 Thijs A. van Schaik
Thijs A. van Schaik Kok-Siong Chen
Kok-Siong Chen Khalid Shah
Khalid Shah- 1Center for Stem Cell Therapeutics and Imaging (CSTI), Harvard Medical School, Boston, MA, United States
- 2Department of Neurosurgery, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, United States
- 3Harvard Stem Cell Institute, Harvard University, Cambridge, MA, United States
Combinatory treatments using surgery, radiotherapy and/or chemotherapy together with immunotherapy have shown encouraging results for specific subsets of tumors, but a significant proportion of tumors remains unsusceptible. Some of these inconsistencies are thought to be the consequence of an immunosuppressive tumor microenvironment (TME) caused by therapy-induced tumor cell death (TCD). An increased understanding of the molecular mechanisms governing TCD has provided valuable insights in specific signaling cascades activated by treatment and the subsequent effects on the TME. Depending on the treatment variables of conventional chemo-, radio- and immunotherapy and the genetic composition of the tumor cells, particular cell death pathways are activated. Consequently, TCD can either have tolerogenic or immunogenic effects on the local environment and thereby affect the post-treatment anti-tumor response of immune cells. Thus, identification of these events can provide new rationales to increase the efficacy of conventional therapies combined with immunotherapies. In this review, we sought to provide an overview of the molecular mechanisms initiated by conventional therapies and the impact of treatment-induced TCD on the TME. We also provide some perspectives on how we can circumvent tolerogenic effects by adequate treatment selection and manipulation of key signaling cascades.
Introduction
In the last decade, incredible steps forward have been made in the advances of cancer treatment. Although combinatory treatments using surgery, radiotherapy and/or chemotherapy together with immunotherapy have shown encouraging results for specific subsets of tumors, a significant proportion of tumors remains unsusceptible or becomes resistant to therapy. Therefore, more efforts are required to understand the underlying mechanisms that influence the outcome of these treatments. One possible explanation for these inconsistencies could be the dynamics of the tumor microenvironment (TME) post treatment (1, 2). The TME has been shown to play a pivotal role in cancer development, progression, immunity and treatment (3). These events are governed by tumor cells and a variety of non-malignant cells such as immune cells, fibroblasts and vasculature cells, which are hijacked by the tumor cells. Consequently, the non-malignant cells in the TME display altered phenotypes (4) and cause an immunosuppressive environment (5, 6). The balance of physical and chemical interactions regulating hijacking and immunosuppression is tightly regulated (3), but this equilibrium shifts when major physiological changes occur.
One process that appears to be a major contributor to these biochemical changes is treatment-induced tumor cell death (TCD), as dying cancer cells have been shown to release a wide variety of regulatory signals that affect both tumor and stromal cells (7). For example, a combination of two chemotherapeutic agents increases immune suppression in the TME as a result of secreted factors from dying tumor cells. These molecules stimulate regulatory T (Treg) cell proliferation and increase the number of immune-suppressive dendritic cells (DCs) (8). In this review, we discuss the effects of different types of TCD induced by conventional therapies on the immune cells in the TME. Additionally, we provide perspectives on how this knowledge can help in the optimization of conventional therapies and the design of new combinatory treatments for cancer.
Based on the morphological changes and biochemical features, there are five major types of cell death (9, 10). Regulating pathways of these distinct forms are complexly intertwined and are summarized in Box 1 and Figure 1. The TME is influenced by each type of cell death differently and depending on the pathway of activation, these effects range from tolerogenic to immunogenic properties, which are considered as ‘silent’ or ‘loud’ respectively. The latter attracts immune cells via the secretion of damage associated molecular patterns (DAMPs) and allows immune cells to provoke an anti-tumor response, which is clinically beneficial (29). Tolerogenic stimuli facilitate the eradication of dead cells without provoking an immune response and are often accompanied by the release of immunosuppressive chemo- and cytokines (30). The generalized effects of the five types of cell death on the TME and their subsequent influence on immune cells in this environment are summarized in Table 1.
Box 1. Overview Molecular Mechanisms Regulating Cell Death
Necroptosis
Necroptosis can be initiated by both intrinsic and extrinsic stimuli that activate receptor-interacting protein (RIP)-kinases (11, 12). These can act together or interact with the caspase pathways, but necroptosis is only activated when RIP-kinases act independently of caspase-8 (13). Subsequently, RIP-kinases activate mixed lineage kinase domain like pseudokinases (MLKL), which form protein-complex, resulting in permeabilization of the cell membrane (14, 15).
Senescence
Cell death can also occur via senescence, which is often activated by p53 and cyclin dependent kinase, and results in a permanent cell-cycle arrest (16). This signaling cascade was found to induce both senescence and senescence associated secretory phenotype (SASP). Furthermore, activation of the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway results in the transformation to the SASP in both cancer cells and healthy cells (17, 18).
Autophagy
Autophagy is tightly regulated by mammalian target of rapamycin (mTOR), members of the pro-apoptotic BCL-2 family (19, 20) and caspase-8 (21) which is essential for homeostasis in healthy cells (22). After autophagy is initiated, an autophagosome is formed where intracellular structures are degraded (23). Consistent activation of the autophagy machinery eventually leads to autophagic mediated cell death.
Apoptosis
Apoptosis is induced via two distinct pathways. The caspase-dependent pathway is either activated via ligand receptor binding of a death receptor complex (24) or though high levels of intracellular stress. Death receptors cleave caspase-8, which can interact with other members of the caspase cascade or initiate apoptosis via the BCL-2 family. Activation of the proapoptotic BCL-2 protein family members also occurs through intracellular stress. These proteins induce mitochondrial outer membrane permeabilization (MOMP) (25), which leads to the secretion of both apoptosis inducing factors (AIFs) and cytochrome C. The latter activates the caspase-cascade, but AIFs induce caspase-independent apoptosis.
Mitotic Catastrophe
Aberrant mitosis during the M-phase can lead to a specific mode of cell death termed mitotic catastrophe. This process is frequently caused by defects in the cell-cycle (26). Once a certain threshold of lethal signals is reached, the cell will die via pathways that regulate senescence, necroptosis and apoptosis (27), and relies on caspase-dependent and independent pathways (28).
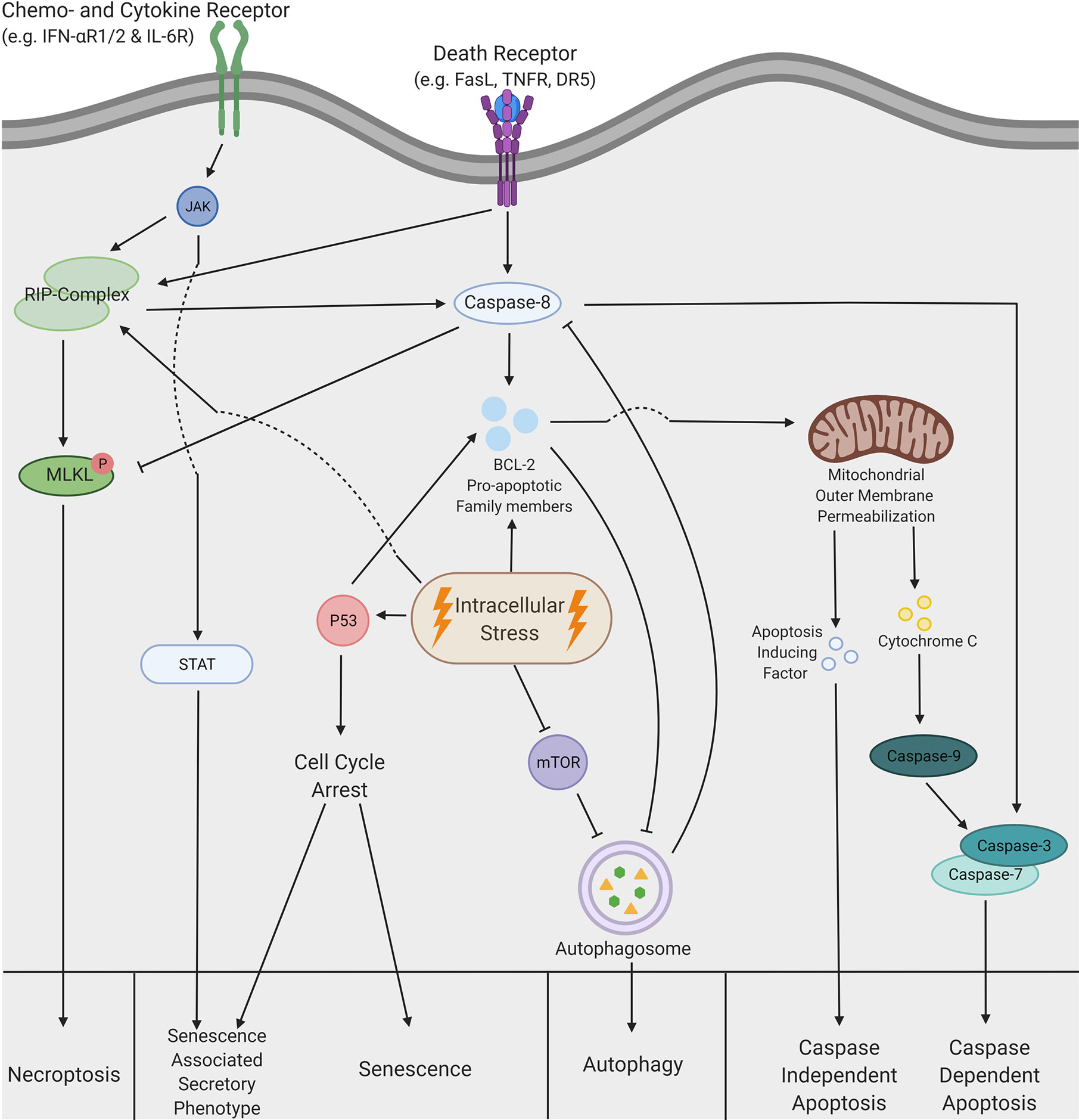
Figure 1 A schematic overview of the complex pathways regulating cell death. Necroptosis can be triggered by both extrinsic and intrinsic stimuli. The extrinsic pathway can be activated by chemokine, cytokine and death receptors, which leads to the activation of receptor interacting protein (RIP)-kinases. Similar activation is also achieved via intracellular stress. Subsequently, activation of the RIP-complex leads to phosphorylation of mixed lineage kinase domain like pseudokinases (MLKL), which is inhibited by caspase-8 under normal circumstances. MLKL phosphorylation induces a translocation to the cell membrane, which leads to permeabilization of the cell and consequently necroptotic cells death. Senescence is predominantly induced via a cell cycle arrest initiated by p53, but this process can also lead to the senescence associated secretory phenotype (SASP). Moreover, SASP can be induced via extrinsic stimuli that activate the Janus kinase- signal transducer and activator of transcription (JAK-STAT) pathway. Intracellular stress can inhibit the mammalian target of rapamycin (mTOR) pathway, which allows formation of the autophagosome. This formation can be inhibited by pro-apoptotic members of the BCL-2 family but is also able to inhibit caspase cleavage and thereby displays the crosstalk between autophagy and apoptosis. Eventually, after formation of the autophagosome, autophagic cell death is induced. Additionally, apoptosis can be induced by both intrinsic and extrinsic stimuli. Extrinsic stimuli allow cleavage of caspase-8, which can either induce cleavage of the caspase3/7 complex or activate members of the pro-apoptotic BCL-2 family. Activation of caspase3/7 results in caspase dependent apoptosis. However, the BCL-2 proteins can also be activated by intrinsic stimuli or intracellular stress and lead to the mitochondrial outer membrane permeabilization (MOMP), which in turn releases both cytochrome C and apoptosis inducing factors (AIF). Cytochrome C interacts with caspase-9 and subsequently induces caspase-dependent apoptosis via the activation of caspase 3/7. In contrast, AIF facilitates caspase-independent apoptosis by initiation of chromatin condensation and DNA fragmentation. This figure was created by authors using Biorender tools (biorender.com).
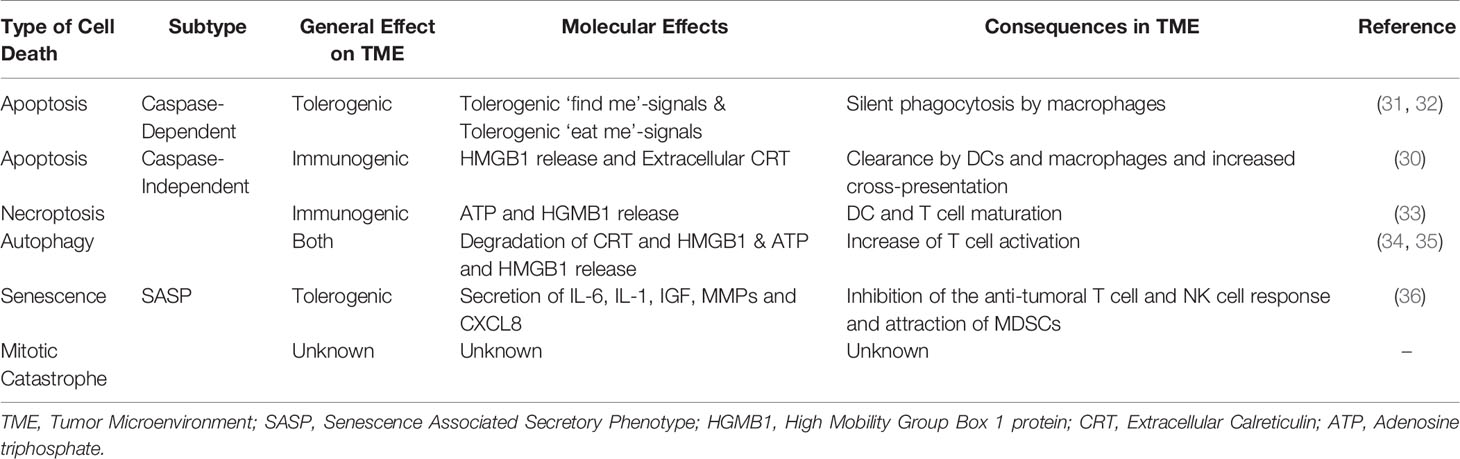
Table 1 Overview of effects of tumor cell death to the tumor microenvironment.
Consequences of Therapy-Induced TCD
Conventional treatments exploit the molecular mechanisms of cell death and therefore have a type-specific effect on the TME. In this section, we focus on the immunogenic or tolerogenic effects on the TME elicited by chemotherapy, radiotherapy and immunotherapy. Comprehension of the molecular mechanisms governing therapy-induced TCD and their consequences can lead to a better understanding of treatment effects, and is therefore crucial for the design of effective anti-cancer therapies.
Chemotherapy
Chemotherapy is the most widely used cancer treatment and induces all five types of TCD depending on the activated molecular mechanisms (37). Most chemotherapeutic agents act on the genomic content of tumor cells to inhibit proliferation and thereby induce cytotoxicity (38). After interfering with processes such as DNA synthesis, microtubule formation and cell division, numerous TCD cascades can be activated. Chemotherapeutic agents induce apoptosis either via the caspase-dependent and independent pathway or both simultaneously (39–41). For example, caspase-dependent apoptosis has been observed after treatment of lymphoma cells with paclitaxel (42), whereas the DNA-damaging agent cisplatin and protein synthesis inhibitor doxycycline activate both the caspase-dependent and -independent pathways (43, 44).
Caspase-independent apoptosis stimulates the upregulation of immune-activating molecules or DAMPs, such as high mobility group box 1 (HMGB1), heat shock proteins (HSPs) and extracellular calreticulin (CRT) (45, 46). These stimuli initiate an immune response via DCs antigen presentation. Mutations in the DAMP-recognizing receptors on DCs were associated with earlier relapse after chemotherapy in breast cancer patients (47). This emphasizes the fact that an effective immune response is necessary for a successful treatment outcome. However, the majority of conventional chemotherapeutic agents act on the caspase-dependent pathways, which results in silent phagocytosis by anti-inflammatory phagocytes that are recruited by tolerogenic ‘find-me’ signals (31) and facilitated by immune-suppressive ‘eat-me’ signals (32). The induction of tolerogenic TCD and thus an improper immune response could therefore be an explanation for the lower progression-free survival in patients treated with caspase-dependent apoptosis inducing agents (48–50).
Autophagy can be initiated by various chemotherapeutic agents such as temozolomide and oxaliplatin (51, 52), but the contribution of autophagy to the therapeutic effects of chemotherapy remains unclear. For example, defects in the autophagic pathways impede the release of HMGB1, as well as the secretion of tolerogenic cytokines such as IL-1β and IL-6 (34, 53, 54). Moreover, an increase in ATP and HMGB1 has been found after chemotherapy-induced autophagic TCD in immune-competent mice (55). These results suggest that induction of autophagic TCD by chemotherapeutic agents can cause an immunogenic transformation of the TME, but also emphasize the need for a better understanding of the autophagic machinery and the interaction with other forms of cell death. A frequently suggested justification for the discrepancies is that some tumors rely on autophagy more heavily in terms of metabolism due to genetic alteration or nutrient deprivation in the TME (56), which could affect the degradation and secretion of immunomodulatory signals.
Conventional chemotherapies such as 5-FU and cisplatin also induce necroptosis in tumor cells (57, 58). For instance, disruption of the necroptotic pathway reduces chemotherapy-induced TCD and significantly decreases the release of DAMPs in mice (59, 60). The DAMPs released after necroptosis mainly consist of HMGB1 and ATP, which promote DC and T cell infiltration and maturation via antigen cross-presentation (13). This leads to a potent anti-tumor response and efficient long-term immunity (33, 61). Unfortunately, various cancer types have displayed silencing of the key regulators in the necroptotic pathway by DNA hypermethylation, which diminishes the chemosensitivity and the immunogenic response, and results in a lower overall survival in patients with various forms of cancer (62, 63). Together, necroptosis has been found to induce an immunogenic effect after chemotherapy, but co-occurrence with apoptosis post-chemotherapies minimized this immunostimulatory effect.
Another substantial consequence of chemotherapy is senescence, which is induced by DNA-damage due to cellular stress (64). Although senescence can contribute to the efficacy of chemotherapy (65), the transformation of tumor cells into the senescence associated secretory phenotype (SASP) negatively affects the clinical outcome (66). This transformation is mainly caused by DNA damage-induced senescence and activated via both p53-dependent and -independent pathways (67). Relative to other senescent tumor cells, SASP tumor cells have a distinct secretome including interleukins, inflammatory cytokines and growth factors such as IL-6, IL-1, IGF, MMPs and CXCL-8 (68, 69). These molecules promote tumor growth (70) and have predominantly tolerogenic effects, such as the inhibition of the anti-tumoral T cell response (36) and natural killer (NK) cell (71), as well as the attraction of myeloid derived suppressor cells (MDSCs) (72). For instance, a strong senescent response has been observed in breast cancer patients after docetaxel and doxorubicin treatment, which is caused by an increase in MDSC infiltration and a decrease in T cell activation and infiltration (66, 73). This data suggests that DNA-damaging chemotherapeutics induce senescence and the SASP simultaneously in tumor cells, which results in a tolerogenic effect on the TME (74).
In summary, chemotherapy predominantly induces caspase-dependent apoptosis complemented with caspase-independent apoptosis, autophagy, senescence and/or necroptosis and can therefore lead to a wide range of effects on the TME. This intimate relationship is further emphasized by the findings that different forms of cell death occur in a time-dependent manner (75). For example, temozolomide used in glioblastoma treatment induces autophagy, senescence and apoptosis in sequential order (76), providing a possible framework for time-dependent administration of adjuvant therapy. Taken together, these studies accentuate the complexity of these intertwined pathways and demonstrate that the treatment efficacy relies on the type of chemotherapy, the induced signaling cascades and consequently the type of cell death. Moreover, in-depth knowledge of the effects of chemotherapy on the TME facilitates a decent fundament to design new combinatory strategies for chemoimmunotherapy.
Radiotherapy
Treatment with ionizing radiation led to the discovery of the apoptotic pathway and was first thought to predominantly rely on the intrinsic apoptotic pathway (77). These highly energetic radio waves induce double-stranded breaks in the DNA and consequently activate the caspase-cascade of apoptosis (78). However, recent studies have shown that cellular stress induced by radiation can lead to activation of various types of TCD (79). Various parameters are thought to influence the activation of TCD pathways such as dose, number of fractions and type of radiation. For example, distinct forms of ionizing radiation such as UV-B and UV-C radiation induce caspase-independent cell death which partially contributes to the cellular susceptibility to radiation-induced TCD (80–82). These results demonstrate the variability induced by the type of radiation. However, direct associations between apoptosis, type of radiotherapy, DAMP release and the subsequent immune response are unexplored.
Although apoptosis is the most commonly examined form of cell death after radiotherapy, high levels of radiation have been shown to induce a substantial amount of necroptosis and subsequently HMGB1 release, which was not observed in a low dose treatment (83, 84). Furthermore, knock-down of the RIP-kinase pathway results in a decrease in extracellular HMGB1 and expression levels of RIP-kinases significantly correlates with progression-free survival in patients with non-small cell lung cancer after high dose radiotherapy (85). This demonstrates the pivotal role of necroptosis in effective radiotherapy and suggests an important contribution of released immunogenic factors, but very little data is available regarding its direct effects on the immune system.
An association between radiation and autophagy has also been demonstrated, but opposing outcomes in terms of clinical efficacy have been found due to the discrepancy between cytoprotecting and cytotoxic autophagy (86). For example, autophagy is a radioprotective mechanism as the inhibition of the machinery sensitized radioresistant cell lines for low radiation doses (87). However, superficial amounts of radiation induce cytotoxic autophagy (88), which is linked to the release of DAMPs in a dose-dependent manner. High doses of radiation increase the induction of cytotoxic autophagy and DAMP release, and thereby transform the TME towards a more immunogenic environment (89). These findings display that only high levels of radiation can exceed the cytoprotective effects of autophagy, which consequently activates immunogenic cell death. Yet, most studies have been performed in vitro and therefore, more clinical or in vivo evidence is necessary to verify these findings.
Radiation also induces mitotic catastrophe if cells have malfunctioning cell cycle checkpoints or DNA damage response (90). The consequences of mitotic catastrophe TCD induced by radiation are similar to either apoptosis or necroptosis depending on the pathway of activation, but specific effects of mitotic catastrophe on the TME remain understudied.
Another understudied type of TCD induced by radiation is senescence since subsequent effects on the TME remain unknown (91). However, high levels of radiation induce SASP in malignant cells, which leads to increased secretion of tolerogenic chemo- and cytokines (92). Furthermore, the amount of SASP cells and consequently levels of SASP-associated cytokines is associated with genetic deficiencies in regular senescent pathways such as p53 and PTEN (93, 94). This demonstrates the strong molecular interaction between regular senescence and SASP and stresses the need for tumor-specific treatments. In general, high levels of radiation induce SASP; but no direct correlations in mouse models or patients have hitherto been found between radiotherapy, these secreted factors and the recruitment of suppressive immune cells.
In conclusion, radiotherapy can induce all forms of TCD. The immune responses of radiation induced TCD in TME are impacted by the types and doses of radiotherapy, as well as the genetic alterations in tumor cells. Doses of highly energetic forms of radiation are linked to more immunogenic forms of TCD such as caspase-independent apoptosis, necroptosis and cytotoxic autophagy. Currently, stereotactic body radiation therapy (SBRT) is of high clinical interest due to the possibility to deliver extremely precise high doses of radiation. The hypothesis is that SBRT delivers considerably higher biological effective doses of radiation, which induces more immunogenic TCD and has been shown to work synergistically with PD-1 checkpoint inhibition (95, 96). Ongoing studies have yet to determine the optimal radiation and fractionation dose, but the abovementioned studies demonstrate the possibilities for improvement of the conventional way of treatment.
Although a wide variety of studies have been published in the field of radiotherapy and tried to address the complexity of radiotherapy-induced cell death; direct interactions, underlying mechanisms of TCD and the interplay with the immune system remain obscure. A deeper understanding of the molecular mechanisms involved in radiotherapy is necessary to develop new radioimmunotherapy strategies.
Immunotherapy
The most common immunotherapies are checkpoint inhibitors and chimeric antigen receptor (CAR) T cells, which both rely on cytotoxic T cells to induce TCD (97, 98). Three major mechanisms are involved in cell death induced by cytotoxic T cells, namely FasL-dependent, Granzyme-mediated and chemokine-dependent cell lysis (99).
Activation of the FasL-pathway results in caspase cleavage and thus leads to tolerogenic apoptosis. However, if the caspase pathway is defective in tumor cells, necroptosis will be activated (100). For granzyme-mediated cell death, both the caspase-dependent and independent apoptotic pathway are involved depending on the type of granzyme secreted (101, 102). Cytotoxic T cells express a wide variety of granzymes and thus initiate both immunogenic and tolerogenic TCD simultaneously. Lastly, the cytokine-dependent lysis of cytotoxic T cells is primarily triggered by TNF-a, which normally activates the caspase-cascade (103). Similar to previously mentioned FasL-dependent necroptosis, TNF-a dependent necroptosis has been observed in tumor cells with deficiencies in the caspase pathway (104).
Although the immediate effects of T cell mediated TCD are known, implications of induced cytotoxicity on the TME remain understudied. More insights into the consequences of T cell-mediated TCD could result in new strategies to enhance immunotherapy and therefore increase the efficacy of this treatment in solid tumors. In contrast, CAR T cells are found to transform the TME after encountering their target epitopes. For example, activated CAR T cells secrete immunomodulatory cytokines such as GM-CSF and IFN-y (105). This results in the transformation of suppressive M2-macrophages to activated M1-macrophages and therefore a more immunogenic environment, which consequently increases macrophage mediated killing and recruitment of host T cells. Thus, tolerogenic forms of apoptosis exploited by T cells might contribute to the immunosuppressive TME, but T cells are also found to directly modulate this environment. Therefore, more evidence is needed to explain the disappointing results in the treatment of solid tumors with immunotherapy (106).
Recently, another form of immunotherapy has been approved by the FDA, namely an oncolytic virus targeting melanoma cells (107). These viruses are capable of replicate and destroy tumor cells selectively and thus spare healthy cells. Furthermore, various forms of immunogenic TCD are observed after oncolysis depending on the type of virus used.
For example, most adenoviruses (AVs) induce autophagic and necroptotic TCD (108, 109), and are found to induce a T cell-mediated anti-tumor response through the release of DAMPs such as ATP and HGMB1 (110, 111). Moreover, herpes simplex virus (HSV)-mediated cytotoxicity relies on autophagy and both caspase-dependent and -independent apoptosis, but inhibits necroptotic TCD (112–114). Various DAMPs are released after induction of these pathways such as HSPs, HMGB1 and CRT (115) and infection of tumor cells has been shown to improve the T cell-mediated anti-tumor immune response in tumor-bearing mice (116). Evidence for this immune response has also been found in HSV-treated patients with various solid tumors (117). Thus, oncolytic viruses initiate immunogenic TCD and display the potential to provoke a potent anti-tumor immune reaction.
Additionally, lysis of virally infected cells adds the advantage of released viral particles or pathogen associated molecular patterns (PAMPs) (118), which are also extremely immunogenic. On the other hand, PAMPs also induce viral clearance and emphasize the importance of adequate viral delivery to the tumor bulk. In conclusion, both the DAMP and PAMP release facilitate an effective immune reaction, which can be exploited to increase the effectiveness of anti-cancer treatment with for example the combination of adjuvant immunotherapy. Furthermore, the activation of multiple cell death pathways after infection again highlights a close relationship between apoptosis, autophagy and necroptosis, which was also found to be exploited by wild type viruses to increase replication (119). This property is often used to increase the effectiveness of treatments by upregulation of certain genes but should be considered carefully to ensure the maximum release of DAMPs via immunogenic forms of cell death.
In essence, chemotherapy, radiotherapy and immunotherapy are capable of inducing both tolerogenic and immunogenic cell death. These effects depend on the treatment type and the genetic alterations in the tumor cells. Henceforth, genetic and treatment variables should be taken into account when designing novel treatment strategies, especially when these standard therapies are combined with immunotherapy.
Strategies to Avoid Tolerogenic TCD
Multiple studies have demonstrated the beneficial effects of immunogenic forms of cell death, caused by the release of DAMPs in the TME and the subsequent activation of immune cells. This provokes an immune reaction against tumor cells, which leads to long-lasting anti-tumor immunity and a decreased risk of tumor recurrence or metastasis (120). In contrast, tolerogenic TCD increases pro-tumor effects by inhibiting the anti-tumor immune response (36), which leads to a lower progression-free and overall clinical survival in cancer patients (121). Therefore, immunological TCD via necroptosis and caspase-independent apoptosis is more desirable than tolerogenic TCD via caspase-dependent apoptosis. Various strategies can be used to increase the immunogenic effects of TCD and establish reliable treatments. Here, we will discuss four strategies: avoiding therapies that induce tolerogenic effects, converting tolerogenic TCD to immunogenic TCD, boosting immunogenetic effects and regulation of the tolerogenic stimuli. Some of these strategies have already been exploited in the clinic, but most are demonstrated in pre-clinical models and are currently under clinical investigation.
Avoiding Tolerogenic Therapies
As described above, most chemotherapies induce TCD via the tolerogenic caspase-dependent pathway and should be avoided. Given that some drugs could induce different forms of TCD, Wang and colleagues (122) summarized multiple chemotherapeutics that provoked an immune response via the upregulation of DAMPs. These drugs are found to act via various cytotoxic mechanisms, such as alkylating agents, antimetabolites and cytotoxic antibiotics, and could be exploited in a wide range of cancer types.
For example, oxaliplatin induces a more potent immunogenic TCD by the upregulation of extracellular CRT relative to cisplatin (123), which resulted in a significantly higher progression-free and overall survival in patients with gastric cancer (124). These results display that chemotherapeutic agents with similar chemical structures and acting mechanisms can have different outcomes and highlights the possibility to switch from a tolerogenic to immunogenic chemotherapy (Figure 2A.1). Hence, understanding the immunogenic properties of cytotoxic agents is necessary to improve the efficacy of combinatory treatments, especially for immunotherapy.
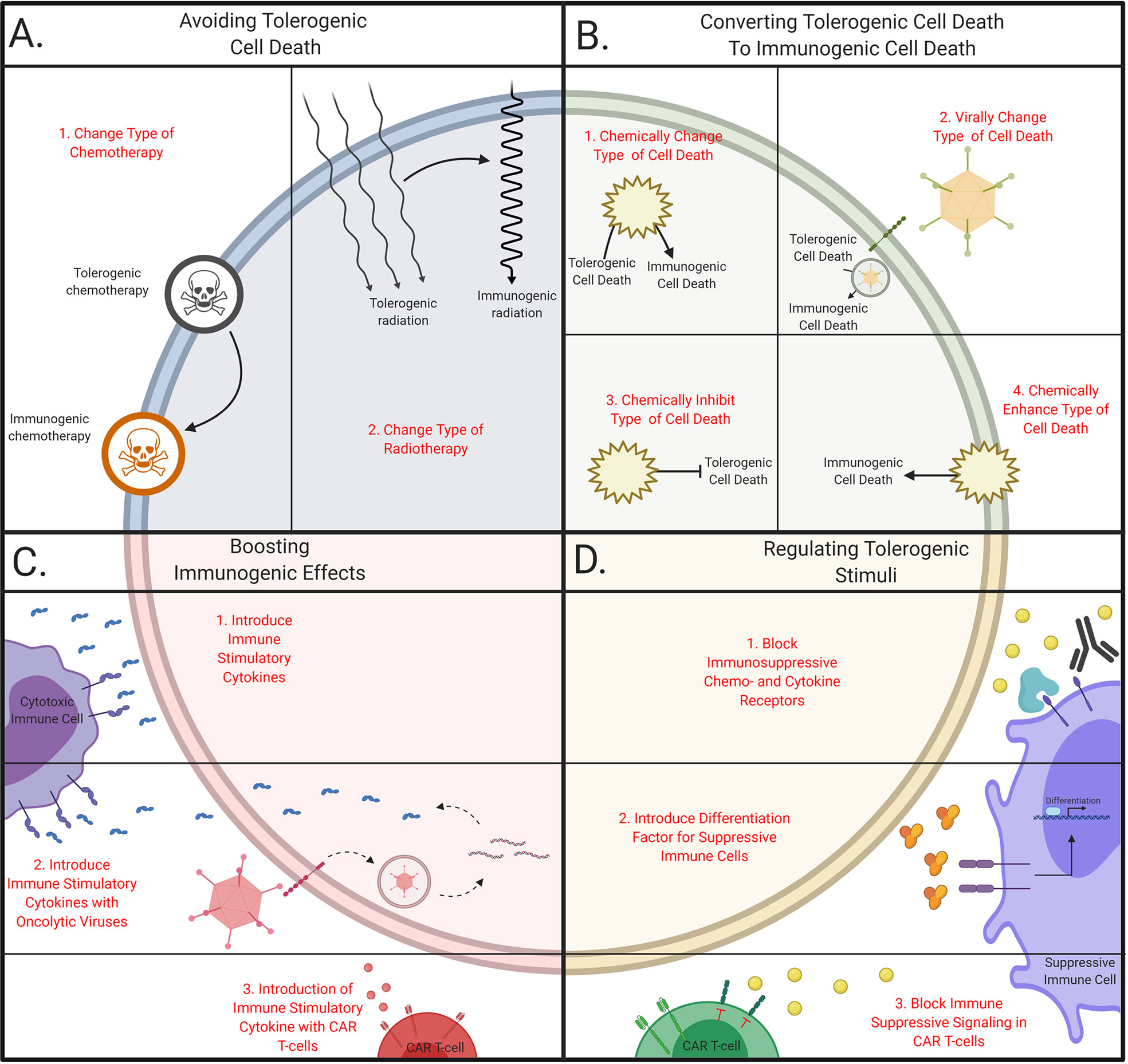
Figure 2 Schematic overview of tailoring therapeutic strategies to avoid tolerogenic cell death. (A) Avoiding tolerogenic cell death. Immunogenic cell death can be induced by (1) changing chemotherapeutic agents or (2) changing dose and regimes in radiotherapy. (B) Converting tolerogenic cell death to immunogenic cell death. Immunogenic cell death can be initiate by (1) chemically or (2) virally changing the cell death pathway, (3) chemically inhibiting tolerogenic pathways or (4) chemically stimulate immunogenic cell death. (C) Boosting immunogenic effects. Immunogenicity can be boosted via (1) the introduction of cytokines directly or via the delivery with (2) oncolytic viruses or (3) CAR T cells. (D) Regulating tolerogenic stimuli. Tolerogenic stimuli can be inhibited with (1) antibodies, (2) immune suppressive cells can be forced into differentiation or (3) T cells can be engineered to become resistant to tolerogenic stimuli. This figure was created by authors using Biorender tools (biorender.com).
Similar to the wide variety of chemotherapy used, radiotherapy is given in different doses, regimes, and radiation types (125). These parameters influence the immune-activating capabilities of the dying cells. For example, a close association between the dose of radiation and the upregulation of DAMPs has been observed in breast cancer cells (126). This is substantiated with increased survival in patients treated with a higher dose of radiation in small cell lung cancer patients (127). Furthermore, multiple rounds of moderate-dose radiation induce a more potent immune response relative to a single round with a similar total amount of Gray (128). This implies that the optimal radiation dose and fractionation regime are crucial to increase the immunogenic effects of radiotherapy (Figure 2A.2). Together, optimization of types and methods used in conventional treatments to increase the immunogenic effect is feasible. This has been shown to increase the survival of cancer patients, presumable by the increase of infiltrating immune cells due to reduced immunosuppression in TME (129). Yet, more research is necessary to determine the optimal effects for specific cancer types and particular genetic alterations.
Converting Tolerogenic TCD to Immunogenic TCD
Shifting TCD from a tolerogenic type to an immunogenic type of TCD could be an approach to increase the anti-tumor immune responses. One proposed method is to chemically shift from cell death pathway, by blocking the conventional pathway (Figure 2B.1). For example, necroptosis and caspase-independent apoptosis are induced after combining an apoptosis-inducing agent along with caspase-inhibiting drugs (Figure 3A) (7, 130). In murine models, TNF-a has been found to induce necroptosis after the incubation with the pan-caspase inhibitor Z-VAD-fmk (131). Furthermore, combining caspase inhibitors with chemotherapy leads to an increase in extracellular DAMPs (132) and results in a decrease of intertumoral Treg cells and an increase in IFN-y secreting cytotoxic T cells.
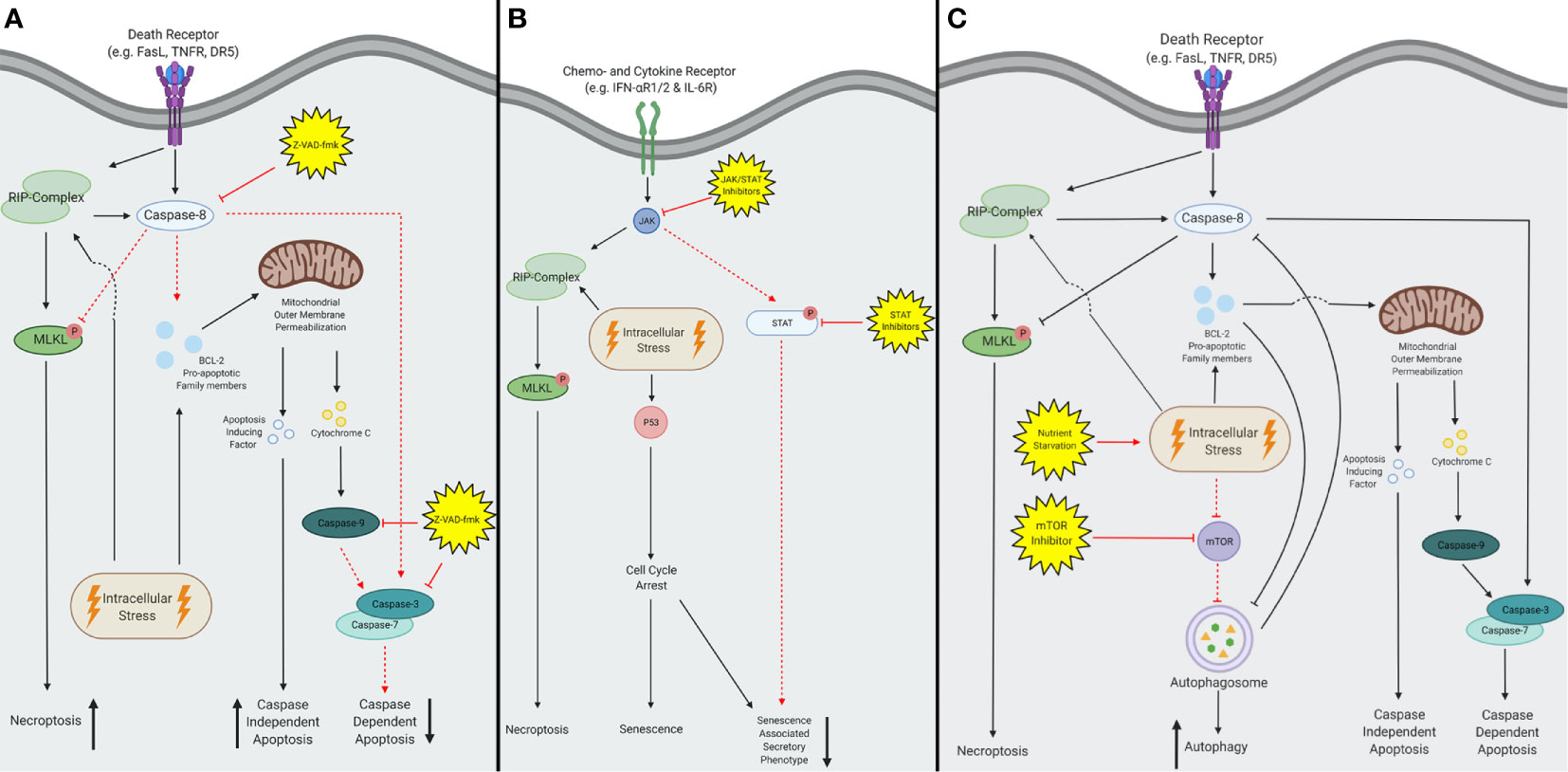
Figure 3 A schematic overview of inhibitors converting tolerogenic cell death to immunogenic cell death. (A) The pan-caspase inhibitor Z-VAD-fmk inhibits all caspase activity, the subsequent effects are depicted with red dotted lines. Z-VAD-fmk forces extrinsic stimuli to activate the RIP-kinase complex and thereby initiates necroptosis instead of caspase-dependent apoptosis. Intrinsic stimuli remain able to activate the pro-apoptotic BCL-2 family members and subsequently facilitate mitochondrial outer membrane permeabilization (MOMP). Consequently, both apoptosis inducing factors (AIF) and cytochrome C are released, but only AIF is able to induce apoptosis. Therefore, caspase-dependent apoptosis will be inhibited and decreased, and caspase-independent apoptosis will increase. (B) Both Janus kinase- signal transducer and activator of transcription (JAK-STAT) and JAK inhibitors are capable of inhibiting the senescent pathway, depicted in red dotted lines. This reduces the activation of the senescent associated secretory phenotype (SASP) after activation of the extrinsic pathway. In contrast, the intrinsic pathway remains able to induce the SASP via activation of p53. (C) Both nutrient starvation and mammalian target of rapamycin (mTOR) inhibition increase autophagy, by upregulation of the autophagosome. This figure was created by authors using Biorender tools (biorender.com).
Additionally, caspase inhibitors are also used along with radiation therapy, which increases TCD (133), but also results in the upregulation of PD-L1 and tumor relapse (134). This effect was effectively controlled by combining radiotherapy, caspase inhibitors and anti-PD-L1. Others have found that the combination of mammalian target of rapamycin (mTOR) inhibitors with radiotherapy and caspase inhibitors results in a potent anti-tumor effect due to an increase of autophagic TCD (135). Additionally, apoptosis induced by IFN-β is converted to necroptosis when either RIP1 or the caspase pathway was inhibited (136). These studies display the possibility to shift from tolerogenic TCD to immunogenic TCD, but clinical efficacy has yet to be proven.
As mentioned above, viruses contain genes that are capable of interfering with cell death pathways. For example, AVs contain a set of genes that are capable of regulating TCD by the sequestering of the pro-apoptotic BCL-2 family (137) and p53 (138), or downregulation of death receptors (139). Moreover, HSVs express proteins that interact with the formation of the autophagosome (140) and inhibit apoptosis (141), some of these genes are deleted during treatment design, to ensure rapid killing of cancer cells (142). These properties can be exploited to convert the mechanisms of cell lysis by oncolytic viruses (Figure 2B.2). For example, the upregulation of E3-11.6K instead of the deletion of E1B-19K in AVs demonstrates comparable cytotoxicity, but the pathway of cell death activation shifts from caspase-dependent to caspase-independent (143). This shows the possibility of changing cell death pathways directly, which can potentially be used in combination with other cell death-inducing agents. For instance, vaccinia viruses (VVs) express proteins that inhibit caspases directly (144), which can potentially be exploited in a similar manner as the caspase inhibitor Z-VAD-fmk. Furthermore, induction of specific cell death pathways via modifications in the autologous genetic composition of the virus saves the limited genomic space and thus provides a possible strategy to convert tolerogenic to immunogenic TCD.
Other strategies have been proposed to target cells with the SASP, where the transformation to this phenotype is blocked (Figure 2B.3) (145). For example, inhibition of the Janus kinase- signal transducer and activator of transcription (JAK-STAT) -pathways decreases the number of SASP cells after radiotherapy and chemotherapy in mice (17, 73). These combinatory treatments reduce the levels of tolerogenic chemokines and cytokines, and these positive results lead to the enrolment of a phase II clinical trial combining paclitaxel and ruxolitinib (146). Thus, the transformation of cells to the SASP can be blocked which results in a less tolerogenic TME (Figure 3B). Current research will prove whether this enhances the immune infiltration in human tumors and yields beneficial clinical results.
Moreover, the inhibition of autophagy is beneficial as it prevents tumor growth in mice (147) and the tumor cells are more susceptible to radiation therapy (148) as well as various chemotherapies such as cisplatin (149) and temozolomide (150). However, most of these studies were performed using immunodeficient mice and studies in immunocompetent models displayed little effect (151). These discrepancies led to the development of a novel strategy where autophagy is enhanced to increase the immunogenicity of the TME (Figure 2B.4) (152–154). Various agents are currently used that mimic effects of fastening in combination with chemotherapy, which has been found to enhance T cell infiltration and depletes Treg cells (155). These studies also demonstrate a significant increase of survival in mice relative to treatment with chemotherapy alone, which resulted in multiple clinical trials (156, 157). Future studies will eventually elucidate the underlying molecular association between autophagy, treatment, and the subsequent immune response (Figure 3C).
Taken together, these studies exhibit the possibilities to convert tolerogenic TCD towards immunogenic TCD with the use of specific pathway inhibitors or oncolytic viruses. Some of these inhibitors have already been used in clinical trials for other diseases and have been proven to be safe (158, 159). Thus, current and future research in combining these converting strategies with conventional treatments will provide data about the efficacy of this rationale. If the balance in the TME is found to shift towards an immunogenic environment, new combinations with immunotherapy can be utilized.
Boosting Immunogenic Effects
The third strategy is boosting the immunogenic effects of treatments using (neo)adjuvant therapies (160). In the past decade, various therapies have been combined with immune checkpoint inhibitors and are effective in some types of cancer (161, 162). This emphasizes the potential to block immune suppressive effects and paved the way for novel strategies that boost immunogenic effects. For example, enhanced immunogenicity can be achieved by the addition of immune stimulatory chemokines and cytokines (Figure 2C.1) (163) such as IL-12, which has been shown to increase the overall survival in mammary tumor-bearing mice (164). When IL-12 is administered in combination with an anti-PD-L1 Ab, a synergistic effect is observed. Moreover, chemokines such as GM-CSF are also used along with monoclonal antibodies (mAbs) to boost the immune system, which is shown to increase the overall survival in cancer patients relative to solely mAbs treatment (165, 166). Although initial pre-clinical results often look promising, admission of these immunomodulatory molecules can have a lot of side effects in cancer patients (167). A potential strategy to overcome these side effects could be local administration or delivery of these immunostimulatory molecules (168, 169). Further development of these delivery methods could lead to novel approaches, which allow local modulation of the TME and thereby enhance the clinical results of therapies boosting the immunogenic effects.
One frequently applied method is the delivery of genes expressing immunostimulatory agents with oncolytic viruses (Figure 2C.2). This appears to enhance the immune infiltration after the immunogenic lysis by viruses. For example, patients treated with AVs loaded with a gene encoding for GM-CSF show an increase of T cell trafficking to the tumor and induction of T cell-mediated immunity via DC cross-presentation (170). Various clinical studies confirmed these results, which led to a successful phase III clinical trial and FDA approval of this therapy (107). Only a minor increase in clinical efficacy was demonstrated, which was suggested to be caused by the upregulation of PD-L1 after treatment with oncolytic viruses (171). These undesired effects are indeed found to be counteracted when this oncolytic virus is combined with an α-PD-L1 antibody since a phase I clinical trial demonstrated improved tumor infiltration of T cells (172). Similar strategies with viruses expressing IL-12 either alone or in combination with other immune modulatory agents have been found to activate NK and T cells and prolong the survival of mice (173, 174). Consequently, a phase I clinical trial is currently being conducted with HSVs expressing IL-12 (175), which will ensure the safety of local IL-12 administration. Together, these findings highlight the possibility to modulate the TME locally with oncolytic viruses together with effective tumor cell lysis.
Comparable strategies are also exploited in CAR T cells, which are genetically modified to secrete immunoenhancing agents (Figure 2C.3). For example, CAR T cells secreting IL-12 have shown enhanced anti-tumor efficacy relative to non-secreting CAR T cells, due to increased immune infiltration and activation (176, 177). These promising results led to the enrolment of a phase I clinical trial in patients with ovarian cancer (178, 179). If these clinical studies prove effective immune modulation of the TME, the road will be paved for novel studies testing the efficacy of other immunomodulatory chemo- and cytokines.
In summary, boosting the immunogenicity of treatments was shown to be effective and can result in potent anti-tumor responses. Strategies using immunotherapy with immune-boosting capacities hold great potential for future treatments. Alternative delivery methods of chemo- and cytokines are being developed and will broaden the spectrum of applicable treatments and possible combinations.
Regulating Tolerogenic Stimuli
Regulating the anticipated tolerogenic stimuli could also be a promising strategy to decrease the immune suppressive effect in the TME. Two major components can be identified as targets which are soluble chemokines and cytokines, or suppressive immune cells. Inhibition of chemo- and cytokines can be achieved by blocking their receptors with inhibitory drugs or antibodies (Figure 2D.1).
For example, the CXCL12/CXCR4 axis inhibits the infiltration of cytotoxic T cells, increases the level of Treg cells and promotes metastasis (180). High levels of CXCL12 are secreted by SASP cells, which results in immunosuppression in the TME (181). Blocking these signaling pathways increases the levels of infiltrating cytotoxic T cells and increased survival in mice when combined with immunotherapy (182). Recently, promising results were obtained from a phase I clinical trial in breast cancer patients, where a CXCR4 antagonist was combined with chemotherapy (183). These results provide new rationales to combine these treatments with immunotherapy.
Furthermore, elevated levels of IL-6 have been linked to tumors containing SASP cells (184) and the regulation of IL-6 level has been shown to affect the anti-tumor immunity (185). Multiple studies demonstrated the possibility to interfere IL-6-mediated signaling pathways by either neutralizing the cytokine or blocking the receptor with monoclonal antibodies (186–188). These antibodies are safe for use in human patients (189) and decrease the infiltration of MDSCs in murine solid tumors (190). Currently, multiple clinical trials are examining the efficacy of IL-6 blockade in combination with conventional treatments in cancer patients (191, 192).
Other forms of TCD have also been reported to recruit the immune-suppressive MDSCs (72), and limiting their numbers has been shown to improve the overall survival in patients (193). Another strategy to decrease the tolerogenic effects induced by MDSCs is by differentiating these cells into less immune suppressive cells (Figure 2D.2). For example, the delivery of all-trans retinoic acid (ATRA) forces MDSCs to differentiate into dendritic cells or macrophages (194). This results in a significant decrease in circulating MDSCs and consequently enhances the immune response in cancer patients (195). Delivery of MDSC differentiating agents together with conventional immunotherapies is suggested to enhance the anti-tumor immune response and is currently clinically investigated (196).
Moreover, radiotherapy-induced SASP cells are found to attract immunosuppressive effects of MDSCs, which resulted in radiotherapy resistance and decreased T cell efficacy (197, 198). This was suggested to be partially regulated via the secretion of TGF-b (199). Two strategies have been found to counteract tolerogenic stimuli with immunotherapy (Figure 2D.3), namely T cell transduced with a dormant TGF-b receptor and the use of a specific subset of T cells resistant to TGF-b mediated apoptosis (200, 201). Both strategies demonstrated increased T cell infiltration and effective tumor cell eradication in high TGF-b environments in both mice and humans, which highlights the possibility to produce T cells resistant to tolerogenic stimuli. Thus, selective depletion of suppressive immune stimuli and/or cells is suggested to be feasible and safe. These treatments could be further developed to increase the immunogenic effects in the TME.
Together, these studies underly the possibility to regulate and dampen the tolerogenic effects induced by TCD. Clinical studies that are currently being conducted will show whether these strategies are effective in humans and will provide more knowledge of the tolerogenic effects in the TME.
Conclusions and Perspectives
Although the mechanisms governing immunogenic TCD are extremely complicated and vary greatly between patients and cancer types, we sought to provide a broad and comprehensive overview. Notably, caspase-independent apoptosis and necroptosis are the most prevalent forms of immunogenic TCD and can direct the balance of the TME towards an immunostimulatory and activating environment. These forms of TCD are therefore suggested to be more beneficial in terms of anti-tumor immunity, which presents an appealing rationale to combine conventional therapies with strategies that promote these types of TCD. Despite the astonishing steps forward in the field of combinatory treatments with immunotherapy, we still have a long way ahead of us before we can achieve effective treatments for all types of cancer. We sought to present four substantiated methods to tailor therapeutics more effectively based on the current knowledge; avoiding tolerogenic TCD, converting tolerogenic TCD to immunogenic TCD, boosting the immunogenic effects and regulating tolerogenic effects. Some of these strategies have been proven to be effective and safe in patients and others are presently under clinical investigation. Whereas current combinatory therapies already provide substantial benefits in patients, combining one or more of these four strategies with immunotherapy will ensure a foundation for a novel era of treatments.
The recent advances in gene editing techniques open up a new discipline of research and allow us to modulate the TME more easily and precisely through sophisticated delivery. Although the delivery of immunomodulatory agents with CAR T cells or oncolytic viruses is in its infancy, these strategies have the potential to provide vigorous methods to disrupt the TME together with the induction of TCD. Increasing knowledge enables opportunities to expand the immunomodulatory possibilities of cell-based therapies. For example, inducible CAR T cell delivery systems have been proven to be robust (177) and might be employed to exploit time-dependent effects of conventional treatments or modulate the TME over time. Furthermore, cellular delivery systems are currently designed to become resistant to the suppressive TME (200) and are also used to deliver (biological) cytotoxic substances with immunomodulatory cells (202). Such immunological-based therapies have yet to be proven to be effective in cancer patients but hold great potential for the future of cancer treatment.
A major hurdle in the pursuit of novel therapy design based on therapy-induced TCD is the inter-patient heterogeneity because consequences of treatment on the TME are patient-specific. For instance, the genetic composition of tumor cells has a considerable impact on the signaling cascade governing TCD and defects in these pathways have been observed frequently (203, 204). These genetic alterations can potentially be used as biomarkers for the design of patient-specific combinatory therapies (205). Besides predictive biomarkers, recent studies have hypothesized the correlation between the immunogenicity of therapy and the level of circulating HMGB1 in various tumor types (206, 207). This method holds the potential to measure the impact of a certain therapy in patients in real-time, but more evidence is needed to uncover the relationship between circulating biomarkers and types of cell death. Development of more accurate methods to assess immunogenicity in patients could also shed a light on the effects of other patient-specific facets such as mutational burden and tumor site, which are thought to also play a huge role in the anti-tumor immune response (208, 209). Moreover, other varying parameters within the tumor such as acidity, hypoxia and nutrient deprivation are also thought to affect pathways of TCD and release of DAMPs (210–212).
Similarly, the cellular composition of the tumor stroma plays a key role in treatment efficacy (213) and should therefore not be overlooked. A growing interest in stroma cells has led to the development of treatments targeting neovascular and fibroblasts (214, 215). This top-down approach focuses on perturbation of the TME instead of designing therapies to overcome environmental challenges. These efforts might lead to resources that allow uniformization of inter-patient TMEs pre-treatment by disrupting certain suppressive structures and enable the use of universal therapies.
For the sake of clarity, this review has made a clear distinction between tolerogenic and immunogenic effects of TCD, but it should be noted that these are two extremes on a wide spectrum. A vast number of interactions determine the balance in the tumor microenvironment and therefore, generalization of immunoregulatory concepts continues to be difficult.
Contributing to this challenge is the focus on a small subset of DAMPs, namely ATP, HMGB1 and CRT. Although it is widely recognized that currently known immunogenic and tolerogenic molecules are only a fraction of actual regulatory signals, progression in the field is slow. Garg and Agostinis (216) summarized all known DAMPs currently known, but the effects of most of these molecules remain understudied. The effects of DAMP release and immunogenic TCD on maturation and activation of immune cells in distant organs such as the lymph nodes and the bone marrow remain yet to be studied. Although it has been shown that inhibiting the release of tolerogenic signals after chemotherapy-induced TCD induces vast changes in the tumor draining lymph nodes, little is known about the relationship between therapy-induced TCD, systemic immune alterations and therapeutical outcome (217, 218). Novel research strategies can change the perception of the immunogenic and tolerogenic effects of TCD and can result in clarification of poorly understood mechanisms and unaccountable results. Furthermore, the relationship of DAMPs with non-immune stromal cells and tumor cells should be investigated more thoroughly, because some studies suggest extensive crosstalk mediated by these molecules (219, 220). This again underscores the relatively unknown effects of anti-cancer therapy on the non-malignant stromal cells but highlights the overwhelming complexity of interactions in the TME.
Furthermore, a clearer picture of the effects of certain types of TCD and immunomodulatory agents on the TME is necessary to increase our knowledge of this complicated system. Multiple studies have shown opposing results, which could be addressed with novel high-throughput techniques such as single cell RNA-seq (221). More thorough discrimination between forms of cell death has resulted in a broader spectrum of known cell death pathways, which can be implemented in future research (222). Moreover, adequate immune-competent models will shine a light on conventional and novel treatments and will pave the way for quicker translation to the clinic. Especially the gap of knowledge in the field of molecular radiotherapy holds potential for improvement. With our growing understanding of biomolecular mechanisms, clinical research is obliged to focus on the aspects underlying treatment efficacy. An increasing number of clinical trials are enrolled with combinatory immunomodulatory therapies, which could provide tremendous amounts of valuable information if properly examined. Therefore, the collaboration between fundamental, translational and clinical sciences is fundamental for comprehension of this ever-growing field of biomedical research. In the end, cancer therapies will only be fully successful if the mechanisms governing TCD and immune interactions in the TME are fully understood.
Author Contributions
TS, K-SC and KS researched data for the article. All authors contributed to writing the article the article and approved the submitted version.
Funding
This work was supported by NIH grants R01-NS121096 (KS) and R01-NS107857 (KS).
Conflict of Interest
KS owns equity in and is a member of the Board of Directors of AMASA Therapeutics, a company developing stem cell based therapies for cancer. KS’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Jarosz-Biej M, Smolarczyk R, Cichoń T, Kułach N. Tumor Microenvironment as a “Game Changer” in Cancer Radiotherapy. Int J Mol Sci (2019) 20(13):18–20. doi: 10.3390/ijms20133212
2. Brown JM. Tumor Microenvironment and the Response to Anticancer Therapy. Cancer Biol Ther (2002) 1(5):453–8. doi: 10.4161/cbt.1.5.157
3. Pietras K, Östman A. Hallmarks of Cancer: Interactions With the Tumor Stroma. Exp Cell Res (2010) 316(8):1324–31. doi: 10.1016/j.yexcr.2010.02.045
4. Hill R, Song Y, Cardiff RD, Van Dyke T. Selective Evolution of Stromal Mesenchyme With P53 Loss in Response to Epithelial Tumorigenesis. Cell (2005) 123(6):1001–11. doi: 10.1016/j.cell.2005.09.030
5. Harper J, Sainson RCA. Regulation of the Anti-Tumour Immune Response by Cancer-Associated Fibroblasts. Semin Cancer Biol (2014) 25:69–77. doi: 10.1016/j.semcancer.2013.12.005
6. Lepique AP, Daghastanli KRP, Cuccovia IM, Villa LL. Hpv16 Tumor Associated Macrophages Suppress Antitumor T Cell Responses. Clin Cancer Res (2009) 15(13):4391–400. doi: 10.1158/1078-0432.CCR-09-0489
7. Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and Tolerogenic Cell Death. Nat Rev Immunol (2009) 23(1):1–7. doi: 10.1038/nri2545
8. Lecciso M, Ocadlikova D, Sangaletti S, Trabanelli S, De Marchi E, Orioli E, et al. ATP Release From Chemotherapy-Treated Dying Leukemia Cells Elicits an Immune Suppressive Effect by Increasing Regulatory T Cells and Tolerogenic Dendritic Cells. Front Immunol (2017) 8:1918. doi: 10.3389/fimmu.2017.01918
9. Okada H, Mak TW. Pathways of Apoptotic and non-Apoptotic Death in Tumour Cells. Nat Rev Cancer (2004) 4(8):592–603. doi: 10.1038/nrc1412
10. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular Definitions of Cell Death Subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ (2012) 19(1):107–20. doi: 10.1038/cdd.2011.96
11. Hur GM, Kim Y-S, Won M, Choksi S, Liu Z. The Death Domain Kinase Rip Has an Important Role in DNA Damage-Induced, P53-Independent Cell Death. J Biol Chem (2006) 281(35):25011–7. doi: 10.1074/jbc.M605577200
12. Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas Triggers an Alternative, caspase-8–independent Cell Death Pathway Using the Kinase RIP as Effector Molecule. Nat Immunol (2000) 1(6):489–95. doi: 10.1038/82732
13. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity (2013) 38(2):209–23. doi: 10.1016/j.immuni.2013.02.003
14. Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed Lineage Kinase Domain-Like is a Key Receptor Interacting Protein 3 Downstream Component of TNF-induced Necrosis. Proc Natl Acad Sci USA (2012) 109(14):5322–7. doi: 10.1073/pnas.1200012109
15. Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the Pseudokinase MLKL Unleashes the Four-Helix Bundle Domain to Induce Membrane Localization and Necroptotic Cell Death. Proc Natl Acad Sci U.S.A. (2014) 111(42):15072–7. doi: 10.1073/pnas.1408987111
16. Muñoz-Espín D, Serrano M. Cellular Senescence: From Physiology to Pathology. Nat Rev Mol Cell Biol (2014) 15(7):482–96. doi: 10.1038/nrm3823
17. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK Inhibition Alleviates the Cellular Senescence-Associated Secretory Phenotype and Frailty in Old Age. Proc Natl Acad Sci USA (2015) 112(46):E6301–10. doi: 10.1073/pnas.1515386112
18. Hubackova S, Novakova Z, Krejcikova K, Kosar M, Dobrovolna J, Duskova P, et al. Regulation of the PML Tumor Suppressor in Drug-Induced Senescence of Human Normal and Cancer Cells by JAK/STAT-mediated Signaling. Cell Cycle (2010) 9(15):3157–71. doi: 10.4161/cc.9.15.12521
19. Pattingre S, Tassa A, Qu X, Garuti R, Xiao HL, Mizushima N, et al. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell (2005) 122(6):927–39. doi: 10.1016/j.cell.2005.07.002
20. Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-Dependent Mtorc1 Association With the ULK1–Atg13–FIP200 Complex Required for Autophagy. Schmid SL Ed Mol Biol Cell (2009) 20(7):1981–91. doi: 10.1091/mbc.e08-12-1248
21. Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic Degradation of Active Caspase-8. Autophagy (2010) 6(7):891–900. doi: 10.4161/auto.6.7.13038
22. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-Eating and Self-Killing: Crosstalk Between Autophagy and Apoptosis. Nat Rev Mol Cell Biol (2007) 8(9):741–52. doi: 10.1038/nrm2239
23. Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: Shaping the Tumor Microenvironment and Therapeutic Response. Trends Mol Med (2013) 19(7):428–46. doi: 10.1016/j.molmed.2013.04.005
24. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 Interacting Protein, Mediates Cytochrome C Release From Mitochondria in Response to Activation of Cell Surface Death Receptors. Cell (1998) 94(4):481–90. doi: 10.1016/S0092-8674(00)81589-5
25. Tait SWG, Parsons MJ, Llambi F, Bouchier-Hayes L, Connell S, Muñoz-Pinedo C, et al. Resistance to Caspase-Independent Cell Death Requires Persistence of Intact Mitochondria. Dev Cell (2010) 18(5):802–13. doi: 10.1016/j.devcel.2010.03.014
26. Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic Catastrophe: A Mechanism for Avoiding Genomic Instability. Nat Rev Mol Cell Biol (2011) 12(6):385–92. doi: 10.1038/nrm3115
27. Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death Through a Tragedy: Mitotic Catastrophe. Cell Death Differ (2008) 15(7):1153–62. doi: 10.1038/cdd.2008.47
28. Mansilla S, Priebe W, Portugal J. Mitotic Catastrophe Results in Cell Death by Caspase-Dependent and Caspase-Independent Mechanisms. Cell Cycle (2006) 5(1):53–60. doi: 10.4161/cc.5.1.2267
29. Kmiecik J, Poli A, Brons NHC, Waha A, Eide GE, Enger PØ, et al. Elevated CD3+ and CD8+ Tumor-Infiltrating Immune Cells Correlate With Prolonged Survival in Glioblastoma Patients Despite Integrated Immunosuppressive Mechanisms in the Tumor Microenvironment and At the Systemic Level. J Neuroimmunol (2013) 264(1–2):71–83. doi: 10.1016/j.jneuroim.2013.08.013
30. Garg AD, Romano E, Rufo N, Agostinis P. Immunogenic Versus Tolerogenic Phagocytosis During Anticancer Therapy: Mechanisms and Clinical Translation. Cell Death Differ (2016) 23(6):938–51. doi: 10.1038/cdd.2016.5
31. Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal to Promote Phagocytic Clearance. Nature (2009) 461(7261):282–6. doi: 10.1038/nature08296
32. Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, et al. Control of Signaling-Mediated Clearance of Apoptotic Cells by the Tumor Suppressor P53. Sci (80) (2015) 349(6247). doi: 10.1126/science.1261669
33. Aaes TL, Kaczmarek A, Delvaeye T, De Craene B, De Koker S, Heyndrickx L, et al. Vaccination With Necroptotic Cancer Cells Induces Efficient Anti-Tumor Immunity. Cell Rep (2016) 15(2):274–87. doi: 10.1016/j.celrep.2016.03.037
34. Thorburn J, Horita H, Redzic J, Hansen K, Frankel AE, Thorburn A. Autophagy Regulates Selective HMGB1 Release in Tumor Cells That are Destined to Die. Cell Death Differ (2009) 16(1):175–83. doi: 10.1038/cdd.2008.143
35. Garg AD, Dudek AM, Ferreira GB, Verfaillie T, Vandenabeele P, Krysko DV, et al. ROS-Induced Autophagy in Cancer Cells Assists in Evasion From Determinants of Immunogenic Cell Death. Autophagy (2013) 9(9):1292–307. doi: 10.4161/auto.25399
36. Ruhland MK, Loza AJ, Capietto AH, Luo X, Knolhoff BL, Flanagan KC, et al. Stromal Senescence Establishes an Immunosuppressive Microenvironment That Drives Tumorigenesis. Nat Commun (2016) 7. doi: 10.1038/ncomms11762
37. Ricci MS, Zong W. Chemotherapeutic Approaches for Targeting Cell Death Pathways. Oncologist (2006) 11(4):342–57. doi: 10.1634/theoncologist.11-4-342
38. Pan ST, Li ZL, He ZX, Qiu JX, Zhou SF. Molecular Mechanisms for Tumour Resistance to Chemotherapy. Clin Exp Pharmacol Physiol (2016) 43(8):723–37. doi: 10.1111/1440-1681.12581
39. Seitz SJ, Schleithoff ES, Koch A, Schuster A, Teufel A, Staib F, et al. Chemotherapy-Induced Apoptosis in Hepatocellular Carcinoma Involves the p53 Family and is Mediated Via the Extrinsic and the Intrinsic Pathway. Int J Cancer (2010) 126(9):2049–66. doi: 10.1002/ijc.24861
40. Stevenson L, Allen WL, Proutski I, Stewart G, Johnston L, McCloskey K, et al. Calbindin 2 (CALB2) Regulates 5-Fluorouracil Sensitivity in Colorectal Cancer by Modulating the Intrinsic Apoptotic Pathway. PloS One (2011) 6(5). doi: 10.1371/journal.pone.0020276
41. Friesen C, Herr I, Krammer P, Debatin K-M. Involvement of the CD95 (Apo-1/FAS) Receptor/Ligand System in Drug-Induced Apoptosis in Leukemia Cells. Nat Med (1996) 2(5):574–7. doi: 10.1038/nm0596-574
42. Wang YF, Chen CY, Chung SF, Chiou YH, Lo HR. Involvement of Oxidative Stress and Caspase Activation in Paclitaxel-Induced Apoptosis of Primary Effusion Lymphoma Cells. Cancer Chemother Pharmacol (2004) 54(4):322–30. doi: 10.1007/s00280-004-0831-0
43. Jong SK, Ji HL, Won WJ, Dae HC, Hee JC, Do HK, et al. Reactive Oxygen Species-Dependent EndoG Release Mediates Cisplatin-Induced Caspase-Independent Apoptosis in Human Head and Neck Squamous Carcinoma Cells. Int J Cancer (2008) 122(3):672–80. doi: 10.1002/ijc.23158
44. Onoda T, Ono T, Dhar DK, Yamanoi A, Nagasue N. Tetracycline Analogues (Doxycycline and COL-3) Induce Caspase-Dependent and -Independent Apoptosis in Human Colon Cancer Cells. Int J Cancer (2006) 118(5):1309–15. doi: 10.1002/ijc.21447
45. Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-Like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat Med (2007) 13(9):1050–9. doi: 10.1038/nm1622
46. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells Through Trans-Activation of LRP on the Phagocyte. Cell (2005) 123(2):321–34. doi: 10.1016/j.cell.2005.08.032
47. Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The Interaction Between HMGB1 and TLR4 Dictates the Outcome of Anticancer Chemotherapy and Radiotherapy. Immunol Rev (2007) 220(1):47–59. doi: 10.1111/j.1600-065X.2007.00573.x
48. Kim WS, Kim H, Kwon KW, Im SH, Lee BR, Ha SJ, et al. Cisplatin Induces Tolerogenic Dendritic Cells in Response to TLR Agonists Via the Abundant Production of IL-10, Thereby Promoting Th2- and Tr1-biased T-Cell Immunity. Oncotarget (2016) 7(23):33765–82. doi: 10.18632/oncotarget.9260
49. Panis C, Lemos LGT, Victorino VJ, Herrera ACSA, Campos FC, Colado Simão AN, et al. Immunological Effects of Taxol and Adryamicin in Breast Cancer Patients. Cancer Immunol Immunother (2012) 61(4):481–8. doi: 10.1007/s00262-011-1117-0
50. Paridaens R, Biganzoli L, Bruning P, Klijn JGM, Gamucci T, Houston S, et al. Paclitaxel Versus Doxorubicin as First-Line Single-Agent Chemotherapy for Metastatic Breast Cancer: A European Organization for Research and Treatment of Cancer Randomized Study With Cross-Over. J Clin Oncol (2000) 18(4):724–4. doi: 10.1200/JCO.2000.18.4.724
51. Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of Autophagy in Temozolomide-Induced Cytotoxicity for Malignant Glioma Cells. Cell Death Differ (2004) 11(4):448–57. doi: 10.1038/sj.cdd.4401359
52. Selvakumaran M, Amaravadi RK, Vasilevskaya IA, O’Dwyer PJ. Autophagy Inhibition Sensitizes Colon Cancer Cells to Antiangiogenic and Cytotoxic Therapy. Clin Cancer Res (2013) 19(11):2995–3007. doi: 10.1158/1078-0432.CCR-12-1542
53. Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, et al. Autophagy Mediates the Mitotic Senescence Transition. Genes Dev (2009) 23(7):798–803. doi: 10.1101/gad.519709
54. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-Based Unconventional Secretory Pathway for Extracellular Delivery of IL-1β. EMBO J (2011) 30(23):4701–11. doi: 10.1038/emboj.2011.398
55. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-Dependent Anticancer Immune Responses Induced by Chemotherapeutic Agents in Mice. Sci (80) (2011) 334(December):1573–8. doi: 10.1126/science.1208347
56. Guo JY, Chen H-Y, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras Requires Autophagy to Maintain Oxidative Metabolism and Tumorigenesis. Genes Dev (2011) 25(5):460–70. doi: 10.1101/gad.2016311
57. Xu Y, Lin Z, Zhao N, Zhou L, Liu F, Cichacz Z, et al. Receptor Interactive Protein Kinase 3 Promotes Cisplatin-Triggered Necrosis in Apoptosis-Resistant Esophageal Squamous Cell Carcinoma Cells. PloS One (2014) 9(6):1–13. doi: 10.1371/journal.pone.0100127
58. Brown MF, Leibowitz BJ, Chen D, He K, Zou F, Sobol RW, et al. Loss of Caspase-3 Sensitizes Colon Cancer Cells to Genotoxic Stress Via RIP1-dependent Necrosis. Cell Death Dis (2015) 6(4):e1729. doi: 10.1038/cddis.2015.104
59. Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, et al. A Role for Tubular Necroptosis in Cisplatin-Induced Aki. J Am Soc Nephrol (2015) 26(11):2647–58. doi: 10.1681/ASN.2014080741
60. Yang H, Ma Y, Chen G, Zhou H, Yamazaki T, Klein C, et al. Contribution of RIP3 and MLKL to Immunogenic Cell Death Signaling in Cancer Chemotherapy. Oncoimmunology (2016) 5(6):1–13. doi: 10.1080/2162402X.2016.1149673
61. Snyder AG, Hubbard NW, Messmer MN, Kofman SB, Hagan CE, Orozco SL, et al. Intratumoral Activation of the Necroptotic Pathway Components RIPK1 and RIPK3 Potentiates Anti-Tumor Immunity. Sci Immunol (2019) 176(3):139–48. doi: 10.1126/sciimmunol.aaw2004
62. Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, et al. Methylation-Dependent Loss of RIP3 Expression in Cancer Represses Programmed Necrosis in Response to Chemotherapeutics. Cell Res (2015) 25(6):707–25. doi: 10.1038/cr.2015.56
63. Hu B, Shi D, Lv X, Chen S, Huang Q, Xie M, et al. Prognostic and Clinicopathological Significance of MLKL Expression in Cancer Patients: A Meta-Analysis. BMC Cancer (2018) 18(1):736. doi: 10.1186/s12885-018-4655-4
64. te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA Damage is Able to Induce Senescence in Tumor Cells In Vitro and In Vivo. Cancer Res (2002) 62(6):1876–83.
65. Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A Senescence Program Controlled by p53 and p16INK4a Contributes to the Outcome of Cancer Therapy. Cell (2002) 109(3):335–46. doi: 10.1016/S0092-8674(02)00734-1
66. Jackson JG, Pant V, Li Q, Chang LL, Quintás-Cardama A, Garza D, et al. P53-Mediated Senescence Impairs the Apoptotic Response to Chemotherapy and Clinical Outcome in Breast Cancer. Cancer Cell (2012) 21(6):793–806. doi: 10.1016/j.ccr.2012.04.027
67. Chang BD, Swift ME, Shen M, Fang J, Broude EV, Roninson IB. Molecular Determinants of Terminal Growth Arrest Induced in Tumor Cells by a Chemotherapeutic Agent. Proc Natl Acad Sci U S A (2002) 99(1):389–94. doi: 10.1073/pnas.012602599
68. Coppé J-P, Desprez P-Y, Krtolica A, Campisi J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu Rev Pathol Mech Dis (2010) 5(1):99–118. doi: 10.1146/annurev-pathol-121808-102144
69. Ohanna M, Cheli Y, Bonet C, Bonazzi VF, Allegra M, Giuliano S, et al. Secretome From Senescent Melanoma Engages the STAT3 Pathway to Favor Reprogramming of Naive Melanoma Towards a Tumor-Initiating Cell Phenotype. Oncotarget (2013) 4(12):2212–24. doi: 10.18632/oncotarget.1143
70. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent Fibroblasts Promote Epithelial Cell Growth and Tumorigenesis: A Link Between Cancer and Aging. Proc Natl Acad Sci U S A (2001) 98(21):12072–7. doi: 10.1073/pnas.211053698
71. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell (2016) 30(4):533–47. doi: 10.1016/j.ccell.2016.09.003
72. Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, et al. Tumour-Infiltrating Gr-1 + Myeloid Cells Antagonize Senescence in Cancer. Nature (2014) 515(7525):134–7. doi: 10.1038/nature13638
73. Toso A, Revandkar A, DiMitri D, Guccini I, Proietti M, Sarti M, et al. Enhancing Chemotherapy Efficacy in Pten-Deficient Prostate Tumors by Activating the Senescence-Associated Antitumor Immunity. Cell Rep (2014) 9(1):75–89. doi: 10.1016/j.celrep.2014.08.044
74. Sidi R, Pasello G, Opitz I, Soltermann A, Tutic M, Rehrauer H, et al. Induction of Senescence Markers After Neo-Adjuvant Chemotherapy of Malignant Pleural Mesothelioma and Association With Clinical Outcome: An Exploratory Analysis. Eur J Cancer (2011) 47(2):326–32. doi: 10.1016/j.ejca.2010.09.044
75. Pawlowska E, Szczepanska J, Szatkowska M, Blasiak J. An Interplay Between Senescence, Apoptosis and Autophagy in Glioblastoma Multiforme—Role in Pathogenesis and Therapeutic Perspective. Int J Mol Sci (2018) 19(3). doi: 10.3390/ijms19030889
76. Knizhnik AV, Roos WP, Nikolova T, Quiros S, Tomaszowski KH, Christmann M, et al. Survival and Death Strategies in Glioma Cells: Autophagy, Senescence and Apoptosis Triggered by a Single Type of Temozolomide-Induced Dna Damage. PloS One (2013) 8(1):1–12. doi: 10.1371/journal.pone.0055665
77. Kerr JFR, Wyllie AH, Currie AR. Apoptosis: A Basic Biological Phenomenon With Wide-Ranging Implications in Human Disease. Br J Cancer (1972) 26:239. doi: 10.1038/bjc.1972.33
78. Ogura A, Oowada S, Kon Y, Hirayama A, Yasui H, Meike S, et al. Redox Regulation in Radiation-Induced Cytochrome C Release From Mitochondria of Human Lung Carcinoma A549 Cells. Cancer Lett (2009) 277(1):64–71. doi: 10.1016/j.canlet.2008.11.021
79. Sia J, Szmyd R, Hau E, Gee HE. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front Cell Dev Biol (2020) 8:41/full(February). doi: 10.3389/fcell.2020.00041/full
80. Takasawa R, Nakamura H, Mori T, Tanuma S. Differential Apoptotic Pathways in Human Keratinocyte HaCaT Cells Exposed to UVB and UVC. Apoptosis (2005) 10(5):1121–30. doi: 10.1007/s10495-005-0901-8
81. Scoltock AB, Cidlowski JA. Activation of Intrinsic and Extrinsic Pathways in Apoptotic Signaling During UV-C-induced Death of Jurkat Cells: The Role of Caspase Inhibition. Exp Cell Res (2004) 297(1):212–23. doi: 10.1016/j.yexcr.2004.03.025
82. Murahashi H, Azuma H, Zamzami N, Furuya K, Ikebuchi K, Yamaguchi M, et al. Possible Contribution of Apoptosis-Inducing Factor (AIF) and Reactive Oxygen Species (ROS) to UVB-induced Caspase-Independent Cell Death in the T Cell Line Jurkat. J Leukoc Biol (2003) 73(3):399–406. doi: 10.1189/jlb.0702335
83. Nehs MA, Lin CI, Kozono DE, Whang EE, Cho NL, Zhu K, et al. Necroptosis is a Novel Mechanism of Radiation-Induced Cell Death in Anaplastic Thyroid and Adrenocortical Cancers. Surgery (2011) 150(6):1032–9. doi: 10.1016/j.surg.2011.09.012
84. Wang L, He L, Bao G, He X, Fan S, Wang H. Ionizing Radiation Induces HMGB1 Cytoplasmic Translocation and Extracellular Release. Int J Radiat Med Nucl Med (2016) 40(2):91–9.
85. Wang HH, Wu ZQ, Qian D, Zaorsky NG, Qiu MH, Cheng JJ, et al. Ablative Hypofractionated Radiation Therapy Enhances Non-Small Cell Lung Cancer Cell Killing Via Preferential Stimulation of Necroptosis in Vitro and In Vivo. Int J Radiat Oncol Biol Phys (2018) 101(1):49–62. doi: 10.1016/j.ijrobp.2018.01.036
86. Jo GH, Bögler O, Chwae YJ, Yoo H, Lee SH, Park JB, et al. Radiation-Induced Autophagy Contributes to Cell Death and Induces Apoptosis Partly in Malignant Glioma Cells. Cancer Res Treat (2015) 47(2):221–41. doi: 10.4143/crt.2013.159
87. Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP. Autophagy Contributes to Resistance of Tumor Cells to Ionizing Radiation. Radiother Oncol (2011) 99(3):287–92. doi: 10.1016/j.radonc.2011.06.002
88. Fukumoto M, Kuwahara Y, Oikawa T, Ochiai Y, Roudkenar MH, Fukamoto M, et al. Enhancement of Autophagy is a Potential Modality for Tumors Refractory to Radiotherapy. Cell Death Dis (2011) 2(6). doi: 10.1038/cddis.2011.56
89. Ohshima Y, Tsukimoto M, Takenouchi T, Harada H, Suzuki A, Sato M, et al. γ-Irradiation Induces P2X7 Receptor-Dependent ATP Release From B16 Melanoma Cells. Biochim Biophys Acta Gen Subj (2010) 1800(1):40–6. doi: 10.1016/j.bbagen.2009.10.008
90. Ianzini F, Bertoldo A, Kosmacek EA, Phillips SL, Mackey MA. Lack of p53 Function Promotes Radiation-Induced Mitotic Catastrophe in Mouse Embryonic Fibroblast Cells. Cancer Cell Int (2006) 6:1–8. doi: 10.1186/1475-2867-6-11
91. Suzuki K, Mori I, Nakayama Y, Miyakoda M, Kodama S, Watanabe M. Radiation-Induced Senescence-like Growth Arrest Requires TP53 Function But Not Telomere Shortening. Radiat Res (2001) 155(1):248–53. doi: 10.1667/0033-7587(2001)155[0248:RISLGA]2.0.CO;2
92. Rodier F, Coppé JP, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, et al. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat Cell Biol (2009) 11(8):973–9. doi: 10.1038/ncb1909
93. Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. Downward J, Editor. PloS Biol (2008) 6(12):e301. doi: 10.1371/journal.pbio.0060301
94. Jeon HY, Kim JK, Ham SW, Oh SY, Kim J, Park JB, et al. Irradiation Induces Glioblastoma Cell Senescence and Senescence-Associated Secretory Phenotype. Tumor Biol (2016) 37(5):5857–67. doi: 10.1007/s13277-015-4439-2
95. Theelen WSME, Peulen HMU, Lalezari F, van der Noort V, de Vries JF, Aerts JGJV, et al. Effect of Pembrolizumab After Stereotactic Body Radiotherapy vs Pembrolizumab Alone on Tumor Response in Patients With Advanced non–Small Cell Lung Cancer. JAMA Oncol (2019) 5(9):1276. doi: 10.1001/jamaoncol.2019.1478
96. Brown JM, Brenner DJ, Carlson DJ. Dose Escalation, Not “New Biology,” Can Account for the Efficacy of Stereotactic Body Radiation Therapy With non-Small Cell Lung Cancer. Int J Radiat Oncol (2013) 85(5):1159–60. doi: 10.1016/j.ijrobp.2012.11.003
97. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol (2017) 8(AUG):1–15. doi: 10.3389/fphar.2017.00561
98. Gill S, June CH. Going Viral: Chimeric Antigen Receptor T-cell Therapy for Hematological Malignancies. Immunol Rev (2015) 263(1):68–89. doi: 10.1111/imr.12243
99. Andersen MH, Schrama D, thor Straten P, Becker JC. Cytotoxic T Cells. J Invest Dermatol (2006) 126(1):32–41. doi: 10.1038/sj.jid.5700001
100. Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic Activity of the caspase-8–FLIPL Complex Inhibits RIPK3-dependent Necrosis. Nature (2011) 471(7338):363–7. doi: 10.1038/nature09852
101. Shresta S, Graubert TA, Thomas DA, Raptis SZ, Ley TJ. Granzyme A Initiates an Alternative Pathway for Granule-Mediated Apoptosis. Immunity (1999) 10(5):595–605. doi: 10.1016/S1074-7613(00)80059-X
102. Adrain C, Murphy BM, Martin SJ. Molecular Ordering of the Caspase Activation Cascade Initiated by the Cytotoxic T Lymphocyte/Natural Killer (CTL/NK) Protease Granzyme B. J Biol Chem (2005) 280(6):4663–73. doi: 10.1074/jbc.M410915200
103. Monney L, Olivier R, Otter I, Jansen B, Poirier GG, Borner C. Role of an Acidic Compartment in Tumor-Necrosis-Factor-α-Induced Production of Ceramide, Activation of Caspase-3 and Apoptosis. Eur J Biochem (1998) 251(1–2):295–303. doi: 10.1046/j.1432-1327.1998.2510295.x
104. He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-α. Cell (2009) 137(6):1100–11. doi: 10.1016/j.cell.2009.05.021
105. Spear P, Barber A, Rynda-Apple A, Sentman CL. Chimeric Antigen Receptor T Cells Shape Myeloid Cell Function Within the Tumor Microenvironment Through IFN-γ and GM-CSF. J Immunol (2012) 188(12):6389–98. doi: 10.4049/jimmunol.1103019
106. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (Her2) –Specific Chimeric Antigen Receptor–Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi: 10.1200/JCO.2014.58.0225
107. Andtbacka RHI, Ross M, Puzanov I, Milhem M, Collichio F, Delman KA, et al. Patterns of Clinical Response With Talimogene Laherparepvec (T-VEC) in Patients With Melanoma Treated in the OPTiM Phase III Clinical Trial. Ann Surg Oncol (2016) 23(13):4169–77. doi: 10.1245/s10434-016-5286-0
108. Hassan MAIAE, van der Meulen-Muileman I, Abbas S, Kruyt FAE. Conditionally Replicating Adenoviruses Kill Tumor Cells Via a Basic Apoptotic Machinery-Independent Mechanism That Resembles Necrosis-Like Programmed Cell Death. J Virol (2004) 78(22):12243–51. doi: 10.1128/JVI.78.22.12243-12251.2004
109. Rodriguez-Rocha H, Gomez-Gutierrez JG, Garcia-Garcia A, Rao X, Chen L, McMasters KM, et al. Adenoviruses Induce Autophagy to Promote Virus Replication and Oncolysis. Virology (2011) 416(1–2):9–15. doi: 10.1016/j.virol.2011.04.017
110. Liikanen I, Ahtiainen L, Hirvinen ML, Bramante S, Cerullo V, Nokisalmi P, et al. Oncolytic Adenovirus With Temozolomide Induces Autophagy and Antitumor Immune Responses in Cancer Patients. Mol Ther (2013) 21(6):1212–23. doi: 10.1038/mt.2013.51
111. Jiang H, Clise-Dwyer K, Ruisaard KE, Fan X, Tian W, Gumin J, et al. Delta-24-RGD Oncolytic Adenovirus Elicits Anti-Glioma Immunity in an Immunocompetent Mouse Model. Castro MG, Editor. PloS One (2014) 9(5):e97407. doi: 10.1371/journal.pone.0097407
112. Colunga AG, Laing JM, Aurelian L. The HSV-2 Mutant ΔPK Induces Melanoma Oncolysis Through Nonredundant Death Programs and Associated With Autophagy and Pyroptosis Proteins. Gene Ther (2010) 17(3):315–27. doi: 10.1038/gt.2009.126
113. Galvan V, Brandimarti R, Roizman B. Herpes Simplex Virus 1 Blocks Caspase-3-Independent and Caspase-Dependent Pathways to Cell Death. J Virol (1999) 73(4):3219–26. doi: 10.1128/JVI.73.4.3219-3226.1999
114. Huang Z, Wu S, Liang Y, Zhou X, Chen W, Li L, et al. Necroptosis to Restrict Virus Propagation in Mice Article Initiates Necroptosis to Restrict Virus Propagation in Mice. Cell Host Microbe (2015) 17(2):229–42. doi: 10.1016/j.chom.2015.01.002
115. Takasu A, Masui A, Hamada M, Imai T, Iwai S, Yura Y. Immunogenic Cell Death by Oncolytic Herpes Simplex Virus Type 1 in Squamous Cell Carcinoma Cells. Cancer Gene Ther (2016) 23(4):107–13. doi: 10.1038/cgt.2016.8
116. Workenhe ST, Simmons G, Pol JG, Lichty BD, Halford WP, Mossman KL. Immunogenic HSV-mediated Oncolysis Shapes the Antitumor Immune Response and Contributes to Therapeutic Efficacy. Mol Ther (2014) 22(1):123–31. doi: 10.1038/mt.2013.238
117. Streby KA, Geller JI, Currier MA, Warren PS, Racadio JM, Towbin AJ, et al. Intratumoral Injection of HSV1716, an Oncolytic Herpes Virus, is Safe and Shows Evidence of Immune Response and Viral Replication in Young Cancer Patients. Clin Cancer Res (2017) 23(14):3566–74. doi: 10.1158/1078-0432.CCR-16-2900
118. Tada S, Hamada M, Yura Y. Proteomic Analysis of Secretomes of Oncolytic Herpes Simplex Virus-Infected Squamous Cell Carcinoma Cells. Cancers (Basel) (2018) 10(2):28. doi: 10.3390/cancers10020028
119. Guo H, Omoto S, Harris PA, Finger JN, Bertin J, Gough PJ, et al. Herpes Simplex Virus Suppresses Necroptosis in Human Cells. Cell Host Microbe (2015) 17(2):243–51. doi: 10.1016/j.chom.2015.01.003
120. Zappasodi R, Pupa SM, Ghedini GC, Bongarzone I, Magni M, Cabras AD, et al. Improved Clinical Outcome in Indolent B-cell Lymphoma Patients Vaccinated With Autologous Tumor Cells Experiencing Immunogenic Death. Cancer Res (2010) 70(22):9062–72. doi: 10.1158/0008-5472.CAN-10-1825
121. Wu DH, Jia CC, Chen J, Lin ZX, Ruan DY, Li X, et al. Autophagic LC3B Overexpression Correlates With Malignant Progression and Predicts a Poor Prognosis in Hepatocellular Carcinoma. Tumor Biol (2014) 35(12):12225–33. doi: 10.1007/s13277-014-2531-7
122. Wang YJ, Fletcher R, Yu J, Zhang L. Immunogenic Effects of Chemotherapy-Induced Tumor Cell Death. Genes Dis (2018) 5(3):194–203. doi: 10.1016/j.gendis.2018.05.003
123. Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, et al. Restoration of the Immunogenicity of Cisplatin-Induced Cancer Cell Death by Endoplasmic Reticulum Stress. Oncogene (2011) 30(10):1147–58. doi: 10.1038/onc.2010.500
124. Montagnani F, Turrisi G, Marinozzi C, Aliberti C, Fiorentini G. Effectiveness and Safety of Oxaliplatin Compared to Cisplatin for Advanced, Unresectable Gastric Cancer: A Systematic Review and Meta-Analysis. Gastric Cancer (2011) 14(1):50–5. doi: 10.1007/s10120-011-0007-7
125. Baskar R, Lee KA, Yeo R, Yeoh KW. Cancer and Radiation Therapy: Current Advances and Future Directions. Int J Med Sci (2012) 9(3):193–9. doi: 10.7150/ijms.3635
126. Golden EB, Frances D, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC. Radiation Fosters Dose-Dependent and Chemotherapy-Induced Immunogenic Cell Death. Oncoimmunology (2014) 3(4). doi: 10.4161/onci.28518
127. Rutter CE, Park HS, Corso CD, Yeboa DN, Mancini BR, Lester-Coll NH, et al. Comparison of Survival Outcomes Among Standard Radiotherapy Regimens in Limited-Stage Small Cell Lung Cancer Patients Receiving Concurrent Chemoradiation. Lung Cancer (2015) 90(2):243–8. doi: 10.1016/j.lungcan.2015.08.002
128. Schaue D, Ratikan JA, Iwamoto KS, McBride WH. Maximizing Tumor Immunity With Fractionated Radiation. Int J Radiat Oncol Biol Phys (2012) 83(4):1306–10. doi: 10.1016/j.ijrobp.2011.09.049
129. Sharma P, Shen Y, Wen S, Yamada S, Jungbluth AA, Gnjatic S, et al. CD8 Tumor-Infiltrating Lymphocytes are Predictive of Survival in Muscle-Invasive Urothelial Carcinoma. Proc Natl Acad Sci USA (2007) 104(10):3967–72. doi: 10.1073/pnas.0611618104
130. Krysko O, Aaes TL, Kagan VE, D’Herde K, Bachert C, Leybaert L, et al. Necroptotic Cell Death in Anti-Cancer Therapy. Immunol Rev (2017) 280(1):207–19. doi: 10.1111/imr.12583
131. Wu YT, Tan HL, Huang Q, Sun XJ, Zhu X, Shen HM. ZVAD-Induced Necroptosis in L929 Cells Depends on Autocrine Production of Tnfα Mediated by the PKC-MAPKs-AP-1 Pathway. Cell Death Differ (2011) 18(1):26–37. doi: 10.1038/cdd.2010.72
132. Werthmöller N, Frey B, Wunderlich R, Fietkau R, Gaipl US. Modulation of Radiochemoimmunotherapy-Induced B16 Melanoma Cell Death by the Pan-Caspase Inhibitor zVAD-fmk Induces Anti-Tumor Immunity in a HMGB1-, Nucleotide- and T-cell-dependent Manner. Cell Death Dis (2015) 6(5):1–11. doi: 10.1038/cddis.2015.129
133. Kim KW, Moretti L, Lu B. M867, a Novel Selective Inhibitor of Caspase-3 Enhances Cell Death and Extends Tumor Growth Delay in Irradiated Lung Cancer Models. PloS One (2008) 3(5). doi: 10.1371/journal.pone.0002275
134. Han C, Liu Z, Zhang Y, Shen A, Dong C, Zhang A, et al. Tumor Cells Suppress Radiation-Induced Immunity by Hijacking Caspase 9 Signaling. Nat Immunol (2020) 21(5):546–54. doi: 10.1038/s41590-020-0641-5
135. Kim KW, Hwang M, Moretti L, Jaboin JJ, Cha YI, Lu B. Autophagy Upregulation by Inhibitors of Caspase-3 and mTOR Enhances Radiotherapy in a Mouse Model of Lung Cancer. Autophagy (2008) 4(5):659–68. doi: 10.4161/auto.6058
136. Yang D, Liang Y, Zhao S, Ding Y, Zhuang Q, Shi Q, et al. ZBP1 Mediates Interferon-Induced Necroptosis. Cell Mol Immunol (2020) 17(4):356–68. doi: 10.1038/s41423-019-0237-x
137. Perez D, White E. TNF-a Signals Apoptosis Through a Bid-Dependent Conformational Change in Bax That Is Inhibited by E1B 19k. Mol Cell (2000) 6(1):53–63. doi: 10.1016/S1097-2765(05)00013-4
138. Marcellus C, Teodoro JG, Charbonneau R, Shore GC, Branton PE. Expression Apoptosis Adenovirus of p53 in Saos-2 Osteosarcoma Which can Be Inhibited by BcI-2 Kda Protein1 Cells Induces or the. Cell Growth Differ (1996) 7(December):1643–50.
139. Benedict CA, Norris PS, Prigozy TI, Bodmer J, Mahr JA, Garnett CT, et al. Three Adenovirus E3 Proteins Cooperate to Evade Apoptosis by Tumor Necrosis Factor-related Apoptosis-Inducing Ligand Receptor-1 and -2. J Biol Chem (2001) 276(5):3270–8. doi: 10.1074/jbc.M008218200
140. Orvedahl A, Alexander D, Tallóczy Z, Sun Q, Wei Y, Zhang W, et al. Hsv-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe (2007) 1(1):23–35. doi: 10.1016/j.chom.2006.12.001
141. Jerome KR, Fox R, Chen Z, Sears AMYE, Lee H, Corey L. Herpes Simplex Virus Inhibits Apoptosis Through the Action of Two Genes , Us5 and Us3. J Virol (1999) 73(11):8950–7. doi: 10.1128/JVI.73.11.8950-8957.1999
142. Dambach MJ, Trecki J, Martin N, Markovitz NS. Oncolytic Viruses Derived From the γ34.5-Deleted Herpes Simplex Virus Recombinant R3616 Encode a Truncated Ul3 Protein. Mol Ther (2006) 13(5):891–8. doi: 10.1016/j.ymthe.2006.02.006
143. Zou A, Atencio I, Huang WM, Horn M, Ramachandra M. Overexpression of Adenovirus E3-11.6K Protein Induces Cell Killing by Both Caspase-Dependent and Caspase-Independent Mechanisms. Virology (2004) 326(2):240–9. doi: 10.1016/j.virol.2004.06.007
144. Dobbelstein M, Shenk T, Hughes H, Biology M. Protection Against Apoptosis by the Vaccinia Virus SPI-2 ( B13R ) Gene Product. J Virol (1996) 70(9):6479–85. doi: 10.1128/JVI.70.9.6479-6485.1996
145. Velarde MC, Demaria M, Campisi J. Senescent Cells and Their Secretory Phenotype as Targets for Cancer Therapy. In: . Interdisciplinary Topics in Gerontology (2013). p. 17–27. Available at: https://www.karger.com/Article/FullText/343572.
146. NCT02876302. Available at: https://clinicaltrials.gov/ct2/show/NCT02876302.
147. Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy Sustains Pancreatic Cancer Growth Through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discovery (2018) 8(3):276–87. doi: 10.1158/2159-8290.CD-17-0952
148. Chen Y, Li X, Guo L, Wu X, He C, Zhang S, et al. Combining Radiation With Autophagy Inhibition Enhances Suppression of Tumor Growth and Angiogenesis in Esophageal Cancer. Mol Med Rep (2015) 12(2):1645–52. doi: 10.3892/mmr.2015.3623
149. Zhao XG, Sun RJ, Yang XY, Liu DY, Lei DP, Jin T, et al. Chloroquine-Enhanced Efficacy of Cisplatin in the Treatment of Hypopharyngeal Carcinoma in Xenograft Mice. PloS One (2015) 10(4):1–12. doi: 10.1371/journal.pone.0126147
150. Golden EB, Cho HY, Jahanian A, Hofman FM, Louie SG, Schönthal AH, et al. Chloroquine Enhances Temozolomide Cytotoxicity in Malignant Gliomas by Blocking Autophagy. Neurosurg Focus (2014) 37(6):1–11. doi: 10.3171/2014.9.FOCUS14504
151. Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, et al. Antitumor Adaptive Immunity Remains Intact Following Inhibition of Autophagy and Antimalarial Treatment. J Clin Invest (2016) 126(12):4417–29. doi: 10.1172/JCI85705
152. Galluzzi L, Pedro JMBS, Demaria S, Formenti SC, Kroemer G. Activating Autophagy to Potentiate Immunogenic Chemotherapy and Radiation Therapy. Nat Rev Clin Oncol (2017) 14(4):247–58. doi: 10.1038/nrclinonc.2016.183
153. Alirezaei M, Kemball CC, Flynn CT, Wood MR, Whitton JL, Kiosses WB. Short-Term Fasting Induces Profound Neuronal Autophagy. Autophagy (2010) 6(6):702–10. doi: 10.4161/auto.6.6.12376
154. Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng C-W, et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell (2016) 30(1):136–46. doi: 10.4161/auto.6.6.12376
155. Pietrocola F, Pol J, Vacchelli E, Rao S, Enot DP, Baracco EE, et al. Caloric Restriction Mimetics Enhance Anticancer Immunosurveillance. Cancer Cell (2016) 30(1):147–60. doi: 10.1016/j.ccell.2016.05.016
156. de Groot S, Pijl H, van der Hoeven JJM, Kroep JR. Effects of Short-Term Fasting on Cancer Treatment. J Exp Clin Cancer Res (2019) 38(1):209. doi: 10.1186/s13046-019-1189-9
157. NCT02126449. Clinical Trial. Available at: https://clinicaltrials.gov/ct2/show/NCT02126449.
158. MacKenzie SH, Schipper JL, Clark AC. The Potential for Caspases in Drug Discovery. Curr Opin Drug Discovery Devel (2010) 13(5):568–76.
159. Fragoulis GE, Mcinnes IB, Siebert S. JAK-Inhibitors. New Players in the Field of Immune-Mediated Diseases, Beyond Rheumatoid Arthritis. Rheumatol (United Kingdom) (2019) 58:i43–54. doi: 10.1093/rheumatology/key276
160. Liu WM, Fowler DW, Smith P, Dalgleish AG. Pre-Treatment With Chemotherapy can Enhance the Antigenicity and Immunogenicity of Tumours by Promoting Adaptive Immune Responses. Br J Cancer (2010) 102(1):115–23. doi: 10.1038/sj.bjc.6605465
161. Addeo A, Banna GL, Metro G, DI Maio M. Chemotherapy in Combination With Immune Checkpoint Inhibitors for the First-Line Treatment of Patients With Advanced non-Small Cell Lung Cancer: A Systematic Review and Literature-Based Meta-Analysis. Front Oncol (2019) 9(MAR). doi: 10.3389/fonc.2019.00264
162. Manukian G, Bar-Ad V, Lu B, Argiris A, Johnson JM. Combining Radiation and Immune Checkpoint Blockade in the Treatment of Head and Neck Squamous Cell Carcinoma. Front Oncol (2019) 9(MAR):1–14. doi: 10.3389/fonc.2019.00122
163. Wu J, Waxman DJ. Immunogenic Chemotherapy: Dose and Schedule Dependence and Combination With Immunotherapy. Cancer Lett (2018) 419(1):210–21. doi: 10.1016/j.canlet.2018.01.050
164. Xu C, Zhang Y, Alexander Rolfe P, Hernández VM, Guzman W, Kradjian G, et al. Combination Therapy With NHS-muIL12 and Avelumab (anti-PD-L1) Enhances Antitumor Efficacy in Preclinical Cancer Models. Clin Cancer Res (2017) 23(19):5869–80. doi: 10.1158/1078-0432.CCR-17-0483
165. Kushner BH, Kramer K, Cheung NV. Phase II Trial of the Anti-G D2 Monoclonal Antibody 3F8 and Granulocyte-Macrophage Colony-Stimulating Factor for Neuroblastoma. J Clin Oncol (2001) 19(22):4189–94. doi: 10.1200/JCO.2001.19.22.4189
166. Chen P, Chen F, Zhou B. Comparisons of Therapeutic Efficacy and Safety of Ipilimumab Plus GM-CSF Versus Ipilimumab Alone in Patients With Cancer: A Meta-Analysis of Outcomes. Drug Des Devel Ther (2018) 12:2025–38. doi: 10.2147/DDDT.S154258
167. Phan GQ, Attia P, Steinberg SM, White DE, Rosenberg SA. Factors Associated With Response to High-Dose Interleukin-2 in Patients With Metastatic Melanoma. J Clin Oncol (2001) 19(15):3477–82. doi: 10.1200/JCO.2001.19.15.3477
168. Seo SH, Han HD, Noh KH, Kim TW, Son SW. Chitosan Hydrogel Containing GMCSF and a Cancer Drug Exerts Synergistic Anti-Tumor Effects Via the Induction of CD8+ T Cell-Mediated Anti-Tumor Immunity. Clin Exp Metastasis (2009) 26(3):179–87. doi: 10.1007/s10585-008-9228-5
169. Kwong B, Liu H, Irvine DJ. Induction of Potent Anti-Tumor Responses While Eliminating Systemic Side Effects Via Liposome-Anchored Combinatorial Immunotherapy. Biomaterials (2011) 32(22):5134–47. doi: 10.1016/j.biomaterials.2011.03.067
170. Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, et al. Antiviral and Antitumor T-Cell Immunity in Patients Treated With GM-CSF-Coding Oncolytic Adenovirus. Clin Cancer Res (2013) 19(10):2734–44. doi: 10.1158/1078-0432.CCR-12-2546
171. Liu Z, Ravindranathan R, Kalinski P, Guo ZS, Bartlett DL. Rational Combination of Oncolytic Vaccinia Virus and PD-L1 Blockade Works Synergistically to Enhance Therapeutic Efficacy. Nat Commun (2017) 8(1):14754. doi: 10.1038/ncomms14754
172. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell (2017) 170(6):1109–1119.e10. doi: 10.1016/j.cell.2017.08.027
173. Choi I, Lee J, Zhang S, Park J, Lee K, Sonn CH, et al. Oncolytic Adenovirus Co-Expressing IL-12 and IL-18 Improves Tumor-Specific Immunity Via Differentiation of T Cells Expressing IL-12R B 2 or IL-18R a. Gene Ther (2011), 898–909. doi: 10.1038/gt.2011.37
174. Lee Y-S, Kim J-H, Choi K-J, Choi I-K, Kim H, Cho S, et al. Enhanced Antitumor Effect of Oncolytic Adenovirus Expressing Interleukin-12 and B7-1 in an Immunocompetent Murine Model. Clin Cancer Res (2006) 12(19):5859–68. doi: 10.1158/1078-0432.CCR-06-0935
175. Patel DM, Foreman PM, Nabors LB, Riley KO, Gillespie GY, Markert JM. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients With Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum Gene Ther Clin Dev (2016) 27(2):69–78. doi: 10.1089/humc.2016.031
176. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. Il-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors In Vivo. Oncoimmunology (2015) 4(3):e994446. doi: 10.4161/2162402X.2014.994446
177. Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, et al. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J Immunol (2019) 203(1):198–207. doi: 10.4049/jimmunol.1800033
178. Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A Phase I Clinical Trial of Adoptive T Cell Therapy Using IL-12 Secreting MUC-16ecto Directed Chimeric Antigen Receptors for Recurrent Ovarian Cancer. J Transl Med (2015) 13(1):102. doi: 10.1186/s12967-015-0460-x
179. NCT02498912. Available at: https://clinicaltrials.gov/ct2/show/NCT02498912.
180. Mirisola V, Zuccarino A, Bachmeier BE, Sormani MP, Falter J, Nerlich A, et al. CXCL12/SDF1 Expression by Breast Cancers is an Independent Prognostic Marker of Disease-Free and Overall Survival. Eur J Cancer (2009) 45(14):2579–87. doi: 10.1016/j.ejca.2009.06.026
181. Kim K, Bae S, Capece T, Nedelkovska H, De Rubio RG, Smrcka AV, et al. Targeted Calcium Influx Boosts Cytotoxic T Lymphocyte Function in the Tumour Microenvironment. Nat Commun (2017) 8(May):1–10. doi: 10.1038/ncomms15365
182. Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking CXCR4 Alleviates Desmoplasia, Increases T-lymphocyte Infiltration, and Improves Immunotherapy in Metastatic Breast Cancer. Proc Natl Acad Sci USA (2019) 116(10):4558–66. doi: 10.1073/pnas.1815515116
183. Pernas S, Martin M, Kaufman PA, Gil-Martin M, Gomez Pardo P, Lopez-Tarruella S, et al. Balixafortide Plus Eribulin in HER2-negative Metastatic Breast Cancer: A Phase 1, Single-Arm, Dose-Escalation Trial. Lancet Oncol (2018) 19(6):812–24. doi: 10.1016/S1470-2045(18)30147-5
184. Na YR, Lee JS, Lee SJ, Seok SH. Interleukin-6-Induced Twist and N-cadherin Enhance Melanoma Cell Metastasis. Melanoma Res (2013) 23(6):434–43. doi: 10.1097/CMR.0000000000000021
185. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-Tumor Immunity. Cell Metab (2016) 24(5):672–84. doi: 10.1016/j.cmet.2016.10.010
186. Chen B-A, Chen R. Siltuximab (CNTO 328): A Promising Option for Human Malignancies. Drug Des Devel Ther (2015) 9:3455. doi: 10.2147/DDDT.S86438
187. Tanaka T, Narazaki M, Kishimoto T. Anti-Interleukin-6 Receptor Antibody, Tocilizumab, for the Treatment of Autoimmune Diseases. FEBS Lett (2011) 585(23):3699–709. doi: 10.1016/j.febslet.2011.03.023
188. Ando K, Takahashi F, Kato M, Kaneko N, Doi T, Ohe Y, et al. Tocilizumab, a Proposed Therapy for the Cachexia of interleukin6-expressing Lung Cancer. PloS One (2014) 9(7):1–10. doi: 10.1371/journal.pone.0102436
189. van Rhee F, Fayad L, Voorhees P, Furman R, Lonial S, Borghaei H, et al. Siltuximab, a Novel Anti–Interleukin-6 Monoclonal Antibody, for Castleman’s Disease. J Clin Oncol (2010) 28(23):3701–8. doi: 10.1200/JCO.2009.27.2377
190. Oh K, Lee O-Y, Shon SY, Nam O, Ryu PM, Seo MW, et al. A Mutual Activation Loop Between Breast Cancer Cells and Myeloid-Derived Suppressor Cells Facilitates Spontaneous Metastasis Through IL-6 Trans-Signaling in a Murine Model. Breast Cancer Res (2013) 15(5):R79. doi: 10.1186/bcr3473
191. NCT02767557. Available at: https://clinicaltrials.gov/ct2/show/NCT02767557.
192. NCT04258150. Available at: https://clinicaltrials.gov/ct2/show/NCT04258150.
193. Jordan KR, Amaria RN, Ramirez O, Callihan EB, Gao D, Borakove M, et al. Myeloid-Derived Suppressor Cells are Associated With Disease Progression and Decreased Overall Survival in Advanced-Stage Melanoma Patients. Cancer Immunol Immunother (2013) 62(11):1711–22. doi: 10.1007/s00262-013-1475-x
194. Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of All-Trans Retinoic Acid Effect on Tumor-Associated Myeloid-Derived Suppressor Cells. Cancer Res (2007) 67(22):11021–8. doi: 10.1158/0008-5472.CAN-07-2593
195. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-Trans-Retinoic Acid Improves Differentiation of Myeloid Cells and Immune Response in Cancer Patients. Cancer Res (2006) 66(18):9299–307. doi: 10.1158/0008-5472.CAN-06-1690
196. Tobin RP, Jordan KR, Robinson WA, Davis D, Borges VF, Gonzalez R, et al. Targeting Myeloid-Derived Suppressor Cells Using All-Trans Retinoic Acid in Melanoma Patients Treated With Ipilimumab. Int Immunopharmacol (2018) 63(August):282–91. doi: 10.1016/j.intimp.2018.08.007
197. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E, et al. Host STING-dependent MDSC Mobilization Drives Extrinsic Radiation Resistance. Nat Commun (2017) 8(1):1736. doi: 10.1038/s41467-017-01566-5
198. Chen H, Xu L, Li L, Liu X, Gao J, Bai Y. Inhibiting the CD8+ T Cell Infiltration in the Tumor Microenvironment After Radiotherapy is an Important Mechanism of Radioresistance. Sci Rep (2018) 8(1):11934. doi: 10.1038/s41598-018-30417-6
199. Thomas DA, Massagué J. Tgf-β Directly Targets Cytotoxic T Cell Functions During Tumor Evasion of Immune Surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
200. Bollard CM, Tripic T, Cruz CR, Dotti G, Gottschalk S, Torrano V, et al. Tumor-Specific T-Cells Engineered to Overcome Tumor Immune Evasion Induce Clinical Responses in Patients With Relapsed Hodgkin Lymphoma. J Clin Oncol (2018) 36(11):1128–39. doi: 10.1200/JCO.2017.74.3179
201. Nguyen HH, Kim T, Song SY, Park S, Cho HH, Jung S-H, et al. Naïve CD8+ T Cell Derived Tumor-Specific Cytotoxic Effectors as a Potential Remedy for Overcoming TGF-β Immunosuppression in the Tumor Microenvironment. Sci Rep (2016) 6(1):28208. doi: 10.1038/srep28208
202. Spano C, Grisendi G, Golinelli G, Rossignoli F, Prapa M, Bestagno M, et al. Soluble TRAIL Armed Human Msc As Gene Therapy for Pancreatic Cancer. Sci Rep (2019) 9(1):1788. doi: 10.1038/s41598-018-37433-6
203. Kim HS, Lee JW, Soung YH, Park WS, Kim SY, Lee JH, et al. Inactivating Mutations of Caspase-8 Gene in Colorectal Carcinomas. Gastroenterology (2003) 125(3):708–15. doi: 10.1016/S0016-5085(03)01059-X
204. Nam JY, Jong WL, Yun JK, Young HS, Su YK, Suk WN, et al. Loss of caspase-2, -6 and -7 Expression in Gastric Cancers. Apmis (2004) 112(6):330–5. doi: 10.1111/j.1600-0463.2004.t01-1-apm1120602.x
205. Razaghi A, Heimann K, Schaeffer PM, Gibson SB. Negative Regulators of Cell Death Pathways in Cancer: Perspective on Biomarkers and Targeted Therapies. Apoptosis (2018) 23(2):93–112. doi: 10.1007/s10495-018-1440-4
206. Exner R, Sachet M, Arnold T, Zinn-Zinnenburg M, Michlmayr A, Dubsky P, et al. Prognostic Value of HMGB 1 in Early Breast Cancer Patients Under Neoadjuvant Chemotherapy. Cancer Med (2016) 5(9):2350–8. doi: 10.1002/cam4.827
207. Huang C-Y, Chiang S-F, Ke T-W, Chen T-W, Lan Y-C, You Y-S, et al. Cytosolic High-Mobility Group Box Protein 1 (HMGB1) and/or PD-1+ Tils in the Tumor Microenvironment may be Contributing Prognostic Biomarkers for Patients With Locally Advanced Rectal Cancer Who Have Undergone Neoadjuvant Chemoradiotherapy. Cancer Immunol Immunother (2018) 67(4):551–62. doi: 10.1007/s00262-017-2109-5
208. Thomas A, Routh ED, Pullikuth A, Jin G, Su J, Chou JW, et al. Tumor Mutational Burden is a Determinant of Immune-Mediated Survival in Breast Cancer. Oncoimmunology (2018) 7(10):e1490854. doi: 10.1080/2162402X.2018.1490854
209. Maleki Vareki S. High and Low Mutational Burden Tumors Versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J Immunother Cancer (2018) 6(1):157. doi: 10.1186/s40425-018-0479-7
210. Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, et al. Hypoxia Induces Autophagic Cell Death in Apoptosis-Competent Cells Through a Mechanism Involving BNIP3. Autophagy (2008) 4(2):195–204. doi: 10.4161/auto.5278
211. Parks SK, Mazure NM, Counillon L, Pouysségur J. Hypoxia Promotes Tumor Cell Survival in Acidic Conditions by Preserving ATP Levels. J Cell Physiol (2013) 228(9):1854–62. doi: 10.1002/jcp.24346
212. Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an Energy Metabolism Regulator That Switches TNF-induced Cell Death From Apoptosis to Necrosis. Sci (80) (2009) 325(5938):332–6. doi: 10.1126/science.1172308
213. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat Med (2018) 24(5):541–50. doi: 10.1038/s41591-018-0014-x
214. Yu F, Hong B, Song X. 620. Cancer Associated Fibroblasts-Targeted Oncolytic Virus Results in Enhanced Antitumor Activity in Mouse Model. Mol Ther (2015) 23(May):S246. doi: 10.1016/S1525-0016(16)34229-0
215. Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J, et al. Targeting Tumor Vasculature With an Oncolytic Virus. Mol Ther [Internet] (2011) 19(5):886–94. doi: 10.1038/mt.2011.26
216. Garg AD, Agostinis P. Cell Death and Immunity in Cancer: From Danger Signals to Mimicry of Pathogen Defense Responses. Immunol Rev (2017) 280(1):126–48. doi: 10.1111/imr.12574
217. Hayashi K, Nikolos F, Lee YC, Jain A, Tsouko E, Gao H, et al. Tipping the Immunostimulatory and Inhibitory DAMP Balance to Harness Immunogenic Cell Death. Nat Commun (2020) 11(1):6299. doi: 10.1038/s41467-020-19970-9
218. Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The PD-1/PD-L1-Checkpoint Restrains T Cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell (2020) 38(5):685–700.e8. doi: 10.1016/j.ccell.2020.09.001
219. Kang R, Tang D, Schapiro NE, Loux T, Livesey KM, Billiar TR, et al. The HMGB1/RAGE Inflammatory Pathway Promotes Pancreatic Tumor Growth by Regulating Mitochondrial Bioenergetics. Oncogene (2014) 33(5):567–77. doi: 10.1038/onc.2012.631
220. Abe A, Kuwata T, Yamauchi C, Higuchi Y, Ochiai A. High Mobility Group Box1 (HMGB1) Released From Cancer Cells Induces the Expression of Pro-Inflammatory Cytokines in Peritoneal Fibroblasts. Pathol Int (2014) 64(6):267–75. doi: 10.1111/pin.12167
221. Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Sci (80 ) (2016) 352(6282):189–96. doi: 10.1126/science.aad0501
Keywords: immunotherapy, tumor cell death, tolerogenic, immunogenic, tumor microenvironment (TME), damage associated molecular pattern (DAMP), caspase-dependent apoptosis, therapy-induced senescence
Citation: van Schaik TA, Chen K-S and Shah K (2021) Therapy-Induced Tumor Cell Death: Friend or Foe of Immunotherapy? Front. Oncol. 11:678562. doi: 10.3389/fonc.2021.678562
Received: 25 March 2021; Accepted: 03 May 2021;
Published: 01 June 2021.
Edited by:
Oliver Kepp, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Udo S. Gaipl, University Hospital Erlangen, GermanyLucillia Bezu, Gustave Roussy Cancer Campus, France
Copyright © 2021 van Schaik, Chen and Shah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Khalid Shah, a3NoYWhAYndoLmhhcnZhcmQuZWR1