 Fernán Gómez-Valenzuela
Fernán Gómez-Valenzuela Enrico Escobar
Enrico Escobar Ricardo Pérez-Tomás3
Ricardo Pérez-Tomás3- 1Department of Hematology‐Oncology, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Department of Oral Pathology and Medicine, Faculty of Dentistry, University of Chile, Santiago, Chile
- 3Department of Pathology and Experimental Therapy - Bellvitge, Faculty of Medicine and Health Sciences, University of Barcelona, Barcelona, Spain
The tumor microenvironment (TME) corresponds to a complex and dynamic interconnection between the extracellular matrix and malignant cells and their surrounding stroma composed of immune and mesenchymal cells. The TME has constant cellular communication through cytokines that sustain an inflammatory profile, which favors tumor progression, angiogenesis, cell invasion, and metastasis. Although the epithelial-mesenchymal transition (EMT) represents a relevant metastasis-initiating event that promotes an invasive phenotype in malignant epithelial cells, its relationship with the inflammatory profile of the TME is poorly understood. Previous evidence strongly suggests that cyclooxygenase-2 (COX-2) overexpression, a pro-inflammatory enzyme related to chronic unresolved inflammation, is associated with common EMT-signaling pathways. This review article summarizes how COX-2 overexpression, within the context of the TME, orchestrates the EMT process and promotes initial metastatic-related events.
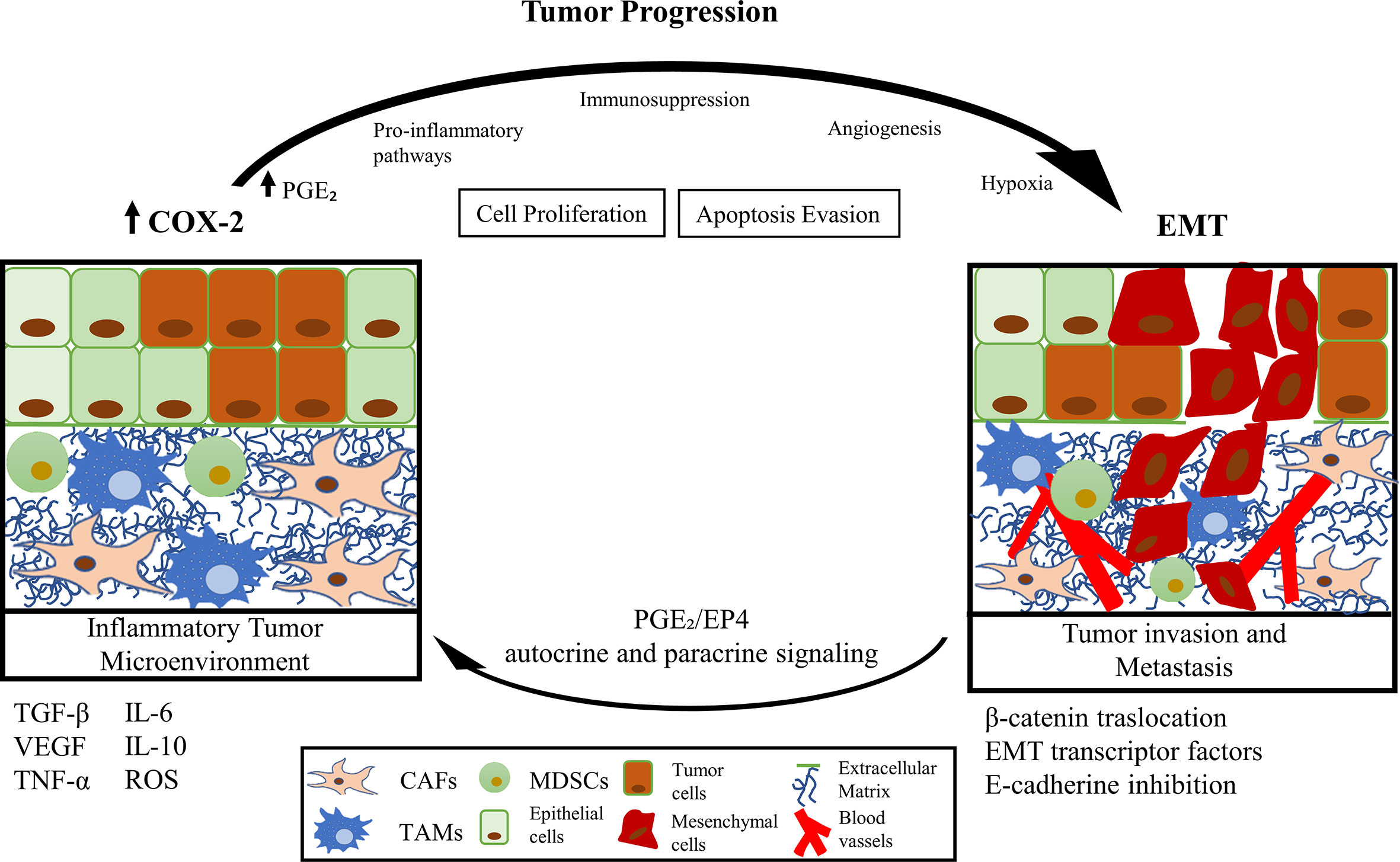
Graphical Abstract
Introduction
The Epithelial-Mesenchymal Transition (EMT) is a transient and reversible cellular modification program (1), essential for various physiological and pathophysiological processes, like embryonic development, stem cell differentiation, wound repair, and healing (2). EMT is characterized by the transition from an epithelial to mesenchymal cellular phenotype, allowing cell migration and invasion (3). Recent researches showed that EMT corresponds to a partial and transitory cellular event composed of multiple stages (4, 5). The main phenotypic changes of epithelial cells that undergo the EMT phenomenon, are the loss of adherens junctions based on E-cadherin and the disorganization of the basal-apical polarity (6, 7). These phenotypic changes favor the expression of classic mesenchymal cell markers, such as vimentin and N-cadherin, together with the increased expression of various transcription factors related to EMT, which are identified both in vitro and in vivo (8, 9). Besides the phenotypic changes described, structural and functional extracellular matrix (ECM) changes, mainly caused by cytokines secretion as the transforming growth factor-beta (TGF-β), promote modifications of EMT-related cellular markers expression (1, 9). Therefore, the EMT program strongly depends on the microenvironment properties (2). Nonetheless, the high heterogeneity of the cellular context limits to unravel of the full spectrum of transcription factors that support all the cancer cell modifications on gene expression and their specific biological consequences (1, 4, 10). The main approaches have proposed the existence of master zinc-finger transcription factors of the Snail family, likewise, Twist and Zeb, as the central directors of EMT, which, along with other transcription factors such as the YAP/TAZ pathway, allow the repression of the epithelial phenotype (1, 2).
In the cancer context, tumor cells appropriate the EMT program, which confers metastatic potential (2, 11). Previous works proposed that one of the fundamental characteristics for EMT development could correspond to their inflammatory profile (12–14). Also, the unresolved-chronic inflammation might increase the risk of cancer (10, 12, 15, 16). The presence of TGF-β and other pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and IL-8, induce the expression of proteins associated with tumor promotion. The overexpression of cyclooxygenase-2 (COX-2), the enzyme responsible for prostanoids synthesis [both thromboxanes and prostaglandins (PGs)], has been indicated as the leading promoter of the inflammatory profile of the TME (17, 18). COX-2 is strongly related to chronic inflammatory events without resolution (19, 20), tumor growth, angiogenesis, cell invasion, metastasis, and chemoresistance, which lead to a low patient survival rate (17, 21, 22). The inducible COX-2 gene is the master switch that activates the inflammatory response (23). COX-2 protein is a critical mediator of inflammation that valuably influences cell proliferation and migration, apoptosis evasion, immunosuppression, tumor angiogenesis, invasion, and metastasis (23–25) (Figure 1).
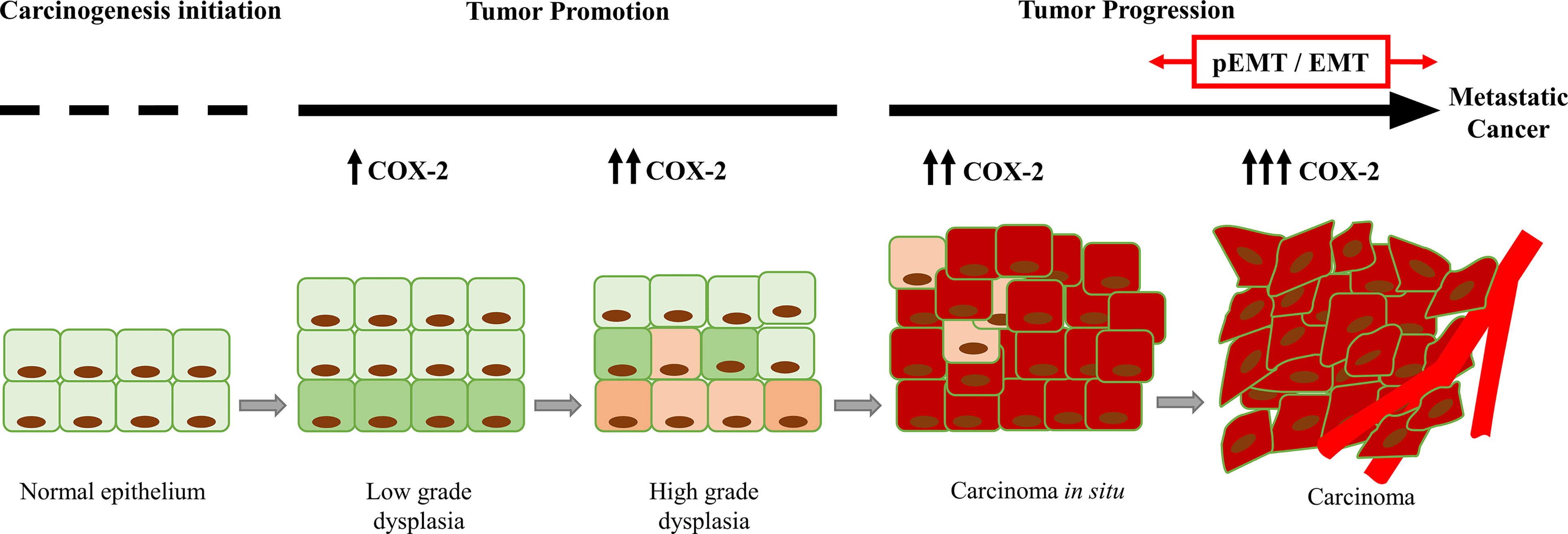
Figure 1 Increase in the COX-2 expression during tumor progression. COX-2 favors tumor progression from the initial stages of tumor promotion (dysplasia). In advanced stages of tumor progression (carcinoma), COX-2 is overexpressed, and it is associated with partial EMT (pEMT) and EMT events.
Currently, it is comprehended that tumor cells can transit through a hybrid EMT phenotype, named partial EMT (pEMT) (26). This pEMT phenotype harbors epithelial and mesenchymal characteristics, promoting a vaster invasive capacity of tumor cells and their collective migration through the keeping of cell-cell adherens junctions. Also, pEMT promotes cancer stemness and drug resistance (27, 28). Cancer cells experiencing pEMT are recognized in the early stages of tumor progression (28) and strongly depend on the tumor context (29). Various microenvironment components, like TGF-β, vascular endothelial growth factor (VEGF), and the hypoxia-inducible factor 1 (HIF-1), are responsible for promoting pEMT (30). We suggest consulting the currently published review by Aggarwal et al. (28) to detail analyze the relationship of the TME with the induction and retention of pEMT.
Surprisingly, the mechanisms proposed for the plasticity and maintenance of pEMT are related to the inflammatory cascade resulting from COX-2 activity (30). An approximation of this possible association was described by Lu et al. (31). They suggested that the overexpression of COX-2 in the MCF-10F breast epithelial cell line would induce a partial transformation towards a mesenchymal phenotype. However, the relationship between COX-2 and the maintenance of pEMT state has not been elucidated.
Inflammation is one of the main contributors to the TME (32). Various tumor promoters, oncogenes, and growth factors mediate the COX-2 up-regulation. The COX-2 overexpression has been reported in several human cancers that include breast (23), lung (33), skin (34), esophagus (35), pancreas (36), prostate (37), bladder (38), stomach (39), oral cavity (40, 41), and colorectal adenocarcinoma (19, 42) (Table 1). In those studies, COX-2 expression was higher in patients with metastases. For example, Harris et al. indicated that 87% of patients with breast cancer metastases present high expression of COX-2 (23). Furthermore, Huang et al. reported that COX-2 expression is higher in tumor stages T3 and T4 in esophageal cancer, concluding that COX-2 is associated with tumor invasion (35). However, the most specific data that support a cause-and-effect connection between COX-2 and tumorigenesis come from genetic studies in animal models (47). Transgenic mice over-expressing COX-2 in epithelial mammary glands, skin, and stomach develop malignancies and metastatic tumors (48–50). Further, COX-2 overexpression can induce the synthesis of proteases and integrins strongly associated with cancer cell invasion in in different tumor types (51). Thus, COX-2 emerges as a crucial factor for metastatic progression in different types of tumors. Also, COX-2 expression is related to the intracellular pathways activated by phosphatidylinositol 3-kinase (PI3K)/Akt, Wnt/β-catenin, and nuclear factor-kappaB (NF-κB) that are associated with tumor progression. The above suggests COX-2 is a relevant element in promoting EMT by modulating the TGF-β pathway (17, 52) and inducing the decrease or complete loss of E-cadherin expression. Therefore, we propose COX-2 regulates E-cadherin expression indirectly by encouraging NF-κB nuclear translocation, which induces the down-regulation of E-cadherin gene and the expression of EMT transcription factors (5).
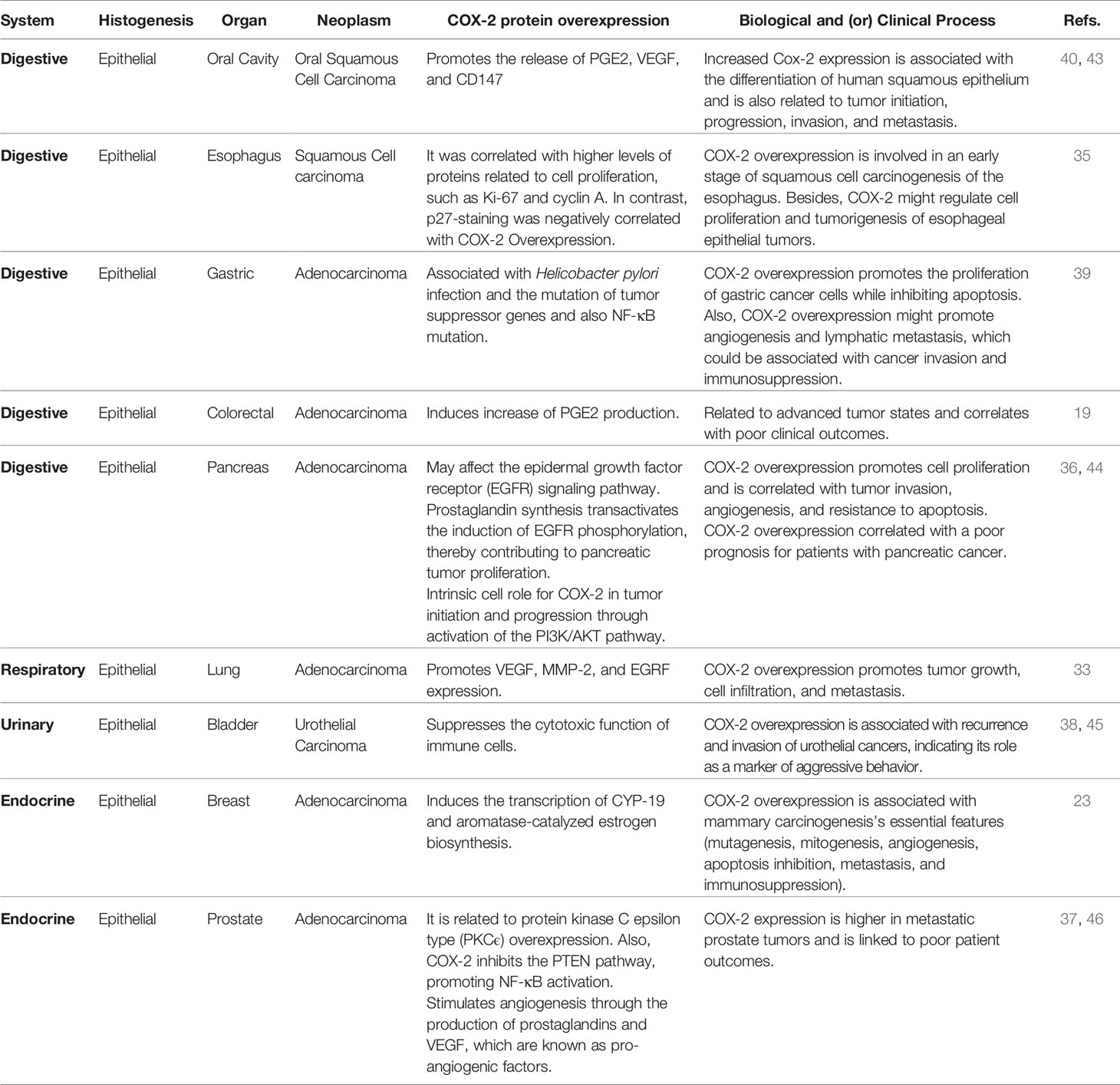
Table 1 COX‐2 protein overexpression related to tumorigenesis and pro-tumoral activity of epithelial cancers (Refs, references).
Although are known various consequences of COX-2 overexpression in the TME, its relationship with the inhibition of E-cadherin expression and EMT phenomenon is still poorly understood. Throughout this review article, we propose signaling pathways that could dominate the relationship between COX-2 (together with its metabolite PGE2, named COX-2/PGE2 axis) and the promotion of EMT in the TME context.
The Tumor Microenvironment (TME) and Inflammation
The TME of solid malignant tumors is a complex and dynamic set of cancer cells, the ECM, surrounding blood vessels, tumor-associated stromal cells (comprising many types of immune cells and fibroblasts), and their secreted soluble factors (53, 54). The interactions within the TME are essential for the heterogeneity of cancer, clonal evolution, and the increase of multidrug resistance, which leads to tumor progression and metastasis (55). During tumor progression, cancer cells elude signals associated with recovering the tissue homeostasis (21). The angiogenic nature of TME is a relevant factor in tumor growth. Folkman described that carcinomas in situ (less than 0.5 to 1 mm in diameter) would have a nonangiogenic profile (56). However, during tumor growth, a shift towards an inflammatory and hypoxic profile of TME causes a strong dependence on angiogenic activity (21), associated with metastatic growth (56). Thus, the TME represents a complex scaffolding of multiple pro- and anti-inflammatory signals without homeostatic balance.
A wide variety of cells participate in this pathologically inflammatory process, which acquire different behaviors and phenotypes in the TME. Among these cell types, tumor-associated macrophages (TAMs) and carcinoma-associated fibroblasts (CAFs) stand out. Within the TME, the COX-2 overexpression orchestrates this inflammatory profile in various types of solid malignant tumors such as gastric cancer (57), colorectal cancer (58), hepatocellular carcinoma (59), melanoma (60), pancreatic cancer (36), endometrial cancer (61), and squamous cell carcinoma of the neck and head (43). Notably, colorectal adenocarcinoma research has helped understand how the COX-2/PGE2 axis affects carcinogenesis and tumor progression. Sada et al. indicates that COX-2/PGE2 overexpression can be observed early in benign lesions like adenomas (62). Therefore, it seems like the sustained overexpression of the COX-2/PGE2 axis favors adenocarcinoma progression in the context of unresolved chronic inflammation (19).
Current investigations have demonstrated a decrease in tumor progression and normalization of the TME through a selective inhibition of COX-2 using the drug celecoxib (CXB) (51, 63). CXB is an anti-inflammatory drug recognized by the World Health Organization (WHO), which can inhibit COX-2 by blocking its functional activity. Based on the action of CXB, it is possible to propose a dual treatment capable of modulating the inflammatory nature of TME, and concomitant, promote the immune response in anti-tumor therapies. Interestingly, this observation was support by Evrard et al., who indicated that COX-2 co-localizes with PD-1 ligand (PD-L1) in peripheral regions of the tumor and its surrounding inflammatory stroma (64). These observations correlate with previous works that demonstrated a positive relationship between the expression of COX-2 and PD-L1 and a high number of metastatic events to lymph nodes in lung adenocarcinoma (65), which could be crucial to understand immunotherapy resistance (64, 66–68). Furthermore, the development of synergic strategies targeting the immune system and the inflammatory nature of the TME is crucial, considering that EMT dramatically influences the response to anti-tumor therapies (69, 70).
COX-2 Overexpression in the Non-Tumoral Cells of the TME: TAMs, Cancer Stem Cells (CSCs), Myeloid-Derived Suppressor Cells (MDSCs) and CAFs
Here, we describe the effects of the COX-2/PGE2 axis activity in the TME, and how would induce the EMT program in cancer cells (Figure 2).
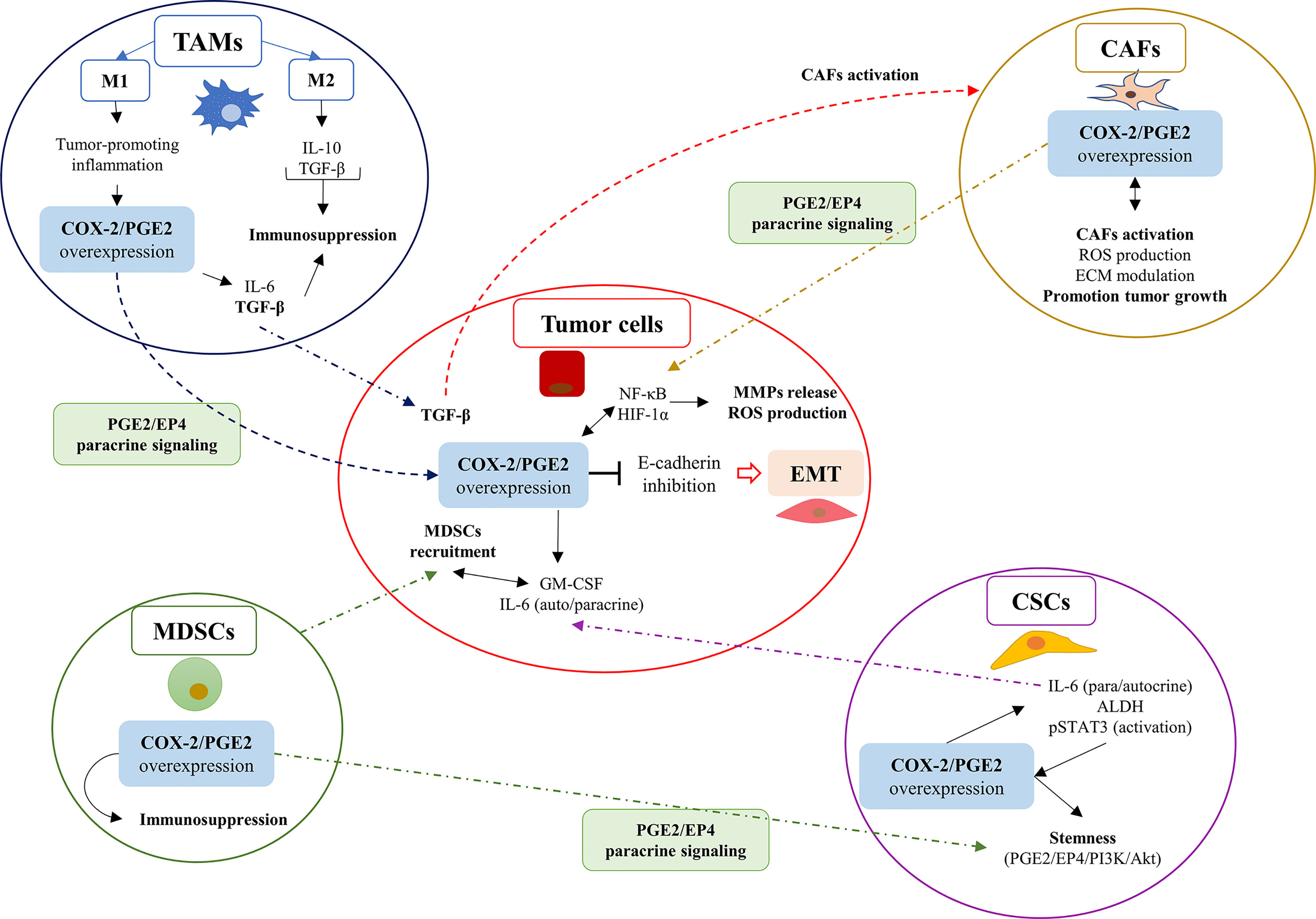
Figure 2 The overexpression of the COX-2/PGE2 axis in cells within the tumor microenvironment (TME) promotes EMT in tumor cells through PGE2/EP4 paracrine signaling. The overexpression of the COX-2/PGE2 axis in cells within the TME promotes EMT in tumor cells through PGE2/EP4 paracrine signaling. The COX-2/PGE2 axis overexpression favors the EMT phenomenon in tumor cells as a consequence of the inhibition of E-cadherin expression. The COX-2/PGE2 overexpression promotes signaling related to ROS and hypoxia, which explains the inflammatory profile of the tumor microenvironment (TME) in solid tumors. Carcinoma-associated fibroblasts (CAFs); cancer stem cells (CSCs); myeloid-derived suppressor cells (MDSCs); and tumor-associated macrophages (TAMs).
COX-2 Overexpression in TAMs
TAMs, originated by the extravasation of monocytes attracted due to inflammatory cytokines secreted by tumor cells (54, 71), stimulate tumor growth, promote EMT and metastasis (54, 72). COX-2 overexpression occurs in different phenotypes of TAMs, and it is accompanied by the expression of COX-2 in tumor cells (73, 74). Gan et al. showed that TAMs co-cultivated with the breast cancer cell line MDA-MB-231, overexpressed COX-2 and induced cancer cell proliferation through the Akt pathway activation. This effect caused the release of matrix metallopeptidases (MMPs) to the TME and the expression of transcription factors associated with EMT, which down-regulate E-cadherin expression (75). In another study, Han et al. showed that TAMs co-cultured with the osteosarcoma cell lines MG-63 and K-HOS promoted cancer cell migration and invasion in vitro and in vivo (76). Also, these cell lines showed up-regulation of COX-2 expression, activation of the STAT3 pathway, and increased release of MMP-9 (76). The overexpression of COX-2 in TAMs also up-regulates IL-6, which perpetuates high levels of COX-2 in tumor cells (77). Recent studies proposed that the expression of IL-6 in TAMs could increase the presence CSCs in esophageal cancer, which is associated with an increase of aldehyde dehydrogenases (ALDHs) expression. This mechanism would promote autocrine signaling of IL-6, together with the activation of the STAT3 pathway in CSCs and tumor cells (15, 78).
Nonetheless, one of the biggest challenges for analyzing the nature of TAMs corresponds to the modulation of their polarity (that is, the differentiation between M1 and M2 phenotypes). Despite the association of the M2 phenotype of TAMs with anti-inflammatory signals, subtypes of alternatively activated M2 endotype (AAM) were associate with tumorigenesis and tumor progression (79). One of the M2 subtypes, known as M2a, has been related to the wound healing process and could be associated with EMT through TGF-β signaling (80). This TAMs-M2 subtype also has been associated with an immunosuppressive profile characterized by increased levels and activation of STAT3 (81). Previous works suggested that TAMs polarization signaling is orchestrated by STAT3/6, TGF-β, and NF-κB, along with other pro-inflammatory pathways closely related to the COX-2/PGE2 axis. Therefore, we hypothesized that in cancers with a high inflammatory profile, the coexistence of different polarities of TAMs could generate cellular communications that favor EMT. Interestingly, Zhao et al. informed that the COX-2 overexpression in TAMs causes up-regulation of TGF-β1 on HCT-116 colon cancer cell line through paracrine pathways (82). This observation was associated with increased expression of the response gene to complement 32 (RGC-32) and the promotion of cell proliferation and migration (82). Several studies propose that the phenotype of TAMs represents a crucial element in the reprogramming profile of their immunosuppressive behavior (83). Hence, we emphasized the necessity of new efforts to explain the mechanisms underlying TAMs acquire different phenotypes in cancer. Also, the discovery of new TAMs biomarkers might be helpful for its modulation in immuno-oncology therapies.
COX-2 Overexpression in CSCs
COX-2 is overexpressed in CSCs from different types of tumors, which include head and neck squamous cell carcinoma (HNSCC) and non-small cell lung cancer (NSCLC), among others (17). Wu et al. showed that aberrant activation of the COX-2/PGE2 axis promotes stemness through the activation of the Wnt/β-catenin pathway in glioblastoma cells (84). Besides, the presence of CSCs was related to radiotherapy resistance and poor survival outcome (85). Interestingly, Majumder et al. exhibited in a preclinical mouse model of breast cancer cell line MCF-7, CSC markers associated with COX-2 expression and EMT (86). PI3K and Akt pathways were common pathways between COX-2 overexpression and EMT promotion. Moreover, the authors established that the PGE2 receptor EP4 protected these cells from apoptosis (86). This result suggests that COX-2 overexpression would induce genes related to progression of CSCs phenotype during EMT.
Consequently, Tong et al. proposed that this COX-2 overexpression was associated with an increase in the CSCs population in various types of tumors (15). Therefore, it has been proposed that selective inhibition of the EP4 receptor combined with other chemotherapeutic strategies, like endocrine therapies, could be used synergistically against chemo resistant cancers (87). Moreover, Lin et al. showed, in a breast cancer model, that the inhibition of the PGE2/EP4 pathway reduces the chemoresistance and the CSCs population in the TME (88). However, until now, the particular role of EP4 receptor in the EMT program has not been established. Terzuoli et al. demonstrated in metastatic melanoma cell line WM266-4 and the metastatic NSCLC cells HCC4006 a large number of CSCs that regulate the redox state of TME (89). Moreover, these authors reported that ALDH expression was related to a higher expression of the EMT markers, NF-κB factor, and COX-2/PGE2 axis. Also, the authors proposed the ALDH3 blockade could be a helpful therapy for cancers strongly inflamed and associated with immunosuppression and stemness. Moreover, they showed that this strategy could be beneficial when therapies based on anti-PD-1/PD-L1 treatment did not have positive outcomes (78, 89). Lastly, it is relevant to recognize that in cancer cell lines from diverse origins, microRNAs (miRNAs) could promote COX-2 overexpression in CSCs (86, 90–92). This information considers possible alternative pathways beyond those traditionally presented, such as stimulation with lipopolysaccharides (LPS) and TNF-α, that could promote COX-2 overexpression in CSCs. Therefore, new studies examining how the inflammatory nature of TME modulates COX-2 activity in CSCs would complement the currently association between EMT and stemness (16, 93).
COX-2 Overexpression in MDSCs
Interestingly, MDSCs, generally described as HLA-DRlow/-CD33+CCD11b+, are not present in normal tissue. Their presence is strongly associated with tumor development and the promotion of immuno-resistance in anti-tumor therapies (94, 95). Similarly, MDSCs are an essential cell type that promotes tumor neovascularization and TME immunosuppression based on CD4+ and CD8+ T cell imbalance (91, 96). Li et al. demonstrated in the nasopharyngeal cancer cell lines TW03 and CNE1, co-cultured with MDSCs, that cell-cell contact stimulated the expression of TGF-β and N2O cytokines in tumor cells (91). Furthermore, the authors illustrated that PGE2 acted on MDSCs EP4 receptors, activating p38-MAPK/extracellular-signal-regulated kinase (ERK) pathways and inducing TGF-β secretion, which causes positive feedback that stimulated EMT. Also, these signals promoted COX-2/PGE2 axis overexpression, as well as β-catenin nuclear translocation (91, 97). On the other hand, Yan et al. confirmed in models of colorectal cancer, that the homeostatic imbalance of TME affected the downstream signal of receptor-interacting protein kinase 3 (RIPK3). RIPK3 was associated with necroptosis promotion, causing the overexpression of the COX-2/PGE2 axis and the induction of the immunosuppressive profile in MDSCs (98). Lastly, Sangaletti et al. suggested that COX-2 overexpression in mammary carcinoma tumor cells, SN25A and SN25ASP activates the IL-6 specifically in rich-collagen stromal areas, inducing the MDSCs recruitment. Further, it was proposed that COX-2 may induce the expression of tumor-associated myelopoiesis factor expression called granulocyte-macrophage colony-stimulating factor (GM-CSF), which allowed cell-cell contact between MDSCs and tumor cells that also favored EMT (99).
COX-2 Overexpression in CAFs
Solid tumors which present CAFs in the tumor stroma have a worse prognosis (100). CAFs differ from normal fibroblasts for the expression of smooth muscle α-actin (α-SMA). CAFs have a phenotype characterized by an over-activated proliferative, secretory, and migratory behavior and can perpetuate an enabling framework for EMT development (13, 21, 54, 101) and the progression of solid tumors (102). CAFs impact the structural and secretory profile of the TME, resulting in the loss of tissue homeostasis (13, 21, 102), along with the release of chemo-attractant and immuno-escaping cytokines. Also, CAFs promote the recruitment of myeloid cells, activation and differentiation of MDSCs, and the polarization of TAMs towards the M2 phenotype (13). Despite the notable advance in understanding how CAFs may induce tumor progression, their contribution in the modulation of the COX-2/PGE2 axis is still not fully elucidated. These questions represent a relevant challenge in the understanding of how CAFs modulate the EMT process. Dudás et al. exposed the ability of CAFs to modify the tumor stroma, co-cultivating oral squamous cell carcinoma cell line SCC-25 with fibroblasts derived from the periodontal ligament. This co-culture generated the transformation of fibroblasts to CAFs after stimulation with IL-1β, which also showed a marked nuclear translocation of NF-κB in CAFs. These effects resulted in transcriptomic overexpression of COX-2 in tumor cells (103). Surprisingly, IL-1β stimulation did not generate an increase in the protein expression of inflammatory mediators like IL-6 (103).
Giannoni et al. demonstrated that CAFs represented the primary source of reactive oxygen species (ROS) in TME, which might be related to their marked COX-2 activity. CAFs would activate pro-inflammatory pathways in tumor cells associated with NF-kB/COX-2/PGE2 and HIF-1 activation and, along with it, raising the levels of small GTPase Rac1b. Besides, the authors proposed ROS as the main EMT-inducing factor (102). Alluringly, these events might explain the origin of COX-2 overexpression in TAMs presented by Gan et al., where CAFs might be one of the principal sources of pro-inflammatory agents (75).
Further, Zhu et al. indicated CAFs from samples of nasopharyngeal carcinoma (NPC) present COX-2/PGE2 axis overexpression and correlate positively with TNF-α (104), which together would promote metastasis. However, EMT markers were unfortunately not evaluated in this model (104). Therefore, further studies are needed to propose CAFs, the primary source of COX-2/PGE2 activity of TME, as the main inductor of EMT.
Repercussions of the COX-2 Overexpression on the EMT Process of Tumor Cells
COX-2/PGE2 axis overexpression corresponds to a transversal and critical event in the inflammatory nature of TME. Consequently, the stromal cells that orchestrate tumor development promote the over-activation of pro-inflammatory signaling pathways in tumor cells, serving for tumor progression and EMT (Figure 3). The evidence suggests a regulation of COX-2 overexpression through a battery of dynamic pro-inflammatory signals emanated from the TME. These pro-inflammatory factors, such as TNF-α and ROS, would stimulate tumor cells directly, affecting COX-2/PGE2 axis overexpression and activation. Furthermore, the same inflammatory context would generate the nuclear translocation of NF-κB and HIF-1, causing an upward rise in these pro-inflammatory factors. The rise of the ROS levels in the TME causes the activation of the interferon regulatory factor 1 (IRF1) in tumor cells and may promote COX-2 activity and PGE2 synthesis (95). In addition, the bidirectional relationship between IL-6 with COX-2/PGE2 overexpression would perpetuate inflammatory events in tumor cells. PGE2 and its receptors play a predominant role in promoting cancer progression. IL-6, also known as interferon-beta2 (IFN-β2), is a pro-inflammatory cytokine that plays a role in inflammation, immune response, hematopoiesis, and cell differentiation. IL-6 production in macrophages is directly stimulated by the COX2/PGE2 axis and TGF-β, while IL-1β and LPS indirectly stimulate IL-6 production via NF-κB activation. A positive association exists between endogenous COX-2 metabolites and IL-6 synthesis in both in vitro and in vivo models of several types of malignant neoplasms (17, 105). IL-6 is the major cytokine in the TME with an extensive range of biological activities. However, the IL-6 expression is deregulated in cancer (106). The IL-6 overexpression is has reported in various cancers, especially from epithelial histogenesis (107) and hematological malignancies (108).
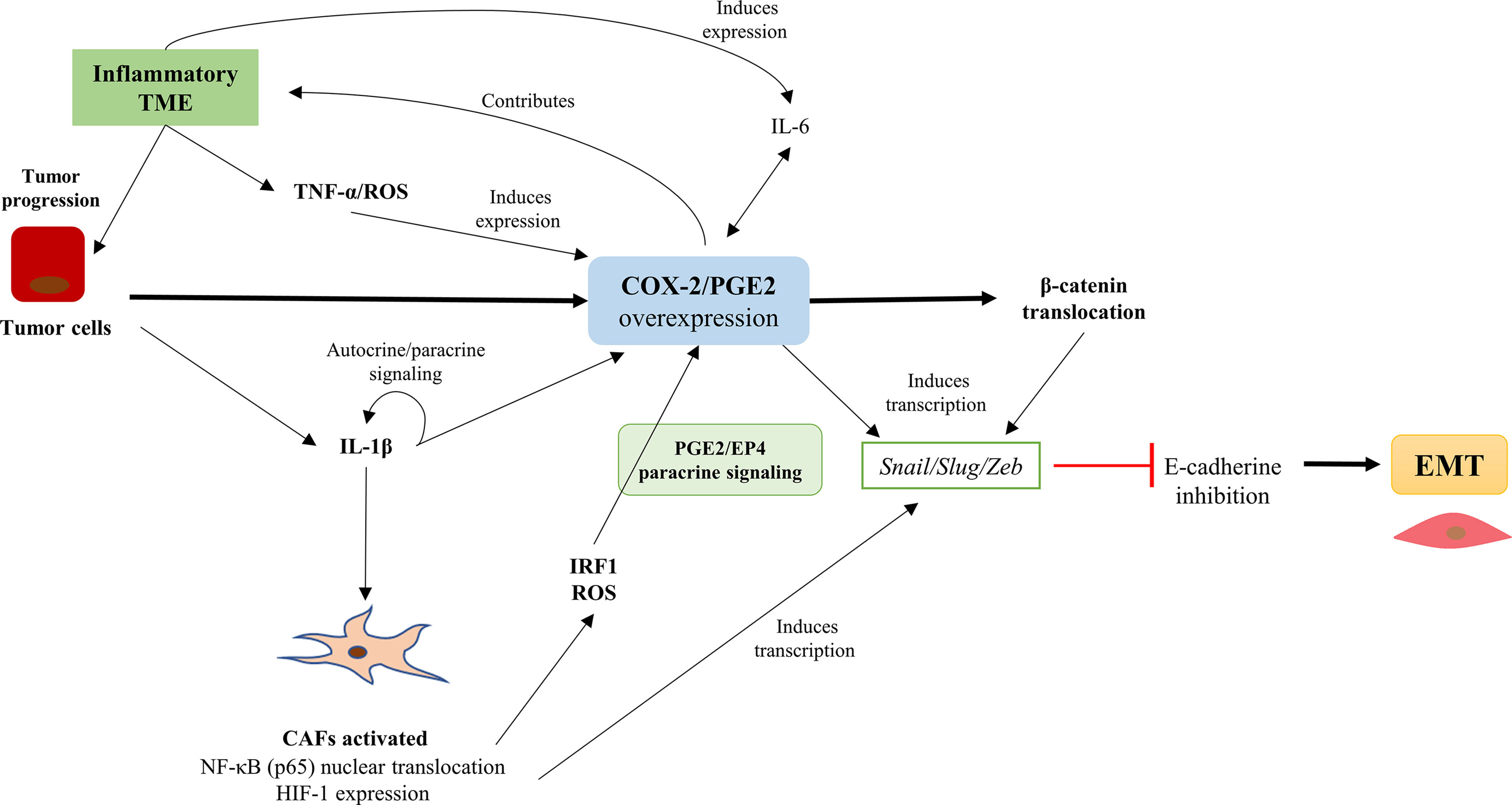
Figure 3 The COX-2/PGE2 axis overexpression is associated with EMT promotion in tumor cells and the presence of cancer-associated fibroblasts (CAFs) in the tumor microenvironment (TME). The COX-2/PGE2 axis overexpression is associated with EMT promotion in cancer cells and the presence of CAFs in the TME. The inflammatory profile of the TME favors the overexpression of the COX-2/PGE2 axis and promotes CAFs activation, which maintain the COX-2/PGE2 axis over-activation. Also, the COX-2/PGE2 axis causing nuclear translocation of β-catenin and the induction of Snail/Slug/Zeb transcription factors expression that repress the E-cadherin and promotes EMT.
Interestingly, the high IL-6 levels in the TME serve to associate chronic inflammation and cancer progression. IL-6 regulates various cancer hallmarks and multiple signaling pathways, such as JAK/STAT3, Ras/MAPK, and PI3K– PKB/Akt. IL-6 regulates many gene products that cause tumor cell growth, evasion of apoptosis, angiogenesis, metastasis, and immunomodulation of the TME (105). Moreover, IL-6 protects cancer cells from therapy-induced DNA damage, oxidative stress, and apoptosis by facilitating the repair and induction of counter signaling (antioxidant and anti-apoptotic/pro-survival) pathways (105). Thus, IL-6 leads to the dysregulation of a plethora of cellular activities that generally promote tumor progression. Patients with high circulating IL-6 levels are generally associated with a poor prognosis and shorter survival (105), mainly in breast carcinoma (105), stomach adenocarcinoma (109), and ovarian epithelial cancer (110).
Activation of Pro-Inflammatory Pathways in Tumor Cells in Response to COX-2 Overexpression
As discussed previously, TGF-β is the principal inducer of EMT. However, previous studies have shown that TGF-β can modulate the expression of VEGF, connective tissue growth factor (CTGF), hepatocyte grow factor (HGF), fibroblast growth factor (FGF), and TNF-α. These factors promote the up-regulation of the COX-2/PGE2 axis, resulting in the induction of β-catenin nuclear translocation in cancer cell lines (51, 95, 111–114) and the expression of EMT transcription factors (75, 77, 95, 113). Moreover, COX-2 overexpression increases the invasive capacity of BGC-823 and SGC-7901 gastric cancer cell lines and may be associated with gastric cancer metastasis (114). In addition, COX-2 regulates the mammalian target of Rapamycin (mTOR) factor and could be a helpful prognostic marker in gastric cancer (114)). In summary, Figure 4 illustrates how the COX-2/PGE2 axis can modulate the classic pro-inflammatory pathways that converge in E-cadherin inhibition and promote the EMT program.
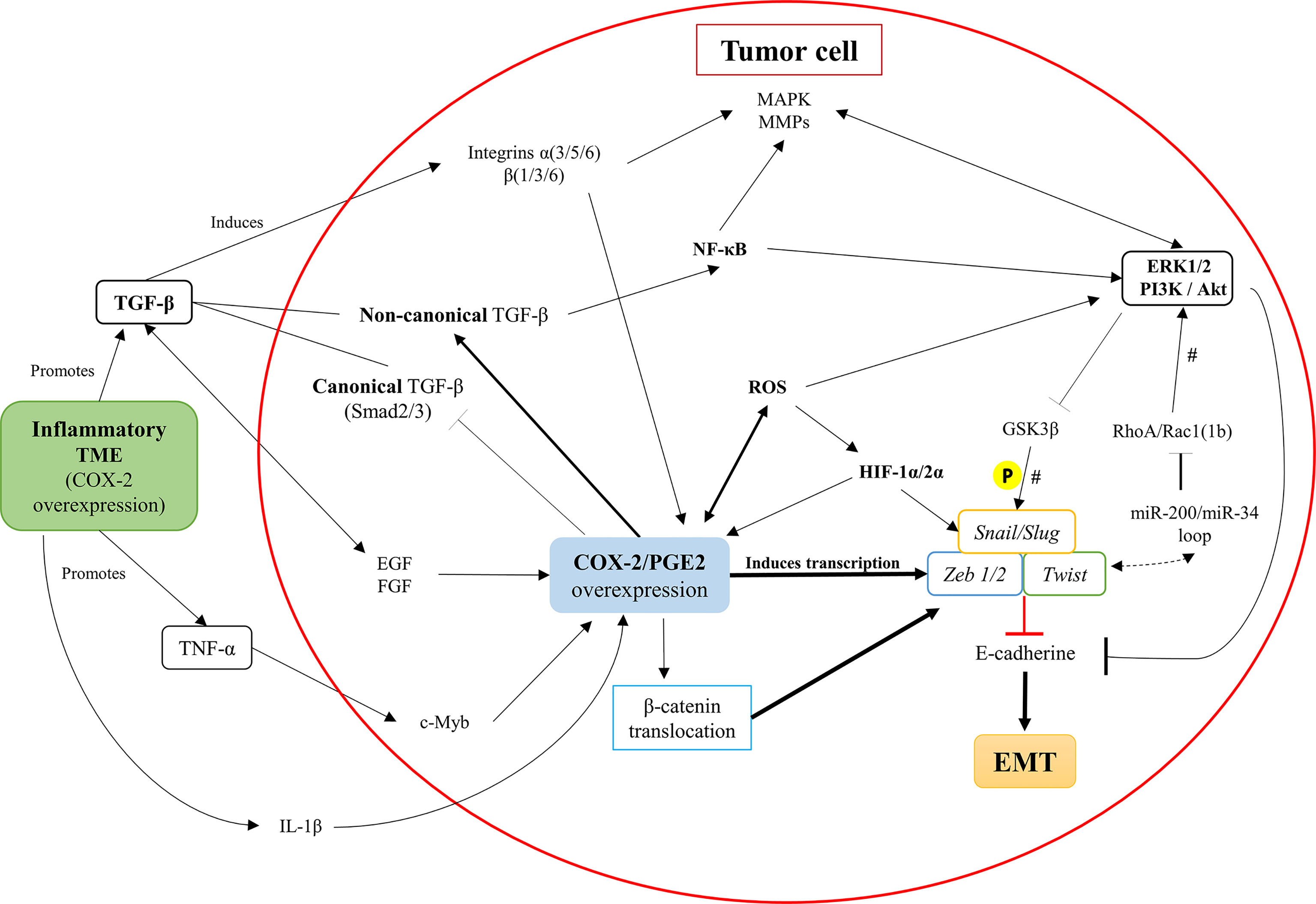
Figure 4 Intracellular pathways in tumor cells that associate COX-2/PGE2 axis overexpression with the promotion of EMT. Inflammatory components of the tumor microenvironment (TME), such as TNF-a and TGF-b, promote the up-regulation of the COX-2/PGE2 axis. Besides, the COX-2/PGE2 axis maintains its autocrine regulation through the activation of the PGE2 EP receptor. The COX-2/PGE2 axis overexpression activates the TGF-β non-canonical pathway (Smad-independent) at the expense of inhibiting the canonical-Smad dependent signaling, along with activation of integrins and secretion of MMPs. Further, the COX-2/PGE2 axis overexpression increases the intracellular ROS levels and activates NF-κB oncogenic functions, which promotes pro-inflammatory signaling pathways and inhibits the GSK3βfactor. The inhibition of GSK3β triggers the activation of master-EMT transcription factors (snail, slug, zeb1/2, and twist), which inhibit E-cadherin expression and finally facilitate the initiation and continuity of the EMT phenomenon. There is a potential EMT modulation associated with RhoA/Rac1, according to the presence of microRNAs that would maintain a dynamic context-dependent role activating EMT transcription factors. #, their participation is established, but their presence does not determine the process; P, phosphorylation.
Neil et al. demonstrated in the murine mammary epithelial cell line NMuMG that, after the EMT induction, the COX-2/PGE2 axis overexpression decouples the canonical TGF-β pathway (dependent on Smad2 and Smad3 factors) through glycogen synthase kinase-3 beta (GSK3β). Interestingly, this COX-2/PGE2 axis overexpression promotes its non-canonical (Smad-independent) pathway, determining the aberrant binding of TGF-β with the transcription factor NFκB (52). This activity may equally be related to the stability of β-catenin through GSK3β (15) and transcription factors of EMT (1). Also, it is proposed that the increase of PGE2 expression PI3K/Akt and ERK1/2 pathways, were strongly associated with oncogenic signaling and EMT promotion (15, 52).
These observations turn out to agree with the results of Chen et al. and Liu et al. (115, 116). They demonstrated NFκB translocation in the gastric cancer cell line SGC-7901 due to COX-2 overexpression, which down.regulates E-cadherin expression via Snail. Along with this, they observed that the silencing of COX-2 increased the mRNA and protein expression of E-cadherin and Snail (115, 116). Furthermore, their results were emphatic for proposing that the functional expression of COX-2 was necessary to regulate NFκB and Snail signals in gastric cancer (116). These results support the idea proposed by García de Herreros et al., who identified NF-κB translocation as a relevant inductor of EMT transcription factors expression and E-cadherin repression (5). However, these authors did not provide a discussion about the origins of this phenomenon beyond the presence of TGF-β (5). Dinicola et al. demonstrated in Caco-2 and HCT-8 colon cancer cells that COX-2/PGE2 axis overexpression induces the activation of PI3K/Akt pathway, and the concomitant NF-κB nuclear translocation, promoting invasiveness (113). In addition, they evinced that after selective COX-2 inhibition, E-cadherin levels normalize consistently with previous studies (113). Furthermore, there is a close relationship between NF-κB nuclear translocation and the redox metabolism of cells within the TME, emphasizing its association in tumor progression, EMT, and metastasis (88, 95).
As previously mentioned, another relevant event associated with COX-2 overexpression corresponds to the presence of a high ROS index within the TME. Concordantly, Giannoni et al. showed high ROS levels induce NF-kB nuclear translocation that activates the HIF-1 and Rac1b factors to promote a TME favorable for tumor progression (102). Current studies indicate that HIF-1 is a relevant factor in the activation of EMT and the promotion of the hypoxic and inflammatory profile of the TME (117). One of the central rationales indicates that this association could promote the inflammatory cytokine-induced c-Myb association towards areas of the promoter-type I collagen gene (COL1A2). This event could generate an increase in the stromal component of the TME and promote hypoxic areas with high expression of HIF1-α, proposing the independent analysis of the COX-2 and c-Myb expression (110).
Various studies have described the role of microenvironmental factors such as hypoxia in COX-2 regulation (118). HIF-1alpha (HIF-1α) is a dimeric protein complex that plays an integral role in low oxygen concentrations (119). Likewise, HIF-1α is a significant regulator of oxygen homeostasis within cells (119), activating multiple genes involved in vasodilatation, angiogenesis, neovascularization, cell survival, invasion, and tumor metastasis (120). HIF-1α protein is overexpressed in multiple types of human cancer, including lung, prostate, breast, pancreas, colon carcinoma, and regional and distant metastases (120, 121). Also, overexpression of HIF-1α can occur very early in carcinogenesis (120, 121). COX-2 up-regulation may facilitate adaptation to cellular stress imposed by hypoxia (118).
During hypoxia, COX-2 upregulation results in higher levels of PGE2 (118). PGE2 can enhance HIF-1 transcriptional activity and VEGF induction under hypoxic conditions (119). For example, in hypoxic colorectal tumor cells, high levels of PGE2 enhance VEGF expression and HIF-1 transcriptional activity by activating the mitogen-activated protein kinase (MAPK) pathway, showing a potential positive feedback loop that contributes to COX-2 up-regulation during hypoxia (118). Further, HIF-1α activates the transcription of genes encoding transferrin, VEGF, endothelin-1, and inducible nitric oxide synthase (NOS2), which are implicated in vasodilation, neovascularization, and tumor metastasis (121).
Interestingly, Basudhar et al. showed a NOS2/COX2 crosstalk during tumor promotion and progression (122). Thereby, the TME promotes various inflammatory pathways that induce NOS2 activation and the increase of nitric oxide (NO) levels. This increase of NO lead to COX-2 activity and the synthesis of PGE2, which also induces NOS2 activity and promotes angiogenesis, immunosuppression, and escape from immunosurveillance (122). Therefore, the products of both enzymes mutually regulate each other in such a way that low and moderate amounts of NO are associated with tumor invasion and EMT (123).
Besides, hypoxia promotes the inflammatory cytokine-induced c-Myb expression towards areas of the promoter-type I collagen gene (COL1A2). This event could generate an excessive increase of the stromal component in the TME and promote hypoxic areas with high expression of HIF1-α (124). Therefore, we suggest to analyzing in detail the association between c-Myb expression and COX-2.
Lastly, Echizen et al., proposed that the COX-2/PGE2 axis overexpression in macrophages and bone marrow-derived cells (BMDCs) up-regulates the transcription factor noxo1 in tumor cells from colon adenocarcinoma. This signaling was due to the TNF-a pathway and may generate an increase in ROS production and stemness in gastrointestinal cancer cell lines (125). Notably, Huang et al. demonstrated in the oxaliplatin-resistant HT29 colorectal cancer cell line that the alteration of PGE2/EP4 signaling reduces intracellular ROS level and inhibits stemness. Therefore, the overexpression of the COX-2/PGE2 axis could promote chemoresistance and the cellular capacity to counteract the potential damages by and elevated level of intracellular ROS (126). Nevertheless, more studies are needed to evaluate this adaptative mechanism against redox state with the EMT program in tumor cells.
Epigenetic Modulation of COX-2/PGE2 Axis and the EMT Transcription Factors
The COX-2/PGE2 axis represents a target for epigenetic modifications that allow its over-activation and perpetuates the inflammatory nature of TME (104, 127, 128). Previous studies suggested various miRNAs that can influence the generation of a dynamic and favorable context for EMT development (128). Surprisingly, several studies highlighted a considerable diversity of influences of these miRNAs on the COX-2/PGE2 axis, which is heavily cancer- and cell context-dependent. It is possible to group these results according to how they finally affect the EMT process. For example, there is evidence of EMT inhibition in bladder cancer (129), oral cancer (130), esophageal squamous cell carcinoma (131), lung cancer (132) and NSCLC (133), cervical cancer (134), melanoma (135), and pancreatic cancer (136), among others. Majumder et al., using the breast cancer cell line MCF-7 observed stimulation of EMT by the miR-526b (86). Therefore, it is necessary to recognize its implications according to each case (89). Consistently, robust evidence indicates epigenetic regulation of the COX-2/PGE2 axis, based on miRNAs presence. Moreover, it is crucial to consider that miRNAs also regulate EMT by inflammatory pathways, directly affecting the canonical and non-canonical signaling of TGF-βand indirectly modulating EMT (127).
According to Mishan et al., there are various routes by which miRNAs could promote the EMT program. Concordantly, MiR-145, miR-335, miR-222, miR-150, and miR-34a have been evidence as EMT promoters. Along with this, miR-200 and miR-34 would be part of an EMT regulatory network (128). These miRNAs are strongly associated with the maintenance of the epithelial phenotype. However, as a result of signals such as NF-κB, TGF-β, and HIF-1 in the cancer context, they also promote the activation of the snail and Zeb transcription factors families. This activation allows the up-regulation of the Rac1/RhoA and PI3K pathways (137), which would indicate the promotion of the mesenchymal phenotype necessary for cell migration. Therefore, it is necessary to evaluate epigenetic elements as possible therapeutic targets, notably considering modulate the COX-2/PGE2 axis and could perpetuate the EMT or pEMT program.
Discussion
Throughout this work, we have highlighted that Epithelial-Mesenchymal Transition is a complex process regulated by microenvironmental demands rather than the internal signaling of each tumor cell. The evidence collected in this work recognizes the COX-2/PGE2 axis as the main driver of EMT and pEMT. The COX-2/PGE2 axis overexpression has an essential role in activating signaling pathways associated with most processes that induce tumor growth and metastasis. Moreover, we consider the COX-2/PGE2 axis not only as a great promoter of tumor progression but metastasis-initiating processes, like EMT. Consequently, it is necessary to carry out clinical studies that consider the potential inhibition of the COX-2/PGE2 axis and EMT markers as metastasis inhibitory treatments in oncology.
Current therapeutic strategies directed to repress the COX-2 overexpression are focused on the inhibition of metabolites resulting from the activity of the COX-2/PGE2 axis. Thus, the specific blockade of the EP4 receptor, has been proposed, showing better results when is with chemotherapeutic regimens or selective COX-2 blocking through CXB (88, 127). Despite this, the selective COX-2 inhibitors have been extensively investigated in diverse types of cancer, generating promising effects as adjuvant therapy. To be specific, it was recognized that CXB reduces the risk of the malignant transformation of colon adenomas towards adenocarcinomas in familial adenomatous polyposis (138). Interestingly, Egashira et al. demonstrated in the HCT-116 colon cancer cell line that CXB inhibits the Wnt/β-catenin signaling pathway, further reducing recurrence after colectomy (139). However, COX-2 inhibitors have been questioned for their potential risk of cardiovascular adverse events. Auspiciously, recent studies showed that CXB has better results according to their gastrointestinal and renal security than other NSAIDs such as ibuprofen and naproxen (140, 141). Also, CXB is associated with fewer cardiovascular adverse events than other COX-2 selective inhibitors, such as rofecoxib (142).
Hashemi Goradel et al. illustrated factors that can modulate the COX-2 targeted therapy, such as type of cancer, type of COX-2 inhibitor, the dose of COX-2 inhibitor, among others (17). Surprisingly, these factors might be associated with the heterogeneous results shown by clinical trials that used CXB as an anti-tumor adjuvant. For example, Hamy et al., based on the exploratory analysis of the REMAGUS02 Trial, indicated that CXB improves the overall survival (OS) and event-free progression (EFP) in breast cancer patients only with high COX-2 expression. Additionally, they indicated the importance of estrogen-receptor status previously the CXB administration (142). Guo et al. demonstrated that the platinum and fluorouracil therapy combined with CXB improved the OS and EFP in patients with advanced and recurrent gastric cancer with COX-2 overexpression (143). However, Kelly et al. evidenced in BOXIT randomized phase III Clinical Trial that CXB does not improve recurrence-free rates of the standard treatment against transitional bladder cell carcinoma (144). Similar results were presented by Bi et al. in Phase II randomized Clinical Trial among patients with NSCLC, without evidence differences between concurrent chemoradiation with or without CXB administration (145). The heterogeneous results of CXB effects in these Clinical Trials could be related to distinct signaling pathways altered in each type of cancer. For example, Zhang et al. showed that the crucial signaling altered in NSCLC was EGF receptor (EGFR). Also, they demonstrated that the use of CXB improves the overall response rate (ORR) (146). Therefore, more studies are required to describe the best therapeutic regimen according to the potential synergism between chemotherapeutic or immunotherapeutic strategies with CXB as an adjuvant.
Other therapeutic strategies with promising prospects are using the association of COX-2 inhibitors and immune checkpoint blockade as a potential choice (147). Several clinical trials of COX-2 inhibitor combination therapy with immune checkpoint inhibitors in cancers have been described. Specifically, the combinations of COX-2 inhibitor inhibitors and anti-programmed cell death protein-1 (anti-PD-1) immunotherapy can promote tumor regression. Wang et al. demonstrated that using COX-2 inhibitor with anti-PD-1 antibody, the objective response rate at six months significantly improved in patients with metastatic melanoma and NSCLC, compared with the anti-PD-1 strategy (148). Furthermore, inhibitor use appears to reverse the unfavorable prognostic effect of a high neutrophil-lymphocyte ratio by prolonging time-to-progression in patients with melanoma (148). Concordantly, Shimizu et al. demonstrated in resected tissue specimens of lung adenocarcinoma that PD-L1 correlated with COX-2 expression, and most cancer cells that express PD-L1 also co-expressed COX-2. However, COX-2 inhibition did not impact PD-L1 expression in NSCLC cell lines as assessed in vitro. These experimental results can be interpreted as suggesting that COX-2 inhibition does not affect PD-L1 expression (149). Therefore, COX-2 inhibition possesses little influence of the efficacy of immune checkpoint inhibitors in lung cancer treatment. Despite this, other studies have shown encouraging results for this combined therapeutic strategy in breast cancer and melanoma (68, 150). News perspectives for the combined use (COX-2 inhibitor and immune checkpoint inhibitors) as a practical therapeutic strategy should be sustained with more evidence in the future.
In conclusion, we propose that COX-2/PGE2 axis overexpression should be recognized as an initial promoter of metastasis. The COX-2/PGE2 axis corresponds to the main promotor of the inflammatory profile of the tumor microenvironment and tumor progression. Hence, its inhibition associated with other therapies may provide beneficial results, especially for those tumors that present overexpression of COX-2. Its potential inhibition could mean the reversal of the EMT or pEMT phenomenon and the impediment of metastases.
Author Contributions
All authors contributed equally to the conception of the work. FG-V drafted and organized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by Project FONDECYT 1150397 (VM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
FG-V acknowledges partial support from CONICYT-PFCHA/Doctorado Nacional 2019-Folio 21190421.
Glossary

References
1. Georgakopoulos-Soares I, Chartoumpekis DV, Kyriazopoulou V, Zaravinos A. Emt Factors and Metabolic Pathways in Cancer. Front Oncol (2020) 10:499. doi: 10.3389/fonc.2020.00499
2. Singh M, Yelle N, Venugopal C, Singh SK. Emt: Mechanisms and Therapeutic Implications. Pharmacol Ther (2018) 182:80–94. doi: 10.1016/j.pharmthera.2017.08.009
3. Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell (2019) 24:65–78. doi: 10.1016/j.stem.2018.11.011
4. Cook DP, Vanderhyden BC. Context Specificity of the EMT Transcriptional Response. Nat Commun (2020) 11:1–9. doi: 10.1038/s41467-020-16066-2
5. García de Herreros A. Epithelial to Mesenchymal Transition in Tumor Cells as Consequence of Phenotypic Instability. Front Cell Dev Biol (2014) 2:71. doi: 10.3389/fcell.2014.00071
6. Bruner HC, Derksen PWB. Loss of E-Cadherin-Dependent Cell–Cell Adhesion and the Development and Progression of Cancer. Cold Spring Harb Perspect Biol (2018) 10:a029330. doi: 10.1101/cshperspect.a029330
7. Mendonsa AM, Na TY, Gumbiner BM. E-Cadherin in Contact Inhibition and Cancer. Oncogene (2018) 37:4769–80. doi: 10.1038/s41388-018-0304-2
8. Plzák J, Bouček J, Bandúrová V, Kolář M, Hradilová M, Szabo P, et al. The Head and Neck Squamous Cell Carcinoma Microenvironment as a Potential Target for Cancer Therapy. Cancers. (2019) 11:440. doi: 10.3390/cancers11040440
9. Derynck R, Weinberg RA. EMT and Cancer: More Than Meets the Eye. Dev Cell (2019) 49:313–6. doi: 10.1016/j.devcel.2019.04.026
10. Ansieau S, Collin G, Hill L. EMT or EMT-promoting Transcription Factors, Where to Focus the Light? Front Oncol (2014) 4:353. doi: 10.3389/fonc.2014.00353
11. Genna A, Vanwynsberghe AM, Villard AV, Pottier C, Ancel J, Polette M, et al. Emt-Associated Heterogeneity in Circulating Tumor Cells: Sticky Friends on the Road to Metastasis. Cancers. (2020) 12:1632. doi: 10.3390/cancers12061632
12. Dominguez C, David JM, Palena C. Epithelial-Mesenchymal Transition and Inflammation at the Site of the Primary Tumor. Semin Cancer Biol (2017) 47:177–84. doi: 10.1016/j.semcancer.2017.08.002
13. Monteran L, Erez N. The Dark Side of Fibroblasts: Cancer-associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol (2019) 10:1835. doi: 10.3389/fimmu.2019.01835
14. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and Inflammation: Inseparable Actors of Cancer Progression. Mol Oncol (2017) 11:805–23. doi: 10.1002/1878-0261.12095
15. Tong D, Liu Q, Wang L, Xie Q, Pang J, Huang Y, et al. The Roles of the COX2/PGE2/EP Axis in Therapeutic Resistance. Cancer Metastasis Rev (2018) 37:355–68. doi: 10.1007/s10555-018-9752-y
16. Jolly MK, Boareto M, Huang B, Jia D, Lu M, Onuchic JN, et al. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front Oncol (2015) 5:155. doi: 10.3389/fonc.2015.00155
17. Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K. Cyclooxygenase-2 in Cancer: A Review. J Cell Physiol (2019) 234:5683–99. doi: 10.1002/jcp.27411
18. Thomas N, Krishnapillai R, Bindhu RP, Thomas T. Immunohistochemical Expression of Cyclooxygenase-2 in Oral Squamous Cell Carcinoma. Indian J Dent Res (2019) 30:102–6. doi: 10.4103/ijdr.IJDR_362_17
19. Sheng J, Sun H, Yu FB, Li B, Zhang Y, Zhu YT. The Role of Cyclooxygenase-2 in Colorectal Cancer. Int J Med Sci (2020) 17:1095–101. doi: 10.7150/ijms.44439
20. Tallima H, El Ridi R. Arachidonic Acid: Physiological Roles and Potential Health Benefits – A Review. J Adv Res (2018) 11:33–41. doi: 10.1016/j.jare.2017.11.004
21. Quail DF, Joyce JA. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat Med (2013) 19:1423–37. doi: 10.1038/nm.3394
22. Fanelli MF, Chinen LTD, Begnami MD, Costa WLJ, Fregnami JHT, Soares FA, et al. The Influence of Transforming Growth Factor-α, Cyclooxygenase-2, Matrix Metalloproteinase (Mmp)-7, MMP-9 and CXCR4 Proteins Involved in Epithelial-Mesenchymal Transition on Overall Survival of Patients With Gastric Cancer. Histopathology. (2012) 61:153–61. doi: 10.1111/j.1365-2559.2011.04139.x
23. Harris RE, Casto BC, Harris ZM. Cyclooxygenase-2 and the Inflammogenesis of Breast Cancer. World J Clin Oncol (2014) 5:677–92. doi: 10.5306/wjco.v5.i4.677
24. Krishnamachary B, Stasinopoulos I, Kakkad S, Penet MF, Jacob D, Wildes F, et al. Breast Cancer Cell Cyclooxygenase-2 Expression Alters Extracellular Matrix Structure and Function and Numbers of Cancer Associated Fibroblasts. Oncotarget. (2017) 8:17981–94. doi: 10.18632/oncotarget.14912
25. Pang LY, Hurst EA, Argyle DJ. Cyclooxygenase-2: A Role in Cancer Stem Cell Survival and Repopulation of Cancer Cells During Therapy. Stem Cells Int (2016) 2016:2048731. doi: 10.1155/2016/2048731
26. Pastushenko I, Blanpain C. Emt Transition States During Tumor Progression and Metastasis. Trends Cell Biol (2019) 29:212–26. doi: 10.1016/j.tcb.2018.12.001
27. Revenco T, Nicodeme A, Pastushenko I, Sznurkowska MK, Latil M, Sotiropoulou PA, et al. Context Dependency of Epithelial-to-Mesenchymal Transition for Metastasis. Cell Rep (2019) 29:1458–68. doi: 10.1016/j.celrep.2019.09.081
28. Aggarwal V, Montoya CA, Donnenberg VS, Sant S. Interplay Between Tumor Microenvironment and Partial EMT as the Driver of Tumor Progression. iScience. (2021) . 24:102113. doi: 10.1016/j.isci.2021.102113
29. Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. Emt, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol (2020) 30:764–76. doi: 10.1016/j.tcb.2020.07.003
30. Saxena K, Jolly MK, Balamurugan K. Hypoxia, Partial EMT and Collective Migration: Emerging Culprits in Metastasis. Transl Oncol (2020) 13:100845. doi: 10.1016/j.tranon.2020.100845
31. Lu S, Yu G, Zhu Y, Archer MC. Cyclooxygenase-2 Overexpression in MCF-10F Human Breast Epithelial Cells Inhibits Proliferation, Apoptosis and Differentiation, and Causes Partial Transformation. Int J Cancer (2005) 116:847–52. doi: 10.1002/ijc.21142
32. Dixon DA, Blanco FF, Bruno A, Patrignani P. Mechanistic Aspects of COX-2 Expression in Colorectal Neoplasia. Recent Results Cancer Res (2013) 191:7–37. doi: 10.1007/978-3-642-30331-9_2
33. Li W, Yue W, Wang H, Lai B, Yang X, Zhang C, et al. Cyclooxygenase-2 is Associated With Malignant Phenotypes in Human Lung Cancer. Oncol Lett (2016) 12:3836–44. doi: 10.3892/ol.2016.5207
34. Shujiao L, Lilin H, Yong S. Cyclooxygenase-2 Expression and Association With Skin Cancer: A Meta-Analysis Based on Chinese Patients. J Cancer Res Ther (2016) 12:C288–90. doi: 10.4103/0973-1482.200762
35. Huang JX, Xiao W, Chen WC, Lin MS, Song ZX, Chen P, et al. Relationship Between COX-2 and Cell Cycle-Regulatory Proteins in Patients With Esophageal Squamous Cell Carcinoma. World J Gastroenterol (2010) 16:5975–81. doi: 10.3748/wjg.v16.i47.5975
36. Wang D, Guo XZ, Li HY, Zhao JJ, Shao XD, Wu CY. Prognostic Significance of Cyclooxygenase-2 Protein in Pancreatic Cancer: A Meta-Analysis. Tumour Biol (2014) 35:10301–7. doi: 10.1007/s13277-014-2260-y
37. Garg R, Blando JM, Perez CJ, Lal P, Feldman MD, Smyth EM, et al. COX-2 Mediates Pro-Tumorigenic Effects of Pkcϵ in Prostate Cancer. Oncogene. (2018) 37:4735–49. doi: 10.1038/s41388-018-0318-9
38. Gakis G. The Role of Inflammation in Bladder Cancer. Adv Exp Med Biol (2014) 816:183–96. doi: 10.1007/978-3-0348-0837-8_8
39. Cheng J, Fan XM. Role of Cyclooxygenase-2 in Gastric Cancer Development and Progression. World J Gastroenterol (2013) 19:7361–8. doi: 10.3748/wjg.v19.i42.7361
40. Renkonen J, Wolff H, Paavonen T. Expression of Cyclo-Oxygenase-2 in Human Tongue Carcinoma and its Precursor Lesions. Virchows Arch (2002) 440:594–7. doi: 10.1007/s00428-002-0616-y
41. Byatnal AA, Byatnal A, Sen S, Guddattu V, Solomon MC. Cyclooxygenase-2–an Imperative Prognostic Biomarker in Oral Squamous Cell Carcinoma–an Immunohistochemical Study. Pathol Oncol Res (2015) 21:1123–31. doi: 10.1007/s12253-015-9940-9
42. Liu Y, Sun H, Hu M, Zhang Y, Chen S, Tighe S, et al. The Role of Cyclooxygenase-2 in Colorectal Carcinogenesis. Clin. Colorectal Cancer (2017) 16:165–72. doi: 10.1016/j.clcc.2016.09.012
43. Nasry WHS, Rodriguez-Lecompte JC, Martin CK. Role of COX-2/PGE2 Mediated Inflammation in Oral Squamous Cell Carcinoma. Cancers. (2018) 10:348. doi: 10.3390/cancers10100348
44. Hill R, Li Y, Tran LM, Dry S, Calvopina JH, Garcia A, et al. Cell Intrinsic Role of COX-2 in Pancreatic Cancer Development. Mol Cancer Ther (2012) 11:2127–37. doi: 10.1158/1535-7163.MCT-12-0342
45. Agrawal U, Kumari N, Vasudeva P, Mohanty NK, Saxena S. Overexpression of COX2 Indicates Poor Survival in Urothelial Bladder Cancer. Ann Diagn Pathol (2018) 34:50–5. doi: 10.1016/j.anndiagpath.2018.01.008
46. Tkacz VL, Tohnya TM, Figg WD. Cyclooxygenase-2 and Angiogenesis in Prostate Cancer. Cancer Biol Ther (2005) 4:813–4. doi: 10.4161/cbt.4.8.2089
47. Subbaramaiah K, Dannenberg AJ. Cyclooxygenase 2: A Molecular Target for Cancer Prevention and Treatment. Trends Pharmacol Sci (2003) 24:96–102. doi: 10.1016/S0165-6147(02)00043-3
48. Oshima H, Oshima M, Inaba K, Taketo MM. Hyperplastic Gastric Tumors Induced by Activated Macrophages in COX-2/mPGES-1 Transgenic Mice. EMBO J (2004) 23:1669–78. doi: 10.1038/sj.emboj.7600170
49. Neufang G, Furstenberger G, Heidt M, Marks F, Müller-Decker K. Abnormal Differentiation of Epidermis in Transgenic Mice Constitutively Expressing Cyclooxygenase-2 in Skin. Proc Natl Acad Sci USA (2001) 98:7629–34. doi: 10.1073/pnas.121574098
50. Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, et al. Overexpression of Cyclooxygenase-2 is Sufficient to Induce Tumorigenesis in Transgenic Mice. J Biol Chem (2001) 276:18563–9. doi: 10.1074/jbc.M010787200
51. Li W, Zhang B, Li H, Zhao C, Zhong Y, Sun J, et al. Tgf β1 Mediates Epithelial Mesenchymal Transition Via β6 Integrin Signaling Pathway in Breast Cancer. Cancer Invest (2014) 32:409–15. doi: 10.3109/07357907.2014.933235
52. Neil JR, Johnson KM, Nemenoff RA, Schiemann WP. Cox-2 Inactivates Smad Signaling and Enhances EMT Stimulated by TGF-b Through a PGE2-dependent Mechanisms. Carcinogenesis. (2008) 29:2227–35. doi: 10.1093/carcin/bgn202
53. Duan Q, Zhang H, Zheng J, Zhang L. Turning Cold Into Hot: Firing Up the Tumor Microenvironment. Trends Cancer (2020) 6:605–18. doi: 10.1016/j.trecan.2020.02.022
54. De Palma M, Biziato D, Petrova TV. Microenvironmental Regulation of Tumour Angiogenesis. Nat Rev Cancer (2017) 17:457–74. doi: 10.1038/nrc.2017.51
55. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun Signal (2020) 18:1–19. doi: 10.1186/s12964-020-0530-4
56. Folkman J. Role of Angiogenesis in Tumor Growth and Metastasis. Semin Oncol (2002) 29:15–8. doi: 10.1053/sonc.2002.37263
57. Echizen K, Hirose O, Maeda Y, Oshima M. Inflammation in Gastric Cancer: Interplay of the COX-2/prostaglandin E2 and Toll-like Receptor/MyD88 Pathways. Cancer Sci (2016) 107:391–7. doi: 10.1111/cas.12901
58. Mizuno R, Kawada K, Sakai Y. Prostaglandin E2/EP Signaling in the Tumor Microenvironment of Colorectal Cancer. Int J Mol Sci (2019) 20:6254. doi: 10.3390/ijms20246254
59. Huang M, Wang L, Chen J, Bai M, Zhou C, Liu S, et al. Regulation of COX-2 Expression and Epithelial-to-Mesenchymal Transition by Hypoxia-Inducible Factor-1α is Associated With Poor Prognosis in Hepatocellular Carcinoma Patients Post TACE Surgery. Int J Oncol (2016) 48:2144–54. doi: 10.3892/ijo.2016.3421
60. Tudor DV, Bâldea I, Lupu M, Kacso T, Kutasi E, Hopârtean A, et al. COX-2 as a Potential Biomarker and Therapeutic Target in Melanoma. Cancer Biol Med (2020) 17:20–31. doi: 10.20892/j.issn.2095-3941.2019.0339
61. Deng L, Liang H, Han Y. Cyclooxygenase-2 and β-Catenin as Potential Diagnostic and Prognostic Markers in Endometrial Cancer. Front Oncol (2020) 10:56. doi: 10.3389/fonc.2020.00056
62. Sada O, Ahmed K, Jeldo A, Shafi M. Role of Anti-Inflammatory Drugs in the Colorectal Cancer. Hosp Pharm (2020) 55:168–80. doi: 10.1177/0018578718823736
63. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE2-induced Tumour Repopulation Abrogates Bladder Cancer Chemoresistance. Nature. (2015) 517:209–13. doi: 10.1038/nature14034
64. Evrard D, Szturz P, Tijeras-Raballand A, Astorgues-Xerri L, Abitbol C, Paradis V, et al. Macrophages in the Microenvironment of Head and Neck Cancer: Potential Targets for Cancer Therapy. Oral Oncol (2019) 88:29–38. doi: 10.1016/j.oraloncology.2018.10.040
65. Shimizu K, Okita R, Saisho S, Maeda A, Nojima Y, Nakata M. Prognostic Value of Cox-2 and PD-L1 Expression and its Relationship With Tumor-Infiltrating Lymphocytes in Resected Lung Adenocarcinoma. Cancer Manage Res (2017) 9:741–50. doi: 10.2147/CMAR.S146897
66. Soundararajan R, Fradette JJ, Konen JM, Moulder S, Zhang X, Gibbons DL, et al. Targeting the Interplay Between Epithelial-to-Mesenchymal-Transition and the Immune System for Effective Immunotherapy. Cancers. (2019) 11:714. doi: 10.3390/cancers11050714
67. Negrao MV, Lam VK, Reuben A, Rubin ML, Landry LL, Roarty EB, et al. Pd-L1 Expression, Tumor Mutational Burden, and Cancer Gene Mutations are Stronger Predictors of Benefit From Immune Checkpoint Blockade Than HLA Class I Genotype in Non–Small Cell Lung Cancer. J Thorac Oncol (2019) 14:1021–31. doi: 10.1016/j.jtho.2019.02.008
68. Botti G, Fratangelo F, Cerrone M, Liguori G, Cantile M, Anniciello AM, et al. COX-2 Expression Positively Correlates With PD-L1 Expression in Human Melanoma Cells. J Transl Med (2017) 15:1–12. doi: 10.1186/s12967-017-1150-7
69. Saloura V, Izumchenko E, Zuo Z, Bao R, Korzinkin M, Ozerov I, et al. Immune Profiles in Primary Squamous Cell Carcinoma of the Head and Neck. Oral Oncol (2019) 96:77–88. doi: 10.1016/j.oraloncology.2019.06.032
70. De Matteis S, Canale M, Verlicchi A, Bronte G, Delmonte A, Crinò L, et al. Advances in Molecular Mechanisms and Immunotherapy Involving the Immune Cell-Promoted Epithelial-to-Mesenchymal Transition in Lung Cancer. J Oncol (2019) 2019:7475364. doi: 10.1155/2019/7475364
71. Joyce JA, Pollard JW. Microenvironmental Regulation of Metastasis. Nat Rev Cancer (2009) 9:239–52. doi: 10.1038/nrc2618
72. Aras S, Raza Zaidi M. Tameless Traitors: Macrophages in Cancer Progression and Metastasis. Br J Cancer (2017) 117:1583–91. doi: 10.1038/bjc.2017.356
73. Bocca C, Ievolella M, Autelli R, Motta M, Mosso L, Torchio B, et al. Expression of Cox-2 in Human Breast Cancer Cells as a Critical Determinant of Epithelial-to-Mesenchymal Transition and Invasiveness. Expert Opin Ther Targets (2014) 18:121–35. doi: 10.1517/14728222.2014.860447
74. Hou Z, Falcone DJ, Subbaramaiah K, Dannenberg AJ. Macrophages Induce COX-2 Expression in Breast Cancer Cells: Role of IL-1β Autoamplification. Carcinogenesis. (2011) 32:695–702. doi: 10.1093/carcin/bgr027
75. Gan L, Qiu Z, Huang J, Li Y, Huang H, Xiang T, et al. Cyclooxygenase-2 in Tumor-Associated Macrophages Promotes Metastatic Potential of Breast Cancer Cells Through Akt Pathway. Int J Biol Sci (2016) 12:1533–43. doi: 10.7150/ijbs.15943
76. Han Y, Guo W, Ren T, Huang Y, Wang S, Liu K, et al. Tumor-Associated Macrophages Promote Lung Metastasis and Induce Epithelial-Mesenchymal Transition in Osteosarcoma by Activating the COX-2/STAT3 Axis. Cancer Lett (2019) 440:116–25. doi: 10.1016/j.canlet.2018.10.011
77. Che D, Zhang S, Jing Z, Shang L, Jin S, Liu F, et al. Macrophages Induce EMT to Promote Invasion of Lung Cancer Cells Through the IL-6-mediated Cox-2/PGE 2 /β-Catenin Signalling Pathway. Mol Immunol (2017) 90:197–210. doi: 10.1016/j.molimm.2017.06.018
78. Chen MF, Chen PT, Lu MS, Chen WC. Role of ALDH1 in the Prognosis of Esophageal Cancer and its Relationship With Tumor Microenvironment. Mol Carcinog (2018) 57:78–88. doi: 10.1002/mc.2273
79. Poh AR, Ernst M. Targeting Macrophages in Cancer: From Bench to Bedside. Front Oncol (2018) 8:49. doi: 10.3389/fonc.2018.00049
80. Sharifi L, Nowroozi MR, Amini E, Arami MK, Ayati M, Mohsenzadegan M. A Review on the Role of M2 Macrophages in Bladder Cancer; Pathophysiology and Targeting. Int Immunopharmacol (2019) 76:105880. doi: 10.1016/j.intimp.2019.105880
81. Prenen H, Mazzone M. Tumor-Associated Macrophages: A Short Compendium. Cell Mol Life Sci (2019) 76:1447–58. doi: 10.1007/s00018-018-2997-3
82. Zhao P, Wang B, Zhang Z, Zhang W, Liu Y. Response Gene to Complement 32 Expression in Macrophages Augments Paracrine Stimulation-Mediated Colon Cancer Progression. Cell Death Dis (2019) 10:1–11. doi: 10.1038/s41419-019-2006-2
83. Beltraminelli T, De Palma M. Biology and Therapeutic Targeting of Tumour-Associated Macrophages. J Pathol (2020) 250:573–92. doi: 10.1002/path.5403
84. Wu M, Guan J, Li C, Gunter S, Nusrat L, Ng S, et al. Aberrantly Activated Cox-2 and Wnt Signaling Interact to Maintain Cancer Stem Cells in Glioblastoma. Oncotarget. (2017) 8:82217. doi: 10.18632/oncotarget.19283
85. Zhao J. Cancer Stem Cells and Chemoresistance: The Smartest Survives the Raid. Pharmacol Ther (2016) 160:145–58. doi: 10.1016/j.pharmthera.2016.02.008
86. Majumder M, Landman E, Liu L, Hess D, Lala PK. Cox-2 Elevates Oncogenic miR-526b in Breast Cancer by EP4 Activation. Mol Cancer Res (2015) 13:1022–33. doi: 10.1158/1541-7786.MCR-14-0543
87. Ching MM, Reader J, Fulton AM. Eicosanoids in Cancer: Prostaglandin E2 Receptor 4 in Cancer Therapeutics and Immunotherapy. Front Pharmacol (2020) 11:819. doi: 10.3389/fphar.2020.00819
88. Lin MC, Chen SY, He PL, Herschman H, Li HJ. PGE2/EP4 Antagonism Enhances Tumor Chemosensitivity by Inducing Extracellular Vesicle-Mediated Clearance of Cancer Stem Cells. Int J Cancer (2018) 143:1440–55. doi: 10.1002/ijc.31523
89. Terzuoli E, Bellan C, Aversa S, Ciccone V, Morbidelli L, Giachetti A, et al. Aldh3a1 Overexpression in Melanoma and Lung Tumors Drives Cancer Stem Cell Expansion, Impairing Immune Surveillance Through Enhanced Pd-L1 Output. Cancers. (2019) 11:1963. doi: 10.3390/cancers11121963
90. Senthil Kumar KJ, Gokila Vani M, Hsieh HW, Lin CC, Liao JW, Chueh PJ, et al. MicroRNA-708 Activation by Glucocorticoid Receptor Agonists Regulate Breast Cancer Tumorigenesis and Metastasis Via Downregulation of NF-κb Signaling. Carcinogenesis. (2019) 40:335–48. doi: 10.1093/carcin/bgz011
91. Li ZL, Ye SB, OuYang LY, Zhang H, Chen YS, He J, et al. COX-2 Promotes Metastasis in Nasopharyngeal Carcinoma by Mediating Interactions Between Cancer Cells and Myeloid-Derived Suppressor Cells. Oncoimmunology. (2015) 4:e1044712. doi: 10.1080/2162402X.2015.1044712
92. Bitarte N, Bandres E, Boni V, Zarate R, Rodriguez J, Gonzalez-Huarriz M, et al. MicroRNA-451 is Involved in the Self-Renewal, Tumorigenicity, and Chemoresistance of Colorectal Cancer Stem Cells. Stem Cells (2011) 29:1661–71. doi: 10.1002/stem.741
93. Liao TT, Yang MH. Revisiting Epithelial-Mesenchymal Transition in Cancer Metastasis: The Connection Between Epithelial Plasticity and Stemness. Mol Oncol (2017) 11:792–804. doi: 10.1002/1878-0261.12096
94. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
95. Hugo HJ, Saunders C, Ramsay RG, Thompson EW. New Insights on COX-2 in Chronic Inflammation Driving Breast Cancer Growth and Metastasis. J Mammary Gland Biol Neoplasia (2015) 20:109–19. doi: 10.1007/s10911-015-9333-4
96. Talmadge JE, Gabrilovich DI. History of Myeloid-Derived Suppressor Cells. Nat Rev Cancer (2013) 13:739–52. doi: 10.1038/nrc3581
97. Li B, Lu Y, Yu L, Han X, Wang H, Mao J, et al. miR-221/222 Promote Cancer Stem-Like Cell Properties and Tumor Growth of Breast Cancer Via Targeting PTEN and Sustained Akt/NF-κb/COX-2 Activation. Chem Biol Interact (2017) 277:33–42. doi: 10.1016/j.cbi.2017.08.014
98. Yan G, Zhao H, Zhang Q, Zhou Y, Wu L, Lei J, et al. A RIPK3-PGE2 Circuit Mediates Myeloid-Derived Suppressor Cell–Potentiated Colorectal Carcinogenesis. Cancer Res (2018) 78:5586–99. doi: 10.1158/0008-5472.CAN-17-3962
99. Sangaletti S, Tripodo C, Santangelo A, Castioni N, Portararo P, Gulino A, et al. Mesenchymal Transition of High-Grade Breast Carcinomas Depends on Extracellular Matrix Control of Myeloid Suppressor Cell Activity. Cell Rep (2016) 17:233–48. doi: 10.1016/j.celrep.2016.08.075
100. Liu L, Liu L, Yao HH, Zhu ZQ, Ning ZL, Huang Q. Stromal Myofibroblasts are Associated With Poor Prognosis in Solid Cancers: A Meta-Analysis of Published Studies. PloS One (2016) 11:1–16. doi: 10.1371/journal.pone.0159947
101. Laplagne C, Domagala M, Le Naour A, Quemerais C, Hamel D, Fournié JJ, et al. Latest Advances in Targeting the Tumor Microenvironment for Tumor Suppression. Int J Mol Sci (2019) 20:4719. doi: 10.3390/ijms20194719
102. Giannoni E, Bianchini F, Calorini L, Chiarugi P. Cancer Associated Fibroblasts Exploit Reactive Oxygen Species Through a Pro-Inflammatory Signature Leading to Epithelial Mesenchymal Transition and Stemness. Antioxidants Redox Signal (2011) 14:2361–71. doi: 10.1089/ars.2010.3727
103. Dudás J, Fullár A, Bitsche M, Schartinger V, Kovalszky I, Sprinzl GM, et al. Tumor-Produced, Active Interleukin-1βregulates Geneexpression in Carcinoma-Associated Fibroblasts. Exp Cell Res (2011) 317:2222–9. doi: 10.1016/j.yexcr.2011.05.023
104. Zhu Y, Shi C, Zeng L, Liu G, Jiang W, Zhang X, et al. High COX-2 Expression in Cancer-Associated Fibiroblasts Contributes to Poor Survival and Promotes Migration and Invasiveness in Nasopharyngeal Carcinoma. Mol Carcinogen (2020) 59:265–80. doi: 10.1002/mc.23150
105. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of Interleukin-6 in Cancer Progression and Therapeutic Resistance. Tumour Biol (2016) 37:11553–72. doi: 10.1007/s13277-016-5098-7
106. Kaur S, Bansal Y, Kumar R, Bansal G. A Panoramic Review of IL-6: Structure, Pathophysiological Roles and Inhibitors. Bioorg Med Chem (2020) 28:115327. doi: 10.1016/j.bmc.2020.115327
107. Zeng J, Tang ZH, Liu S, Guo SS. Clinicopathological Significance of Overexpression of Interleukin-6 in Colorectal Cancer. W. J Gastroenterol (2017) 23:1780–6. doi: 10.3748/wjg.v23.i10.1780
108. Li Y, Du Z, Wang X, Wang G, Li W. Association of IL-6 Promoter and Receptor Polymorphisms With Multiple Myeloma Risk: A Systematic Review and Meta-Analysis. Genet Test Mol Biomarkers (2016) 20:587–96. doi: 10.1089/gtmb.2015.0169
109. Ashizawa T, Okada R, Suzuki Y, Takagi M, Yamazaki T, Sumi T, et al. Clinical Significance of Interleukin-6 (IL-6) in the Spread of Gastric Cancer: Role of IL-6 as a Prognostic Factor. Gastric Cancer (2005) 8:124–31. doi: 10.1007/s10120-005-0315-x
110. Guo Y, Nemeth J, O’Brien C, Susa M, Liu X, Zhang Z, et al. Effects of Siltuximab on the IL-6–Induced Signaling Pathway in Ovarian Cancer. Clin Cancer Res (2010) 16:5759–69. doi: 10.1158/1078-0432.CCR-10-1095
111. Guan X. Cancer Metastases: Challenges and Opportunities. Acta Pharm Sin B (2015) 5:402–18. doi: 10.1016/j.apsb.2015.07.005
112. Ogunwobi OO, Wang T, Zhang L, Liu C. Cyclooxygenase-2 and Akt Mediate Multiple Growth-Factor-Induced Epithelial-Mesenchymal Transition in Human Hepatocellular Carcinoma. J Gastroenterol Hepatol (2012) 27:566–78. doi: 10.1111/j.1440-1746.2011.06980.x
113. Dinicola S, Masiello MG, Proietti S, Coluccia P, Fabrizi G, Catizone A, et al. Nicotine Increases Colon Cancer Cell Migration and Invasion Through Epithelial to Mesenchymal Transition (EMT): COX-2 Involvement. J Cell Physiol (2018) 233:4935–48. doi: 10.1002/jcp.26323
114. Xiang L, Wang W, Zhou Z, Lv M, Tao L, Ni T, et al. COX-2 Promotes Metastasis and Predicts Prognosis on Gastric Cancer Via Regulating Mtor. Biomark Med (2020) 14:421–32. doi: 10.2217/bmm-2019-0357
115. Chen Z, Liu M, Liu X, Huang S, Li L, Song B, et al. COX-2 Regulates E-cadherin Expression Through the NF-κb/Snail Signaling Pathway in Gastric Cancer. Int J Mol Med (2013) 32:93–100. doi: 10.3892/ijmm.2013.1376
116. Liu XJ, Chen ZF, Li HL, Hu ZN, Liu M, Tian AP, et al. Interaction Between Cyclooxygenase-2, Snail, and E-cadherin in Gastric Cancer Cells. World J Gastroenterol (2013) 19:6265–71. doi: 10.3748/wjg.v19.i37.6265
117. Wei Q, Qian Y, Yu J, Wong CC. Metabolic Rewiring in the Promotion of Cancer Metastasis: Mechanisms and Therapeutic Implications. Oncogene (2020) 39:1–18. doi: 10.1038/s41388-020-01432-7
118. Kaidi A, Qualtrough D, Williams AC, Paraskeva C. Direct Transcriptional Up-Regulation of Cyclooxygenase-2 by Hypoxia-Inducible Factor (HIF)-1 Promotes Colorectal Tumor Cell Survival and Enhances HIF-1 Transcriptional Activity During Hypoxia. Cancer Res (2006) 66:6683–91. doi: 10.1158/0008-5472.CAN-06-0425
119. Masoud GN, Li W. Hif-1alpha Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm Sin B (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
120. Jin X, Dai L, Ma Y, Wang J, Liu Z. Implications of HIF-1α in the Tumorigenesis and Progression of Pancreatic Cancer. Cancer Cell Int (2020) 20:273. doi: 10.1186/s12935-020-01370-0
121. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, et al. Overexpression of Hypoxia-Inducible Factor 1a in Common Human Cancers and Their Metastases. Cancer Res (1999) 59:5830–5.
122. Basudhar D, Glynn SA, Greer M, Somasundaram V, No JH, Scheiblin DA, et al. Coexpression of NOS2 and COX2 Accelerates Tumor Growth and Reduces Survival in Estrogen Receptor-Negative Breast Cancer. PNAS (2017) 114:13030–35. doi: 10.1073/pnas.1709119114
123. Khan FH, Dervan E, Bhattacharyya DD, McAuliffe JD, Miranda KM, Glynn SA. The Role of Nitric Oxide in Cancer: Master Regulator or Not? Int J Mol Sci (2020) 21:9393. doi: 10.3390/ijms21249393
124. Xie R, Yang Y, Zhang H, Liu H, Guo J, Qin H, et al. C-Myb and its Effector COX-2 as an Indicator Associated With Prognosis and Therapeutic Outcome in Colorectal Cancer. J Cancer (2019) 10:1601–10. doi: 10.7150/jca.27261
125. Echizen K, Oshima H, Nakayama M, Oshima M. The Inflammatory Microenvironment That Promotes Gastrointestinal Cancer Development and Invasion. Adv Biol Regul (2018) 68:39–45. doi: 10.1016/j.jbior.2018.02.001
126. Huang H, Aladelokun O, Ideta T, Giardina C, Ellis LM, Rosenberg DW. Inhibition of PGE 2/EP4 Receptor Signaling Enhances Oxaliplatin Efficacy in Resistant Colon Cancer Cells Through Modulation of Oxidative Stress. Sci Rep (2019) 9:1–11. doi: 10.1038/s41598-019-40848-4
127. Cebola I, Peinado MA. Epigenetic Deregulation of the COX Pathway in Cancer. Prog Lipid Res (2012) 51:301–13. doi: 10.1016/j.plipres.2012.02.005
128. Mishan MA, Khazeei Tabari MA, Zargari M, Bagheri A. MicroRNAs in the Anticancer Effects of Celecoxib: A Systematic Review. Eur J Pharmacol (2020) 882:173325. doi: 10.1016/j.ejphar.2020.173325
129. Liu X, Wu Y, Zhou Z, Huang M, Deng W, Wang Y, et al. Celecoxib Inhibits the Epithelial-to-Mesenchymal Transition in Bladder Cancer Via the miRNA-145/TGFBR2/Smad3 Axis. Int J Mol Med (2019) 44:683–93. doi: 10.3892/ijmm.2019.4241
130. D’Souza W, Kumar A. microRNAs in Oral Cancer: Moving From Bench to Bed as Next Generation Medicine. Oral Oncol (2020) 111:104916. doi: 10.1016/j.oraloncology.2020.104916
131. Shao Y, Li P, Zhu ST, Yue JP, Ji XJ, Ma D, et al. MiR-26a and miR-144 Inhibit Proliferation and Metastasis of Esophageal Squamous Cell Cancer by Inhibiting Cyclooxygenase-2. Oncotarget. (2016) 7:15173–86. doi: 10.3892/or.2014.3648
132. Monteleone NJ, Lutz CS. miR-708-5p Targets Oncogenic Prostaglandin E2 Production to Suppress a Pro-Tumorigenic Phenotype in Lung Cancer Cells. Oncotarget. (2020) 11:2464–83. doi: 10.18632/oncotarget.27614
133. Lai XN, Li J, Tang LB, Chen WT, Zhang L, Xiong LX. MiRNAs and LncRNAs: Dual Roles in TGF-β Signaling-Regulated Metastasis in Lung Cancer. Int J Mol Sci (2020) 21:1193. doi: 10.3390/ijms21041193
134. Miao J, Regenstein JM, Xu D, Zhou D, Li H, Zhang H, et al. The Roles of microRNA in Human Cervical Cancer. Arch Biochem Biophys (2020) 690:108480. doi: 10.1016/j.abb.2020.108480
135. Panza E, Ercolano G, De Cicco P, Armogida C, Scognamiglio G, Botti G, et al. MicroRNA-143-3p Inhibits Growth and Invasiveness of Melanoma Cells by Targeting Cyclooxygenase-2 and Inversely Correlates With Malignant Melanoma Progression. Biochem Pharmacol (2018) 156:52–9. doi: 10.1016/j.bcp.2018.08.008
136. Gong R, Jiang Y. Non-Coding RNAs in Pancreatic Ductal Adenocarcinoma. Front Oncol (2020) 10:309. doi: 10.3389/fonc.2020.00309
137. Gkretsi V, Stylianopoulos T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front Oncol (2018) 8:145. doi: 10.3389/fonc.2018.00145
138. Kim B, Giardiello FM. Chemoprevention in Familial Adenomatous Polyposis. Best Pract Res Clin Gastroenterol (2011) 25:607–22. doi: 10.1016/j.bpg.2011.08.002
139. Egashira I, Takahashi-Yanaga F, Nishida R, Arioka M, Igawa K, Tomooka K, et al. Celecoxib and 2, 5-Dimethylcelecoxib Inhibit Intestinal Cancer Growth by Suppressing the Wnt/β-Catenin Signaling Pathway. Cancer Sci (2017) 108:108–15. doi: 10.1111/cas.13106
140. Varrassi G, Pergolizzi JV, Dowling P, Paladini A. Ibuprofen Safety at the Golden Anniversary: Are All NSAIDs the Same? A Narrative Review. Adv Ther (2020) 37:61–82. doi: 10.6084/m9.figshare.10075727
141. Li M, Yu C, Zeng X. Comparative Efficacy of Traditional non-Selective NSAIDs and Selective Cyclo-Oxygenase-2 Inhibitors in Patients With Acute Gout: A Systematic Review and Meta-Analysis. BMJ Open (2020) 10:e036748. doi: 10.1136/bmjopen-2019-036748
142. Hamy AS, Tury S, Wang X, Gao J, Pierga JY, Giacchetti S, et al. Celecoxib With Neoadjuvant Chemotherapy for Breast Cancer Might Worsen Outcomes Differentially by COX-2 Expression and ER Status: Exploratory Analysis of the REMAGUS02 Trial. J Clin Oncol (2019) 37:624–35. doi: 10.1200/JCO.18.00636
143. Guo Q, Li Q, Wang J, Liu M, Wang Y, Chen Z, et al. A Comprehensive Evaluation of Clinical Efficacy and Safety of Celecoxib in Combination With Chemotherapy in Metastatic or Postoperative Recurrent Gastric Cancer Patients: A Preliminary, Three-Center, Clinical Trial Study. Med (Baltimore) (2019) 98:e16234. doi: 10.1097/MD.0000000000016234
144. Kelly JD, Tan WS, Porta N, Mostafid H, Huddart R, Protheroe A, et al. BOXIT—a Randomised Phase III Placebo-Controlled Trial Evaluating the Addition of Celecoxib to Standard Treatment of Transitional Cell Carcinoma of the Bladder (CRUK/07/004). Eur Urol (2019) 75:593–601. doi: 10.1016/j.eururo.2018.09.020
145. Bi N, Liang J, Zhou Z, Chen D, Fu Z, Yang X, et al. Effect of Concurrent Chemoradiation With Celecoxib vs Concurrent Chemoradiation Alone on Survival Among Patients With Non–Small Cell Lung Cancer With and Without Cyclooxygenase 2 Genetic Variants: A Phase 2 Randomized Clinical Trial. JAMA Netw Open (2019) 2:e1918070–e1918070. doi: 10.1001/jamanetworkopen.2019.18070
146. Zhang W, Yi L, Shen J, Zhang H, Luo P, Zhang J. Comparison of the Benefits of Celecoxib Combined With Anticancer Therapy in Advanced Non-Small Cell Lung Cancer: A Meta-Analysis. J Cancer (2020) 11:1816. doi: 10.7150/jca.35003
147. Pu D, Yin L, Huang L, Qin C, Zhou Y, Wu Q, et al. Cyclooxygenase-2 Inhibitor: A Potential Combination Strategy With Immunotherapy in Cancer. Front Oncol (2021) 26:637504. doi: 10.3389/fonc.2021.637504
148. Wang SJ, Khullar K, Kim S, Yegya-Raman N, Malhotra J, Groisberg R, et al. Effect of Cyclo-Oxygenase Inhibitor Use During Checkpoint Blockade Immunotherapy in Patients With Metastatic Melanoma and Non-Small Cell Lung Cancer. J Immunother Cancer (2020) 8:e000889. doi: 10.1136/jitc-2020-000889
149. Shimizu K, Okita R, Saisho S, Maeda AI, Nojima Y, Nakata M. Impact of COX2 Inhibitor for Regulation of PD-L1 Expression in Non-Small Cell Lung Cancer. Anticancer Res (2018) 38:4637–44. doi: 10.21873/anticanres.12768
Keywords: tumor microenvironment, cyclooxygenase-2, epithelial-mesenchymal transition, cancer, inflammation
Citation: Gómez-Valenzuela F, Escobar E, Pérez-Tomás R and Montecinos VP (2021) The Inflammatory Profile of the Tumor Microenvironment, Orchestrated by Cyclooxygenase-2, Promotes Epithelial-Mesenchymal Transition. Front. Oncol. 11:686792. doi: 10.3389/fonc.2021.686792
Received: 27 March 2021; Accepted: 13 May 2021;
Published: 10 June 2021.
Edited by:
Daniele Vergara, University of Salento, ItalyReviewed by:
Eric Hervouet, Université Bourgogne Franche-Comté, FranceSara Ricardo, Universidade do Porto, Portugal
Copyright © 2021 Gómez-Valenzuela, Escobar, Pérez-Tomás and Montecinos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernán Gómez-Valenzuela, ZmRnb21lejJAdWMuY2w=; Viviana P. Montecinos, dml2aWFuYS5tb250ZWNpbm9zQHVjLmNs