also enabled the bypassing of the construction of these costly strains. In this review, we describe the Somatically Engineered Mouse Models (SEMMs) as a new category of models where a specific oncogenic signature is introduced in somatic cells of an intended organ in a post-natal animal. In addition, SEMMs represent a novel platform to perform in vivo functional genomics studies, here defined as DIVoS (Direct In Vivo Screening).
Introduction
Integrative molecular analysis of human and cancer genomes has thrown a spotlight on the immense complexity of tumor biology (1). The ability to identify driver mutations and discriminate them from passenger events is key for the design of effective therapies to induce tumor regression. Moreover, being able to understand how tumors interact with the surrounding microenvironment and in particular with the immune system, is paramount for the preclinical design of therapeutic approaches combining targeted and immuno-therapy (2, 3). In this particular context, Genetically Engineered Mouse Models (GEMMs) of cancer can be invaluable in defining mechanisms driving tumor initiation, progression, and response to therapy (4). However, despite the wealth of information, they could provide a few challenges that have so far limited the use of GEMMs for preclinical drug development. First, the generation of GEMMs is time-consuming because it requires several steps such as precise gene targeting in Embryonic Stem Cells (ESCs), implantation, germline transmission, and colony expansion before achieving an experimental cohort. Timelines of this process would be further expanded when multiple allele engineering is required. Another limitation is that only a fraction of the animals employed in the generation process will eventually be used as experimental models, resulting in high husbandry costs and animal waste. Moreover, in many cases the phenotype penetrance of some oncogenes is only partial, requiring additional mice to have conclusive studies, and the tumor latency could be over a year, like in the case of animal models of squamous cell lung carcinoma (SCLC) (5). Last but not least, the ideal GEMM would develop tumors in a specific tissue as a consequence of mutations occurring in somatic cells rather than the germline. Even if this last point has been partially addressed by generating conditional models leveraging the Cre-recombinase or Tet-on inducible systems, the amount of breeding and crossing required makes the process tedious and expensive. A valid alternative to the GEMMs has been represented in the last few years by animal models where somatic cells would be directly engineered. We define as SEMMs (Somatically Engineered Mouse Models) in vivo models were a given genetic modification is induced in a specific somatic cell of an intended organ. Typically, somatic editing would be performed on animals with a wild type genotype using genome editing enzymes such as Cas9 or its derivatives or the Cas12a/Cpf1 delivered directly in vivo along with a set of specific sgRNAs. More in general, the concept of SEMM is not formally limited to rodents but can be extended to other species. The immediate advantage of SEMMs over GEMMs is the time efficiency, as almost no breeding is needed, the drastic reduction of animals required to achieve an experimental cohort as well as the versatility since multiple sgRNAs can be delivered simultaneously to mimic complex oncogenic events (for a comparison between GEMMs and SEMMs refer to Table 1).
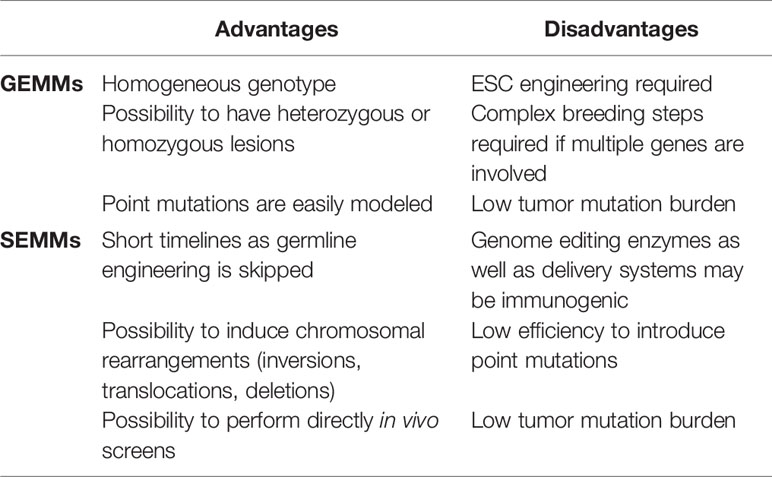
Table 1 Comparison of GEMMs and SEMMs.
SEMMs: Development and Applications
Achieving precise, efficient, and consistent genome editing directly in vivo is feasible due to the successful combination of the CRISPR/Cas9 system with efficient delivery systems that vary according to the target organ (6). Generation of a desired oncogenic signature in a SEMM is dependent on a two-step process: first, the DNA damage induced by the endonuclease and second the consequent repair event. Introduction of a double-strand break triggers two distinct and competing repair mechanisms: the Non-Homologous-End-Joining (NHEJ) pathway results in the insertion or deletion (indels) of nucleotides at the cutting side, resulting in frameshifts (7); the Homologous Recombination (HR) pathway uses a foreign DNA template that can be incorporated at the site of damage (7). It has to be noted however, that especially in vivo, the NHEJ pathway is dominant over the HR mechanism and therefore strategies involving the latter should be carefully designed. Based on which mechanism is leveraged, in vivo delivery of the Cas9 can inactivate tumor suppressors and/or trigger mutations at hotspots on oncogenic drivers. Also, CRISPR-based models offer the unique advantage of generating large chromosomal rearrangements such as inversions, deletions, and translocations (8). It was indeed not possible to trigger such events in somatic cells in vivo, apart from approaches where multiple rounds of ESC engineering were required. Last but not least, SEMMs offer the unique advantage of screening directly in vivo for genes contributing to tumor development, and resistance in a specific organ of interest.
SEMMs Simplify Genetic Mouse Modeling
The possibility of delivering the CRISPR/Cas9 system in vivo with viral and non-viral methods (summarized in Figure 1) along with the generation of animal models expressing constitutively or conditionally the Cas9 endonuclease, has opened the possibility of generating various animal models in a cost- and time-efficient way. Notably, patient-derived genomic information plays a two-fold role: while on one hand it guides the generation of murine models of disease, on the other hand it is the main source of information for designing specific libraries to interrogate the genome directly in vivo.
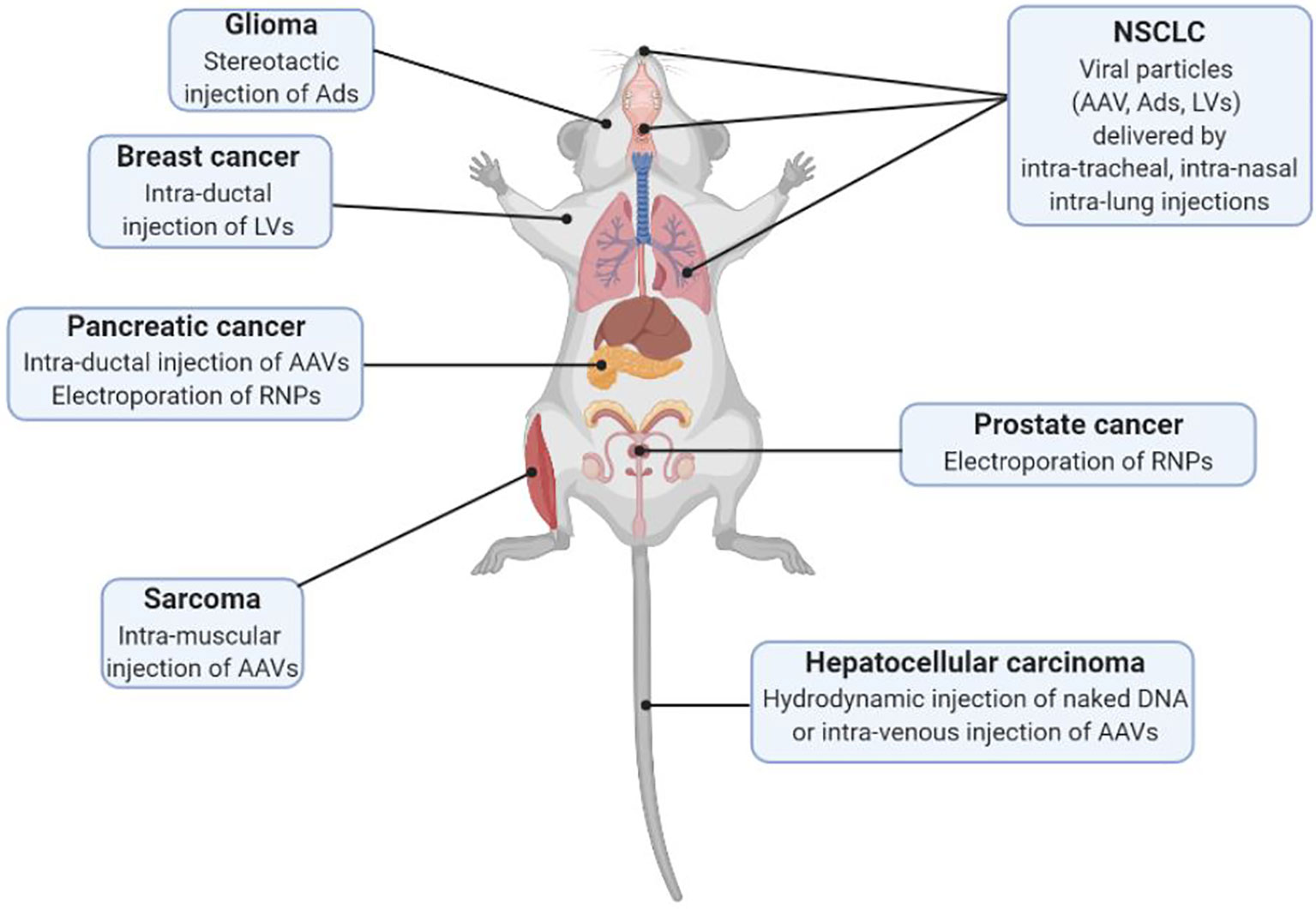
Figure 1 Schematic representation of different delivery modalities of the CRISPR/Cas9 system in vivo for SEMM generation (AAV, Adeno Associated Virus; Ads, Adenoviruses; LVs, Lentiviruses; RNPs, Ribonucleoproteins).
At this stage the main limitation for the generation of a SEMM is the tissue tropism of a given delivery system and the possibility of editing a specific cell population. In the section below we report as proof of concept limited examples of SEMMs organized by target organ generated in the past few years (summarized in Table 2).
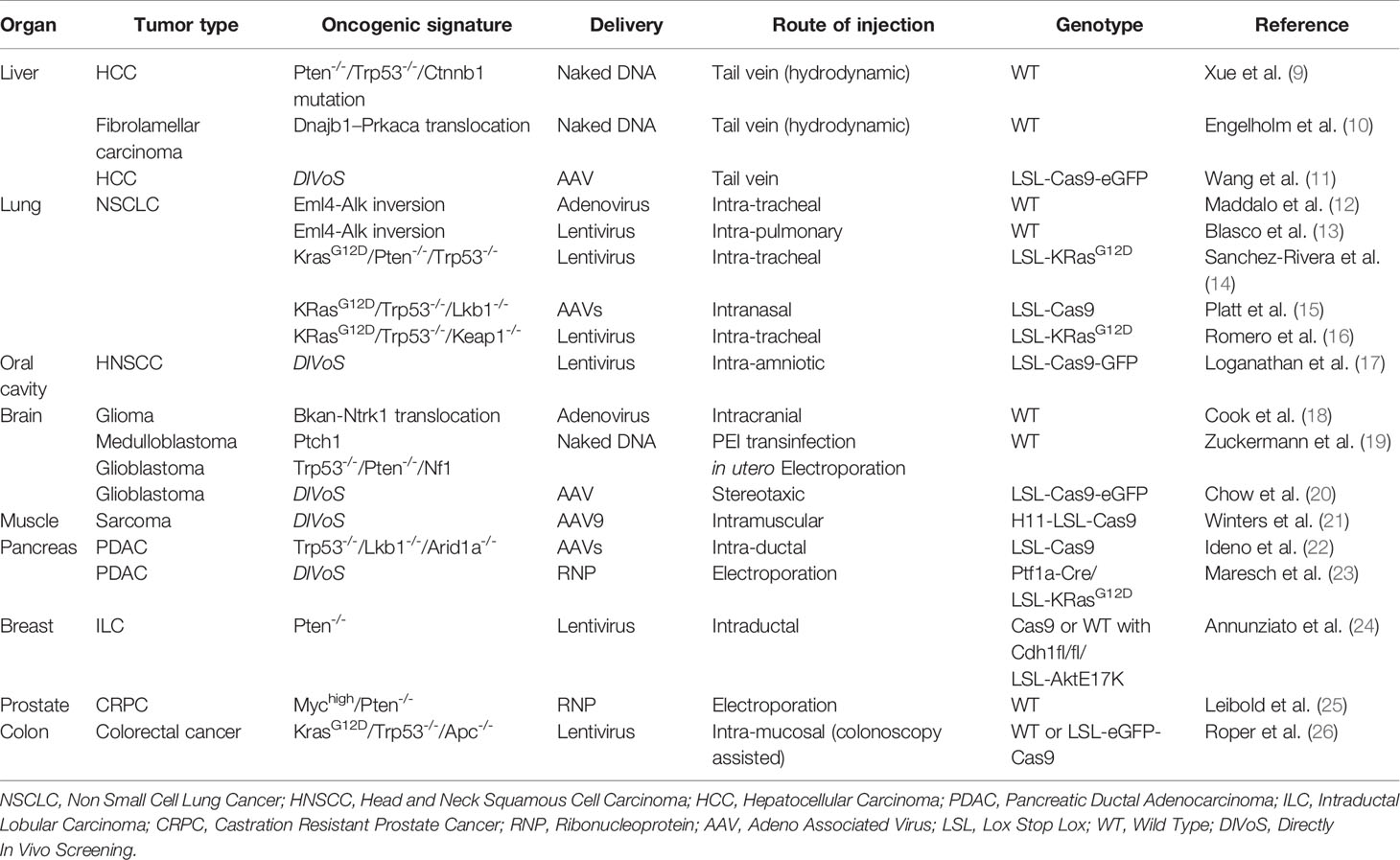
Table 2 Summary of SEMMs and application as DIVoS.
Liver
It is not surprising that the liver is among the first organs to be targeted to generate SEMMs as delivery of plasmid DNA can be easily achieved with the use of hydrodynamic injection (HDI) (27). While not a practical delivery method for clinical use, many groups found they could generate human-relevant disease mouse models using the technique (28). One of these groups combined a plasmid with an AAV integration sequence to reverse fumarylacetoacetate hydrolase (FAH) deficiency in mice and the same delivery technique was successfully used to deliver Cas9 and an sgRNA to accomplish the same treatment (a similar deficiency occurs in individuals with hereditary tyrosinemia type 1 (HT1) leading to liver failure and hepatocellular carcinoma (28). Clinical procedures for human HDI are being developed which mimic the delivery of therapeutic agents to humans with safer lower volumes of fluid and in more direct routes that target the liver or portions of the liver (29). Delivering plasmid DNA by HDI presents several advantages over a viral-based platform: first, viral packaging is not required making it safer while also making gene delivery more cost and time effective. Second, depending on the construct delivered, the CRISPR/Cas9 system is only transiently expressed, minimizing off-target effects and immunogenicity; third, it can be easily combined with other systems, such as the Sleeping Beauty or the PiggyBac transposase, for concomitant gene overexpression.
Researchers have demonstrated that CRISPR mediated knockdown of tumor suppressor genes commonly found in human cholangiocarcinoma such as Trp53, and Pten, results in tumor development within 3 months. All animals developed liver tumors with bile duct differentiation features consistent with tumors that arise from Cre-mediated double conditional knockout mice. Additionally, they demonstrated that an sgRNA targeting β-catenin (Ctnnb1) co-delivered with a 200-nucleotide single stranded DNA oligonucleotide could induce a gain-of-function mutation (9). Such an approach demonstrated that in vivo genome editing could not only induce gene knockouts but also very precise mutations by homologous recombination. However, it has to be noted that the donor integration efficiency is still very low as only around 0.4% of hepatocytes showed nuclear β-catenin 7 days after injection. Similarly, SEMMs were generated to investigate the synergistic effect of knocking out a tumor suppressor such as Pten and/or Trp53 together with the expression of an oncogene such as NRas or cMet (mutated or wild type) as well as the impact on tumor latency in HBV-driven models (9).
Another approach to combine gene KO and overexpression was shown in two models of intrahepatic cholangiocarcinoma (ICC) driven by the loss of the Trp53 gene and the expression of the oncogenes KRasG12D or HRasG12V. In this approach gene integration was achieved by a dual sgRNA approach: one sgRNA linearized the donor plasmid while the second one targeted the integration site on the genome, the 3’-UTR region of the strongly expressed β-Actin gene. Even if this method has shown some success in tumor induction, it is however limited by events such as random integration of the plasmid donor, misorientation of the construct and inconsistency in the tumor penetrance (30).
The generation of SEMMs is also an efficient method to determine the oncogenic potential of uncharacterized genetic lesions such as fusions resulting from chromosomal rearrangements. For example, a SEMM of fibrolamellar hepatocellular carcinoma (FL-HCC) was generated by triggering the intra-chromosomal deletion Dnajb1-Prkaca. This phenotype highlighted the oncogenic potential, in vivo, of the fusion DNAJB1-PRKACA and its role in the initiation of FL-HCC (10). Induction of oncogenic gene fusions in vivo with the CRISPR/Cas9 system has several implications. First, it simplifies remarkably disease modeling as previously resembling gene rearrangements relied upon transgene expression (31, 32) or sequential introduction of loxP sites (33, 34). Second, it represented a clear warning concerning the safety of future Cas9-based therapies where sgRNA off targets may induce chromosomal aberrations.
Lung
SEMMs of lung cancer have been among the first models generated as the lung epithelium is easily accessible without the need for invasive surgery and highly infective by viral vectors. Lung cancer models are of particular clinical relevance since adenocarcinomas of the lung epithelium are the most frequently occurring cancers. Estimated new cases for the U.S. in 2020 alone are 228,820 or 13% of total new cases with 135,720 or 23% estimated number of U.S. deaths for 2020 (35). Sequencing data from 188 cancers has shown that the most common changes found in lung adenocarcinoma are mutations in genes such as TP53, STK11, PTEN, CDKN2A, KRAS, BRAF, EGFR, ERBB2, PIK3CA (35). The first SEMMs of NSCLC reported modeled the oncogenic mutation KRasG12D with the inactivation of tumor suppressors such as Trp53 and Lkb1/Stk11 (15). Platt and colleagues generated KrasG12D/Trp53KO/Lkb1KO lung tumors by delivering an AAV expressing the Cre recombinase to induce Cas9 expression, a donor template targeting exon 2 of the Kras gene to introduce the point mutation G12D and three sgRNA targeting Trp53, Lkb1, and KRas (15). When crossed with a β-actin Cre driver line, progeny mice with constitutive Cas9 expression were reported to be phenotypically normal down to a type of particularly sensitive CA1 pyramidal neuronal cells (15). This approach was the first example of a murine model expressing the Cas9 enzyme, resulting in increased versatility as viral particle capacity would otherwise be impacted by the genetic size of the endonuclease (approximately 4 kilobases). It has to be noted that such approach also highlighted the limitation of the CRISPR/Cas9 system to induce specific point mutations as the HDR pathway is significantly disadvantaged compared to the NHEJ, resulting in prolonged tumor latency or due to indels and/or incorrect inclusion of the DNA donor template. A more consistent experimental outcome was obtained when heterozygous LSL-KrasG12D mice were infected with lentivirus containing expression vectors for Cas9, Cre, and a sgRNA targeting Trp53. The resulting adenocarcinomas were consistent with NSCLC features demonstrated by Cre-activated lung tumors of KrasLSLG12D/WTxTrp53flox/flox established tumor models (15). The versatility of such a model has been shown in a parallel study investigating the role of the KEAP1 (16), PTEN, NKX1 and APC (14) in the same GEMM of KRAS-driven lung cancer. Manipulation of somatic cells by CRISPR-Cas9 mediated genetic loss can alter oncogenic pathways in an already well-established mouse genetic tumor model such as KrasLSLG12D/WTxTrp53flox/flox. This mouse model was used to study the effect and possible vulnerabilities of Keap1 loss in human LUAD (16). This demonstrates the many combinations of techniques, mouse strains and targeting vectors that can be used to create useful and clinically-relevant models without the need for further genetic targeting and breeding.
Chromosomal aberrations such as deletions, inversions and translocations are frequent drivers of lung cancer and this particular type of genetic perturbation has been difficult to recapitulate in germline genetic models as it required multiple rounds of ESC engineering. For the first time genetic aberrations have been generated directly in vivo with the CRISPR/cas9 system. In this instance the genetic inversion EML4-ALK, found in approximately 5% of NSCLC patients, was modeled in a lung SEMM where two sgRNAs targeted the intronic region of Eml4 and Alk. While in vitro the two double strand breaks resulted in a variety of combinations including indels, deletions and inversions, in vivo the fusion Eml4-Alk was positively selected and drove the formation of NSCL with histopathological feature overlapping with the human disease. There are two examples of generation of a SEMM of EML4-ALK inversion in the lung epithelium. In one case two sgRNAs and the Cas9 enzyme were delivered by intra-tracheal injection to the lung epithelium by an all-in-one adenoviral system, resulting in tumor formation as early as four weeks (12). In another case the same lesion was induced by two distinct lentiviral particles each expressing an sgRNA directly injected into the lung (13). Notably, in this case tumor incidence and latency were significantly less efficient, suggesting that viral tropism, vector design and relative sgRNA cutting efficiency play a crucial role in the successful design of a SEMM of chromosomal rearrangements.
Brain
Identification of genetic lesions driving tumor formation in the brain has been elusive due to the high number and the low frequency of mutations making the use of germline models for driver and therapeutic screening time consuming and expensive. An extensive effort at characterizing 543 patients with a subset contributing to whole-exome sequencing data identifying 71 significantly mutated genes (36). Additionally, a number of fusion gene products have been identified by recently developed heuristics investigating potentially dysregulating gene altering events (37). Generation of SEMMs via transformed Trp53 null adult neural stem cells implanted into immunodeficient NCr-Foxn1nu mice helped characterize four potential gene-fusion drivers: Fgfr3-Tacc3, Sec61g-Egfr, Bcan-Ntrk1 and Gga2-Prkcb, but only cells harboring the fusion BCAN-NTRK1 generated high-grade gliomas (18). These tumors also responded to therapeutic inhibition with entrectinib, an inhibitor specific to the NTRK1 kinase but interestingly not through caspase mediated apoptosis.
Cook and group went on to create an autochthonous model generated by inducing an intra-chromosomal deletion via stereotactic intracranial injection of adenoviral particles expressing the Cas9 and two sgRNAs. Upon infection, wild type animals developed high grade glioma within 3 months with a penetrance of roughly 50%, demonstrating the ability of the fusion gene to drive tumor formation. Another approach interrogated multiple tumor suppressor genes by a low throughput loss-of-function screening to identify oncogenic events driving the initiation of medulloblastoma and glioblastoma. In this study the CRISPR/Cas9 system was delivered as expression plasmids either by in utero electroporation of (E13.5) embryos or by polyethylenimine (PEI) mediated intracranial transfection of P0 postnatal mice. These methods of transfection overcome many technical hurdles such as size limitation, immunogenicity, and costs involved in viral vector delivery (19). The blood-brain barrier (BBB) is a major obstacle to the delivery of viral and plasmid vectors that must be overcome as in previous examples by direct injection into the target area or mediated by surgery and specialized transfection techniques such as electroporation. Made up of tight-junctions in endothelial cells and other anatomical components, systemic delivery of transforming vectors to various cell types of the CNS requires overcoming this obstacle. Groups in the field of gene therapy are making progress in this area by the use of recombinant AAV of various serotypes with demonstrated capability of traversing the BBB (38). For example AAV9 has shown the ability to penetrate endothelial cell junctions in an in vitro model of the BBB and infect many CNS cell types, albeit at a lower transduction efficiency (39).
Investigation into the possibility of delivering a retroviral vector capable of expressing MYC to induce medulloblastoma tumors similar to MYC expressing pediatric tumors demonstrated a low penetrance but metastasizing model when coupled with either Trp53 loss or Bcl-2 expression (40). The group injected a cell line capable of constitutively producing virus to the brains of RCAS-TVA animals. This approach uses replication-competent avian leukosis virus splice-acceptor (RCAS) vectors to target individual cells engineered to express the cell surface receptor TVA. Cells transduced with the retrovirus will express MYC and either lose tumor suppressor expression or express the apoptosis inhibitor BCL-2 and results in a low penetrance but highly metastatic model, a marker of poor prognosis in the clinic.
Muscle
Point mutations can be introduced into tissues using libraries of viral vectors to discover how different point mutations affect the mutant gene’s tumorigenicity in practically any tissue type (21). This platform utilized a multiplexed set of AAV targeting vectors that uniquely barcoded single-nucleotide point mutations of Kras exon2 and 3 to discover how different point mutations are expressed in tumors. Winters et al. used Cas9-mediated homology-directed recombination (HDR) to introduce this diverse set of uniquely barcoded Kras point mutations in muscle to demonstrate how different point mutants of Kras affect the formation of soft tissue sarcomas. This technique closely models two types of pediatric sarcoma found to harbor Kras mutations. Mice were injected with AAV-KRas donor/sgKras/Cre into the gastrocnemii of PT;H11LSL-Cas9 mice to initiate tumors. 3-7 months later 5/7 mice subjected to this procedure produced invasive sarcomas with oncogenic KrasG12D, KrasG12A, or KrasG13R alleles but few tumors harbored other point mutations. Viral delivery via intramuscular injection is a simple procedure that allows for the relatively rapid transformation of normal muscle cells to facilitate multiplexed functional studies for the rapid identification of driver mutations in vivo. This group was able to couple this system with next-generation sequencing in models that produce multiple tumor foci and metastasizes to distant locations to enable the determination of lesion size, location and even the tracking of distant metastasis to the originating tumor (21).
Pancreas
Over 48,000 new cases of pancreatic ductal adenocarcinoma (PDAC) in the United States and nearly as many deaths from this aggressive disease in 2015, PDAC is predicted to become the second leading cause of cancer mortality by 2030 (41). Models for this target organ would require survival surgery but groups with experience in the technique report high rates of success with little training of personnel (42). The production of three complex PDAC models was accomplished by AAV-mediated delivery of sgRNA into the murine pancreas of p48-Cre/LSL-Cas9 mice (22).These models recapitulate the phenotype of KrasG12D driven tumors with cooperating Trp53, Lkb1 and Arid1A lesions in a single workhorse mouse by the injection of different AAV packaged sgRNAs that direct the pancreatic epithelium-specific p48 promoter driven Cas9. This study demonstrated how a single germline engineered mouse could produce a wide range of tumor types. Since fewer stains of mice could cover a wider panel of specific tumor types and indications, mice are more likely to be available shortening the timelines to perform in vivo studies. The ability to multiplex gene editing CRISPR/Cas9 sgRNAs was performed in a carefully optimized set of experiments where precisely controlled electroporation of the pancreas facilitated the uptake of injected plasmid preparations (23). The group, interestingly, discovered a negative-selection of Brca2 inactivation in cancer cell lines derived from CRISPR/Cas9 derived PDAC tumors, demonstrating the feasibility of using this approach to explore therapeutic vulnerabilities in vivo (22).
Hormone Dependent Cancers
Somatic engineering to induce malignancies driven by hormone receptors such as breast and prostate cancer has allowed deeper understanding of pathways key to tumor initiation and to the interaction with the microenvironment. Breast cancer patients are molecularly stratified based on their estrogen, progesterone and HER-2 status (43). Estrogen receptor status has been a diagnostic marker since the 1970’s and closely recorded since 1990 but is absent in mice (44). This, among other differences, between mice and human breast tissue partially explains the difficulty of engineering a genetic mouse model that recapitulates human indications even if subgroups of breast cancer make this a promising area of development for SEMMs. Breast cancers that arise in mice not only require fewer oncogenic events to form and have general chromosomal stability with rare telomere dysfunction but do contain structural rearrangement albeit fewer than human breast tumors (45). The development of new SEMMs will need to address these differences when translating results to the clinic. While a large number of effective treatments are available (46) resistance remains a major contributing factor to death from this disease. To address this problem many groups are turning to CRISPR/Cas9 systems to interrogate potential targets involved in resistance to current therapies. This approach could have benefits to patients that have developed multi-drug resistance to re-sensitize tumors. The path to this therapy requires knowledge of the precise mechanism of resistance and if that mechanism would be amenable to gene editing therapy. For example, the PI3K pathway has been shown to confer partial resistance to anti-HER2 therapy in a HER2 positive and PIK3CAH1047R mouse model and organoid (mammosphere) system derived from the strain (47). This system lends itself to discovery of the resistance mechanism in organoids using a CRISPR/Cas9 discovery platform to find novel resistance marker first in the organoids then verify hits in vivo in the mouse strain using a targeted sgRNA (47). In another report it was also shown how intra-ductal lobular breast carcinoma (ILC) formation was impacted by Cas9-driven immunogenicity, in a mouse model where Cre mediates inactivation of E-cadherin1 (Cdh1) and activation of AktE17K. While intraductal injection of lentiviruses encoding Cre, Cas9 and an sgRNA targeting Pten resulted in highly infiltrated tumors not resembling the human disease, delivery of a lentiviral vector encoding the Cre and the same sgPten in a model expressing constitutively the Cas9 efficiently induced ILC (24). In another work it has been shown that electroporation of the prostate gland with the complex Cas9/sgRNAs is able to induce prostate cancer in vivo with tumor latency and penetrance comparable to the traditional GEMMs (25). Once again, this SEM model, defined by the authors as EPO-GEMM, provided a time- and cost- efficient approach to generate complex genotypes and identify the Wnt pathway as a key regulator of metastases.
Colon
Many GEMM models of colorectal cancer (CRC) suffer from lack of tumor foci location and frequency control. APC GEMM models only develop tumors in the small intestine quickly occluding the intestinal tract before any develop to carcinomas and metastasize. Colorectal cancers are currently best recapitulated with their low number of foci of primary tumors, location in the colonic submucosa, and metastatic behavior to distant organs, most commonly, liver and the lung with or without the use of GEMM model mouse strains by utilizing colonoscopy (26). In a detailed paper Jatin et al. (2018) lay out procedures to study colon cancer in mouse model systems. CRISPR/Cas9 is used to edit organoids grown and edited in vitro for colonoscopy guided implantation into the colon submucosa. This technique could also be used to directly insert CRISPR/Cas9 viral or DNA vectors capable of editing the genome of target cells. Colonoscopy allows researchers to reach the distal colon and avoid transplantation or induction of the rectum, which is more difficult and traumatic to the mouse if attempted with rectal prolapse. Colonoscopy while costly in equipment investment is indispensable for CRC research as it allows for the targeted induction of models, minimally invasive surveillance of tumor development and tumor sampling and avoids mortality of subject mice experienced from survival surgery techniques. Coupling colonoscopy and somatic gene editing can greatly reduce the number of animals used for CRC research. Research looking at the most common mutated genes in CRC APC, TP53, SMAD4, PIK3CA and KRAS can be easily augmented with more recently discovered mutated genes including ARID1A, SOX9 and PAM123B (48) using the rapid generation of relevant models.
SEMMs as a Platform for DIVoS (Direct In Vivo Screening)
In addition to the immediate advantage of time and cost savings over the GEMMs, SEMMs hold the promise of playing an important role to perform Direct In Vivo Screening (DIVoS) enabling the discovery of genes and/or gene networks relevant only in a specific context and environment. Ideally, genes identified with DIVoS could be ultimately validated in vivo with a dedicated SEMM (Figure 2). Compared to classic ex-vivo screening, an approach of screening directly in vivo has several advantages. First, it allows the interrogation of genes (especially tumor suppressors) key to tumor initiation and influencing sensitivity to therapy. In addition, the in vivo target cell population is surrounded by its original microenvironment compared to cell cultures, making data interpretation more straight forward as it is not influenced by culture conditions such as media and 2D vs 3D cell seeding. Moreover, since the screening takes place concomitantly to tumor formation, specific oncogenic signatures can be chosen upfront. In addition, DIVoS performed using SEMMs as a platform are clearly distinguished from previous efforts to perform screening in vivo using transposon systems such as Sleeping Beauty or PiggyBac, as in this case genome interrogation is random and limited to specific sequences (49, 50). As one of the major hurdles for this approach is the limited take rate due to immune-rejection of the viral vectors utilized to deliver the libraries, it is not surprising that only a specific pool of genes can be investigated for any given screening campaign and that gain-of-function approaches are highly favored compared to drop out screens. Also, organ accessibility and viral tropism play a key role in determining which tumor types can be easily screened. The generation of animal models expressing the Cas9 endonuclease and the refinement of viral vectors has built the core of a platform to screen, in vivo, hundreds of genes by sgRNA infection. The typical workflow for a DiVoS contemplates the identification of specific genetic modifications from a given patient population and the subsequent interrogation of such a gene pool in an in vivo model. One of the first example of DiVoS was the screening of 13 tumor suppressor genes electroporated into the murine pancreas where a negative selection was shown when Brca2 gene was lost. Other experiments were performed in autochthonous models of hepatocellular carcinoma (11) and glioblastoma (20) where AAV-CRISPR vectors were used to map tumor suppressor genes contributing to cancer development. In both instances it is also clear that a few technical hurdles are still limiting the screening efficiency, such as partial integration of viral vector sequences, rejection by the immune-system and generation of a multifocal lesions sometimes difficult to dissect. An elegant strategy to circumvent immune rejection used ultra-sound guided intra-amniotic injection into the surface epithelium of mice to identify long tail genes having a tumor suppressor function in head and neck squamous cell carcinoma (HNSCC) (17). Another interesting approach is also represented by a hybrid system where sgRNA are delivered to the liver by using the PiggyBac transposon system (51). This example of hybrid approach Cas9/Transposon allows in vivo screening campaigns requiring a significant lower amount of resources compared to viral-based technology, even if the number of genes that can be screened remains limited. It has to be noted that even if DIVoS provide an invaluable source for novel target identification, partial vector integration as well as limited take rate due to immune rejection still represent a hurdle for the development of the field. In addition, with DIVoS it is not possible to control the multiplicity of infection (MOI) as tightly as in vitro and a translation from mouse to human of each finding is always required (but not always possible). A summary of advantages and disadvantages of DIVoS is shown in Table 3. Next generation viral vectors unable to trigger an immune response will probably be a valuable tool to refine quality and resolution of these experiments.
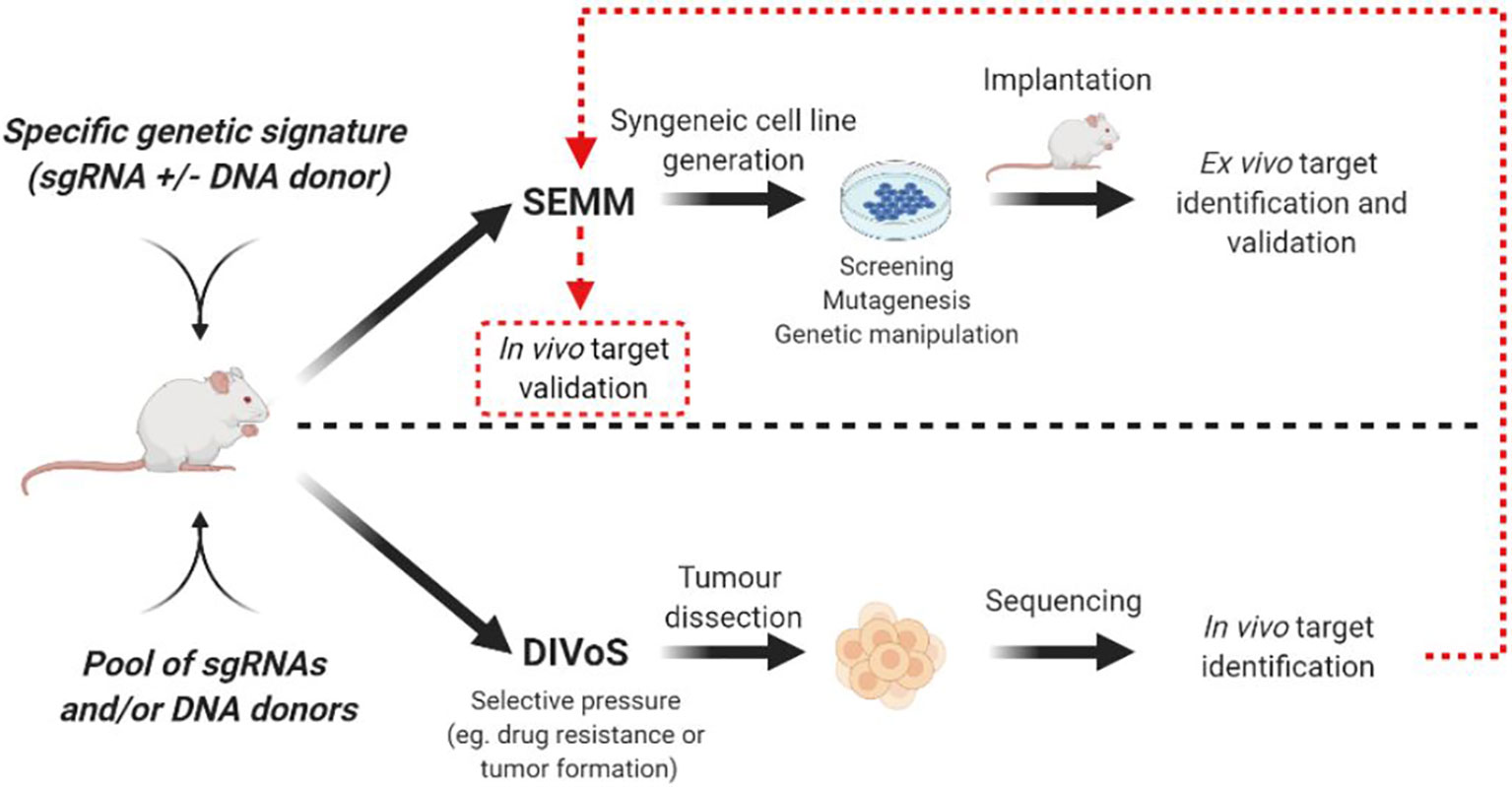
Figure 2 Representative workflow for SEMM generation and DIVoS design starting from CRISPR/Cas9 in vivo delivery. Targets identified with DIVoS are eventually validated with the generation of a SEMM (red dotted line).

Table 3 DIVoS: advantages and disadvantages.
Future Perspective and Limitations
The possibility of efficiently delivering the CRISPR/Cas9 tool in vivo together with the development of additional Cas9 variants to modulate gene expression and/or epigenetic status, is already offering the unprecedented opportunity to quickly test and validate biological questions and hypotheses. Such democratization will have an immediate impact on three main aspects: a) reduction of number of animals needed to generate an experimental cohort; b) reduction of the turnaround time to validate gene function in complex disease models; c) possibility to have a multiplexed approach for the interrogation of genes directly in vivo. We foresee that especially the last aspect will enable the discovery of novel targets, highly dependent on the context where tumors are generated and not always and necessarily supported by simple genetic analysis.
In spite of many clear advantages, it has however to be noted that SEMMs show also some limitations. First, the expression of the Cas9 protein or any endonuclease could be a source of immunogenicity, making challenging the interpretation of studies investigating the interaction between the tumor and the immune system. In addition, even when animals expressing the endonuclease are used, delivery of sgRNAs by viral vectors would again induce an immune response. Such limitation, common also in gene therapy experiments, can be overcome by alternative delivery systems using hypotonic solutions or electroporation to deliver the ribonucleoprotein complex (52). Moreover, while the CRISPR/Cas9 allows control over the induction of double strand breaks in precise areas of the genome, not much leverage is however given in terms of controlling the pathways involved in the subsequent DNA repair. Since non-homologous end joining is significantly dominant over homologous recombination, attempts to introduce specific point mutations by providing an oligonucleotide donor have proven challenging. While such option is formally viable, the low frequency and the mixed outcome in terms of editing are still a clear hurdle making germline engineering still a preferred strategy over somatic editing. In addition, with the SEMM approach is not possible to control how many alleles to edit, a crucial aspect in some studies investigating heterozygous versus homozygous gene loss. It has to be considered that, just like the GEMMs, SEMMs do not develop tumors harboring a high number of mutations and are therefore not fully representative of cancer complexity and heterogeneity. Moreover, it is sometimes difficult to engineer tissues because of the limited tropism of some viral vectors, even if evolution of AAV serotypes along with the use of tissue specific promoters can help circumventing this problem. In addition it has to be considered that each organ generally requires a specific delivery method/procedure, each of them having advantages and limitations. Last but not least, due to tumor accessibility and complexity, SEMMs will not replace (at least fully) classic xenograft models to study pharmacokinetic, pharmacodynamics and efficacy correlation of candidate molecules but could however represent a valid source of novel, more representative syngeneic murine cancer lines.
In conclusion, with increasing delivering technologies as well as the discovery of alternative endonucleases, the field of the SEMMs has the potential to expand beyond disease modeling and support/address key scientific questions in the field of functional genomics and target identification.
Author Contributions
DM designed and conceived the original idea of the article. DM and AL wrote the article and reviewed the relevant literature. All authors contributed to the article and approved the submitted version.
Conflict of Interest
AL was employed by Genentech, Inc. and DM was employed by Genentech, Inc. and Roche Pharmaceuticals.
Acknowledgments
We thank Mark Merchant and Benjamin Haley for the scientific advice and suggestions. All the figures were generated with BioRender (www.biorender.com).
References
1. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell (2018) 173:291–304.e6. doi: 10.1016/j.cell.2018.03.022
2. Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res (2020) 30:507–19. doi: 10.1038/s41422-020-0337-2
3. Petitprez F, Meylan M, Reyniès A, Sautès-Fridman C, Fridman WH. The Tumor Microenvironment in the Response to Immune Checkpoint Blockade Therapies. Front Immunol (2020) 11:784. doi: 10.3389/fimmu.2020.00784
4. Gould SE, Junttila MR, de Sauvage FJ. Translational value of mouse models in oncology drug development. Nat Med (2015) 21:431–9. doi: 10.1038/nm.3853
5. Meuwissen R, Linn SC, Linnoila I, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell (2003) 4:181–9. doi: 10.1016/S1535-6108(03)00220-4
6. Glass Z, Lee M, Li Y, Xu Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends Biotechnol (2018) 36:173–85. doi: 10.1016/j.tibtech.2017.11.006
7. Zhao B, Rothenberg E, Ramsden DA, Lieber MR. The molecular basis and disease relevance of non-homologous DNA end joining. Nat Rev Mol Cell Bio (2020) 21:765–81. doi: 10.1038/s41580-020-00297-8
8. Maddalo D, Ventura A. Somatic Engineering of Oncogenic Chromosomal Rearrangements: A Perspective. Cancer Res (2016) 76:4918–23. doi: 10.1158/0008-5472.CAN-16-0726
9. Xue W, Chen S, Yin H, Tammela T, Papagiannakopoulos T, Joshi NS, et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature (2014) 514:380–4. doi: 10.1038/nature13589
10. Engelholm LH, Riaz A, Serra D, Dagnæs-Hansen F, Johansen JV, Santoni–Rugiu E, et al. CRISPR/Cas9 Engineering of Adult Mouse Liver Demonstrates That the Dnajb1–Prkaca Gene Fusion Is Sufficient to Induce Tumors Resembling Fibrolamellar Hepatocellular Carcinoma. Gastroenterology (2017) 153:1662–73.e10. doi: 10.1111/j.1440-1746.2009.06156.x
11. Wang G, Chow RD, Ye L, Guzman CD, Dai X, Dong MB, et al. Mapping a functional cancer genome atlas of tumor suppressors in mouse liver using AAV-CRISPR–mediated direct in vivo screening. Sci Adv (2018) 4:eaao5508. doi: 10.1126/sciadv.aao5508
12. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature (2014) 516:423–7. doi: 10.1038/nature13902
13. Blasco RB, Karaca E, Ambrogio C, Cheong TC, Karayol E, Minero VG, et al. Simple and Rapid In Vivo Generation of Chromosomal Rearrangements using CRISPR/Cas9 Technology. Cell Rep (2014) 9:1219–27. doi: 10.1016/j.celrep.2014.10.051
14. Sánchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, et al. Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature (2014) 516:428–31. doi: 10.1038/nature13906
15. Platt RJ, Chen S, Zhou Y, Yim MK, Swiech L, Kempton HR, et al. CRISPR-Cas9 Knockin Mice for Genome Editing and Cancer Modeling. Cell (2014) 159:440–55. doi: 10.1016/j.cell.2014.09.014
16. Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med (2017) 23:1362–8. doi: 10.1038/nm.4407
17. Loganathan SK, Schleicher K, Malik A, Quevedo R, Langille E, Teng K, et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science (2020) 367:1264–9. doi: 10.1126/science.aax0902
18. Cook PJ, Thomas R, Kannan R, de Sanchez EL, Drilon A, Rosenblum MK, et al. Somatic chromosomal engineering identifies BCAN-NTRK1 as a potent glioma driver and therapeutic target. Nat Commun (2017) 8:15987. doi: 10.1038/ncomms15987
19. Zuckermann M, Hovestadt V, Knobbe-Thomsen CB, Sapatka M, Northcott PA, Shramm K, et al. Somatic CRISPR/Cas9-mediated tumour suppressor disruption enables versatile brain tumour modelling. Nat Commun (2015) 6:7391. doi: 10.1038/ncomms8391
20. Chow RD, Guzman G, Wang G, Schmidt F, Youngblood MW, Ye L, et al. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat Neurosci (2017) 20:1329–41. doi: 10.1038/nn.4620
21. Winters IP, Chiou SH, Paulk NK, McFarland CD, Lalgudi PV, Ma RK, et al. Multiplexed in vivo homology-directed repair and tumor barcoding enables parallel quantification of Kras variant oncogenicity. Nat Commun (2017) 8:2053. doi: 10.1038/s41467-017-01519-y
22. Ideno N, Yamaguchi H, Okumura T, Huang J, Brun MJ, Ho ML, et al. A pipeline for rapidly generating genetically engineered mouse models of pancreatic cancer using in vivo CRISPR-Cas9-mediated somatic recombination. Lab Invest (2019) 99:1233–44. doi: 10.1038/s41374-018-0171-z
23. Maresch R, Mueller S, Veltkamp C, Öllinger R, Friedrich M, Heid I, et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Nat Commun (2016) 7:10770. doi: 10.1038/ncomms10770
24. Annunziato S, Lutz C, Henneman L, Bhin J, Wong K, Siteur B, et al. In situ CRISPR-Cas9 base editing for the development of genetically engineered mouse models of breast cancer. EMBO J (2020) 39:e102169. doi: 10.15252/embj.2019102169
25. Leibold J, Ruscetti M, Cao Z, Ho YJ, Baslan T, Zou M, et al. Somatic Tissue Engineering in Mouse Models Reveals an Actionable Role for WNT Pathway Alterations in Prostate Cancer Metastasis. Cancer Discovery (2020) 10:1038–57. doi: 10.1158/2159-8290.CD-19-1242
26. Roper J, Tammela T, Akkad A, Almeqdadi M, Santos SB, Jacks T, et al. Colonoscopy-based colorectal cancer modeling in mice with CRISPR–Cas9 genome editing and organoid transplantation. Nat Protoc (2018) 13:217–34. doi: 10.1038/nprot.2017.136
27. Zhang G, Budker V, Wolff JA. High Levels of Foreign Gene Expression in Hepatocytes after Tail Vein Injections of Naked Plasmid DNA. Hum Gene Ther (1999) 10:1735–7. doi: 10.1089/10430349950017734
28. Eggenhofer E, Doenecke A, Renner P, Slowik P, Piso P, Geissler E, et al. High volume naked DNA tail-vein injection restores liver function in Fah-knock out mice. J Gastroen Hepatol (2010) 25:1002–8. doi: 10.1111/j.1440-1746.2009.06156.x
29. Suda T, Liu D. Hydrodynamic Gene Delivery: Its Principles and Applications. Mol Ther (2007) 15:2063–9. doi: 10.1038/sj.mt.6300314
30. Mou H, Ozata DM, Smith JL, Sheel A, Kwan SY, Hough S, et al. CRISPR-SONIC: targeted somatic oncogene knock-in enables rapid in vivo cancer modeling. Genome Med (2019) 11:21. doi: 10.1186/s13073-019-0627-9
31. Pyo KH, Lim SM, Kim HR, Sung YH, Yun MR, Kim S, et al. Establishment of a Conditional Transgenic Mouse Model Recapitulating EML4-ALK–Positive Human Non–Small Cell Lung Cancer. J Thorac Oncol (2017) 12:491–500. doi: 10.1016/j.jtho.2016.10.022
32. Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci (2008) 105:19893–7. doi: 10.1073/pnas.0805381105
33. Buchholz F, Refaeli Y, Trumpp A, Bishop JM. Inducible chromosomal translocation of AML1 and ETO genes through Cre/loxP-mediated recombination in the mouse. EMBO Rep (2000) 1:133–9. doi: 10.1093/embo-reports/kvd027
34. Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q, et al. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell (2005) 7:445–55. doi: 10.1016/j.ccr.2005.03.037
35. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
36. Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The Somatic Genomic Landscape of Glioblastoma. Cell (2013) 155:462–77. doi: 10.1016/j.cell.2013.09.034
37. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun (2014) 5:4846. doi: 10.1038/ncomms5846
38. Zhang H, Yang B, Mu X, Ahmed SS, Su Q, He R, et al. Several rAAV Vectors Efficiently Cross the Blood–brain Barrier and Transduce Neurons and Astrocytes in the Neonatal Mouse Central Nervous System. Mol Ther (2011) 19:1440–8. doi: 10.1038/mt.2011.98
39. Merkel SF, Andrews AM, Lutton EM, Mu D, Hudry E, Hyman BT, et al. Trafficking of adeno-associated virus vectors across a model of the blood–brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. J Neurochem (2017) 140:216–30. doi: 10.1111/jnc.13861
40. Jenkins NC, Rao G, Eberhart CG, Pedone CA, Bubuc AM, Fults DW, et al. Somatic cell transfer of c-Myc and Bcl-2 induces large-cell anaplastic medulloblastomas in mice. J Neuro-oncol (2016) 126:415–24. doi: 10.1007/s11060-015-1985-9
41. Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell (2017) 32:185–203.e13. doi: 10.1016/j.ccell.2017.07.007
42. Chiou S-H, Winters IP, Wang J, Naranjo S, Dudgeon C, Tamburini FB, et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Gene Dev (2015) 29:1576–85. doi: 10.1101/gad.264861.115
43. Turkoz FP, Solak M, Petekkaya I, Keskin O, Kertmen N, Sarici F, et al. Association between common risk factors and molecular subtypes in breast cancer patients. Breast (2013) 22:344–50. doi: 10.1016/j.breast.2012.08.005
44. Visvader JE, Smith GH. Murine Mammary Epithelial Stem Cells: Discovery, Function, and Current Status. Csh Perspect Biol (2010) 3:a004879–a004879. doi: 10.1101/cshperspect.a004879
45. Varela I, Klijn C, Stephens PJ, Mudie LJ, Stebbings L, Galappaththige D, et al. Somatic structural rearrangements in genetically engineered mouse mammary tumors. Genome Biol (2010) 11:R100. doi: 10.1186/gb-2010-11-10-r100
46. Chen Y, Zhang Y. Application of the CRISPR/Cas9 System to Drug Resistance in Breast Cancer. Adv Sci (2018) 5:1700964. doi: 10.1002/advs.201700964
47. Hanker AB, Cook RS, Arteaga CL. Mouse models and anti-HER2 therapies. Oncotarget (2013) 4:1866–7. doi: 10.18632/oncotarget.1481
48. Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487:330–7. doi: 10.1038/nature11252
49. Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature (2005) 436:221–6. doi: 10.1038/nature03691
50. Rad R, Rad L, Wang W, Cadinanos J, Vassiliou G, Rice S, et al. PiggyBac Transposon Mutagenesis: A Tool for Cancer Gene Discovery in Mice. Science (2010) 330:1104–7. doi: 10.1126/science.1193004
51. Xu C, Qi X, Du X, Zou H, Gao F, Feng T, et al. piggyBac mediates efficient in vivo CRISPR library screening for tumorigenesis in mice. Proc Natl Acad Sci (2017) 114:722–7. doi: 10.1073/pnas.1615735114
Keywords: clustered regularly interspaced short palindromic repeat/CRISPR associated protein 9-mediated genome editing, mouse models, genetically engineered mouse models, somatically engineered mouse models, translational research, animal models
Citation: Lima A and Maddalo D (2021) SEMMs: Somatically Engineered Mouse Models. A New Tool for In Vivo Disease Modeling for Basic and Translational Research. Front. Oncol. 11:667189. doi: 10.3389/fonc.2021.667189
Received: 12 February 2021; Accepted: 23 March 2021;
Published: 23 April 2021.
Edited by:
Attila A. Seyhan, Brown University, United StatesCopyright © 2021 Lima and Maddalo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danilo Maddalo, ZGFuaWxvLm1hZGRhbG9Acm9jaGUuY29t