 Ntombikayise Xelwa
Ntombikayise Xelwa Geoffrey Patrick Candy
Geoffrey Patrick Candy John Devar
John Devar Jones Omoshoro-Jones
Jones Omoshoro-Jones Martin Smith
Martin Smith Ekene Emmanuel Nweke*
Ekene Emmanuel Nweke*- Department of Surgery, University of Witwatersrand, Johannesburg, South Africa
Pancreatic cancer is one of the most deadly cancers, ranking amongst the top leading cause of cancer related deaths in developed countries. Features such as dense stroma microenvironment, abnormal signaling pathways, and genetic heterogeneity of the tumors contribute to its chemoresistant characteristics. Amongst these features, growth factors have been observed to play crucial roles in cancer cell survival, progression, and chemoresistance. Here we review the role of the individual growth factors in pancreatic cancer chemoresistance. Importantly, the interplay between the tumor microenvironment and chemoresistance is explored in the context of pivotal role played by growth factors. We further describe current and future potential therapeutic targeting of these factors.
Introduction
In developed countries, pancreatic cancer (PC) is poised to become amongst the top three causes of deaths in cancer patients by 2030 (1, 2). PC is characterized by non-specific symptoms especially at the early stages of the cancer enabling the progression of the disease. Due to late diagnosis and the biology of PC, current drug treatments usually lead to poor tumor response rates and early relapse (3, 4). Pancreatic cancer cells produce growth factors that play various key roles in propagating tumorigenesis, one of which is in chemoresistance (5). In PC treatment and management, chemotherapy remains a well-known mode of treatment. The lack of effective chemotherapeutic agents to treat PC contribute significantly in making it a lethal condition.
Chemoresistance in PDAC: Role of the Tumor Microenvironment
Many features contribute to chemoresistance and tumor progression, with the presence of a pro-tumoral microenvironment being the dominant factor (6, 7). It has been observed that 90% of the total tumor volume comprises the desmoplastic stroma arising due to the pro-fibrotic state initiated by the PC cells and microenvironment. This composition ensures that PC cells are chemoresistant by reducing drug delivery into the tumor microenvironment (8). Due to this, PC cells can develop resistance towards standard of care chemotherapeutic drugs such as gemcitabine used for PC treatment (9). Gemcitabine is a deoxycytidine analog, that functions by intercalating with the DNA, consequently blocks the cell cycle at the G1/S phase, and inhibits cellular proliferation. However, this blockage may be impaired by changes in drug transporters, activation and inactivation of enzymes and their targets that are characteristic of chemoresistance (10).
The tumor microenvironment of PC which consists of cells such as tumor-associated macrophages (TAMs), myofibroblasts, stromal cells, and cancer-associated fibroblasts (CAFs), is a key player in inducing chemoresistance (11–19). For example, CAFs have been implicated in drug resistance in pancreatic cancer via several mechanisms such as upregulating cytokine levels, blocking adequate delivery of drugs, activating tumor-promoting signaling pathways, increasing cell proliferation, promoting metastasis, and dysregulating cellular metabolism. Recently, novel strategies to target them are being developed (20–24). Most proteins within the intricate extracellular matrix (ECM) of PC are secreted by CAFs (20, 25); thus, the dominance and heterogeneity of CAF within the TME guarantees that it plays a pivotal role in therapeutic resistance (20, 26). Drug delivery to tumor cells is essential for drug sensitivity and therapeutic efficacy. In PC, the dense TME architecture ensures that drug delivery is impeded. Our group recently showed using SWATH-MS, the intricate network of signaling pathways within the ECM of PC tumors highlighting different known and potentially novel associating proteins (27). Furthermore, mouse models of PC showed that CAFs accumulate gemcitabine and this may contribute to drug resistance (28). Also, CAFs originate from pancreatic stellate cells (PSCs) and these cells can secrete metabolites such as deoxycytidine which confers resistance on PC cells (29).
TAMs can either function as inflammatory macrophages known as M1-like inflammatory macrophages which are tumor inhibiting or as the tumor-promoting M2-like immunosuppressive macrophages, both of which play important roles in solid tumors such as pancreatic cancer (30–32). The M1 and M2-like macrophages can be described as either tumor killing or promoting, respectively. Chemoresistance in PC has also been linked to pancreatic cancer stem cells which have enhanced epithelial-mesenchymal transition (EMT), altered metabolism, altered key genes (such as KRAS, TP53, CCND1, BIRC5, and BCL-2), dysregulated signaling pathways (including Notch, PI3K/AKT, Hedgehog, and NF-κB), reduced apoptosis, and increased cell cycle (33, 34).
Growth Factors and Chemoresistance
Growth factors are associated in the progression of several cancers including pancreatic cancer, leading to the development of several drugs to target them (5, 35). The functions of the various growth factors in promoting chemoresistance in pancreatic cancer, current drugs targeting them, and clinical trials are further elaborated on.
Insulin-Like Growth Factor (IGF)
The IGF pathway is dysregulated in pancreatic cancer and its secretion has been linked to the tumor microenvironment (36, 37). Recent studies have shown its dysregulation in both tumors and blood samples of pancreatic cancer patients (38, 39). Furthermore, the upregulation of the insulin/IGF-1R pathway in PDAC occurs in about 72% of patients and is associated with an increase in the number of CD163+TAMs (12). The Ireland et al. (2016) study reported on the role of TAMs and myofibroblasts in promoting drug resistance in pancreatic cancer by the secretion of IGF 1 and 2. The secretion of these factors desensitized pancreatic cancer cells to gemcitabine as they showed that by blocking the IGF pathway, gemcitabine became effective. Also, fibroblast exposed to pancreatic cancer cells has also been shown to secrete the IGF contributing to survival and proliferation (36). When IGF-1R was silenced in PC cells, cell growth, proliferation, and metastasis were inhibited (40). Supporting evidence revealed that blocking IGF-1R and ErbB3 sensitized tumor cells to nab-Paclitaxel and gemcitabine (41). The efficacy of another IGF-1R inhibitor, ganitumab, was evaluated in a phase II clinical trial and found to improve the overall survival of metastatic PC patients over a period of 6 months while ensuring manageable toxicity levels (42). In another study, although combination of ganitumab and gemcitabine resulted in admissible toxicity levels, it did not improve overall survival (43). In in vivo models, the simultaneous inhibition of IGF-1R and ErbB3 using istiratumab (MM-141) blocked the PI3K/AKT/mTOR axis thus increasing the efficacy of blocking IGF-1R alone or in combination with other agents such as gemcitabine (44). On the other hand, a combinatorial treatment of istiratumab with gemcitabine and nab-paclitaxel did not improve outcomes in metastatic pancreatic cancer patients compared to standard of care chemotherapy (45). However there are currently 136 ongoing clinical trial investigating the efficacy of nab-paclitaxel alone or in combination with other drugs such as FOLFIRINOX (Table 1).
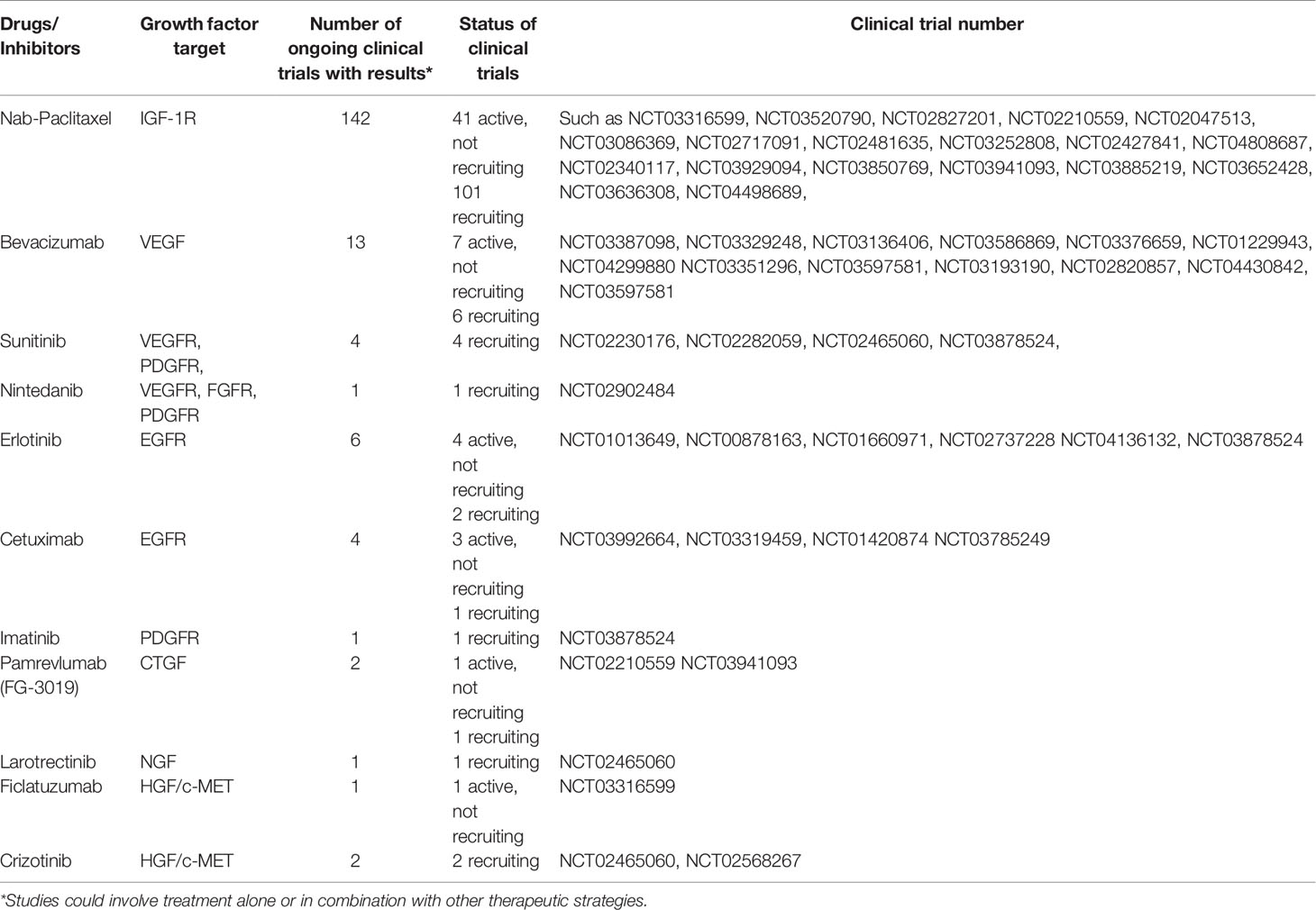
Table 1 Ongoing clinical trials evaluating growth factor inhibition in pancreatic cancer.
Vascular Endothelial Growth Factor (VEGF)
Angiogenesis is required for solid tumor development and progression (46). The heparin-binding glycoprotein, VEGF, functions as an endothelial cell mitogen and is strongly linked to angiogenesis in different tumors, including pancreatic cancer (47). VEGF is highly expressed in PC and many studies have determined that its overexpression, correlated to greater tumor size, increased liver metastases, and a reduced patient survival (48–54). Several preclinical and clinical studies have evaluated the efficacy of inhibiting VEGF and its receptors in PC (54–57). For example, the inhibition of VEGF/VEGFR by foretinib blocked angiogenesis and cell proliferation and resulted in increased apoptosis (58). Bevacizumab (an anti-VEGF antibody) and Sunitinib [an anti-tumor and anti-angiogenic tyrosine kinase (TKI) inhibitor] were observed to inhibit PC cell motility and migration (54). Sunitinib blocks VEGFR, allowing angiogenesis to be inhibited in pancreatic cancer cells (59) (Figure 1A). Treatment with cedirinib, a VEGF inhibitor, was also found to reduce the expression of key epithelial-to-mesenchymal transition (EMT) markers such as ZEB1, Snail, and N-cadherin (60), suggesting potential roles in inhibiting cancer cell migration and metastasis. The combination of VEGF inhibitors such as bevacizumab and chemotherapeutic agents like gemcitabine and 5-FU have shown promising potential in treating PC (61).
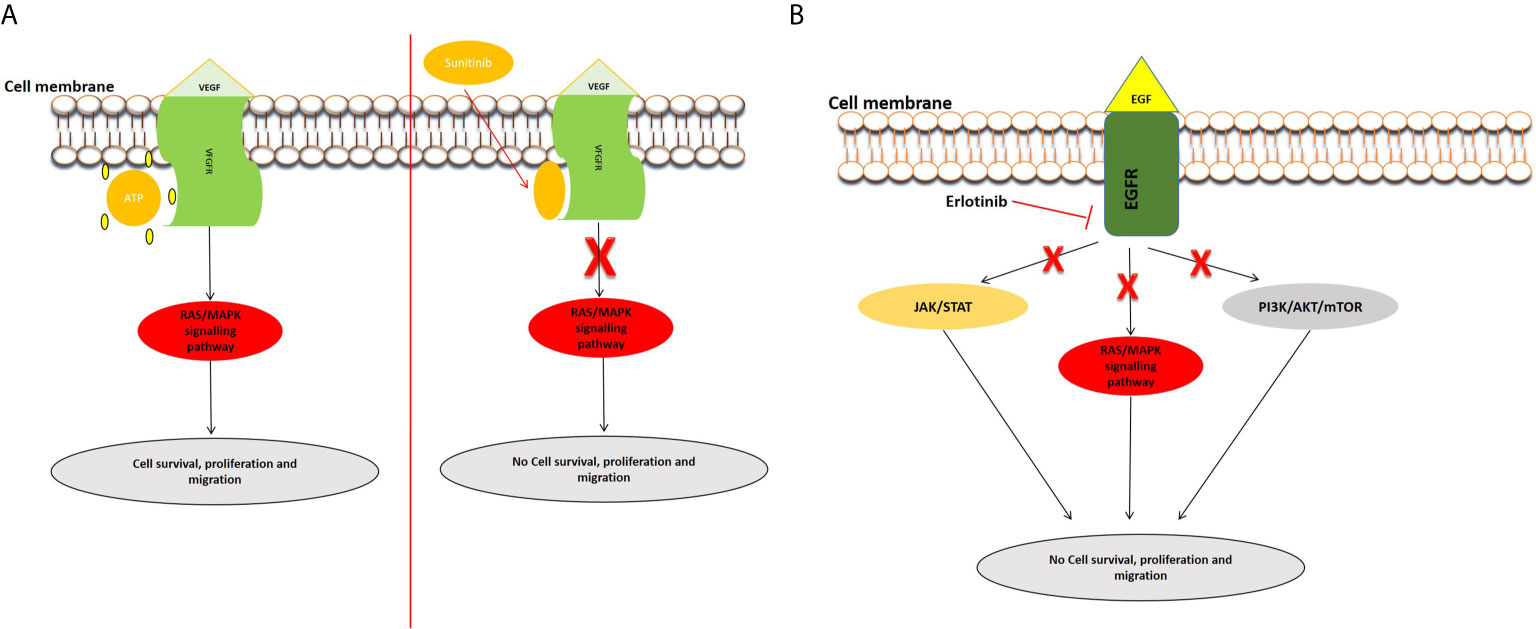
Figure 1 Examples of inhibition of the VEGFR and EGFR signaling pathways. (A) Inhibition of VEGFR signal transduction by sunitinib. After the entry of sunitinib into the cytoplasm, it competitively binds at the ATP site of VEGFR, consequently inhibiting the activation of the pathway. (B) Mechanism of action of Tyrosine kinase inhibitor, erlotinib. Erlotinib is a small molecule that acts as an ATP analogue and inhibits EGF signaling by binding to receptor tyrosine kinases (RTKs), and inhibits the activation of downstream signaling pathways.
Epidermal Growth Factor (EGF)
Epidermal growth factor receptor (EGFR) is expressed in up to 60–90% of pancreatic cancers and is involved inducing cell growth and migration. Targeted anti-EGFR molecular strategies have been employed in the treatment of PDAC especially to circumvent chemoresistance (62, 63). Erlotinib, one of the most studied EGFR inhibitors, has been used to block the EGFR signaling pathway (Figure 1B). A phase I and II clinical trial concluded that erlotinib might be an effective and safe treatment option in PC (64). Combinational treatment of erlotinib with gemcitabine prolonged survival in pancreatic cancer patients in a phase III study (65). Simultaneous treatment of erlotinib, gemcitabine, and capecitabine also showed efficacy in metastatic PC patients (66). Additionally, studies have demonstrated the use of statins in combination with anti-EGFR agents in pancreatic cancer treatment (67). Statins, known to lower lipid concentrations, block the production of intermediates needed for prenylation and RAS/mitogen-activated protein kinase 1 signaling activation. They appear to alter resistance to anti-EGFR agents, such as erlotinib and show efficacy when combined with drugs like gemcitabine (68, 69).
Fibroblast Growth Factor (FGF)
The fibroblast growth factors (FGFs) include about 23 known proteins and their receptors are associated with PC playing a role in tissue hyperplasia, transition of EMT, tumor metastasis, and angiogenesis (70). FGF is overexpressed in PC and promotes cell growth, proliferation, and invasion (71, 72). The overexpression of FGF and FGFR can lead to oxidative stress evident through increased nitric oxide synthase (iNOS) (73). FGF10 is a molecule involved in mesenchymal-epithelial signaling and is crucial in development of multiple organs including the pancreas (74). However, when alterations occur, the FGF10 can induce migration and invasion in the PC cells. As the mesenchyme is essential for the pancreas growth, its absence can result in lack of islet cells and hypoplasia. Furthermore, the crosstalk between the FGF10 and TGF-β pathway can promote EMT and cancer cell invasion (75, 76). A recent study observed that a high expression of FGF8 was independently associated with diminished overall patient survival, indicative of poor prognosis (77). The FGF can also play a role in chemoresistance. It was determined that, acquired drug resistance observed when tumors were treated with VEGF inhibitors was induced by several factors including hypoxia and FGF expression (78). FGF2 targeting agents and inhibitors can aid in preventing TAM-associated cell migration and chemoresistance (11, 73). One study evaluated the use of dovitinib, an inhibitor of FGFR/VEGFR pathway in combination with gemcitabine and capacitabine and determined improved efficacy in patients with advanced PC (79). Mastinib, another inhibitor of FGFR and PDGFR, was shown to decrease inflammation in PC patients (80). Combining mastinib and gemcitabine in both in vitro and in vivo PC models showed that mastinib sensitized tumor cells to gemcitabine (81). This was further supported by a phase III clinical trial which indicated the use of the combination of mastinib and gemcitabine in patients that overexpress ACOX1 (82).
Transforming Growth Factor-Beta (TGF‐β)
Mutations of key genes such as SMAD4 are a major characteristic of PC initiation and progression. These mutations can be found in more than half of pancreatic cancer patients and play an important role in TGF-β signaling (83, 84). In the TME, TGF‐β signaling pathway is involved in the regulation of several cell types. For instance, it promotes differentiation of myofibroblasts, recruitment of immune cells and influences epithelial and endothelial cell differentiation (85). This pathway also has contradictory functions as a tumor suppressor and promoter (86). During early tumorigenesis, TGF-β signaling can inhibit cell cycle and induce apoptosis, but could also promote tumor growth via enhancing EMT, cancer stem cells formation, cellular migration, invasion, and immune response evasion by inhibiting Th1 immune response (87). Cancer cells can secrete both PDGF-BB and TGF-β leading to promotion of fibroblasts transformation thereby resulting in the low expression of the anti-cancer Pigment Epithelium-Derived Factor (PEDF) (88).
Another study determined that silencing TGF-βRII, the protein that begins the TGF-β signaling, promoted tumor growth and resistance to gemcitabine (89). On the other hand, several inhibitors of the TGF-β signaling pathway such as trabedersen and galunisertib have been developed and tested in pancreatic cancer with the aim of inhibiting tumor growth (90, 91). LY2109761, an inhibitor of TGF-β receptors I and II, was used alone and in combination with gemcitabine and revealed to inhibit cell survival, migration, and metastases (92). Cisplatin (Platin) resistance can also be promoted through altering TGF-β/SMAD4 signaling and up-regulation of EMT-markers by tumor-derived exosomes (TDEs), known to function in development and progression of several biological processes in cancer (93). It was shown that the release of TGF‐β and FGF5 from CAFs causes myofibroblast reprogramming in cancer stem cells (CSCs), which is needed to protect the cells from external influences and to acquire a chemoresistance characteristic within the cells (94).
Connective Tissue Growth Factor
Connective Tissue Growth Factor (CTGF/CCN2) is a protein found in the extracellular matrix that functions in regulating diverse cellular processes such as cell survival, proliferation, migration, and apoptosis (95). CTGF has also been linked to other growth factor signaling pathways including TGF-β and FGF. Studies showed that TGF-β can prompt CTGF production in PC cell lines and it can induce the expression of FGFR2 (96, 97). Over recent years, CTGF has been increasingly investigated as a target for PC therapies (98). In vivo silencing of CTGF resulted in the reduction of tumor growth and its expression was closely associated with hypoxia and density of tumor-surrounding stromal cells (99). Treatment with an antagonist of CTGF, mAbFG-3019, using murine models of pancreatic ductal adenocarcinoma revealed that gemcitabine-based chemotherapy was enhanced by increasing levels of gemcitabine and leading to reduction of tumor size (100). The efficacy of pamrevlumab (FG-3019) against pancreatic tumors was further confirmed by another study that showed its effectiveness in selectively targeting pancreatic tumor cells and inhibiting metastases (101). In a phase I and II study, pamrevlumab, enhanced gemcitabine activity in locally advanced pancreatic cancer patients (102). Similarly, another study also demonstrated the use of peptides targeting CTGF alone and in combination with gemcitabine in reducing tumor size (103).
Platelet-Derived Growth Factor (PDGF)
The PDGF family can bind to the tyrosine kinase receptors, PDGFRα and PDGFRβ, and interacts with different cell types within the tumor microenvironment to enhance tumor progression and chemoresistance (104, 105). PDGF regulates PC progression by mediating pathways such as HIPPO/Yes and via its interaction with DUSP28 (106, 107). Inhibiting the phosphorylation of PDGFR by using Gleevec (Imatinib) with gemcitabine led to a reduction in pancreatic tumor growth in nude mice models (108). The tyrosine kinase inhibitor (Sunitinib) targets both VEGFR1-3 and PDGFR pathways (109). Sunitinib appears to be a promising drug in instances where patients did not respond well to gemcitabine-based treatments (110). In another study, the use of a multi-kinase inhibitor, nintedanib, which simultaneously targets VEGFR, FGFR, and PDGFR signaling was investigated alone, and in combination with gemcitabine in xenograft models and this drug displayed a strong antitumor activity (111).
Nerve Growth Factor (NGF)
Nerve growth factor (NGF) is present in sympathetic and neural crest-derived sensory neurons, as well as in the central nervous system (CNS) (112). Pancreatic cancer cell can invade surrounding nerve cell spaces leading to perineural invasion. Perineural invasion is common in pancreatic cancer cells correlating to poor prognosis and can be facilitated by nerve growth factor (113–115). Overexpression of NGF and BDNF induces noradrenaline accumulation consequently inducing pancreatic cancer cell growth (116). Due to its role in inhibiting apoptosis, NGF has also been implicated in chemoresistance (117). Time-dependent treatment of PC mouse models determined that depleting NGF inhibits inflammation and metastasis (118). The inhibition of NGF by blocking STAT3 resulted in decreased pancreatic cancer migration and reduced perineural invasion (119). Similarly, another study showed that NGF knockdown prevented pancreatic cancer cell proliferation, invasion, and migration (120).
Hepatocyte Growth Factor (HGF)
Together with its receptor, c-MET, HGF is overexpressed in PC and has been linked to cancer cell invasion, metastasis, and chemoresistance via tumor-promoting pathways such as P13/Akt and neuropilin (121–125). HGF levels predicted overall survival in locally advanced PC patients that underwent neoadjuvant therapy (126). One study demonstrated the promotion of PC cells by pancreatic stellate cells through the HGF/c-MET pathway; this pathway required survivin expression and was regulated by the p53/p21 pathway (127). Several inhibitors such as rilotumumab, ficlatuzumab onartuzumab, crizotinib, tivantinib, foretinib, and cabozantinib can be used against HGF/c-MET (128). For example, blocking HGF using rilotumumab in vivo led to decreased metastasis compared to treatment with gemcitabine (129). Patients treated with the combinatorial therapy of ficlatuzumab nab-paclitaxel and gemcitabine had favorable treatment response albeit with an observed significant decrease in albumin levels and increased body swelling (130). A synergistic effect was observed when PC cell cultures were treated with tivantinib and gemcitabine, suggesting their possible use in patient treatment (131).
Conclusion and Future Prospects
Chemoresistance is rampant in pancreatic cancer. Growth factors play a crucial role in chemoresistance. Efficient elucidation of growth factor signaling and associated pathways in initiating and propagating chemoresistance is crucial in targeting these pathways. In this instance, the investigation of the interplay of growth factor signaling pathways and other upstream/downstream pathways might provide us with better understanding of chemoresistance in pancreatic cancer and how to circumvent it. Of importance is the elucidation of the diverse roles these growth factors play in the tumor microenvironment. Targeting these growth factors by the use of pharmaceutical agents and inhibitors individually may prove difficult. Perhaps the use of combination therapy targeting the various growth factors may be more effective. For example, blocking FGFR/PDGFR/VEGFR increased survival in mouse models, slowed tumor growth and increased effectiveness of gemcitabine on pancreatic cancer cells (35). Additionally, combining growth factor inhibitors with chemotherapeutic drugs such as gemcitabine may facilitate synergistic drug action and improve chemosensitivity. Although, several preclinical and clinical trials are needed as it is possible that some drug combination may be antagonistic as shown in one study where HGF inhibitor and gemcitabine was combined (132). Toxicity levels are also of concern with combinatorial treatments, while efficacy towards tumor treatment might be achieved, the tolerable toxicity levels might be exceeded. Another promising strategy would be the combination of growth factor inhibitors with peptide inhibitors, statins, or non-coding RNAs inhibitors. This type of strategy was demonstrated when the simultaneous silencing of miR-21 and sunitnib treatment resulted in a synergistic increase in anti-tumor effects (133). In conclusion, growth factor inhibition is promising in pancreatic cancer treatment and management which is evident from the many studies discussed here and the various ongoing clinical trials evaluating their efficacies.
Author Contributions
NX, GC, MS, and EN drafted and conceptualized the article. NX, GC, JD, JO-J, and MS critically edited the article. All authors contributed to the article and approved the submitted version.
Funding
These laboratories of the authors are supported by the National Research Foundation (NRF), the Cancer Association of South Africa (CANSA), and the South African Medical Research Council grant awarded to the Wits Epithelial Cancer thrust.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rawla P, Sunkara T, Gaduputi V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J Oncol (2019) 10:10–27. doi: 10.14740/wjon1166
2. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
3. Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX Versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med (2011) 364:1817–25. doi: 10.1056/NEJMoa1011923
4. Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, et al. Gemcitabine Plus Nab-Paclitaxel is an Active Regimen in Patients With Advanced Pancreatic Cancer: A Phase I/II Trial. J Clin Oncol Off J Am Soc Clin Oncol (2011) 29:4548–54. doi: 10.1200/JCO.2011.36.5742
5. Juhász M, Nitsche B, Malfertheiner P, Ebert MP. Implications of Growth Factor Alterations in the Treatment of Pancreatic Cancer. Mol Cancer (2003) 2:5. doi: 10.1186/1476-4598-2-5
6. De Palma M, Lewis CE. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell (2013) 23:277–86. doi: 10.1016/j.ccr.2013.02.013
7. Junttila MR, de Sauvage FJ. Influence of Tumour Micro-Environment Heterogeneity on Therapeutic Response. Nature (2013) 501:346–54. doi: 10.1038/nature12626
8. Cascetta P, Cavaliere A, Piro G, Torroni L, Santoro R, Tortora G, et al. Pancreatic Cancer and Obesity: Molecular Mechanisms of Cell Transformation and Chemoresistance. Int J Mol Sci (2018) 19:3331. doi: 10.3390/ijms19113331
9. Amrutkar M, Gladhaug IP. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers (2017) 9:157. doi: 10.3390/cancers9110157
10. Zeng S, Pöttler M, Lan B, Grützmann R, Pilarsky C, Yang H. Chemoresistance in Pancreatic Cancer. Int J Mol Sci (2019) 20:4504. doi: 10.3390/ijms20184504
11. Ireland LV, Mielgo A. Macrophages and Fibroblasts, Key Players in Cancer Chemoresistance. Front Cell Dev Biol (2018) 6:131. doi: 10.3389/fcell.2018.00131
12. Ireland L, Santos A, Ahmed MS, Rainer C, Nielsen SR, Quaranta V, et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res (2016) 76:6851–63. doi: 10.1158/0008-5472.CAN-16-1201
13. Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The Pancreas Cancer Microenvironment. Clin Cancer Res (2012) 18:4266. doi: 10.1158/1078-0432.CCR-11-3114
14. Shibuya KC, Goel VK, Xiong W, Sham JG, Pollack SM, Leahy AM, et al. Pancreatic Ductal Adenocarcinoma Contains an Effector and Regulatory Immune Cell Infiltrate That is Altered by Multimodal Neoadjuvant Treatment. PloS One (2014) 9:e96565. doi: 10.1371/journal.pone.0096565
15. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and Cathepsin Proteases Blunt Chemotherapeutic Response in Breast Cancer. Genes Dev (2011) 25:2465–79. doi: 10.1101/gad.180331.111
16. Jena BC, Das CK, Bharadwaj D, Mandal M. Cancer Associated Fibroblast Mediated Chemoresistance: A Paradigm Shift in Understanding the Mechanism of Tumor Progression. Biochim Biophys Acta BBA - Rev Cancer (2020) 1874:188416. doi: 10.1016/j.bbcan.2020.188416
17. Schnittert J, Bansal R, Prakash J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer (2019) 5:128–42. doi: 10.1016/j.trecan.2019.01.001
18. Elebo N, Fru P, Omoshoro−Jones J, Patrick Candy G, Nweke EE. Role of Different Immune Cells and Metabolic Pathways in Modulating the Immune Response in Pancreatic Cancer (Review). Mol Med Rep (2020) 22:4981–91. doi: 10.3892/mmr.2020.11622
19. Nsingwane Z, Candy G, Devar J, Omoshoro-Jones J, Smith M, Nweke E. Immunotherapeutic Strategies in Pancreatic Ductal Adenocarcinoma (PDAC): Current Perspectives and Future Prospects. Mol Biol Rep (2020) 47:6269–80. doi: 10.1007/s11033-020-05648-4
20. Han X, Zhang W-H, Wang W-Q, Yu X-J, Liu L. Cancer-Associated Fibroblasts in Therapeutic Resistance of Pancreatic Cancer: Present Situation, Predicaments, and Perspectives. Biochim Biophys Acta Rev Cancer (2020) 1874:188444. doi: 10.1016/j.bbcan.2020.188444
21. Sun Q, Zhang B, Hu Q, Qin Y, Xu W, Liu W, et al. The Impact of Cancer-Associated Fibroblasts on Major Hallmarks of Pancreatic Cancer. Theranostics (2018) 8:5072–87. doi: 10.7150/thno.26546
22. Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The Role of Stromal Cancer-Associated Fibroblasts in Pancreatic Cancer. J Hematol OncolJ Hematol Oncol (2017) 10:76. doi: 10.1186/s13045-017-0448-5
23. Wang S, Li Y, Xing C, Ding C, Zhang H, Chen L, et al. Tumor Microenvironment in Chemoresistance, Metastasis and Immunotherapy of Pancreatic Cancer. Am J Cancer Res (2020) 10:1937–53.
24. Thomas D, Radhakrishnan P. Tumor-Stromal Crosstalk in Pancreatic Cancer and Tissue Fibrosis. Mol Cancer (2019) 18:14. doi: 10.1186/s12943-018-0927-5
25. Kalluri R. The Biology and Function of Fibroblasts in Cancer. Nat Rev Cancer (2016) 16:582–98. doi: 10.1038/nrc.2016.73
26. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J Exp Med (2017) 214:579–96. doi: 10.1084/jem.20162024
27. Nweke EE, Naicker P, Aron S, Stoychev S, Devar J, Tabb DL, et al. Swath-MS Based Proteomic Profiling of Pancreatic Ductal Adenocarcinoma Tumours Reveals the Interplay Between the Extracellular Matrix and Related Intracellular Pathways. PloS One (2020) 15:e0240453. doi: 10.1371/journal.pone.0240453
28. Hessmann E, Patzak MS, Klein L, Chen N, Kari V, Ramu I, et al. Fibroblast Drug Scavenging Increases Intratumoural Gemcitabine Accumulation in Murine Pancreas Cancer. Gut (2018) 67:497–507. doi: 10.1136/gutjnl-2016-311954
29. Dalin S, Sullivan MR, Lau AN, Grauman-Boss B, Mueller HS, Kreidl E, et al. Deoxycytidine Release From Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res (2019) 79:5723–33. doi: 10.1158/0008-5472.CAN-19-0960
30. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
31. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
32. Ruffell B, Affara NI, Coussens LM. Differential Macrophage Programming in the Tumor Microenvironment. Trends Immunol (2012) 33:119–26. doi: 10.1016/j.it.2011.12.001
33. Di Carlo C, Brandi J, Cecconi D. Pancreatic Cancer Stem Cells: Perspectives on Potential Therapeutic Approaches of Pancreatic Ductal Adenocarcinoma. World J Stem Cells (2018) 10:172–82. doi: 10.4252/wjsc.v10.i11.172
34. Gzil A, Zarębska I, Bursiewicz W, Antosik P, Grzanka D, Szylberg Ł. Markers of Pancreatic Cancer Stem Cells and Their Clinical and Therapeutic Implications. Mol Biol Rep (2019) 46:6629–45. doi: 10.1007/s11033-019-05058-1
35. Taeger J, Moser C, Hellerbrand C, Mycielska ME, Glockzin G, Schlitt HJ, et al. Targeting FGFR/PDGFR/VEGFR Impairs Tumor Growth, Angiogenesis, and Metastasis by Effects on Tumor Cells, Endothelial Cells, and Pericytes in Pancreatic Cancer. Mol Cancer Ther (2011) 10:2157–67. doi: 10.1158/1535-7163.MCT-11-0312
36. Thomas D, Radhakrishnan P. Role of Tumor and Stroma-Derived IGF/Igfbps in Pancreatic Cancer. Cancers (2020) 12:1228. doi: 10.3390/cancers12051228
37. Mutgan AC, Besikcioglu HE, Wang S, Friess H, Ceyhan GO, Demir IE. Insulin/IGF-Driven Cancer Cell-Stroma Crosstalk as a Novel Therapeutic Target in Pancreatic Cancer. Mol Cancer (2018) 17. doi: 10.1186/s12943-018-0806-0
38. Nweke E, Ntwasa M, Brand M, Devar J, Smith M, Candy G. Increased Expression of Plakoglobin is Associated With Upregulated MAPK and PI3K/AKT Signalling Pathways in Early Resectable Pancreatic Ductal Adenocarcinoma. Oncol Lett (2020) 19(6):4133–41. doi: 10.3892/ol.2020.11473
39. Nweke EE, Brand M. Downregulation of the Let-7 Family of microRNAs may Promote Insulin Receptor/Insulin-Like Growth Factor Signalling Pathways in Pancreatic Ductal Adenocarcinoma. Oncol Lett (2020) 20:2613–20. doi: 10.3892/ol.2020.11854
40. Subramani R, Lopez-Valdez R, Arumugam A, Nandy S, Boopalan T, Lakshmanaswamy R. Targeting Insulin-Like Growth Factor 1 Receptor Inhibits Pancreatic Cancer Growth and Metastasis. PloS One (2014) 9:e97016. doi: 10.1371/journal.pone.0097016
41. Camblin AJ, Pace EA, Adams S, Curley MD, Rimkunas V, Nie L, et al. Dual Inhibition of IGF-1R and ErbB3 Enhances the Activity of Gemcitabine and Nab-Paclitaxel in Preclinical Models of Pancreatic Cancer. Clin Cancer Res (2018) 24:2873–85. doi: 10.1158/1078-0432.CCR-17-2262
42. Kindler HL, Richards DA, Garbo LE, Garon EB, Stephenson JJ, Rocha-Lima CM, et al. A Randomized, Placebo-Controlled Phase 2 Study of Ganitumab (AMG 479) or Conatumumab (AMG 655) in Combination With Gemcitabine in Patients With Metastatic Pancreatic Cancer. Ann Oncol Off J Eur Soc Med Oncol (2012) 23:2834–42. doi: 10.1093/annonc/mds142
43. Fuchs CS, Azevedo S, Okusaka T, Van Laethem J-L, Lipton LR, Riess H, et al. A Phase 3 Randomized, Double-Blind, Placebo-Controlled Trial of Ganitumab or Placebo in Combination With Gemcitabine as First-Line Therapy for Metastatic Adenocarcinoma of the Pancreas: The GAMMA Trial. Ann Oncol Off J Eur Soc Med Oncol (2015) 26:921–7. doi: 10.1093/annonc/mdv027
44. Fitzgerald JB, Johnson BW, Baum J, Adams S, Iadevaia S, Tang J, et al. Mm-141, an IGF-IR- and ErbB3-directed Bispecific Antibody, Overcomes Network Adaptations That Limit Activity of IGF-IR Inhibitors. Mol Cancer Ther (2014) 13:410–25. doi: 10.1158/1535-7163.MCT-13-0255
45. Kundranda M, Gracian AC, Zafar SF, Meiri E, Bendell J, Algül H, et al. Randomized, Double-Blind, Placebo-Controlled Phase II Study of Istiratumab (MM-141) Plus Nab-Paclitaxel and Gemcitabine Versus Nab-Paclitaxel and Gemcitabine in Front-Line Metastatic Pancreatic Cancer (CARRIE). Ann Oncol Off J Eur Soc Med Oncol (2020) 31:79–87. doi: 10.1016/j.annonc.2019.09.004
46. Sipos B, Weber D, Ungefroren H, Kalthoff H, Zühlsdorff A, Luther C, et al. Vascular Endothelial Growth Factor Mediated Angiogenic Potential of Pancreatic Ductal Carcinomas Enhanced by Hypoxia: An In Vitro and In Vivo Study. Int J Cancer (2002) 102:592–600. doi: 10.1002/ijc.10753
47. Nusrat O, Belotte J, Fletcher NM, Memaj I, Saed MG, Diamond MP, et al. The Role of Angiogenesis in the Persistence of Chemoresistance in Epithelial Ovarian Cancer. Reprod Sci Thousand Oaks Calif (2016) 23:1484–92. doi: 10.1177/1933719116645191
48. Ikeda N. Prognostic Significance of Angiogenesis in Human Pancreatic Cancer. Br J Cancer (1999) 79:1533–63. doi: 10.1038/sj.bjc.6690248
49. Itakura J. Concomitant Over-Expression of Vascular Endothelial Growth Factor and its Receptors in Pancreatic Cancer. Int J Cancer (2000) 85. doi: 10.1002/(SICI)1097-0215(20000101)85:1<27::AID-IJC5>3.0.CO;2-8
50. Itakura J. Enhanced Expression of Vascular Endothelial Growth Factor in Human Pancreatic Cancer Correlates With Local Disease Progression. Clin Cancer Res (1997) 3:1309–16.
51. Seo Y. High Expression of Vascular Endothelial Growth Factor is Associated With Liver Metastasis and a Poor Prognosis for Patients With Ductal Pancreatic Adenocarcinoma. Cancer (2000) 88:2239–45. doi: 10.1002/(SICI)1097-0142(20000515)88:10<2239::AID-CNCR6>3.0.CO;2-V
52. Niedergethmann M. High Expression of Vascular Endothelial Growth Factor Predicts Early Recurrence and Poor Prognosis After Curative Resection for Ductal Adenocarcinoma of the Pancreas. Pancreas (2002) 25:122–29. doi: 10.1097/00006676-200208000-00002
53. Luo J. Pancreatic Cancer Cell-Derived Vascular Endothelial Growth Factor is Biologically Active In Vitro and Enhances Tumorigenicity In Vivo. Int J Cancer (2001) 92:361–9. doi: 10.1002/ijc.1202
54. Doi Y, Yashiro M, Yamada N, Amano R, Noda S, Hirakawa K. Vegf-A/VEGFR-2 Signaling Plays an Important Role for the Motility of Pancreas Cancer Cells. Ann Surg Oncol (2012) 19:2733–43. doi: 10.1245/s10434-011-2181-6
55. Craven KE, Gore J, Korc M. Overview of Pre-Clinical and Clinical Studies Targeting Angiogenesis in Pancreatic Ductal Adenocarcinoma. Cancer Lett (2016) 381:201–10. doi: 10.1016/j.canlet.2015.11.047
56. Tsuzuki Y. Pancreas Microenvironment Promotes VEGF Expression and Tumor Growth: Novel Window Models for Pancreatic Tumor Angiogenesis and Microcirculation. Lab Invest (2001) 81:1439–51. doi: 10.1038/labinvest.3780357
57. Costache MI, Ioana M, Iordache S, Ene D, Costache CA, Săftoiu A. Vegf Expression in Pancreatic Cancer and Other Malignancies: A Review of the Literature. Romanian J Intern Med Rev Roum Med Interne (2015) 53:199–208. doi: 10.1515/rjim-2015-0027
58. Chen H-M, Tsai C-H, Hung W-C. Foretinib Inhibits Angiogenesis, Lymphangiogenesis and Tumor Growth of Pancreatic Cancer In Vivo by Decreasing VEGFR-2/3 and TIE-2 Signaling. Oncotarget (2015) 6:14940–52. doi: 10.18632/oncotarget.3613
59. Delbaldo C, Faivre S, Dreyer C, Raymond E. Sunitinib in Advanced Pancreatic Neuroendocrine Tumors: Latest Evidence and Clinical Potential. Ther Adv Med Oncol (2012) 4:9–18. doi: 10.1177/1758834011428147
60. Momeny M, Alishahi Z, Eyvani H, Esmaeili F, Zaghal A, Ghaffari P, et al. Anti-Tumor Activity of Cediranib, a Pan-Vascular Endothelial Growth Factor Receptor Inhibitor, in Pancreatic Ductal Adenocarcinoma Cells. Cell Oncol (2020) 43:81–93. doi: 10.1007/s13402-019-00473-9
61. Martin LK, Li X, Kleiber B, Ellison EC, Bloomston M, Zalupski M, et al. VEGF Remains an Interesting Target in Advanced Pancreas Cancer (APCA): Results of a Multi-Institutional Phase II Study of Bevacizumab, Gemcitabine, and Infusional 5-Fluorouracil in Patients With APCA. Ann Oncol (2012) 23:2812–20. doi: 10.1093/annonc/mds134
62. Bloomston M, Bhardwaj A, Ellison EC, Frankel WL. Epidermal Growth Factor Receptor Expression in Pancreatic Carcinoma Using Tissue Microarray Technique. Dig Surg (2006) 23:74–9. doi: 10.1159/000093497
63. Oliveira-Cunha M, Newman WG, Siriwardena AK. Epidermal Growth Factor Receptor in Pancreatic Cancer. Cancers (2011) 3:1513–26. doi: 10.3390/cancers3021513
64. Adamska A, Elaskalani O, Emmanouilidi A, Kim M, Abdol Razak NB, Metharom P, et al. Molecular and Cellular Mechanisms of Chemoresistance in Pancreatic Cancer. Adv Biol Regul (2018) 68:77–87. doi: 10.1016/j.jbior.2017.11.007
65. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib Plus Gemcitabine Compared With Gemcitabine Alone in Patients With Advanced Pancreatic Cancer: A Phase III Trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol Off J Am Soc Clin Oncol (2007) 25:1960–6. doi: 10.1200/JCO.2006.07.9525
66. Oh D-Y, Lee KW, Lee K-H, Sohn C-H, Park YS, Zang DY, et al. A Phase II Trial of Erlotinib in Combination With Gemcitabine and Capecitabine in Previously Untreated Metastatic/Recurrent Pancreatic Cancer: Combined Analysis With Translational Research. Invest New Drugs (2012) 30:1164–74. doi: 10.1007/s10637-011-9651-3
67. Gong J, Sachdev E, Robbins LA, Lin E, Hendifar AE, Mita MM. Statins and Pancreatic Cancer. Oncol Lett (2017) 13:1035–40. doi: 10.3892/ol.2017.5572
68. Dulak J, Józkowicz A. Anti-Angiogenic and Anti-Inflammatory Effects of Statins: Relevance to Anti-Cancer Therapy. Curr Cancer Drug Targets (2005) 5:579–94. doi: 10.2174/156800905774932824
69. Hong JY, Nam EM, Lee J, Park JO, Lee S-C, Song S-Y, et al. Randomized Double-Blinded, Placebo-Controlled Phase II Trial of Simvastatin and Gemcitabine in Advanced Pancreatic Cancer Patients. Cancer Chemother Pharmacol (2014) 73:125–30. doi: 10.1007/s00280-013-2328-1
70. Gnatenko DA, Kopantsev EP, Sverdlov ED. The Role of the Signaling Pathway FGF/FGFR in Pancreatic Cancer. Biochem Mosc Suppl Ser B BioMed Chem (2017) 11:101–10. doi: 10.1134/S1990750817020032
71. Kornmann M. Fibroblast Growth Factor-5 Stimulates Mitogenic Signaling and is Overexpressed in Human Pancreatic Cancer: Evidence for Autocrine and Paracrine Actions. Oncogene (1997) 15:1417–24. doi: 10.1038/sj.onc.1201307
72. Kornmann MH, Beger G, Korc M. Role of Fibroblast Growth Factors and Their Receptors in Pancreatic Cancer and Chronic Pancreatitis. Pancreas (1998) 17:169–75. doi: 10.1097/00006676-199808000-00010
73. Kang X, Lin Z, Xu M, Pan J, Wang Z. Deciphering Role of FGFR Signalling Pathway in Pancreatic Cancer. Cell Prolif (2019) 52:e12605. doi: 10.1111/cpr.12605
74. Itoh N, Ohta H. Fgf10: A Paracrine-Signaling Molecule in Development, Disease, and Regenerative Medicine. Curr Mol Med (2014) 14:504–9. doi: 10.2174/1566524014666140414204829
75. Clayton NS, Grose RP. Emerging Roles of Fibroblast Growth Factor 10 in Cancer. Front Genet (2018) 9:499. doi: 10.3389/fgene.2018.00499
76. Ndlovu R, Deng L-C, Wu J, Li X-K, Zhang J-S. Fibroblast Growth Factor 10 in Pancreas Development and Pancreatic Cancer. Front Genet (2018) 9:482. doi: 10.3389/fgene.2018.00482
77. Jomrich G, Wilfing L, Radosavljevic S, Parak A, Winkler D, Timelthaler G, et al. Fibroblast Growth Factor 8 Overexpression is Predictive of Poor Prognosis in Pancreatic Ductal Adenocarcinoma. Eur Surg (2020) 52:282–9. doi: 10.1007/s10353-020-00669-6
78. Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug Resistance by Evasion of Antiangiogenic Targeting of VEGF Signaling in Late-Stage Pancreatic Islet Tumors. Cancer Cell (2005) 8:299–309. doi: 10.1016/j.ccr.2005.09.005
79. Ma WW, Xie H, Fetterly G, Pitzonka L, Whitworth A, LeVea C, et al. A Phase Ib Study of the FGFR/VEGFR Inhibitor Dovitinib With Gemcitabine and Capecitabine in Advanced Solid Tumor and Pancreatic Cancer Patients. Am J Clin Oncol (2019) 42:184–9. doi: 10.1097/COC.0000000000000492
80. Waheed A, Purvey S, Saif MW. Masitinib in Treatment of Pancreatic Cancer. Expert Opin Pharmacother (2018) 19:759–64. doi: 10.1080/14656566.2018.1459566
81. Humbert M, Castéran N, Letard S, Hanssens K, Iovanna J, Finetti P, et al. Masitinib Combined With Standard Gemcitabine Chemotherapy: In Vitro and In Vivo Studies in Human Pancreatic Tumour Cell Lines and Ectopic Mouse Model. PloS One (2010) 5:e9430. doi: 10.1371/journal.pone.0009430
82. Deplanque G, Demarchi M, Hebbar M, Flynn P, Melichar B, Atkins J, et al. A Randomized, Placebo-Controlled Phase III Trial of Masitinib Plus Gemcitabine in the Treatment of Advanced Pancreatic Cancer. Ann Oncol Off J Eur Soc Med Oncol (2015) 26:1194–200. doi: 10.1093/annonc/mdv133
84. Li M, Li A, Zhao X, Hou S, Lu S, Mou Y, et al. Alteration of Transforming Growth Factor β Signaling Pathway Predicts Worse Prognosis in Pancreatic Ductal Adenocarcinoma. Pancreas (2020) 49:534–42. doi: 10.1097/MPA.0000000000001522
85. Ma X, Cui Z, Du Z, Lin H. Transforming Growth Factor-β Signaling, a Potential Mechanism Associated With Diabetes Mellitus and Pancreatic Cancer? J Cell Physiol (2020) 235:5882–92. doi: 10.1002/jcp.29605
86. Zhou Q, Xia S, Guo F, Hu F, Wang Z, Ni Y, et al. Transforming Growth Factor-β in Pancreatic Diseases: Mechanisms and Therapeutic Potential. Pharmacol Res (2019) 142:58–69. doi: 10.1016/j.phrs.2019.01.038
87. Vivekanandhan S, Mukhopadhyay D. Genetic Status of KRAS Influences Transforming Growth Factor-beta (Tgf-β) Signaling: An Insight Into Neuropilin-1 (NRP1) Mediated Tumorigenesis. Semin Cancer Biol (2019) 54:72–9. doi: 10.1016/j.semcancer.2018.01.014
88. Nwani NG, Deguiz ML, Jimenez B, Vinokour E, Dubrovskyi O, Ugolkov A, et al. Melanoma Cells Block PEDF Production in Fibroblasts to Induce the Tumor-Promoting Phenotype of Cancer-Associated Fibroblasts. Cancer Res (2016) 76:2265–76. doi: 10.1158/0008-5472.CAN-15-2468
89. Drubay V, Skrypek N, Cordiez L, Vasseur R, Schulz C, Boukrout N, et al. Tgf-βrii Knock-Down in Pancreatic Cancer Cells Promotes Tumor Growth and Gemcitabine Resistance. Importance of STAT3 Phosphorylation on S727. Cancers (2018) 10:254. doi: 10.3390/cancers10080254
90. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical Development of Galunisertib (LY2157299 Monohydrate), a Small Molecule Inhibitor of Transforming Growth Factor-Beta Signaling Pathway. Drug Des Devel Ther (2015) 9:4479–99. doi: 10.2147/DDDT.S86621
91. Schlingensiepen K-H, Jaschinski F, Lang SA, Moser C, Geissler EK, Schlitt HJ, et al. Transforming Growth Factor-Beta 2 Gene Silencing With Trabedersen (AP 12009) in Pancreatic Cancer. Cancer Sci (2011) 102:1193–200. doi: 10.1111/j.1349-7006.2011.01917.x
92. Melisi D, Ishiyama S, Sclabas GM, Fleming JB, Xia Q, Tortora G, et al. LY2109761, a Novel Transforming Growth Factor Beta Receptor Type I and Type II Dual Inhibitor, as a Therapeutic Approach to Suppressing Pancreatic Cancer Metastasis. Mol Cancer Ther (2008) 7:829–40. doi: 10.1158/1535-7163.MCT-07-0337
93. Mashouri L, Yousefi H, Aref AR, Ahadi Mohammad A, Molaei F, Alahari SK. Exosomes: Composition, Biogenesis, and Mechanisms in Cancer Metastasis and Drug Resistance. Mol Cancer (2019) 18:75. doi: 10.1186/s12943-019-0991-5
94. Najafi M, Farhood B, Mortezaee K. Cancer Stem Cells (Cscs) in Cancer Progression and Therapy. J Cell Physiol (2019) 234:8381–95. doi: 10.1002/jcp.27740
95. Charrier A, Brigstock DR. Regulation of Pancreatic Function by Connective Tissue Growth Factor (CTGF, CCN2). Cytokine Growth Factor Rev (2013) 24:59–68. doi: 10.1016/j.cytogfr.2012.07.001
96. Aoyama E, Kubota S, Takigawa M. CCN2/CTGF Binds to Fibroblast Growth Factor Receptor 2 and Modulates Its Signaling. FEBS Lett (2012) 586:4270–5. doi: 10.1016/j.febslet.2012.10.038
97. Kwon S, Munroe X, Crawley SC, Lee HY, Spong S, Bradham D, et al. Expression of Connective Tissue Growth Factor in Pancreatic Cancer Cell Lines. Int J Oncol (2007) 31:693–703. doi: 10.3892/ijo.31.4.693
98. Chen Z, Zhang N, Chu HY, Yu Y, Zhang Z-K, Zhang G, et al. Connective Tissue Growth Factor: From Molecular Understandings to Drug Discovery. Front Cell Dev Biol (2020) 8:1239. doi: 10.3389/fcell.2020.593269
99. Bennewith KL, Huang X, Ham CM, Graves EE, Erler JT, Kambham N, et al. The Role of Tumor Cell-Derived Connective Tissue Growth Factor (CTGF/CCN2) in Pancreatic Tumor Growth. Cancer Res (2009) 69:775–84. doi: 10.1158/0008-5472.CAN-08-0987
100. Neesse A, Frese KK, Bapiro TE, Nakagawa T, Sternlicht MD, Seeley TW, et al. CTGF Antagonism With Mab FG-3019 Enhances Chemotherapy Response Without Increasing Drug Delivery in Murine Ductal Pancreas Cancer. Proc Natl Acad Sci USA (2013) 110:12325–30. doi: 10.1073/pnas.1300415110
101. Dornhöfer N, Spong S, Bennewith K, Salim A, Klaus S, Kambham N, et al. Connective Tissue Growth Factor-Specific Monoclonal Antibody Therapy Inhibits Pancreatic Tumor Growth and Metastasis. Cancer Res (2006) 66:5816–27. doi: 10.1158/0008-5472.CAN-06-0081
102. Picozzi VJ, Pishvaian MJ, Mody K, Winter JM, Glaspy JA, Larson T, et al. Effect of anti-CTGF Human Recombinant Monoclonal Antibody Pamrevlumab on Resectability and Resection Rate When Combined With Gemcitabine/Nab-Paclitaxel in Phase 1/2 Clinical Study for the Treatment of Locally Advanced Pancreatic Cancer Patients. J Clin Oncol (2018) 36:4016–6. doi: 10.1200/JCO.2018.36.15_suppl.4016
103. Resovi A, Borsotti P, Ceruti T, Passoni A, Zucchetti M, Berndt A, et al. Ccn-Based Therapeutic Peptides Modify Pancreatic Ductal Adenocarcinoma Microenvironment and Decrease Tumor Growth in Combination With Chemotherapy. Cells (2020) 9:952. doi: 10.3390/cells9040952
104. Hoffmann A-C, Mori R, Vallbohmer D, Brabender J, Klein E, Drebber U, et al. High Expression of HIF1a is a Predictor of Clinical Outcome in Patients With Pancreatic Ductal Adenocarcinomas and Correlated to PDGFA, VEGF, and Bfgf. Neoplasia N Y N (2008) 10:674–9. doi: 10.1593/neo.08292
105. Bartoschek M, Pietras K. PDGF Family Function and Prognostic Value in Tumor Biology. Biochem Biophys Res Commun (2018) 503:984–90. doi: 10.1016/j.bbrc.2018.06.106
106. Lee J, Lee J, Yun JH, Choi C, Cho S, Kim SJ, et al. Autocrine DUSP28 Signaling Mediates Pancreatic Cancer Malignancy Via Regulation of PDGF-a. Sci Rep (2017) 7:12760. doi: 10.1038/s41598-017-13023-w
107. Li T, Guo T, Liu H, Jiang H, Wang Y. Platelet-Derived Growth factor-BB Mediates Pancreatic Cancer Malignancy Via Regulation of the Hippo/Yes-associated Protein Signaling Pathway. Oncol Rep (2021) 45:83–94. doi: 10.3892/or.2020.7859
108. Hwang RF, Yokoi K, Bucana CD, Tsan R, Killion JJ, Evans DB, et al. Inhibition of Platelet-Derived Growth Factor Receptor Phosphorylation by STI571 (Gleevec) Reduces Growth and Metastasis of Human Pancreatic Carcinoma in an Orthotopic Nude Mouse Model. Clin Cancer Res Off J Am Assoc Cancer Res (2003) 9:6534–44.
109. Christensen JG. A Preclinical Review of Sunitinib, a Multitargeted Receptor Tyrosine Kinase Inhibitor With Anti-Angiogenic and Antitumour Activities. Ann Oncol (2007) x3–x10. doi: 10.1093/annonc/mdm408
110. O’Reilly EM, Niedzwiecki D, Hall M, Hollis D, Bekaii-Saab T, Pluard T, et al. A Cancer and Leukemia Group B Phase II Study of Sunitinib Malate in Patients With Previously Treated Metastatic Pancreatic Adenocarcinoma (Calgb 80603). Oncol (2010) 15:1310–9. doi: 10.1634/theoncologist.2010-0152
111. Awasthi N, Hinz S, Brekken RA, Schwarz MA, Schwarz RE. Nintedanib, a Triple Angiokinase Inhibitor, Enhances Cytotoxic Therapy Response in Pancreatic Cancer. Cancer Lett (2015) 358:59–66. doi: 10.1016/j.canlet.2014.12.027
112. Skaper SD. Nerve Growth Factor: A Neuroimmune Crosstalk Mediator for All Seasons. Immunology (2017) 1–15. doi: 10.1111/imm.12717
113. Gu J, Xu W, Peng C, Zhu Y, Wang D, Wang X, et al. Perineural Invasion is Related to p38 Mitogen-Activated Protein Kinase Pathway Activation and Promotes Tumor Growth and Chemoresistance in Pancreatic Cancer. J Cell Biochem (2019) 120:11775–83. doi: 10.1002/jcb.28457
114. Ceyhan GO, Demir IE, Altintas B, Rauch U, Thiel G, Müller MW, et al. Neural Invasion in Pancreatic Cancer: A Mutual Tropism Between Neurons and Cancer Cells. Biochem Biophys Res Commun (2008) 374:442–7. doi: 10.1016/j.bbrc.2008.07.035
115. Hirai I, Kimura W, Ozawa K, Kudo S, Suto K, Kuzu H, et al. Perineural Invasion in Pancreatic Cancer. Pancreas (2002) 24:15–25. doi: 10.1097/00006676-200201000-00003
116. Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell (2018) 33:75–90.e7. doi: 10.1016/j.ccell.2017.11.007
117. Liu H, Nowak R, Chao W, Bloch KD. Nerve Growth Factor Induces Anti-Apoptotic Heme Oxygenase-1 in Rat Pheochromocytoma PC12 Cells. J Neurochem (2003) 86:1553–63. doi: 10.1046/j.1471-4159.2003.01978.x
118. Saloman JL, Singhi AD, Hartman DJ, Normolle DP, Albers KM, Davis BM. Systemic Depletion of Nerve Growth Factor Inhibits Disease Progression in a Genetically Engineered Model of Pancreatic Ductal Adenocarcinoma. Pancreas (2018) 47:856–63. doi: 10.1097/MPA.0000000000001090
119. Guo K, Ma Q, Li J, Wang Z, Shan T, Li W, et al. Interaction of the Sympathetic Nerve With Pancreatic Cancer Cells Promotes Perineural Invasion Through the Activation of STAT3 Signaling. Mol Cancer Ther (2013) 12:264–73. doi: 10.1158/1535-7163.MCT-12-0809
120. Bapat AA, Munoz RM, Von Hoff DD, Han H. Blocking Nerve Growth Factor Signaling Reduces the Neural Invasion Potential of Pancreatic Cancer Cells. PloS One (2016) 11:e0165586. doi: 10.1371/journal.pone.0165586
121. Matsushita A, Götze T, Korc M. Hepatocyte Growth Factor–Mediated Cell Invasion in Pancreatic Cancer Cells is Dependent on Neuropilin-1. Cancer Res (2007) 67:10309–16. doi: 10.1158/0008-5472.CAN-07-3256
122. Gafarli S, Tian M, Rückert F. Hepatocyte Growth Factor Signaling Pathway as a Potential Target in Ductal Adenocarcinoma of the Pancreas. JOP J Pancreas (2017) 18:448–57.
123. Watanabe S, Kishimoto T, Yokosuka O. Hepatocyte Growth Factor Inhibits Anoikis of Pancreatic Carcinoma Cells Through Phosphatidylinositol 3-Kinase Pathway. Pancreas (2011) 40:608–14. doi: 10.1097/MPA.0b013e318214fa6c
124. Modica C, Tortarolo D, Comoglio PM, Basilico C, Vigna E. Met/Hgf Co-Targeting in Pancreatic Cancer: A Tool to Provide Insight Into the Tumor/Stroma Crosstalk. Int J Mol Sci (2018) 19:3920. doi: 10.3390/ijms19123920
125. Pothula SP, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. Targeting HGF/c-MET Axis in Pancreatic Cancer. Int J Mol Sci (2020) 21:9170. doi: 10.3390/ijms21239170
126. Roberts HJ, Wo JY-L, Yeap BY, Ulysse CA, Murphy JE, Weekes CD, et al. The Use of Elevated Circulating Hepatocyte Growth Factor (HGF) Level as a Potential Prognostic Biomarker in Locally Advanced Pancreatic Cancer. J Clin Oncol (2021) 39:429–9. doi: 10.1200/JCO.2021.39.3_suppl.429
127. Yang X-P, Liu S-L, Xu J-F, Cao S-G, Li Y, Zhou Y-B. Pancreatic Stellate Cells Increase Pancreatic Cancer Cells Invasion Through the Hepatocyte Growth Factor /c-Met/survivin Regulated by P53/P21. Exp Cell Res (2017) 357:79–87. doi: 10.1016/j.yexcr.2017.04.027
128. Demkova L, Kucerova L. Role of the HGF/c-MET Tyrosine Kinase Inhibitors in Metastasic Melanoma. Mol Cancer (2018) 26:1–14. doi: 10.1186/s12943-018-0795-z
129. Jin H, Yang R, Zheng Z, Romero M, Ross J, Bou-Reslan H, et al. MetMAb, the One-Armed 5d5 Anti-c-Met Antibody, Inhibits Orthotopic Pancreatic Tumor Growth and Improves Survival. Cancer Res (2008) 68:4360–8. doi: 10.1158/0008-5472.CAN-07-5960
130. Perez K, Weekes CD, Cleary JM, Abrams TA, Blaszkowsky LS, Enzinger PC, et al. Phase Ib Study of Gemcitabine, Nab-Paclitaxel, and Ficlatuzumab in Patients With Advanced Pancreatic Cancer. J Clin Oncol (2020) 38:693–3. doi: 10.1200/JCO.2020.38.4_suppl.693
131. Rolfo CD, Avan A, Leon LG, Castiglia M, Honeywell R, Pauwels P, et al. Tivantinib-Gemcitabine: Pharmacological Rational for a New Combination in Pancreatic Cancer. Ann Oncol (2014) 25:iv158. doi: 10.1093/annonc/mdu331.34
132. Pothula SP, Xu Z, Goldstein D, Biankin AV, Pirola RC, Wilson JS, et al. Hepatocyte Growth Factor Inhibition: A Novel Therapeutic Approach in Pancreatic Cancer. Br J Cancer (2016) 114:269–80. doi: 10.1038/bjc.2015.478
Keywords: pancreatic cancer, chemoresistance, growth factors, chemotherapy, signaling pathways
Citation: Xelwa N, Candy GP, Devar J, Omoshoro-Jones J, Smith M and Nweke EE (2021) Targeting Growth Factor Signaling Pathways in Pancreatic Cancer: Towards Inhibiting Chemoresistance. Front. Oncol. 11:683788. doi: 10.3389/fonc.2021.683788
Received: 30 March 2021; Accepted: 24 May 2021;
Published: 14 June 2021.
Edited by:
Kwong Tsang, Precision Biologics, Inc., United StatesReviewed by:
Massimo Fantini, Precision Biologics, Inc., United StatesPaul B. Fisher, Virginia Commonwealth University, United States
Copyright © 2021 Xelwa, Candy, Devar, Omoshoro-Jones, Smith and Nweke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ekene Emmanuel Nweke, ZWtlbmUubndla2VAd2l0cy5hYy56YQ==