 Wenting Du1
Wenting Du1 Yaqing Zhang
Yaqing Zhang- 1Department of Surgery, University of Michigan, Ann Arbor, MI, United States
- 2Rogel Cancer Center, University of Michigan, Ann Arbor, MI, United States
- 3Department of Cell and Developmental Biology, University of Michigan, Ann Arbor, MI, United States
The stroma-rich, immunosuppressive microenvironment is a hallmark of pancreatic ductal adenocarcinoma (PDA). Tumor cells and other cellular components of the tumor microenvironment, such as cancer associated fibroblasts, CD4+ T cells and myeloid cells, are linked by a web of interactions. Their crosstalk not only results in immune evasion of PDA, but also contributes to pancreatic cancer cell plasticity, invasiveness, metastasis, chemo-resistance, immunotherapy-resistance and radiotherapy-resistance. In this review, we characterize several prevalent populations of stromal cells in the PDA microenvironment and describe how the crosstalk among them drives and maintains immune suppression. We also summarize therapeutic approaches to target the stroma. With a better understanding of the complex cellular and molecular networks in PDA, strategies aimed at sensitizing PDA to chemotherapy or immunotherapy through re-programing the tumor microenvironment can be designed, and in turn lead to improved clinical treatment for pancreatic cancer patients.
Introduction
Pancreatic ductal adenocarcinoma (PDA) is the most common form of pancreatic cancer, the third leading cause of cancer related death in the United States, with a 5-year survival rate of around 10% (1, 2). Over 80% of PDA patients are diagnosed at a late stage when the tumor is already locally advanced or metastatic to distant organs and therefore do not qualify for surgery (1). Standard chemotherapy, such as Gemcitabine alone or in combination of Albumin-bound paclitaxel (Abraxane) has long been the standard of care for PDA. However, it provides only modest survival benefit since a large percentage of patients are either intrinsically resistant or develop resistance soon after treatment starts (3). Multidrug regimens such as FOLFIRINOX (combination of oxaliplatin, irinotecan, leucovorin, and fluorouracil) have become standard of care for those patients that can tolerate it, as they increase patient median overall survival to 11.1 months. However, FOLFIRINOX is associated with high toxicity (4). Therefore, there is urgent need for the development of novel therapeutic strategies for PDA patients. Immune checkpoint blockade has achieved significant therapeutic success for a subset of cancer patients. Unfortunately, single agent immunotherapy has been ineffective in PDA (5). The reasons for this failure are complex, and likely stem from the nature of the stroma-rich tumor microenvironment (TME) in PDA, with abundant immunosuppressive cells such as cancer associated fibroblasts (CAFs) (6), CD4+ T cells (7) and myeloid cells including tumor associated macrophages (TAMs) and myeloid derived suppressor cells (MDSCs) (8–11). The stromal and immune compartments are linked by a web of interactions that promotes immune evasion of PDA cancer cells and contributes to the onset and progression of pancreatic carcinogenesis, affecting cell plasticity, metastasis, chemo-resistance and radiotherapy-resistance (3, 7, 12–18). This review summarizes the crosstalk between several key cell types that are dominant within the immunosuppressive TME of pancreatic cancer and discusses the most promising immune regulatory approaches to activate anti-tumor immune responses in PDA.
The Immunologically “Cold” TME Is Modulated by Oncogenic Pathways in PDA
PDA initiates with activating mutation of oncogenes such as KRAS (mutant in over 90% of tumors, and present in the majority of precursor lesions as well) (19) and followed by inactivation of tumor suppressors such as CDKN2A or P53 (altered in 90% and 70% of PDAs, respectively) (20–23). Pancreatic cancer develops from precursor lesions such as pancreatic intraepithelial neoplasia (PanIN) that over time progress to advanced and metastatic stage (24). Other types of precursor lesions such as intraductal papillary mucinous neoplasms and mucinous cystic neoplasms (25) are less common and have been reviewed elsewhere (20). Genetically engineered mouse models (GEMMs) that harbor pancreas-specific expression of oncogenic Kras, such as KC (LSL-KrasG12D; p48/Pdx-1-Cre) (26) and iKras* model (p48-Cre; R26-rtTa-IRES-EGFP; TetO-KrasG12D) (27), recapitulate the stepwise carcinogenesis process of human PDA. PanIN occurs spontaneously in these models and can progress to metastatic cancer with long latency. Using the KC model, researchers discovered that immunosuppressive cells, including CD4+ FOXP3+ regulatory T cells (Tregs), TAMs, and MDSCs, accumulate both in PanIN and PDA stages compared to normal pancreas (28). CD8+ cytotoxic T cells are scarce in PanIN and only present in a subset of PDA; even when they are present, they lack effector function (28). Similar kinetics of leukocytic infiltration were also described in the more aggressive KPC (KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx-1-Cre) GEMM (29). Reduced infiltration of CD8+ cytotoxic T cells and increased infiltration of CD4+FOXP3+CD25+ Tregs in PDA have also been shown in human patient samples (30, 31). The evidence from both GEMMs and patient samples indicate an immunologically ‘cold’ TME of PDA. Even when CD8+ T cells are present within the tumor nest in a small cohort of PDA patients, they are dysfunctional or exhausted (32). Recently, our laboratory defined an exhausted CD8+ T cell phenotype in human PDA by expression of T cell immunoglobulin and ITIM domains (TIGIT), an immune checkpoint that is relatively understudied (11). Using a combination of mass cytometry, single-cell RNA sequencing (scRNA-seq) and multiplex immunohistochemistry, we found increased markers of CD8+ T cell dysfunction with an up-regulation of TIGIT in PDA compared to non-malignant pancreas samples; further, the dysfunctional status of CD8+ T cells was more pronounced at later stages of carcinogenesis (11).
Oncogenic KRAS is a key mediator of immune suppression in PDA. A recent study using scRNA-seq approaches and TCGA data analysis suggest greater immune infiltration in KRAS independent and KRAS-low tumors compared to KRAS dependent and KRAS-high groups (33). In this model, inactivation of mutant Kras in PDA cells did not affect their tumorigenic capacity, but led to failure to evade the host immune system (33). The authors determined that KRAS knockout (KO) PDA cells had a striking up-regulation of major histocompatibility complex I (MHC I) genes compared with KRAS intact control cells, underlying increased susceptibility to anti-tumor immunity. M1-like TAMs, CD8+ cytotoxic T cells and natural killer T (NK T) cells dominated in KRAS KO tumors. Mechanistically, this study identified BRAF and MYC as key downstream regulators of KRAS-driven tumor immune suppression for PDA maintenance (33). MHC I accumulation in the cell is also negatively regulated by autophagy, which is in turn activated by oncogenic KRAS (34).
KRAS activates essential pathways to control the expression and secretion of cytokines and chemokines from tumor cells, thereby regulating the recruitment and development of immune cells. For example, granulocyte–macrophage colony-stimulating factor (GM-CSF) produced by pancreatic cancer cells carrying the KRASG12D mutation recruits immunosuppressive myeloid cells (35, 36). A key downstream effector of KrasG12D is the mitogen−activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway. Mitogen-activated protein kinases (MAPK)/ERK targets include interleukin-10 (IL-10) and transforming growth factor beta (TGF-β), which in turn induce Treg differentiation (37). MAPK/ERK signaling also induces expression of intercellular adhesion molecule (ICAM-1), which acts as chemoattractant for macrophages (38). Besides, growth and differentiation factor 15 (GDF-15), a direct target of nuclear factor kappa B (NF-κB) in tumor cells, suppresses the pro-apoptotic activity of macrophages by inhibiting tumor necrosis factor (TNF) and nitric oxide (NO) production (39). Depletion of GDF-15 in the KPC mouse model delayed tumor development and was accompanied by increased infiltrating antitumor macrophages (39). Extracellular KrasG12D in tumor-derived exosomes directly promotes alternatively activated or M2-like macrophage polarization via signal transducer and activator of transcription 3 (STAT3)-dependent fatty acid oxidation (40). Blocking KrasG12D release from tumor cells and uptake by macrophages suppresses macrophage-mediated pancreatic tumor growth in vivo (40). Other inflammatory mediators secreted by PDA cells include granulocyte colony-stimulating factor (G-CSF) (41), IL-6 (42), IL-1α (43), IL-1β (44, 45), ubiquitin specific peptidase 22 (USP22) (46), C-X-C motif chemokine ligand 8 (CXCL8) (47), matrix metallopeptidase 9 (MMP-9) and indoleamine-2,3-dioxygenase (IDO) (48), which all contribute to the establishment of immunosuppressive TME in pancreatic cancer (Figure 1).
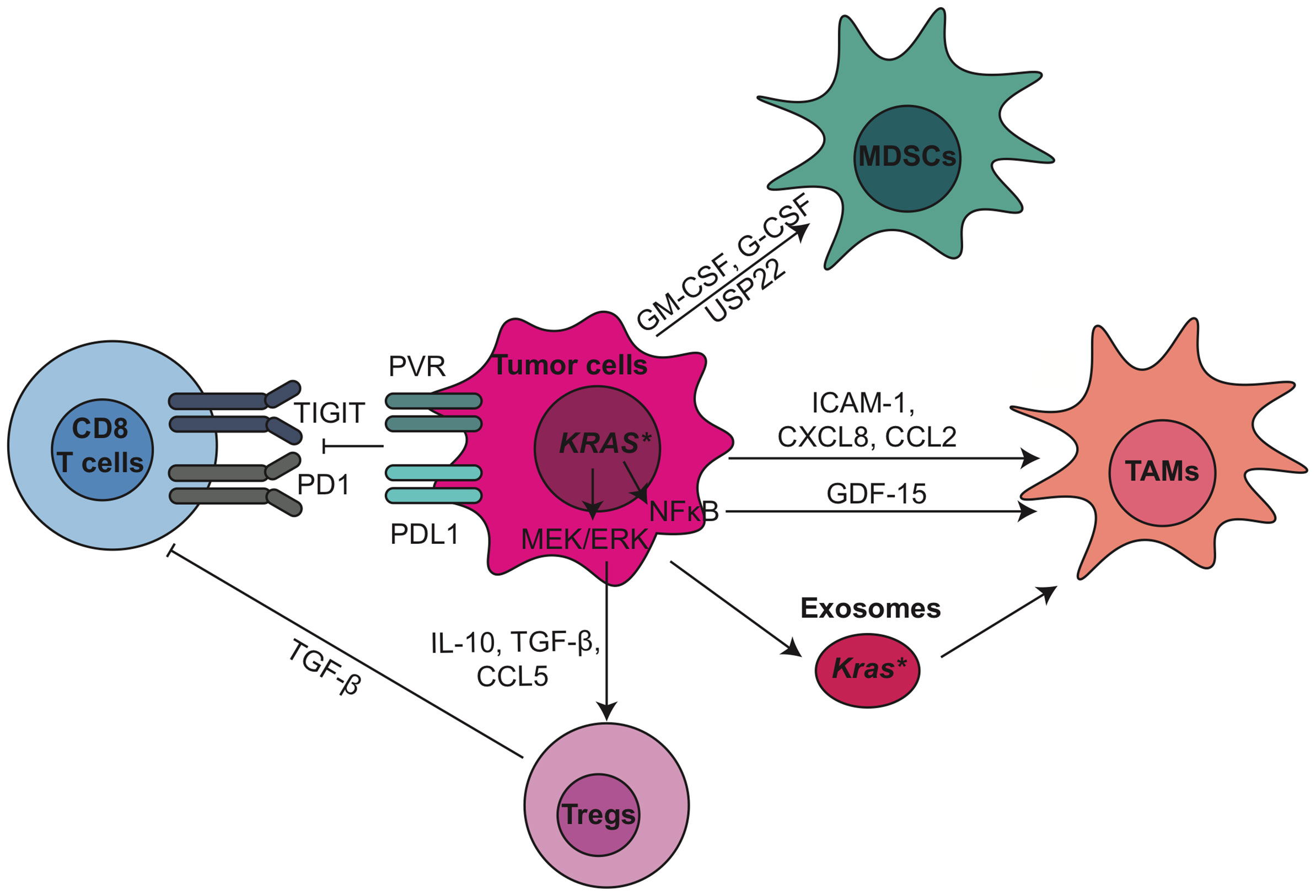
Figure 1 Tumor epithelial cells modulate immunosuppressive tumor microenvironment through oncogenic pathways and immune checkpoint pathways in PDA. CCL, C-C motif chemokine ligand; CXCL, C-X-C motif chemokine ligand; ERK, extracellular signal-regulated kinase; G-CSF, granulocyte-colony stimulating factor; GDF-15, growth/differentiation factor-15; GM-CSF, granulocyte-macrophage colony-stimulating factor; ICAM-1, intercellular adhesion molecule 1; MDSCs, myeloid-derived suppressor cells; MEK, mitogen−activated protein kinase kinase; NF-κB, nuclear factor kappa B; PD-1, anti-programmed cell death 1; PD-L1, programmed cell death ligand 1; PVR, poliovirus receptor; TAMs, tumor associated macrophages; TGF-β, transforming growth factor β; TIGIT, T cell immunoreceptor with Ig and ITIM domains; Tregs, regulatory T cells; USP22, ubiquitin specific peptidase 22.
PDA cells can also induce immune suppression by engaging critical immune checkpoint pathways such as programmed cell death protein 1 (PD1)/programmed death-ligand 1 (PD-L1). In addition, we recently discovered that Poliovirus receptor (PVR), one of the checkpoint TIGIT ligands, is expressed by tumor epithelial cells (11). Previously we showed that expression of PD-L1 in PDA cancer cells is regulated by epidermal growth factor receptor (EGFR)/MAPK signaling cascade (49). PD-L1 expressed by PDA cells directly induced the apoptosis of PD-1-expressing T cells, and its expression positively correlated with poor prognosis and inversely correlated with tumor-infiltrating T cells, particularly CD8+ T cells, in pancreatic cancer (50). These pre-clinical data have encouraged clinical trials targeting PD-1 and PD-L1, either as monotherapy or in combination with radiation or chemotherapy, in pancreatic cancer (Table 1, current active clinical trials, and Table 2, past clinical trials targeting pancreatic cancer TME). However, single agent immune checkpoint blockade has thus far been unsuccessful in PDA patients. One potential reason for this failure is the exclusion of active T cells from TME, which reveals an urgent need for strategies transforming the immunologically ‘cold’ microenvironment into ‘hot’. Targeting oncogenic signaling pathways such as KRAS, BRAF and MEK potentially provides an opportunity to alter the TME and sensitize PDA to immune checkpoint blockade (33, 51).
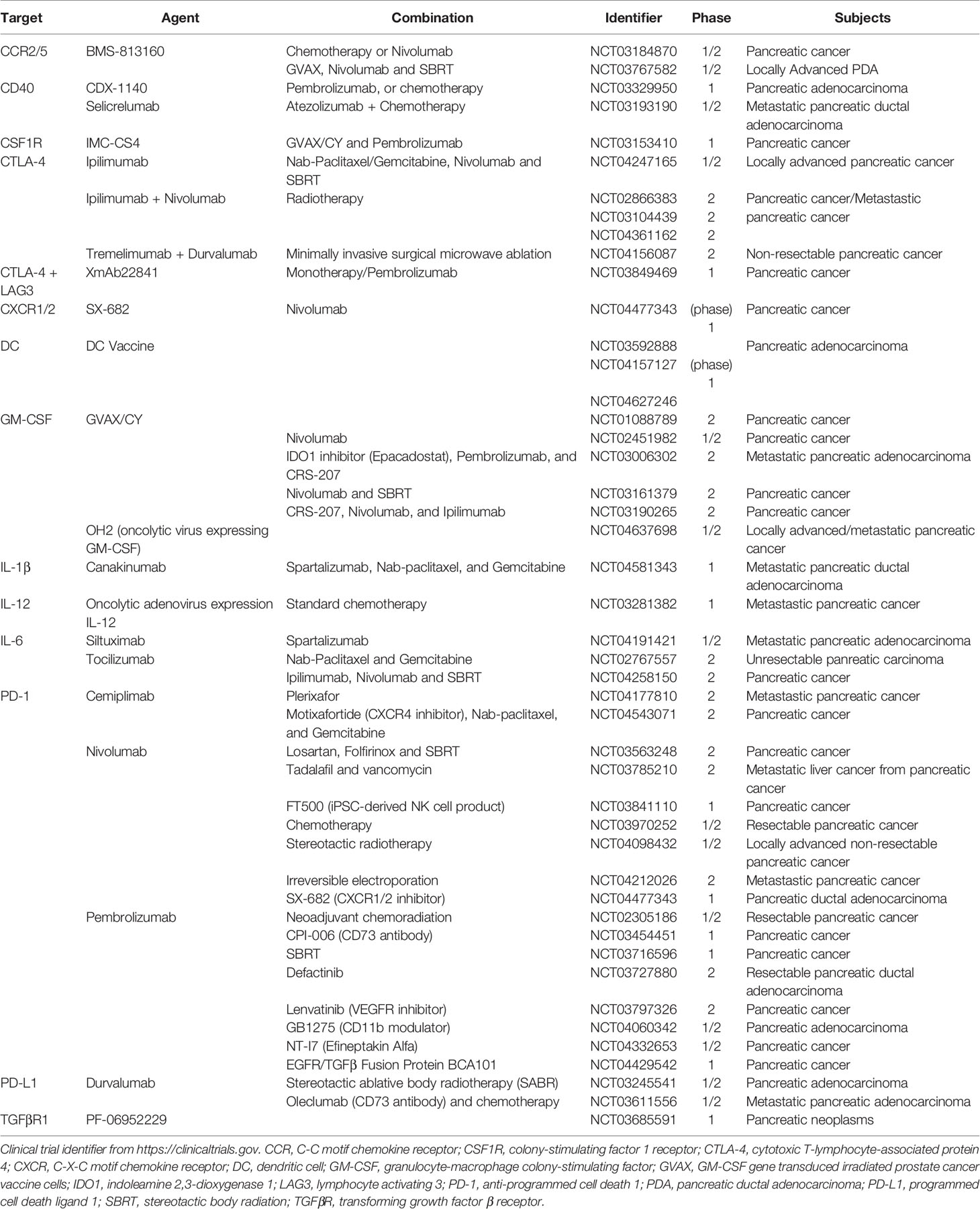
Table 1 Currently active clinical trials targeting the microenvironment of PDA.
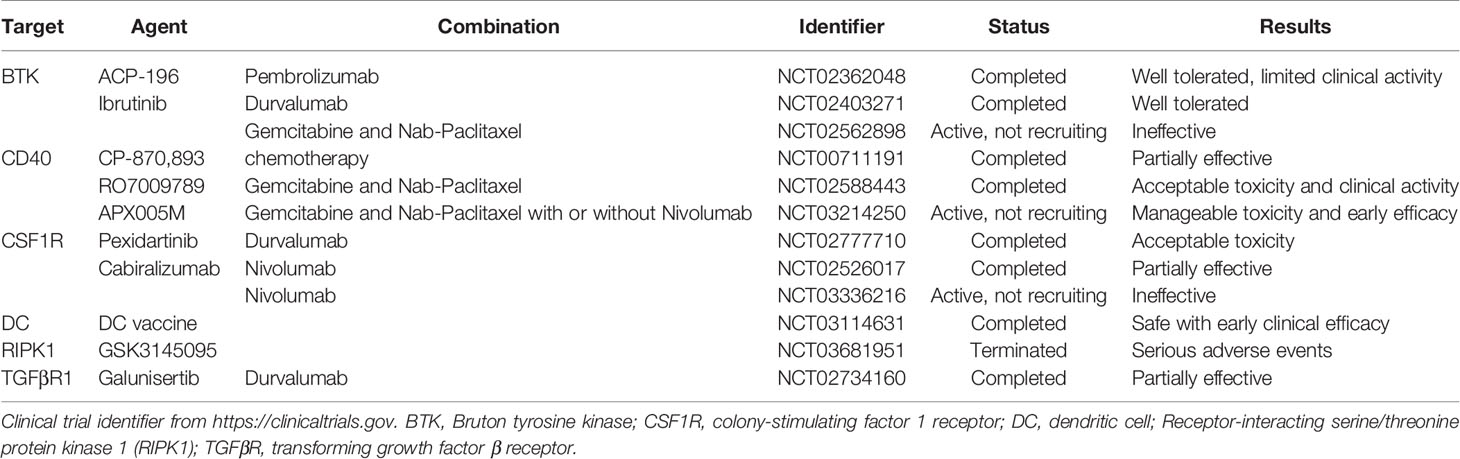
Table 2 Past clinical trials targeting the microenvironment of PDA.
CD4+ T Cells and Their Crosstalk With Stromal Cells Negatively Regulate the Tumor Immunity in PDA
CD4+ T cells infiltrate into the pancreas starting at early stages (PanINs) of carcinogenesis (28). Genetic depletion of CD4+ T cells increased tumor infiltrating CD8+ T cells and up-regulated their capacity to produce IFN-γ and granzyme B, therefore inhibiting tumorigenesis in a GEMM of PDA in a CD8+ T cell-dependent manner (7). This highlights that the formation of immunosuppressive microenvironment occurs even at the onset of pancreatic tumorigenesis, and shows that CD8+ T cells mediated anti-tumor immunity during PDA initiation is negatively regulated by CD4+ T cells.
CD4+ T cells include several subtypes, such as T helper 1 (Th1) cells, T helper 2 (Th2) cells, IL-17-producing T helper (Th17) cells, and Tregs (10). Th1 cells secrete pro-inflammatory cytokines such as Interferon gamma (IFN-γ), IL-2, TNF-α, IL-8, and IL-1β and can have anti-tumor effects (52). In contrast, Th2 cells secrete anti-inflammatory cytokines, such as IL-4, IL-5, and IL-10, and are tumor-promoting (53, 54). In human PDA, Th2 (GATA-3+) cells are predominant over Th1 (T-bet+) cells and the ratio of Th2/Th1 is an independent predictive marker of reduced patient survival (54). CD25+ Th17 cells express high levels of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and mediate CD8+ T cell suppression in an immune checkpoint dependent manner (55). IL-17 secreted by Th17 cells accelerates PanIN initiation and progression by acting directly on epithelial cells that express the IL17 receptor (56). IL17 also recruits neutrophils, triggers neutrophil extracellular traps and excludes cytotoxic CD8+ T cells from tumors (57). Thus, pharmacological and genetical inhibition of IL17/IL17RA signaling in the KPC model increased immune checkpoint blockade sensitivity (57).
Tregs, defined as CD4+FOXP3+CD25+ T cells, are the most abundant CD4+ T cell subpopulation in PDA TME (28). High number of Tregs positively correlates with the progression and poor prognosis of PDA patients (31, 58). Tregs can be recruited by C-C chemokine ligand 5 (CCL5) (59). Disrupting CCL5/C-C chemokine receptor 5 (CCR5) signaling inhibited Treg migration to tumor (60). Tregs promoted the development of PDA through the suppression of IFN-γ-producing-CD8+ T cells in an orthotopic implantation model with primary KrasG12D-expressing pancreatic ductal epithelial cells (61). In this model, intratumoral Tregs directly interacted with tumor associated CD11c+ dendritic cells (DCs) and reduced their expression of costimulatory molecules necessary for CD8+ T cell activation such as CD40, CD80 and CD86 (61). Ablation of Tregs led to the restoration of immunogenic tumor-associated CD11c+ DCs and increased CD8+ T cell-dependent antitumor immunity, which resulted in an inhibition of tumor growth (61). Interactions between T cells and myeloid cell subsets are summarized in Figure 2, and we will further discuss their crosstalk within pancreatic cancer TME in Section 4.
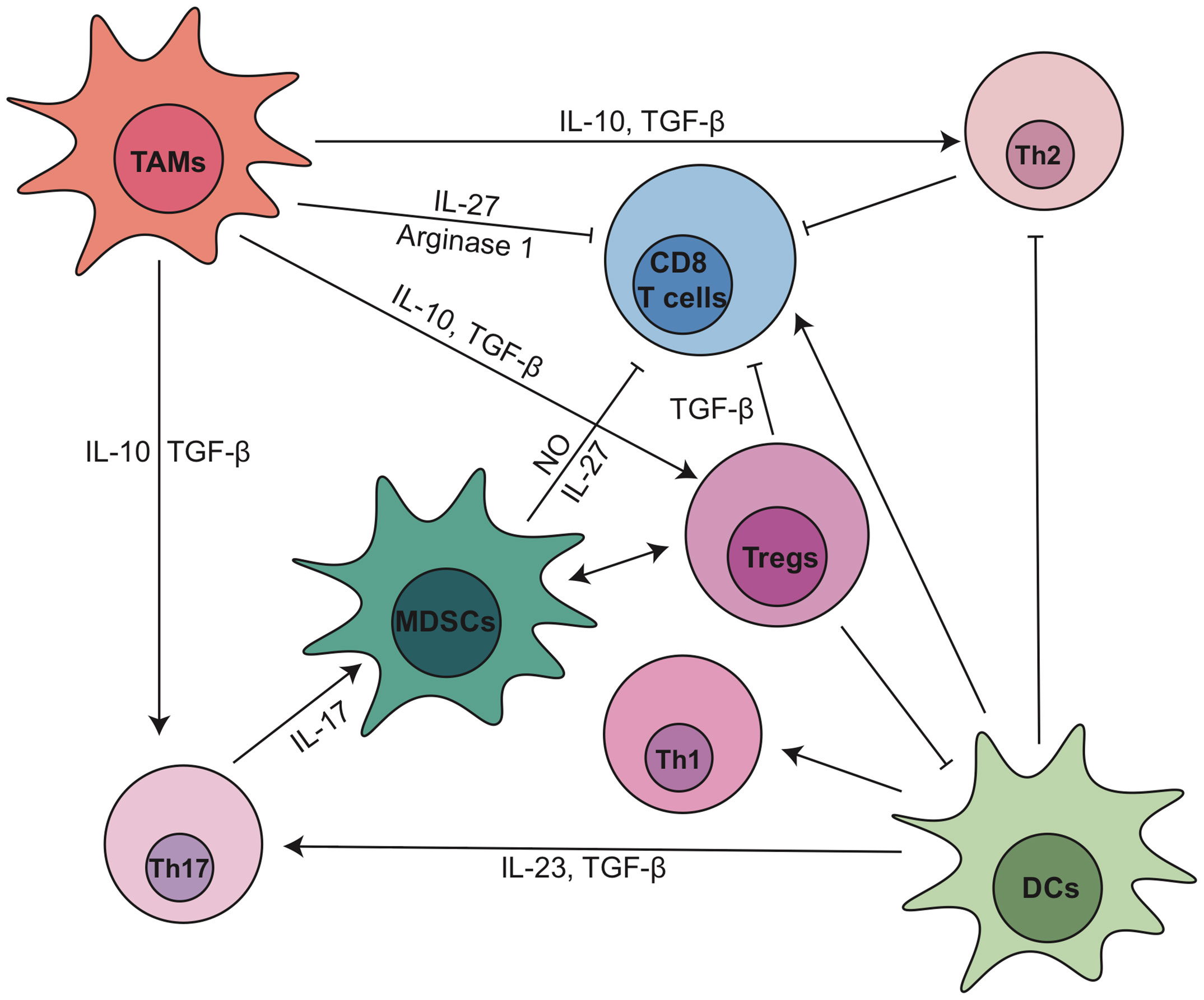
Figure 2 The dynamic cellular and molecular interactions between T cells and myeloid cell subsets in pancreatic cancer. DC, dendritic cell; IL, interleukin; MDSCs, myeloid-derived suppressor cells; NO, nitric oxide; TAMs, tumor associated macrophages; TGF-β, transforming growth factor β; Th, T helper; Tregs, regulatory T cells.
Recently, a new study from our laboratory showed that Treg depletion failed to relieve immunosuppression and accelerated tumor progression in the KC and KPC GEMMs (62). Our study suggests that Tregs are a key source of TGFβ which facilitates the expansion of α-smooth muscle actin (αSMA)+ CAFs. Depletion of Tregs reprogramed the fibroblast populations inducing loss of tumor-restraining αSMA+ CAFs. The reprogramed fibroblasts secreted increased level of chemokines such as CCL3, CCL6, and CCL8 that act as chemoattractant for suppressive myeloid cells. Therefore, Treg depletion resulted in increased Arginase1 (Arg1)+ and PD-L1+ TAMs, restoring the immunosuppressive TME and promoting carcinogenesis. This effect was inhibited by an inhibitor for the common CCL3/6/8 receptor CCR1. Treg depletion also led to an increase in Th2 cytokine profile, indicating that the absence of Tregs fails to restore immune surveillance, likely because of compensation driven by other sub-populations of CD4+ T cells and immunosuppressive myeloid cells. Thus, a better approach might be reprogramming Tregs rather than depleting them altogether.
Targeting Tumor Myeloid Cells as a Crucial Therapeutic Strategy To Relieve Immunosuppression in PDA
Myeloid cells, including immature myeloid cells (also commonly referred to as MDSCs), TAMs and tumor associated neutrophils (TANs) accumulate during the progression of pancreatic cancer (10). Myeloid cells directly promote acinar cell dedifferentiation during the earliest stages of pancreatic cancer (63). Our group has shown that myeloid cells are required for the establishment of an immunosuppressive environment in pancreatic cancer (49). EGFR ligands secreted by tumor infiltrating myeloid cells stimulated EGFR/MAPK signaling and increased the expression of PD-L1 on the epithelial cells to activate the PD-1/PD-L1 checkpoint (49). Depletion of myeloid cells by administration of Diphtheria Toxin (DT) to CD11b-diphtheria toxin receptor (DTR) mice reversed immune suppression and enabled CD8+ T cell activity, thus preventing PanIN formation in the iKras∗; CD11b-DTR GEMM and inhibiting tumor growth in CD11b-DTR mice transplanted with PDA cells (49). Therefore, controlled regulation of myeloid cells is an essential avenue for improvement of clinical efficacy against PDA.
Myeloid-Derived Suppressor Cells in Cooperation With Tregs to Suppress T Cell Activation in PDA
MDSCs are Gr-1+CD11b+ myeloid cells that suppress T cell activation. Inducible nitric oxide synthase (64) enzyme in MDSCs regulates the release of nitric oxide from MDSCs, which subsequently causes DNA damage in CD8+ T cells (65). A recent study demonstrated that in vivo depletion of MDSCs led to a reduction in Tregs in pancreatic tumors (66). Through light sheet fluorescent microscopy and ex vivo functional assays, the authors showed that MDSCs induced Tregs by cell-cell direct interaction, which was lost in the Transwell system, and Tregs in turn affected the survival and/or proliferation of MDSCs (66). GM-CSF is necessary and sufficient to drive the development of Gr-1+CD11b+ cells (35, 36). GM-CSF blockade resulted in reduced MDSC infiltration and higher number of active CD8+ T cells in KPC tumors (36). Further, stroma-derived Dickkopf-1 (DKK1) activates β-catenin in MDSCs and regulates the recruitment and immunosuppressive effects of MDSCs (67). The two main categories of MDSCs are monocytic-MDSCs (Mo-MDSCs), characterized by the surface markers CD11b+Ly6G-Ly6CHi, and granulocyte-derived MDSCs (Gr-MDSCs) by CD11b+Ly6G+Ly6CLow. Selective targeting of Gr-MDSCs was sufficient to induce the activation and proliferation of systemic and intratumoral CD8+ T cells (8).
CD11b+Ly6G+ cells are also identified as tumor associated neutrophils (TANs). In PDA, the abundance of TANs is strongly associated with poor prognosis (68). High level of CXCL5, a chemokine for the recruitment of neutrophils, as well as its receptor C-X-C chemokine receptor 2 (CXCR2), which is highly expressed on TANs, has been associated with poor outcome in PDA patients (68). CXCR2 inhibition prevented TAN accumulation in PDA TME, potently suppressed tumor growth and metastasis and sensitized PDA tumors to anti-PD-1 therapy (12, 13). The absence of TANs correlated with significant infiltration of activated T cells in the TME (13).Thus, inhibition of trafficking or depletion of MDSCs may offer a potential strategy to enhance the efficacy of immunotherapy for PDA. The role of CXCR2 ligands/CXCR2 biological axis in pancreatic cancer has been studied in a clinical trial (NCT00851955, results are pending). An ongoing clinical trial will evaluate the safety and tolerability of a CXCR1/CXCR2 inhibitor in combination with anti-PD1 as a maintenance therapy in patients with metastatic pancreatic ductal adenocarcinoma and will also assess the immunophenotypic and stromal changes to the tumor microenvironment after treatment (NCT04477343).
Reprogramming of Tumor-Associated Macrophages in PDA as a Strategy to Restore Anti-Tumor Immune Responses
TAMs originate from inflammatory monocytes and tissue resident macrophages with different functions (69). Monocyte-derived TAMs function in antigen presentation whereas embryonically derived TAMs exhibit a pro-fibrotic transcriptional profile (9). TAMs can be recruited by cytokines and chemokines such as colony-stimulating factor-1 (CSF1), CCL2 and CCL5, and polarized into different states (70–76). Pro-inflammatory cytokines such as IFN-γ, TNF-α, and IL-12 secreted by classically activated M1-like macrophages recruit Th1 cells and stimulate anti-tumor activity (77), while alternatively activated M2-like TAMs produce anti-inflammatory cytokines such as IL-10 and TGF-β to promote the expansion of Th2/Th17 cells and Tregs (77–79). M2-like TAMs also produce Arg1 which in turn depletes L-arginine that is necessary for T cell function (80, 81). TAMs can exert opposite roles in cancer, either promoting or restraining tumorigenesis based on their polarization (82, 83). In PDA, TAMs have a highly dynamic and heterogeneous status, although as a whole they are inclined to be M2-like and have a profound influence on tumorigenesis and metastasis, as well as on immunosuppression and chemotherapeutic resistance. Currently, a growing interest in the field is to disrupt TAM recruitment or to reprogram TAMs to hinder tumor development, boost antitumor immunity and improve clinical therapy.
The CSF1 receptor (CSF1R) is expressed on F4/80+ macrophages and on monocytes in mice. Targeting TAMs through CSF1R inhibitor (CSF1Ri) or a CSF1 neutralizing antibody inhibited tumor growth, reduced metastatic burden and prolonged survival in KPC mice (84, 85). CSF1Ri treatment decreased the number of CD11b+Ly6G-Ly6CLoF4/80HiMHCII+ macrophages and CD11b+Ly6G-Ly6CHi Mo-MDSCs (86). The expression of immunosuppressive molecules, including Pdcd1lg2, Il10, Arg1, Tgfb1, and Ccl22, was reduced in macrophages while proinflammatory genes, such as Il12a, Ifna, Ifnb1, Ifng, Cxcl10, and Nos2, were upregulated, indicative of reprogramming of TAMs toward a M1-like phenotype (86). Consistently, CD3+CD8+ cytotoxic T cells and CD3+CD4+FOXP3- effector T (Teff) cells were significantly up-regulated upon treatment with CSF1Ri, while CD4+FOXP3+ Tregs were down-regulated, ending up with a significantly improved Teff/Treg ratio (86). Ex vivo assays also revealed that CSF1 blockade alleviated immunosuppressive activities and enhanced antigen-presenting potential in both TAMs and DCs (86). Moreover, CSF1Ri upregulated PD-1 and CTLA-4 expression on T cells and sensitized PDA to immune checkpoint blockade. CSF1Ri-treated tumors also displayed less prominent αSMA+ stromal expansion, which was partly due to reduction of granulin, a secreted glycoprotein that stimulates fibroblast activation and migration. The expression of granulin is mediated by CSF1/CSF1R signaling in TAMs (87). CSF1R inhibitors Pexidartinib and Cabiralizumab have been tested in clinical trials with standard therapies or immune checkpoint blockade in advanced pancreatic cancer patients (NCT02777710, NCT03336216, NCT02526017). Although the toxicity of CSF1R inhibitor combined with immune checkpoint blockade was generally consistent with monotherapy, and the combination resulted in dose-related reduction of circulating monocytes1 (88), unfortunately, in phase II study (NCT03336216) the combination of Cabiralizumab and nivolumab (anti-PD1) with or without chemotherapy failed to improve progression-free survival of patients with advanced pancreatic cancer2. One possible reason of the unresponsiveness to these immunomodulatory approaches could still be the lack of active T cells in the ‘cold’ tumors. CSF1R inhibitor IMC-CS4 is currently being tested in combination with pancreatic cancer vaccine and immune checkpoint blockade in pancreatic cancer patients (NCT03153410).
Other approaches developed to reprogram TAMs include targeting Receptor-interacting serine/threonine protein kinase 1 (RIPK1), a critical receptor kinase on TAMs. Targeting RIPK1 with a small molecule GSK3145095 up-regulated STAT1 signaling in TAMs and reprogrammed intratumoral TAMs toward an MHCIIhiTNFα+IFNγ+ immunogenic phenotype with a reduction in CD206, IL-10, TGF-β and Arg1 (89). RIPK1-inhibited TAMs induced cytotoxic T cell activation and the differentiation of T helper cells toward a mixed Th1/Th17 phenotype. RIPK1 inhibition thus led to active innate and adaptive immunity in both orthotopic KPC tumors and in organotypic models of human PDA. RIPK1 inhibition also synergized with anti-PD-1 treatment (89). However, the clinical trial of GSK3145095 was terminated because 50% of patients (4/8) involved in part 1 of this phase I/II study developed serious adverse events (NCT03681951).
While targeting TAMs emerges as a potential therapeutic strategy in pancreatic cancer, tumor-associated neutrophils might compensate for the loss of TAMs in PDA. Treatment with a CCR2 inhibitor to target CCR2+ TAMs resulted in a compensatory influx of CXCR2+ TANs in PDA patients (90). Accordingly, combination targeting of both CCR2+ TAMs and CXCR2+ TANs further augmented the anti-tumor immunity and enhanced the efficacy of chemotherapy in PDA. The CCR2/5 inhibitor BMS-813160 is under investigation in combination with chemotherapy or immune checkpoint blockade in advanced PDA patients (NCT03184870, NCT03767582).
Dendritic Cell Scarcity and Insufficient T Cell Priming Contribute to the Cold Tumor of PDA
Increasing evidence points to the possibility that insufficient T cell priming due to lack of dendritic cells in PDA is a root cause of its nature as an immunologically cold tumor. Conventional dendritic cells (cDCs) have been recognized as one of the antigen-presenting cells that mediate T cell priming and cytotoxic T cell activity. A recent study showed that endogenous antigen-specific responses in PDA were aberrant due to a scarcity of DCs and an expansion of Th2/Th17 responses (91). Moreover, dysfunction of type 1 conventional dendritic cells (cDC1s) occurred in the earliest stages of tumorigenesis in KPC mice due to elevated apoptosis induced by IL-6 (92). Neutralization of IL-6 or combination treatment of CD40 agonist and Flt3 ligand rescued cDC1 abundance, leading to the control of tumor outgrowth (92). Restoring cDCs in KPC mice also blocked Th2 and Th17 cells and enhanced Th1 and CD8+ T cell activity, which ultimately resulted in reduced and lower-grade PanIN lesions accompanied by decreased collagen deposition and αSMA+ fibroblast density (91). Another study found that a distinct subset of DCs (CD11b+CD103-) predominated in PDA and induced tumor-promoting FOXP3-IL-10+IL-17+IFNγ+ regulatory CD4+ T cells through the secretion of IL-23 and TGF-β (93). This DC mediated-CD4+ T-cell differentiation was modulated by retinoic acid signaling (93).
Increasing attention has turned toward restoring T cell priming to overcome checkpoint unresponsiveness. CD40 is a cell surface molecule that regulates dendritic cells to promote T cell activation. CD40 on DCs binds to CD154 on CD4+ T helper cells and enables DCs to prime cytotoxic T cells (94). Activation of CD40 reprograms macrophages to destroy tumor stroma (95). Combination of CD40 activating antibody and anti-PD-1/CTLA-4 resulted in tumor regression and immunological memory in KPC mice (96). The T cell activating effect of this combination therapy was dependent on CD103+ DCs without the need for innate immune sensing pathways such as TLR, stimulator of interferon genes (STING) or interferon-α receptor (IFNAR) pathways, indicating that the CD40 pathway represents a distinct and alternative bridge between DCs and adaptive immunity in PDA (96). A previous clinical trial showed CD40 agonist monoclonal antibody (mAb) selicrelumab (formally named as CP-870,893 or RO7009789) with gemcitabine was well tolerated and therapeutic efficacy was observed in a cohort of patients with metastatic PDA (95) (NCT00711191). Another phase 1b study combining agonistic CD40 APX005M (sotigalimab) with gemcitabine plus nab-paclitaxel, with and without nivolumab, in 30 patients with metastatic PDA showed encouraging clinical activity and manageable toxicity (97). A recent phase 1b study used selicrelumab with or without gemcitabine and nab-paclitaxel in 16 resectable PDA patients prior to surgery followed by adjuvant chemotherapy and selicrelumab. The results showed CD40 agonist induced T cell immune response both at the tumor site and systemically in those early-stage PDA patients3. Currently, there’re more clinical trials exploring the combination of CD40 agonist with immune checkpoint blockade and/or chemotherapy in advanced pancreatic cancer (NCT03193190, NCT03329950).
In addition, there are encouraging results of dendritic cell-based immunotherapy to activate cytolytic T cell responses in pancreatic cancer from preclinical and clinical pilot studies (98, 99). In one study (NCT03114631), DCs generated from blood monocytes and pulsed with tumor lysates or tumor antigens MUC1 and WT1 were injected subcutaneously to 26 patients with stage II–IV pancreatic cancer. The preliminary results indicate DC-based immunotherapy is safe and provides immediate favorable outcome in pancreatic cancer patients (100). More clinical trials of DC vaccines in PDA patients are on-going (NCT04627246, NCT04157127, NCT03592888).
B Cells Contribute to the Disfunction of T Cell-Dependent Antitumor Immune Responses in PDA
B cells are another immune cell population that plays a significant role in PDA progression, although some controversy regarding their precise function remains. Depletion of B cells using a CD20-specific mAb reduced PanIN formation in KC mice (101). An IL-35 expressing CD1dhiCD5+ B cell subset is required for the pro-tumorigenic effect of B cells in PDA (102). The growth of orthotopic KC cells in B cell-deficient (μMT) mice was significantly inhibited, a phenotype that was rescued by the reconstitution of CD1dhiCD5+ B cells through IL-35 mediated promotion of tumor cell proliferation (102). Bruton tyrosine kinase (BTK), a key B cell and macrophage kinase, contributes to the regulation of T cell-dependent anti-tumor immune responses in PDA (103). Phosphatidylinositol 3-kinase-gamma (PI3Kγ) activated BTK on B cells and Fc receptor γ-chain (FcRγ)+ TAMs, resulting in M2-type macrophage programming that suppressed CD8+ T cell cytotoxicity (103). BTK inhibitors Ibrutinib and ACP-196 were relatively well tolerated in metastatic PDA patients with the combination of chemotherapy or immune checkpoint blockade (NCT02403271, NCT02362048, NCT02562898). However, in the phase III trial patients with metastatic pancreatic cancer treated with Ibrutinib in combination with gemcitabine and nab-paclitaxel didn’t show improved progression free survival and overall survival (NCT02562898) (104). Besides, either monotherapy of ACP-196 or combined with pembrolizumab showed limited clinical activity in phase II study despite consistent reduction of MDSCs in peripheral blood (105). Only in two patients treated with combination therapy profound anti-tumor responses were observed, highlighting the necessity of targeting multiple TME components to improve efficacy as well as the need to better understand the complex human pancreatic tumor microenvironment, which may in part contributed to the failure of BTK inhibitors in this disease despite its success in hematologic malignancies (106).
Extensive Network of Cancer-Associated Fibroblasts to Regulate Immune Suppression in PDA
CAFs are the major contributor to the desmoplastic stroma in PDA (107, 108). Extracellular matrix (ECM) and soluble factors secreted by CAFs are believed to activate key signaling pathways in cancer cells leading to cancer progression, cell survival, metastasis and drug resistance (107, 109, 110). ECM can also act as a physical barrier that prevents drug delivery (111). Sub-populations of CAFs have been noticed by several independent groups (112–115). A subpopulation of CAFs, myofibroblastic CAFs (myCAFs), are found adjacent to cancer cells. They have high expression of αSMA and have been hypothesized to restrict tumor progression. Inflammatory CAFs (iCAFs) are located in the desmoplastic stromal areas of the tumor. They express low level of αSMA but high levels of cytokines and chemokines such as IL-6, IL-11 and leukemia inhibitory factor (LIF) and promote tumor growth. A third sub-population of CAFs is antigen presenting CAFs (apCAFs), also described as mesothelial cells (116, 117). These CAFs express MHC class II related genes and can present antigens to CD4+ T cells. While these subpopulations of CAFs are spatially separated and phenotypically distinct, they still show some dynamic feature since myCAFs and iCAFs are interconvertible and apCAFs can also convert into myCAFs under certain conditions (113, 114).
Due to the heterogeneity of CAFs, they play a complex role in the regulation of PDA progression and TME (Figure 3). Depletion of αSMA+ myofibroblasts starting at either the PanIN or the PDA stage led to invasive, undifferentiated, hypoxic tumors with diminished survival (118). Myofibroblast depletion also decreased overall immune infiltration in PDA but increased CD4+FOXP3+ Tregs, resulting in a reduction in both the Teff/Treg ratio and the cytotoxic CD8+/Treg ratio (118). A similar effect was observed when the Collagen 1 gene Col1a1 was inactivated in a mouse model of pancreatic cancer (118). Due to the increased CTLA-4 expression following myofibroblast depletion, anti-CTLA4 immunotherapy reversed the disease acceleration caused by myofibroblast depletion and prolonged animal survival in p48-Cre; LSL-KrasG12D; Tgfbr2flox/flox (PKT) GEMM (118). On the other hand, the depletion of fibroblast activation protein (FAP)+ CAFs reduced the tumor growth and improved the efficacy of anti-CTLA-4 and anti-PD-L1 in KPC GEMM (6). FAP+ CAFs is the main source of CXCL12 in PDA, which coats and protects the cancer cells. Inhibiting CXCR4, a CXCL12 receptor, induced T cell accumulation among cancer cells and synergized with anti-PD-L1 to cause cancer regression (6). Pancreatic stellate cells (PSCs), characterized by lipid droplets in the cytoplasm, were found as a subset of pancreatic CAFs that correlates with increased suppressive immune cell populations and decreased T cells, natural killer (NK) cells, NK T cells and M1-type TAMs in the PDA tumor tissues (119, 120). The infiltration of CD8+ T cells was regulated through NFκB-mediated expression of CXCL12 in PSCs (121). PSCs also enhanced the differentiation and function of MDSCs through the production of MDSC-promoting cytokines IL-6, vascular endothelial growth factor (VEGF), CSF1 and chemokines CXCL12 and CCL2 (122). IL-6 secreted from PSCs led to the phosphorylation of STAT3 in peripheral blood mononuclear cells (PBMCs), which promoted the differentiation of PBMCs into MDSCs (122). Finally, when we inhibited Hedgehog (Hh) signaling, thus shifting the CAF population to a predominant iCAF phenotype, we also observed a decrease in cytotoxic T cells and an expansion of Tregs, indicating increased immunosuppression (123).
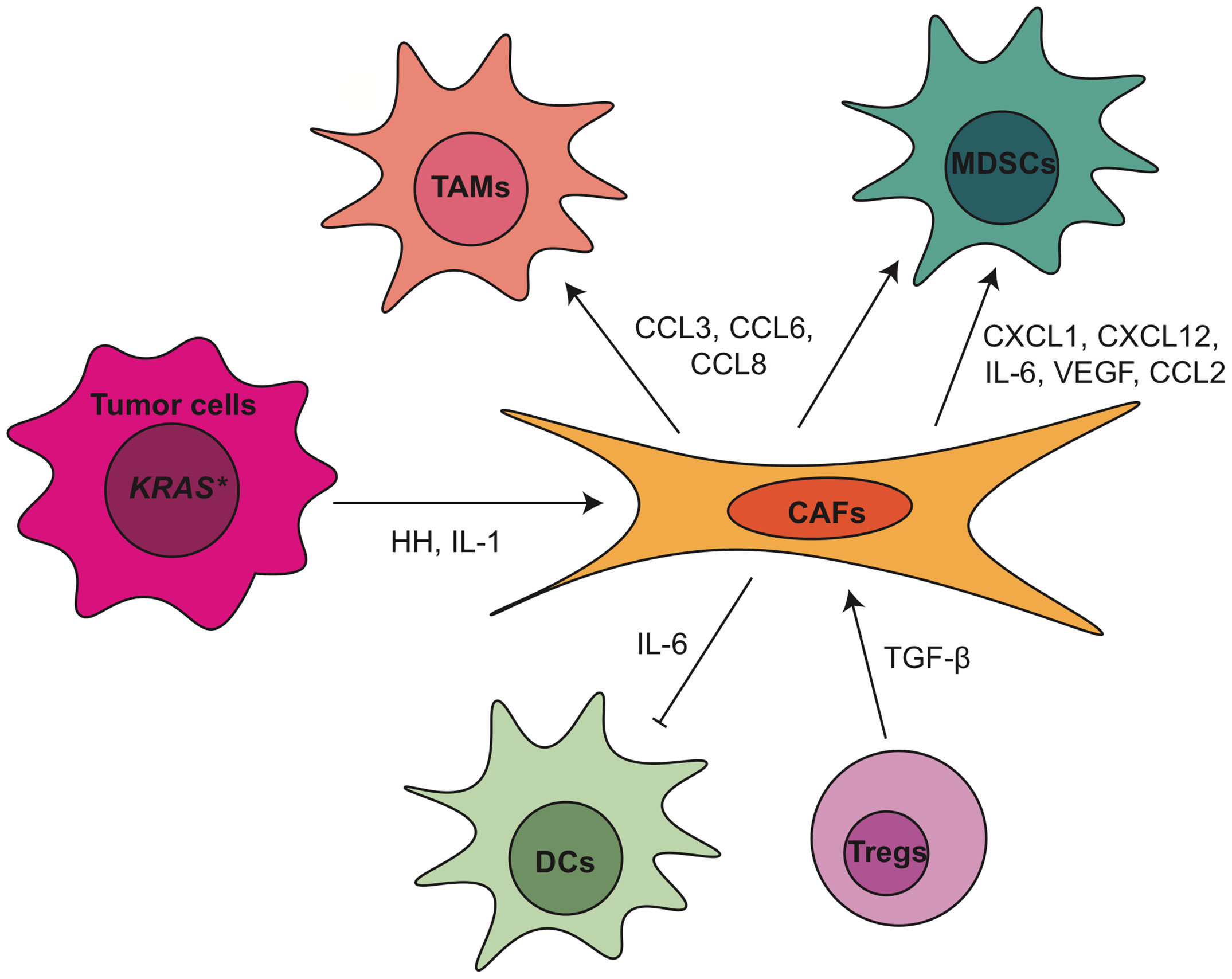
Figure 3 Crosstalk between tumor associated fibroblasts and other TME components within pancreatic cancer. CAF, cancer associated fibroblasts; CCL, C-C motif chemokine ligand; CXCL, C-X-C motif chemokine ligand; DC, dendritic cell; HH, hedgehog; IL, interleukin; MDSCs, myeloid-derived suppressor cells; TAMs, tumor associated macrophages; TGF-β, transforming growth factor β; Tregs, regulatory T cells; VEGF, vascular endothelial growth factor.
As mentioned above, CAFs secrete a variety of soluble factors that in turn shape the PDA TME, including IL-6, which plays multiple roles in the modulation of the immune response in PDA. Il6 not only is responsible for DC disfunction and MDSCs differentiation, as described earlier, but also regulates NK cell activity and Treg infiltration in PDA (124, 125). Combination blockade of IL-6 and PD-L1 led to increased Th1 T cell infiltration and decreased tumor growth (126). In iKras*; IL-6-/- model, we observed deficiency of IL-6 resulted in reduction of tumor infiltrating macrophages and MDSCs (127). Clinical trials targeting IL-6 in PDA patients include IL-6 antibodies Tocilizumab and Siltuximab (NCT02767557, NCT04258150, NCT04191421). Other proteins secreted by CAFs as messenger to crosstalk with immune cells in PDA include but are not limited to CXCL1 (128), CXCL10 (129), IL-33 (130), ETS2 (131), galectin-1 (132), thymic stromal lymphopoietin (TSLP) (54) and βig-h3 (133).
The phenotype and function of CAFs in PDA TME is under control of epithelial cells as well as immune cells. For example, inactivation of oncogenic KRAS in the epithelial cells at the PanIN stage reduced α-SMA expression and inhibited CAF proliferation (14). Hedgehog ligands from tumor cells activated the Hh signaling in CAFs and promoted their expansion (134–136). Further study identified the Hh signaling effector glioma-associated oncogene homolog 1 (GLI1) as a critical transcriptional effector in this process (137). Deletion of a single allele of Gli1 in iKras∗ GEMM was enough to disrupt the recruitment of immune cells by activated fibroblasts (137). Recently, our laboratory demonstrated that hedgehog signaling inhibition with smoothened antagonist LDE225 altered fibroblast composition with reduced myCAF and increased iCAF numbers in the KPC model (123). Immune cells, such as myeloid cells and Tregs, also participate in the regulation of CAFs. Stromal inactivation and remodeling of ECM were observed in both myeloid cell-depleted PanINs (49) and in CSF1Ri-treated PDA tumors (87). IL-1 and TGF-β have also been identified as ligands to promote CAF heterogeneity (125, 138). Recently, our lab showed that the loss of TGFβ1 upon Treg depletion reprogramed the fibroblast population with loss of αSMA+ myCAFs (62). Notably, TGFβ receptor inhibitor Galunisertib has been investigated in clinical trial in combination with durvalumab (anti-PD-L1) for metastatic PDA patients (NCT02734160). Newly published results of this trial showed phase II dose of galunisertib co-administered with durvalumab was tolerable and the disease control rate was 25% (8 patients had partial response or stable disease among 32 patients enrolled). The limited clinical benefit might be due in part to the aggressive nature of the advanced stage of disease (139). TGFβ receptor inhibitor PF-06952229 is currently under investigation in advanced solid tumors including pancreatic cancer patients (NCT03685591).
Concluding Remarks
The stroma-rich, immunosuppressive microenvironment is a hallmark of pancreatic cancer. Tumor evasion of immune surveillance happens at the very early stages of tumorigenesis. Abundant immunosuppressive cells such as macrophages, Tregs and activated fibroblasts are evident even at the onset of acinar-ductal metaplasia (ADM), a key event for PDA initiation (140, 141). In contrast, antitumor effector cells such as CD8+ T cells are either scarce or excluded from the tumor nests. When intratumorally CD8+ T cells are present they are usually exhausted and express checkpoints such as TIGIT (11), lymphocyte-activation gene 3 (LAG-3) and PD-1 (32, 142). Recent research identified intratumoral exhausted T cells (PD-1+Lag3+Tox+) as induced by myeloid cell derived IL-27 in an orthotopic model of PDA (143). Those intratumoral T cells not only produced less IFNγ and Granzyme B but also expressed more IL-10, thus contributing to immune suppression in an autocrine manner. T cell exhaustion in cancer can be self-regulated through cell intrinsic mechanisms, however, the interaction between other cells or cytokines in the TME play an essential role in inducing T cell dysfunction. The TME in pancreatic cancer is composed of various types of cells that secrete abundant cytokines, including tumor cells, immunosuppressive cells, CAFs, inhibitory cytokines such as IL-6, IL-10 and TGF-β. The TME collectively form a complex and integrated immunosuppressive network to limit T cell differentiation, priming and drive T cell exhaustion. Therefore, when tumors have more CD8+ T cells they often also have increased granulocytes, immunosuppressive macrophages, and Tregs, and thus remain immune suppressive (32).
NK cells also play an important role in immune defense and immune regulation in cancer. In addition to their cytolytic activity, NK cells produce cytokines to modulate adaptive immune responses (144). In PDA, NK cells are reported being dysfunctional. NK cells from PDA patients exhibited a significant decrease in cytotoxic degranulation compared with those from healthy controls, a phenomenon that was associated with increased TGF-β1 expression in tumors (145). Future studies are needed to fully understand the mechanisms adopted by the TME to restrain NK cell activity in PDA, which might potentially provide new opportunities to devise new combination treatments for enhanced cancer immunotherapy response.
Significant progress has been made in the application of active immunotherapies including cytokines, immunomodulatory mAbs, and cancer vaccines or passive immunotherapies such as cell‐based therapies in cancer (146, 147). Mono-immunotherapies such as single immune checkpoint inhibitor anti-CTLA4 (ipilimumab), anti-PD1 (nivolumab, pembrolizumab) or anti-PD-L1 (duravalumab) have very limited benefits for PDA patients. It is now widely accepted that due to the complicated cellular crosstalk in PDA, targeting one immune-modulating pathway or a single population of stromal cells has very limited efficacy on reactivating immune system and restraining tumor progression. Therefore, simultaneously targeting multiple immunosuppressive components may acquire therapeutic benefits or improve the efficacy of immunomodulating anticancer therapeutics in PDA patients. In fact, a large number of clinical trials have explored the possibility of combination strategy such as the combination of multiple immunotherapy-based treatments, or combining immunotherapy with chemotherapy, radiation, and other cancer targeted therapies. So far, encouraging results from preclinical and clinical studies have demonstrated that combining an immunostimulatory approach, such as T cell priming via CD40 activation, with immune checkpoint blockade to prevent negative feedback signals on activated T cells represents the most promising treatment strategy to achieve clinical therapeutic benefit in this immunologically “cold” disease. We summarized a number of promising TME-targeting approaches for pancreatic cancer that are currently under clinical investigation in Figure 4 and Table 1.
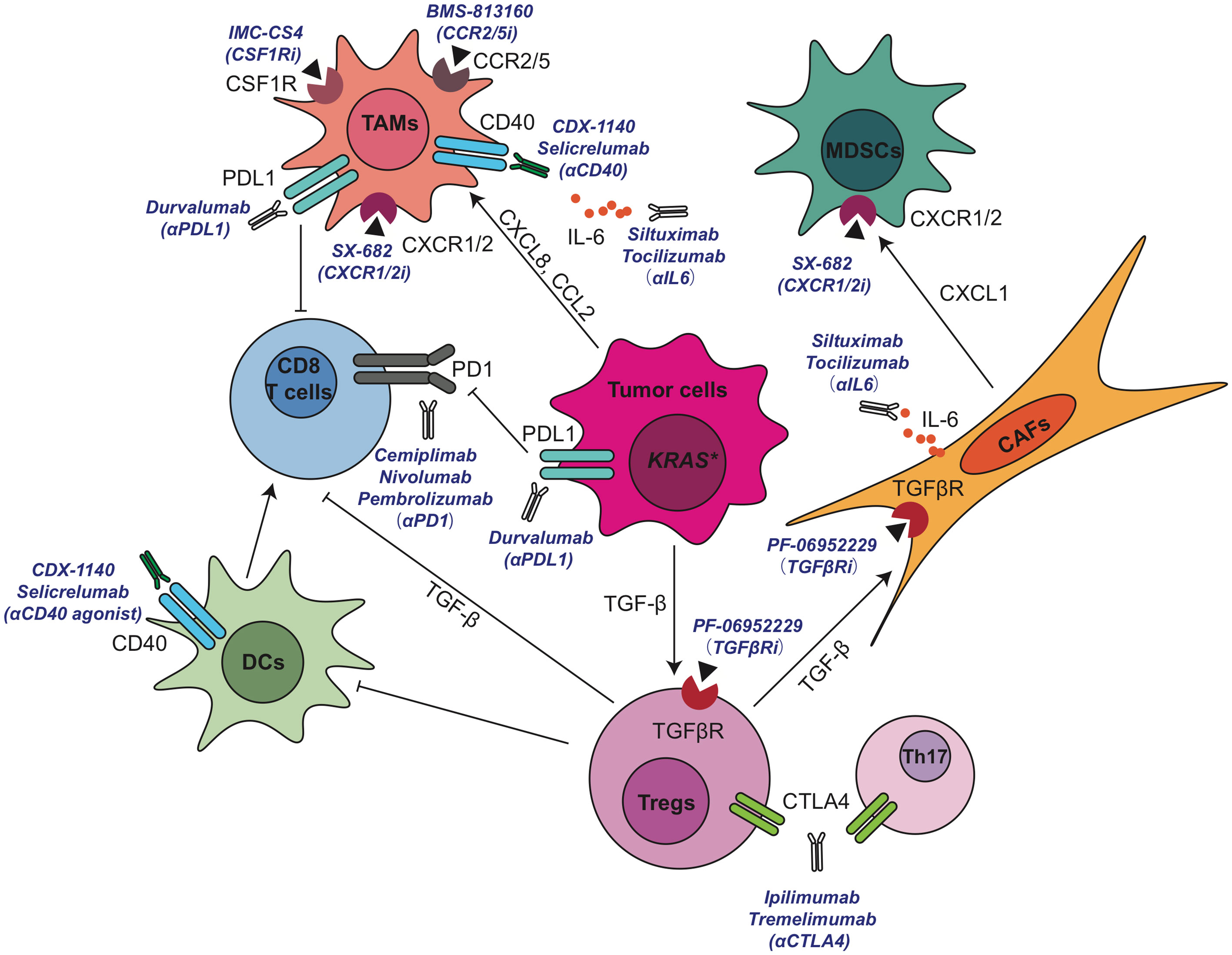
Figure 4 Current active clinical trials targeting immunosuppressive TME in pancreatic cancer. CAF, cancer associated fibroblasts; CCL, C-C motif chemokine ligand; CCR, C-C motif chemokine receptor; CSF1R, colony-stimulating factor 1 receptor; CXCL, C-X-C motif chemokine ligand; DC, dendritic cell; IL, interleukin; MDSCs, myeloid-derived suppressor cells; PD-1, anti-programmed cell death 1; PD-L1, programmed cell death ligand 1; TAMs, tumor associated macrophages; TGF-β, transforming growth factor β; Th, T helper; Tregs, regulatory T cells.
The advent of next-generation sequencing technology and large-scale tumor molecular profiling has shed light on the heterogeneous immune infiltration and tumor microenvironment in human PDA both across and within tumors and the heterogeneity in the expression levels of checkpoints on tumor infiltrating T cells (11, 148–150). Based on these considerations, it is important to understand the variety and individual differences in immune response for future translational studies and clinical trials, including personalized immunotherapy approaches.
With a better dissection of cell heterogeneity and their crosstalk involving cancer and stromal cells within the TME, strategies aimed at targeting multiple mechanisms with synergistic effects may sensitize PDA tumors to chemotherapy or immunotherapy through re-programing the tumor microenvironment of PDA.
Author Contributions
WD wrote the manuscript. YZ and MP reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH/NCI grants R50 CA232985 to YZ; R01CA151588, R01CA198074, and U01CA224145 to MP. This work was also supported by the University of Michigan Cancer Center support grant (P30CA046592), including an Administrative Supplement to MP.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the colleagues whose work could not be included due to space restrictions. We thank the members of the Pasca di Magliano laboratory for critical discussion.
Footnotes
- ^ https://www.businesswire.com/news/home/20171109005417/en/Bristol-Myers-Squibb-and-Five-Prime-Present-Phase-1a1b-Data-Evaluating-Cabiralizumab-anti-CSF-1-receptor-antibody-with-Opdivo-nivolumab-in-Patients-with-Advanced-Solid-Tumors
- ^ https://www.businesswire.com/news/home/20200218005144/en/Five-Prime-Therapeutics-Provides-Update-on-Phase-2-Trial-of-Cabiralizumab-Combined-with-Opdivo®-in-Pancreatic-Cancer
- ^ https://www.abstractsonline.com/pp8/#!/9325/presentation/5136
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
2. Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review, (1975-2018). Bethesda, MD: National Cancer Institute. Available at: https://seer.cancer.gov/csr/1975_2018/, based on November 2020 SEER data submission, posted to the SEER web site, April 2021.
3. Liu Q, Liao Q, Zhao Y. Chemotherapy and Tumor Microenvironment of Pancreatic Cancer. Cancer Cell Int (2017) 17:68. doi: 10.1186/s12935-017-0437-3
4. Vaccaro V, Sperduti I, Milella M. FOLFIRINOX Versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med (2011) 365(8):768–9. doi: 10.1056/NEJMc1107627
5. Martinez-Bosch N, Vinaixa J, Navarro P. Immune Evasion in Pancreatic Cancer: From Mechanisms to Therapy. Cancers (Basel) (2018) 10(1):6. doi: 10.3390/cancers10010006
6. Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 From FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes With Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc Natl Acad Sci USA (2013) 110(50):20212–7. doi: 10.1073/pnas.1320318110
7. Zhang Y, Yan W, Mathew E, Bednar F, Wan S, Collins MA, et al. CD4+ T Lymphocyte Ablation Prevents Pancreatic Carcinogenesis in Mice. Cancer Immunol Res (2014) 2(5):423–35. doi: 10.1158/2326-6066.CIR-14-0016-T
8. Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted Depletion of an MDSC Subset Unmasks Pancreatic Ductal Adenocarcinoma to Adaptive Immunity. Gut (2014) 63(11):1769–81. doi: 10.1136/gutjnl-2013-306271
9. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate From Embryonic Hematopoiesis and Promote Tumor Progression. Immunity (2017) 47(3):597. doi: 10.1016/j.immuni.2017.08.018
10. Zhang Y, Crawford HC, Pasca di Magliano M. Epithelial-Stromal Interactions in Pancreatic Cancer. Annu Rev Physiol (2019) 81:211–33. doi: 10.1146/annurev-physiol-020518-114515
11. Steele NG, Carpenter ES, Kemp SB, Sirihorachai VR, The S, Delrosario L, et al. Multimodal Mapping of the Tumor and Peripheral Blood Immune Landscape in Human Pancreatic Cancer. Nat Cancer (2020) 1(11):1097–112. doi: 10.1038/s43018-020-00121-4
12. Sano M, Ijichi H, Takahashi R, Miyabayashi K, Fujiwara H, Yamada T, et al. Blocking CXCLs-CXCR2 Axis in Tumor-Stromal Interactions Contributes to Survival in a Mouse Model of Pancreatic Ductal Adenocarcinoma Through Reduced Cell Invasion/Migration and a Shift of Immune-Inflammatory Microenvironment. Oncogenesis (2019) 8(2):8. doi: 10.1038/s41389-018-0117-8
13. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell (2016) 29(6):832–45. doi: 10.1016/j.ccell.2016.04.014
14. Zhang Y, Yan W, Mathew E, Kane KT, Brannon A, Adoumie M, et al. Epithelial-Myeloid Cell Crosstalk Regulates Acinar Cell Plasticity and Pancreatic Remodeling in Mice. Elife (2017) 6:e27388. doi: 10.7554/eLife.27388
15. Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab (2019) 29(6):1390–9.e6. doi: 10.1016/j.cmet.2019.02.001
16. Weizman N, Krelin Y, Shabtay-Orbach A, Amit M, Binenbaum Y, Wong RJ, et al. Macrophages Mediate Gemcitabine Resistance of Pancreatic Adenocarcinoma by Upregulating Cytidine Deaminase. Oncogene (2014) 33(29):3812–9. doi: 10.1038/onc.2013.357
17. Azad A, Yin Lim S, D’Costa Z, Jones K, Diana A, Sansom OJ, et al. PD-L1 Blockade Enhances Response of Pancreatic Ductal Adenocarcinoma to Radiotherapy. EMBO Mol Med (2017) 9(2):167–80. doi: 10.15252/emmm.201606674
18. Griesmann H, Drexel C, Milosevic N, Sipos B, Rosendahl J, Gress TM, et al. Pharmacological Macrophage Inhibition Decreases Metastasis Formation in a Genetic Model of Pancreatic Cancer. Gut (2017) 66(7):1278–85. doi: 10.1136/gutjnl-2015-310049
19. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, et al. Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology (2012) 142(4):730–3 e9. doi: 10.1053/j.gastro.2011.12.042
20. Ying H, Dey P, Yao W, Kimmelman AC, Draetta GF, Maitra A, et al. Genetics and Biology of Pancreatic Ductal Adenocarcinoma. Genes Dev (2016) 30(4):355–85. doi: 10.1101/gad.275776.115
21. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature (2016) 531(7592):47–52. doi: 10.1038/nature16965
22. Cancer Genome Atlas Research Network. Electronic Address Aadhe, Cancer Genome Atlas Research N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell (2017) 32(2):185–203.e13. doi: 10.1016/j.ccell.2017.07.007
23. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature (2015) 518(7540):495–501. doi: 10.1038/nature14169
24. Storz P, Crawford HC. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology (2020) 158(8):2072–81. doi: 10.1053/j.gastro.2020.02.059
25. Wilson C, Ye X, Pham T, Lin E, Chan S, McNamara E, et al. AXL Inhibition Sensitizes Mesenchymal Cancer Cells to Antimitotic Drugs. Cancer Res (2014) 74(20):5878–90. doi: 10.1158/0008-5472.CAN-14-1009
26. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and Invasive Ductal Pancreatic Cancer and its Early Detection in the Mouse. Cancer Cell (2003) 4(6):437–50. doi: 10.1016/s1535-6108(03)00309-x
27. Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, et al. Oncogenic Kras is Required for Both the Initiation and Maintenance of Pancreatic Cancer in Mice. J Clin Invest (2012) 122(2):639–53. doi: 10.1172/JCI59227
28. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the Immune Reaction to Pancreatic Cancer From Inception to Invasion. Cancer Res (2007) 67(19):9518–27. doi: 10.1158/0008-5472.CAN-07-0175
29. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell (2005) 7(5):469–83. doi: 10.1016/j.ccr.2005.04.023
30. Bernard V, Semaan A, Huang J, San Lucas FA, Mulu FC, Stephens BM, et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin Cancer Res (2019) 25(7):2194–205. doi: 10.1158/1078-0432.CCR-18-1955
31. Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, et al. An Increased Abundance of Tumor-Infiltrating Regulatory T Cells is Correlated With the Progression and Prognosis of Pancreatic Ductal Adenocarcinoma. PloS One (2014) 9(3):e91551. doi: 10.1371/journal.pone.0091551
32. Stromnes IM, Hulbert A, Pierce RH, Greenberg PD. Hingorani SR. T-Cell Localization, Activation, and Clonal Expansion in Human Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res (2017) 5(11):978–91. doi: 10.1158/2326-6066.CIR-16-0322
33. Ischenko I, D’Amico S, Rao M, Li J, Hayman MJ, Powers S, et al. KRAS Drives Immune Evasion in a Genetic Model of Pancreatic Cancer. Nat Commun (2021) 12(1):1482. doi: 10.1038/s41467-021-21736-w
34. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature (2020) 581(7806):100–5. doi: 10.1038/s41586-020-2229-5
35. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell (2012) 21(6):836–47. doi: 10.1016/j.ccr.2012.04.024
36. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell (2012) 21(6):822–35. doi: 10.1016/j.ccr.2012.04.025S1535-6108(12)00167-5[pii
37. Cheng H, Fan K, Luo G, Fan Z, Yang C, Huang Q, et al. Kras(G12D) Mutation Contributes to Regulatory T Cell Conversion Through Activation of the MEK/ERK Pathway in Pancreatic Cancer. Cancer Lett (2019) 446:103–11. doi: 10.1016/j.canlet.2019.01.013
38. Liou GY, Doppler H, Necela B, Edenfield B, Zhang L, Dawson DW, et al. Mutant KRAS-Induced Expression of ICAM-1 in Pancreatic Acinar Cells Causes Attraction of Macrophages to Expedite the Formation of Precancerous Lesions. Cancer Discovery (2015) 5(1):52–63. doi: 10.1158/2159-8290.CD-14-0474
39. Ratnam NM, Peterson JM, Talbert EE, Ladner KJ, Rajasekera PV, Schmidt CR, et al. NF-kappaB Regulates GDF-15 to Suppress Macrophage Surveillance During Early Tumor Development. J Clin Invest (2017) 127(10):3796–809. doi: 10.1172/JCI91561
40. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-Dependent Ferroptosis Drives Tumor-Associated Macrophage Polarization via Release and Uptake of Oncogenic KRAS Protein. Autophagy (2020) 16(11):2069–83. doi: 10.1080/15548627.2020.1714209
41. Pickup MW, Owens P, Gorska AE, Chytil A, Ye F, Shi C, et al. Development of Aggressive Pancreatic Ductal Adenocarcinomas Depends on Granulocyte Colony Stimulating Factor Secretion in Carcinoma Cells. Cancer Immunol Res (2017) 5(9):718–29. doi: 10.1158/2326-6066.CIR-16-0311
42. Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, et al. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-Tumor Immunity. Cell Metab (2016) 24(5):672–84. doi: 10.1016/j.cmet.2016.10.010
43. Wiedemann GM, Knott MM, Vetter VK, Rapp M, Haubner S, Fesseler J, et al. Cancer Cell-Derived IL-1alpha Induces CCL22 and the Recruitment of Regulatory T Cells. Oncoimmunology (2016) 5(9):e1175794. doi: 10.1080/2162402X.2016.1175794
44. Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. Tumor Cell-Derived IL1beta Promotes Desmoplasia and Immune Suppression in Pancreatic Cancer. Cancer Res (2020) 80(5):1088–101. doi: 10.1158/0008-5472.CAN-19-2080
45. Takahashi R, Macchini M, Sunagawa M, Jiang Z, Tanaka T, Valenti G, et al. Interleukin-1beta-Induced Pancreatitis Promotes Pancreatic Ductal Adenocarcinoma. Via B Lymphocyte-Mediated Immune Suppression Gut (2021) 70(2):330–41. doi: 10.1136/gutjnl-2019-319912
46. Li J, Yuan S, Norgard RJ, Yan F, Yamazoe T, Blanco A, et al. Tumor Cell-Intrinsic USP22 Suppresses Antitumor Immunity in Pancreatic Cancer. Cancer Immunol Res (2020) 8(3):282–91. doi: 10.1158/2326-6066.CIR-19-0661
47. Zhang M, Huang L, Ding G, Huang H, Cao G, Sun X, et al. Interferon Gamma Inhibits CXCL8-CXCR2 Axis Mediated Tumor-Associated Macrophages Tumor Trafficking and Enhances Anti-PD1 Efficacy in Pancreatic Cancer. J Immunother Cancer (2020) 8(1):e000308. doi: 10.1136/jitc-2019-000308
48. Peng YP, Zhang JJ, Liang WB, Tu M, Lu ZP, Wei JS, et al. Elevation of MMP-9 and IDO Induced by Pancreatic Cancer Cells Mediates Natural Killer Cell Dysfunction. BMC Cancer (2014) 14:738. doi: 10.1186/1471-2407-14-738
49. Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid Cells are Required for PD-1/PD-L1 Checkpoint Activation and the Establishment of an Immunosuppressive Environment in Pancreatic Cancer. Gut (2017) 66(1):124–36. doi: 10.1136/gutjnl-2016-312078
50. Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin Cancer Res (2007) 13(7):2151–7. doi: 10.1158/1078-0432.CCR-06-2746
51. Ruscetti M, Morris J, Mezzadra R, Russell J, Leibold J, Romesser PB, et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell (2020) 181(2):424–41 e21. doi: 10.1016/j.cell.2020.03.008
52. Geginat J, Paroni M, Maglie S, Alfen JS, Kastirr I, Gruarin P, et al. Plasticity of Human CD4 T Cell Subsets. Front Immunol (2014) 5:630. doi: 10.3389/fimmu.2014.00630
53. Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R, et al. MyD88 Inhibition Amplifies Dendritic Cell Capacity to Promote Pancreatic Carcinogenesis. Via Th2 Cells J Exp Med (2012) 209(9):1671–87. doi: 10.1084/jem.20111706
54. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T Helper Type 2 Cell Infiltrate Correlates With Cancer-Associated Fibroblast Thymic Stromal Lymphopoietin Production and Reduced Survival in Pancreatic Cancer. J Exp Med (2011) 208(3):469–78. doi: 10.1084/jem.20101876
55. Lang C, Wang J, Chen L. CD25-Expressing Th17 Cells Mediate CD8(+) T Cell Suppression in CTLA-4 Dependent Mechanisms in Pancreatic Ductal Adenocarcinoma. Exp Cell Res (2017) 360(2):384–9. doi: 10.1016/j.yexcr.2017.09.030
56. McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic Kras Activates a Hematopoietic-to-Epithelial IL-17 Signaling Axis in Preinvasive Pancreatic Neoplasia. Cancer Cell (2014) 25(5):621–37. doi: 10.1016/j.ccr.2014.03.014
57. Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, et al. Interleukin-17-Induced Neutrophil Extracellular Traps Mediate Resistance to Checkpoint Blockade in Pancreatic Cancer. J Exp Med (2020) 217(12):e20190354. doi: 10.1084/jem.20190354
58. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ Regulatory T Cells Increases During the Progression of Pancreatic Ductal Adenocarcinoma and its Premalignant Lesions. Clin Cancer Res (2006) 12(18):5423–34. doi: 10.1158/1078-0432.CCR-06-0369
59. Wang X, Lang M, Zhao T, Feng X, Zheng C, Huang C, et al. Cancer-FOXP3 Directly Activated CCL5 to Recruit FOXP3(+)Treg Cells in Pancreatic Ductal Adenocarcinoma. Oncogene (2017) 36(21):3048–58. doi: 10.1038/onc.2016.458
60. Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-Dependent Homing of Regulatory T Cells Inhibits Tumor Growth in a Murine Model of Pancreatic Cancer. J Immunol (2009) 182(3):1746–55. doi: 10.4049/jimmunol.182.3.1746
61. Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk Between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-Tumor Immunity in Pancreatic Cancer. Cell Rep (2017) 20(3):558–71. doi: 10.1016/j.celrep.2017.06.062
62. Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, et al. Regulatory T-Cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov (2020) 10(3):422–39. doi: 10.1158/2159-8290.CD-19-0958
63. Liou GY, Doppler H, Necela B, Krishna M, Crawford HC, Raimondo M, et al. Macrophage-Secreted Cytokines Drive Pancreatic Acinar-to-Ductal Metaplasia Through NF-kappaB and MMPs. J Cell Biol (2013) 202(3):563–77. doi: 10.1083/jcb.201301001
64. O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, et al. Axl, a Transforming Gene Isolated From Primary Human Myeloid Leukemia Cells, Encodes a Novel Receptor Tyrosine Kinase. Mol Cell Biol (1991) 11(10):5016–31. doi: 10.1128/MCB.11.10.5016
65. Cartwright ANR, Suo S, Badrinath S, Kumar S, Melms J, Luoma A, et al. Immunosuppressive Myeloid Cells Induce Nitric Oxide-Dependent DNA Damage and P53 Pathway Activation in CD8(+) T Cells. Cancer Immunol Res (2021) 9(4):470–85. doi: 10.1158/2326-6066.CIR-20-0085
66. Siret C, Collignon A, Silvy F, Robert S, Cheyrol T, Andre P, et al. Deciphering the Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells in Pancreatic Ductal Adenocarcinoma. Front Immunol (2019) 10:3070. doi: 10.3389/fimmu.2019.03070
67. D’Amico L, Mahajan S, Capietto AH, Yang Z, Zamani A, Ricci B, et al. Dickkopf-Related Protein 1 (Dkk1) Regulates the Accumulation and Function of Myeloid Derived Suppressor Cells in Cancer. J Exp Med (2016) 213(5):827–40. doi: 10.1084/jem.20150950
68. Chao T, Furth EE, Vonderheide RH. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-Cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res (2016) 4(11):968–82. doi: 10.1158/2326-6066.CIR-16-0188
69. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
70. Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 Recruits Inflammatory Monocytes to Facilitate Breast-Tumour Metastasis. Nature (2011) 475(7355):222–5. doi: 10.1038/nature10138
71. Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 Promotes Breast Cancer Recurrence Through Macrophage Recruitment in Residual Tumors. Elife (2019) 8:e43653. doi: 10.7554/eLife.43653
72. Keophiphath M, Rouault C, Divoux A, Clement K, Lacasa D. CCL5 Promotes Macrophage Recruitment and Survival in Human Adipose Tissue. Arterioscler Thromb Vasc Biol (2010) 30(1):39–45. doi: 10.1161/ATVBAHA.109.197442
73. Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin Cancer Res (2013) 19(13):3404–15. doi: 10.1158/1078-0432.CCR-13-0525
74. Van Overmeire E, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, et al. M-CSF and GM-CSF Receptor Signaling Differentially Regulate Monocyte Maturation and Macrophage Polarization in the Tumor Microenvironment. Cancer Res (2016) 76(1):35–42. doi: 10.1158/0008-5472.CAN-15-0869
75. Wood GW, De M, Sanford T, Choudhuri R. Macrophage Colony Stimulating Factor Controls Macrophage Recruitment to the Cycling Mouse Uterus. Dev Biol (1992) 152(2):336–43. doi: 10.1016/0012-1606(92)90140-c
76. Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, et al. Macrophage Migration Inhibitory Factor Induces Macrophage Recruitment via CC Chemokine Ligand 2. J Immunol (2006) 177(11):8072–9. doi: 10.4049/jimmunol.177.11.8072
77. Arango Duque G, Descoteaux A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front Immunol (2014) 5:491. doi: 10.3389/fimmu.2014.00491
78. Savage ND, de Boer T, Walburg KV, Joosten SA, van Meijgaarden K, Geluk A, et al. Human Anti-Inflammatory Macrophages Induce Foxp3+ GITR+ CD25+ Regulatory T Cells, Which Suppress. Via Membrane-bound TGFbeta-1 J Immunol (2008) 181(3):2220–6. doi: 10.4049/jimmunol.181.3.2220
79. Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina Propria Macrophages and Dendritic Cells Differentially Induce Regulatory and Interleukin 17-Producing T Cell Responses. Nat Immunol (2007) 8(10):1086–94. doi: 10.1038/ni1511
80. Ellyard JI, Quah BJ, Simson L, Parish CR. Alternatively Activated Macrophage Possess Antitumor Cytotoxicity That is Induced by IL-4 and Mediated by Arginase-1. J Immunother (2010) 33(5):443–52. doi: 10.1097/CJI.0b013e3181cd8746
81. Bronte V, Zanovello P. Regulation of Immune Responses by L-Arginine Metabolism. Nat Rev Immunol (2005) 5(8):641–54. doi: 10.1038/nri1668
82. Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, et al. Macrophage Polarization in Tumour Progression. Semin Cancer Biol (2008) 18(5):349–55. doi: 10.1016/j.semcancer.2008.03.004
83. Sica A, Mantovani A. Macrophage Plasticity and Polarization: In Vivo Veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/JCI59643
84. Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, et al. CSF1R(+) Macrophages Sustain Pancreatic Tumor Growth Through T Cell Suppression and Maintenance of Key Gene Programs That Define the Squamous Subtype. Cell Rep (2018) 23(5):1448–60. doi: 10.1016/j.celrep.2018.03.131
85. Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res (2013) 73(3):1128–41. doi: 10.1158/0008-5472.CAN-12-27310008-5472.CAN-12-2731[pii
86. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-Cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res (2014) 74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723
87. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage-Secreted Granulin Supports Pancreatic Cancer Metastasis by Inducing Liver Fibrosis. Nat Cell Biol (2016) 18(5):549–60. doi: 10.1038/ncb3340
88. Cassier PA, Garin G, Eberst L, Delord J-P, Chabaud S, Terret C, et al. MEDIPLEX: A Phase 1 Study of Durvalumab (D) Combined With Pexidartinib (P) in Patients (Pts) With Advanced Pancreatic Ductal Adenocarcinoma (PDAC) and Colorectal Cancer (CRC). Am Soc Clin Oncol (2019) 37(15_suppl):2579. doi: 10.1200/JCO.2019.37.15_suppl.2579
89. Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell (2018) 34(5):757–74 e7. doi: 10.1016/j.ccell.2018.10.006
90. Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting Both Tumour-Associated CXCR2(+) Neutrophils and CCR2(+) Macrophages Disrupts Myeloid Recruitment and Improves Chemotherapeutic Responses in Pancreatic Ductal Adenocarcinoma. Gut (2018) 67(6):1112–23. doi: 10.1136/gutjnl-2017-313738
91. Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell (2020) 37(3):289–307 e9. doi: 10.1016/j.ccell.2020.02.008
92. Lin JH, Huffman AP, Wattenberg MM, Walter DM, Carpenter EL, Feldser DM, et al. Type 1 Conventional Dendritic Cells are Systemically Dysregulated Early in Pancreatic Carcinogenesis. J Exp Med (2020) 217(8):e20190673. doi: 10.1084/jem.20190673
93. Barilla RM, Diskin B, Caso RC, Lee KB, Mohan N, Buttar C, et al. Specialized Dendritic Cells Induce Tumor-Promoting IL-10(+)IL-17(+) FoxP3(neg) Regulatory CD4(+) T Cells in Pancreatic Carcinoma. Nat Commun (2019) 10(1):1424. doi: 10.1038/s41467-019-09416-2
94. Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med (2020) 71:47–58. doi: 10.1146/annurev-med-062518-045435
95. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science (2011) 331(6024):1612–6. doi: 10.1126/science.1198443
96. Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH. Sufficiency of CD40 Activation and Immune Checkpoint Blockade for T Cell Priming and Tumor Immunity. Proc Natl Acad Sci USA (2020) 117(14):8022–31. doi: 10.1073/pnas.1918971117
97. O’Hara MH, O’Reilly EM, Varadhachary G, Wolff RA, Wainberg ZA, Ko AH, et al. CD40 Agonistic Monoclonal Antibody APX005M (Sotigalimab) and Chemotherapy, With or Without Nivolumab, for the Treatment of Metastatic Pancreatic Adenocarcinoma: An Open-Label, Multicentre, Phase 1b Study. Lancet Oncol (2021) 22(1):118–31. doi: 10.1016/S1470-2045(20)30532-5
98. Deicher A, Andersson R, Tingstedt B, Lindell G, Bauden M, Ansari D. Targeting Dendritic Cells in Pancreatic Ductal Adenocarcinoma. Cancer Cell Int (2018) 18:85. doi: 10.1186/s12935-018-0585-0
99. Bauer C, Dauer M, Saraj S, Schnurr M, Bauernfeind F, Sterzik A, et al. Dendritic Cell-Based Vaccination of Patients With Advanced Pancreatic Carcinoma: Results of a Pilot Study. Cancer Immunol Immunother (2011) 60(8):1097–107. doi: 10.1007/s00262-011-1023-5
100. Hancharou A, Timohina O, Prokhorov A, Romanovskaya S, Dubuske L. Efficacy of Dendritic Cell Based Immunotherapy for Patients With Pancreatic Cancer. J Allergy Clin Immunol (2020) 145(2, Supplement):AB242. doi: 10.1016/j.jaci.2019.12.145
101. Lee KE, Spata M, Bayne LJ, Buza EL, Durham AC, Allman D, et al. Hif1a Deletion Reveals Pro-Neoplastic Function of B Cells in Pancreatic Neoplasia. Cancer Discovery (2016) 6(3):256–69. doi: 10.1158/2159-8290.CD-15-0822
102. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discovery (2016) 6(3):247–55. doi: 10.1158/2159-8290.CD-15-0843
103. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov (2016) 6(3):270–85. doi: 10.1158/2159-8290.CD-15-0827
104. Tempero M, Oh DY, Tabernero J, Reni M, Van Cutsem E, Hendifar A, et al. Ibrutinib in Combination With Nab-Paclitaxel and Gemcitabine for First-Line Treatment of Patients With Metastatic Pancreatic Adenocarcinoma: Phase III RESOLVE Study. Ann Oncol (2021) 32(5):600–8. doi: 10.1016/j.annonc.2021.01.070
105. Overman M, Javle M, Davis RE, Vats P, Kumar-Sinha C, Xiao L, et al. Randomized Phase II Study of the Bruton Tyrosine Kinase Inhibitor Acalabrutinib, Alone or With Pembrolizumab in Patients With Advanced Pancreatic Cancer. J Immunother Cancer (2020) 8(1):e000587. doi: 10.1136/jitc-2020-000587
106. Wen T, Wang J, Shi Y, Qian H, Liu P. Inhibitors Targeting Bruton’s Tyrosine Kinase in Cancers: Drug Development Advances. Leukemia (2021) 35(2):312–32. doi: 10.1038/s41375-020-01072-6
107. Whittle MC, Hingorani SR. Fibroblasts in Pancreatic Ductal Adenocarcinoma: Biological Mechanisms and Therapeutic Targets. Gastroenterology (2019) 156(7):2085–96. doi: 10.1053/j.gastro.2018.12.044
108. Thomas D, Radhakrishnan P. Pancreatic Stellate Cells: The Key Orchestrator of The Pancreatic Tumor Microenvironment. Adv Exp Med Biol (2020) 1234:57–70. doi: 10.1007/978-3-030-37184-5_5
109. Castells M, Thibault B, Delord JP, Couderc B. Implication of Tumor Microenvironment in Chemoresistance: Tumor-Associated Stromal Cells Protect Tumor Cells From Cell Death. Int J Mol Sci (2012) 13(8):9545–71. doi: 10.3390/ijms13089545
110. Neesse A, Bauer CA, Ohlund D, Lauth M, Buchholz M, Michl P, et al. Stromal Biology and Therapy in Pancreatic Cancer: Ready for Clinical Translation? Gut (2019) 68(1):159–71. doi: 10.1136/gutjnl-2018-316451
111. Stylianopoulos T, Munn LL, Jain RK. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer (2018) 4(4):292–319. doi: 10.1016/j.trecan.2018.02.005
112. Hosein AN, Huang H, Wang Z, Parmar K, Du W, Huang J, et al. Cellular Heterogeneity During Mouse Pancreatic Ductal Adenocarcinoma Progression at Single-Cell Resolution. JCI Insight (2019) 5:e129212. doi: 10.1172/jci.insight.129212
113. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov (2019) 9(8):1102–23. doi: 10.1158/2159-8290.CD-19-0094
114. Ohlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J Exp Med (2017) 214(3):579–96. doi: 10.1084/jem.20162024
115. Helms E, Onate MK, Sherman MH. Fibroblast Heterogeneity in the Pancreatic Tumor Microenvironment. Cancer Discov (2020) 10(5):648–56. doi: 10.1158/2159-8290.CD-19-1353
116. Huang H, Wang Z, Zhang Y, Brekken RA. Mesothelial Cell-Derived Antigen-Presenting Cancer-Associated Fibroblasts Induce Expansion of Regulatory T Cells in Pancreatic Cancer. bioRxiv (2021), 2021.02.04.429827. doi: 10.1101/2021.02.04.429827
117. Dominguez CX, Muller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution Into LRRC15(+) Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discovery (2020) 10(2):232–53. doi: 10.1158/2159-8290.CD-19-0644
118. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer With Reduced Survival. Cancer Cell (2014) 25(6):719–34. doi: 10.1016/j.ccr.2014.04.005
119. Li C, Cui L, Yang L, Wang B, Zhuo Y, Zhang L, et al. Pancreatic Stellate Cells Promote Tumor Progression by Promoting an Immunosuppressive Microenvironment in Murine Models of Pancreatic Cancer. Pancreas (2020) 49(1):120–7. doi: 10.1097/MPA.0000000000001464
120. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated Pancreatic Stellate Cells Sequester CD8+ T Cells to Reduce Their Infiltration of the Juxtatumoral Compartment of Pancreatic Ductal Adenocarcinoma. Gastroenterology (2013) 145(5):1121–32. doi: 10.1053/j.gastro.2013.07.025
121. Garg B, Giri B, Modi S, Sethi V, Castro I, Umland O, et al. NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells. Via Up-Regulation CXCL12 Gastroenterol (2018) 155(3):880–91 e8. doi: 10.1053/j.gastro.2018.05.051
122. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res (2013) 73(10):3007–18. doi: 10.1158/0008-5472.CAN-12-4601
123. Steele NG, Biffi G, Kemp SB, Zhang Y, Drouillard D, Syu L, et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin Cancer Res (2021) 27(7):2023–37. doi: 10.1158/1078-0432.CCR-20-3715
124. Feurino LW, Zhang Y, Bharadwaj U, Zhang R, Li F, Fisher WE, et al. IL-6 Stimulates Th2 Type Cytokine Secretion and Upregulates VEGF and NRP-1 Expression in Pancreatic Cancer Cells. Cancer Biol Ther (2007) 6(7):1096–100. doi: 10.4161/cbt.6.7.4328
125. Huang H, Zhang Y, Gallegos V, Sorrelle N, Zaid MM, Toombs J, et al. Targeting TGFbetaR2-Mutant Tumors Exposes Vulnerabilities to Stromal TGFbeta Blockade in Pancreatic Cancer. EMBO Mol Med (2019) 11(11):e10515. doi: 10.15252/emmm.201910515
126. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 Antibody Blockade Combination Therapy Reduces Tumour Progression in Murine Models of Pancreatic Cancer. Gut (2018) 67(2):320–32. doi: 10.1136/gutjnl-2016-311585
127. Zhang Y, Yan W, Collins MA, Bednar F, Rakshit S, Zetter BR, et al. Interleukin-6 is Required for Pancreatic Cancer Progression by Promoting MAPK Signaling Activation and Oxidative Stress Resistance. Cancer Res (2013) 73(20):6359–74. doi: 10.1158/0008-5472.CAN-13-1558-T
128. Wen Z, Liu Q, Wu J, Xu B, Wang J, Liang L, et al. Fibroblast Activation Protein Alpha-Positive Pancreatic Stellate Cells Promote the Migration and Invasion of Pancreatic Cancer by CXCL1-Mediated Akt Phosphorylation. Ann Transl Med (2019) 7(20):532. doi: 10.21037/atm.2019.09.164
129. Lunardi S, Lim SY, Muschel RJ, Brunner TB. IP-10/CXCL10 Attracts Regulatory T Cells: Implication for Pancreatic Cancer. Oncoimmunology (2015) 4(9):e1027473. doi: 10.1080/2162402X.2015.1027473
130. Andersson P, Yang Y, Hosaka K, Zhang Y, Fischer C, Braun H, et al. Molecular Mechanisms of IL-33-Mediated Stromal Interactions in Cancer Metastasis. JCI Insight (2018) 3(20):e122375. doi: 10.1172/jci.insight.122375
131. Pitarresi JR, Liu X, Sharma SM, Cuitino MC, Kladney RD, Mace TA, et al. Stromal ETS2 Regulates Chemokine Production and Immune Cell Recruitment During Acinar-To-Ductal Metaplasia. Neoplasia (2016) 18(9):541–52. doi: 10.1016/j.neo.2016.07.006
132. Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H, et al. High Expression of Galectin-1 in Pancreatic Stellate Cells Plays a Role in the Development and Maintenance of an Immunosuppressive Microenvironment in Pancreatic Cancer. Int J Cancer (2012) 130(10):2337–48. doi: 10.1002/ijc.26290
133. Goehrig D, Nigri J, Samain R, Wu Z, Cappello P, Gabiane G, et al. Stromal Protein Betaig-H3 Reprogrammes Tumour Microenvironment in Pancreatic Cancer. Gut (2019) 68(4):693–707. doi: 10.1136/gutjnl-2018-317570
134. Spivak-Kroizman TR, Hostetter G, Posner R, Aziz M, Hu C, Demeure MJ, et al. Hypoxia Triggers Hedgehog-Mediated Tumor-Stromal Interactions in Pancreatic Cancer. Cancer Res (2013) 73(11):3235–47. doi: 10.1158/0008-5472.CAN-11-1433
135. Hwang RF, Moore TT, Hattersley MM, Scarpitti M, Yang B, Devereaux E, et al. Inhibition of the Hedgehog Pathway Targets the Tumor-Associated Stroma in Pancreatic Cancer. Mol Cancer Res (2012) 10(9):1147–57. doi: 10.1158/1541-7786.MCR-12-0022
136. Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, et al. Sonic Hedgehog Promotes Desmoplasia in Pancreatic Cancer. Clin Cancer Res (2008) 14(19):5995–6004. doi: 10.1158/1078-0432.CCR-08-0291
137. Mathew E, Collins MA, Fernandez-Barrena MG, Holtz AM, Yan W, Hogan JO, et al. The Transcription Factor GLI1 Modulates the Inflammatory Response During Pancreatic Tissue Remodeling. J Biol Chem (2014) 289(40):27727–43. doi: 10.1074/jbc.M114.556563
138. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov (2019) 9(2):282–301. doi: 10.1158/2159-8290.CD-18-0710
139. Melisi D, Oh DY, Hollebecque A, Calvo E, Varghese A, Borazanci E, et al. Safety and Activity of the TGFbeta Receptor I Kinase Inhibitor Galunisertib Plus the Anti-PD-L1 Antibody Durvalumab in Metastatic Pancreatic Cancer. J Immunother Cancer (2021) 9(3):e002068. doi: 10.1136/jitc-2020-002068
140. Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, et al. Chronic Pancreatitis is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell (2007) 11(3):291–302. doi: 10.1016/j.ccr.2007.01.012
141. Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris J, et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell (2012) 22(6):737–50. doi: 10.1016/j.ccr.2012.10.025
142. Burrack AL, Spartz EJ, Raynor JF, Wang I, Olson M, Stromnes IM. Combination PD-1 and PD-L1 Blockade Promotes Durable Neoantigen-Specific T Cell-Mediated Immunity in Pancreatic Ductal Adenocarcinoma. Cell Rep (2019) 28(8):2140–55 e6. doi: 10.1016/j.celrep.2019.07.059
143. Burrack AL, Rollins MR, Spartz EJ, Mesojednik TD, Schmiechen ZC, Raynor JF, et al. CD40 Agonist Overcomes T Cell Exhaustion Induced by Chronic Myeloid Cell IL-27 Production in a Pancreatic Cancer Preclinical Model. J Immunol (2021) 206(6):1372–84. doi: 10.4049/jimmunol.2000765
144. Ben-Shmuel A, Biber G, Barda-Saad M. Unleashing Natural Killer Cells in the Tumor Microenvironment-The Next Generation of Immunotherapy? Front Immunol (2020) 11:275. doi: 10.3389/fimmu.2020.00275
145. Jun E, Song AY, Choi JW, Lee HH, Kim MY, Ko DH, et al. Progressive Impairment of NK Cell Cytotoxic Degranulation Is Associated With TGF-Beta1 Deregulation and Disease Progression in Pancreatic Cancer. Front Immunol (2019) 10:1354. doi: 10.3389/fimmu.2019.01354
146. Drake CG. Combination Immunotherapy Approaches. Ann Oncol (2012) 23 Suppl 8:viii41–6. doi: 10.1093/annonc/mds262
147. Morrissey KM, Yuraszeck TM, Li CC, Zhang Y, Kasichayanula S. Immunotherapy and Novel Combinations in Oncology: Current Landscape, Challenges, and Opportunities. Clin Transl Sci (2016) 9(2):89–104. doi: 10.1111/cts.12391
148. Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity (2018) 49(1):178–93.e7. doi: 10.1016/j.immuni.2018.06.006
149. Liudahl SM, Betts CB, Sivagnanam S, Morales-Oyarvide V, da Silva A, Yuan C, et al. Leukocyte Heterogeneity in Pancreatic Ductal Adenocarcinoma: Phenotypic and Spatial Features Associated With Clinical Outcome. Cancer Discov (2021). doi: 10.1158/2159-8290.Cd-20-0841
Keywords: pancreatic cancer, tumor microenvironment, immune suppression, T cells, myeloid cells, cancer-associated fibroblasts
Citation: Du W, Pasca di Magliano M and Zhang Y (2021) Therapeutic Potential of Targeting Stromal Crosstalk-Mediated Immune Suppression in Pancreatic Cancer. Front. Oncol. 11:682217. doi: 10.3389/fonc.2021.682217
Received: 18 March 2021; Accepted: 21 June 2021;
Published: 05 July 2021.
Edited by:
Takatsugu Ishimoto, Kumamoto University, JapanReviewed by:
Joanna Napp, University Medical Center Göttingen, GermanyDavide Melisi, University of Verona, Italy
Copyright © 2021 Du, Pasca di Magliano and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yaqing Zhang, eWFxaW5nemhAdW1pY2guZWR1; Marina Pasca di Magliano, bWFyaW5hcGFAdW1pY2guZWR1