 Martina Addeo1,2†
Martina Addeo1,2† Giuseppina Di Paola1†
Giuseppina Di Paola1† Henu Kumar Verma1,3
Henu Kumar Verma1,3 Simona Laurino4
Simona Laurino4 Sabino Russi4
Sabino Russi4 Pietro Zoppoli4
Pietro Zoppoli4 Geppino Falco1,2,3,4*
Geppino Falco1,2,3,4* Pellegrino Mazzone1*
Pellegrino Mazzone1*- 1Istituto di Ricerche Genetiche Gaetano Salvatore Biogem Scarl, Ariano Irpino, Italy
- 2Department of Biology, University of Naples Federico II, Naples, Italy
- 3IEOS-CNR, Institute of Experimental Endocrinology and Oncology “G. Salvatore” – National Research Council, Naples, Italy
- 4Laboratory of Pre-Clinical and Translational Research, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS)-centro di riferimento oncologico della basilicata (CROB), Referral Cancer Center of Basilicata, Rionero in Vulture, Italy
Gastric cancer (GC) is one of the most widespread causes of cancer-related death worldwide. Recently, emerging implied that gastric cancer stem cells (GCSCs) play an important role in the initiation and progression of GC. This subpopulation comprises cells with several features, such as self-renewal capability, high proliferating rate, and ability to modify their metabolic program, which allow them to resist current anticancer therapies. Metabolic pathway intermediates play a pivotal role in regulating cell differentiation both in tumorigenesis and during normal development. Thus, the dysregulation of both anabolic and catabolic pathways constitutes a significant opportunity to target GCSCs in order to eradicate the tumor progression. In this review, we discuss the current knowledge about metabolic phenotype that supports GCSC proliferation and we overview the compounds that selectively target metabolic intermediates of CSCs that can be used as a strategy in cancer therapy.
Introduction
Gastric cancer (GC) is recognized as the fifth most common cancer in the world (5.7% of all cancers) with a higher prevalence in males compared to females, and it is the fourth leading cause of cancer death (1). Lifestyle variations, especially in dietary habits such as high salt intake, iron depletion and alcohol consumption, along with genetic background, have led to a discrepancy in GC incidence in different regions of the world with Europeans and Latinos less affected than Asians (2–4). The high mortality rate could be accounted by the absence of early-stage symptomatology, by the lack of early diagnosis and poorly effective treatments (5). Gastric cancer is classified as a multifactorial disease that results from a combination of specific genetic alterations such as gene mutations, somatic copy number alterations (sCNAs), epigenetic changes (6–8), and environmental factors (9). The latter has a critical role in the GC onset, with major risk factors being Helicobacter pylori infection (10). At the present, H. pylori infection is a strong risk factor for the adenocarcinoma that arises within the stomach (11), and it was classified as a class I carcinogen by the International Agency for Research on Cancer (12). H. pylori colonizes the gastric mucosa, where it expresses an array of proteins that lead to persistent inflammation (13, 14). All these factors together promote cancer stem cells insurgence even if it is unclear whether this mechanism is due to a somatic cancer cell that acquires stemness feature, or to a normal stem cell which acquires cancer properties.
Hallmarks of Cancer Stem Cells (CSCs)
Cancer stem cells (CSCs) represent a small subpopulation within the tumor that is involved in the initiation and progression of carcinomas (15–17). According to the CSCs theory, the tumor bulk is composed of a plethora of heterogeneous and differentiated cancer cells which is fueled by a rare population of CSCs characterized by self-renewal and differentiation capabilities (18, 19).
The molecular features displayed by CSCs are not universal and many of the genes, discovered as markers of these cells, were first identified in embryonic stem cells (ESCs). The core regulatory network for embryonic stem cell maintenance and self-renewal OCT4, SOX2, KLF4, NANOG, and SALL4 are abnormally expressed in human tumor samples suggesting the presence of cancer stem cells (20). The overexpression of these pluripotency genes in gastric cancer tumor tissues versus the paired adjacent normal tissues positively correlate with tumor size, tumor grade, TNM stage, and shortened overall survival time (21). Zscan4, a transcription factor (TF) firstly identified as exclusive of murine 2-cell embryos (22) and murine ESCs (mESCs) (23), is also been associated with stem cell phenotype in human head and neck squamous cell carcinoma (HNSCC) (24). ZSCAN4 is indeed enriched in HNSCC cells which are able to form tumorspheres, and its overexpression is associated with elevated histone 3 hyperacetylation at NANOG and OCT4 promoters. The TF c-MYC is one of the most studied oncogenes, and originally part of Yamanaka cocktail together with OCT4, SOX2 and KLF4 (OSKM), to reprogram somatic cell to a pluripotent cell (25). The reactivation of MYC in mammary epithelial cells is able to downregulate lineage specific TFs to reprogram the cell to a stem cell-like state favoring tumor initiation and progression (26). As the CSCs could be considered normal stem cells which have lost control over regulation mechanisms, they can also take advantage of other protective mechanisms typical of stem/progenitor cells. The adenosine triphosphate–binding cassette (ABC) transporters are efflux pumps expressed at high level on progenitor cells membrane and are responsible for the protection of the stem cell population from toxic molecules (27). Although the expression of ABC transporters strongly fosters multidrug resistance (MDR) (28), the capacity of CSCs to resist chemotherapeutic treatment is a multifactorial feature achieved by a highly efficient DNA repair machinery that is employed to overcome the DNA damage induced by therapeutic treatment (28) as well as increasing the autophagic process to obtain nutrients necessary to support cell survival (28, 29). Also, upon drug treatment, other molecular pathways may take action in sustaining the survival of CSCs. VEGF/VEGFR-1(Flt) autocrine signaling is activated in a subpopulation of highly tumorigenic cells in response to cisplatin treatment (CDDP) and is characterized by the expression of the pluripotency genes OCT4, NANOG and BMI1 (30). The drug exposure can therefore enhance the tumorigenic potential of the CSCs hence revealing the need to use combined therapies in order to target different molecular pathways and reduce the chance of cancer relapse.
Cancer Stem Cells Metabolism
The metabolic hallmarks of the CSCs have been the subject of intensive investigation in different types of tumors (31, 32). A common feature of CSCs is the reprogramming of cellular metabolism with glycolysis preferred over oxidative phosphorylation (OXPHOS) as the primary source of ATP molecules even in presence of oxygen. This metabolic switch, known as the “Warburg effect” or aerobic glycolysis, provides ready-to-use energy that is essential to meet the need of high energy demand associated with a high proliferative state (33). Although this mechanism is energetically unfavorable, as the amount of ATP generated by glycolysis is lower than the quantity deriving from OXPHOS, CSCs overcome the energy limitation enhancing glucose uptake and upregulating some intermediates of the glycolytic pathway. On the other hand, in normal cells or quiescent somatic cells, mitochondria produce the primary energy through the tricarboxylic acid cycle (TCA) deriving from glucose via glycolysis, or fatty acid via β-oxidation associated with OXPHOS (Figure 1) (35). All these metabolic changes allow the CSCs adaptation to the tumor microenvironment leading to tumor progression, metastases formation and chemo-resistance (36).
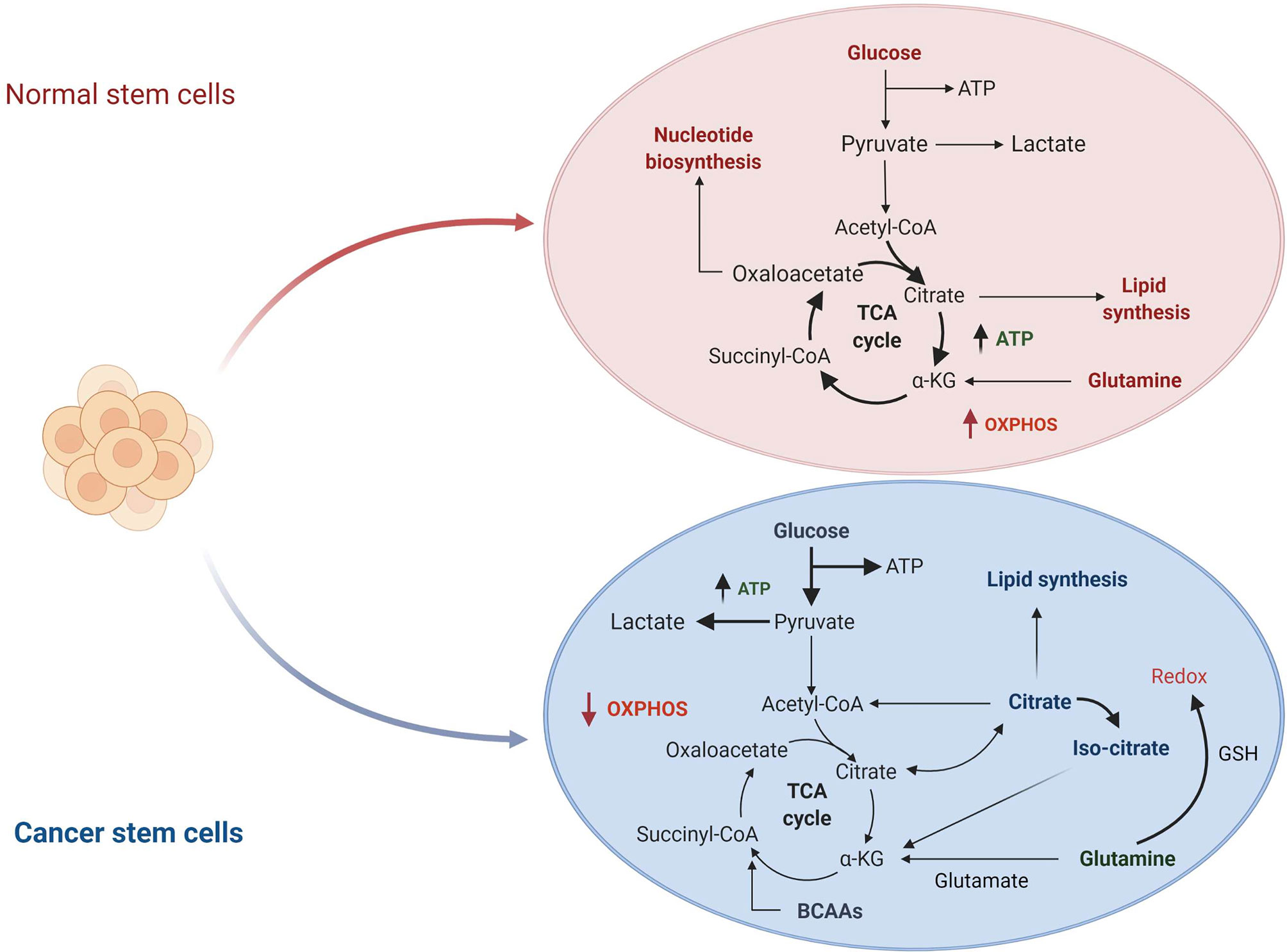
Figure 1 Comparison of normal stem cells versus cancer stem cells metabolism. In normal cells, glucose is converted to pyruvate and then oxidized to Acetyl-CoA in the mitochondrial TCA cycle. Other substrates, including lipids and nucleotides, are produced from these catabolic reactions. The majority of ATP is produced by oxidative phosphorylation (OXPHOS) (not shown in the figure). In cancer cells, glucose uptake and glycolysis are strongly increased. Part of pyruvate is converted to lactate, while the remaining is converted to Acetyl-CoA to enter in the TCA cycle where it is converted to citrate. The produced citrate is secreted from the mitochondria for fatty acid synthesis that are necessary for structural maintenance of the membrane. TCA, tricarboxylic acid cycle; αKG, α-KetoGlutarate; GSH, glutathione; BCAAs, branched-chain amino acids. Figure modified from De Berardinis et al. (34).
In T-cell leukemia the activation of MYC leads to HIF2α induction that is facilitated by the pluripotency factors NANOG and SOX2. This results in a change of redox balance of CSCs as HIF2α negatively regulates p53 activation and positively regulates glutathione (GSH) production (37). Furthermore, several studies reported that an aberrant activation of WNT signaling plays an important role in the metabolic switch from an oxidative metabolism to aerobic glycolysis supporting cancer stem cells. This shift could be partly explained by the production of reactive oxygen species (ROS) as they are capable of altering the self-renewal capacity of cancer stem cells (38). As a matter of fact, WNT has been shown to regulate pyruvate dehydrogenase kinase, PDK1, by phosphorylating and inactivating the pyruvate dehydrogenase (PDH) enzyme complex, responsible for the conversion of pyruvate into Acetyl-CoA (39). A decreased entry of Acetyl-CoA into TCA and oxidative phosphorylation leads to a reduced mitochondrial respiration and, therefore, to lower ROS levels. The role of WNT in supporting the stemness of CSCs was further confirmed by the use of a WNT antagonist, secreted frizzled-related protein 4 (sFRP4). This protein, through its binding to WNT receptor, is able to reduce the CSCs viability even in the presence of a variable concentration of glucose (40).
Moreover, Notch1 signaling plays a role in the regulation of metabolism in CSCs. In particular, upon binding to Jagged1 ligand, Notch1 activates a downstream cascade that, through the interaction with PTEN-induced kinase 1 (PINK1), results in mTORC2/AKT activation (41). In CSCs, mTORC2 regulates different mechanisms through the AKT activation which, in turn, phosphorylates SOX2 and OCT4 and, therefore, it positively contributes to the stemness maintenance. Furthermore, mTORC2 activation is also implied in metabolic regulation as it is involved in the repression of the FoxO3, a transcriptional factor that is responsible for the inhibition of the glycolytic pathway (42).
Besides, Hedgehog signaling (Hh) plays an important role in CSCs sustenance. A recent study describes a role of this pathway in the activation of SOX2, NANOG and OCT4 stemness genes following a stress caused by depletion of folate in colon cancer cells (43). Furthermore, in breast cancer, Hh signaling regulates the ability of stem like cells to generate tumor bulk (44).
The activation of this pathway is further associate with lipid metabolism. Indeed, defects in cholesterol biosynthesis results in Hh signaling arrest in embryonic development (44).
Metabolic Profile of Gastric Cancer Stem Cells (GCSCs)
Gastric cancer stem cells (GCSCs) show a distinct expression of several surface markers. These include: CD44 (cluster of differentiation 44), EpCAM (epithelial cell adhesion molecule), LGR5 (leucine-rich, repeat-containing, G-protein–coupled receptor 5), ALDH1 (aldehyde dehydrogenase 1), CD133 (cluster of differentiation 133), and SOX2 (sex-determining region Y-box 2) (45–47). The positivity to these markers is associated with vascular and lymph node invasion, tumor size and response to chemotherapeutic drugs (48). Interestingly, CSCs metabolism has become an active field of innovative research to target cancer progression (32). Here, we reviewed the current knowledge about GCSCs metabolism and the therapeutic strategies that can be employed to target metabolic pathways.
Glycolytic Metabolism
Glycolysis is the central pathway for glucose catabolism which can occur both in the presence of oxygen (aerobic) and in the absence of oxygen (anaerobic) (49, 50). In a hypoxic microenvironment, GCSCs reprogram their metabolism to adapt to lower oxygen levels through the upregulation of hypoxia-inducible factors (HIFs) (51). These factors regulate the expression of genes involved in glucose uptake and in the glycolytic pathway (52). Among these, GLUT transporters permit glucose entry through the plasma membrane allowing the cells to survive in low oxygen conditions (53, 54). Indeed, Yamada A. et al. reported that high Glut1 expression occurred at an early stage of gastric cancer where it facilitates 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) uptake (55), and it is responsible for gastric cancer progression by activating the AKT pathway (56), which plays a key role in glucose metabolism. However, the employment of specific inhibitors of Glut1 transporters, such as WZB117, has been demonstrated to affect tumor insurgence through the downregulation of stemness-associated genes, such as SOX2, NANOG and BMI1 in different cancers including pancreatic, ovarian and glioblastoma cancer (57). This allows them to be utilized as a possible therapeutic strategy even in gastric cancer treatment. Subsequently to the higher intracellular glucose levels, Hexokinase 2 (HK-2), the first enzyme of the glycolytic pathway, is consistently overexpressed in gastric tumors and is associated with poor survival in patients with digestive system malignancies (58) (Figure 2). Recently, a novel molecular mechanism of metabolic regulation linking a key embryonic stem cells (ESCs) pluripotency factor SALL4 to HK-2 has been described in GC cells (59). Particularly, Shao et al. demonstrated that knockdown of SALL4 results in inhibition of glucose uptake and HK-2 activity. Conversely, SALL4 overexpression promotes cancer metabolic phenotype which can be reversed by HK-2 knockdown suggesting that this glycolytic enzyme is a downstream effector of the transcriptional factor SALL4. Indeed, targeting HK-2 in leukemic cells, with 3-bromopyruvate, leads to the dissociation of HK2 from mitochondrial membrane which is responsible for the enhanced sensitivity to the first-line chemotherapeutic drug (60). This approach could be used as a potential strategy to target GCSCs with high HK-2 activity.
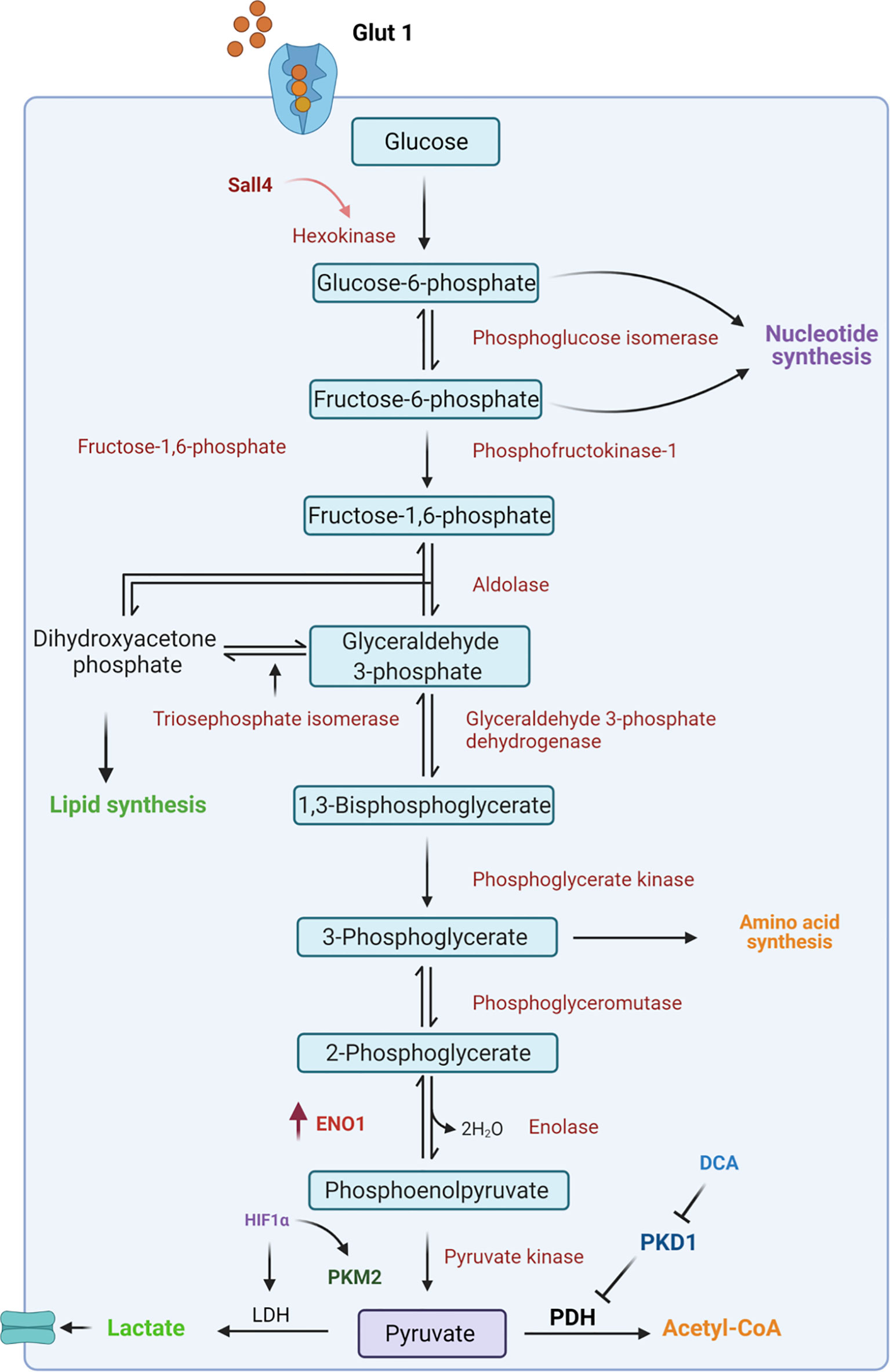
Figure 2 Schematic representation of glycolytic pathway. In CSCs, glucose from the extracellular environment is mainly metabolized through glycolysis. In particular, upregulation of several intermediates of this catabolic pathway, such as Hexokinase 2 and ENO1, leads to an increase in lactate production. Moreover, the inactivation of pyruvate dehydrogenase (PDH) by PDK1 is responsible for failure in the conversion of pyruvate to Acetyl-CoA and this results in increased lactate production. ENO1, Enolase1; HIF1α, hypoxia-inducible factor; LDH, Lactate Dehydrogenase; PKM2, Pyruvate kinase isozyme M2; PDH, Pyruvate dehydrogenase; PDK1, Pyruvate dehydrogenase kinase 1; DCA, dicloroacetate.
Additionally, the overexpression of glycolytic enzyme Enolase 1 (ENO1) regulates stem cell-like characteristics of tumor cells and it is related to poor prognosis of GC (61). ENO1 overexpression promotes cisplatin resistance through a higher glycolytic activity. Moreover, the glycolytic phenotype can be modulated by Pyruvate kinase isozyme M2 (PKM2), an enzyme that has been proposed to be crucial for maintaining cell homeostasis. In GC, GCSCs, expressing CD44 surface marker, show an upregulation of PKM2 upon induction with CagA deriving from H. Pylori infection (62). It has been demonstrated that PKM2 overexpression plays a role in the stabilization of the transcriptional factor NF-κB that, through its binding to Bcl-xL anti-apoptotic protein, promotes GC development and progression (63). Inhibition of PKM2 achieved by shikonin hinders glycolysis in breast cancer cell lines making this compound a good candidate for other tumors (64).
The final step of glycolysis provides pyruvate molecules which can be converted in lactate through lactate dehydrogenase (LDH) or in Acetyl-CoA by pyruvate dehydrogenase (PDH) enzymes. The activity of PDH is negatively regulated by pyruvate dehydrogenase kinase 1 (PDK1) which is responsible for the interruption of the link between glycolysis and the TCA cycle, enhancing the conversion of pyruvate in lactate (65).
Alteration in the glucose metabolism by PDK1 activity is associated with a poor prognosis in gastric cancer. Targeting PDK1 with dichloroacetate (DCA) has been reported to induce metabolic changes that increase the cell sensitivity to chemotherapy (Figure 2) (66). As previously highlighted, although CSCs mostly utilize the glycolytic pathway, part of the energy is still produced by the OXPHOS pathway. To date, several drugs are available to reduce energy production via OXPHOS such as Metformin and Phenformin treatment (67, 68). These compounds inhibit the mitochondrial respiratory complex I delaying cancer growth in vivo and inducing apoptosis of CSCs (68). Furthermore, mitochondrial metabolism can be impaired by targeting mitochondrial protein biosynthesis and maturation. Indeed, treatments with tetracyclines, which hamper ribosomes, and Gamitrinib, an inhibitor of TRAP1 chaperone, result in OXPHOS reduction (69, 70). However, due to the metabolic plasticity of CSCs, dual inhibition of the glycolytic and OXPHOS pathways may represent a promising approach for CSCs targeting and tumor treatment.
Lipid Metabolism
Lipids are essential components of the cell, both structurally and functionally, as they regulate the fluidity of the plasmatic membrane and they are involved in different cellular activities including cell-cell recognition, energy supplies and signaling transduction (71). Indeed, their metabolism is finely regulated by the cell through anabolic or catabolic pathways modulation, according to the availability of carbon source for de novo lipogenesis (44). Although an increase in lipid metabolism is already attributed to stem cells during somatic cell reprogramming (72), several reports describe the lipid metabolism rewiring of CSCs as a mechanism necessary to avoid death under unfavorable conditions (35). Recent studies show that exogenous absorption or endogenous synthesis of lipids plays a primary role in supporting CSCs’ self-renewal during the tumorigenesis process. Indeed, many of the enzymes responsible for lipid synthesis, such as ATP-citrate lyase (ACYL), acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN) are highly up-regulated in cancer (Figure 3) (44, 71). Likewise, inhibition of ATP-citrate lyase, the enzyme responsible for the conversion of cytosolic citrate to acetyl-CoA results in the downregulation of the transcriptional factor Snail, a key regulator of stemness phenotype in cancer stem cells (73). In particular, FASN, a key lipogenic enzyme that converts Acetyl-CoA and malonyl-CoA to palmitate, is drastically upregulated in many cancers such as breast, colon, lung, bladder, gastric, endometrial, ovary, kidney, pancreatic, head and neck, prostate, brain and melanoma (74–76). Inhibition of FASN activity with cerulenin induces regression in the formation of tumorspheres with a reduction in the expression of stemness markers, such as Nestin and CD133, and an increase in the expression of differentiation markers (77). Furthermore, comparative studies of metabolomic profiles between CSCs and non-tumor counterparts have shown that CSCs support their stemness by synthesizing a greater amount of monounsaturated lipids (MUFAs) (78). Lipid desaturation, or the conversion of saturated lipids into unsaturated lipids, is mediated by specific enzymes, such as stearoyl-CoA desaturase-1 (SCD1). In gastric cancer, SCD1 has found to be upregulated and this results in poor survival rates. Furthermore, this enzyme is able to induce tumorigenesis, drug resistance and metastasis by regulating GCSCs proliferation through the Hippo pathway (79). Hence CSCs, compared to their differentiated tumor counterparts, show an enhancement of lipid synthesis de novo, which is also reflected in the accumulation of intracellular lipids. An important role of lipid droplets (LD) located at the cytoplasmic level in supporting the excessive metabolic demand of CSCs has also been advanced. LD, a source of accumulation of fatty acids in the form of triglycerides, act as a reservoir of energy for the sustenance of CSCs in glucose deprivation condition (80). Moreover, another strategy that CSCs activate in a low glucose condition is represented by the β-oxidation of free fatty acids (44). Thus, Nanog, a stem cell marker, reprograms the metabolism of tumor-initiating stem-like cells (TIC) by repressing the expression of OXPHOS genes and activating the fatty acids oxidation (FAO). This allows tumor-initiating stem-like cells to maintain self-renewal and drug resistance (81). It has been demonstrated that, in GC, mesenchymal stem cells (MSCs) promote the synthesis of MACC1-ASI, a lncRNA that is responsible for FAO-dependent stemness and chemo-resistance insurgence. Furthermore, it has been found that the downregulation of CPT1, the FAO rate-limiting enzyme, reduced stemness and resistance to 5-FU and oxaliplatin. Accordingly, inhibition of CPT1 with Etomoxir allows reversing the resistance that mesenchymal stem cells show to the FOLFOX therapeutic regimen (Figure 3 and Table 1) (82). However, a recent report describes a lipid metabolism shift that occurs when stem cells switch from a quiescent state to a proliferative state (88). This represents a paradox because, in a quiescent condition, cells rely on β-oxidation to produce energy for the cell viability and stemness phenotype maintenance as the citrate is first converted to succinate and, then, to malate. The selective inhibition of this catabolic pathway resulted in a loss of the quiescence state and in the acquisition of a differentiated phenotype (88).
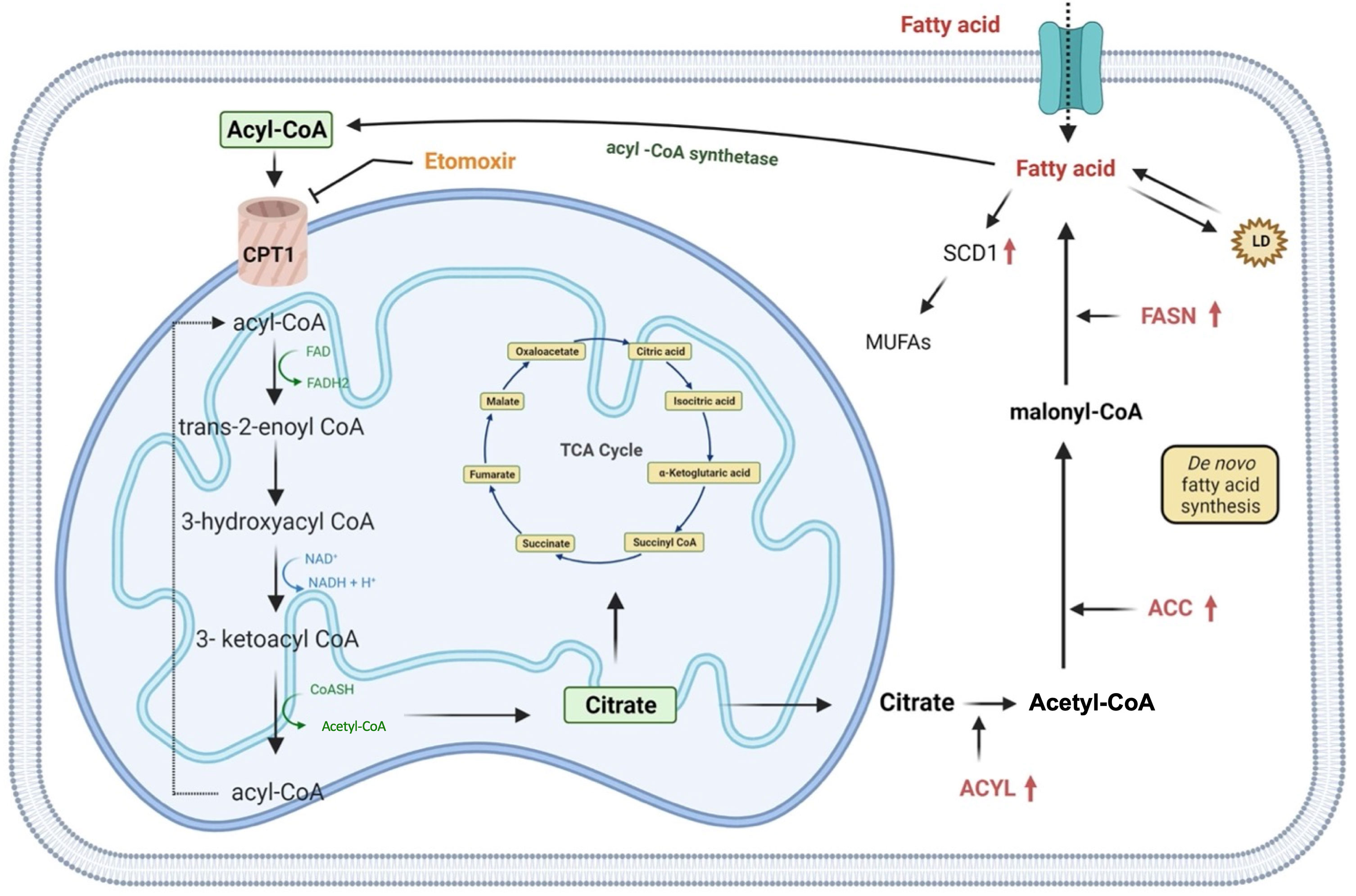
Figure 3 Representation of lipid metabolism. Rewiring of lipid metabolism including increased lipid uptake, lipid desaturation, de novo lipogenesis, lipolysis and FAO is necessary for supply GCSCs with energy. Extracellular FFAs are transported into cells and catabolized in mitochondria to produce acetyl-CoA which is converted in citrate and it enters in TCA cycle for the oxidation. Alternatively, de novo fatty acids synthesis begins with acetyl-CoA that is converted in malonyl-CoA and then in palmitate. Lipid catabolism can even occur through fatty acids esterification that allow them to be stored as triglycerides in lipid droplets. In addition, saturated fatty acids can also be converted into mono-unsaturated fatty acids by SCD1, an enzyme upregulated in gastric cancer. ACC, acetyl-CoA carboxylase; ACYL, ATP citrate lyase; FASN, fatty acid synthase; MUFAs, mono-unsaturated fatty acids; SCD1, stearoyl-CoA desaturase 1; CPT1, carnitine palmitoyltransferase 1; TCA, cycle, tricarboxylic acid cycle; LD, lipid droplet.

Table 1 List of compounds targeting CSCs metabolism.
Along with the metabolism of fatty acids, cholesterol metabolism is also a hallmark of cancer. In particular, an increase in the levels of SREBP2, a transcription factor responsible for cholesterol biosynthesis and homeostasis, is correlated with an increase in the tumor stem cell bulk population (89). De novo lipidic synthesis and the oxidation of fatty acids represent the new metabolic targets to inhibit CSCs self-renewal and identifying new inhibitors of the key players involved could slow down the growth of the tumor mass (90).
Exploring the Role of Glutamine Metabolism
Glutamine metabolism is an essential mechanism that CSCs display to produce macromolecules, such as lipids, proteins and nucleic acids, that are indispensable for proliferation (91). To date, no evidence describes the role of this metabolism in gastric cancer stem cells, however, given its importance as the carbon source in other cancer stem cells, further investigation should be carried out to understand the metabolic function of glutamine in gastric stemness. In particular, in cancer stem cells glutamine is used as a pivotal source of nitrogenous as it represents a good donor of reduced nitrogen for building both purine and pyrimidine bases as well as proteins (91). Furthermore, glutamine acts as a carbon source for the TCA cycle through its conversion to alpha-ketoglutarate (α-KG) (92). Specifically, glutamine is first converted to N-acetyl-glucosamine by glutaminase (GLS), a MYC regulated enzyme, then converted to α-KG by glutamate dehydrogenase (GDH) and then it enters in TCA cycle for energy production. This mechanism is defined as oxidative glutaminolysis (93). Targeting these enzymes could represent a good pharmacological strategy for potential therapy in different types of cancer. Indeed, the use of a specific inhibitor targeting GLS leads to an inhibition of MYC-induced B-cell lymphoma and MYC-induced hepatocarcinoma (83), while, GDH inhibition results in lower α-KG levels and high ROS production, resulting in hindering of cancer cell proliferation and tumor progression (94). Thus, this emerging evidence shows that increasing glutamine metabolism promotes tumor growth despite the regulation of redox homeostasis (84). However, beyond the oxidative glutaminolysis, glutamine can follow a reductive carboxylation pathway. Indeed, in a hypoxic microenvironment, glutamine can also be converted to citrate by the reductive activity of NADP+-dependent isocitrate dehydrogenase 1 (IDH1) leading to Acetyl-CoA production that is used for fatty acid synthesis and it is necessary to guarantee substrates for cell proliferation (91). This makes glutamine metabolism a crucial point for the regulation of survival, proliferation and differentiation of cancer stem cells. Recent studies describe glutamine metabolism as a possible target in cancer therapy. Several compounds are described being able to modulate different intermediates of this metabolic pathway. The first-line strategy to regulate glutamine metabolism could be the inhibition of glutamine transporter ASCT2 with benzylserine to prevent the uptake of this amino acid and its employment in anabolic and catabolic pathways (85). Subsequently, intracellular glutamine is converted into glutamate through glutaminase (GLS) activity; the inhibition of GLS with bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide (BPTES) is associated with DNA replication arrest leading to cell death in the B lymphoma cell line (83). Another targetable step is represented by the conversion of glutamate into α -ketoglutarate acid through the activity of glutamate dehydrogenase 1 (GDH1). Targeting GDH1 with R162 prevents glutamine entry in the TCA cycle increasing ROS levels in several cancer cell lines (84). Also, ketone-bodies are used as a substrate for energy production in cancer stem cells as they are converted in Acetyl-CoA which enters in the TCA cycle providing carbons for energy production. A new class of compound, named “mitoketoscins”, has been found to impair this conversion leading to the inhibition of breast cancer stem cells proliferation. However, the molecular mechanism of mitoketoscins still remains unclear (Figure 4) (86).
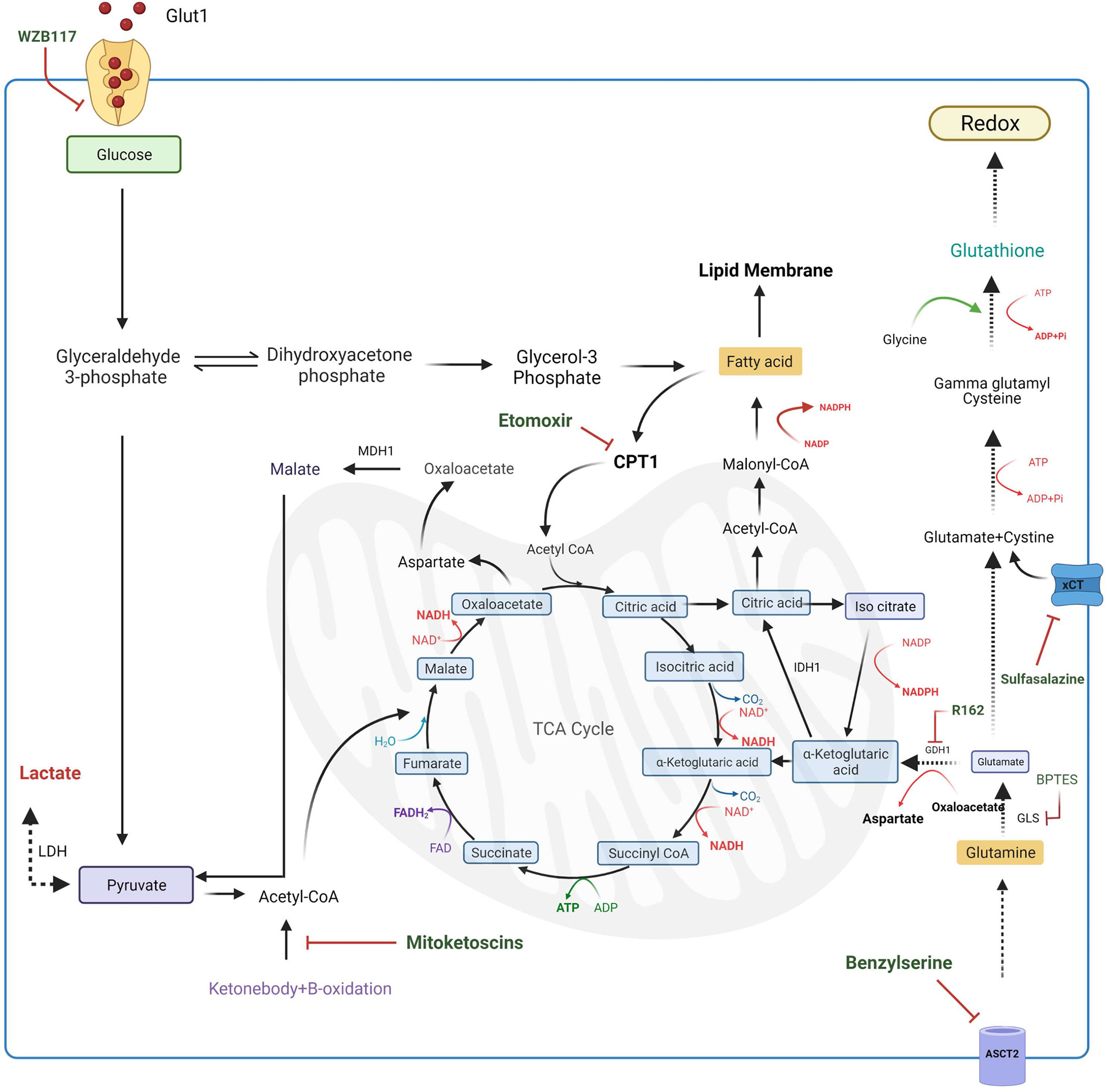
Figure 4 Targeting of metabolic pathways in CSCs. Several compounds inhibit different metabolic intermediates in CSCs. Glut1 uptake can be inhibited by WZB11 treatment resulting in a lower energy production. Also targeting the glutamine metabolism represents a potential strategy to apply in cancer treatment. Indeed, inhibition of ASCT2 transporter with Benzylserine prevents the glutamine uptake while blocking the metabolic intermediates, such as GLS with BTES, hinders glutaminolysis. Moreover, glutamate deriving from processed glutamine, together with intracellular cysteine, can participate in glutathione synthesis that is responsible for low ROS maintenance. Another strategy to increase CSCs death is the reduction of cysteine uptake by inhibition of xCT transporter with Sulfasalazine. CSCs survival can be also hampered by targeting the conversion of ketonebodies into Acetyl-CoA, a substrate necessary for TCA cycle. Moreover, also lipid entering in TCA cycle can be inhibited using selective inhibitors of enzymes involved in lipid metabolism. Indeed, treatment of CSCs with Etomoxir, results in the CPT1 enzyme inactivation resulting in the cytosolic accumulation of lipidic intermediates. TCA, tricarboxylic acid cycle; G6PD, Glucose-6-phosphate dehydrogenase; LDH, lactate dehydrogenase; GLS, glutaminase; GDH1, glutamate dehydrogenase 1; ASCT2, Alanine/Serine/Cysteine-preferring Transporter 2; IDH1, isocitrate dehydrogenase; xCT, cysteine/glutamate transporter; CPT1, carnitine palmitoyltransferase 1; MDH1, malate dehydrogenase; BPTES, bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl) ethyl sulfide.
Regulation of Redox State in GCSCs
GCSCs like normal stem cells, exhibit a tightly regulated metabolism that governs their function. It is well known that, in stem cells, intracellular levels of Reactive Oxygen Species (ROS) play a pivotal role in the regulation of the balance between self-renewal and differentiation (95, 96). Indeed, while mitochondrial ROS production has been implied to have a role in muscle and adipocyte differentiation (97, 98), low levels of ROS are actively maintained in stem cells via aerobic glycolysis (99), and high activity of antioxidant machinery. Also CSCs, like normal stem cells, display strictly regulated ROS production through the metabolic switch and upregulation of the radical scavenging system including the members of the superoxide dismutase family (SODs) or glutathione peroxidase family (GPXs) (100–102). Furthermore, it has been demonstrated that increased ROS levels may enhance the cell sensibility to drugs, reducing chemo-resistance, while, on the other hand, low intracellular ROS levels may have a protective function in tumor bulk (103, 104). A recent study demonstrated that CD13, a CSCs marker, negatively regulates ROS levels, resulting in increased stemness in liver CSCs (105). Further, in breast cancer, CD24-/low/CD44+ initiating cells show low levels of radiation-induced ROS that confer higher tumorigenicity and resistance to radiation (106). This evidence endorses the association between low levels of intracellular ROS and cancer stemness even if, as for normal stem cells, the redox status of CSCs is not well-defined. Glutathione (GSH) is an antioxidant peptide with a high abundance in mitochondria of eukaryotic cells. GSH is involved in the maintenance of redox balance through ROS detoxification, and in the protection of phospholipids in the mitochondrial membrane (107). Interestingly, high glutathione levels are found in embryonic stem cells (108) and mesenchymal stem cells where they are responsible for the maintenance of stemness (109). Likewise, high GSH levels together with GSH-related enzymes are found in CSCs from gastric (110, 111), liver (105) and breast cancer (112). Conversely, little is known about the glutathione pathway in CSCs. It has been demonstrated that pancreatic CSCs show high levels of GSH content and upregulation of several genes involved in the GSH signaling (111). As for CSCs of other carcinomas, stem cells from gastric cancer, marked by upregulation of a variant of CD44 receptor (CD44v) (113, 114), display an enhanced capacity of glutathione synthesis and defense against ROS. In particular, high levels of intracellular GSH are due to the high activity of the plasma membrane transporter xCT, a subunit of the cystine–glutamate exchange transporter which is involved in the cysteine uptake, essential for GSH synthesis (110, 115). It is well established that cancer stem cells activate the p38 MAPK pathway to face oxidative stress (116). In particular, ROS production induces ASK1 kinase activation, which, through MAPK3/4/6 activation (117) leads to phosphorylation and activation of p38. Phosphorylated p38 is responsible for apoptosis activation and growth arrest, having so, a negative role on tumorigenesis (118). In GCSCs, the interaction of CD44v with xCT leads to an increased intracellular GSH which results in ROS-p38 MAPK suppression and enhanced tumor development (110, 115). Then, a specific therapy targeted to the CD44v-xCT pathway may impair the ability of GCSCs to protect themselves from oxidative stress, increasing the sensitivity to cancer available treatments. Indeed, inhibition of xCT transporter by sulfasalazine sensitizes gastric cancer stem cells to the drugs with a positive effect on the clinical efficacy of chemotherapy (Figure 4 and Table 1) (87).
Role of miRNAs in the Metabolic Reprogramming of GCSCs
MiRNAs are small single-stranded RNAs capable of binding to the 3’UTR of mRNAs inducing inhibitory signals to the ribosome which detaches from the mRNA blocking the translation of the protein (119). It is already known that these small non-coding RNAs are able to regulate the expression of many genes involved in cellular homeostasis such as progression, cell cycle, migration, apoptosis and cell differentiation (120). In cancer cells, the expression of miRNAs is deregulated through genetic and epigenetic modifications. Indeed, it has been found that the overexpression of some miRNAs (oncomiR), due to genomic amplification of their coding region, negatively regulates the levels of tumor suppressor genes. Conversely, deletions or loss of function mutations in miRNAs coding regions, which regulate proto-oncogenes, leads to reduced control over cell growth and differentiation, unlocking their tumorigenic potential (121). The analysis of stem-like gastro-spheres highlights deregulation of several miRNAs such as miR‐21, miR‐10b, and miR‐146a which are responsible for upregulation stem cell-related genes, clonogenicity ability and chemotherapeutic resistance (122–124). Indeed, the comparison between GCSCs and their non-stem cancer cell counterparts, through RT-PCR analysis, has shown a differential expression in miRNA, suggesting that the pathways governing these two cell populations are different (125).
Little is known about the role of miRNAs in the metabolic reprogramming of GCSCs. In CSCs, as already discussed, the glycolytic pathway plays a pivotal role in energy production and several miRNAs are described to be involved in this process. HK-2, the first rate-limiting enzyme of glycolysis, can be modulated by miR-181b which has a binding site in the 3′-untranslated region of HK-2 transcript and found to be down-regulated in gastric cancer tissues (126). Likewise, a comparison between gastric cancer tissues and adjacent noncancerous gastric mucosa tissues revealed the down-regulation of miR-422a expression level which inversely correlates with tumor size and depth of infiltration. He et al. pointed out how miR-422a is linked to metabolism, since it represses PDK2 activity, restoring pyruvate conversion in Acetyl-CoA through the PDH enzyme (127). A different role is attributed to miR-200 which appears to have conflicting effects in promoting stemness associated with oxidative stress (128). Also in GC were found non-canonical miRNAs that are independent from Drosha, an enzyme responsible for miRNA processing. In particular, it has been found that miR-6778-5p maintains the stem properties of GCSCs by blocking the YWHAE/c-MYC axis responsible for the reduced expression of SHMT1, a cytosolic isoenzyme involved in the metabolism of one-carbon folate-dependent. Thus, an increase in the expression of SHMT1 dependent on miR-6778-5p promotes the maintenance of stemness and enrichment of GCSCs (129). Furthermore, several studies show that lncRNAs can be involved in the post-transcriptional regulation process by interacting with miRNAs (130). As already mentioned, in GC, lnc-MSCC1-AS1 promotes stemness and chemo-resistance through the reprogramming of lipid metabolism. This role has been attributed to antagonism with miR-145-5p which promotes apoptosis of tumor cells through an increase in ROS levels and drug-associated toxicity (82). Thus, given the growing importance that has been attributed to miRNAs, it is necessary to improve the current knowledge of these biomolecules and their involvement in the metabolic reprogramming processes.
Conclusion
Over the course of this decade, substantial evidence suggested that CSCs are accountable for the progression of various types of cancer, including GC. These cells have been shown to have a distinct metabolic phenotype that depends on the microenvironment and genetic background. Several reports suggest that the metabolic rewiring is responsible for the conventional chemotherapy and molecularly targeted therapy failure in GC patients, resulting in poor clinical outcomes. As described, the OXPHOS and glycolysis are the primary sources of energy even if several regulatory pathways participate in this metabolic cycle for CSCs. Numerous factors are involved in the regulation of metabolic shifting, and for this reason, it is challenging to establish a targeted cancer therapy. Therefore, revealing the molecular mechanisms accountable for genetic mutation and epigenetic changes in GCSCs or the discovery of new metabolic targets that regulate tumor development and drug resistance might be a possible therapeutic opening for GC. However, focusing on alternative approaches should be the best way for patients’ benefits. These approaches include: to find out the specific CSCs markers to establish an early diagnosis of GC; to define the metabolic profile of tumor-inducing CSCs; to target the metabolic pathways, which play a crucial role in triggering the transitional states of normal to gastric cancer stem cells. We are just at the beginning of understanding metabolic reprogramming from normal stem cells to GCSCs. More studies should be done to increase the knowledge about this mechanism in order to improve the quality of life of GC patients.
Author Contributions
Conceptualization, PM and GF. Writing—original draft preparation, MA, GDP, HV and SL. Writing—review and editing, SR, PZ and PM. Supervision, GF and PM. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by PRIN 2017 (ENTI DI RICERCA DI RILEVANTE INTERESSE NAZIONALE) -Prot.2017XJ38A4.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 3(71):209–49. doi: 10.3322/caac.21660
2. Khazaei S, Rezaeian S, Soheylizad M, Khazaei S, Biderafsh A. Global Incidence and Mortality Rates of Stomach Cancer and the Human Development Index: An Ecological Study. Asian Pac J Cancer Prev (2016) 17(4):1701–4. doi: 10.7314/APJCP.2016.17.4.1701
3. Brenner H, Rothenbacher D, Arndt V. Epidemiology of Stomach Cancer. Methods Mol Biol (2009) 472:467–77. doi: 10.1007/978-1-60327-492-0_23
4. Bonequi P, Meneses-Gonzalez F, Correa P, Rabkin CS, Camargo MC. Risk Factors for Gastric Cancer in Latin America: A Meta-Analysis. Cancer Causes Control (2013) 24(2):217–31. doi: 10.1007/s10552-012-0110-z
5. Matsuzaka M, Tanaka R, Sasaki Y. High Mortality Rate of Stomach Cancer Caused Not by High Incidence But Delays in Diagnosis in Aomori Prefecture, Japan. Asian Pac J Cancer Prev (2016) 17(10):4723–7. doi: 10.22034/apjcp.2016.17.10.4723
6. Sitarz R, Skierucha M, Mielko J, Offerhaus GJA, Maciejewski R, Polkowski WP. Gastric Cancer: Epidemiology, Prevention, Classification, and Treatment. Cancer Manag Res (2018) 10:239–48. doi: 10.2147/CMAR.S149619
7. Tan P, Yeoh KG. Genetics and Molecular Pathogenesis of Gastric Adenocarcinoma. Gastroenterology (2015) 149(5):1153–62.e3. doi: 10.1053/j.gastro.2015.05.059
8. Chia NY, Tan P. Molecular Classification of Gastric Cancer. Ann Oncol (2016) 27(5):763–9. doi: 10.1093/annonc/mdw040
9. Yin J, Wu X, Li S, Li C, Guo Z. Impact of Environmental Factors on Gastric Cancer: A Review of the Scientific Evidence, Human Prevention and Adaptation. J Environ Sci (China) (2020) 89:65–79. doi: 10.1016/j.jes.2019.09.025
10. Cover TL, Peek RM Jr. Diet, Microbial Virulence, and Helicobacter Pylori-Induced Gastric Cancer. Gut Microbes (2013) 4(6):482–93. doi: 10.4161/gmic.26262
11. Herrera V, Parsonnet J. Helicobacter Pylori and Gastric Adenocarcinoma. Clin Microbiol Infect (2009) 15(11):971–6. doi: 10.1111/j.1469-0691.2009.03031.x
12. Forman D, Newell DG, Fullerton F, Yarnell JW, Stacey AR, Wald N, et al. Association Between Infection With Helicobacter Pylori and Risk of Gastric Cancer: Evidence From a Prospective Investigation. BMJ (1991) 302(6788):1302–5. doi: 10.1136/bmj.302.6788.1302
13. Oleastro M, Menard A. The Role of Helicobacter Pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biol (Basel) (2013) 2(3):1110–34. doi: 10.3390/biology2031110
14. Lamb A, Chen LF. Role of the Helicobacter Pylori-Induced Inflammatory Response in the Development of Gastric Cancer. J Cell Biochem (2013) 114(3):491–7. doi: 10.1002/jcb.24389
15. O’Connor ML, Xiang D, Shigdar S, Macdonald J, Li Y, Wang T, et al. Cancer Stem Cells: A Contentious Hypothesis Now Moving Forward. Cancer Lett (2014) 344(2):180–7. doi: 10.1016/j.canlet.2013.11.012
16. Bao B, Ahmad A, Azmi AS, Ali S, Sarkar FH. Overview of Cancer Stem Cells (CSCs) and Mechanisms of Their Regulation: Implications for Cancer Therapy. Curr Protoc Pharmacol (2013) Chapter 14:Unit 14 25. doi: 10.1002/0471141755.ph1425s61
17. Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer Stem Cells. Int J Biochem Cell Biol (2012) 44(12):2144–51. doi: 10.1016/j.biocel.2012.08.022
18. Ayob AZ, Ramasamy TS. Cancer Stem Cells as Key Drivers of Tumour Progression. J BioMed Sci (2018) 25(1):20. doi: 10.1186/s12929-018-0426-4
19. Lathia JD, Liu H. Overview of Cancer Stem Cells and Stemness for Community Oncologists. Target Oncol (2017) 12(4):387–99. doi: 10.1007/s11523-017-0508-3
20. Zhao W, Li Y, Zhang X. Stemness-Related Markers in Cancer. Cancer Transl Med (2017) 3(3):87–95. doi: 10.4103/ctm.ctm_69_16
21. Basati G, Mohammadpour H, Emami Razavi A. Association of High Expression Levels of SOX2, NANOG, and OCT4 in Gastric Cancer Tumor Tissues With Progression and Poor Prognosis. J Gastrointest Cancer (2020) 51(1):41–7. doi: 10.1007/s12029-018-00200-x
22. Falco G, Lee SL, Stanghellini I, Bassey UC, Hamatani T, Ko MS. Zscan4: A Novel Gene Expressed Exclusively in Late 2-Cell Embryos and Embryonic Stem Cells. Dev Biol (2007) 307(2):539–50. doi: 10.1016/j.ydbio.2007.05.003
23. Tagliaferri D, Mazzone P, Noviello TMR, Addeo M, Angrisano T, Del Vecchio L, et al. Retinoic Acid Induces Embryonic Stem Cells (ESCs) Transition to 2 Cell-Like State Through a Coordinated Expression of Dux and Duxbl1. Front Cell Dev Biol (2019) 7:385. doi: 10.3389/fcell.2019.00385
24. Portney BA, Arad M, Gupta A, Brown RA, Khatri R, Lin PN, et al. ZSCAN4 Facilitates Chromatin Remodeling and Promotes the Cancer Stem Cell Phenotype. Oncogene (2020) 39(26):4970–82. doi: 10.1038/s41388-020-1333-1
25. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of Pluripotent Stem Cells From Adult Human Fibroblasts by Defined Factors. Cell (2007) 131(5):861–72. doi: 10.1016/j.cell.2007.11.019
26. Poli V, Fagnocchi L, Fasciani A, Cherubini A, Mazzoleni S, Ferrillo S, et al. MYC-Driven Epigenetic Reprogramming Favors the Onset of Tumorigenesis by Inducing a Stem Cell-Like State. Nat Commun (2018) 9(1):1024. doi: 10.1038/s41467-018-03264-2
27. Moitra K, Lou H, Dean M. Multidrug Efflux Pumps and Cancer Stem Cells: Insights Into Multidrug Resistance and Therapeutic Development. Clin Pharmacol Ther (2011) 89(4):491–502. doi: 10.1038/clpt.2011.14
28. Zhou HM, Zhang JG, Zhang X, Li Q. Targeting Cancer Stem Cells for Reversing Therapy Resistance: Mechanism, Signaling, and Prospective Agents. Signal Transduct Target Ther (2021) 6(1):62. doi: 10.1038/s41392-020-00430-1
29. Wei MF, Chen MW, Chen KC, Lou PJ, Lin SY, Hung SC, et al. Autophagy Promotes Resistance to Photodynamic Therapy-Induced Apoptosis Selectively in Colorectal Cancer Stem-Like Cells. Autophagy (2014) 10(7):1179–92. doi: 10.4161/auto.28679
30. Tsuchida R, Das B, Yeger H, Koren G, Shibuya M, Thorner PS, et al. Cisplatin Treatment Increases Survival and Expansion of a Highly Tumorigenic Side-Population Fraction by Upregulating VEGF/Flt1 Autocrine Signaling. Oncogene (2008) 27(28):3923–34. doi: 10.1038/onc.2008.38
31. De Francesco EM, Sotgia F, Lisanti MP. Cancer Stem Cells (CSCs): Metabolic Strategies for Their Identification and Eradication. Biochem J (2018) 475(9):1611–34. doi: 10.1042/BCJ20170164
32. Sancho P, Barneda D, Heeschen C. Hallmarks of Cancer Stem Cell Metabolism. Br J Cancer (2016) 114(12):1305–12. doi: 10.1038/bjc.2016.152
33. Zhu X, Chen HH, Gao CY, Zhang XX, Jiang JX, Zhang Y, et al. Energy Metabolism in Cancer Stem Cells. World J Stem Cells (2020) 12(6):448–61. doi: 10.4252/wjsc.v12.i6.448
34. DeBerardinis RJ, Chandel NS. Fundamentals of Cancer Metabolism. Sci Adv (2016) 2(5):e1600200. doi: 10.1126/sciadv.1600200
35. Yadav UP, Singh T, Kumar P, Sharma P, Kaur H, Sharma S, et al. Metabolic Adaptations in Cancer Stem Cells. Front Oncol (2020) 10:1010. doi: 10.3389/fonc.2020.01010
36. Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, Yeger H. Hypoxia Enhances Tumor Stemness by Increasing the Invasive and Tumorigenic Side Population Fraction. Stem Cells (2008) 26(7):1818–30. doi: 10.1634/stemcells.2007-0724
37. Das B, Pal B, Bhuyan R, Li H, Sarma A, Gayan S, et al. MYC Regulates the HIF2alpha Stemness Pathway via Nanog and Sox2 to Maintain Self-Renewal in Cancer Stem Cells Versus Non-Stem Cancer Cells. Cancer Res (2019) 79(16):4015–25. doi: 10.1158/0008-5472.CAN-18-2847
38. El-Sahli S, Xie Y, Wang L, Liu S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers (Basel) (2019) 11(7). doi: 10.3390/cancers11070904
39. Peng F, Wang JH, Fan WJ, Meng YT, Li MM, Li TT, et al. Glycolysis Gatekeeper PDK1 Reprograms Breast Cancer Stem Cells Under Hypoxia. Oncogene (2018) 37(8):1062–74. doi: 10.1038/onc.2017.368
40. Deshmukh A, Deshpande K, Arfuso F, Newsholme P, Dharmarajan A. Cancer Stem Cell Metabolism: A Potential Target for Cancer Therapy. Mol Cancer (2016) 15(1):69. doi: 10.1186/s12943-016-0555-x
41. Hossain F, Sorrentino C, Ucar DA, Peng Y, Matossian M, Wyczechowska D, et al. Notch Signaling Regulates Mitochondrial Metabolism and NF-kappaB Activity in Triple-Negative Breast Cancer Cells via IKKalpha-Dependent Non-Canonical Pathways. Front Oncol (2018) 8:575. doi: 10.3389/fonc.2018.00575
42. Robles-Flores M, Moreno-Londono AP, Castaneda-Patlan MC. Signaling Pathways Involved in Nutrient Sensing Control in Cancer Stem Cells: An Overview. Front Endocrinol (Lausanne) (2021) 12:627745. doi: 10.3389/fendo.2021.627745
43. Feng HC, Lin JY, Hsu SH, Lan WY, Kuo CS, Tian YF, et al. Low Folate Metabolic Stress Reprograms DNA Methylation-Activated Sonic Hedgehog Signaling to Mediate Cancer Stem Cell-Like Signatures and Invasive Tumour Stage-Specific Malignancy of Human Colorectal Cancers. Int J Cancer (2017) 141(12):2537–50. doi: 10.1002/ijc.31008
44. Li H, Feng Z, He ML. Lipid Metabolism Alteration Contributes to and Maintains the Properties of Cancer Stem Cells. Theranostics (2020) 10(16):7053–69. doi: 10.7150/thno.41388
45. Brungs D, Aghmesheh M, Vine KL, Becker TM, Carolan MG, Ranson M. Gastric Cancer Stem Cells: Evidence, Potential Markers, and Clinical Implications. J Gastroenterol (2016) 51(4):313–26. doi: 10.1007/s00535-015-1125-5
46. Bekaii-Saab T, El-Rayes B. Identifying and Targeting Cancer Stem Cells in the Treatment of Gastric Cancer. Cancer (2017) 123(8):1303–12. doi: 10.1002/cncr.30538
47. Yang L, Levi E, Zhu S, Du J, Majumdar AP. Cancer Stem Cells Biomarkers in Gastric Carcinogenesis. J Gastrointest Cancer (2013) 44(4):428–35. doi: 10.1007/s12029-013-9534-2
48. Russi S, Verma HK, Laurino S, Mazzone P, Storto G, Nardelli A, et al. Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells. Int J Mol Sci (2019) 20(15). doi: 10.3390/ijms20153736
49. Koukourakis MI, Pitiakoudis M, Giatromanolaki A, Tsarouha A, Polychronidis A, Sivridis E, et al. Oxygen and Glucose Consumption in Gastrointestinal Adenocarcinomas: Correlation With Markers of Hypoxia, Acidity and Anaerobic Glycolysis. Cancer Sci (2006) 97(10):1056–60. doi: 10.1111/j.1349-7006.2006.00298.x
50. Yuan LW, Yamashita H, Seto Y. Glucose Metabolism in Gastric Cancer: The Cutting-Edge. World J Gastroenterol (2016) 22(6):2046–59. doi: 10.3748/wjg.v22.i6.2046
51. Yang SW, Zhang ZG, Hao YX, Zhao YL, Qian F, Shi Y, et al. HIF-1alpha Induces the Epithelial-Mesenchymal Transition in Gastric Cancer Stem Cells Through the Snail Pathway. Oncotarget (2017) 8(6):9535–45. doi: 10.18632/oncotarget.14484
52. Zhang Q, Han Z, Zhu Y, Chen J, Li W. Role of Hypoxia Inducible Factor-1 in Cancer Stem Cells (Review). Mol Med Rep (2021) 23(1):1. doi: 10.3892/mmr.2020.11655
53. Watanabe Y, Suefuji H, Hirose Y, Kaida H, Suzuki G, Uozumi J, et al. 18F-FDG Uptake in Primary Gastric Malignant Lymphoma Correlates With Glucose Transporter 1 Expression and Histologic Malignant Potential. Int J Hematol (2013) 97(1):43–9. doi: 10.1007/s12185-012-1225-4
54. Thorens B, Mueckler M. Glucose Transporters in the 21st Century. Am J Physiol Endocrinol Metab (2010) 298(2):E141–5. doi: 10.1152/ajpendo.00712.2009
55. Yamada A, Oguchi K, Fukushima M, Imai Y, Kadoya M. Evaluation of 2-Deoxy-2-[18F]fluoro-D-Glucose Positron Emission Tomography in Gastric Carcinoma: Relation to Histological Subtypes, Depth of Tumor Invasion, and Glucose Transporter-1 Expression. Ann Nucl Med (2006) 20(9):597–604. doi: 10.1007/BF02984657
56. Zhou D, Jiang L, Jin L, Yao Y, Wang P, Zhu X. Glucose Transporter-1 Cooperating With AKT Signaling Promote Gastric Cancer Progression. Cancer Manag Res (2020) 12:4151–60. doi: 10.2147/CMAR.S251596
57. Shibuya K, Okada M, Suzuki S, Seino M, Seino S, Takeda H, et al. Targeting the Facilitative Glucose Transporter GLUT1 Inhibits the Self-Renewal and Tumor-Initiating Capacity of Cancer Stem Cells. Oncotarget (2015) 6(2):651–61. doi: 10.18632/oncotarget.2892
58. Wu J, Hu L, Wu F, Zou L, He T. Poor Prognosis of Hexokinase 2 Overexpression in Solid Tumors of Digestive System: A Meta-Analysis. Oncotarget (2017) 8(19):32332–44. doi: 10.18632/oncotarget.15974
59. Shao M, Zhang J, Zhang J, Shi H, Zhang Y, Ji R, et al. SALL4 Promotes Gastric Cancer Progression via Hexokinase II Mediated Glycolysis. Cancer Cell Int (2020) 20:188. doi: 10.1186/s12935-020-01275-y
60. Rai Y, Yadav P, Kumari N, Kalra N, Bhatt AN. Hexokinase II Inhibition by 3-Bromopyruvate Sensitizes Myeloid Leukemic Cells K-562 to Anti-Leukemic Drug, Daunorubicin. Biosci Rep (2019) 39(9). doi: 10.1042/BSR20190880
61. Yang T, Shu X, Zhang HW, Sun LX, Yu L, Liu J, et al. Enolase 1 Regulates Stem Cell-Like Properties in Gastric Cancer Cells by Stimulating Glycolysis. Cell Death Dis (2020) 11(10):870. doi: 10.1038/s41419-020-03087-4
62. Shiroki T, Yokoyama M, Tanuma N, Maejima R, Tamai K, Yamaguchi K, et al. Enhanced Expression of the M2 Isoform of Pyruvate Kinase Is Involved in Gastric Cancer Development by Regulating Cancer-Specific Metabolism. Cancer Sci (2017) 108(5):931–40. doi: 10.1111/cas.13211
63. Kwon OH, Kang TW, Kim JH, Kim M, Noh SM, Song KS, et al. Pyruvate Kinase M2 Promotes the Growth of Gastric Cancer Cells via Regulation of Bcl-xL Expression at Transcriptional Level. Biochem Biophys Res Commun (2012) 423(1):38–44. doi: 10.1016/j.bbrc.2012.05.063
64. Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its Analogs Inhibit Cancer Cell Glycolysis by Targeting Tumor Pyruvate Kinase-M2. Oncogene (2011) 30(42):4297–306. doi: 10.1038/onc.2011.137
65. Yao S, Shang W, Huang L, Xu R, Wu M, Wang F. The Oncogenic and Prognostic Role of PDK1 in the Progression and Metastasis of Ovarian Cancer. J Cancer (2021) 12(3):630–43. doi: 10.7150/jca.47278
66. Hur H, Xuan Y, Kim YB, Lee G, Shim W, Yun J, et al. Expression of Pyruvate Dehydrogenase Kinase-1 in Gastric Cancer as a Potential Therapeutic Target. Int J Oncol (2013) 42(1):44–54. doi: 10.3892/ijo.2012.1687
67. Mayer MJ, Klotz LH, Venkateswaran V. Metformin and Prostate Cancer Stem Cells: A Novel Therapeutic Target. Prostate Cancer Prostatic Dis (2015) 18(4):303–9. doi: 10.1038/pcan.2015.35
68. Snyder V, Reed-Newman TC, Arnold L, Thomas SM, Anant S. Cancer Stem Cell Metabolism and Potential Therapeutic Targets. Front Oncol (2018) 8:203. doi: 10.3389/fonc.2018.00203
69. Chae YC, Angelin A, Lisanti S, Kossenkov AV, Speicher KD, Wang H, et al. Landscape of the Mitochondrial Hsp90 Metabolome in Tumours. Nat Commun (2013) 4:2139. doi: 10.1038/ncomms3139
70. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, et al. Antibiotics That Target Mitochondria Effectively Eradicate Cancer Stem Cells, Across Multiple Tumor Types: Treating Cancer Like an Infectious Disease. Oncotarget (2015) 6(7):4569–84. doi: 10.18632/oncotarget.3174
71. Santos CR, Schulze A. Lipid Metabolism in Cancer. FEBS J (2012) 279(15):2610–23. doi: 10.1111/j.1742-4658.2012.08644.x
72. Wang L, Zhang T, Wang L, Cai Y, Zhong X, He X, et al. Fatty Acid Synthesis is Critical for Stem Cell Pluripotency via Promoting Mitochondrial Fission. EMBO J (2017) 36(10):1330–47. doi: 10.15252/embj.201695417
73. Hanai JI, Doro N, Seth P, Sukhatme VP. ATP Citrate Lyase Knockdown Impacts Cancer Stem Cells In Vitro. Cell Death Dis (2013) 4:e696. doi: 10.1038/cddis.2013.215
74. Menendez JA, Lupu R. Fatty Acid Synthase and the Lipogenic Phenotype in Cancer Pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi: 10.1038/nrc2222
75. Jiang L, Wang H, Li J, Fang X, Pan H, Yuan X, et al. Up-Regulated FASN Expression Promotes Transcoelomic Metastasis of Ovarian Cancer Cell Through Epithelial-Mesenchymal Transition. Int J Mol Sci (2014) 15(7):11539–54. doi: 10.3390/ijms150711539
76. Walter K, Hong SM, Nyhan S, Canto M, Fedarko N, Klein A, et al. Serum Fatty Acid Synthase as a Marker of Pancreatic Neoplasia. Cancer Epidemiol Biomarkers Prev (2009) 18(9):2380–5. doi: 10.1158/1055-9965.EPI-09-0144
77. Yasumoto Y, Miyazaki H, Vaidyan LK, Kagawa Y, Ebrahimi M, Yamamoto Y, et al. Inhibition of Fatty Acid Synthase Decreases Expression of Stemness Markers in Glioma Stem Cells. PloS One (2016) 11(1):e0147717. doi: 10.1371/journal.pone.0147717
78. Song M, Lee H, Nam MH, Jeong E, Kim S, Hong Y, et al. Loss-of-Function Screens of Druggable Targetome Against Cancer Stem-Like Cells. FASEB J (2017) 31(2):625–35. doi: 10.1096/fj.201600953
79. Gao Y, Li J, Xi H, Cui J, Zhang K, Zhang J, et al. Stearoyl-CoA-desaturase-1 Regulates Gastric Cancer Stem-Like Properties and Promotes Tumour Metastasis Via Hippo/YAP Pathway. Br J Cancer (2020) 122(12):1837–47. doi: 10.1038/s41416-020-0827-5
80. Visweswaran M, Arfuso F, Warrier S, Dharmarajan A. Aberrant Lipid Metabolism as an Emerging Therapeutic Strategy to Target Cancer Stem Cells. Stem Cells (2020) 38(1):6–14. doi: 10.1002/stem.3101
81. Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. Nanog Metabolically Reprograms Tumor-Initiating Stem-Like Cells Through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab (2016) 23(1):206–19. doi: 10.1016/j.cmet.2015.12.004
82. He W, Liang B, Wang C, Li S, Zhao Y, Huang Q, et al. MSC-Regulated Lncrna MACC1-AS1 Promotes Stemness and Chemoresistance Through Fatty Acid Oxidation in Gastric Cancer. Oncogene (2019) 38(23):4637–54. doi: 10.1038/s41388-019-0747-0
83. Xiang Y, Stine ZE, Xia J, Lu Y, O’Connor RS, Altman BJ, et al. Targeted Inhibition of Tumor-Specific Glutaminase Diminishes Cell-Autonomous Tumorigenesis. J Clin Invest (2015) 125(6):2293–306. doi: 10.1172/JCI75836
84. Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, et al. Glutamate Dehydrogenase 1 Signals Through Antioxidant Glutathione Peroxidase 1 to Regulate Redox Homeostasis and Tumor Growth. Cancer Cell (2015) 27(2):257–70. doi: 10.1016/j.ccell.2014.12.006
85. Ye J, Huang Q, Xu J, Huang J, Wang J, Zhong W, et al. Targeting of Glutamine Transporter ASCT2 and Glutamine Synthetase Suppresses Gastric Cancer Cell Growth. J Cancer Res Clin Oncol (2018) 144(5):821–33. doi: 10.1007/s00432-018-2605-9
86. Ozsvari B, Sotgia F, Simmons K, Trowbridge R, Foster R, Lisanti MP. Mitoketoscins: Novel Mitochondrial Inhibitors for Targeting Ketone Metabolism in Cancer Stem Cells (CSCs). Oncotarget (2017) 8(45):78340–50. doi: 10.18632/oncotarget.21259
87. Miyoshi S, Tsugawa H, Matsuzaki J, Hirata K, Mori H, Saya H, et al. Inhibiting xCT Improves 5-Fluorouracil Resistance of Gastric Cancer Induced by CD44 Variant 9 Expression. Anticancer Res (2018) 38(11):6163–70. doi: 10.21873/anticanres.12969
88. Coller HA. The Paradox of Metabolism in Quiescent Stem Cells. FEBS Lett (2019) 593(20):2817–39. doi: 10.1002/1873-3468.13608
89. Li X, Wu JB, Li Q, Shigemura K, Chung LW, Huang WC. SREBP-2 Promotes Stem Cell-Like Properties and Metastasis by Transcriptional Activation of c-Myc in Prostate Cancer. Oncotarget (2016) 7(11):12869–84. doi: 10.18632/oncotarget.7331
90. Mukha A, Dubrovska A. Metabolic Targeting of Cancer Stem Cells. Front Oncol (2020) 10:537930. doi: 10.3389/fonc.2020.537930
91. Zhou T, Yang Y, Chen Q, Xie L. Glutamine Metabolism Is Essential for Stemness of Bone Marrow Mesenchymal Stem Cells and Bone Homeostasis. Stem Cells Int (2019) 2019:8928934. doi: 10.1155/2019/8928934
92. Wise DR, Thompson CB. Glutamine Addiction: A New Therapeutic Target in Cancer. Trends Biochem Sci (2010) 35(8):427–33. doi: 10.1016/j.tibs.2010.05.003
93. Zhang J, Pavlova NN, Thompson CB. Cancer Cell Metabolism: The Essential Role of the Nonessential Amino Acid, Glutamine. EMBO J (2017) 36(10):1302–15. doi: 10.15252/embj.201696151
94. Jin L, Alesi GN, Kang S. Glutaminolysis as a Target for Cancer Therapy. Oncogene (2016) 35(28):3619–25. doi: 10.1038/onc.2015.447
95. Bigarella CL, Liang R, Ghaffari S. Stem Cells and the Impact of ROS Signaling. Development (2014) 141(22):4206–18. doi: 10.1242/dev.107086
96. Schieber M, Chandel NS. ROS Function in Redox Signaling and Oxidative Stress. Curr Biol (2014) 24(10):R453–62. doi: 10.1016/j.cub.2014.03.034
97. Tormos KV, Anso E, Hamanaka RB, Eisenbart J, Joseph J, Kalyanaraman B, et al. Mitochondrial Complex III ROS Regulate Adipocyte Differentiation. Cell Metab (2011) 14(4):537–44. doi: 10.1016/j.cmet.2011.08.007
98. Lee S, Tak E, Lee J, Rashid MA, Murphy MP, Ha J, et al. Mitochondrial H2O2 Generated From Electron Transport Chain Complex I Stimulates Muscle Differentiation. Cell Res (2011) 21(5):817–34. doi: 10.1038/cr.2011.55
99. Troiano A, Pacelli C, Ruggieri V, Scrima R, Addeo M, Agriesti F, et al. ZSCAN4(+) Mouse Embryonic Stem Cells Have an Oxidative and Flexible Metabolic Profile. EMBO Rep (2020) 21(6):e48942. doi: 10.15252/embr.201948942
100. Peng G, Tang Z, Xiang Y, Chen W. Glutathione Peroxidase 4 Maintains a Stemness Phenotype, Oxidative Homeostasis and Regulates Biological Processes in Panc1 Cancer Stemlike Cells. Oncol Rep (2019) 41(2):1264–74. doi: 10.3892/or.2018.6905
101. Zhou D, Shao L, Spitz DR. Reactive Oxygen Species in Normal and Tumor Stem Cells. Adv Cancer Res (2014) 122:1–67. doi: 10.1016/B978-0-12-420117-0.00001-3
102. Ding S, Li C, Cheng N, Cui X, Xu X, Zhou G. Redox Regulation in Cancer Stem Cells. Oxid Med Cell Longev (2015) 2015:750798. doi: 10.1155/2015/750798
103. Yang H, Villani RM, Wang H, Simpson MJ, Roberts MS, Tang M, et al. The Role of Cellular Reactive Oxygen Species in Cancer Chemotherapy. J Exp Clin Cancer Res (2018) 37(1):266. doi: 10.1186/s13046-018-0909-x
104. Zaidieh T, Smith JR, Ball KE, An Q. ROS as a Novel Indicator to Predict Anticancer Drug Efficacy. BMC Cancer (2019) 19(1):1224. doi: 10.1186/s12885-019-6438-y
105. Kim HM, Haraguchi N, Ishii H, Ohkuma M, Okano M, Mimori K, et al. Increased CD13 Expression Reduces Reactive Oxygen Species, Promoting Survival of Liver Cancer Stem Cells via an Epithelial-Mesenchymal Transition-Like Phenomenon. Ann Surg Oncol (2012) 19 Suppl 3:S539–48. doi: 10.1245/s10434-011-2040-5
106. Phillips TM, McBride WH, Pajonk F. The Response of CD24(-/low)/CD44+ Breast Cancer-Initiating Cells to Radiation. J Natl Cancer Inst (2006) 98(24):1777–85. doi: 10.1093/jnci/djj495
107. Mari M, Morales A, Colell A, Garcia-Ruiz C, Fernandez-Checa JC. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxid Redox Signal (2009) 11(11):2685–700. doi: 10.1089/ars.2009.2695
108. Shaban S, El-Husseny MWA, Abushouk AI, Salem AMA, Mamdouh M, Abdel-Daim MM. Effects of Antioxidant Supplements on the Survival and Differentiation of Stem Cells. Oxid Med Cell Longev (2017) 2017:5032102. doi: 10.1155/2017/5032102
109. Lim J, Heo J, Ju H, Shin JW, Kim Y, Lee S, et al. Glutathione Dynamics Determine the Therapeutic Efficacy of Mesenchymal Stem Cells for Graft-Versus-Host Disease via CREB1-NRF2 Pathway. Sci Adv (2020) 6(16):eaba1334. doi: 10.1126/sciadv.aba1334
110. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 Variant Regulates Redox Status in Cancer Cells by Stabilizing the xCT Subunit of System xc(-) and Thereby Promotes Tumor Growth. Cancer Cell (2011) 19(3):387–400. doi: 10.1016/j.ccr.2011.01.038
111. Jagust P, Alcala S, Sainz B, Heeschen C, Sancho P. Glutathione Metabolism is Essential for Self-Renewal and Chemoresistance of Pancreatic Cancer Stem Cells. World J Stem Cells (2020) 12(11):1410–28. doi: 10.4252/wjsc.v12.i11.1410
112. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of Reactive Oxygen Species Levels and Radioresistance in Cancer Stem Cells. Nature (2009) 458(7239):780–3. doi: 10.1038/nature07733
113. Zavros Y. Initiation and Maintenance of Gastric Cancer: A Focus on CD44 Variant Isoforms and Cancer Stem Cells. Cell Mol Gastroenterol Hepatol (2017) 4(1):55–63. doi: 10.1016/j.jcmgh.2017.03.003
114. Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, et al. Identification of Gastric Cancer Stem Cells Using the Cell Surface Marker CD44. Stem Cells (2009) 27(5):1006–20. doi: 10.1002/stem.30
115. Nagano O, Okazaki S, Saya H. Redox Regulation in Stem-Like Cancer Cells by CD44 Variant Isoforms. Oncogene (2013) 32(44):5191–8. doi: 10.1038/onc.2012.638
116. Gutierrez-Uzquiza A, Arechederra M, Bragado P, Aguirre-Ghiso JA, Porras A. p38alpha Mediates Cell Survival in Response to Oxidative Stress via Induction of Antioxidant Genes: Effect on the p70S6K Pathway. J Biol Chem (2012) 287(4):2632–42. doi: 10.1074/jbc.M111.323709
117. Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, et al. Mechanism of P38 MAP Kinase Activation In Vivo. Genes Dev (2003) 17(16):1969–78. doi: 10.1101/gad.1107303
118. Kennedy NJ, Cellurale C, Davis RJ. A Radical Role for P38 MAPK in Tumor Initiation. Cancer Cell (2007) 11(2):101–3. doi: 10.1016/j.ccr.2007.01.009
119. Sun X, Jiao X, Pestell TG, Fan C, Qin S, Mirabelli E, et al. MicroRNAs and Cancer Stem Cells: The Sword and the Shield. Oncogene (2014) 33(42):4967–77. doi: 10.1038/onc.2013.492
120. Khan AQ, Ahmed EI, Elareer NR, Junejo K, Steinhoff M, Uddin S. Role of miRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells (2019) 8(8). doi: 10.3390/cells8080840
121. Takahashi RU, Miyazaki H, Ochiya T. The Role of microRNAs in the Regulation of Cancer Stem Cells. Front Genet (2014) 4:295. doi: 10.3389/fgene.2013.00295
122. Bakhshi M, Asadi J, Ebrahimi M, Moradi AV, Hajimoradi M. Increased Expression of miR-146a, miR-10b, and miR-21 in Cancer Stem-Like Gastro-Spheres. J Cell Biochem (2019) 120(10):16589–99. doi: 10.1002/jcb.28918
123. Ruggieri V, Russi S, Zoppoli P, La Rocca F, Angrisano T, Falco G, et al. The Role of MicroRNAs in the Regulation of Gastric Cancer Stem Cells: A Meta-Analysis of the Current Status. J Clin Med (2019) 8(5). doi: 10.3390/jcm8050639
124. Verma HK, Ratre YK, Mazzone P, Laurino S, Bhaskar LVKS. Micro-RNA Facilitated Chemoresistance in Gastric Cancer: A Novel Biomarkers and Potential Therapeutics. Alexandria J Med (2020) 56(1):81–92. doi: 10.1080/20905068.2020.1779992
125. Golestaneh AF, Atashi A, Langroudi L, Shafiee A, Ghaemi N, Soleimani M. miRNAs Expressed Differently in Cancer Stem Cells and Cancer Cells of Human Gastric Cancer Cell Line MKN-45. Cell Biochem Funct (2012) 30(5):411–8. doi: 10.1002/cbf.2815
126. Li LQ, Yang Y, Chen H, Zhang L, Pan D, Xie WJ. MicroRNA-181b Inhibits Glycolysis in Gastric Cancer Cells via Targeting Hexokinase 2 Gene. Cancer Biomark (2016) 17(1):75–81. doi: 10.3233/CBM-160619
127. He Z, Li Z, Zhang X, Yin K, Wang W, Xu Z, et al. MiR-422a Regulates Cellular Metabolism and Malignancy by Targeting Pyruvate Dehydrogenase Kinase 2 in Gastric Cancer. Cell Death Dis (2018) 9(5):505. doi: 10.1038/s41419-018-0564-3
128. Bao B, Azmi AS, Li Y, Ahmad A, Ali S, Banerjee S, et al. Targeting CSCs in Tumor Microenvironment: The Potential Role of ROS-Associated miRNAs in Tumor Aggressiveness. Curr Stem Cell Res Ther (2014) 9(1):22–35. doi: 10.2174/1574888X113089990053
129. Zhao M, Hou Y, Du YE, Yang L, Qin Y, Peng M, et al. Drosha-Independent miR-6778-5p Strengthens Gastric Cancer Stem Cell Stemness via Regulation of Cytosolic One-Carbon Folate Metabolism. Cancer Lett (2020) 478:8–21. doi: 10.1016/j.canlet.2020.02.040
Keywords: gastric cancer, cancer stem cell (CSC), metabolism, reprogramming, therapy
Citation: Addeo M, Di Paola G, Verma HK, Laurino S, Russi S, Zoppoli P, Falco G and Mazzone P (2021) Gastric Cancer Stem Cells: A Glimpse on Metabolic Reprogramming. Front. Oncol. 11:698394. doi: 10.3389/fonc.2021.698394
Received: 22 April 2021; Accepted: 31 May 2021;
Published: 16 June 2021.
Edited by:
Kanjoormana Aryan Manu, Amala Cancer Research Centre, IndiaCopyright © 2021 Addeo, Di Paola, Verma, Laurino, Russi, Zoppoli, Falco and Mazzone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geppino Falco, Z2VwcGluby5mYWxjb0B1bmluYS5pdA==; Pellegrino Mazzone, cGVsbGVncmluby5tYXp6b25lQGJpb2dlbS5pdA==
†These authors share first authorship