 Sumana Shrestha
Sumana Shrestha Alaide Morcavallo1
Alaide Morcavallo1 Chiara Gorrini
Chiara Gorrini- 1Division of Clinical Studies, Institute of Cancer Research (ICR), London and Royal Marsden NHS Trust, Sutton, United Kingdom
- 2Division of Cancer Therapeutics, The Institute of Cancer Research (ICR), and The Royal Marsden NHS Trust, Sutton, United Kingdom
The constitutive and dysregulated expression of the transcription factor MYCN has a central role in the pathogenesis of the paediatric brain tumour medulloblastoma, with an increased expression of this oncogene correlating with a worse prognosis. Consequently, the genomic and functional alterations of MYCN represent a major therapeutic target to attenuate tumour growth in medulloblastoma. This review will provide a comprehensive synopsis of the biological role of MYCN and its family components, their interaction with distinct signalling pathways, and the implications of this network in medulloblastoma development. We will then summarise the current toolbox for targeting MYCN and highlight novel therapeutic avenues that have the potential to results in better-tailored clinical treatments.
Introduction
The MYC family of transcription factors, including c-MYC (MYC), MYCL and MYCN are amongst the most commonly altered genes in cancer, including paediatric cancers (1). Tumorigenic activity of the MYC family can result from constitutive activation of associated mitogenic signalling pathways e.g., Wingless (WNT), Hedgehog (SHH), Transforming growth factor beta (TGF-β), or through direct genetic alterations from amplification or chromosomal aberrations. Sequence homology between the two proteins, MYC and MYCN remain relatively high, and similarities remain between organisation of loci, and protein binding sites (2–4). Pioneering developmental studies have been integral in illustrating the interchangeable nature of the MYC proteins, in particular between MYC and MYCN (5). These studies assessing the phenotypic consequences of Myc or Mycn deficiency in mouse development identified expression divergence during the early developmental stages. Whilst null homozygosity for both Myc and Mycn resulted in embryonic lethality (approximately E10.5-E11.5), Myc-null embryos were associated with marked reduction in size and a general delay in primitive development of the heart (6). Mycn-null embryos also exhibited delayed development and stunted growth, as well as diminished cellularity in organs that normally express abundant Mycn transcripts, most notably the cranial and spinal ganglia (7–9). Significantly, despite a compensatory Myc increase observed in Mycn deficient embryos (8), developmental defects occurred that suggested a unique and essential role for Mycn during CNS development. Conversely, replacement of endogenous Myc-coding sequences with Mycn-coding sequences showed that Mycn is capable of performing most of the essential functions of Myc required for embryonic development and proliferation (5). Whilst the proteins share similarities in structure and binding partner MAX (10), the differences remain in their spatial and temporal expression patterns, with MYCN showing a preference to the early hindbrain development (11–14). Overall, both proteins at the transcriptional level can orchestrate the cell cycle machinery and stimulate cell growth, division, and regulate the differentiation states of cells throughout development.
In this review, we will focus on the role of the MYC family proteins, specifically MYCN, in different subgroups of the childhood brain tumour medulloblastoma (MB). MYC proteins play an important role in MB biology and often are dysregulated in all MB tumours, with MYC, MYCN and MYCL1 each showing commitment to specific subgroup (15). MYC and MYCN amplifications especially are prominent in MB due to the highly aggressive nature of tumours associated with these aberrations (16). MYC and MYCN have been considered undruggable for many years as they carry out essential functions in proliferative tissues, suggesting that their inhibition could cause severe side effects. Only recently has there been an improvement in making their protein surfaces amenable to binding small molecules, further accelerating their use in therapeutics (17). We will highlight the potential application of several new therapeutic strategies targeting MYCN and its signalling partners to tackle the overarching obstacles.
Medulloblastoma
Clinical and Molecular Diversity of Medulloblastoma
Medulloblastoma is one of the most prevalent malignant paediatric brain tumours (WHO grade IV) (18). MB arises from the posterior fossa and features a heterogeneous tumour landscape. MB accounts for ~63% of childhood intracranial embryonal tumours, and has an incidence of 4.9 per 1 million children, peaking at ~7 years of age (19, 20). The tumour is usually proximal to the fourth ventricle, making metastasis through cerebrospinal fluid (CSF) flow common (21). The current standard of care consists of maximal surgical resection followed by cranio-spinal irradiation (CSI) in patients >3 years, and multi-agent chemotherapy. The overall survival rate ranges from 40-90%, depending on the molecular subtype and other factors such as extent of dissemination and degree of resection. Whilst survival rates have improved overtime due to better understanding and implementation of CSI, ~1/3 of patients still succumb to the disease, and survivors often experience debilitating neurologic, endocrinologic, and cognitive sequelae from treatment (22).
Different Subgroups and Subtypes
Initially, using gene expression analysis techniques, MB was segregated into four distinct molecular subgroups, with differences in genomic drivers, mutational events, methylation patterns and clinical characteristics (23). These groups are known as: wingless (WNT), Sonic hedgehog (SHH), and, group 3 (Grp3), and group 4 (Grp4), also known as non-WNT/non-SHH (24–27). Due to the well-defined developmental pathways of WNT and SHH, many studies have been able to dissect the mechanism of these two groups, whereas Grp3 and Grp4 have recently raised attention owing to next-generation sequencing techniques (15, 28–31). Further gene expression and DNA-methylation analysis of the subgroups by Cavalli et al. introduced additions layers of heterogeneity within the four main subgroups, these are as follows: WNT; WNT-α and WNT-β, SHH; SHH-α, SHH-β, SHH-χ, SHH-δ, Group 3; Grp3-α, Grp3-β, Grp3-χ and Group 4; Grp4-α, Grp4-β, Grp4-χ (32). Adding to this, other variations of subtypes emerged during the same time as several research groups utilised different sample analysis methods (33, 34). Whilst the use of separate analytic techniques provides a more dynamic and richer dataset, it is now essential to compare and combine these differences to produce a single, streamline set of subtypes.
Comparing the clinical features of the subgroups, the WNT subgroup has the most favourable clinical outcomes, with the overall survival standing at >90% (35). However, this is the least common MB subtype, accounting for only 10% of MBs (23). The favourable prognosis associated with activation of the WNT signalling pathway is now being exploited for other subgroups of this cancer (36). The majority of these tumours (86%) harbour activating mutations in β-catenin (CTNNB1), a central orchestrator of the canonical WNT pathway (33, 37, 38), or mutations in the tumour suppressor gene APC (71%) (39). Further prominent genes identified from whole genome sequencing include DDX3X (7.6%), SMARCA4 (3.4%), TP53 (~10%) and KMT2D (~7%) (40).
The SHH-subgroup, despite its heterogeneity, is the best clinically and molecularly characterised MB-subgroup. The age of the patient is especially important here as each age group has a distinct transcriptomic profile; adult patients (SHH-δ) show frequent mutation in the TERT promoter, whilst younger patients show enrichment in focal amplifications of MYCN, GLI2, and YAP1, frequent germline or somatic TP53 mutations, and more recently discovery of germline variants in ELP1 (32, 41–44). ELP1 encodes the scaffold protein elongator complex protein 1 (ELP1) which is involved in neuronal migration and is responsible for transcriptional elongation (45, 46). Furthermore, PTCH1 mutation is highly frequent in this subgroup, the only distinction of this within the subtypes is the number of additional aberrations accommodating this mutation; with a higher aberrational load seen in the adult subtype (SHH-δ). More recent research shows a prominent role for TP53 dysregulation. This frequently arises in conjunction with chromothripsis, a catastrophic genomic rearrangement commonly occurring due to micronucleus formations (29, 42, 47).
In contrast to WNT and SHH, both Grp3, and Grp4 have very few prominent driver genes. Nonetheless, they both show distinct genetic events which define each subgroup as separate entities. Grp3 MB primarily occurs during infancy and childhood and is associated with a high rate of disseminated disease. This subgroup is defined by high levels of MYC amplification and a particular genetic signature related to increased transcription and translation (48). Important genes dysregulated at a somatic level include SMARCA4, KBTBD4, and KMT2D. Furthermore, recent studies have shown increased activation of genes representing the Notch and TGFβ signalling pathways, and a particular inclination for activation of GFI1/GFI1B through enhancer hijacking (49). Grp4 MB is the most prevalent MB subgroup. With a lack of single gene mutations, this subgroup features a higher frequency of disposition to somatic mutations, with notable mutations in histone-modifying genes such as KDM6A, ZMYM3 and KMT2C. The recent discovery of ERBB4-SRC signalling in Grp4 tumours has highlighted this pathway as a hallmark of Grp4 MB (50). Both Grp3 and Grp4 groups have recurrent mutations in KBTBD4, underpinning the gene as a common candidate tumour driver (33).
Cancer predisposition syndromes remain a risk factors for the development of MB and account for approximately 5-6% of MBs (39). Germline mutations in WNT signalling pathway genes such as APC mutations, found in Turcot Syndrome, can lead to the WNT subgroup. SHH MB can be initiated through various germline mutations such as PTCH1, occurring in the autosomal dominant condition Gorlin syndrome (known as nevoid basal cell carcinoma syndrome), or aberration in germline TP53 as seen in Li-Fraumeni syndrome (33). Furthermore, germline mutation in SMO from Curry-Jones syndrome is also associated with SHH MB (39). With the advancement in stem cell research and patient-derived iPSC culture systems, this information will inevitably allow for more accurate modelling and prediction of the development of particular subgroups of MB (51).
The innate differences within this highly heterogeneous cancer provides an insight into the putative cells of origin residing in different regions of the cerebellum (32, 52). The embryonic nature of MB makes it difficult to pinpoint the exact cell of origin. Therefore, it is essential to investigate the complete genetic aetiology of these tumours in order to build more accurate and robust models of the disease. The normal development of the cerebellum serves as a healthy control for the development of MB. MB cells are thought to arise from progenitor cell populations from early hindbrain development. This has been investigated for WNT and SHH MB, with WNT tumours thought to arise from the extracerebellar lower rhombic lip, and SHH from cerebellar granule cell precursors (GCPs) (53). More recently, it was postulated that for Grp4, the cellular origin consisted of more differentiated neuronal population, with glutamatergic cells including residues of unipolar brush cells and glutamatergic cerebellar nuclei (30, 54). Whilst for Grp3 origin remains quite vague, with studies referring to a potential origin of undifferentiated progenitor-like lineage with high MYC activity (54).
The Role of MYCN in the Origin of Medulloblastoma
MYCN in Cerebellar Development
When looking at the development of MB, the environment in which the tumour grows should be treated almost as a crime scene, as Bailey and Cushing once wrote "the histogenesis of the brain furnishes the indispensable background for an understanding of its tumours'' (55). A defect in the normal expansion of the cerebellar precursor population can lead to uncontrolled proliferation, resulting in the development of MB.
The developing cerebellum is moulded by three distinct pools of progenitor cells; these consist of GCPs from the deep nuclei (emerging from rhombic lip), GABAergic Purkinje cells (arising from multipotent precursors of the primary germinal epithelium in the roof of the 4th ventricle), and CD133/Nestin+ cells (the white matter of the postnatal cerebellum). During postnatal development, GCPs rapidly proliferate and expand in response to SHH secreted by Purkinje cells (56), and mature to become cerebellar granule neurons – the largest neuronal population in the brain (57), as shown in Figure 1.
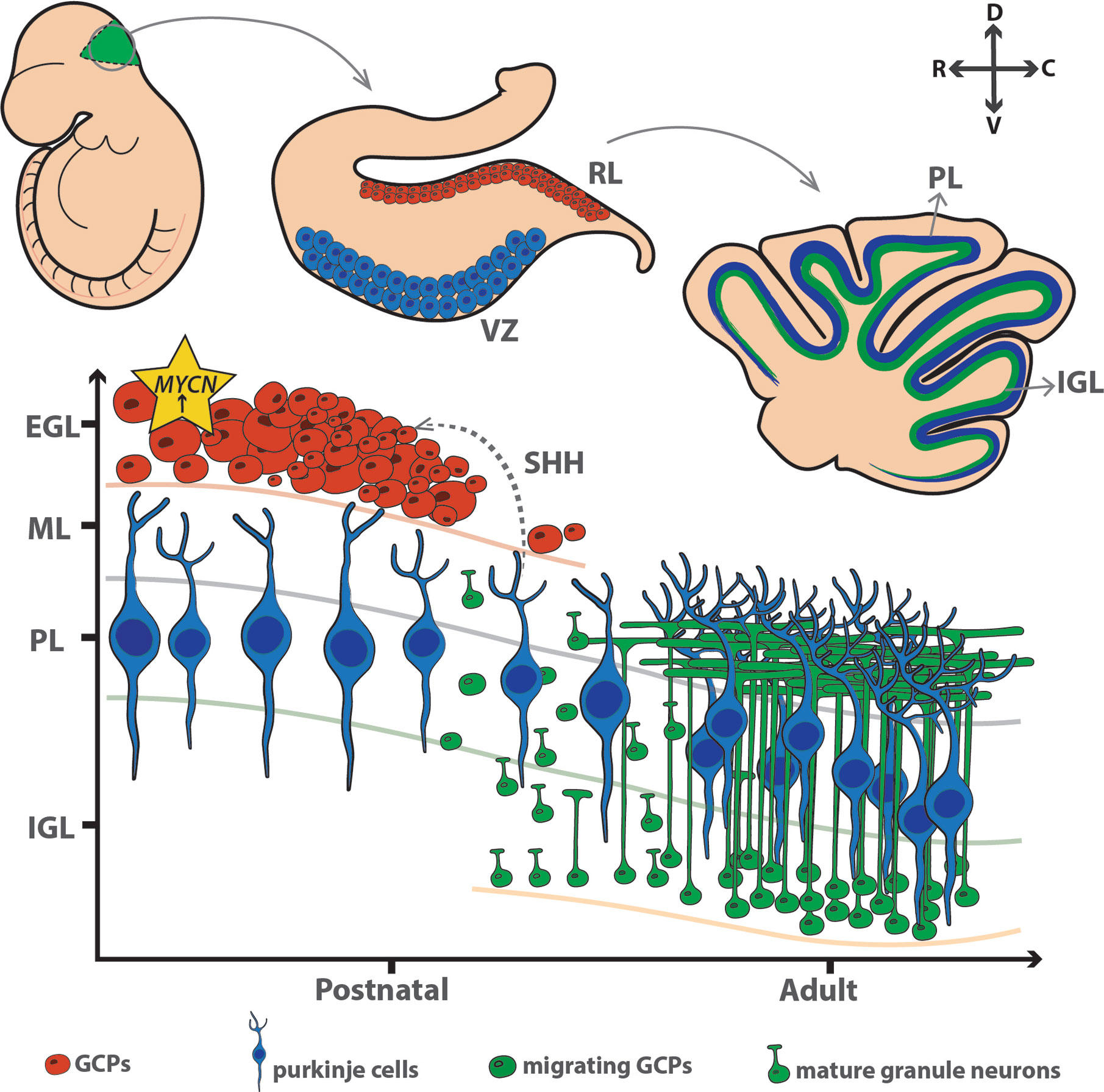
Figure 1 MYCN maintains the proliferation of granule cell progenitors in the external granule layer during early development. VZ, ventricular zone; RL, rhombic lip; PL, purkinje layer; IGL, internal granule layer; EGL, external granule layer; ML, molecular layer; GCP, granule cell precursors. D, dorsal; C, caudal; V, ventral; R, rostral.
MYCN plays a fundamental role in orchestrating both normal and abnormal development of the cerebellum, with critical functions in precursor growth and maturation (Figure 1). MYCN is present at low levels in many neonatal tissues and expressed at particularly high levels in the hindbrain (8, 58). MYCN expression persists during differentiation stages where MYC is downregulated (8). MYCN expression is predominant in neural stem cells and progenitor cell populations, with its expression diminishing after the cells become committed to more differentiated states. One putative mechanism employed by MYCN to sustain CNS development is the conservation of large domains of chromatin in an euchromatic state (59). This is demonstrated through double knockout animal models of MYC and MYCN, which showed gene alterations in chromatin structure (60).
MYCN also contributes to cerebellum development downstream of SHH. SHH is an extracellular signalling molecule with a critical role in regulating growth and differentiation in the developing brain (61, 62). SHH signalling upregulates MYCN through activation of PI3K, a corollary of this is glycogen synthase kinase 3 beta (GSK-3β) inhibition, preventing destabilisation of the MYCN protein by GSK-3β and halting skp-cullin-F-box (SCF)-FBXW7 induced proteasomal degradation (63). Several studies have demonstrated SHH as a primary driver of the expansion of GCPs through direct upregulation of MYCN, highlighting the importance of MYCN in the proliferation of GCP cells and for their responses to SHH (64, 65). As a mitogen, SHH induces genes involved in cell cycle progression and DNA replication, mainly during the period of post-natal hindbrain expansion. In parallel, the role of MYCN is most critical during this phase. Dysfunctional MYCN can prime the progenitor cells by altering internal regulatory mechanisms, making them more susceptible to defects in cerebellar development. Studies have shown MYCN null neural precursors have high levels of specific cyclin dependent kinase inhibitors (CDKI), p18Ink4c and p27KIP1 which induces differentiation programmes in cells, supported by reduced levels of cyclin-D2 (66, 67). More specific to its structure, preventing phosphorylation of the MYCN amino-terminal impedes cell cycle exit of GCPs (68); phosphorylation of the S62 priming site of MYCN by CDK1/cyclin A/B prevents this continuous activation of the cell cycle and allows cell cycle exit (69). MYCN has a vital role in the early cerebellum development. By orchestrating a time-dependent expansion of progenitor cells to form the EGL, it indicates a window of high activity, after which it is downregulates to allow cell cycle arrest and subsequent differentiation and maturation of the cells.
The Role of MYCN in MB Groups
Collectively, this family of oncogene is of particular interest as MYC and MYCN are each committed to specific subtypes of MB. MYC is often found to be overexpressed in WNT tumours, despite a lack of MYC amplification, whereas amplification of MYC is commonly detected in G3 tumours (31). Both MYCN amplification and overexpression is observed in SHH MB. MYCN amplifications are also present in G4 tumours, however, this is generally at a much lower level compared with the SHH subgroup (31). MYC and MYCN amplifications in particular have been the main focus in MB due to the highly aggressive nature of tumours associated with these aberrations (16). Alongside genetic abnormalities, dysregulated epigenetic modifiers are also frequently observed in more aggressive medulloblastoma tumours, including those harbouring MYCN/MYCN abnormalities (70, 71). The preference of MYC to a distinct subtype suggests potential ideas about MB tumorigenesis.
The WNT Group
Moderately high levels of MYCN and MYCL1 are observed in this subtype compared to Grp3 and Grp4 (72). Furthermore, MYC is also highly expressed with comparable levels to those seen in Grp3 (13). This high level MYC expression could be explained by MYC being a downstream target of WNT signalling (73). Whilst MYC expression usually correlates with poor prognosis in other MB subgroups, the WNT subtype displays the most positive prognosis within the subgroups, regardless of MYC levels (74).
The SHH Group
Gene amplification is a very common event in SHH MB. The most prevalent amplifications include MYCN and MYCL1, as well as other important genes such as GLI2, MDM4, PPM1D and YAP1 (15, 75). Patients with SHH subtype MB have a frequent gain of chromosome 2, which harbours MYCN, this may also explain the resulting MYCN amplification event seen regularly in this subgroup. In subtypes where MYCN amplification co-occurs with TP53-mutations, there is a worsening of the overall outcome (32, 76). An event linking the two together is chromothripsis. Tumours with high levels of this complex genome arrangement show a positive correlation in the frequency of MYC/MYCN amplifications (77). The chronological order of TP53 mutation, MYCN amplification and chromothripsis is largely unknown and yet to be explored. Pursuing this further will inevitably shed light on novel DNA repair mechanisms which can be utilised for therapeutic targeting.
Studies have shown that MYCN has dual-capacity to produce either SHH-dependent (63) or SHH-independent MB (78). Formation of either is highly dependent on the temporal expression of MYCN, either during embryonic or postnatal development (58). This further highlights MYCN's dynamic role in CNS development. Novel pathways fuelling MB growth include the evolutionarily conserved signalling pathway known as the Salvador-Warts-Hippo (Hippo) pathway (79). SHH signalling may have cross talk with the Hippo pathway to regulate important downstream efforts such as the Yes-associated protein (YAP), an oncoprotein shown to promote proliferation of CGPs (80). Indeed, genomic profiling of OLIG2-expressing glial progenitors as transit amplifying cells of SHH-MB revealed that these cells activate oncogenic networks including HIPPO-YAP/TAZ and AURORA-A/MYCN (81).
Group 3
The majority of Grp3 tumours are characterised by high protein levels of MYC, either induced by MYC amplifications or by aberrant MYC expression (41). Differential analysis of super-enhancers has identified MYC as a prominent target in Grp3. Thus, MYC is noted as the key driver of Grp3 MB (30). Plasmacytoma Variant Translocation 1 (PVT1) gene fusion in Grp3 is linked to chromothripsis and MYC amplification on chromosome 8q24 (82). New molecular stratification of this subtype into different subgroups, has shown that subgroup II and III harbour amplifications of the MYC oncogene and are associated with poor outcomes (83). Interestingly, subgroup V, characterised by amplification of both MYC and MYCN, is associated with moderate clinical outcomes (83). Moreover, the increased abundance of many proteins involved in mRNA processing, transcription and translation observed in Grp3 MB is associated with high MYC expression (48, 50). Interestingly, whilst this subgroup is mainly associated with MYC amplifications, MYCN amplifications is also seen in a minority of patients (5%) (33).
Group 4
The most frequent somatic copy-number alterations (SCNAs) in this group target the gene SNCAIP (synuclein, alpha interacting protein) (15). While MYCN amplification also occur in this subtype, they are mutually exclusive with SNCAIP duplications (31).
Current Management of MYCN-Associated MB
Since the identification and isolation of MB as a distinct entity in 1926 by Cushing and Bailey, the prognosis of patients has alleviated from no survival to now, the most positive outcome of 80% 5-year overall survival (OS). This improvement was led through continuous progression in understanding the biological mechanisms behind this cancer and strengthened by emerging technologies and treatments. Although this OS sounds very positive, the reality of the age of these patients, coupled with the harsh quality of life (QOL) observed after the treatments (84, 85) pushes this scientific field to develop more novel and targeted therapies which can ameliorate the dismal QOL.
While our understanding of MB biology and molecular features has greatly improved over the last decade, current treatment regimens for MB have been relatively unchanged. These strategies are principally tailored based on clinico-radiological risk criterion, used to define the standard-risk (SR) or high-risk (HR) group (86). Children who are >3 years with no evidence of metastatic disease (M0), post-surgical residual tumour <1.5 cm2, and histologically non-anaplastic are categorized as SR, while the remaining are considered HR. Children >3 years who have significant residual disease following surgery, large cell/anaplastic (LC/A) histology and metastatic disease have a worse prognosis with poor survival outcome (87).
Medulloblastomas are typically more radiosensitive than other paediatric brain tumours, including glioblastomas (GBM). Therefore, radiotherapy is an essential element in the multidisciplinary management of children with MB, and postoperative craniospinal axis radiotherapy is considered a curative treatment. Commonly, children >3 years, receive surgery, external beam radiation to the spine and brain, combined with multidrug chemotherapy (cisplatin, vincristine, and cyclophosphamide). While both SR and HR children are treated with radiation, HR patients are given larger boosts of radiation. Children <3 years are treated postoperatively with high-dose chemotherapy as an irradiation-avoiding strategy or with non-high-dose chemotherapy during induction followed by a reduced dose of conformal radiotherapy (CRT) to the tumour bed (88, 89). Radiation is generally avoided in children <3 years due to the adverse effects on the developing brain. Radiation in older children has been linked to reduced IQ and induction of secondary cancers, vasculopathy, hearing loss, and future strokes (90–92). The standardised treatment of MB solely based on histopathology and clinico-radiological risk stratification can lead to unpredictable relapses and therapeutic failures. Disease relapse is the most adverse prognostic factor in MB, occurring in approximately 30% of patients (93). Children with tumour relapse receive various strategies, including continuous administration of low doses of chemotherapeutic, high-dose chemotherapy, intrathecal medication, and re-irradiation, but these approaches are commonly unsuccessful (94, 95).
It is evident from the review that MYCN is a very attractive therapeutic target. Nevertheless, it has proven challenging to target, with current techniques unsuccessful in exploiting the molecule for therapeutic gain (10). Structurally, MYC proteins lack any enzymatic activity/globular functional domains, which makes it unapproachable for structure-based virtual screening, and undesirable for the long-established enzyme inhibitor design (96). Adding to this, MYCN is also known as an intrinsically disordered (ID) oncoprotein, meaning the protein structure of MYCN in isolation fails to adopt a defined three-dimensional structure (97). This is advantageous for its role as a transcription factor as the ID structure allows MYCN to hold a larger surface area for increased interaction with numerous other proteins – the disordered domains mean it can be "re-used" in multiple pathways (98). However, this makes it challenging to target MYCN directly.
Further to its structure and function, the widespread expression of MYCN by all early proliferating cells also poses a concern. The numerous target genes of MYCN makes it difficult to define critical oncogenic effector pathways for precise drugging. Thus, targeting this oncogene may present with unacceptable toxicities (99). However, as most "normal" CNS cells spend the majority of their life in quiescence, the adverse effects may be more negligible than expected (100). Limited direct targeting of MYCN has motivated strategies to look at indirect or MYCN-dependent interactions instead. Due to the high growth-inducing activity of MYCN, its mechanism is controlled at multiple steps.
Emerging Therapeutic Opportunities
Targeting MYCN Stability
The stability of the MYCN protein itself is a critical level of regulation. MYCN is controlled by phosphorylation of specific residues, most of which takes place within Myc box I (MBI). First, phosphorylation mediated by CDK1-CyclinA/B1 complexes occurs at S62, which permits recognition. Second, phosphorylation activity occurs via serine/threonine kinase GSK3β on T58. The phosphorylating activity of GSK3β causes degradation of MYCN. MYCN protein has an ephemeral half-life (20-30 minutes), and is tightly regulated by E3 ubiquitin ligase (E3 ligase) through recruitment and proteasomal degradation (97, 101). Whilst there is also a role played by calpains for the turnover of MYC, the majority of degradation is carried out by the ubiquitin-proteasome system (UPS) (102, 103). The ligases FBXW7 and TRUSS have essential roles in restricting MYCN functions via the UPS. FBXW7 recognises MYCN upon phosphorylation at both S62 and T58, causing MYCN to be specifically degraded during mitosis, providing a mechanism which induces cell cycle exit and differentiation of neural progenitor cells. Thus, increasing the level of FBXW7 would be especially attractive to MB with MYCN overexpression. More recently, in a study by Skowron et al. looking at the transcriptome of 250 human SHH MB, they discovered missense mutation within the tryptophan-aspartic acid motif (WD40) of FBXW7 in SHH MBs (42). This supports the idea of targeting this important ligase to alter MYCN activity. Another ligase responsible for restricting MYCN function is the HECT (Homologous to the E6-AP Carboxy Terminus)-domain ubiquitin ligase, HUWE1. This degradation system acts by priming the protein through addition of Lys 48-mediated linkages. HUWE1 carries this out for both MYC and MYCN, but shows a greater efficiency for the latter (104). Interfering with this mechanism, at the MYCN protein level, remains a potential strategy of intervention.
The degradation of MYCN can be further inhibited by the activation of PI3K/AKT/mTOR axis, as active AKT can phosphorylate and inactivate GSK-3β, leading to MYCN stabilisation. Targeting PI3K may therefore be valuable to control the level of MYCN (105) (Figure 2). This has been attempted through various candidate inhibitors such as taselisib, copanlisib, pictilisib, buparlisib, dacotilisib and idelasib, only to find the emergence of resistance to be common, and usually associated with upregulation of MYCN. To overcome this, studies have tried to utilise a combination therapy method with compounds such as SF2523 (106). This compound was investigated in a study by Andrews et al. in which it was able to inhibit both the MYC transcriptional co-factor, BRD4 and PI3K with increased efficacy and reduced toxicity to animals (106). More recent compounds targeting this pathway, and are ongoing clinical trials include LY3023414 [NCT03155620] and AZD2014 [NCT02813135]. In addition to the PI3K/AKT/mTOR pathway, WNT and SHH also play a role in inactivating GSK-3β, leading to upregulation and stabilisation of MYCN. Both pathways hold potential targets to regulate this stabilisation.
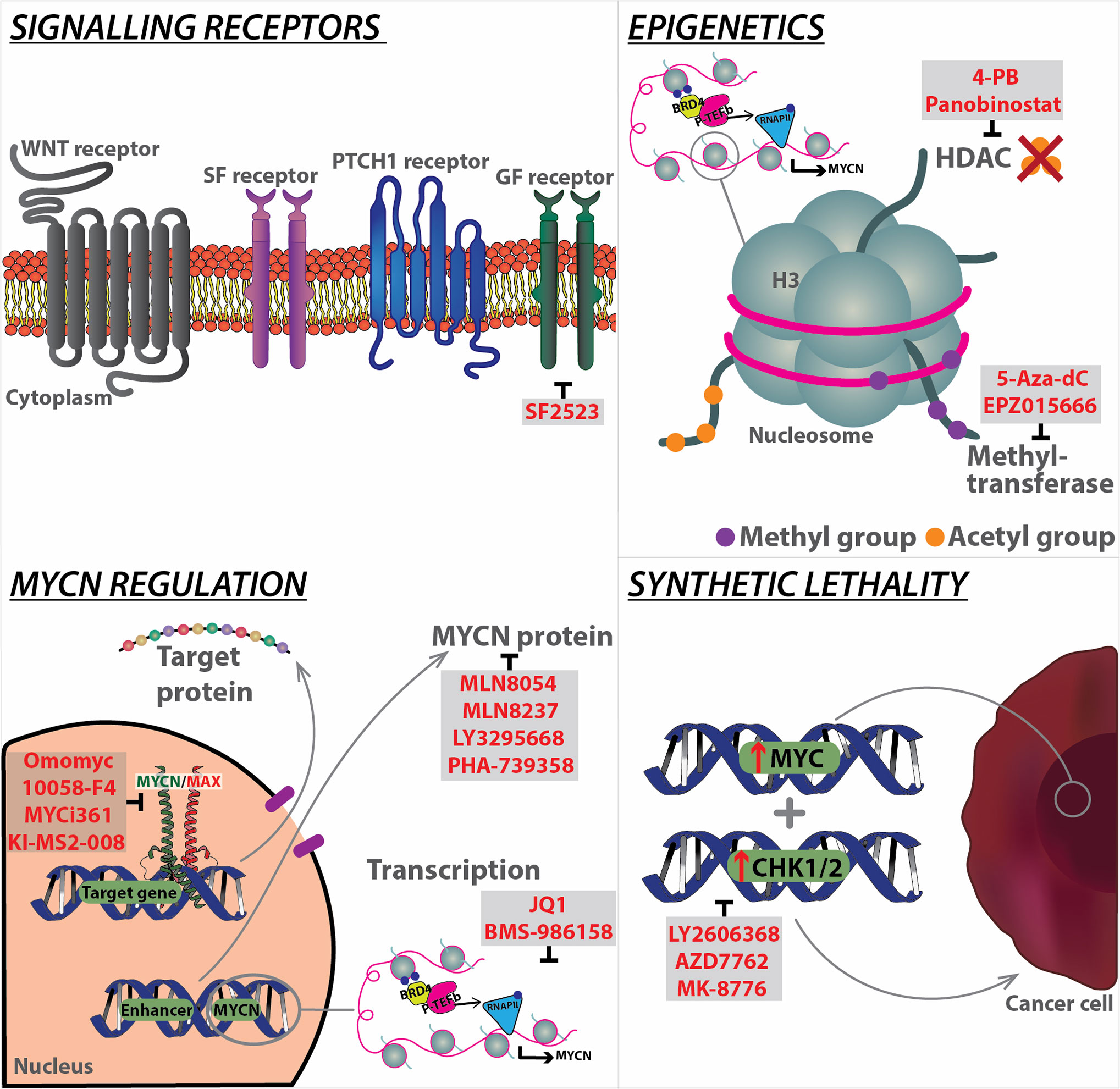
Figure 2 Overview of current strategies targeting MYCN at different levels, from signalling receptors to downstream regulation, as well as epigenetics, and synthetic lethality. Specific modalities (highlighted in red) have been developed to target the mechanistic components of each pathway. BRD4, Bromodomain-containing protein 4; GF, Growth Factor; HDAC, Histone deacetylases; MAX, MYC-associated factor X; P-TEFb, Positive transcription elongation factor b; PTCH1, Patched 1; RNA pol II, RNA polymerase II; SF, Survival factor; WNT, Wingless.
Recently, Aurora kinase A (AURKA), a member of the Aurora family of mitotic regulators, has been shown to form a complex with MYCN, to prevent its degradation by FBXW7. AURKA stabilises MYCN via a direct interaction with a protein binding site flanking MYCN's MBI sequence (107), this stabilisation of MYCN exacerbates its oncogenic functions, and prevents differentiation of neuroblasts in MYCN-driven neuroblastoma (NB) cell lines, leading to aberrant proliferation (108). It is likely that AURKA exerts similarly deleterious effects in MYCN-driven MB, with evidence for this provided by significantly decreased tumour volumes, and a tendency towards increased survival in Ptch1+/-;p53-/- mice treated with the AURKA inhibitor CD535 (109). Other inhibitors which target the AURKA complex with MYCN include MLN8054 and MLN8237. Upon destabilisation of the complex by these small molecule inhibitors, AURKA is no longer able to protect MYCN from proteasomal degradation. Using MLN8237, which blocks the interaction between AURKA and MYCN, our group was able to demonstrate that AURKA inhibition is effective against NB in a MYCN-driven transgenic mouse model (TH-MYCN), in which high-level expression of MYCN is driven in neural crest by a tyrosine hydroxylase (TH) promoter (110). Correlating with this finding, MLN8237 also significantly impaired the growth of MB allografts derived from GTML (Glt1-tTA/TRE-MYCN-Luc) tumour-derived neurosphere cell lines (111, 112). Additionally, MLN8237's in vivo activity was positively confirmed using a panel of human NB xenografts (113). Another AURKA inhibitor, namely PHA-739358, suppress proliferation of human SHH MB models, including allografts of Patched mutant tumour cells and patient-derived xenografts (114) (Figure 2).
These findings have led to the investigation of AURKA inhibitors for the treatment of MYCN-dependent paediatric cancers. However, despite the encouraging results in pre-clinical studies, clinical trials with different Aurora Kinase inhibitors showed a limited efficacy against solid tumours (115). This has been attributed to mechanisms of resistance triggered by strong upregulation of ATP-binding cassette transporters, such as ABCB1, ABCG2 and ABCC2, and the emergence of AURKA mutations, impairing the efficient binding of the inhibitor in the ATP pocket of the enzyme and functional single nucleotide polymorphisms (SNP) (116, 117). In an analysis of a highly specific AURKA inhibitor LY3295668 in 560 cancer cell lines, NB was among the most sensitive tumour type tested, with MYC/MYCN amplification identified as among the strongest predictors of sensitivity to this agent (Figure 2). Phase I trial of alisertib with irinotecan and temozolomide showed promising results prompting to a phase 2 study in children with relapsed/refractory NB. While these clinical studies supported a potential role for AURKA inhibition in the management of patients with advanced NB, patients with MYCN/MYC-driven tumours still showed poor outcomes despite treatment with this regimen (118). Inhibition of another component of AURK family, AURKB, has been found to sensitize MYC overexpressed Grp3 MB cells to cell death both in vitro and in vivo (119).
Targeting MYCN Transcriptional Activity
Another therapeutic opportunity against MYCN-dependent MB is the use of drugs that effect the transcriptional activity of MYCN. For instance, MYC family gene expression depends on the activity of the co-factor bromodomain and extra-terminal (BET) family member BRD4. The bromodomain and extra-terminal (BET) family contain bromodomains (BRD), acetyl-lysine-specific protein interaction modules that play a key role in regulating gene transcription and are evolutionarily conserved and present in diverse nuclear proteins (120).
The BET family member BRD4 is of particular relevance to MYC-driven MB (Figure 2). MYC gene expression is dependent on the activity of the BET family members (121). BRD4 preferentially binds to acetylated lysine residues K9/14 of histone H3, and deacetylated lysine residues K5/K12 of histone H4 (122, 123), following this BRD4 interacts with the positive transcription elongation factor b (P-TEFb) complex, leading to RNA polymerase II transcriptional activity (124). BRD4 is recruited to a wide range of promoter regions, including those for G1 cell cycle regulators (125, 126), and MYC (121, 127, 128), following this BRD4 co-recruits P-TEFb leading to gene transcription, which in the case of MYC is essential for MYC-dependent stimulation of its target genes (129). Taken together this highlights the potential of BET proteins, in particular BRD4, in driving cell cycle and MYC dysregulation, a corollary of which may be aberrant proliferation and tumorigenesis. By using a cell permeable BET inhibitor (BETi) called JQ1, a thieno-triazolo-1,4-diazepine, which displaces BET bromodomains from chromatin by competitively binding to the acetyl lysine recognition pocket, different research teams have demonstrated that tumours with deregulated MYC are susceptible to JQ1 inhibition both in vitro and in vivo (121, 127, 130, 131). In an unbiased screen of a collection of 673 genetically characterized tumour-derived cellular models, NB cell lines were identified as among the most JQ1 sensitive and MYCN amplification as the most predictive marker of sensitivity (132). Additional studies have demonstrated that JQ1 also suppresses MYC/MYCN expression and MYC/MYCN-associated transcriptional activity in MB, resulting in an overall decrease in MB cell viability (132–134). JQ1 treatment has been shown to be effective in MYC- and MYCN-driven MB by targeting cancer dependency genes driven by super-enhancers. More recently, the pan-BETi clinical compound Molibresib (GSK525762) shows positive outcome in Phase I and awaits further clinical trial result (135) (Figure 2).
Cyclin-dependent kinases, especially CDK1 and CDK2, are key players in stabilizing phosphorylation of MYC proteins at S62 upon activation (69, 136, 137). Mechanistically, the response to BET inhibitors in MB is regulated by the suppression of genes involved in neuronal differentiation and progression through the cell cycle. In particular, the upregulation of the cell-cycle regulator CCND2 is a key mediator of sensitivity or resistance to BET inhibitors. Indeed, cells that tolerate BET inhibition do not terminally differentiate, maintain high expression of CCND2, that allows them to cycle through the S-phase. More recently it has been shown that JQ1 combined with Milciclib, an inhibitor of the MYC-stabilising enzyme CDK2, results in synergistic anti-tumoral effects. Mice xenograft of the human MB MB002 cell line showed prolonged survival when treated with JQ1 and Milciclib compared to vehicle and individual JQ1 or Milciclib treatment (138). This provides a strategy by which MYC, an 'undruggable' protein, may be indirectly targeted for therapeutic gain. Several small molecule BET inhibitors, structurally related to JQ1, are in clinical development and have shown preliminary clinical activity in solid tumours and blood cancers (139, 140). A phase I clinical trial with the BET inhibitor, BMS-986158, is currently ongoing in patients with paediatric cancers [NCT03936465].
Cyclin dependent kinases such as CDK7 and CDK9 play a key role in regulating transcriptional activity of MYCN (141–143). We have identified strong enrichment for CDK9 at both the MYCN promoter and the distal super enhancer and shown that pharmacologic blockade of CDK9 using Fadraciclib targeted MYCN-dependent transcriptional landscape (143). Several small molecule inhibitors targeting CDK7 and CDK9 (such as fadraciclib, dinaciclib and BAY1143572) have been shown to inhibit MYCN transcription and selectively kill MYCN amplified or expressing neuroblastoma, medulloblastoma and other cancer cells and are currently in early phase clinical trial.
Targeting MYCN-Associated Epigenetic Molecules
MYC family genes also regulate transcription via epigenetic modifications, suggesting that epigenetic drugs could be used in the clinic to successfully treat MYC/MYCN-amplified tumours. Epigenetic alterations and aberrant expression of genes controlling epigenetic mechanisms have been identified in several cancers, including NB and MB. In this regard, numerous in vitro and in vivo evidence indicate that histone deacetylase inhibitors (HDACi) suppress MYCN expression and are promising candidates for novel treatment strategies of paediatric cancers (144–146). Selective inhibition of HDAC8 by small-molecule inhibitors kills tumour growth in xenograft mouse models of MYCN-amplified NB (147). The combination of the HDAC inhibitor, 4-phenylbutyrate (4-PB) and the demethylation agent, 5-Aza-2'deoxycytidine (5-Aza-dC) reduces DNA methyltransferase activity, global methylation and induces apoptosis in MB cell lines (148). Ecker and colleagues found HDAC2 to be overexpressed in MB subgroups with poor prognosis (SHH, Grp3 and Grp4) harbouring a MYC amplification compared to normal brain and the WNT subgroup. Indeed, increased sensitivity to HDACi is specifically observed in MYC amplified cells (149). HDACi further enhances the anticancer efficacy of other therapeutic regimens, such as ionizing radiation (IR) and can synergize with PI3K or MAPK/ERK inhibitors to impair tumour growth in vivo (150–152). Mechanistically, HDACi have been associated with different biological activities in MB, including the dissipation of mitochondrial membrane potential, changes in cell stemness, increased expression of the FOXO1 tumour suppressor gene, enhancing mitochondrial apoptosis in a p53-dependent manner and inhibition of the Hedgehog signalling (150, 152–154). Taken together, these data provide strong support for clinical testing of HDACi in the treatment of paediatric brain cancer patients, particularly those with MB. Further studies supporting this include a phase I trial and pharmacokinetic study of SAHA in children with solid tumours found to be well-tolerated (155), and a phase-I consortium clinical study recommending vorinostat in combination with the proteasome inhibitor bortezomib for future phase 2 studies in children with recurrent or refractory solid tumours (156).
HDACs represent an important epigenetic mechanism by which MYCN exerts its transcriptional effects. Treatment of murine and human PDX medulloblastoma cell lines with the pan-HDAC inhibitor Panobinostat lead to significant decreases in cell viability, with the lowest IC50 (14.4nM) seen in Grp3 MYC-driven PDX cells, and a marginally higher IC50 (25.27nM) in Grp4 MYCN-driven PDX (152). Exploration of the mechanism of HDAC inhibition using mouse MYC-driven medulloblastoma cell lines and Grp3 MYC-driven PDX cells revealed that HDAC inhibition significantly alters gene expression in treated MB cells. Having particularly notable effects on BRD4 target genes, MYC target genes, and stem cell proliferation genes, which were all downregulated following Panobinostat treatment. Further analysis of differential gene expression changes in these cells identified increased FOXO1 expression, and its subsequent interactions, as a key driver of the efficacy of HDAC inhibitors in MYC-driven medulloblastoma cells. This increase in FOXO1 expression may also be synergistically increased via combination therapy of Panobinostat with the PI3K inhibitor buparlisib (BKM-120) (152). Whilst the potential efficacy of HDAC inhibitors such as Panobinostat has been relatively neglected in MYCN-driven Grp4 and SHH MB, data from studies of MYCN-driven NB may provide evidence for HDAC inhibitors having similar effects on MYCN activity to those seen in MYC-driven cell lines. In one study of particular note, Panobinostat and the BRD4 inhibitor JQ1 acted synergistically to increase apoptosis and inhibit growth in human Kelly and SK-N-BE (2) MYCN-driven NB cells, whilst also synergistically reducing MYCN protein levels, but not mRNA levels (157). Together these studies in Grp3 MB and NB suggest that inhibition of HDACs may also be efficacious in MYCN-driven medulloblastoma. Future studies utilising Grp4 MB PDX cells will be required to confirm this hypothesis (Figure 2).
MYC-driven primary medulloblastoma tumours have high expression of the arginine methyltransferase PRMT5 compared to non-MYC medulloblastoma tumours and adjacent normal tissues (158). PRMT5 is the major symmetric arginine methylase of histone tails and this histone modification is associated to both transcription activation and repression (159). PRMT5-mediated arginine methylation modulates a variety of cellular processes including cell growth, metastasis, ribosome biogenesis, cellular differentiation, gene transcription, germ cell specification, alternative splicing, and Golgi apparatus formation. Interestingly, the PRMT5 inhibitor EPZ015666 significantly suppressed cell growth and induced apoptosis in MYC-driven medulloblastoma cells (159) (Figure 2). A variety of PRMT5 enzymatic inhibitors are currently applied in clinical trials of myelodysplastic syndrome, acute myeloid leukaemia, breast cancer and B cell non-Hodgkin lymphoma, prompting for further investigation in medulloblastoma [NCT03614728, NCT03573310, NCT02783300] (160).
Targeting MYC-MAX Complexes
The bHLH-LZ structure of MYCN allows it to dimerise with various proteins, this is particularly relevant for its obligate partner MAX, with this interaction forming a stable four-helix bundle. MYCN/MAX heterodimers are required for non-consensus binding, as well as binding to the E-box sequences (161). Once bound to promoters of target genes, the complex can recruit transcriptional coactivators, elongation factors, and histone modifying enzymes to initiate gene transcription. Antagonists of this heterodimer complex represent strong candidates for MYCN-specific inhibitors. MAX forms homodimers, and heterodimers with other partners such as MNT and the MAD family members MAD1-4; these compete with MYCN for MAX. Complexes with MAD predominantly occur in resting or differentiated cells, whilst MYCN/MAX complexes are common in proliferating cells (162, 163). A potent inhibitor of the complex is Omomyc. This is a dominant-negative Myc peptide which facilitates the binding with the MYC protein through four specifically designed amino acid substitutions, thus disrupting the binding between MYC/MAX. This molecule has shown to promote apoptosis is many cancers retaining high MYC activity (133, 164), in particular with a preference for MB tumours (165). Other notable small-molecule MYCN/MAX inhibitors include 10058-F4, MYCi361, MYCi975 and KI-MS2-008 (17, 166, 167) (Figure 2).
Synthetic Lethal Targets of MYCN
A revolutionary method of indirectly targeting MYCN is to use the approach of synthetic lethal interactions. This term is defined as the extreme form of negative genetic interaction wherein the combination of two genes leads to cell death, whilst the two genes alone have no effect on viability of the cell. Synthetic lethal screens for MYCN amplification/overexpression have been more extensively investigated in NB, and more recently in MB due to identification of specific cell cycle checkpoint kinases (Chk1/2). In particular for Grp3 MB, Endersby et al. showed increased sensitivity of MYC amplified Grp3 MB cells to these check inhibitors (Chki) Prexasertib (LY2606368, (Chk1/2i), AZD7762 (Chk1/2i), and MK-8776 (Chk1i), with LY2606368 showing superior activity over the other compounds (Figure 2). Ongoing basket trial for this compound is further investigating its anti-tumour activity [NCT02873975]. Furthermore, when used in combination with typical cancer drugs, LY2606368/gemcitabine combination showed specific activity for Grp3 MB subgroup alone. Whilst SHH MB showed reduced sensitivity to the LY2606368 compound alone, better outcome was seen when used in combination as LY2606368/cyclophosphamide (168). Current ongoing trial for these combinations include SHH and Grp3/4 patients [NCT04023669].
Future Areas of Research for Innovative Therapies
MYCN-Driven Cancer Metabolism
Being a high-grade tumour, MB must balance energy metabolism with the need to synthesize the macromolecules essential for its rapid proliferation. This contrasts with lower grade tumours that do not require constant accumulation of biomass and can therefore prioritize ATP production. Likewise, the neural progenitors from which it derives, MB cellular metabolism is characterized by increased lipogenesis and aerobic glycolysis. Indeed, both normal and malignant neuronal cells face similar challenges: they need the largest amount of ATP to support electrical activity and intercellular communication, but this requirement must be in balance with the additional metabolic requirements of rapid proliferation (169).
During the early stage of development, the rapid expansion of cerebellar GCPs (CGCP), fuelled by SHH signalling, compete for intermediates for the synthesis of lipids, nucleic acids and proteins with the downstream generation of ATP. SHH induces lipogenesis in CGCPs through a mechanism dependent on E2F1 transcriptional activity, involving the up-regulation of fatty acid synthase (FASN) and acetyl-CoA carboxylase 1 (ACC1). In parallel, it down-regulates fatty acid catabolism enzymes, including acyl-CoA oxidase 1 (ACOX1) and medium chain acyl-CoA dehydrogenase (MCAD) (170–172). SHH signalling also induces aerobic glycolysis in CGCPs and tumour cells to support biosynthesis (173). Hexokinase-2 (Hk2) is a key metabolic regulator induced by SHH, its importance is highlighted upon deletion, which leads to impairment in CGCP development and reduced tumorigenesis in the MB-prone SmoM2 mouse model (174). The nutrient sensor peroxisome proliferator-activated receptor γ (PPARγ) is also involved in SHH-mediated regulation of glycolysis; pharmacological blockade of PPARγ inhibits CGCP proliferation and extends animal survival in the NeuroD2-SmoA1 mouse model of MB by inducing cell death (170).
The activation of MYC family is a key point of convergence of the metabolic features of many different cancer types. Similar to its family members, MYCN is a potent regulator of cellular metabolism, through controlled expression of amino acid transporters and other proteins involved in aerobic glycolysis, oxidative phosphorylation, detoxification of reactive oxygen species (ROS), and fatty acid oxidation (175). While numerous studies have demonstrated a key role of MYCN in NB and GBM metabolism (176–180), its metabolic function in medulloblastoma still remains elusive. It is likely that in these tumours, as in other cancers, MYCN reconfigures metabolism to favour aerobic glycolysis and a dependency on the serine-glycine-one-carbon (SGOC) to generate metabolic products starting from serine and glycine amino acids (181).
Selective targeting of tumour glucose metabolism has long been considered as an attractive therapeutic strategy. MYC invariably promotes expression of critical enzymes involved in aerobic glycolysis, such as HK2 and LDHA, making cancer cells more vulnerable to glycolysis inhibition. 2-Deoxyglucose, an analogue of glucose that binds and inhibits HK2, has yielded promising antitumour activity in vitro and in vivo. Aerobic glycolysis produces excessive lactate that is toxic to tumour cells. MYC modulates lactate export by inducing MCT1/MCT2 expression to shift toxic levels of lactate within tumour cells. Therefore, a potential, effective strategy is to block MYC-driven lactate export by MCT1/MCT2 inhibitors. Of note, clinical trials of the MCT1 inhibitor AZD3965 in diffuse large B cell lymphoma and Burkitt's lymphoma, two typical MYC-driven cancer types, are currently ongoing [NCT01791595] (Figure 3).
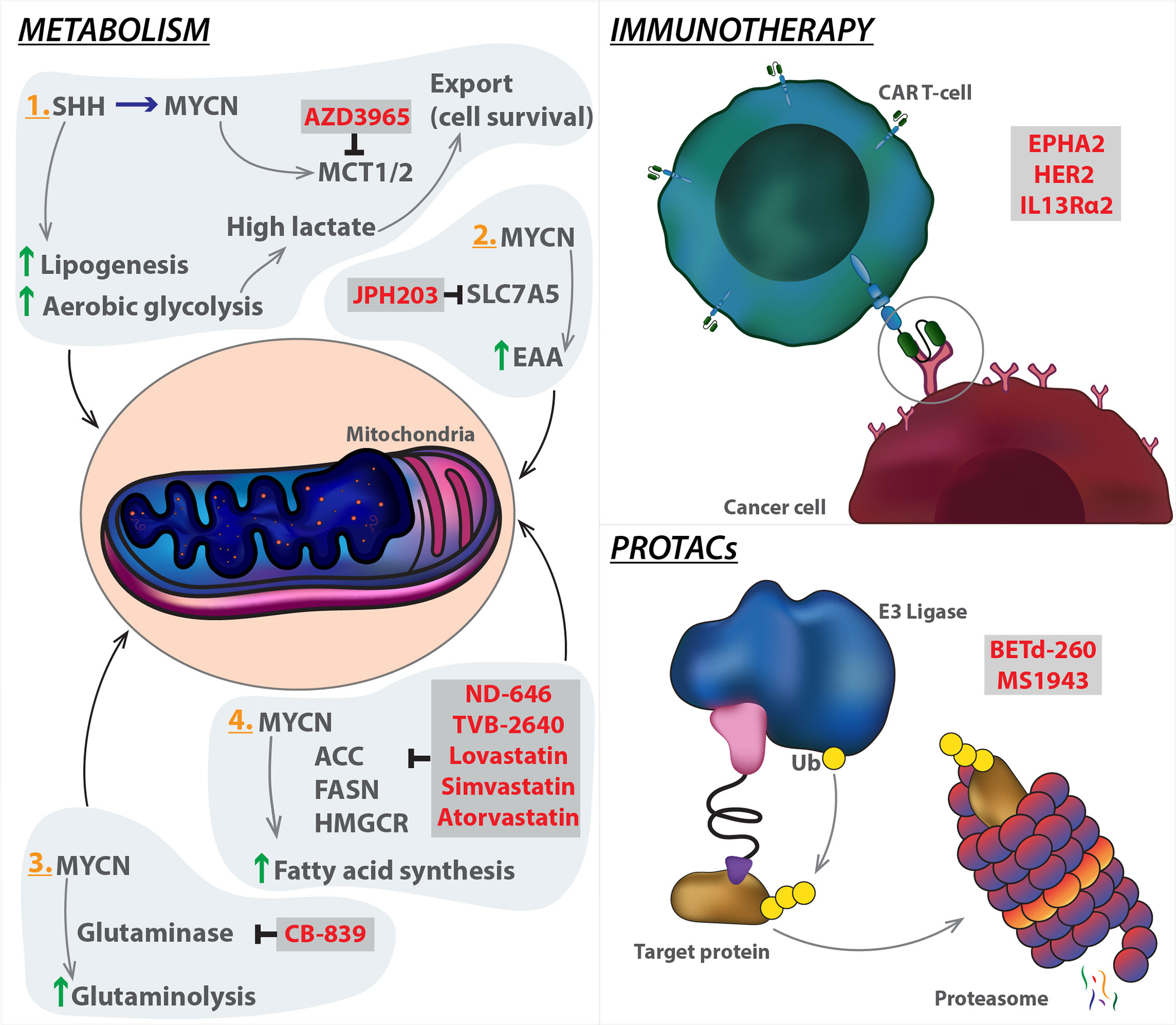
Figure 3 Emerging strategies targeting MYCN. Innate systems regulating metabolism and immune response can be manipulated in cancer models to hijack the tumorigenic mechanism. PROTAC technology can be used to target MYCN (the target protein) by enhancing protein degradation through coupling with E3 ligase, which ubiquitinates the protein leading to degradation via the proteasome. Specific modalities (highlighted in red) have been developed to target the mechanistic components of each pathway. SHH, Sonic hedgehog; MCT1/2, Monocarboxylate transporter 1; SLC7A5, Solute carrier family 7 member 5; EAA, Essential amino acid; CAR T-cell, Chimeric antigen receptor T cells; ACC, Acetyl-CoA carboxylase; FASN, Fatty acid synthase; HMGCR, 3-hydroxy-3-methylglutaryl-CoA reductase; PROTACs, proteolysis targeting chimeras.
Inhibitors of glutaminase or transaminase have shown the therapeutic efficacy in multiple MYC-driven tumour models, and a representative glutaminase inhibitor, CB-839, is currently under clinical trials for patient treatment. MYC and SLC7A5 constitute a feedback loop to amplify MYC transcriptional program, and sustain essential amino acid (EAA) metabolism in tumour cells (182). In principle, therapeutic targeting of SLC7A5 would offer an opportunity to unleash the functional association between MYC and SLC7A5, leading to tumour suppression. JPH203 (also known as KYT-0353), a specific SLC7A5 inhibitor (183) can be evaluated as a MYC-selective cancer therapeutics in the future clinical trials (Figure 3). MYC is a key player in regulation of lipid metabolic reprogramming. ACC, FASN, and 3-Hydroxy-3-Methylglutaryl-CoA reductase (HMGCR), three key enzymes for lipid metabolism, are significantly activated by MYC. ND-646, an allosteric inhibitor of ACC that prevents ACC dimerization and subsequently suppresses fatty acid synthesis, has shown efficacy in mouse models of lung cancer (184). TVB-2640 is a highly potent, selective, and reversible first-in-class inhibitor of FASN. Its monotherapy and in combination with paclitaxel have entered the clinical trial stage [NCT03179904]. Lovastatin, simvastatin, and atorvastatin are specific HMGCR inhibitors that have been FDA approved to lower cholesterol (185). Targeting these enzymes may be a therapeutic alternative for MYC-driven cancers (Figure 3). However, caution should also be taken because it remains unclear as to which aspects of cell metabolism could represent a realistic, targetable vulnerability of tumour cells in comparison with normal counterparts. It should be noted that cancer cells acquire metabolic adaptations in response to a variety of cell-extrinsic and cell-intrinsic cues, thus, MYC effects on cellular metabolism depend both on the tissue of tumour origin and on interaction with tumour microenvironment. A better understanding of these metabolic diversities will improve our ability to define their contribution to aggressive tumour progression.
Immunotherapy in Medulloblastoma
The last decade has seen a tremendous progress in our understanding of how cancer cell evade the immune system and how to harness these mechanisms to develop new therapies. Cancer immunotherapy has proven successful in the so-called "hot" tumours, such as lung cancer and melanoma, characterized by high infiltrating immune cells, while "cold" tumour with low infiltrates still represents a therapeutic challenge for immunotherapy.
The MB microenvironment inhabits reduced numbers of infiltrating immune cells and have been generally considered as immunologically "cold". This is largely backed by a limited amount of information existing on the immune microenvironment. Whilst this is the current understanding, the paracrine signalling between the tumour microenvironment suggests the existence of a more intricate interaction (186). More recently, an increasing number of studies have shed light on the immune profiling of MB, in an attempt to use these data as diagnostic and prognostic tools. Grabovska and colleagues have mapped the tumour immune microenvironment of >6000 primarily paediatric tumour of the CNS, by using methylCIBERSORT, an algorithm derived from CIBERSORT and based on genome-wide DNA methylation data (187, 188). By combining methyCIBERSORT data with pre-existing clinico-pathological and parallel multi-omics, the study exhibited varying proportions of infiltrates within the four classic subgroups of MB. CD8+ T cells (27% of all non-cancer cells), B cells (16%), and eosinophils (15%) (187) were the most abundantly estimated non-cancer cell infiltrates within all MB tumours. The distribution of cell types within the subgroups showed Grp3 MB holding the highest proportion of CD8+ T cells, Grp4 MB homing the natural killer (NK) cells, and SHH MB, the B cells. MYC amplification in Grp3 MB is associated with a significantly higher frequency of tumour infiltrating lymphocytes, CD8+ T cells, and B cells and a lower infiltration of regulatory T cells (Treg). Interestingly, this immune infiltrate analysis further supports the recent refinement of the Grp3/Grp4 MB subgroups into eight subtypes I–VIII (83).
In all cases, the methyCIBERSORT estimates of TILs aligns with the "Cytolytic score", derived from the expression of granzyme A (GZMA) and perforin 1 (PRF1), secreted by effector cytotoxic T cells and NK cells. In another gene expression study, a smaller cohort of SHH MB tumours show high content of fibroblasts, T cells and macrophages, whilst Grp4 MB expresses markers of cytotoxic lymphocytes. SHH MB subgroup has increased expression of inflammation-related genes (CD14, PTX3, CD4, CD163, CSF1R, and TGFB2) and significantly higher infiltration of tumour-associated fibroblasts than Grp3 MB and Grp4 MB (189). In another cohort, cytotoxic T-cells, with variable activation status, showed no correlation with overall survival of the patients (190). While these studies prove that immune profiles are specific to the different molecular subgroups of MB, their applicability in the clinical settings is still unclear. Moreover, data on the immune checkpoint proteins, PD-1 and PD-L1 are limited and controversial, due both to technical challenges to detect these markers or discrepancy between in vitro and in vivo results (191–193).
Another reason why MB are considered "cold" is the relatively low expression of cancer-specific antigens on their cell surface. Orlando and colleagues have recently reported expression of the tumour-associated antigen PRAME in 82% of MB tumour tissues. However, its levels only showed correlation with the worst overall survival groups. Moreover, MB cells targeted using genetically modified T cells carrying a PRAME-specific TCR controlled tumour growth in an orthotopic mouse model of MB (194). Intrathecal delivery of T cells engineered to express EPHA2, HER2 and interleukin 13 receptor α2 (IL13Rα2) chimeric antigen receptors showed efficacy in the treatment for primary, metastatic, and recurrent Grp3-MB xenografts in mouse models. Administration of these chimeric antigen receptor T (CAR-T) cells into the CSF, alone or in combination with the epigenetic modifier azacytidine, was highly effective against different metastatic mouse models of Grp3 MB, thereby providing a rationale for CAR-T approaches in the clinic (195) (Figure 3).
While computational analyses are advancing our general knowledge of MB-tumour microenvironment (TME), the direct link between MYCN expression and TME profiling is still under investigation. What has emerged in neuroblastoma and other malignancies (small cell lung cancer, rhabdomyosarcoma, Wilms' tumour, retinoblastoma, acute myeloid leukaemia, and T-acute lymphoid leukaemia) is that MYCN has a great role in dysregulating the immune network. In neuroblastoma patients for instance, gene set enrichment analysis has shown that MYCN levels negatively correlate with genes involved in different immune system pathways, especially those associated to interferon gamma and phagocytosis (196). Overall, MYCN suppresses the immune landscape, through dysregulation of immune checkpoints, CD4+ helper T (Th) cytokines, major histocompatibility complex (MHC) genes, and Toll-like receptors (TLRs) (197). Apart from tumour cases with MYCN gene amplification, the immune system dysregulation can occur as a consequence of other events leading to increased MYCN activity (mRNA and protein stabilization, mi-RNA alteration). Based on these considerations, it is imperative to have a better understanding of the mechanistic components linking MYCN to MB-TME functions. The blockage of MYCN or specific MYCN dependencies could ameliorate the immune suppression by restoring the responsiveness of the immune system, opening the way to combinatorial treatments with immunotherapies. In this context, the link between MYCN and polycomb repressive complex 2 (PRC2) may offer a promising therapeutic opportunity via a mechanism that alters TME immunogenicity (198, 199).
Use of PROTACs
Proteolysis targeting chimeras (PROTAC) and hydrophobic tagging are successful technologies/strategies for selective degradation of the target protein (200, 201). Although PROTAC technology has been rapidly gaining momentum in the drug discovery field, the hydrophobic tagging approach has received considerably less attention from the biomedical community. This approach utilizes a bulky and hydrophobic group attaching to a small-molecule binder of the target protein. The binding of this bivalent compound to the target protein leads to misfolding of the target protein and its subsequent degradation by the proteasome (202). Targeting oncogenic proteins for degradation using PROTACs recently gained an increased momentum in the field of cancer research. Compared with BET inhibitors HJB-97 and JQ1, the activity of the PROTAC BET degrader BETd-260 increased over 1000 times (203). The degrader complex showed stability through cooperative binding between AURKA and CEREBLON (204). The enhancer of zeste homolog 2 (EZH2) is the main enzymatic subunit of the polycomb repressive complex 2, which catalyses tri-methylation of lysine 27 on histone H3 (H3K27me3) to promote transcriptional silencing. PRC2 complex has important roles in tissue development, primarily to maintain cell identity (205). EZH2 is overexpressed in multiple types of cancer including triple-negative breast cancer (TNBC), and high expression levels correlate with poor prognosis. In MB, studies have shown increased expression of EZH2 in all subgroups, with particularly high levels in G3 and G4 (206, 207). The link between this enzyme and MYC or MYCN remains largely unexplored. Whilst studies have shown correlation between levels of the enzyme and MYC activity (208), the causation behind this is yet to be mapped. Nonetheless, Chen et al. showed in MYCN amplified neuroblastoma, a strong dependency between tumour cells and the PRC2 complex. MYCN was shown to directly activate EZH2 by binding to its promoter, leading to inhibition of neuronal differentiation networks in MYCN-amplified (209). This approach should also be applied to MB to elucidate the underlying mechanism. Several EZH2 inhibitors, which inhibit the methyltransferase activity of EZH2, have shown promising results in treating sarcoma and follicular lymphoma in clinics. However, EZH2 inhibitors are ineffective at blocking proliferation of TNBC cells, even though they effectively reduce the H3K27me3 mark. Using a hydrophobic tagging approach, generation of MS1943, a first-in-class EZH2 selective degrader that effectively reduces EZH2 levels in cells (210) (Figure 3).
Conclusion and Future Outlook
In this review, we have highlighted the relationship between MYCN and the paediatric brain tumour medulloblastoma, with an emphasis on the emerging therapeutic avenues to target this. The ever-increasing advancements in sequencing technologies, coupled with global efforts to improve the disease models through strong collaborations, and the use of more humanised systems, is rapidly dissecting the precise role of MYCN in all MB subgroups. Model systems such as patient-derived iPSCs (51, 211, 212), and human hindbrain-derived neuroepithelial stem cells (213) align with the developmental trajectory of the CNS, therefore are likely to reflect a more authentic evolution of the tumour through targeting of relevant oncogenes such as MYCN. Novel MB targeting strategies using PROTACs, and CAR T-cell therapy offer a selective advantage over the more generic inhibitors. Additional focus on the metabolic dependencies of MB tumours can shed light on the most vulnerable target for tumour growth. Whilst the supporting evidence indicate a practical use for these technologies in MB, these mechanisms remain largely unexplored. Recently, liquid biopsies using CSF to assess circulating tumour DNA have been used to genetically characterise MB (214). This is an important development in the management of MB, one particularly relevant to the potential MYCN-focussed therapeutic approaches discussed here, as liquid biopsies detect the majority of MYCN-effecting mutations such as MYCN and Gli2 amplification, and SUFU loss (214). As CSF may be obtained during hydrocephalus surgery, a common procedure for MB patients, this will open the possibility to personalised medicine approaches for the treatment of this devastating disease. Furthermore, MB is a brain tumour protected from the systematic delivery of cancer drugs by the blood brain barrier (BBB). This defence system is a unique problem which prevents majority of the current therapies from succeeding. Utilising lipid-soluble cargoes such as nanoparticles which disintegrate at the target site (215, 216), or focused ultrasound techniques e.g., pulsed ultrasound (217), can greatly improve the delivery of targeted drugs.
It is certainly evident that MYCN is a phenomenally complex molecule. As illustrated in this review, the multiple downstream signalling pathways directly or indirectly regulated by Myc highlights that targeting this oncogene is a compelling, yet challenging strategy for MB. Our ultimate goal is to increase the proportion of surviving patients, more specifically by reverting the adverse effects of disseminated disease and treatment sequelae. Thus, our expanding knowledge of the mechanisms in this cancer offers the promise to formulate more targeted therapies and translate this to the clinic in the best form.
Author Contributions
SS and CG wrote the review with advice and contribution from AM and LC. SS made the figures with input from AM and CG. All authors contributed to the article and approved the submitted version.
Funding
This review article was financially supported by The Institute of Cancer Research PhD studentship (SS), Higher Education Funding Council of England (LC) and The Institute of Cancer Research funding scheme (CG).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Evon Poon and Colin Kwok for their invaluable advice and support. Many apologies to our colleagues who were unable to be cited due to space constraints. The insightful discussions within the team were instrumental in the planning of this review.
References
1. Dang CV. MYC on the Path to Cancer. Cell Cell Press; (2012) 149p:22–35. doi: 10.1016/j.cell.2012.03.003
2. Westermann F, Muth D, Benner A, Bauer T, Henrich KO, Oberthuer A, et al. Distinct Transcriptional MYCN/C-MYC Activities Are Associated With Spontaneous Regression or Malignant Progression in Neuroblastomas. Genome Biol (2008) 9(10):R150. doi: 10.1186/gb-2008-9-10-r150
3. Beltran H. The N-myc Oncogene: Maximizing its Targets, Regulation, and Therapeutic Potential. Mol Cancer Res (2014) 12(6):815–22. doi: 10.1158/1541-7786.MCR-13-0536
4. Kohl NE, Legouy E, Depinho RA, Nisen PD, Smith RK, Gee CE, et al. Human N-myc is Closely Related in Organization and Nucleotide Sequence to C-Myc. Nature (1986) 319(6048):73–7. doi: 10.1038/319073a0
5. Malynn BA, De Alboran IM, O’Hagan RC, Bronson R, Davidson L, DePinho RA, et al. N-Myc can Functionally Replace C-Myc in Murine Development, Cellular Growth, and Differentiation. Genes Dev [Internet] (2000) 14(11):1390–9. doi: 10.1101/gad.14.11.1390
6. Davis AC, Wims M, Spotts GD, Hann SR, Bradley A. A Null C-Myc Mutation Causes Lethality Before 10.5 Days of Gestation in Homozygotes and Reduced Fertility in Heterozygous Female Mice. Genes Dev [Internet] (1993) 7(4):671–82. doi: 10.1101/gad.7.4.671
7. Charron J, Malynn B, Fisher P, Stewart V, Jeannotte L, Goff S, et al. Embryonic Lethality in Mice Homozygous for a Targeted Disruption of the N-myc Gene. Genes Dev (1992) 6(12A):2248–57. doi: 10.1101/GAD.6.12A.2248
8. Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF. Loss of N-myc Function Results in Embryonic Lethality and Failure of the Epithelial Component of the Embryo to Develop. Genes Dev [Internet] (1992) 6(12):2235–47. doi: 10.1101/gad.6.12a.2235
9. Sawai S, Shimono A, Wakamatsu Y, Palmes C, Hanaoka K, Kondoh H. Defects of Embryonic Organogenesis Resulting From Targeted Disruption of the N-myc Gene in the Mouse. Development (1993) 117(4):1445–55. doi: 10.1242/dev.117.4.1445
10. Meyer N, Penn LZ. Reflecting on 25 Years With MYC [Internet. Nat Rev Cancer (2008) 8:976–90. doi: 10.1038/nrc2231
11. Zimmerman KA, Yancopoulos GD, Collum RG, Smith RK, Kohl NE, Denis KA, et al. Differential Expression of Myc Family Genes During Murine Development. Nature (1986) 319(6056):780–3. doi: 10.1038/319780a0
12. Hatton BA, Knoepfler PS, Kenney AM, Rowitch DH, Moreno De Alborán I, Olson JM, et al. N-Myc is an Essential Downstream Effector of Shh Signaling During Both Normal and Neoplastic Cerebellar Growth. Cancer Res [Internet] (2006) 66(17):8655–61. doi: 10.1158/0008-5472.CAN-06-1621
13. Roussel MF, Robinson GW. Role of MYC in Medulloblastoma. Cold Spring Harb Perspect Med (2013) 3(11):a014308. doi: 10.1101/cshperspect.a014308
14. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med (2013) 3(10):a014415. doi: 10.1101/cshperspect.a014415
15. Northcott PA, Shih DJH, Peacock J, Garzia L, Sorana Morrissy A, Zichner T, et al. Subgroup-Specific Structural Variation Across 1,000 Medulloblastoma Genomes. Nature (2012) 487(7409):49–56. doi: 10.1038/nature11327
16. Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, et al. Outcome Prediction in Pediatric Medulloblastoma Based on DNA Copy-Number Aberrations of Chromosomes 6q and 17q and the MYC and MYCN Loci. J Clin Oncol [Internet] (2009) 27(10):1627–36. doi: 10.1200/JCO.2008.17.9432
17. Han H, Jain AD, Truica MI, Izquierdo-Ferrer J, Anker JF, Lysy B, et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell [Internet] (2019) 36(5):483–97.e15. doi: 10.1016/j.ccell.2019.10.001
18. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol (2016) 131(6):803–20. doi: 10.1007/s00401-016-1545-1
19. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol (2007) 114(2):97–109. doi: 10.1007/s00401-007-0243-4
20. Tsui K, Gajjar A, Li C, Srivastava D, Broniscer A, Wetmore C, et al. Subsequent Neoplasms in Survivors of Childhood Central Nervous System Tumors: Risk After Modern Multimodal Therapy. Neuro Oncol [Internet] (2015) 17(3):448–56. doi: 10.1093/neuonc/nou279
21. Sonabend AM, Ogden AT, Maier LM, Anderson DE, Canoll P, Bruce JN, et al. Medulloblasoma: Challenges for Effective Immunotherapy. J Neuro-Oncol J Neurooncol (2012) 108:1–10. doi: 10.1007/s11060-011-0776-1
22. Polkinghorn WR, Tarbell NJ. Medulloblastoma: Tumorigenesis, Current Clinical Paradigm, and Efforts to Improve Risk Stratification. Nat Clin Pract Oncol (2007) 4:295–304. doi: 10.1038/ncponc0794
23. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular Subgroups of Medulloblastoma: The Current Consensus. Acta Neuropathol (2012) 123(4):465–72. doi: 10.1007/s00401-011-0922-z
24. Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, et al. Genomics Identifies Medulloblastoma Subgroups That are Enriched for Specific Genetic Alterations. J Clin Oncol [Internet] (2006) 24(12):1924–31. doi: 10.1200/JCO.2005.04.4974
25. Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, Van Sluis P, et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes With Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PloS One [Internet] (2008). doi: 10.1371/journal.pone.0003088
26. Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, et al. Integrative Genomic Analysis of Medulloblastoma Identifies a Molecular Subgroup That Drives Poor Clinical Outcome. J Clin Oncol [Internet] (2011) 29(11):1424–30. doi: 10.1200/JCO.2010.28.5148
27. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J Clin Oncol (2011) 29(11):1408–14. doi: 10.1200/JCO.2009.27.4324
28. Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, et al. Medulloblastoma Exome Sequencing Uncovers Subtype-Specific Somatic Mutations. Nature. Nat Publishing Group (2012) 488p:106–10. doi: 10.1038/nature11329
29. Rausch T, Jones DTW, Zapatka M, Stütz AM, Zichner T, Weischenfeldt J, et al. Genome Sequencing of Pediatric Medulloblastoma Links Catastrophic DNA Rearrangements With Tp53 Mutations. Cell (2012) 148(1–2):59–71. doi: 10.1016/j.cell.2011.12.013
30. Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, et al. Active Medulloblastoma Enhancers Reveal Subgroup-Specific Cellular Origins. Nat [Internet] (2016) 530(7588):57–62. doi: 10.1038/nature16546
31. Northcott PA, Jones DTW, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, et al. Medulloblastomics: The End of the Beginning [Internet. Nat Rev Cancer. NIH Public Access (2012) 12:818–34. doi: 10.1038/nrc3410
32. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral Heterogeneity Within Medulloblastoma Subgroups. Cancer Cell [Internet] (2017) 31(6):737–54.e6. doi: 10.1016/j.ccell.2017.05.005
33. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The Whole-Genome Landscape of Medulloblastoma Subtypes. Nature (2017) 547(7663):311–7. doi: 10.1038/nature22973
34. Schwalbe EC, Lindsey JC, Nakjang S, Crosier S, Smith AJ, Hicks D, et al. Novel Molecular Subgroups for Clinical Classification and Outcome Prediction in Childhood Medulloblastoma: A Cohort Study. Lancet Oncol (2017) 18(7):958–71. doi: 10.1016/S1470-2045(17)30243-7
35. Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, et al. β-Catenin Status Predicts a Favorable Outcome in Childhood Medulloblastoma: The United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol (2005) 23(31):7951–7. doi: 10.1200/JCO.2005.01.5479
36. Manoranjan B, Venugopal C, Bakhshinyan D, Adile AA, Richards L, Kameda-Smith MM, et al. Wnt Activation as a Therapeutic Strategy in Medulloblastoma. Nat Commun (2020) 11(1):1–12. doi: 10.1038/s41467-020-17953-4
37. Eberhart CG, Tihan T, Burger PC. Nuclear Localization and Mutation of β-Catenin in Medulloblastomas. J Neuropathol Exp Neurol [Internet] (2000) 59(4):333–7. doi: 10.1093/jnen/59.4.333
38. Polakis P. The Oncogenic Activation of β-Catenin. Curr Opin Genet Dev [Internet] (1999) 9(1):15–21. doi: 10.1016/S0959-437X(99)80003-3
39. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S, et al. Spectrum and Prevalence of Genetic Predisposition in Medulloblastoma: A Retrospective Genetic Study and Prospective Validation in a Clinical Trial Cohort. Lancet Oncol [Internet] (2018) 19(6):785–98. doi: 10.1016/S1470-2045(18)30242-0
40. Northcott PA, Dubuc AM, Pfister S, Taylor MD. Molecular Subgroups of Medulloblastoma. Expert Rev Neurother (2012) 12(7):871–84. doi: 10.1586/ern.12.66
41. Northcott PA, Hielscher T, Dubuc A, Mack S, Shih D, Remke M, et al. Pediatric and Adult Sonic Hedgehog Medulloblastomas are Clinically and Molecularly Distinct. Acta Neuropathol [Internet] (2011) 122(2):231–40. doi: 10.1007/s00401-011-0846-7
42. Skowron P, Farooq H, Cavalli FMG, Morrissy AS, Ly M, Hendrikse LD, et al. The Transcriptional Landscape of Shh Medulloblastoma. Nat Commun (2021) 12(1):1749. doi: 10.1038/s41467-021-21883-0
43. Tikhonov DB, Mellor IR, Usherwood PNR. Modeling Noncompetitive Antagonism of a Nicotinic Acetylcholine Receptor. Biophys J (2004) 87(1):159–70. doi: 10.1529/biophysj.103.037457
44. Waszak SM, Robinson GW, Gudenas BL, Smith KS, Forget A, Kojic M, et al. Germline Elongator Mutations in Sonic Hedgehog Medulloblastoma. Nature (2020) 580(7803):396–401. doi: 10.1038/s41586-020-2164-5
45. Otero G, Fellows J, Yang L, De Bizemont T, Dirac AMG, Gustafsson CM, et al. Elongator, a Multisubunit Component of a Novel RNA Polymerase Ii Holoenzyme for Transcriptional Elongation. Mol Cell (1999) 3(1):109–18. doi: 10.1016/S1097-2765(00)80179-3
46. Hunnicutt BJ, Chaverra M, George L, Lefcort F. Ikap/Elp1 is Required in Vivo for Neurogenesis and Neuronal Survival, But Not for Neural Crest Migration. PloS One (2012) 7(2):e32050. doi: 10.1371/journal.pone.0032050
47. Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJH, Martin DC, et al. Subgroup-Specific Prognostic Implications of TP53 Mutation in Medulloblastoma. J Clin Oncol (2013) 31(23):2927–35. doi: 10.1200/JCO.2012.48.5052
48. Archer TC, Ehrenberger T, Mundt F, Gold MP, Krug K, Mah CK, et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity Within Medulloblastoma Subgroups. Cancer Cell (2018) 34(3):396–410.e8. doi: 10.1016/j.ccell.2018.08.004
49. Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, et al. Enhancer Hijacking Activates GFI1 Family Oncogenes in Medulloblastoma. Nature (2014) 511(7510):428–34. doi: 10.1038/nature13379
50. Forget A, Martignetti L, Puget S, Calzone L, Brabetz S, Picard D, et al. Aberrant ERBB4-SRC Signaling as a Hallmark of Group 4 Medulloblastoma Revealed by Integrative Phosphoproteomic Profiling. Cancer Cell [Internet] (2018) 34(3):379–95.e7. doi: 10.1016/j.ccell.2018.08.002
51. Huang M, Tailor J, Zhen Q, Gillmor AH, Miller ML, Weishaupt H, et al. Engineering Genetic Predisposition in Human Neuroepithelial Stem Cells Recapitulates Medulloblastoma Tumorigenesis. Cell Stem Cell (2019) 25(3):433–46.e7. doi: 10.1016/j.stem.2019.05.013
52. Bihannic L, Ayrault O. Insights Into Cerebellar Development and Medulloblastoma. Bull Cancer (2016) 103(1):30–40. doi: 10.1016/j.bulcan.2015.11.002
53. Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of Medulloblastoma Have Distinct Developmental Origins. Nature (2010) 468(7327):1095–9. doi: 10.1038/nature09587
54. Hovestadt V, Smith KS, Bihannic L, Filbin MG, Shaw MKL, Baumgartner A, et al. Resolving Medulloblastoma Cellular Architecture by Single-Cell Genomics. Nat [Internet] (2019) 572(7767):74–9. doi: 10.1038/s41586-019-1434-6
55. Bailey P, Cushing H. A Classification of the Tumours of the Glioma Group on a Histogenetic Basis, With a Correlated Study of Prognosis. Lippincott [Internet] (1926) 14(55):554–5. doi: 10.1002/bjs.1800145540
56. Wallace VA. Purkinje-Cell-Derived Sonic Hedgehog Regulates Granule Neuron Precursor Cell Proliferation in the Developing Mouse Cerebellum. Curr Biol [Internet] (1999) 9(8):445–8. doi: 10.1016/S0960-9822(99)80195-X
57. Azevedo FAC, Carvalho LRB, Grinberg LT, Farfel JM, Ferretti REL, Leite REP, et al. Equal Numbers of Neuronal and Nonneuronal Cells Make the Human Brain an Isometrically Scaled-Up Primate Brain. J Comp Neurol [Internet] (2009) 513(5):532–41. doi: 10.1002/cne.21974
58. Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, et al. Pleiotropic Role for MYCN in Medulloblastoma. Genes Dev [Internet] (2010) 24(10):1059–72. doi: 10.1101/gad.1907510
59. Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN. Myc Influences Global Chromatin Structure. EMBO J [Internet] (2006) 25(12):2723–34. doi: 10.1038/sj.emboj.7601152
60. Wey A, Knoepfler PS. C-Myc and N-myc Promote Active Stem Cell Metabolism and Cycling as Architects of the Developing Brain. Oncotarget [Internet] (2010) 1(2):120–30. doi: 10.18632/oncotarget.116
61. Dahmane N, Ruiz-i-Altaba A. Sonic Hedgehog Regulates the Growth and Patterning of the Cerebellum. Development (1999) 15126(14):3089–100. doi: 10.1242/dev.126.14.3089
62. Dahmane N, Sánchez P, Gitton Y, Palma V, Sun T, Beyna M, et al. The Sonic Hedgehog-Gli Pathway Regulates Dorsal Brain Growth and Tumorigenesis - Pubmed. Development (2001) p:5201–12. doi: 10.1242/dev.128.24.5201
63. Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, et al. Transcriptional Profiling of the Sonic Hedgehog Response: A Critical Role for N-myc in Proliferation of Neuronal Precursors. Proc Natl Acad Sci U.S.A. [Internet] (2003) 100(12):7331–6. doi: 10.1073/pnas.0832317100
64. Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, et al. Prediction of Central Nervous System Embryonal Tumour Outcome Based on Gene Expression. Nat [Internet] (2002) 415(6870):436–42. doi: 10.1038/415436a
65. Kenney AM, Cole MD, Rowitch DH. Nmyc Upregulation by Sonic Hedgehog Signaling Promotes Proliferation in Developing Cerebellar Granule Neuron Precursors. Development (2003) 130p:15–28. doi: 10.1242/dev.00182
66. Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y. A Role for p27/Kip1 in the Control of Cerebellar Granule Cell Precursor Proliferation. J Neurosci [Internet] (2000) 20(15):5756–63. doi: 10.1523/jneurosci.20-15-05756.2000
67. Gartel AL, Shchors K. Mechanisms of C-Myc-Mediated Transcriptional Repression of Growth Arrest Genes. Exp Cell Res Acad Press Inc (2003) 283:17–21. doi: 10.1016/S0014-4827(02)00020-4
68. Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 Kinase Signaling Converge on Nmyc1 to Promote Cell Cycle Progression in Cerebellar Neuronal Precursors. Dev Company Biologists Ltd (2004) 131:217–28. doi: 10.1242/dev.00891
69. Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The Cdk1 Complex Plays a Prime Role in Regulating N-Myc Phosphorylation and Turnover in Neural Precursors. Dev Cell (2005) Sep 19(3):327–38. doi: 10.1016/j.devcel.2005.07.014
70. Roussel MF, Stripay JL. Epigenetic Drivers in Pediatric Medulloblastoma. Cerebellum (2018) 17(1):28re2–36. doi: 10.1007/s12311-017-0899-9
71. Yi J, Wu J. Epigenetic regulation in medulloblastoma. Mol Cell Neurosci (2018) 87:65re2–76. doi: 10.1016/j.mcn.2017.09.003
72. Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel Mutations Target Distinct Subgroups of Medulloblastoma. Nat [Internet] (2012) 488(7409):43–8. doi: 10.1038/nature11213
73. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, Da Costa LT, et al. Identification of c-MYC as a Target of the APC Pathway. Sci (80- ) [Internet] (1998) 281(5382):1509–12. doi: 10.1126/science.281.5382.1509
74. Park AK, Lee SJ, Phi JH, Wang KC, Kim DG, Cho BK, et al. Prognostic Classification of Pediatric Medulloblastoma Based on Chromosome 17p Loss, Expression of MYCC and MYCN, and Wnt Pathway Activation. Neuro Oncol (2012) 14(2):203–14. doi: 10.1093/neuonc/nor196
75. Roussel MF, Hatten ME. Cerebellum: Development and Medulloblastoma. Curr Topics Dev Biol NIH Public Access (2011) 94:235–82. doi: 10.1016/B978-0-12-380916-2.00008-5
76. Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, et al. Risk Stratification of Childhood Medulloblastoma in the Molecular Era: The Current Consensus. Acta Neuropathol [Internet] (2016) 131(6):821–31. doi: 10.1007/s00401-016-1569-6
77. Ratnaparkhe M, Wong JKL, Wei PC, Hlevnjak M, Kolb T, Simovic M, et al. Defective DNA Damage Repair Leads to Frequent Catastrophic Genomic Events in Murine and Human Tumors. Nat Commun (2018) Dec 19(1):1–13. doi: 10.1038/s41467-018-06925-4
78. Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Northcott PA, et al. Distinct Neural Stem Cell Populations Give Rise to Disparate Brain Tumors in Response to N-MYC. Cancer Cell [Internet] (2012) 21(5):601–13. doi: 10.1016/j.ccr.2012.04.012
79. Fernandez LA, Squatrito M, Northcott P, Awan A, Holland EC, Taylor MD, et al. Oncogenic YAP Promotes Radioresistance and Genomic Instability in Medulloblastoma Through IGF2-mediated Akt Activation. Oncogene [Internet] (2012) 31(15):1923–37. doi: 10.1038/onc.2011.379
80. Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, et al. YAP1 is Amplified and Up-Regulated in Hedgehog-Associated Medulloblastomas and Mediates Sonic Hedgehog-Driven Neural Precursor Proliferation. Genes Dev [Internet] (2009) 23(23):2729–41. doi: 10.1101/gad.1824509
81. Zhang L, He X, Liu X, Zhang F, Huang LF, Potter AS, et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades During Tumorigenesis and Relapse. Cancer Cell (2019) 1636(3):302–18. doi: 10.1016/j.ccell.2019.07.009
82. Onagoruwa OT, Pal G, Ochu C, Ogunwobi OO. Oncogenic Role of PVT1 and Therapeutic Implications [Internet] Front Oncol Front Media SA (2020) 10:17. doi: 10.3389/fonc.2020.00017
83. Sharma T, Schwalbe EC, Williamson D, Sill M, Hovestadt V, Mynarek M, et al. Second-Generation Molecular Subgrouping of Medulloblastoma: An International Meta-Analysis of Group 3 and Group 4 Subtypes. Acta Neuropathol [Internet] (2019) 138(2):309–26. doi: 10.1007/s00401-019-02020-0
84. Medeiros CB, Moxon-Emre I, Scantlebury N, Malkin D, Ramaswamy V, Decker A, et al. Medulloblastoma has a Global Impact on Health Related Quality of Life: Findings From an International Cohort. Cancer Med (2020) 219(2):447–59. doi: 10.1002/cam4.2701
85. Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma [Internet]. Nat Rev Dis Primers Nat Publishing Group (2019) 5p:1–20. doi: 10.1038/s41572-019-0063-6
86. Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, et al. Risk-Adapted Craniospinal Radiotherapy Followed by High-Dose Chemotherapy and Stem-Cell Rescue in Children With Newly Diagnosed Medulloblastoma (St Jude Medulloblastoma-96): Long-Term Results From a Prospective, Multicentre Trial. Lancet Oncol (2006) 7(10):813–20. doi: 10.1016/S1470-2045(06)70867-1
87. Von Bueren AO, Von Hoff K, Pietsch T, Gerber NU, Warmuth-Metz M, Deinlein F, et al. Treatment of Young Children With Localized Medulloblastoma by Chemotherapy Alone: Results of the Prospective, Multicenter Trial Hit 2000 Confirming the Prognostic Impact of Histology. Neuro Oncol (2011) 13(6):669–79. doi: 10.1093/neuonc/nor025
88. Rutkowski S, Gerber NU, Von Hoff K, Gnekow A, Bode U, Graf N, et al. Treatment of Early Childhood Medulloblastoma by Postoperative Chemotherapy and Deferred Radiotherapy. Neuro Oncol (2009) 11(2):201–10. doi: 10.1215/15228517-2008-084
89. Ashley DM, Merchant TE, Strother D, Zhou T, Duffner P, Burger PC, et al. Induction Chemotherapy and Conformal Radiation Therapy for Very Young Children With Nonmetastatic Medulloblastoma: Children’s Oncology Group Study P9934. J Clin Oncol (2012) 30(26):3181–6. doi: 10.1200/JCO.2010.34.4341
90. Nanney AD, El Tecle NE, El Ahmadieh TY, Daou MR, Bit Ivan EN, Marymont MH, et al. Intracranial Aneurysms in Previously Irradiated Fields: Literature Review and Case Report [Internet]. World Neurosurgery Elsevier Inc (2014) 81:511–9. doi: 10.1016/j.wneu.2013.10.044
91. Merchant TE, Kiehna EN, Li C, Shukla H, Sengupta S, Xiong X, et al. Modeling Radiation Dosimetry to Predict Cognitive Outcomes in Pediatric Patients With Cns Embryonal Tumors Including Medulloblastoma. Int J Radiat Oncol Biol Phys (2006) 65(1):210–21. doi: 10.1016/j.ijrobp.2005.10.038
92. Tanyildizi Y, Keweloh S, Neu MA, Russo A, Wingerter A, Weyer-Elberich V, et al. Radiation-Induced Vascular Changes in the Intracranial Irradiation Field in Medulloblastoma Survivors: An Mri Study. Radiother Oncol (2019) 136:50–5. doi: 10.1016/j.radonc.2019.03.017
93. Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, et al. Recurrence Patterns Across Medulloblastoma Subgroups: An Integrated Clinical and Molecular Analysis. Lancet Oncol (2013) 14(12):1200–7. doi: 10.1016/S1470-2045(13)70449-2
94. Sabel M, Fleischhack G, Tippelt S, Gustafsson G, Doz F, Kortmann R, et al. Relapse Patterns and Outcome After Relapse in Standard Risk Medulloblastoma: A Report From the HIT-SIOP-PNET4 Study. J Neurooncol (2016) 129(3):515–24. doi: 10.1007/s11060-016-2202-1
95. Aguilera D, Mazewski C, Fangusaro J, MacDonald TJ, McNall-Knapp RY, Hayes LL, et al. Response to Bevacizumab, Irinotecan, and Temozolomide in Children With Relapsed Medulloblastoma: A Multi-Institutional Experience. Child’s Nerv Syst (2013) 29(4):589–96. doi: 10.1007/s00381-012-2013-4
96. Thaimattam R, Banerjee R, Miglani R, Iqbal J. Protein Kinase Inhibitors: Structural Insights Into Selectivity. Curr Pharm Des [Internet] (2007) 13(27):2751–65. doi: 10.2174/138161207781757042
97. Andresen C, Helander S, Lemak A, Farès C, Csizmok V, Carlsson J, et al. Transient Structure and Dynamics in the Disordered C-Myc Transactivation Domain Affect Bin1 Binding. Nucleic Acids Res (2012) 40(13):6353–66. doi: 10.1093/nar/gks263
98. Madan Babu M, van der Lee R, Sanchez De Groot N, Rg Gsponer J, Gough J, Dunker K. Intrinsically Disordered Proteins: Regulation and Disease This Review Comes From a Themed Issue on Sequences and Topology Edited. Curr Opin Struct Biol [Internet] (2011) 21:1–9. doi: 10.1016/j.sbi.2011.03.011
99. Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, et al. In Vitro Cytotoxicity and In Vivo Efficacy, Pharmacokinetics, and Metabolism of 10074-G5, a Novel Small-Molecule Inhibitor of c-Myc/Max Dimerization. J Pharmacol Exp Ther [Internet] (2010) 335(3):715–27. doi: 10.1124/jpet.110.170555
100. Wang YAZ, Plane JM, Jiang P, Zhou CJ, Deng W. Concise Review: Quiescent and Active States of Endogenous Adult Neural Stem Cells: Identification and Characterization [Internet]. Stem Cells NIH Public Access; (2011) 29:907–12. doi: 10.1002/stem.644
101. Dani C, Blanchard JM, Piechaczyk M, El Sabouty S, Marty L, Jeanteur P. Extreme Instability of Myc mRNA in Normal and Transformed Human Cells. Proc Natl Acad Sci USA [Internet] (1984) 81(22 I):7046–50. doi: 10.1073/pnas.81.22.7046
102. Small GW, Chou TY, Dang CV, Orlowski RZ. Evidence for Involvement of Calpain in C-Myc Proteolysis In Vivo. Arch Biochem Biophys [Internet] (2002) 400(2):151–61. doi: 10.1016/S0003-9861(02)00005-X
103. Thomas LR, Tansey WP. Proteolytic Control of the Oncoprotein Transcription Factor Myc. In: Adv Cancer Res [Internet] Acad Press Inc (2011) 77–106. doi: 10.1016/B978-0-12-386469-7.00004-9
104. Zhao X, Heng JIT, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, et al. The HECT-domain Ubiquitin Ligase Huwe1 Controls Neural Differentiation and Proliferation by Destabilizing the N-Myc Oncoprotein. Nat Cell Biol [Internet] (2008) 10(6):643–53. doi: 10.1038/ncb1727
105. Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, et al. Inhibition of Phosphatidylinositol 3-Kinase Destabilizes Mycn Protein and Blocks Malignant Progression in Neuroblastoma. Cancer Res [Internet] (2006) 66(16):8139–46. doi: 10.1158/0008-5472.CAN-05-2769
106. Andrews FH, Singh AR, Joshi S, Smith CA, Morales GA, Garlich JR, et al. Dual-Activity PI3K-BRD4 Inhibitor for the Orthogonal Inhibition of MYC to Block Tumor Growth and Metastasis. Proc Natl Acad Sci USA [Internet] (2017) 114(7):E1072–80. doi: 10.1073/pnas.1613091114
107. Richards MW, Burgess SG, Poon E, Carstensen A, Eilers M, Chesler L, et al. Structural Basis of N-Myc Binding by Aurora-A and its Destabilization by Kinase Inhibitors. Proc Natl Acad Sci USA [Internet] (2016) 113(48):13726–31. doi: 10.1073/pnas.1610626113
108. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of N-Myc is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell [Internet] (2009) 15(1):67–78. doi: 10.1016/j.ccr.2008.12.005
109. Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, et al. Drugging MYCN Through an Allosteric Transition in Aurora Kinase a. Cancer Cell [Internet] (2014) 26(3):414–27. doi: 10.1016/j.ccr.2014.07.015
110. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell [Internet] (2013) 24(1):75–89. doi: 10.1016/j.ccr.2013.05.005
111. Ahmad Z, Jasnos L, Gil V, Howell L, Hallsworth A, Petrie K, et al. Molecular and in Vivo Characterization of Cancer-Propagating Cells Derived From MYCN-dependent Medulloblastoma. PloS One (2015) 10(3):e0119834. doi: 10.1371/journal.pone.0119834
112. Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, et al. Combined MYC and P53 Defects Emerge at Medulloblastoma Relapse and Define Rapidly Progressive, Therapeutically Targetable Disease. Cancer Cell (2015) 27(1):72–84. doi: 10.1016/j.ccell.2014.11.002
113. Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial Testing of the Aurora Kinase a Inhibitor MLN8237 by the Pediatric Preclinical Testing Program (Pptp). Pediatr Blood Cancer (2010) 55(1):26–34. doi: 10.1002/pbc.22430
114. Markant SL, Esparza LA, Sun J, Barton KL, McCoig LM, Grant GA, et al. Targeting Sonic Hedgehog-Associated Medulloblastoma Through Inhibition of Aurora and Polo-Like Kinases. Cancer Res (2013) 73(20):6310–22. doi: 10.1158/0008-5472.CAN-12-4258
115. Bavetsias V, Linardopoulos S. Aurora Kinase Inhibitors: Current Status and Outlook [Internet]. Front Oncol (2015) 5:278. doi: 10.3389/fonc.2015.00278
116. Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora Kinase Inhibitors: Progress Towards the Clinic [Internet]. Investigational New Drugs Invest New Drugs (2012) 30:2411–32. doi: 10.1007/s10637-012-9798-6
117. Niu H, Shin H, Gao F, Zhang J, Bahamon B, Danaee H, et al. Aurora A Functional Single Nucleotide Polymorphism (Snp) Correlates With Clinical Outcome in Patients With Advanced Solid Tumors Treated With Alisertib, an Investigational Aurora A Kinase Inhibitor. EBioMedicine (2017) 25:50–7. doi: 10.1016/j.ebiom.2017.10.015
118. DuBois SG, Mosse YP, Fox E, Kudgus RA, Reid JM, McGovern R, et al. Phase II Trial of Alisertib in Combination With Irinotecan and Temozolomide for Patients With Relapsed or Refractory Neuroblastoma. Clin Cancer Res (2018) 24(24):6142–9. doi: 10.1158/1078-0432.CCR-18-1381
119. Diaz RJ, Golbourn B, Faria C, Picard D, Shih D, Raynaud D, et al. Mechanism of Action and Therapeutic Efficacy of Aurora Kinase B Inhibition in MYC Overexpressing Medulloblastoma. Oncotarget (2015) 6(5):3359–74. doi: 10.18632/oncotarget.3245
120. Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell [Internet] (2012) 149(1):214–31. doi: 10.1016/j.cell.2012.02.013
121. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. Bet Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell [Internet] (2011) 146(6):904–17. doi: 10.1016/j.cell.2011.08.017
122. Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The Double Bromodomain Protein Brd4 Binds to Acetylated Chromatin During Interphase and Mitosis. Proc Natl Acad Sci USA [Internet] (2003) 100(15):8758–63. doi: 10.1073/pnas.1433065100
123. Nishiyama A, Mochizuki K, Mueller F, Karpova T, McNally JG, Ozato K. Intracellular Delivery of Acetyl-Histone Peptides Inhibits Native Bromodomain-Chromatin Interactions and Impairs Mitotic Progression. FEBS Lett [Internet] (2008) 582(10):1501–7. doi: 10.1016/j.febslet.2008.03.044
124. Sims RJ, Belotserkovskaya R, Reinberg D. Elongation by RNA Polymerase II: The Short and Long of it. Genes Dev Genes Dev (2004) 18p:2437–68. doi: 10.1101/gad.1235904
125. Mochizuki K, Nishiyama A, Moon KJ, Dey A, Ghosh A, Tamura T, et al. The Bromodomain Protein Brd4 Stimulates G1 Gene Transcription and Promotes Progression to S Phase. J Biol Chem [Internet] (2008) 283(14):9040–8. doi: 10.1074/jbc.M707603200
126. Dey A, Nishiyama A, Karpova T, McNally J, Ozato K. Brd4 Marks Select Genes on Mitotic Chromatin and Directs Postmitotic Transcription. Mol Biol Cell [Internet] (2009) 20(23):4899–909. doi: 10.1091/mbc.E09-05-0380
127. Mertz JA, Conery AR, Bryant BM, Sandy P, Balasubramanian S, Mele DA, et al. Targeting MYC Dependence in Cancer by Inhibiting Bet Bromodomains. Proc Natl Acad Sci USA [Internet] (2011) 108(40):16669–74. doi: 10.1073/pnas.1108190108
128. Ba M, Long H, Yan Z, Wang S, Wu Y, Tu Y, et al. Brd4 Promotes Gastric Cancer Progression Through the Transcriptional and Epigenetic Regulation of C-MYC. J Cell Biochem [Internet] (2018) 119(1):973–82. doi: 10.1002/jcb.26264
129. Gargano B, Amente S, Majello B, Lania L. P-TEFb is a Crucial Co-Factor for Myc Transactivation. Cell Cycle [Internet] (2007) 6(16):2031–7. doi: 10.4161/cc.6.16.4554
130. Jake Slavish P, Chi L, Yun MK, Tsurkan L, Martinez NE, Jonchere B, et al. Bromodomain-Selective BET Inhibitors are Potent Antitumor Agents Against MYC-driven Pediatric Cancer. Cancer Res (2020) 80(17):3507–18. doi: 10.1158/0008-5472.CAN-19-3934
131. Henssen A, Thor T, Odersky A, Heukamp L, El-Hindy N, Beckers A, et al. Bet Bromodomain Protein Inhibition is a Therapeutic Option for Medulloblastoma. Oncotarget [Internet] (2013) 4(11):2080–95. doi: 10.18632/oncotarget.1534
132. Puissant A, Frumm SM, Alexe G, Bassil CF, Qi J, Chanthery YH, et al. Targeting MYCN in Neuroblastoma by BET Bromodomain Inhibition. Cancer Discovery (2013) 3(3):309–23. doi: 10.1158/2159-8290.CD-12-0418
133. Alimova I, Pierce A, Danis E, Donson A, Birks DK, Griesinger A, et al. Inhibition of MYC Attenuates Tumor Cell Self-Renewal and Promotes Senescence in SMARCB1-deficient Group 2 Atypical Teratoid Rhabdoid Tumors to Suppress Tumor Growth In Vivo. Int J Cancer [Internet] (2019) 144(8):1983–95. doi: 10.1002/ijc.31873
134. Bandopadhayay P, Bergthold G, Nguyen B, Schubert S, Gholamin S, Tang Y, et al. Bet Bromodomain Inhibition of MYC-amplified Medulloblastoma. Clin Cancer Res [Internet] (2014) 20(4):912–25. doi: 10.1158/1078-0432.CCR-13-2281
135. Piha-Paul SA, Hann CL, French CA, Cousin S, Braña I, Cassier PA, et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr (2020) 4(2):pkz093. doi: 10.1093/jncics/pkz093
136. Hydbring P, Bahram F, Su Y, Tronnersjö S, Högstrand K, Von Der Lehr N, et al. Phosphorylation by Cdk2 is Required for Myc to Repress Ras-Induced Senescence in Cotransformation. Proc Natl Acad Sci USA (2010) 107(1):58–63. doi: 10.1073/pnas.0900121106
137. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent Phosphorylation Pathways Regulate Myc Protein Stability. Genes Dev [Internet] (2000) 14(19):2501–14. doi: 10.1101/gad.836800
138. Bolin S, Borgenvik A, Persson CU, Sundström A, Qi J, Bradner JE, et al. Combined BET Bromodomain and CDK2 Inhibition in MYC-driven Medulloblastoma. Oncogene [Internet] (2018) 37(21):2850–62. doi: 10.1038/s41388-018-0135-1
139. Lewin J, Soria JC, Stathis A, Delord JP, Peters S, Awada A, et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. In: Journal of Clinical Oncology. Am Soc Clin Oncol (2018) p:3007–14. doi: 10.1200/JCO.2018.78.2292
140. Ameratunga M, Braña I, Bono P, Postel-Vinay S, Plummer R, Aspegren J, et al. First-in-Human Phase 1 Open Label Study of the BET Inhibitor ODM-207 in Patients With Selected Solid Tumours. Br J Cancer (2020) 123(12):1730–6. doi: 10.1038/s41416-020-01077-z
141. Buzzetti M, Morlando S, Solomos D, Mehmood A, Cox AWI, Chiesa M, et al. Pre-Therapeutic Efficacy of the CDK Inhibitor Dinaciclib in Medulloblastoma Cells. Sci Rep (2021) 11(1):5374. doi: 10.1038/s41598-021-84082-3
142. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. Cdk7 Inhibition Suppresses Super-Enhancer-Linked Oncogenic Transcription in MYCN-driven Cancer. Cell (2014) 159(5):1126–39. doi: 10.1016/j.cell.2014.10.024
143. Poon E, Liang T, Jamin Y, Walz S, Kwok C, Hakkert A, et al. Orally Bioavailable Cdk9/2 Inhibitor Shows Mechanism-Based Therapeutic Potential in MYCN-driven Neuroblastoma. J Clin Invest (2020) 130(11):5875–92. doi: 10.1172/JCI134132
144. Pak E, Mackenzie EL, Zhao X, Pazyra-Murphy MF, Park PMC, Wu L, et al. A Large-Scale Drug Screen Identifies Selective Inhibitors of Class I Hdacs as a Potential Therapeutic Option for SHH Medulloblastoma. Neuro Oncol (2019) 21(9):1150–63. doi: 10.1093/neuonc/noz089
145. Perla A, Fratini L, Cardoso PS, Nör C, Brunetto AT, Brunetto AL, et al. Histone Deacetylase Inhibitors in Pediatric Brain Cancers: Biological Activities and Therapeutic Potential [Internet]. Vol. 8, Frontiers in Cell and Developmental Biology. Front Media SA (2020). doi: 10.3389/fcell.2020.00546
146. de Andrade P F, Andrade A, de Paula Queiroz R, Trevisan F, Tone L, Valera E. Novel Histone Deacetylase Inhibitors for the Treatment of Pediatric Brain Tumors. Cent Nerv Syst Agents Med Chem (2014) 14(2):90–5. doi: 10.2174/1871524914666141112093541
147. Rettig I, Koeneke E, Trippel F, Mueller WC, Burhenne J, Kopp-Schneider A, et al. Selective Inhibition of HDAC8 Decreases Neuroblastoma Growth In Vitro and In Vivo and Enhances Retinoic Acid-Mediated Differentiation. Cell Death Dis (2015) 6(2):e1657. doi: 10.1038/cddis.2015.24
148. Marino AM, Frijhoff J, Calero R, Baryawno N, Östman A, Johnsen JI. Effects of Epigenetic Modificators in Combination With Small Molecule Inhibitors of Receptor Tyrosine Kinases on Medulloblastoma Growth. Biochem Biophys Res Commun (2014) 450(4):1600–5. doi: 10.1016/j.bbrc.2014.07.042
149. Ecker J, Oehme I, Mazitschek R, Korshunov A, Kool M, Hielscher T, et al. Targeting Class I Histone Deacetylase 2 in MYC Amplified Group 3 Medulloblastoma. Acta Neuropathol Commun (2015) 3:22. doi: 10.1186/s40478-015-0201-7
150. Sonnemann J, Kumar KS, Heesch S, Müller C, Hartwig C, Maass M, et al. Histone Deacetylase Inhibitors Induce Cell Death and Enhance the Susceptibility to Ionizing Radiation, Etoposide, and TRAIL in Medulloblastoma Cells. Int J Oncol (2006) 28(3):755–66. doi: 10.3892/ijo.28.3.755
151. da Cunha Jaeger M, Ghisleni EC, Cardoso PS, Siniglaglia M, Falcon T, Brunetto AT, et al. HDAC and MAPK/ERK Inhibitors Cooperate to Reduce Viability and Stemness in Medulloblastoma. J Mol Neurosci (2020) 70(6):981–92. doi: 10.1007/s12031-020-01505-y
152. Pei Y, Liu KW, Wang J, Garancher A, Tao R, Esparza LA, et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell [Internet] (2016) 29(3):311–23. doi: 10.1016/j.ccell.2016.02.011
153. Häcker S, Karl S, Mader I, Cristofanon S, Schweitzer T, Krauss J, et al. Histone Deacetylase Inhibitors Prime Medulloblastoma Cells for Chemotherapy-Induced Apoptosis by Enhancing P53-Dependent Bax Activation. Oncogene (2011) 30(19):2275–81. doi: 10.1038/onc.2010.599
154. Coni S, Mancuso AB, Di Magno L, Sdruscia G, Manni S, Serrao SM, et al. Corrigendum: Selective Targeting of HDAC1/2 Elicits Anticancer Effects Through Gli1 Acetylation in Preclinical Models of SHH Medulloblastoma [Internet]. Sci Rep NLM (Medline) (2017) 7:46645. doi: 10.1038/srep46645
155. Fouladi M, Park JR, Stewart CF, Gilbertson RJ, Schaiquevich P, Sun J, et al. Pediatric Phase I Trial and Pharmacokinetic Study of Vorinostat: A Children’s Oncology Group Phase I Consortium Report. J Clin Oncol (2010) 28(22):3623–9. doi: 10.1200/JCO.2009.25.9119
156. Muscal JA, Thompson PA, Horton TM, Ingle AM, Ahern CH, McGovern RM, et al. A Phase I Trial of Vorinostat and Bortezomib in Children With Refractory or Recurrent Solid Tumors: A Children’s Oncology Group Phase I Consortium Study (Advl0916). Pediatr Blood Cancer (2013) 60(3):390–5. doi: 10.1002/pbc.24271
157. Shahbazi J, Liu PY, Atmadibrata B, Bradner JE, Marshall GM, Lock RB, et al. The Bromodomain Inhibitor Jq1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin Cancer Res [Internet] (2016) 22(10):2534–44. doi: 10.1158/1078-0432.CCR-15-1666
158. Chaturvedi NK, Mahapatra S, Kesherwani V, Kling MJ, Shukla M, Ray S, et al. Role of Protein Arginine Methyltransferase 5 in Group 3 (Myc-Driven) Medulloblastoma. BMC Cancer (2019) 19(1):1056. doi: 10.1186/s12885-019-6291-z
159. Migliori V, Phalke S, Bezzi M, Guccione E. Arginine/Lysine-Methyl/Methyl Switches: Biochemical Role of Histone Arginine Methylation in Transcriptional Regulation [Internet]. Epigenomics Epigenomics (2010) 2p:119–37. doi: 10.2217/epi.09.39
160. Bonday ZQ, Cortez GS, Grogan MJ, Antonysamy S, Weichert K, Bocchinfuso WP, et al. Lly-283, a Potent and Selective Inhibitor of Arginine Methyltransferase 5, PRMT5, With Antitumor Activity. ACS Med Chem Lett (2018) 9(7):612–7. doi: 10.1021/acsmedchemlett.8b00014
161. Blackwell TK, Huang J, Ma A, Kretzner L, Alt FW, Eisenman RN, et al. Binding of Myc Proteins to Canonical and Noncanonical Dna Sequences. Mol Cell Biol [Internet] (1993) 13(9):5216–24. doi: 10.1128/mcb.13.9.5216
162. Ayer DE, Eisenman RN. A Switch From Myc:Max to Mad:Max Heterocomplexes Accompanies Monocyte/Macrophage Differentiation. Genes Dev [Internet] (1993) 7(11):2110–9. doi: 10.1101/gad.7.11.2110
163. Adhikary S, Eilers M. Transcriptional Regulation and Transformation by Myc Proteins. Nat Rev Mol Cell Biol Nat Publishing Group (2005) 6p:635–45. doi: 10.1038/nrm1703
164. Beaulieu ME, Jauset T, Massó-Vallés D, Martínez-Martín S, Rahl P, Maltais L, et al. Intrinsic Cell-Penetrating Activity Propels Omomyc From Proof of Concept to Viable Anti-Myc Therapy. Sci Transl Med [Internet] (2019) 11(484):5012. doi: 10.1126/scitranslmed.aar5012
165. Jung LA, Gebhardt A, Koelmel W, Ade CP, Walz S, Kuper J, et al. Omomyc Blunts Promoter Invasion by Oncogenic MYC to Inhibit Gene Expression Characteristic of MYC-dependent Tumors. Oncogene [Internet] (2017) 36(14):1911–24. doi: 10.1038/onc.2016.354
166. Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL, et al. Stabilization of the Max Homodimer With a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem Biol [Internet] (2019) 26(5):711–723.e14. doi: 10.1016/j.chembiol.2019.02.009
167. Müller I, Larsson K, Frenzel A, Oliynyk G, Zirath H, Prochownik EV, et al. Targeting of the MYCN Protein With Small Molecule c-MYC Inhibitors. PloS One (2014) 9(5):e97285. doi: 10.1371/journal.pone.0097285
168. Endersby R, Whitehouse J, Pribnow A, Kuchibhotla M, Hii H, Carline B, et al. Small-Molecule Screen Reveals Synergy of Cell Cycle Checkpoint Kinase Inhibitors With DNA-damaging Chemotherapies in Medulloblastoma. Sci Transl Med (2021) 13(577):7401. doi: 10.1126/scitranslmed.aba7401
169. Bélanger M, Allaman I, Magistretti PJ. Brain Energy Metabolism: Focus on Astrocyte-neuron Metabolic Cooperation [Internet]. Cell Metab Cell Metab; (2011) 14:724–38. doi: 10.1016/j.cmet.2011.08.016
170. Bhatia B, Potts CR, Guldal C, Choi SP, Korshunov A, Pfister S, et al. Hedgehog-Mediated Regulation of PPARc Controls Metabolic Patterns in Neural Precursors and Shh-Driven Medulloblastoma. Acta Neuropathol (2012) 123(4):587–600. doi: 10.1007/s00401-012-0968-6
171. Bhatia B, Hsieh M, Kenney AM, Nahlé Z. Mitogenic Sonic Hedgehog Signaling Drives E2F1-dependent Lipogenesis in Progenitor Cells and Medulloblastoma. Oncogene (2011) 30(4):410–22. doi: 10.1038/onc.2010.454
172. Tech K, Deshmukh M, Gershon TR. Adaptations of Energy Metabolism During Cerebellar Neurogenesis are Co-Opted in Medulloblastoma [Internet]. Cancer Letters Elsevier Ireland Ltd (2015) 356:268–72. doi: 10.1016/j.canlet.2014.02.017
173. Di Magno L, Manzi D, D’Amico D, Coni S, Macone A, Infante P, et al. Druggable Glycolytic Requirement for Hedgehog-dependent Neuronal and Medulloblastoma Growth. Cell Cycle (2014) 13(21):3404–13. doi: 10.4161/15384101.2014.952973
174. Gershon TR, Crowther AJ, Tikunov A, Garcia I, Annis R, Yuan H, et al. Hexokinase-2-Mediated Aerobic Glycolysis is Integral to Cerebellar Neurogenesis and Pathogenesis of Medulloblastoma. Cancer Metab (2013) 1(1):2. doi: 10.1186/2049-3002-1-2
175. Yoshida GJ. Beyond the Warburg Effect: N-Myc Contributes to Metabolic Reprogramming in Cancer Cells [Internet]. Front Oncol (2020) 10:791. doi: 10.3389/fonc.2020.00791
176. Oliynyk G, Ruiz-Pérez MV, Sainero-Alcolado L, Dzieran J, Zirath H, Gallart-Ayala H, et al. Mycn-Enhanced Oxidative and Glycolytic Metabolism Reveals Vulnerabilities for Targeting Neuroblastoma. iScience (2019) Nov 2221:188–204. doi: 10.1016/j.isci.2019.10.020
177. Rellinger EJ, Craig BT, Alvarez AL, Dusek HL, Kim KW, Qiao J, et al. Fx11 Inhibits Aerobic Glycolysis and Growth of Neuroblastoma Cells. In: Surg (United States) Mosby Inc (2017) p:747–52. doi: 10.1016/j.surg.2016.09.009
178. Dorneburg C, Fischer M, Barth TFE, Mueller-Klieser W, Hero B, Gecht J, et al. LDHA in Neuroblastoma is Associated With Poor Outcome and its Depletion Decreases Neuroblastoma Growth Independent of Aerobic Glycolysis. Clin Cancer Res (2018) 24(22):5772–83. doi: 10.1158/1078-0432.CCR-17-2578
179. Zhao E, Hou J, Cui H. Serine–Glycine-One-Carbon Metabolism: Vulnerabilities in MYCN-amplified Neuroblastoma. Oncogenesis (2020) 9:14. doi: 10.1038/s41389-020-0200-9
180. Tjaden B, Baum K, Marquardt V, Simon M, Trajkovic-Arsic M, Kouril T, et al. N-Myc-Induced Metabolic Rewiring Creates Novel Therapeutic Vulnerabilities in Neuroblastoma. Sci Rep (2020) 10:7157. doi: 10.1038/s41598-020-64040-1
181. Garcia AR, Arsenian-Henriksson M. Serine-Glycine-One-Carbon Metabolism: The Hidden Achilles Heel of MYCN-amplified Neuroblastoma? Cancer Res (2019) 79(15):3818–9. doi: 10.1158/0008-5472.CAN-19-1816
182. Yue M, Jiang J, Gao P, Liu H, Qing G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep (2017) 21(13):3819–32. doi: 10.1016/j.celrep.2017.12.002
183. Oda K, Hosoda N, Endo H, Saito K, Tsujihara K, Yamamura M, et al. L-Type Amino Acid Transporter 1 Inhibitors Inhibit Tumor Cell Growth. Cancer Sci (2010) 101(1):173–9. doi: 10.1111/j.1349-7006.2009.01386.x
184. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, et al. Inhibition of Acetyl-Coa Carboxylase Suppresses Fatty Acid Synthesis and Tumor Growth of non-Small-Cell Lung Cancer in Preclinical Models. Nat Med (2016) 22(10):1108–19. doi: 10.1038/nm.4181
185. Longo J, van Leeuwen JE, Elbaz M, Branchard E, Penn LZ. Statins as Anticancer Agents in the Era of Precision Medicine. Clin Cancer Res (2020) 26(22):5791–800. doi: 10.1158/1078-0432.ccr-20-1967
186. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A Paracrine Requirement for Hedgehog Signalling in Cancer. Nature (2008) 455(7211):406–10. doi: 10.1038/nature07275
187. Grabovska Y, Mackay A, O’Hare P, Crosier S, Finetti M, Schwalbe EC, et al. Pediatric Pan-Central Nervous System Tumor Analysis of Immune-Cell Infiltration Identifies Correlates of Antitumor Immunity. Nat Commun (2020) 11:4324. doi: 10.1038/s41467-020-18070-y
188. Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, et al. Pan-Cancer Deconvolution of Tumour Composition Using DNA Methylation. Nat Commun (2018) 9:3220. doi: 10.1038/s41467-018-05570-1
189. Margol AS, Robison NJ, Gnanachandran J, Hung LT, Kennedy RJ, Vali M, et al. Tumor-Associated Macrophages in SHH Subgroup of Medulloblastomas. Clin Cancer Res (2015) 21(6):1457–65. doi: 10.1158/1078-0432.CCR-14-1144
190. Vermeulen JF, Van Hecke W, Adriaansen EJM, Jansen MK, Bouma RG, Villacorta Hidalgo J, et al. Prognostic Relevance of Tumor-Infiltrating Lymphocytes and Immune Checkpoints in Pediatric Medulloblastoma. Oncoimmunology (2017) 7(3):e1398877. doi: 10.1080/2162402X.2017.1398877
191. Diao S, Gu C, Zhang H, Yu C. Immune Cell Infiltration and Cytokine Secretion Analysis Reveal a non-Inflammatory Microenvironment of Medulloblastoma. Oncol Lett (2020) 20(6):397. doi: 10.3892/ol.2020.12260
192. Martin AM, Nirschl CJ, Polanczyk MJ, Bell WR, Nirschl TR, Harris-Bookman S, et al. Pd-L1 Expression in Medulloblastoma: An Evaluation by Subgroup. Oncotarget (2018) 9(27):19177–91. doi: 10.18632/oncotarget.24951
193. Aoki T, Hino M, Koh K, Kyushiki M, Kishimoto H, Arakawa Y, et al. Low Frequency of Programmed Death Ligand 1 Expression in Pediatric Cancers. Pediatr Blood Cancer (2016) 63(8):1461–4. doi: 10.1002/pbc.26018
194. Orlando D, Miele E, De Angelis B, Guercio M, Boffa I, Sinibaldi M, et al. Adoptive Immunotherapy Using Prame-Specific T Cells in Medulloblastoma. Cancer Res (2018) 78(12):3337–49. doi: 10.1158/0008-5472.CAN-17-3140
195. Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional Delivery of CAR T Cells to the Cerebrospinal Fluid for Treatment of Metastatic Medulloblastoma and Ependymoma. Nat Med (2020) 26(5):720–31. doi: 10.1038/s41591-020-0827-2
196. Raieli S, Di Renzo D, Lampis S, Amadesi C, Montemurro L, Pession A, et al. Mycn Drives a Tumor Immunosuppressive Environment Which Impacts Survival in Neuroblastoma. Front Oncol (2021) 11:625207. doi: 10.3389/fonc.2021.625207
197. Casey SC, Baylot V, Felsher DW. The MYC Oncogene is a Global Regulator of the Immune Response [Internet]. Blood. Am Soc Hematol (2018) 131:2007–15. doi: 10.1182/blood-2017-11-742577
198. Christofides A, Karantanos T, Bardhan K, Boussiotis VA. Epigenetic Regulation of Cancer Biology and Anti-Tumor Immunity by EZH2 [Internet]. Oncotarget Impact Journals LLC; (2016) 7:85624–40. doi: 10.18632/oncotarget.12928
199. Dai X, Bu X, Gao Y, Guo J, Hu J, Jiang C, et al. Energy Status Dictates PD-L1 Protein Abundance and Anti-Tumor Immunity to Enable Checkpoint Blockade. Mol Cell (2021). doi: 10.1016/j.molcel.2021.03.037
200. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric Molecules That Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation [Internet]. Natl Acad Sci (2001). doi: 10.1073/pnas.141230798
201. Ding Y, Fei Y, Lu B. Emerging New Concepts of Degrader Technologies [Internet. Trends Pharmacol Sci Elsevier Ltd (2020) 41p:464–74. doi: 10.1016/j.tips.2020.04.005
202. Ravid T, Hochstrasser M. Diversity of Degradation Signals in the Ubiquitin-Proteasome System [Internet]. Nat Rev Mol Cell Biol Nat Publishing Group (2008) 9:679–89. doi: 10.1038/nrm2468
203. Shi C, Zhang H, Wang P, Wang K, Xu D, Wang H, et al. Protac Induced-BET Protein Degradation Exhibits Potent Anti-Osteosarcoma Activity by Triggering Apoptosis. Cell Death Dis (2019) 10:815. doi: 10.1038/s41419-019-2022-2
204. Adhikari B, Bozilovic J, Diebold M, Schwarz JD, Hofstetter J, Schröder M, et al. Protac-Mediated Degradation Reveals a non-Catalytic Function of AURORA-A Kinase. Nat Chem Biol (2020) 16(11):1179–88. doi: 10.1038/s41589-020-00652-y
205. Margueron R, Reinberg D. The Polycomb Complex PRC2 and its Mark in Life [Internet]. Nature (2011) 469:343–9. doi: 10.1038/nature09784
206. Zhang H, Zhu D, Zhang Z, Kaluz S, Yu B, Devi NS, et al. Ezh2 Targeting Reduces Medulloblastoma Growth Through Epigenetic Reactivation of the BAI1/p53 Tumor Suppressor Pathway. Oncogene (2020) 39(5):1041–8. doi: 10.1038/s41388-019-1036-7
207. Vo BHT, Li C, Morgan MA, Theurillat I, Finkelstein D, Wright S, et al. Inactivation of Ezh2 Upregulates Gfi1 and Drives Aggressive Myc-Driven Group 3 Medulloblastoma. Cell Rep [Internet] (2017) 18(12):2907–17. doi: 10.1016/j.celrep.2017.02.073
208. Alimova I, Venkataraman S, Harris P, Marquez VE, Northcott PA, Dubuc A, et al. Targeting the Enhancer of Zeste Homologue 2 in Medulloblastoma. Int J Cancer (2012) 131(8):1800–9. doi: 10.1002/ijc.27455
209. Chen L, Alexe G, Dharia NV, Ross L, Iniguez AB, Conway AS, et al. Crispr-Cas9 Screen Reveals a MYCN-amplified Neuroblastoma Dependency on EZH2. J Clin Invest (2018) 128(1):446–62. doi: 10.1172/JCI90793
210. Ma A, Stratikopoulos E, Park KS, Wei J, Martin TC, Yang X, et al. Discovery of a First-in-Class Ezh2 Selective Degrader. Nat Chem Biol (2020) 16(2):214–22. doi: 10.1038/s41589-019-0421-4
211. Čančer M, Hutter S, Holmberg KO, Rosén G, Sundström A, Tailor J, et al. Humanized Stem Cell Models of Pediatric Medulloblastoma Reveal an Oct4/mTOR Axis That Promotes Malignancy. Cell Stem Cell (2019) 25(6):855–70.e11. doi: 10.1016/j.stem.2019.10.005
212. Susanto E, Navarro AM, Zhou L, Sundström A, van Bree N, Stantic M, et al. Modeling SHH-driven Medulloblastoma With Patient Ips Cell-Derived Neural Stem Cells. Proc Natl Acad Sci USA (2020) 117(33):20127–38. doi: 10.1073/PNAS.1920521117
213. Tailor J, Kittappa R, Leto K, Gates M, Borel M, Paulsen O, et al. Stem Cells Expanded From the Human Embryonic Hindbrain Stably Retain Regional Specification and High Neurogenic Potency. J Neurosci (2013) 33(30):12407–22. doi: 10.1523/JNEUROSCI.0130-13.2013
214. Escudero L, Llort A, Arias A, Diaz-Navarro A, Martínez-Ricarte F, Rubio-Perez C, et al. Circulating Tumour Dna From the Cerebrospinal Fluid Allows the Characterisation and Monitoring of Medulloblastoma. Nat Commun (2020) 11(1):1–11. doi: 10.1038/s41467-020-19175-0
215. Wyatt EA, Davis ME. Nanoparticles Containing a Combination of a Drug and an Antibody for the Treatment of Breast Cancer Brain Metastases. Mol Pharm (2020) 17(2):717–21. doi: 10.1021/acs.molpharmaceut.9b01167
216. Kim J, Dey A, Malhotra A, Liu J, Ahn SI, Sei YJ, et al. Engineered Biomimetic Nanoparticle for Dual Targeting of the Cancer Stem-Like Cell Population in Sonic Hedgehog Medulloblastoma. Proc Natl Acad Sci USA (2020) 117(39):24205–12. doi: 10.1073/pnas.1911229117
Keywords: MYCN, medulloblastoma, targeted therapy, metabolism, immunotherapy, PROTACs
Citation: Shrestha S, Morcavallo A, Gorrini C and Chesler L (2021) Biological Role of MYCN in Medulloblastoma: Novel Therapeutic Opportunities and Challenges Ahead. Front. Oncol. 11:694320. doi: 10.3389/fonc.2021.694320
Received: 12 April 2021; Accepted: 19 May 2021;
Published: 14 June 2021.
Edited by:
Christer Einvik, Arctic University of Norway, NorwayReviewed by:
Mariana Maschietto, Centro Infantil Boldrini, BrazilKwok-Ming Yao, The University of Hong Kong, Hong Kong, SAR China
Copyright © 2021 Shrestha, Morcavallo, Gorrini and Chesler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Gorrini, Y2hpYXJhLmdvcnJpbmlAaWNyLmFjLnVr; Louis Chesler, bG91aXMuY2hlc2xlckBpY3IuYWMudWs=