 Aboli Bhingarkar
Aboli Bhingarkar Hima V. Vangapandu
Hima V. Vangapandu Sanjay Rathod
Sanjay Rathod Keito Hoshitsuki
Keito Hoshitsuki Christian A. Fernandez
Christian A. Fernandez- 1Center for Pharmacogenetics and Department of Pharmaceutical Sciences, University of Pittsburgh School of Pharmacy, Pittsburgh, PA, United States
- 2Division of General Internal Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Amino acid (AA) metabolism plays an important role in many cellular processes including energy production, immune function, and purine and pyrimidine synthesis. Cancer cells therefore require increased AA uptake and undergo metabolic reprogramming to satisfy the energy demand associated with their rapid proliferation. Like many other cancers, myeloid leukemias are vulnerable to specific therapeutic strategies targeting metabolic dependencies. Herein, our review provides a comprehensive overview and TCGA data analysis of biosynthetic enzymes required for non-essential AA synthesis and their dysregulation in myeloid leukemias. Furthermore, we discuss the role of the general control nonderepressible 2 (GCN2) and-mammalian target of rapamycin (mTOR) pathways of AA sensing on metabolic vulnerability and drug resistance.
Introduction
Myeloid leukemias are a group of disorders characterized by the presence of increased numbers of immature myeloid cells in the marrow and peripheral blood. These diseases are most common in adults with survival rates and first-line therapy options varying by age. In this review, we focus on the differences of non-essential amino acid (NEAA) metabolism in myeloid leukemias with an emphasis on the enzymes required for amino acid biosynthesis. While normal cells can synthesize NEAAs, cancers, in order to maintain their rapid growth and proliferation, can alter the expression of genes involved in amino acid biosynthesis and therefore lose the ability to synthesize specific NEAAs. This auxotrophy, or the inability of cancer cells to synthesize certain NEAA amino acids required for growth, leads to a dependency on exogenous sources of NEAAs, which can be pharmacologically targeted.
To comprehensively examine potential metabolic targets in myeloid leukemias within this review, we first introduce the disease and currently available pharmacotherapies to demonstrate the need for novel agents and to identify patients most likely to benefit from pharmacological agents targeting NEAA dependencies. We thereafter discuss the biosynthesis of NEAA, explore gene expression data analysis of potentially dysregulated pathways, and consider the supporting evidence for novel vulnerabilities in myeloid leukemias. Lastly, we detail mechanisms of cancer cell adaptation to nutrient stress and considerations for the optimal design of strategies targeting NEAA metabolic vulnerabilities.
Myeloid Leukemias and Need for Novel Treatment Options
Novel Drug Therapies Are Needed to Improve Outcomes in Myeloid Leukemia Patients
Myeloid leukemias are classified as either chronic or acute based on their proliferation rate. Acute and chronic myeloid leukemias make up 14 and 33% of all estimated new 2020 leukemia cases in the United States, respectively (1).
Acute myeloid leukemia (AML) is the most common acute leukemia in adults. As of 2016, the 5-year overall survival (OS) of AML is approximately 24% (2), but the highest rate of AML deaths in the United States is among older patients (~90% for ages ≥ 65)1, underscoring the need for better therapy options. Due to the lower performance status of this demographic and inability to tolerate aggressive chemotherapy, these older patients have more limited curative pharmacotherapy options available.
Younger AML patients with good performance status can receive traditional intensive remission-induction chemotherapy with cytarabine, an anthracycline, and a FLT3 inhibitor depending on FLT3 mutation status. Alternatively, pharmacotherapy options for patients not eligible for intensive induction therapy include targeted approaches such as the hypomethylation agents decitabine or azacitidine and the BCL2 inhibitor venetoclax (3). Current approaches for post-induction therapy for low-risk patients generally include additional therapy using similar induction agents (such as high-dose cytarabine), and higher risk patients proceed to allogenic hematopoietic cell transplantation (HCT).
The main characteristic of chronic myeloid leukemia (CML) is the translocation between chromosomes 22 and 9 (i.e. Philadelphia chromosome) that creates a BCR-ABL fusion oncogene with constitutively activated tyrosine kinase activity (4, 5). The cornerstone of CML treatment is the BCR-ABL tyrosine kinase inhibitor (6). However, there remains a need to develop novel therapies for CML patients with unfavorable prognosis, including those that are negative for the Philadelphia chromosome (7) and the 20–30% of CML patients who fail to achieve treatment milestones or develop TKI resistance. Altogether, current responses to standard AML and CML treatment regimens indicate that while many patients attain responses, there remains a need to develop therapies for those patients with poor prognosis.
Targeting AAs Dependencies in Myeloid Leukemias
Novel agents for the treatment of myeloid leukemias with unfavorable prognosis will most likely rely on certain cancer cell vulnerabilities to specifically inhibit their growth and limit treatment toxicity. Differences between the metabolic status of cancerous and normal cells are one of the primary hallmarks of cancer (8), and changes in AA metabolism, glucose utilization, lipid consumption, and ATP generation are adaptations cancer cells can acquire for meeting their increased energy demands (9–13). These metabolic changes can lead to an “addiction” of particular fuel sources and identification of these vulnerabilities has led to the development of several agents directed toward specific molecular targets (14).
AAs are the building blocks of protein and are necessary for the sustenance of both normal and leukemic cells. There are 20 AAs classified as essential, non-essential, and conditionally essential (15). Essential AAs must be obtained exogenously from diet, whereas the body possesses the metabolic pathways to catalyze the synthesis of NEAAs. Conditionally essential AAs, like arginine and glutamine, are those that can become essential during certain physiological conditions that limit their synthesis (16). AAs can serve as precursors to many biological compounds, including those involved nucleotide synthesis (17), redox balance (18), lipogenesis (19, 20), and molecules that can fuel the TCA cycle (21). Furthermore, AAs can modulate the tumor microenvironment (22), global chromatin structures (23, 24), and epigenetic factors (25–28).
Herein, our discussion concerning myeloid leukemias and amino acid metabolism will focus on identifying potential vulnerabilities in AA biosynthesis that could be exploited for therapeutic purposes. Below, we provide a thorough discussion on NEAA biosynthesis for subsequently identifying dysregulations in potential targets that can lead to a dependency on extracellular NEAA sources for survival. Upon identification of an AA dependency, there are several targets within AA metabolic pathways that can be selectively targeted to exploit the vulnerability, including AA transporters, synthases, and transaminases that regulate biosynthesis (13, 29–31). In addition, enzymatic AA depletion therapy is an effective strategy for preferentially targeting cancer cells dependent on a specific AA for maintaining cell proliferation demands (13).
Biosynthesis of NEAA
Glutamate Plays a Central Role in the Biosynthesis of Several NEAA
The concentration of glutamate in the body is tightly regulated to balance its important role in various biological processes (32). The biosynthesis of several NEAAs is interlinked with each other with glutamate playing a central role in the synthesis of several NEAAs as a precursor of alanine, proline, glutamine, aspartate, and serine (Figure 1). There are several biochemical pathways leading to the synthesis of glutamate, with a majority of this NEAA derived from the hydrolysis of glutamine by the amidohydrolase enzyme, glutaminase (GLS). Glutamate can also be derived from branch-chain amino acids (BCAAs) and proline (Figure 1). BCAA aminotransferase (BCAT) catalyzes the conversion of BCAAs and α-ketoglutarate into glutamate and branched chain α-keto acids, whereas oxidation of proline occurs via two enzymes: proline dehydrogenase (PRODH) and P5C dehydrogenase (P5CDH). In addition, glutamate is metabolized by glutamate dehydrogenase (GLUD1/2) back to α-ketoglutarate (33).
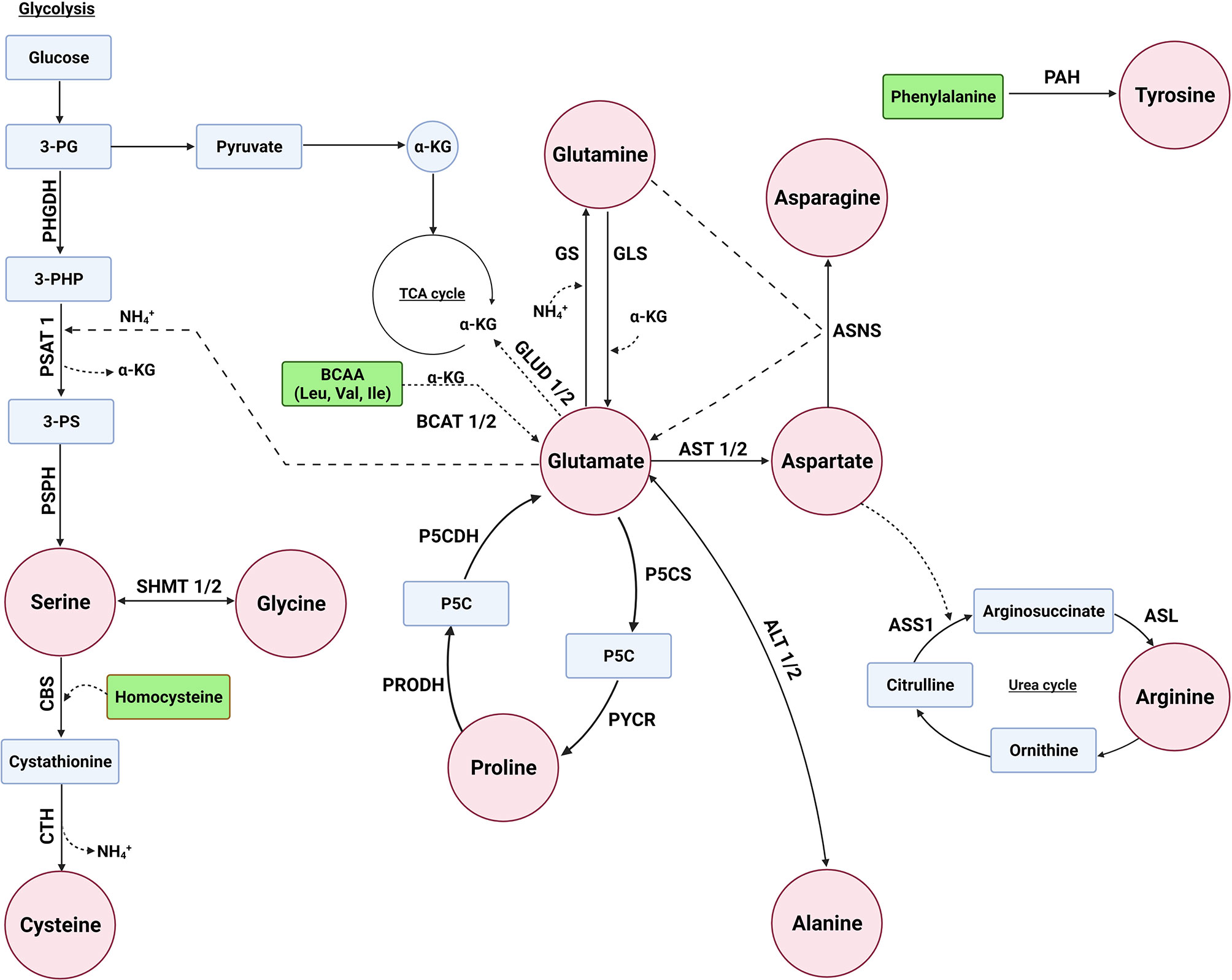
Figure 1 Several enzymes are involved in the biosynthesis of non-essential amino acids. Glutamate can serve as a precursor for the biosynthesis of alanine, aspartate, glutamine, proline, and serine. The enzymes glutaminase (GLS/GLS2), proline dehydrogenase 1 (PRODH), P5C dehydrogenase (P5CDH or ALDH4A1), and branched chain amino acid transaminase (BCAT1/2) are involved in glutamate biosynthesis, whereas glutamate dehydrogenas (GLUD1/2) is involved in its metabolism. The enzymes alanine aminotransaminase (ALT1/2 or GPT/2), aspartate aminotransferase (AST1/2 or GOT1/2), and glutamine synthetase (GS or GLUL) convert glutamate to alanine, aspartate, and glutamine, respectively. Pyrroline-5-carboxylate synthase (P5CS/ALDH18A1) and pyrroline-5-carboxylate reductase 1 (PYCR1) are used for proline biosynthesis from glutamate, whereas serine biosynthesis involves the transamination of 3-phosphohydroxypyruvate to 3-phosphoserine (3-PS) by phosphoserine aminotransferase (PSAT1) with glutamate as the amino donor and phosphoserine phosphatase (PSPH) to convert 3-PS to serine. Asparatate is a precursor of asparagine and arginine. The enzyme asparagine synthetase (ASNS), argininosuccinate synthase 1 (ASS1), and argininosuccinate lyase (ASL) are required for converting aspartate to asparagine and arginine, respectively. Serine is a precursor of glycine and cysteine, where serine is converted to glycine by serine hydroxymethyltransferase (SHMT1/2). Cysteine is synthesized through a process called the reverse transsulfuration pathway involving cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH). Tyrosine biosynthesis not linked to glutamate, but rather the enzyme phenylalanine hydroxylases (PAH) converts phenylalanine to tyrosine.
Alanine, Aspartate, Glutamine, Proline, and Serine Can Be Synthesized From Glutamate
As previously mentioned, glutamate serves as a substrate for several enzymes involved in the synthesis of several NEAAs. For alanine, the enzyme alanine aminotransferase (ALT/GPT) catalyzes the transamination of pyruvate, with glutamate as the amino donor, to synthesize alanine (34). Similar to ALT, aspartate aminotransferase (AST/GOT) transfers an amine group from glutamate to oxaloacetate of the TCA cycle producing aspartate and α‐ketoglutarate (35).
Glutamine is the most abundant amino acid in blood and is indispensable for the survival and growth of cancers. While humans acquire glutamine mostly through diet, glutamine synthetase (GS or GLUL, Figure 1) can synthesize glutamine through the condensation of glutamate and ammonia (36). For proline biosynthesis (Figure 1), pyrroline-5-carboxylate synthase (P5CS) produces P5C from glutamate, which is converted to proline by pyrroline-5-carboxylate reductase (PYCR) (37).
Glutamate is also used for the biosynthesis of serine (Figure 1), where the glycolysis intermediate 3-phosphoglycerate (3-PG) is oxidized by phosphoglycerate dehydrogenase (PHGDH) to generate 3-phosphohydroxypyruvate (3-PHP). Phosphoserine aminotransferase (PSAT) catalyzes the transamination of 3-phosphohydroxypyruvate to 3-phosphoserine (3-PS) with glutamate as the amino donor, and phosphoserine phosphatase (PSPH) hydrolyzes 3-phosphoserine to serine (38).
Aspartate Is a Precursor for the Biosynthesis of Asparagine and Arginine
Similar to glutamate, other NEAAs, such as aspartate, serve as precursors for the biosynthesis of other AAs. Asparagine is synthesized from aspartate and glutamine via asparagine synthetase (ASNS, Figure 1), which transfers an amine group of glutamine to aspartate (39). Similarly, aspartate can be used for the synthesis of arginine through a multi-enzyme pathway in which the enzyme argininosuccinate synthase 1 (ASS1) initiates the process by catalyzing the formation of argininosuccinate from citrulline, which is an intermediate of urea synthesis, and aspartate (Figure 1). Subsequent cleavage of argininosuccinate by argininosuccinate lyase (ASL) produces arginine and fumarate (40).
Serine Is a Precursor of Both Cysteine and Glycine
Serine can serve as a precursor for several AAs, including cysteine and glycine (Figure 1). While cancers can dysregulate cysteine biosynthesis (41–43), typically cysteine is derived from extracellular sources (44–46). The metabolic pathway that leads to the generation of several sulfur metabolites, including cysteine, is called the transsulfuration pathway, where mammalian cells can only obtain cysteine through the reverse transsulfuration pathway (47–49). This pathway for generating cysteine in mammals involves the condensation of homocysteine (derived from methionine) with serine to generate cystathionine through the enzyme cystathionine β-synthase (CBS). Cystathionine γ-lyase (CTH) then converts cystathionine to cysteine (50).
Serine can also be converted to glycine by serine hydroxymethyltransferase (SHMT), which transfers the β-carbon of serine to tetrahydrofolate to produce glycine and 5,10-methylene tetrahydrofolate (51). In humans, there are two SHMT genes: cytoplasmic SHMT1 and mitochondrial SHMT2 (52).
Tyrosine Is Synthesized by Phenylalanine Hydroxylases (PAH) From Phenylalanine
While the biosynthesis of other NEAA is linked to each other, mammals synthesize tyrosine from the essential amino acid phenylalanine. The enzyme phenylalanine hydroxylases (PAH) hydroxylates the aromatic side-chain of phenylalanine to generate tyrosine (53).
Dysregulation of Enzymes Involved in NEAA Biosynthesis in Myeloid Malignancies
Gene Expression Analysis of TCGA AML Samples
The Expression of Enzymes Involved in the Biosynthesis of Glutamate Is Dysregulated in AML
A gene expression analysis of enzymes involved in NEAA biosynthesis was performed using the Gene Expression Profiling Interactive Analysis (GEPIA) web server (54, 55). GEPIA uses RNA-Seq datasets based on the UCSC Xena project2 that has a standard processing pipeline allowing users to compare gene and transcript expression from The Cancer Genome Atlas (TCGA) tumor samples to corresponding Genotype-Tissue Expression (GTEx) normal samples. GEPIA includes over 8,000 normal samples from TCGA and the GTEx projects, albeit from unrelated donors (56, 57). For AML, GEPIA was used to screen for dysregulated enzymes by comparing transcript expression between AML (n = 173) and corresponding normal GTEx samples (n = 70).
Using GEPIA and TCGA AML samples, a gene expression analysis of enzymes involved in the synthesis of glutamate was performed. Consistent with the central role glutamate plays in the biosynthesis of several NEAA, we found that the expression of the glutaminase isoforms GLS and GLS2 in AML is upregulated relative to normal controls (P <0.01, Figures 2A, B). Similar to GLS, PRODH was significantly upregulated in AML samples (P <0.01, Figure 2C), whereas expression levels of P5CDH (i.e., ALDH4A1, data not shown) showed a similar but non-statistically significant trend. Interestingly, the two isoforms of BCAT (cytosolic BCAT1 and mitochondrial BCAT2) were downregulated in AML TCGA samples (P <0.01, Figures 2D, E), indicating a potential strong dependence on glutamine and proline for sources of glutamate. For glutamate dehydrogenase, which is involved in glutamate metabolism, no association was identified for GLUD1, whereas the GLUD2 genes was significantly downregulated (P <0.01, Figure 2F), consistent with a glutamate dependency in AML.
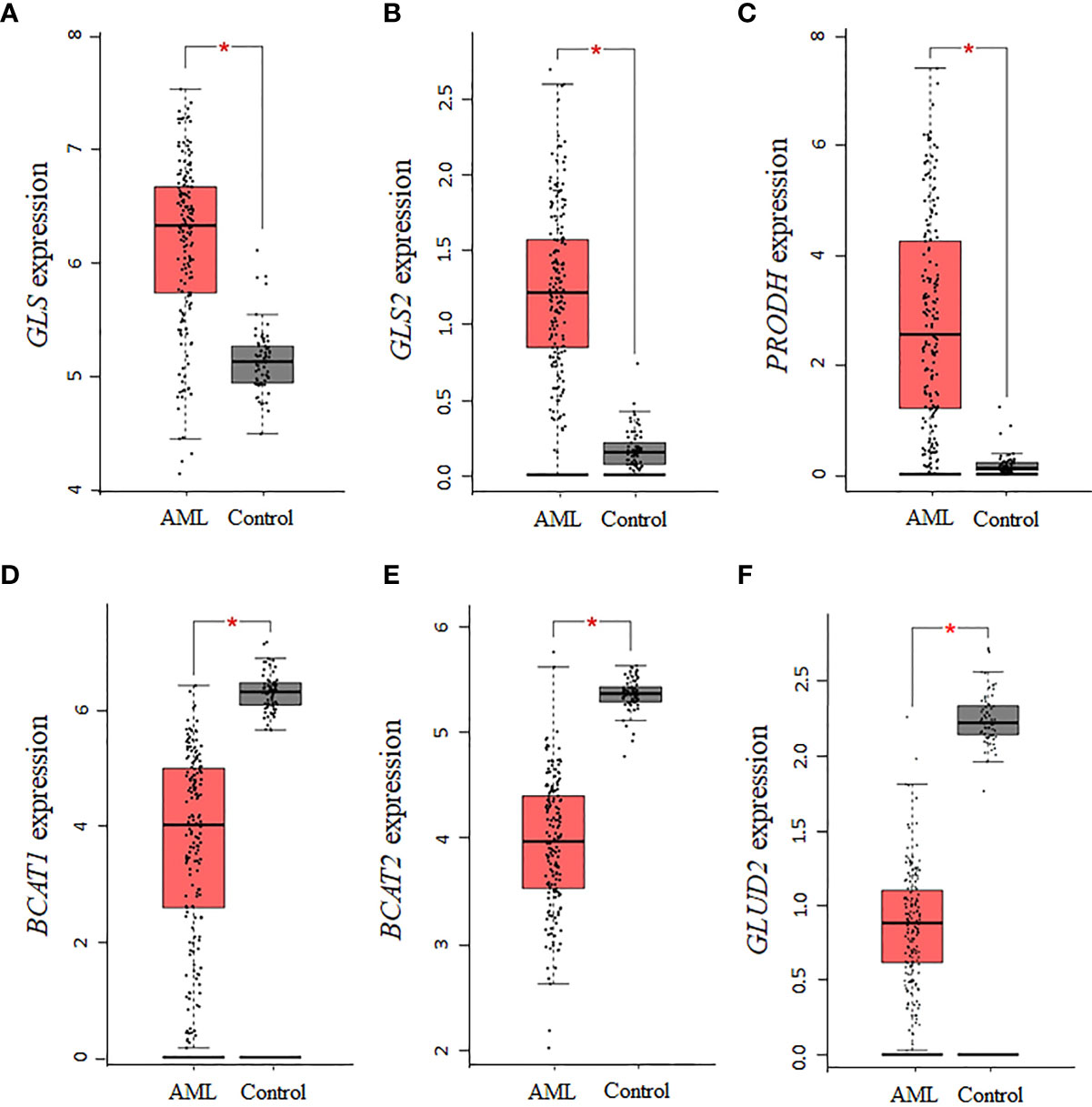
Figure 2 Several enzymes involved in the biosynthesis of glutamate are dysregulated in AML. The expression levels of genes involved in the biosynthesis and metabolism of glutamate were assessed in AML TCGA samples using GEPIA. The data indicate that the expression of (A) GLS, (B) GLS2, and (C) PRODH is upregulated, whereas the expression of (D) BCAT1, (E) BCAT2, and (F) GLUD2 is downregulated. Log2 (TPM + 1) was used for log-scale and P values ≤ 0.01 are denoted with an asterisk (*) and indicate significance (nAML = 173; ncontrol = 70).
The Expression of Enzymes Involved in the Biosynthesis of NEAA Requiring Glutamate as a Precursor Is Decreased in AML
Given that several enzymes of glutamate biosynthesis were dysregulated in AML, it was possible that the corresponding effect on glutamate levels in AML had an influence on other enzymes involved in the biosynthesis of NEAAs requiring glutamate as a precursor. An analysis of the 10 enzymes involved in the biosynthesis of alanine, aspartate, glutamine, proline, and serine identified significant downregulations in AST (GOT1 and GOT2), GLUL, PYCR1, PSAT1, and PHGDH (P <0.01, Figures 3A–F), but no statistical dysregulations in ALT (GPT and GPT2), P5CS (ALDH18A1), or PSPH (data not shown). Overall, there was a downregulation in 6 of 10 enzymes involved in the biosynthesis of these NEAAs, with alanine being the only NEAA with no association. Therefore, while there seems to be a dependence on glutamine and proline for sources of glutamate, many of the pathways involved in the biosynthesis of NEAA requiring glutamate as precursor were repressed.
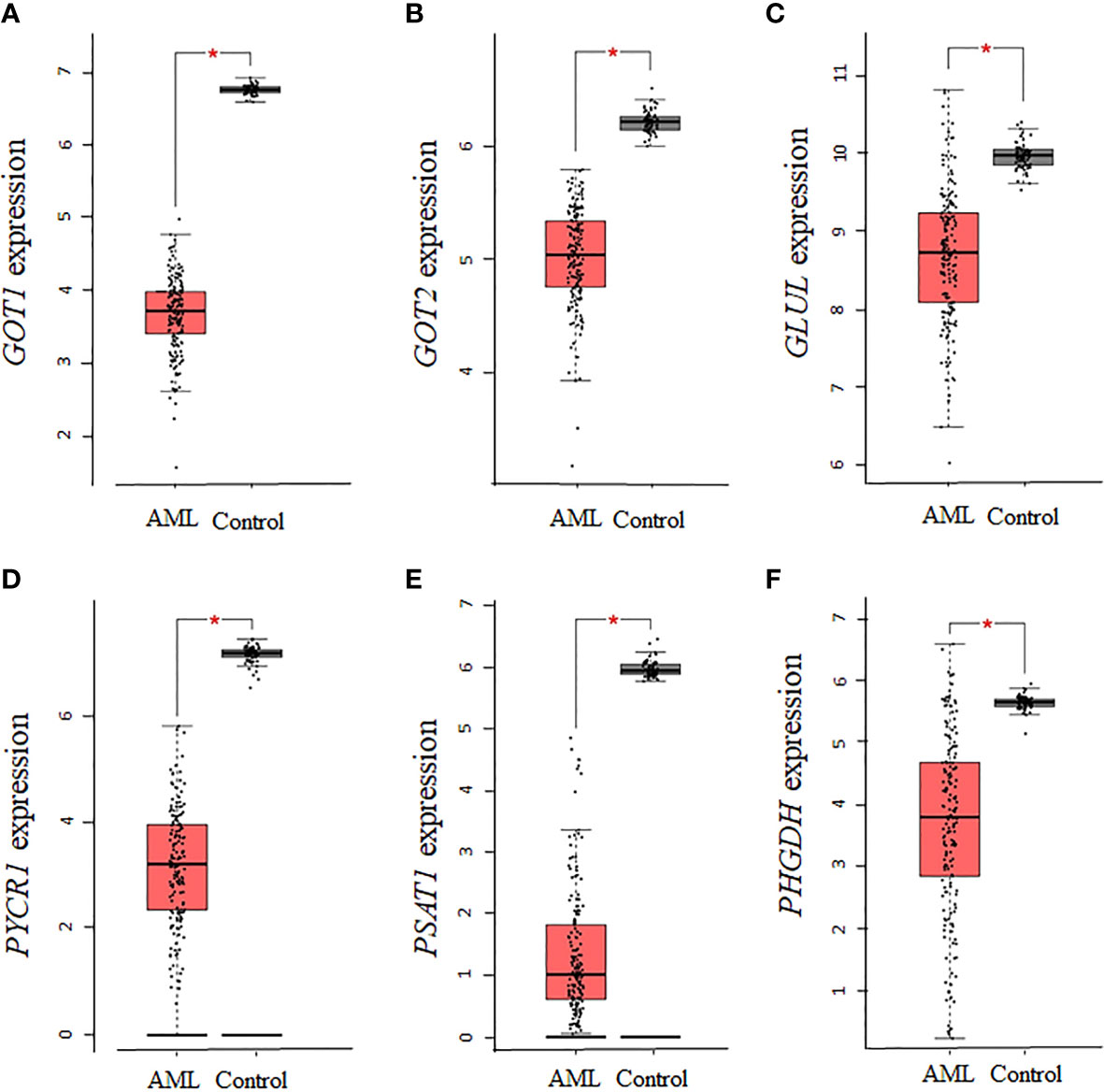
Figure 3 The expression of genes involved in the biosynthesis of alanine, aspartate, glutamine, proline, and serine is decreased in AML. The expression of genes involved in the biosynthesis of alanine, aspartate, glutamine, proline, and serine was analyzed using TCGA AML samples. (A), GOT1, (B) GOT2, (C) GLUL, (D) PYCR1, (E) PSAT1, and (F) PHGDH gene expression was significantly downregulated in AML. Log2 (TPM + 1) was used for log-scale and P values ≤ 0.01 are denoted with an asterisk (*) and indicate significance (nAML = 173; ncontrol = 70).
The Expression of Enzymes Involved in the Biosynthesis of NEAA Requiring Aspartate or Serine as Precursors Is Decreased in AML
Aspartate is a precursor for the biosynthesis of asparagine and arginine, whereas serine is a precursor of cysteine and glycine. Of the seven collective enzymes involved in the biosynthesis of asparagine, arginine, cysteine, and glycine, we found that ASNS, ASS1, ASL, CTH, and SHMT2 were all statistically downregulated in AML samples (Figures 4A–E), whereas CBS and SHMT1 showed non-statistically significant downregulations relative to controls (data not shown). Altogether, our analysis using the AML samples available through TCGA shows that there is a consistent decrease in the expression of enzymes involved in the biosynthesis of NEAAs that are interconnected through glutamate. In contrast, for tyrosine, whose biosynthesis is not linked to glutamate, there was no statistical difference in the expression of PAH between AML and control samples.
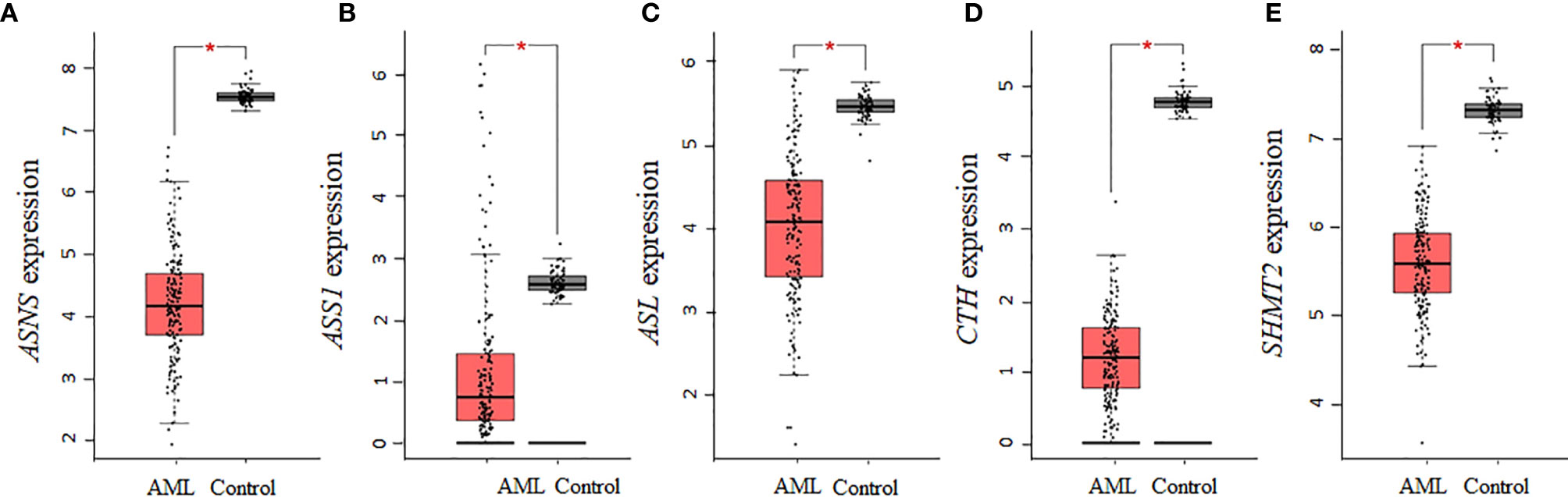
Figure 4 The gene expression of enzymes involved in the biosynthesis of asparagine, arginine, cysteine, and glycine is decreased in AML. AML TCGA analysis of genes involved in the biosynthesis of asparagine and arginine identified a statistical downregulation in (A) ASNS, (B) ASS1, and (C) ASL. Analysis of genes involved in cysteine and glycine biosynthesis identified a statistical downregulation in (D) CTH and (E) SHMT2. Log2 (TPM + 1) was used for log-scale and P values ≤ 0.01 are denoted with an asterisk (*) and indicate significance (nAML = 173; ncontrol = 70).
In summary, using GEPIA and TCGA AML samples we identified statistically significant dysregulations in enzymes involved in the biosynthesis of glutamate, aspartate, glutamine, proline, serine, asparagine, arginine, cysteine, and glycine. Therefore, future efforts should focus on validating these associations across various AML subtypes to best identify specific patient groups with potential NEAA metabolism vulnerabilities, which was not possible using GEPIA.
Gene Expression Analysis of NEAA Biosynthesis in CML
The Expression of Enzymes Involved in Regulating Glutamate, Aspartate, Alanine, Asparagine, and Cysteine Levels Is Upregulated in CML
CML samples were not available through the TCGA database; rather, we used the largest publicly available data set disaggregated by CML phase with gene expression for all 26 genes involved in the biosynthesis of NEAA (Gene Expression Omnibus, GEO accession no. GSE47927)3. Unlike AML, we found few statistical associations for genes encoding enzymes involved in NEAA biosynthesis (Figure 5). Of the 26 genes encoding enzymes involved in the biosynthesis of NEAAs used for our AML analysis, only five statistically significant dysregulated genes were identified in the combined CML cohort. We identified a significant downregulation in PRODH expression in combined and chronic phase CML samples (Figure 5A, PComb = 4.6 × 10−2, PChronic = 9.4 × 10−3), and significant upregulations in AST1 (Figure 5B, PComb = 0.02, PChronic = 0.03), GPT/ALT1 (Figure 5C, PComb = 0.03; PChronic = 0.04), ASNS (Figure 5D, PComb = 0.04; PChronic = 0.02), and CTH (Figure 5E, PComb = 3 × 10−4; PChronic <1 × 10−4) in the combined and chronic phase CML sample analysis. Of the genes that were identified as significant in the combined CML analysis, only AST1 upregulation was significant in blast phase CML samples (Figure 5B, PBlast = 4.8 × 10−2). While no consistent similarities between AML and CML were identified, dysregulation of genes involved in sustaining glutamate (i.e., PRODH), aspartate (i.e., AST1), alanine (i.e., GPT), asparagine (i.e., ASNS), and cysteine (i.e., CTH) levels suggest potential vulnerabilities involving these amino acids.
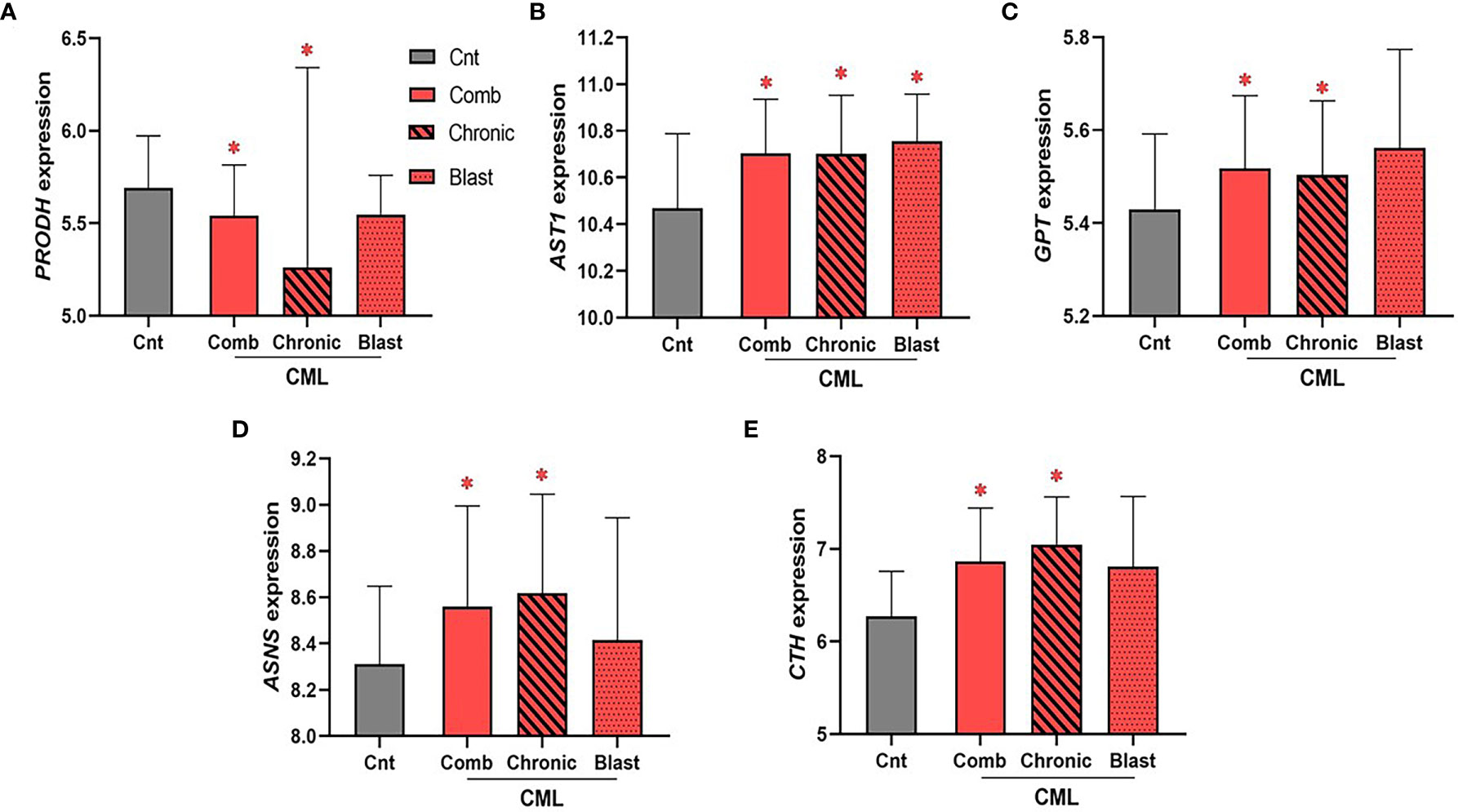
Figure 5 The expression of enzymes involved in glutamate, aspartate, alanine, asparagine, and cysteine biosynthesis is dysregulated in CML. The expression of 26 genes encoding enzymes involved in the biosynthesis of NEAA was analyzed in combined (“comb”) and phase disaggregated (“chronic” and “blast” phase) CML samples. (A) We identified a significant downregulation in PRODH expression in combined (PComb value = 4.6 × 10−2) and chronic (PChronic value = 9.4 × 10−3) CML samples. In contrast, (B) AST1 (PComb value = 0.02; PChronic value = 0.03), (C) GPT/ALT1 (PComb value = 0.03; PChronic value = 0.04), (D) ASNS (PComb value = 0.04; PChronic value = 0.02), and (E) CTH (PComb value = 3 × 10−4; PChronic value < 1 × 10-4) were all upregulated in the combined and chronic CML sample analysis. Only AST1 expression was significantly dysregulated in blast CML samples (PBlast value = 4.8 × 10−2). Log2 was used for log-scale and differential expression was determined using Mann–Whitney testing (nCnt = 15, nComb = 52, nChronic = 24, and nBlast = 10). P values < 0.05 are denoted with an asterisk (*).
Evidence Supporting NEAA Vulnerabilities in AML and CML
Various factors can lead to genetic alterations in cancers that result in the downregulation of an enzyme involved in the biosynthesis of a NEAA, thus rendering the cancer cells dependent on extracellular sources of the NEAA for proliferation and growth. Based on the expression changes observed in AML and CML samples described in the previous section, significant NEAA biosynthesis pathways identified will be reviewed to identify high-priority NEAA vulnerabilities in AML and CML.
There Is Strong Clinical Evidence Supporting Vulnerabilities to Aspartate-Derived NEAAs in Leukemias
The best evidence supporting amino acid vulnerabilities for the treatment of leukemias is based on the efficacy of the chemotherapeutic L-asparaginase (58–63), which is a bacteria-derived enzyme that hydrolyzes asparagine into aspartate and ammonia (64, 65). The enzyme also possesses off-target residual glutaminase activity (66, 67) and several studies have indicated that both its asparaginase and glutaminase activity are required for antileukemic efficacy depending on the expression of ASNS by the cancer (68–72). Generally, asparaginase may be beneficial for malignancies expressing low or no ASNS activity (68, 70, 73, 74), and several studies have investigated the use of asparaginase in non-hematologic malignancies, such as pancreatic, ovarian, and breast cancers (75–79). Furthermore, there is substantial clinical evidence that including asparaginase during the treatment of AML results in better treatment outcomes (80–87). While few studies in CML patients are available to assess the clinical efficacy of asparaginase (88–91), preclinical studies indicate that asparaginase may also be beneficial for CML treatment (92, 93).
Similar to asparaginase, many human cancers, such as melanoma, lymphoma, glioma, and prostate cancer, have low or no detectable expression of ASS1 (94–97). Therefore, several approaches have been investigated for depleting arginine in various cancer types and have led to the development of two pharmacological agents: arginine deiminase derived from bacteria (98–100) and human arginase 1 (101–110). Both are currently undergoing clinical trials (NCT03449901, NCT04587830, NCT02709512 and NCT03455140) and encouraging clinical efficacy has been reported (107, 110–116), including for AML (112, 117).
In our analysis, enzymes involved in the biosynthesis of aspartate (i.e., AST/GOT) were downregulated in AML (Figures 3A, B), where aspartate serves as a precursor of asparagine and arginine (Figure 1). Interestingly, most human cells express low levels of the mitochondrial aspartate/glutamate SLC25A12 transporter, thereby depending on AST-dependent aspartate synthesis (118–120). Furthermore, levels of aspartate has been linked to cancer cell proliferation (118–122) and have been shown to provide a competitive growth advantage to cancer cells under hypoxia by contributing to the formation of oxaloacetate and enabling NADH recycling for glycolysis (119, 120, 123, 124). Aminooxyacetic acid (125, 126), hydrazinosuccinic acid (126–129), and iGOT1-01 (130) have been identified as AST inhibitors. While limited information is available regarding the potential clinical efficacy of targeting the aspartate biosynthesis pathway in cancer, in vitro studies using these inhibitors have demonstrated that they can decreases the proliferation of MDA-MB-231 breast adenocarcinoma cells (125), PaTu-8902 pancreatic cancer cells (125), PaTu-8902 pancreatic cancer cells (130), and DLD1 colon cancer cells (130).
Restriction of Glutamate or Glutamine Metabolism Via Inhibitors of Glutaminase Are Effective Against Myelodysplastic Syndrome
As previously mentioned, glutaminase (GLS) controls the formation of glutamate (Figure 1) and is used for various biosynthetic purposes by cells, in addition to NEAA biosynthesis, including to generate TCA cycle intermediates, glutathione (GSH), NADPH, nucleotides, and fatty acids (131). Therefore, successful strategies targeting the role of glutamate in cancers have focused on restricting glutamine metabolism via glutaminase inhibition (132). Studies investigating the non-competitive allosteric GLS1 inhibitor CB-839 have demonstrated that blocking glutamine metabolism has anticancer activity against triple-negative breast cancer, lung adenocarcinoma, chondrosarcoma, lymphomas, esophageal squamous cell carcinoma, and hepatocellular carcinoma (132–138). Currently, there are 12 ongoing clinical trials evaluating the CB-839 in patients with colorectal cancer (NCT02861300 and NCT03263429), myelodysplastic syndrome (NCT03047993), advanced stage non-small cell lung cancer (NCT04250545 and NCT03831932), diffuse astrocytoma (NCT03528642), ovarian cancer (NCT03944902), refractory multiple myeloma (NCT03798678), advanced or metastatic solid tumors (NCT03965845), metastatic renal cell carcinoma (NCT03428217), malignancies with NF1, KEAP1/NRF2, or STK11/LKB1 mutations (NCT03872427), and non-squamous non-small-cell lung cancer (NCT04265534). Additionally, recent interim results from a phase Ib clinical study of CB-839 in combination with azacitidine in patients with advanced myelodysplastic syndrome demonstrate the possible potential of targeting glutamate sources in leukemias, where the regimen was safely tolerated and 70% of MDS patients achieved a complete response to therapy (139). In addition to CB-839, compound 968 (C968) is another small molecule inhibitor of GLS (140) that has been demonstrated to have anticancer activity in ovarian, brain, pancreatic, and breast cancer (140–142).
Restriction of Other NEAAs Derived From Glutamate Decrease Myeloid Leukemia Proliferation
The biosynthesis of serine and its derivatives were also identified as possible targets in AML and CML (Figures 3E, F and Figure 5E), and consistent with the gene expression analysis, substantial evidence supports that serine, cysteine, and glycine play important roles in cancers, including myeloid malignancies. One-carbon metabolism is required for the synthesis of proteins, lipids, and nucleic acids, with serine being the main source of one-carbon units for methylation reactions that occur through the generation of S-adenosylmethionine (SAM) (143). Glycine is also a major source of methyl groups for one-carbon pools and is required for the biosynthesis of GSH and purines (51), whereas cysteine contributes to redox control and ATP production as a carbon source for biomass and energy production (144). Furthermore, restriction of serine (145–148), glycine (145, 148, 149), or cysteine (150) can decrease cancer cell proliferation. Strategies suppressing PHGDH, SHMT, or cysteine levels via the enzyme cyst(e)inase have demonstrated encouraging results. For AML, there is evidence that serine restriction or PHGDH inhibition can attenuate cell proliferation and that PHGDH expression is prognostic of AML overall survival (151). In addition, cysteine has been demonstrated to be critical for the survival of leukemic stem cells (LSCs) in AML patients, whereas glycine deprivation has been demonstrated to suppress AML cell proliferation (152). For CML, similar to our analysis, several studies have shown that levels of glutamate, serine, which is a precursor of cysteine, and alanine are increased by CML (153–155). Consistent with these NEAAs playing an important role in CML, restriction of serine (156, 157) and its derivatives glycine (156), or cysteine (158, 159) decreases CML proliferation, supporting that targeting these NEAA pathways may be a potential target for CML.
In addition to serine, glycine, and cysteine, several studies support that inhibiting proline biosynthesis in melanoma and breast cancers can impede cell growth (160–162). Furthermore, PYCR1 inhibitors with anticancer activity have been identified to explore the role of proline biosynthesis in AML (163, 164). Nevertheless, limited information is available regarding the effect of inhibiting either enzymes involved in proline biosynthesis or directly restricting proline in AML. Rather AML studies available have indicated that proline uptake is elevated in LSCs isolated from de novo AML patients (165), and that the proline metabolism pathway is significantly impacted by differences in the oncogenic receptor tyrosine kinase FLT3 status of pediatric AML samples (166) or in AML cells overexpressing the proto-oncogene EVI1 (167).
Alanine is secreted by skeletal muscles and provides the carbon source for hepatic gluconeogenesis (168, 169), yet limited information is available about the role of alanine metabolism in myeloid leukemias. Nevertheless, evidence supports that alanine provides a source of α-ketoglutarate for pancreatic and breast cancers that can be used to fuel the TCA cycle (170, 171) or remodel the extracellular matrix for metastasis (172). Additionally, KRAS mutations have been demonstrated to induce the expression of ALT2/GPT2 and drive α-ketoglutarate production and cell growth (173). Few studies have investigated pharmacological strategies for targeting alanine metabolism, yet l-cycloserine has been identified as an inhibitor of ALT/GPT that can attenuate the in vitro and in vivo growth of LLC1 Lewis lung carcinoma cells (173). Taken together, there is substantial evidence that the NEAA biosynthesis pathways dysregulated in AML and CML are promising targets for myeloid leukemias.
GCN2 and mTOR Pathways can Modulate Resistance to Strategies Targeting NEAA Vulnerabilities
Strategies targeting NEAA biosynthesis vulnerabilities can be a promising approach for myeloid malignancies. However, cancer cell adaptation to nutrient stress can lead to drug resistance or treatment failure (174). This adaptation can be due to upregulation of amino acid transporters and/or cell changes leading to increased availability of NEAA pools (8, 174, 175). Therefore, understanding the mechanism by which myeloid leukemias adapt and sense NEAA restriction is important for optimizing therapeutic approaches targeting NEAA metabolic vulnerabilities. Two essential kinases that are involved in the adaptation to nutrient stress are mammalian target of rapamycin (mTOR) and general control nonderepressible-2 kinase (GCN2). Collectively, these pathways can decrease the cellular demand for amino acids while concurrently increasing their synthesis to overcome the nutrient stress and lead to resistance (Figure 6).
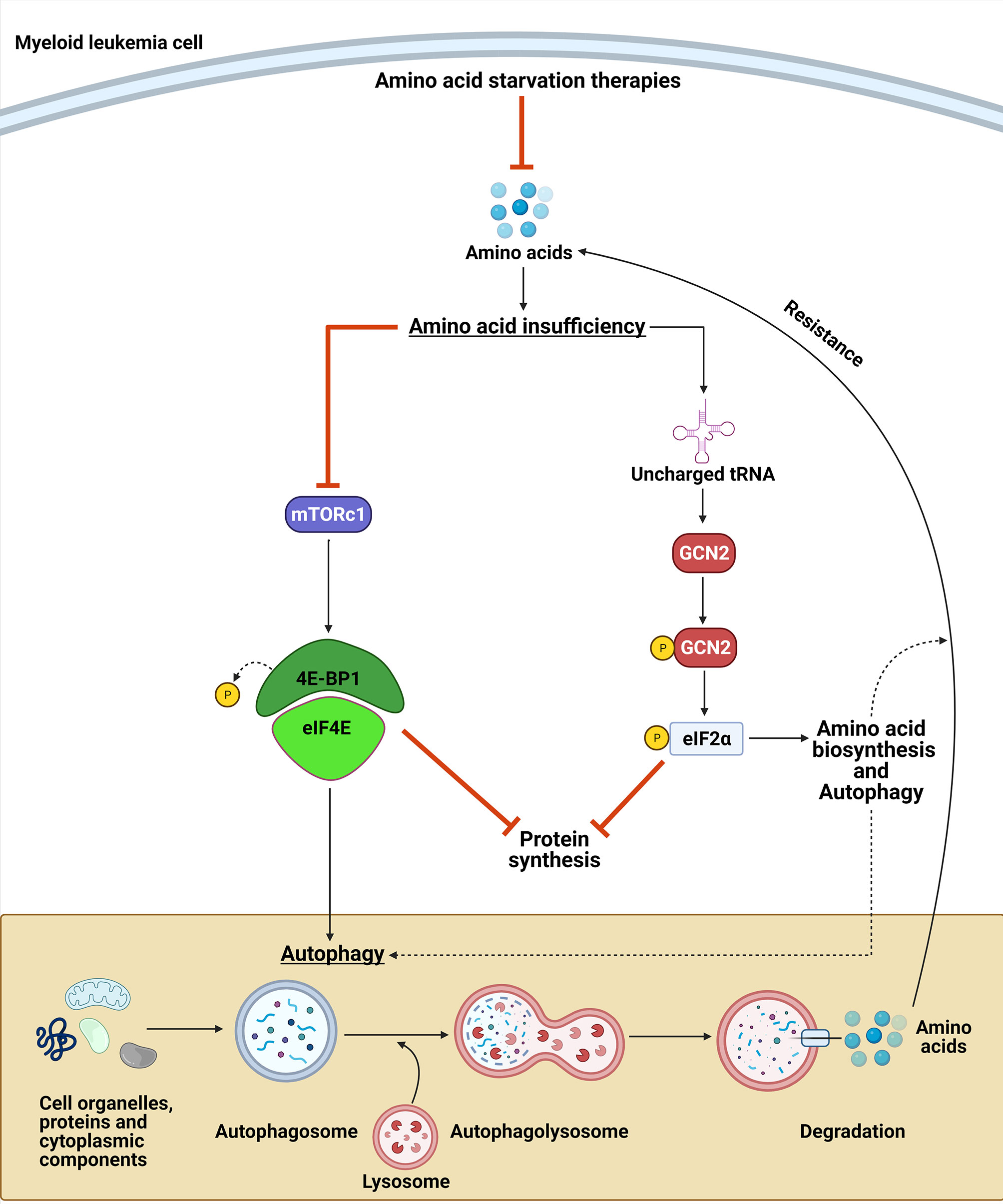
Figure 6 GCN2 and mTORC1 contribute to amino acid deprivation resistance. Upon amino acid deprivation, the accumulation of uncharged tRNA leads to the activation of the GCN2 kinase, which phosphorylates eIF2α. GCN2 activation can lead to a resistance to the amino acid restriction by inhibiting protein synthesis and by upregulating the translation of amino acid transporters, enzymes involved in amino acid biosynthesis, and proteins involved in autophagy. In contrast, amino acid suppression leads to inactivation of mTORC1, which induces autophagy and leads to a decrease in protein synthesis. Collectively, these mechanisms provide amino acid sources to maintain myeloid leukemia cell proliferation upon NEAA deprivation.
Activation of the Amino Acid Response Pathway (AAR) Can Lead to Nutrient Stress Resistance
The GCN2 kinase activates the amino acid response (AAR) pathway by sensing amino acid insufficiency via direct binding to uncharged tRNAs that accumulate during nutrient stress (176). Upon activation via autophosphorylation, GCN2 phosphorylates its only known substrate, the α subunit of the translation eukaryotic initiation factor (eIF2) (177). Phosphorylation of eIF2α has two essential consequences that can affect cancer cell responses to amino acid deprivation (Figure 6). First, it attenuates the general protein translation of most mRNAs by blocking the activity of the eIF2B guanylate exchange factor, thereby limiting the availability of ternary complex required for initiating translation (178–180). Second, it activates the AAR pathway by concomitantly increasing the translation of a subset of mRNAs with upstream ORFs that regulate translation of the downstream main ORF (179, 181–183). The AAR pathway leads to the upregulation of amino acid transporters, aminoacyl-tRNA synthetases, enzymes involved in the biosynthesis of amino acids, and proteins involved in autophagy (29, 184–186).
In contrast to GCN2, which directly senses amino acid depletion, mTORC1 indirectly senses amino acid sufficiency through mechanisms that were recently elucidated (187–190). mTORC1 is a serine/threonine protein kinase that acts as an essential regulator of cell growth and metabolism in response to nutrient changes. SLC38A9, Sestrin1/2, and CASTOR1 have been identified as amino acid sensors upstream of mTORC1 that sense the availability of arginine and leucine (187–191). SLC38A9 stimulates mTORC1 activity through the regulation of Rag proteins, whereas Sestrin1/2 and CASTOR1 dissociate from GATOR2, which is a positive regulator of mTORC1, in the presence of leucine or arginine. Activated mTORC1 phosphorylates 4EBP1 and leads to the dissociation of 4EBP1 from EIF4E, enabling the formation of the translation initiation complex. In contrast, amino acid deficiency inactivates mTORC1 and suppresses 4EBP1 phosphorylation and protein synthesis (192–195). In addition to translation, mTORC1 is a negative regulator of autophagy, which is an intracellular process that allows orderly degradation and recycling of cellular components, including amino acids. Autophagy is regulated by mTORC1 through its phosphorylation of UNC-51-like kinase 1 (ULK) (196–200). Therefore, mTORC1 signaling plays a role in AA deprivation adaptation by limiting protein translation and increasing amino acid pools through autophagy (Figure 6).
Interestingly, there is substantial evidence supporting cross-talk between the GCN2/eIF2α pathway and mTORC1 signaling. Activating transcription factor 4 (ATF4) is induced by phosphorylation of eIF2α and several of its targets are mTORC1 inhibitors, including REDD1, GADD34, and Sestrin2 (201–203). Furthermore, autophagy regulation can be directly affected by ATF4 via the upregulation of many autophagy genes, including ATG16L, ATG12, ATG3, BECN1, LC3B, and p62 (185). Several studies have also demonstrated that inhibiting the GCN2/eIF2α pathway during amino acid deprivation prevents mTORC1 inactivation (204–206), whereas mTORC1 inhibition by rapamycin has been demonstrated to results in activation of the GCN2/eIF2α pathway (207).
Because mTORC1 and the GCN2/eIF2α pathway provide mechanisms of resistance to amino acid deprivation strategies, it is conceivable that blocking these adaptation pathways would further sensitize cancer cells to the nutrient stress. Consistent with that hypothesis, the development of a GCN2 inhibitor has been shown to enhance the antileukemic efficacy of L-asparaginase (208). Other studies have demonstrated similar adaptations and GCN2-dependent sensitizations (174, 209–215). While constitutive activation of mTORC1 can lead to resistance against targeted therapies (216), in the context of amino acid vulnerabilities, blocking the generation of amino acids from autophagic protein degradation can sensitize cancer cells to amino acid deprivation approaches (217). Consistent with this concept, blocking autophagy during asparagine (93, 218–222), arginine (106, 108, 223–229), and glutamine (230) restriction leads to enhanced efficacy.
Conclusions and Future Perspectives
Cancers including myeloid leukemias use various energy sources to maintain their abnormal proliferation rate and therefore can become auxotrophic for particular NEAAs. Identifying these vulnerabilities in myeloid leukemias can therefore lead to new treatment options and improve survival rates. To date, L-asparaginase is the most successful clinical example of exploiting cancer nutrient dependencies. While outside of the scope of this review, there are several fuel sources aside from NEAA not considered here that contribute to metabolic vulnerabilities, including glucose, essential amino acids, fatty acids, lactate, and acetate (8–11). Furthermore, additional factors, including the expression of amino acid transporters (231–236) and the effect of stromal cells on the microenvironment, can contribute to cancer metabolism and proliferation (8). It is also clear that optimal treatment strategies will consider mechanisms of cell adaptation to nutrient stress. Our AML analysis identified that the biosynthesis of glutamate, aspartate, glutamine, proline, serine, asparagine, arginine, cysteine, and glycine are altered in TCGA samples, indicating that targeting multiple NEAAs may have a greater effect than a single agent approach. It is feasible that a combination approach to NEAA metabolic vulnerabilities may require lower drug exposures versus a single agent approach, and therefore can lead to better antileukemic efficacy while decreasing the risk of toxicities. Successful novel regimens with reduced risks of toxicities can lead to the development of new first-line therapy options for myeloid leukemia patients with limited treatment options.
Footnotes
1. SEER*Explorer: An interactive website for SEER cancer statistics. Surveillance Research Program, National Cancer Institute. Available from https://seer.cancer.gov/explorer/. [Cited 2021 April 1]
2. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE47927
Author Contributions
CF and AB performed all data analysis. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the University of Pittsburgh School of Pharmacy, the NIH TL1TR001858 Training Grant, the Rho Chi Society and American Foundation for Pharmaceutical Education, and the NIH Grant RO1 CA216815.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2020. CA Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
2. Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM. Epidemiology of Acute Myeloid Leukemia: Recent Progress and Enduring Challenges. Blood Rev (2019) 36:70–87. doi: 10.1016/j.blre.2019.04.005
3. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med (2020) 383(7):617–29. doi: 10.1056/NEJMoa2012971.Citedin:Pubmed
4. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N Engl J Med (2001) 344(14):1031–7. doi: 10.1056/NEJM200104053441401
5. Tanizawa A. Optimal Management for Pediatric Chronic Myeloid Leukemia. Pediatr Int (2016) 58(3):171–9. doi: 10.1111/ped.12876
6. Chan O, Talati C, Isenalumhe L, Shams S, Nodzon L, Fradley M, et al. Side-Effects Profile and Outcomes of Ponatinib in the Treatment of Chronic Myeloid Leukemia. Blood Adv (2020) 4(3):530–8. doi: 10.1182/bloodadvances.2019000268
7. Issa GC, Kantarjian HM, Gonzalez GN, Borthakur G, Tang G, Wierda W, et al. Clonal Chromosomal Abnormalities Appearing in Philadelphia Chromosome–Negative Metaphases During CML Treatment. Blood (2017) 130(19):2084–91. doi: 10.1182/blood-2017-07-792143%JBlood
8. Vettore L, Westbrook RL, Tennant DA. New Aspects of Amino Acid Metabolism in Cancer. Br J Cancer (2020) 122(2):150–6. doi: 10.1038/s41416-019-0620-5
9. Keenan MM, Chi JT. Alternative Fuels for Cancer Cells. Cancer J (2015) 21(2):49–55. doi: 10.1097/ppo.0000000000000104
10. Vazquez A, Kamphorst JJ, Markert EK, Schug ZT, Tardito S, Gottlieb E. Cancer Metabolism at a Glance. J Cell Sci (2016) 129(18):3367–73. doi: 10.1242/jcs.181016
11. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab (2016) 23(1):27–47. doi: 10.1016/j.cmet.2015.12.006
12. Otten JJ, Hellwig JP, Meyers LD eds. Medicine Io. Dietary Reference Intakes: The Essential Guide to Nutrient Requirements Vol. 1344. Washington, DC: The National Academies Press (2006). Available at: https://www.nap.edu/catalog/11537/dietary-reference-intakes-the-essential-guide-to-nutrient-requirements.
13. Fung MKL, Chan GC. Drug-Induced Amino Acid Deprivation as Strategy for Cancer Therapy. J Hematol Oncol (2017) 10(1):144. doi: 10.1186/s13045-017-0509-9
14. Hanahan D, Weinberg Robert A. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
15. Lopez MJ MS. Biochemistry, Essential Amino Acids (2021). StatPearls Publishing. Available at: https://www.ncbi.nlm.nih.gov/books/NBK557845/?report=classic (Accessed January 2021).
16. Morris CR, Hamilton-Reeves J, Martindale RG, Sarav M, Ochoa Gautier JB. Acquired Amino Acid Deficiencies: A Focus on Arginine and Glutamine. Nutr Clin Pract (2017) 32(1S):30S–47S. doi: 10.1177/0884533617691250
17. Nilsson A, Haanstra JR, Engqvist M, Gerding A, Bakker BM, Klingmüller U, et al. Quantitative Analysis of Amino Acid Metabolism in Liver Cancer Links Glutamate Excretion to Nucleotide Synthesis. PNAS (2020) 117(19):10294–304. doi: 10.1073/pnas.1919250117
18. Asantewaa G, Harris IS. Glutathione and its Precursors in Cancer. Curr Opin Biotechnol (2021) 68:292–9. doi: 10.1016/j.copbio.2021.03.001
19. Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP, et al. Branched-Chain Amino Acid Catabolism Fuels Adipocyte Differentiation and Lipogenesis. Nat Chem Biol (2016) 12(1):15–21. doi: 10.1038/nchembio.1961
20. Sugden MC, Watts DI, West PS, Norman Palmer T. Proline and Hepatic Lipogenesis. Biochim Biophys Acta (BBA) - Gen Subj (1984) 798(3):368–73. doi: 10.1016/0304-4165(84)90111-9
21. Corbet C, Feron O. Cancer Cell Metabolism and Mitochondria: Nutrient Plasticity for TCA Cycle Fueling. Biochim Biophys Acta (BBA) - Rev Cancer (2017) 1868(1):7–15. doi: 10.1016/j.bbcan.2017.01.002
22. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment [Review]. English (2017) 8:248. doi: 10.3389/fimmu.2017.00248
23. Nakanishi S, Cleveland JL. Polyamine Homeostasis in Development and Disease. Med Sci (Basel) (2021) 9(2):28. doi: 10.3390/medsci9020028
24. Pasini A, Caldarera CM, Giordano E. Chromatin Remodeling by Polyamines and Polyamine Analogs. Amino Acids (2014) 46(3):595–603. doi: 10.1007/s00726-013-1550-9
25. Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, et al. Influence of Threonine Metabolism on S-Adenosylmethionine and Histone Methylation. Science (2013) 339(6116):222–6. doi: 10.1126/science.1226603%JScience
26. Sun L, Zhang H, Gao P. Metabolic Reprogramming and Epigenetic Modifications on the Path to Cancer. Protein Cell (2021). doi: 10.1007/s13238-021-00846-7
27. Maddocks ODK, Labuschagne CF, Adams PD, Vousden KH. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation Through De Novo ATP Synthesis in Cancer Cells. Mol Cell (2016) 61(2):210–21. doi: 10.1016/j.molcel.2015.12.014
28. Ulrey CL, Liu L, Andrews LG, Tollefsbol TO. The Impact of Metabolism on DNA Methylation. Hum Mol Genet (2005) 14(suppl_1):R139–47. doi: 10.1093/hmg/ddi100%JHumanMolecularGenetics
29. Pathria G, Ronai ZA. Harnessing the Co-Vulnerabilities of Amino Acid-Restricted Cancers. Cell Metab (2021) 33(1):9–20. doi: 10.1016/j.cmet.2020.12.009
30. Maggi M, Scotti C. Enzymes in Metabolic Anticancer Therapy. Adv Exp Med Biol (2019) 1148:173–99. doi: 10.1007/978-981-13-7709-9_9
31. Tabe Y, Lorenzi PL, Konopleva M. Amino Acid Metabolism in Hematologic Malignancies and the Era of Targeted Therapy. Blood (2019) 134(13):1014–23. doi: 10.1182/blood.2019001034
32. Boyko M, Gruenbaum SE, Gruenbaum BF, Shapira Y, Zlotnik A. Brain to Blood Glutamate Scavenging as a Novel Therapeutic Modality: A Review. J Neural Transm (Vienna) (2014) 121(8):971–9. doi: 10.1007/s00702-014-1181-7
33. Mastorodemos V, Kotzamani D, Zaganas I, Arianoglou G, Latsoudis H, Plaitakis A. Human GLUD1 and GLUD2 Glutamate Dehydrogenase Localize to Mitochondria and Endoplasmic Reticulum. Biochem Cell Biol (2009) 87(3):505–16. doi: 10.1139/o09-008
34. Doebbe A, Keck M, La Russa M, Mussgnug JH, Hankamer B, Tekce E, et al. The Interplay of Proton, Electron, and Metabolite Supply for Photosynthetic H2 Production in Chlamydomonas Reinhardtii. J Biol Chem (2010) 285(39):30247–60. doi: 10.1074/jbc.M110.122812
35. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine Uptake and Metabolism are Coordinately Regulated by ERK/MAPK During T Lymphocyte Activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
36. Rajagopalan KN, DeBerardinis RJ. Role of Glutamine in Cancer: Therapeutic and Imaging Implications. J Nucl Med (2011) 52(7):1005–8. doi: 10.2967/jnumed.110.084244
37. Perez-Arellano I, Carmona-Alvarez F, Martinez AI, Rodriguez-Diaz J, Cervera J. Pyrroline-5-Carboxylate Synthase and Proline Biosynthesis: From Osmotolerance to Rare Metabolic Disease. Protein Sci (2010) 19(3):372–82. doi: 10.1002/pro.340
38. Walsh DA, Sallach HJ. Comparative Studies on the Pathways for Serine Biosynthesis in Animal Tissues. J Biol Chem (1966) 241(17):4068–76. doi: 10.1016/S0021-9258(18)99812-1
39. Chiu M, Taurino G, Bianchi MG, Kilberg MS, Bussolati O. Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Front Oncol (2019) 9:1480. doi: 10.3389/fonc.2019.01480
40. Keshet R, Szlosarek P, Carracedo A, Erez A. Rewiring Urea Cycle Metabolism in Cancer to Support Anabolism. Nat Rev Cancer (2018) 18(10):634–45. doi: 10.1038/s41568-018-0054-z
41. Belalcazar AD, Ball JG, Frost LM, Valentovic MA, Wilkinson JT. Transsulfuration Is a Significant Source of Sulfur for Glutathione Production in Human Mammary Epithelial Cells. ISRN Biochem (2014) 2013:637897. doi: 10.1155/2013/637897
42. Leikam C, Hufnagel A, Walz S, Kneitz S, Fekete A, Muller MJ, et al. Cystathionase Mediates Senescence Evasion in Melanocytes and Melanoma Cells. Oncogene (2014) 33(6):771–82. doi: 10.1038/onc.2012.641
43. Lien EC, Ghisolfi L, Geck RC, Asara JM, Toker A. Oncogenic PI3K Promotes Methionine Dependency in Breast Cancer Cells Through the Cystine-Glutamate Antiporter xCT. Sci Signal (2017) 10(510). doi: 10.1126/scisignal.aao6604.Citedin:Pubmed
44. Bannai S. Transport of Cystine and Cysteine in Mammalian Cells. Biochim Biophys Acta (1984) 779(3):289–306. doi: 10.1016/0304-4157(84)90014-5
45. Iglehart JK, York RM, Modest AP, Lazarus H, Livingston DM. Cystine Requirement of Continuous Human Lymphoid Cell Lines of Normal and Leukemic Origin. J Biol Chem (1977) 252(20):7184–91. doi: 10.1016/S0021-9258(19)66953-X
46. Uren JR, Lazarus H. L-Cyst(E)Ine Requirements of Malignant Cells and Progress Toward Depletion Therapy. Cancer Treat Rep (1979) 63(6):1073–9.
47. McBean GJ. The Transsulfuration Pathway: A Source of Cysteine for Glutathione in Astrocytes. Amino Acids (2012) 42(1):199–205. doi: 10.1007/s00726-011-0864-8
48. Sbodio JI, Snyder SH, Paul BD. Regulators of the Transsulfuration Pathway. Br J Pharmacol (2019) 176(4):583–93. doi: 10.1111/bph.14446
49. Stipanuk MH, Dominy JE Jr., Lee JI, Coloso RM. Mammalian Cysteine Metabolism: New Insights Into Regulation of Cysteine Metabolism. J Nutr (2006) 136(6 Suppl):1652S–9S. doi: 10.1093/jn/136.6.1652S
50. Liu N, Lin X, Huang C. Activation of the Reverse Transsulfuration Pathway Through NRF2/CBS Confers Erastin-Induced Ferroptosis Resistance. Br J Cancer (2020) 122(2):279–92. doi: 10.1038/s41416-019-0660-x
51. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and Glycine Metabolism in Cancer. Trends Biochem Sci (2014) 39(4):191–8. doi: 10.1016/j.tibs.2014.02.004
52. Garrow TA, Brenner AA, Whitehead VM, Chen XN, Duncan RG, Korenberg JR, et al. Cloning of Human cDNAs Encoding Mitochondrial and Cytosolic Serine Hydroxymethyltransferases and Chromosomal Localization. J Biol Chem (1993) 268(16):11910–6. doi: 10.1016/S0021-9258(19)50286-1
53. Fitzpatrick PF. Tetrahydropterin-Dependent Amino Acid Hydroxylases. Annu Rev Biochem (1999) 68:355–81. doi: 10.1146/annurev.biochem.68.1.355
54. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res (2017) 45(W1):W98–W102. doi: 10.1093/nar/gkx247
55. Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: An Enhanced Web Server for Large-Scale Expression Profiling and Interactive Analysis. Nucleic Acids Res (2019) 47(W1):W556–w560. doi: 10.1093/nar/gkz430.Citedin:Pubmed
56. Goldman M, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. The UCSC Xena Platform for Public and Private Cancer Genomics Data Visualization and Interpretation. bioRxiv (2019) 326470. doi: 10.1101/326470%JbioRxiv
57. Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat Biotechnol (2020) 38(6):675–8. doi: 10.1038/s41587-020-0546-8
58. Sallan SE, Hitchcock-Bryan S, Gelber R, Cassady JR, Frei E, Nathan DG. Influence of Intensive Asparaginase in the Treatment of Childhood Non-T-Cell Acute Lymphoblastic Leukemia. Can Res (1983) 43:5601–7.
59. Amylon MD, Shuster J, Pullen J, Berard C, Link MP, Wharam M, et al. Intensive High-Dose Asparaginase Consolidation Improves Survival for Pediatric Patients With T Cell Acute Lymphoblastic Leukemia and Advanced Stage Lymphoblastic Lymphoma: A Pediatric Oncology Group Study. Leukemia (1999) 13(3):335–42. doi: 10.1038/sj.leu.2401310
60. Silverman LB, Gelber RD, Dalton VK, Asselin BL, Barr RD, Clavell LA, et al. Improved Outcome for Children With Acute Lymphoblastic Leukemia: Results of Dana-Farber Consortium Protocol 91-01. Blood (2001) 97(5):1211–8. doi: 10.1182/blood.V97.5.1211
61. Pui CH, Campana D, Pei D, Bowman WP, Sandlund JT, Kaste SC, et al. Treating Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation. N Engl J Med (2009) 360(26):2730–41. doi: 10.1056/NEJMoa0900386
62. Ertel IJ, Nesbit ME, Hammond D, Weiner J, Sather H. Effective Dose of L-Asparaginase for Induction of Remission in Previously Treated Children With Acute Lymphocytic Leukemia: A Report From Childrens Cancer Study Group. Can Res (1979) 39:3893–6.
63. Clavell LA, Gelber RD, Cohen HJ, Hitchcock-Bryan S, Cassady JR, Tarbell NJ, et al. Four-Agent Induction and Intensive Asparaginase Therapy for Treatment of Childhood Acute Lymphoblastic Leukemia. N Engl J Med (1986) 315:657–63. doi: 10.1056/NEJM198609113151101
64. Fernandez CA, Cai X, Elozory A, Liu C, Panetta JC, Jeha S, et al. High-Throughput Asparaginase Activity Assay in Serum of Children With Leukemia. Int J Clin Exp Med (2013) 6(7):478–87.
65. Broome JD. L-Asparaginase: Discovery and Development as a Tumor-Inhibitory Agent. Cancer Treat Rep (1981) 65 Suppl 4:111–4.
66. Miller HK, Balis ME. Glutaminase Activity of L-Asparagine Amidohydrolase. Biochem Pharmacol (1969) 18(9):2225–32. doi: 10.1016/0006-2952(69)90329-3
67. Distasio JA, Salazar AM, Nadji M, Durden DL. Glutaminase-Free Asparaginase From Vibrio Succinogenes: An Antilymphoma Enzyme Lacking Hepatotoxicity. Int J Cancer (1982) 30(3):343–7. doi: 10.1002/ijc.2910300314
68. Chan WK, Horvath TD, Tan L, Link T, Harutyunyan KG, Pontikos MA, et al. Glutaminase Activity of L-Asparaginase Contributes to Durable Preclinical Activity Against Acute Lymphoblastic Leukemia. Mol Cancer Ther (2019) 18(9):1587–92. doi: 10.1158/1535-7163.MCT-18-1329
69. Wu MC, Arimura GK, Yunis AA. Mechanism of Sensitivity of Cultured Pancreatic Carcinoma to Asparaginase. Int J Cancer (1978) 22(6):728–33. doi: 10.1002/ijc.2910220615
70. Chan WK, Lorenzi PL, Anishkin A, Purwaha P, Rogers DM, Sukharev S, et al. The Glutaminase Activity of L-Asparaginase is Not Required for Anticancer Activity Against ASNS-Negative Cells. Blood (2014) 123(23):3596–606. doi: 10.1182/blood-2013-10-535112
71. Offman MN, Krol M, Patel N, Krishnan S, Liu J, Saha V, et al. Rational Engineering of L-Asparaginase Reveals Importance of Dual Activity for Cancer Cell Toxicity. Blood (2011) 117(5):1614–21. doi: 10.1182/blood-2010-07-298422
72. Parmentier JH, Maggi M, Tarasco E, Scotti C, Avramis VI, Mittelman SD. Glutaminase Activity Determines Cytotoxicity of L-Asparaginases on Most Leukemia Cell Lines. Leuk Res (2015) 39(7):757–62. doi: 10.1016/j.leukres.2015.04.008
73. Kiriyama Y, Kubota M, Takimoto T, Kitoh T, Tanizawa A, Akiyama Y, et al. Biochemical Characterization of U937 Cells Resistant to L-Asparaginase: The Role of Asparagine Synthetase. Leukemia (1989) 3(4):294–7.
74. Stams WA, den Boer ML, Beverloo HB, Meijerink JP, Stigter RL, van Wering ER, et al. Sensitivity to L-Asparaginase is Not Associated With Expression Levels of Asparagine Synthetase in T(12;21)+ Pediatric ALL. Blood (2003) 101(7):2743–7. doi: 10.1182/blood-2002-08-2446
75. Yang H, He X, Zheng Y, Feng W, Xia X, Yu X, et al. Down-Regulation of Asparagine Synthetase Induces Cell Cycle Arrest and Inhibits Cell Proliferation of Breast Cancer. Chem Biol Drug Des (2014) 84(5):578–84. doi: 10.1111/cbdd.12348
76. Domenech C, Thomas X, Chabaud S, Baruchel A, Gueyffier F, Mazingue F, et al. L-Asparaginase Loaded Red Blood Cells in Refractory or Relapsing Acute Lymphoblastic Leukaemia in Children and Adults: Results of the GRASPALL 2005-01 Randomized Trial. Br J Haematol (2011) 153(1):58–65. doi: 10.1111/j.1365-2141.2011.08588.x
77. Hammel P, Fabienne P, Mineur L, Metges JP, Andre T, de la Fouchardiere C, et al. Erythrocyte-Encapsulated Asparaginase (Eryaspase) Combined With Chemotherapy in Second-Line Treatment of Advanced Pancreatic Cancer: An Open-Label, Randomized Phase IIb Trial. Eur J Cancer (2020) 124:91–101. doi: 10.1016/j.ejca.2019.10.020
78. Lorenzi PL, Reinhold WC, Rudelius M, Gunsior M, Shankavaram U, Bussey KJ, et al. Asparagine Synthetase as a Causal, Predictive Biomarker for L-Asparaginase Activity in Ovarian Cancer Cells. Mol Cancer Ther (2006) 5(11):2613–23. doi: 10.1158/1535-7163.MCT-06-0447
79. Lorenzi PL, Llamas J, Gunsior M, Ozbun L, Reinhold WC, Varma S, et al. Asparagine Synthetase is a Predictive Biomarker of L-Asparaginase Activity in Ovarian Cancer Cell Lines. Mol Cancer Ther (2008) 7(10):3123–8. doi: 10.1158/1535-7163.MCT-08-0589
80. Capizzi RL, Poole M, Cooper MR, Richards F 2nd, Stuart JJ, Jackson DV Jr., et al. Treatment of Poor Risk Acute Leukemia With Sequential High-Dose ARA-C and Asparaginase. Blood (1984) 63(3):694–700.
81. Capizzi RL, Davis R, Powell B, Cuttner J, Ellison RR, Cooper MR, et al. Synergy Between High-Dose Cytarabine and Asparaginase in the Treatment of Adults With Refractory and Relapsed Acute Myelogenous Leukemia–a Cancer and Leukemia Group B Study. J Clin Oncol (1988) 6(3):499–508. doi: 10.1200/JCO.1988.6.3.499
82. Wells RJ, Woods WG, Lampkin BC, Nesbit ME, Lee JW, Buckley JD, et al. Impact of High-Dose Cytarabine and Asparaginase Intensification on Childhood Acute Myeloid Leukemia: A Report From the Childrens Cancer Group. J Clin Oncol (1993) 11(3):538–45. doi: 10.1200/JCO.1993.11.3.538
83. Horikoshi A, Takei K, Iriyama N, Uenogawa K, Ishizuka H, Shiraiwa H, et al. Effect of L-Asparaginase Combined With Vincristine and Prednisolone on Acute Myeloblastic Leukemia (M0) Associated With non-Hodgkin Lymphoma. Acta Haematol (2009) 122(1):54–7. doi: 10.1159/000243725
84. Buaboonnam J, Cao X, Pauley JL, Pui CH, Ribeiro RC, Rubnitz JE, et al. Sequential Administration of Methotrexate and Asparaginase in Relapsed or Refractory Pediatric Acute Myeloid Leukemia. Pediatr Blood Cancer (2013) 60(7):1161–4. doi: 10.1002/pbc.24470
85. Ahmed T, Holwerda S, Klepin HD, Isom S, Ellis LR, Lyerly S, et al. High Dose Cytarabine, Mitoxantrone and L-Asparaginase (HAMA) Salvage for Relapsed or Refractory Acute Myeloid Leukemia (AML) in the Elderly. Leuk Res (2015) 39(9):945–9. doi: 10.1016/j.leukres.2015.05.010
86. Emadi A, Zokaee H, Sausville EA. Asparaginase in the Treatment of non-ALL Hematologic Malignancies. Cancer Chemother Pharmacol (2014) 73(5):875–83. doi: 10.1007/s00280-014-2402-3
87. Ohnuma T, Holland JF, Nagel G, Arneault GS. Effects of L-Asparaginase in Acute Myelocytic Leukemia. JAMA (1969) 210(10):1919–21. doi: 10.1001/jama.210.10.1919
88. Wadhwa J, Szydlo RM, Apperley JF, Chase A, Bua M, Marin D, et al. Factors Affecting Duration of Survival After Onset of Blastic Transformation of Chronic Myeloid Leukemia. Blood (2002) 99(7):2304–9. doi: 10.1182/blood.v99.7.2304
89. Naqvi K, Cortes JE, Luthra R, O’Brien S, Wierda W, Borthakur G, et al. Characteristics and Outcome of Chronic Myeloid Leukemia Patients With E255K/V BCR-ABL Kinase Domain Mutations. Int J Hematol (2018) 107(6):689–95. doi: 10.1007/s12185-018-2422-6
90. Meyran D, Petit A, Guilhot J, Suttorp M, Sedlacek P, De Bont E, et al. Lymphoblastic Predominance of Blastic Phase in Children With Chronic Myeloid Leukaemia Treated With Imatinib: A Report From the I-CML-Ped Study. Eur J Cancer (2020) 137:224–34. doi: 10.1016/j.ejca.2020.06.024
91. Morita K, Jabbour E, Ravandi F, Borthakur G, Khoury JD, Hu S, et al. Clinical Outcomes of Patients With Chronic Myeloid Leukemia With Concurrent Core Binding Factor Rearrangement and Philadelphia Chromosome. Clin Lymphoma Myeloma Leuk (2021) 21(5):338–44. doi: 10.1016/j.clml.2020.12.025.Citedin:Pubmed
92. Spiro TE, Mattelaer MA, Efira A, Stryckmans P. Sensitivity of Myeloid Progenitor Cells in Healthy Subjects and Patients With Chronic Myeloid Leukemia to Chemotherapeutic Agents. J Natl Cancer Inst (1981) 66(6):1053–9. doi: 10.1093/jnci/66.6.1053
93. Song P, Ye L, Fan J, Li Y, Zeng X, Wang Z, et al. Asparaginase Induces Apoptosis and Cytoprotective Autophagy in Chronic Myeloid Leukemia Cells. Oncotarget (2015) 6(6):3861–73. doi: 10.18632/oncotarget.2869
94. Mussai F, Egan S, Higginbotham-Jones J, Perry T, Beggs A, Odintsova E, et al. Arginine Dependence of Acute Myeloid Leukemia Blast Proliferation: A Novel Therapeutic Target. Blood (2015) 125(15):2386–96. doi: 10.1182/blood-2014-09-600643
95. Sugimura K, Ohno T, Kusuyama T, Azuma I. High Sensitivity of Human Melanoma Cell Lines to the Growth Inhibitory Activity of Mycoplasmal Arginine Deiminase In Vitro. Melanoma Res (1992) 2(3):191–6. doi: 10.1097/00008390-199209000-00007
96. Wheatley DN. Arginine Deprivation and Metabolomics: Important Aspects of Intermediary Metabolism in Relation to the Differential Sensitivity of Normal and Tumour Cells. Semin Cancer Biol (2005) 15(4):247–53. doi: 10.1016/j.semcancer.2005.04.002
97. Wheatley DN, Kilfeather R, Stitt A, Campbell E. Integrity and Stability of the Citrulline-Arginine Pathway in Normal and Tumour Cell Lines. Cancer Lett (2005) 227(2):141–52. doi: 10.1016/j.canlet.2005.01.004
98. Takaku H, Takase M, Abe S, Hayashi H, Miyazaki K. In Vivo Anti-Tumor Activity of Arginine Deiminase Purified From Mycoplasma Arginini. Int J Cancer (1992) 51(2):244–9. doi: 10.1002/ijc.2910510213
99. Sugimura K, Fukuda S, Wada Y, Taniai M, Suzuki M, Kimura T, et al. Identification and Purification of Arginine Deiminase That Originated From Mycoplasma Arginini. Infect Immun (1990) 58(8):2510–5. doi: 10.1128/IAI.58.8.2510-2515.1990
100. Izzo F, Marra P, Beneduce G, Castello G, Vallone P, De Rosa V, et al. Pegylated Arginine Deiminase Treatment of Patients With Unresectable Hepatocellular Carcinoma: Results From Phase I/II Studies. J Clin Oncol (2004) 22(10):1815–22. doi: 10.1200/JCO.2004.11.120
101. Cheng PN, Leung YC, Lo WH, Tsui SM, Lam KC. Remission of Hepatocellular Carcinoma With Arginine Depletion Induced by Systemic Release of Endogenous Hepatic Arginase Due to Transhepatic Arterial Embolisation, Augmented by High-Dose Insulin: Arginase as a Potential Drug Candidate for Hepatocellular Carcinoma. Cancer Lett (2005) 224(1):67–80. doi: 10.1016/j.canlet.2004.10.050
102. Storr JM, Burton AF. The Effects of Arginine Deficiency on Lymphoma Cells. Br J Cancer (1974) 30(1):50–9. doi: 10.1038/bjc.1974.112
103. Savoca KV, Davis FF, van Es T, McCoy JR, Palczuk NC. Cancer Therapy With Chemically Modified Enzymes. II. The Therapeutic Effectiveness of Arginase, and Arginase Modified by the Covalent Attachment of Polyethylene Glycol, on the Taper Liver Tumor and the L5178Y Murine Leukemia. Cancer Biochem Biophys (1984) 7(3):261–8.
104. Cheng PN, Lam TL, Lam WM, Tsui SM, Cheng AW, Lo WH, et al. Pegylated Recombinant Human Arginase (Rharg-Peg5,000mw) Inhibits the In Vitro and In Vivo Proliferation of Human Hepatocellular Carcinoma Through Arginine Depletion. Cancer Res (2007) 67(1):309–17. doi: 10.1158/0008-5472.CAN-06-1945
105. Lam TL, Wong GK, Chow HY, Chong HC, Chow TL, Kwok SY, et al. Recombinant Human Arginase Inhibits the In Vitro and In Vivo Proliferation of Human Melanoma by Inducing Cell Cycle Arrest and Apoptosis. Pigment Cell Melanoma Res (2011) 24(2):366–76. doi: 10.1111/j.1755-148X.2010.00798.x
106. Zeng X, Li Y, Fan J, Zhao H, Xian Z, Sun Y, et al. Recombinant Human Arginase Induced Caspase-Dependent Apoptosis and Autophagy in non-Hodgkin’s Lymphoma Cells. Cell Death Dis (2013) 4:e840. doi: 10.1038/cddis.2013.359
107. Yau T, Cheng PN, Chan P, Chen L, Yuen J, Pang R, et al. Preliminary Efficacy, Safety, Pharmacokinetics, Pharmacodynamics and Quality of Life Study of Pegylated Recombinant Human Arginase 1 in Patients With Advanced Hepatocellular Carcinoma. Invest New Drugs (2015) 33(2):496–504. doi: 10.1007/s10637-014-0200-8
108. Nasreddine G, El-Sibai M, Abi-Habib RJ. Cytotoxicity of [HuArgI (Co)-PEG5000]-Induced Arginine Deprivation to Ovarian Cancer Cells is Autophagy Dependent. Invest New Drugs (2020) 38(1):10–9. doi: 10.1007/s10637-019-00756-w
109. Hsueh EC, Knebel SM, Lo WH, Leung YC, Cheng PN, Hsueh CT. Deprivation of Arginine by Recombinant Human Arginase in Prostate Cancer Cells. J Hematol Oncol (2012) 5:17. doi: 10.1186/1756-8722-5-17
110. Yau T, Cheng PN, Chan P, Chan W, Chen L, Yuen J, et al. A Phase 1 Dose-Escalating Study of Pegylated Recombinant Human Arginase 1 (Peg-Rharg1) in Patients With Advanced Hepatocellular Carcinoma. Invest New Drugs (2013) 31(1):99–107. doi: 10.1007/s10637-012-9807-9
111. Feun LG, Marini A, Walker G, Elgart G, Moffat F, Rodgers SE, et al. Negative Argininosuccinate Synthetase Expression in Melanoma Tumours may Predict Clinical Benefit From Arginine-Depleting Therapy With Pegylated Arginine Deiminase. Br J Cancer (2012) 106(9):1481–5. doi: 10.1038/bjc.2012.106
112. Tsai HJ, Jiang SS, Hung WC, Borthakur G, Lin SF, Pemmaraju N, et al. A Phase II Study of Arginine Deiminase (ADI-PEG20) in Relapsed/Refractory or Poor-Risk Acute Myeloid Leukemia Patients. Sci Rep (2017) 7(1):11253. doi: 10.1038/s41598-017-10542-4
113. Ott PA, Carvajal RD, Pandit-Taskar N, Jungbluth AA, Hoffman EW, Wu BW, et al. Phase I/II Study of Pegylated Arginine Deiminase (ADI-PEG 20) in Patients With Advanced Melanoma. Invest New Drugs (2013) 31(2):425–34. doi: 10.1007/s10637-012-9862-2
114. Glazer ES, Piccirillo M, Albino V, Di Giacomo R, Palaia R, Mastro AA, et al. Phase II Study of Pegylated Arginine Deiminase for Nonresectable and Metastatic Hepatocellular Carcinoma. J Clin Oncol (2010) 28(13):2220–6. doi: 10.1200/JCO.2009.26.7765
115. De Santo C, Cheng P, Beggs A, Egan S, Bessudo A, Mussai F. Metabolic Therapy With PEG-Arginase Induces a Sustained Complete Remission in Immunotherapy-Resistant Melanoma. J Hematol Oncol (2018) 11(1):68. doi: 10.1186/s13045-018-0612-6
116. Curley SA, Bomalaski JS, Ensor CM, Holtsberg FW, Clark MA. Regression of Hepatocellular Cancer in a Patient Treated With Arginine Deiminase. Hepatogastroenterology (2003) 50(53):1214–6.
117. Tanios R, Bekdash A, Kassab E, Stone E, Georgiou G, Frankel AE, et al. Human Recombinant Arginase I(Co)-PEG5000 [HuArgI(Co)-PEG5000]-Induced Arginine Depletion is Selectively Cytotoxic to Human Acute Myeloid Leukemia Cells. Leuk Res (2013) 37(11):1565–71. doi: 10.1016/j.leukres.2013.08.007
118. Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell (2015) 162(3):540–51. doi: 10.1016/j.cell.2015.07.016
119. Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, et al. Aspartate is a Limiting Metabolite for Cancer Cell Proliferation Under Hypoxia and in Tumours. Nat Cell Biol (2018) 20(7):775–81. doi: 10.1038/s41556-018-0118-z
120. Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, et al. Aspartate is an Endogenous Metabolic Limitation for Tumour Growth. Nat Cell Biol (2018) 20(7):782–8. doi: 10.1038/s41556-018-0125-0
121. Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, et al. Diversion of Aspartate in ASS1-Deficient Tumours Fosters De Novo Pyrimidine Synthesis. Nature (2015) 527(7578):379–83. doi: 10.1038/nature15529.Citedin:Pubmed
122. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell (2015) 162(3):552–63. doi: 10.1016/j.cell.2015.07.017
123. Borst P. The Malate-Aspartate Shuttle (Borst Cycle): How it Started and Developed Into a Major Metabolic Pathway. IUBMB Life (2020) 72(11):2241–59. doi: 10.1002/iub.2367
124. Gaude E, Schmidt C, Gammage PA, Dugourd A, Blacker T, Chew SP, et al. NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine With Glycolysis in Cells With Mitochondrial Dysfunction. Mol Cell (2018) 69(4):581–593 e7. doi: 10.1016/j.molcel.2018.01.034.Citedin:Pubmed
125. Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, et al. Targeting Aspartate Aminotransferase in Breast Cancer. Breast Cancer Res (2008) 10(5):R84. doi: 10.1186/bcr2154
126. Antti H, Sellstedt M. Metabolic Effects of an Aspartate Aminotransferase-Inhibitor on Two T-Cell Lines. PloS One (2018) 13(12):e0208025. doi: 10.1371/journal.pone.0208025
127. Yamada RH, Wakabayashi Y, Iwashima A, Hasegawa T. Inhibition of Aspartate Aminotransferase by Hydrazinosuccinate. Biochim Biophys Acta (1984) 801(1):151–4. doi: 10.1016/0304-4165(84)90224-1
128. Yamada R, Wakabayashi Y, Iwashima A, Hasegawa T. Inhibition of Aspartate Aminotransferase by D-Hydrazinosuccinate: Comparison With L-Hydrazinosuccinate. Biochim Biophys Acta (1986) 871(3):279–84. doi: 10.1016/0167-4838(86)90209-8
129. Yamada R, Wakabayashi Y, Iwashima A, Hasegawa T. In Vivo Inhibition of Aspartate Aminotransferase in Mice by L-Hydrazinosuccinate. Biochim Biophys Acta (1987) 911(3):372–5. doi: 10.1016/0167-4838(87)90080-x
130. Holt MC, Assar Z, Beheshti Zavareh R, Lin L, Anglin J, Mashadova O, et al. Biochemical Characterization and Structure-Based Mutational Analysis Provide Insight Into the Binding and Mechanism of Action of Novel Aspartate Aminotransferase Inhibitors. Biochemistry (2018) 57(47):6604–14. doi: 10.1021/acs.biochem.8b00914
131. Yoo HC, Yu YC, Sung Y, Han JM. Glutamine Reliance in Cell Metabolism. Exp Mol Med (2020) 52(9):1496–516. doi: 10.1038/s12276-020-00504-8
132. Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, et al. Targeted Inhibition of Tumor-Specific Glutaminase Diminishes Cell-Autonomous Tumorigenesis. J Clin Invest. (2015) 125(6):2293–306. doi: 10.1172/JCI75836
133. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol Cancer Ther (2014) 13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870
134. Boysen G, Jamshidi-Parsian A, Davis MA, Siegel ER, Simecka CM, Kore RA, et al. Glutaminase Inhibitor CB-839 Increases Radiation Sensitivity of Lung Tumor Cells and Human Lung Tumor Xenografts in Mice. Int J Radiat Biol (2019) 95(4):436–42. doi: 10.1080/09553002.2018.1558299
135. Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming With Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res (2019) 79(13):3251–67. doi: 10.1158/0008-5472.CAN-18-3527
136. Peterse EFP, Niessen B, Addie RD, de Jong Y, Cleven AHG, Kruisselbrink AB, et al. Targeting Glutaminolysis in Chondrosarcoma in Context of the IDH1/2 Mutation. Br J Cancer (2018) 118(8):1074–83. doi: 10.1038/s41416-018-0050-9
137. Qie S, Yoshida A, Parnham S, Oleinik N, Beeson GC, Beeson CC, et al. Targeting Glutamine-Addiction and Overcoming CDK4/6 Inhibitor Resistance in Human Esophageal Squamous Cell Carcinoma. Nat Commun (2019) 10(1):1296. doi: 10.1038/s41467-019-09179-w
138. Jin H, Wang S, Zaal EA, Wang C, Wu H, Bosma A, et al. A Powerful Drug Combination Strategy Targeting Glutamine Addiction for the Treatment of Human Liver Cancer. Elife (2020) 9. doi: 10.7554/eLife.56749.Citedin:Pubmed
139. Guerra V, Dinardo CD, Konopleva M, Burger JA, Borthakur G, Jabbour E, et al. Interim Results From a Phase Ib/II Clinical Study of the Glutaminase Inhibitor Telaglenastat (CB-839) in Combination With Azacitidine in Patients With Advanced Myelodysplastic Syndrome (MDS). J Clin Oncol (2019) 37(15_suppl):7037–7. doi: 10.1200/JCO.2019.37.15_suppl.7037
140. Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, et al. Targeting Mitochondrial Glutaminase Activity Inhibits Oncogenic Transformation. Cancer Cell (2010) 18(3):207–19. doi: 10.1016/j.ccr.2010.08.009
141. Yuan L, Sheng X, Clark LH, Zhang L, Guo H, Jones HM, et al. Glutaminase Inhibitor Compound 968 Inhibits Cell Proliferation and Sensitizes Paclitaxel in Ovarian Cancer. Am J Transl Res (2016) 8(10):4265–77.
142. Katt WP, Antonyak MA, Cerione RA. Simultaneously Targeting Tissue Transglutaminase and Kidney Type Glutaminase Sensitizes Cancer Cells to Acid Toxicity and Offers New Opportunities for Therapeutic Intervention. Mol Pharm (2015) 12(1):46–55. doi: 10.1021/mp500405h
143. Kalhan SC, Hanson RW. Resurgence of Serine: An Often Neglected But Indispensable Amino Acid. J Biol Chem (2012) 287(24):19786–91. doi: 10.1074/jbc.R112.357194
144. Serpa J. Cysteine as a Carbon Source, a Hot Spot in Cancer Cells Survival. Front Oncol (2020) 10:947. doi: 10.3389/fonc.2020.00947
145. Maddocks ODK, Athineos D, Cheung EC, Lee P, Zhang T, van den Broek NJF, et al. Modulating the Therapeutic Response of Tumours to Dietary Serine and Glycine Starvation. Nature (2017) 544(7650):372–6. doi: 10.1038/nature22056
146. Maddocks OD, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, et al. Serine Starvation Induces Stress and P53-Dependent Metabolic Remodelling in Cancer Cells. Nature (2013) 493(7433):542–6. doi: 10.1038/nature11743
147. Tavana O, Gu W. The Hunger Games: P53 Regulates Metabolism Upon Serine Starvation. Cell Metab (2013) 17(2):159–61. doi: 10.1016/j.cmet.2013.01.012
148. Tajan M, Hennequart M, Cheung EC, Zani F, Hock AK, Legrave N, et al. Serine Synthesis Pathway Inhibition Cooperates With Dietary Serine and Glycine Limitation for Cancer Therapy. Nat Commun (2021) 12(1):366. doi: 10.1038/s41467-020-20223-y
149. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, et al. Metabolite Profiling Identifies a Key Role for Glycine in Rapid Cancer Cell Proliferation. Science (2012) 336(6084):1040–4. doi: 10.1126/science.1218595
150. Combs JA, DeNicola GM. The Non-Essential Amino Acid Cysteine Becomes Essential for Tumor Proliferation and Survival. Cancers (Basel) (2019) 11(5):678. doi: 10.3390/cancers11050678.Citedin:Pubmed
151. Nguyen CH, Gluxam T, Schlerka A, Bauer K, Grandits AM, Hackl H, et al. SOCS2 is Part of a Highly Prognostic 4-Gene Signature in AML and Promotes Disease Aggressiveness. Sci Rep (2019) 249(1):9139. doi: 10.1038/s41598-019-45579-0
152. Bjelosevic S, Gruber E, Newbold A, Shembrey C, Devlin JR, Hogg SJ, et al. Serine Biosynthesis is a Metabolic Vulnerability in FLT3-ITD-Driven Acute Myeloid Leukaemia. Cancer Discov (2021) 11(6):1582–99. doi: 10.1158/2159-8290.CD-20-0738.Citedin:Pubmed
153. Yang B, Wang C, Xie Y, Xu L, Wu X, Wu D. Monitoring Tyrosine Kinase Inhibitor Therapeutic Responses With a Panel of Metabolic Biomarkers in Chronic Myeloid Leukemia Patients. Cancer Sci (2018) 109(3):777–84. doi: 10.1111/cas.13500
154. Karlikova R, Siroka J, Friedecky D, Faber E, Hrda M, Micova K, et al. Metabolite Profiling of the Plasma and Leukocytes of Chronic Myeloid Leukemia Patients. J Proteome Res (2016) 15(9):3158–66. doi: 10.1021/acs.jproteome.6b00356
155. Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, et al. Cancer Progression by Reprogrammed BCAA Metabolism in Myeloid Leukaemia. Nature (2017) 545(7655):500–4. doi: 10.1038/nature22314
156. Polet F, Corbet C, Pinto A, Rubio LI, Martherus R, Bol V, et al. Reducing the Serine Availability Complements the Inhibition of the Glutamine Metabolism to Block Leukemia Cell Growth. Oncotarget (2016) 7(2):1765–76. doi: 10.18632/oncotarget.6426
157. Regan JD, Vodopick N, Takeda S, Lee WH, Faulcon FM. Serine Requirement in Leukemic and Normal Blood Cells. Science (1969) 163(3874):1452–3. doi: 10.1126/science.163.3874.1452
158. Onuma T, Waligunda J, Holland JF. Amino Acid Requirements In Vitro of Human Leukemic Cells. Cancer Res (1971) 31(11):1640–4.
159. Wang D, Yang H, Zhang Y, Hu R, Hu D, Wang Q, et al. Inhibition of Cystathionine Beta-Synthase Promotes Apoptosis and Reduces Cell Proliferation in Chronic Myeloid Leukemia. Signal Transduct Target Ther (2021) 6(1):52. doi: 10.1038/s41392-020-00410-5
160. Kardos GR, Wastyk HC, Robertson GP. Disruption of Proline Synthesis in Melanoma Inhibits Protein Production Mediated by the GCN2 Pathway. Mol Cancer Res (2015) 13(10):1408. doi: 10.1158/1541-7786.MCR-15-0048
161. Loayza-Puch F, Rooijers K, Buil LCM, Zijlstra J, F. Oude Vrielink J, Lopes R, et al. Tumour-Specific Proline Vulnerability Uncovered by Differential Ribosome Codon Reading. Nature (2016) 530(7591):490–4. doi: 10.1038/nature16982
162. Ding J, Kuo M-L, Su L, Xue L, Luh F, Zhang H, et al. Human Mitochondrial Pyrroline-5-Carboxylate Reductase 1 Promotes Invasiveness and Impacts Survival in Breast Cancers. Carcinogenesis (2017) 38(5):519–31. doi: 10.1093/carcin/bgx022
163. Christensen EM, Bogner AN, Vandekeere A, Tam GS, Patel SM, Becker DF, et al. In Crystallo Screening for Proline Analog Inhibitors of the Proline Cycle Enzyme PYCR1. J Biol Chem (2020) 295(52):18316–27. doi: 10.1074/jbc.RA120.016106
164. Milne K, Sun J, Zaal EA, Mowat J, Celie PHN, Fish A, et al. A Fragment-Like Approach to PYCR1 Inhibition. Bioorg Med Chem Lett (2019) 29(18):2626–31. doi: 10.1016/j.bmcl.2019.07.047
165. Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell (2019) 35(2):333–5. doi: 10.1016/j.ccell.2019.01.013
166. Stockard B, Garrett T, Guingab-Cagmat J, Meshinchi S, Lamba J. Distinct Metabolic Features Differentiating FLT3-ITD AML From FLT3-WT Childhood Acute Myeloid Leukemia. Sci Rep (2018) 8(1):5534. doi: 10.1038/s41598-018-23863-9
167. Fenouille N, Bassil CF, Ben-Sahra I, Benajiba L, Alexe G, Ramos A, et al. The Creatine Kinase Pathway is a Metabolic Vulnerability in EVI1-Positive Acute Myeloid Leukemia. Nat Med (2017) 23(3):301–13. doi: 10.1038/nm.4283
168. Felig P, Pozefsky T, Marliss E, Cahill GF Jr. Alanine: Key Role in Gluconeogenesis. Science (1970) 167(3920):1003–4. doi: 10.1126/science.167.3920.1003
169. Hewton KG, Johal AS, Parker SJ. Transporters at the Interface Between Cytosolic and Mitochondrial Amino Acid Metabolism. Metabolites (2021) 11(2):112. doi: 10.3390/metabo11020112.Citedin:Pubmed
170. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic Stellate Cells Support Tumour Metabolism Through Autophagic Alanine Secretion. Nature (2016) 536(7617):479–83. doi: 10.1038/nature19084
171. Xu R, Yang J, Ren B, Wang H, Yang G, Chen Y, et al. Reprogramming of Amino Acid Metabolism in Pancreatic Cancer: Recent Advances and Therapeutic Strategies. Front Oncol (2020) 10:572722. doi: 10.3389/fonc.2020.572722
172. Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, et al. Breast Cancer Cells Rely on Environmental Pyruvate to Shape the Metastatic Niche. Nature (2019) 568(7750):117–21. doi: 10.1038/s41586-019-0977-x
173. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial Metabolism and ROS Generation are Essential for Kras-Mediated Tumorigenicity. Proc Natl Acad Sci U S A (2010) 107(19):8788–93. doi: 10.1073/pnas.1003428107
174. Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 Pathway is Critical for Tumour Cell Survival and Proliferation in Response to Nutrient Deprivation. EMBO J (2010) 29(12):2082–96. doi: 10.1038/emboj.2010.81
175. Liu K, Sutter BM, Tu BP. Autophagy Sustains Glutamate and Aspartate Synthesis in Saccharomyces Cerevisiae During Nitrogen Starvation. Nat Commun (2021) 12(1):57. doi: 10.1038/s41467-020-20253-6
176. Hinnebusch AG. Translational Regulation of GCN4 and the General Amino Acid Control of Yeast. Annu Rev Microbiol (2005) 59:407–50. doi: 10.1146/annurev.micro.59.031805.133833
177. Chantranupong L, Wolfson RL, Sabatini DM. Nutrient-Sensing Mechanisms Across Evolution. Cell (2015) 161(1):67–83. doi: 10.1016/j.cell.2015.02.041
178. Carroll B, Korolchuk VI, Sarkar S. Amino Acids and Autophagy: Cross-Talk and Co-Operation to Control Cellular Homeostasis. Amino Acids (2015) 47(10):2065–88. doi: 10.1007/s00726-014-1775-2
179. Kilberg MS, Shan J, Su N. ATF4-Dependent Transcription Mediates Signaling of Amino Acid Limitation. Trends Endocrinol Metab (2009) 20(9):436–43. doi: 10.1016/j.tem.2009.05.008
180. Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG. Tight Binding of the Phosphorylated Alpha Subunit of Initiation Factor 2 (Eif2alpha) to the Regulatory Subunits of Guanine Nucleotide Exchange Factor Eif2b is Required for Inhibition of Translation Initiation. Mol Cell Biol (2001) 21(15):5018–30. doi: 10.1128/MCB.21.15.5018-5030.2001
181. Hinnebusch AG, Ivanov IP, Sonenberg N. Translational Control by 5’-Untranslated Regions of Eukaryotic mRNAs. Science (2016) 352(6292):1413–6. doi: 10.1126/science.aad9868
182. Chikashige Y, Kato H, Thornton M, Pepper W, Hilgers M, Cecil A, et al. Gcn2 Eif2alpha Kinase Mediates Combinatorial Translational Regulation Through Nucleotide Motifs and uORFs in Target mRNAs. Nucleic Acids Res (2020) 48(16):8977–92. doi: 10.1093/nar/gkaa608
183. Leppek K, Das R, Barna M. Functional 5’ UTR mRNA Structures in Eukaryotic Translation Regulation and How to Find Them. Nat Rev Mol Cell Biol (2018) 19(3):158–74. doi: 10.1038/nrm.2017.103
184. Lindqvist LM, Tandoc K, Topisirovic I, Furic L. Cross-Talk Between Protein Synthesis, Energy Metabolism and Autophagy in Cancer. Curr Opin Genet Dev (2018) 48:104–11. doi: 10.1016/j.gde.2017.11.003
185. B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The Eif2alpha/ATF4 Pathway is Essential for Stress-Induced Autophagy Gene Expression. Nucleic Acids Res (2013) 41(16):7683–99. doi: 10.1093/nar/gkt563
186. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, et al. An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol Cell (2003) 11(3):619–33. doi: 10.1016/s1097-2765(03)00105-9
187. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
188. Shimobayashi M, Hall MN. Multiple Amino Acid Sensing Inputs to Mtorc1. Cell Res (2016) 26(1):7–20. doi: 10.1038/cr.2015.146
189. Kitada M, Xu J, Ogura Y, Monno I, Koya D. Mechanism of Activation of Mechanistic Target of Rapamycin Complex 1 by Methionine. Front Cell Dev Biol (2020) 8:715. doi: 10.3389/fcell.2020.00715
190. Wolfson RL, Sabatini DM. The Dawn of the Age of Amino Acid Sensors for the Mtorc1 Pathway. Cell Metab (2017) 26(2):301–9. doi: 10.1016/j.cmet.2017.07.001
191. Takahara T, Amemiya Y, Sugiyama R, Maki M, Shibata H. Amino Acid-Dependent Control of Mtorc1 Signaling: A Variety of Regulatory Modes. J BioMed Sci (2020) 27(1):87. doi: 10.1186/s12929-020-00679-2
192. Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mTOR for Cancer Therapy. J Hematol Oncol (2019) 12(1):71. doi: 10.1186/s13045-019-0754-1
193. Musa J, Orth MF, Dallmayer M, Baldauf M, Pardo C, Rotblat B, et al. Eukaryotic Initiation Factor 4E-Binding Protein 1 (4E-BP1): A Master Regulator of mRNA Translation Involved in Tumorigenesis. Oncogene (2016) 35(36):4675–88. doi: 10.1038/onc.2015.515
194. Martelli AM, Evangelisti C, Chappell W, Abrams SL, Basecke J, Stivala F, et al. Targeting the Translational Apparatus to Improve Leukemia Therapy: Roles of the PI3K/PTEN/Akt/mTOR Pathway. Leukemia (2011) 25(7):1064–79. doi: 10.1038/leu.2011.46
195. Moschetta M, Reale A, Marasco C, Vacca A, Carratu MR. Therapeutic Targeting of the mTOR-Signalling Pathway in Cancer: Benefits and Limitations. Br J Pharmacol (2014) 171(16):3801–13. doi: 10.1111/bph.12749
196. Mugume Y, Kazibwe Z, Bassham DC. Target of Rapamycin in Control of Autophagy: Puppet Master and Signal Integrator. Int J Mol Sci (2020) 21(21):8259. doi: 10.3390/ijms21218259.Citedin:Pubmed
197. Dossou AS, Basu A. The Emerging Roles of Mtorc1 in Macromanaging Autophagy. Cancers (Basel) (2019) 11(10):1422. doi: 10.3390/cancers11101422.Citedin:Pubmed
198. Rabanal-Ruiz Y, Korolchuk VI. Mtorc1 and Nutrient Homeostasis: The Central Role of the Lysosome. Int J Mol Sci (2018) 19(3):818. doi: 10.3390/ijms19030818.Citedin:Pubmed
199. Rabanal-Ruiz Y, Otten EG, Korolchuk VI. Mtorc1 as the Main Gateway to Autophagy. Essays Biochem (2017) 61(6):565–84. doi: 10.1042/EBC20170027
200. Noda T. Regulation of Autophagy Through TORC1 and Mtorc1. Biomolecules (2017) 7(3):52. doi: 10.3390/biom7030052
201. Dennis MD, McGhee NK, Jefferson LS, Kimball SR. Regulated in DNA Damage and Development 1 (REDD1) Promotes Cell Survival During Serum Deprivation by Sustaining Repression of Signaling Through the Mechanistic Target of Rapamycin in Complex 1 (Mtorc1). Cell Signal (2013) 25(12):2709–16. doi: 10.1016/j.cellsig.2013.08.038
202. Minami K, Tambe Y, Watanabe R, Isono T, Haneda M, Isobe K, et al. Suppression of Viral Replication by Stress-Inducible GADD34 Protein via the Mammalian Serine/Threonine Protein Kinase mTOR Pathway. J Virol (2007) 81(20):11106–15. doi: 10.1128/JVI.01063-07
203. Averous J, Lambert-Langlais S, Mesclon F, Carraro V, Parry L, Jousse C, et al. GCN2 Contributes to Mtorc1 Inhibition by Leucine Deprivation Through an ATF4 Independent Mechanism. Sci Rep (2016) 6:27698. doi: 10.1038/srep27698
204. Fukuda T, Sofyantoro F, Tai YT, Chia KH, Matsuda T, Murase T, et al. Tripartite Suppression of Fission Yeast TORC1 Signaling by the GATOR1-Sea3 Complex, the TSC Complex, and Gcn2 Kinase. Elife (2021) 10. doi: 10.7554/eLife.60969.Citedin:Pubmed
205. Ye J, Palm W, Peng M, King B, Lindsten T, Li MO, et al. GCN2 Sustains Mtorc1 Suppression Upon Amino Acid Deprivation by Inducing Sestrin2. Genes Dev (2015) 29(22):2331–6. doi: 10.1101/gad.269324.115
206. Nikonorova IA, Mirek ET, Signore CC, Goudie MP, Wek RC, Anthony TG. Time-Resolved Analysis of Amino Acid Stress Identifies Eif2 Phosphorylation as Necessary to Inhibit Mtorc1 Activity in Liver. J Biol Chem (2018) 293(14):5005–15. doi: 10.1074/jbc.RA117.001625
207. Wengrod J, Wang D, Weiss S, Zhong H, Osman I, Gardner LB. Phosphorylation of Eif2alpha Triggered by Mtorc1 Inhibition and PP6C Activation is Required for Autophagy and is Aberrant in PP6C-Mutated Melanoma. Sci Signal (2015) 8(367):ra27. doi: 10.1126/scisignal.aaa0899.Citedin:Pubmed
208. Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, et al. Inhibition of GCN2 Sensitizes ASNS-Low Cancer Cells to Asparaginase by Disrupting the Amino Acid Response. Proc Natl Acad Sci U S A (2018) 115(33):E7776–85. doi: 10.1073/pnas.1805523115
209. Broer A, Gauthier-Coles G, Rahimi F, van Geldermalsen M, Dorsch D, Wegener A, et al. Ablation of the ASCT2 (SLC1A5) Gene Encoding a Neutral Amino Acid Transporter Reveals Transporter Plasticity and Redundancy in Cancer Cells. J Biol Chem (2019) 294(11):4012–26. doi: 10.1074/jbc.RA118.006378
210. Bohlen J, Harbrecht L, Blanco S, Clemm von Hohenberg K, Fenzl K, Kramer G, et al. DENR Promotes Translation Reinitiation via Ribosome Recycling to Drive Expression of Oncogenes Including ATF4. Nat Commun (2020) 11(1):4676. doi: 10.1038/s41467-020-18452-2
211. Parzych K, Saavedra-Garcia P, Valbuena GN, Al-Sadah HA, Robinson ME, Penfold L, et al. The Coordinated Action of VCP/p97 and GCN2 Regulates Cancer Cell Metabolism and Proteostasis During Nutrient Limitation. Oncogene (2019) 38(17):3216–31. doi: 10.1038/s41388-018-0651-z
212. Kato Y, Kunimasa K, Takahashi M, Harada A, Nagasawa I, Osawa M, et al. GZD824 Inhibits GCN2 and Sensitizes Cancer Cells to Amino Acid Starvation Stress. Mol Pharmacol (2020) 98(6):669–76. doi: 10.1124/molpharm.120.000070
213. Wang SF, Chen MS, Chou YC, Ueng YF, Yin PH, Yeh TS, et al. Mitochondrial Dysfunction Enhances Cisplatin Resistance in Human Gastric Cancer Cells via the ROS-Activated GCN2-Eif2alpha-ATF4-xCT Pathway. Oncotarget (2016) 7(45):74132–51. doi: 10.18632/oncotarget.12356
214. Longchamp A, Mirabella T, Arduini A, MacArthur MR, Das A, Trevino-Villarreal JH, et al. Amino Acid Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell (2018) 173(1):117–129 e14. doi: 10.1016/j.cell.2018.03.001.Citedin:Pubmed
215. Koppula P, Zhang Y, Zhuang L, Gan B. Amino Acid Transporter SLC7A11/xCT at the Crossroads of Regulating Redox Homeostasis and Nutrient Dependency of Cancer. Cancer Commun (Lond) (2018) 38(1):12. doi: 10.1186/s40880-018-0288-x
216. Gremke N, Polo P, Dort A, Schneikert J, Elmshauser S, Brehm C, et al. mTOR-Mediated Cancer Drug Resistance Suppresses Autophagy and Generates a Druggable Metabolic Vulnerability. Nat Commun (2020) 11(1):4684. doi: 10.1038/s41467-020-18504-7
217. Wang Z, Xie Q, Zhou H, Zhang M, Shen J, Ju D. Amino Acid Degrading Enzymes and Autophagy in Cancer Therapy. Front Pharmacol (2020) 11:582587. doi: 10.3389/fphar.2020.582587
218. Takahashi H, Inoue J, Sakaguchi K, Takagi M, Mizutani S, Inazawa J. Autophagy is Required for Cell Survival Under L-Asparaginase-Induced Metabolic Stress in Acute Lymphoblastic Leukemia Cells. Oncogene (2017) 36(30):4267–76. doi: 10.1038/onc.2017.59
219. Polak R, Bierings MB, van der Leije CS, Sanders MA, Roovers O, Marchante JRM, et al. Autophagy Inhibition as a Potential Future Targeted Therapy for ETV6-RUNX1-Driven B-Cell Precursor Acute Lymphoblastic Leukemia. Haematologica (2019) 104(4):738–48. doi: 10.3324/haematol.2018.193631
220. Chen Q, Ye L, Fan J, Zhang X, Wang H, Liao S, et al. Autophagy Suppression Potentiates the Anti-Glioblastoma Effect of Asparaginase In Vitro and In Vivo. Oncotarget (2017) 8(53):91052–66. doi: 10.18632/oncotarget.19409.Citedin:Pubmed
221. Ji Y, Li L, Tao Q, Zhang X, Luan J, Zhao S, et al. Deprivation of Asparagine Triggers Cytoprotective Autophagy in Laryngeal Squamous Cell Carcinoma. Appl Microbiol Biotechnol (2017) 101(12):4951–61. doi: 10.1007/s00253-017-8221-9
222. Zhang B, Fan J, Zhang X, Shen W, Cao Z, Yang P, et al. Targeting Asparagine and Autophagy for Pulmonary Adenocarcinoma Therapy. Appl Microbiol Biotechnol (2016) 100(21):9145–61. doi: 10.1007/s00253-016-7640-3
223. Delage B, Luong P, Maharaj L, O’Riain C, Syed N, Crook T, et al. Promoter Methylation of Argininosuccinate Synthetase-1 Sensitises Lymphomas to Arginine Deiminase Treatment, Autophagy and Caspase-Dependent Apoptosis. Cell Death Dis (2012) 3:e342. doi: 10.1038/cddis.2012.83
224. Savaraj N, You M, Wu C, Wangpaichitr M, Kuo MT, Feun LG. Arginine Deprivation, Autophagy, Apoptosis (AAA) for the Treatment of Melanoma. Curr Mol Med (2010) 10(4):405–12. doi: 10.2174/156652410791316995
225. Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, et al. Arginine Deiminase as a Novel Therapy for Prostate Cancer Induces Autophagy and Caspase-Independent Apoptosis. Cancer Res (2009) 69(2):700–8. doi: 10.1158/0008-5472.CAN-08-3157
226. Wang Z, Shi X, Li Y, Fan J, Zeng X, Xian Z, et al. Blocking Autophagy Enhanced Cytotoxicity Induced by Recombinant Human Arginase in Triple-Negative Breast Cancer Cells. Cell Death Dis (2014) 5:e1563. doi: 10.1038/cddis.2014.503
227. Li Y, Zeng X, Wang S, Fan J, Wang Z, Song P, et al. Blocking Autophagy Enhanced Leukemia Cell Death Induced by Recombinant Human Arginase. Tumour Biol (2016) 37(5):6627–35. doi: 10.1007/s13277-015-4253-x
228. Lin C, Wang Z, Li L, He Y, Fan J, Liu Z, et al. The Role of Autophagy in the Cytotoxicity Induced by Recombinant Human Arginase in Laryngeal Squamous Cell Carcinoma. Appl Microbiol Biotechnol (2015) 99(20):8487–94. doi: 10.1007/s00253-015-6565-6
229. Shen W, Zhang X, Fu X, Fan J, Luan J, Cao Z, et al. A Novel and Promising Therapeutic Approach for NSCLC: Recombinant Human Arginase Alone or Combined With Autophagy Inhibitor. Cell Death Dis (2017) 8(3):e2720. doi: 10.1038/cddis.2017.137
230. Li J, Song P, Zhu L, Aziz N, Zhou Q, Zhang Y, et al. Synthetic Lethality of Glutaminolysis Inhibition, Autophagy Inactivation and Asparagine Depletion in Colon Cancer. Oncotarget (2017) 8(26):42664–72. doi: 10.18632/oncotarget.16844
231. Kandasamy P, Gyimesi G, Kanai Y, Hediger MA. Amino Acid Transporters Revisited: New Views in Health and Disease. Trends Biochem Sci (2018) 43(10):752–89. doi: 10.1016/j.tibs.2018.05.003
232. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of Amino Acids in Cancer. Front Cell Dev Biol (2020) 8:603837. doi: 10.3389/fcell.2020.603837
233. Lopes C, Pereira C, Medeiros R. ASCT2 and LAT1 Contribution to the Hallmarks of Cancer: From a Molecular Perspective to Clinical Translation. Cancers (Basel) (2021) 13(2):203. doi: 10.3390/cancers13020203.Citedin:Pubmed
234. Ganapathy V, Thangaraju M, Prasad PD. Nutrient Transporters in Cancer: Relevance to Warburg Hypothesis and Beyond. Pharmacol Ther (2009) 121(1):29–40. doi: 10.1016/j.pharmthera.2008.09.005
235. Lin W, Wang C, Liu G, Bi C, Wang X, Zhou Q, et al. SLC7A11/xCT in Cancer: Biological Functions and Therapeutic Implications. Am J Cancer Res (2020) 10(10):3106–26.
Keywords: non-essential amino acid, GCN2, general control non-derepressible 2, mTORC1, myeloid leukemias
Citation: Bhingarkar A, Vangapandu HV, Rathod S, Hoshitsuki K and Fernandez CA (2021) Amino Acid Metabolic Vulnerabilities in Acute and Chronic Myeloid Leukemias. Front. Oncol. 11:694526. doi: 10.3389/fonc.2021.694526
Received: 13 April 2021; Accepted: 15 June 2021;
Published: 01 July 2021.
Edited by:
Qiong Yang, Beijing Normal University, ChinaReviewed by:
Hussein A. Abbas, MD Anderson Cancer Center, United StatesDing Xinchun, Purdue University Indianapolis, United States
Copyright © 2021 Bhingarkar, Vangapandu, Rathod, Hoshitsuki and Fernandez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian A. Fernandez, Y2hmNjNAcGl0dC5lZHU=