Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal types of cancer. Despite major advances in defining the molecular mutations driving PDAC, this disease remains universally lethal with an overall 5-year survival rate of only about 7–8%. Genetic alterations in PDAC are exemplified by four critical genes (KRAS, TP53, CDKN2A, and SMAD4) that are frequently mutated. Among these, KRAS mutation ranges from 88% to 100% in several studies. Hippo signaling is an evolutionarily conserved network that plays a key role in normal organ development and tissue regeneration. Its core consists of the serine/threonine kinases mammalian sterile 20-like kinase 1 and 2 (MST1/2) and large tumor suppressor 1 and 2. Interestingly, pancreas-specific MST1/2 double knockout mice have been reported to display a decreased pancreas mass. Many of the genes involved in the Hippo signaling pathway are recognized as tumor suppressors, while the Hippo transducers Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) are identified as oncogenes. By dephosphorylation, YAP and TAZ accumulate in the nucleus and interact with transcription factors such as TEA domain transcription factor-1, 2, 3, and 4. Dysregulation of Hippo signaling and activation of YAP/TAZ have been recognized in a variety of human solid cancers, including PDAC. Recent studies have elucidated that YAP/TAZ play a crucial role in the induction of acinar-to-ductal metaplasia, an initial step in the progression to PDAC, in genetically engineered mouse models. YAP and TAZ also play a key role in the development of PDAC by both KRAS-dependent and KRAS-independent bypass mechanisms. YAP/TAZ have become extensively studied in PDAC and their biological importance during the development and progression of PDAC has been uncovered. In this review, we summarize the biological significance of a dysregulated Hippo signaling pathway or activated YAP/TAZ in PDAC and propose a role for YAP/TAZ as a therapeutic target.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal types of cancer (1). Genetic alterations in PDAC are exemplified by four critical genes that are frequently mutated (KRAS, TP53, CDKN2A, and SMAD4). Some of these mutations occur when the tumors are in a preneoplastic condition (2). Despite major advances in defining the molecular mutations driving PDAC, this disease remains universally lethal, with an overall 5-year survival rate of only about 7–8%. Although recent developments in systemic chemotherapy such as FOLFIRINOX (5-fluorouracil, folinic acid, irinotecan, and oxaliplatin) and GnP (gemcitabine plus nab-paclitaxel) regimens have provided improved survival outcomes of patients with metastatic PDAC (3, 4), chemoresistance to current systemic chemotherapies (FOLFIRINOX and GnP) is a major treatment issue. Furthermore, of the patients who receive surgical treatments, 60% relapse within 12 months; this is most likely due to micro-metastases that were not detected during the diagnostic computed tomography scan (5). Although approximately 25–30% of patients respond to chemotherapeutic drugs, most eventually become resistant. Resistance mechanisms include deficiencies in drug uptake, alteration of drug targets, activation of DNA repair pathways, and resistance to apoptosis (6). Heterogeneity caused by admixture of tumor cells and stromal cells also produces chemoresistance and limits the targeted therapy of PDAC (7). Unfortunately, our knowledge of the genetic and biological backgrounds in this deadly disease has not yet been linked to improved patient survival. Further developments in therapeutic approaches by continued elucidation of the genetics and molecular biology of PDACs may be the next approach to overcoming this poor prognostic disease and improving survival outcomes.
The Hippo signaling pathway was first discovered from studies in Drosophila melanogaster (8–10). Hippo signaling is an evolutionarily conserved network that plays a key role in normal organ development and tissue regeneration (11). Multiple inputs control Hippo signaling, ranging from mechanical cues instructed by the cellular microenvironment (mechano-transduction) to soluble factors and metabolic pathways (12, 13). The Hippo pathway also displays extensive crosstalk with other signaling pathways such as transforming growth factor-beta (14, 15), Wnt (16, 17), Sonic hedgehog (18, 19), and Notch (20, 21). Its core consists of the serine/threonine kinases mammalian sterile 20-like kinase 1 and 2 (MST1 and MST2; Hippo in Drosophila) and large tumor suppressor 1 and 2 (LATS1 and LATS2). MST1/2 cooperate with salvador homolog 1 to phosphorylate and activate LATS1/2 kinases. LATS1/2 kinases then combine with the adaptor MOB kinase activator 1 to phosphorylate the Hippo transducers Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) (9, 22) (Figure 1). Many of the genes involved in the Hippo signaling pathway are recognized as tumor suppressors, while YAP/TAZ are oncogenes. In addition, YAP and TAZ can be phosphorylated at numerous sites (23, 24). Active LATS1/2 kinases phosphorylate YAP at 5 serine residues (S61, S109, S127, S164, and S381) and TAZ at 4 serine residues (S66, S89, S117, and S311) (23, 24). Among these, S127 (S89 in TAZ; as noted below, the two proteins share moderate sequence similarity) and S381 (S311 in TAZ) are key phosphorylation sites in suppressing YAP/TAZ oncogenic activity (24, 25). Phosphorylation of YAP and TAZ results in their cytoplasmic translocation, sequestration by 14-3-3 proteins, and recruitment of the β-TrCP (SCF) ubiquitin ligase complex (24).
Figure 1
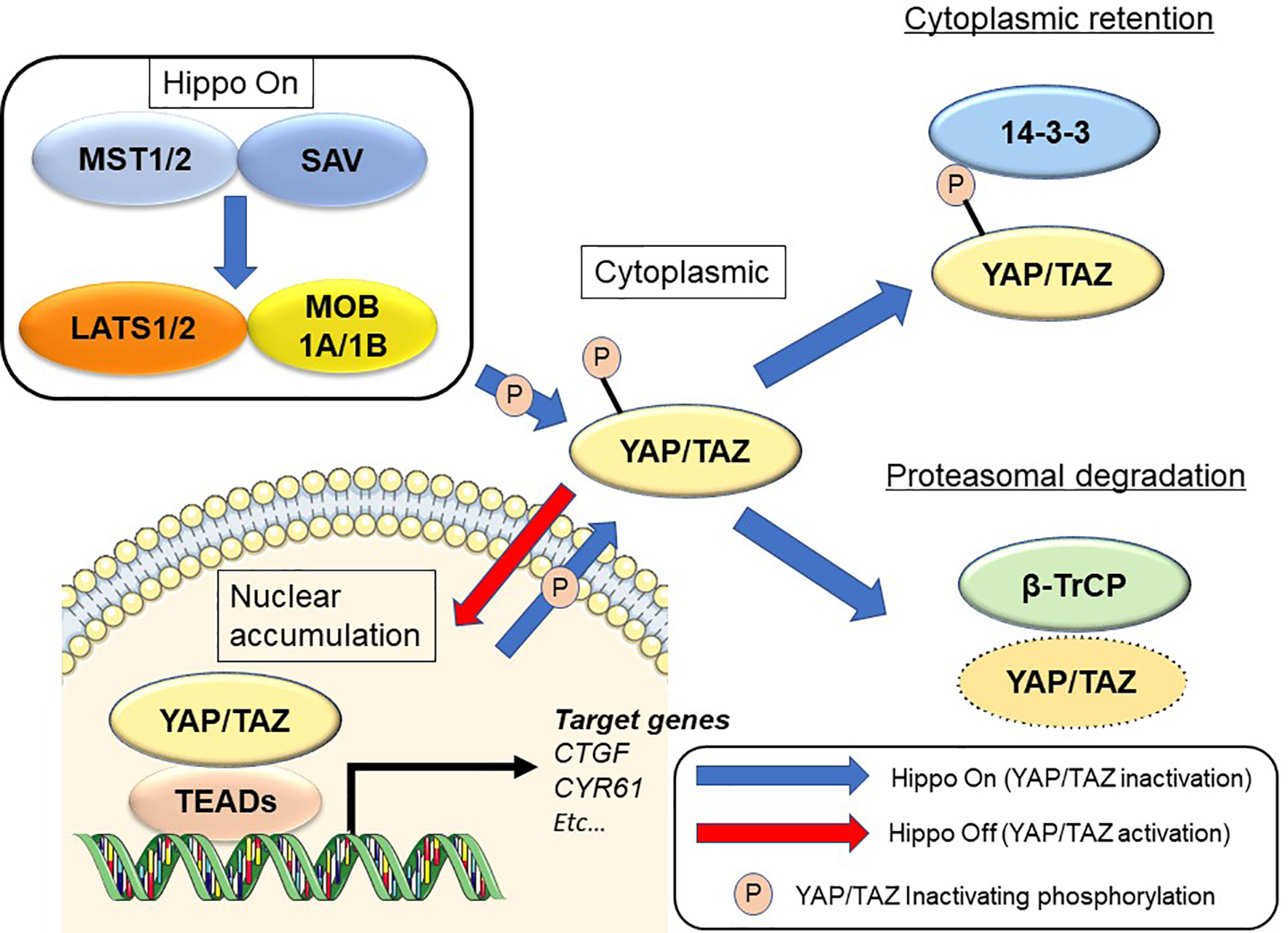
Regulation of the Hippo signaling pathway in mammalian cells. See text for details.
Upon dephosphorylation, YAP and TAZ accumulate in the nucleus and interact with transcription factors such as TEA domain transcriptional factor (TEAD)1, TEAD2, TEAD3, and TEAD4. YAP/TAZ also transcriptionally activate target genes such as connective tissue growth factor (CTGF) and cysteine-rich angiogenic inducer 61 (CYR61) (11). Deregulation of Hippo signaling has been recognized in a variety of human solid cancers, including PDAC (26–28). YAP/TAZ induce the epithelial-to-mesenchymal transition (EMT) and also induce a more undifferentiated state with malignant behavior in cancer cells (25, 29). YAP/TAZ also contribute to the strongly immunosuppressive microenvironment characteristic of mouse and human PDAC (30). Although YAP and TAZ have very similar structural topologies, share nearly half of their overall amino acid sequences, and are thought to be largely redundant, they may differ in their regulation and downstream functions (31, 32).
YAP/TAZ have become extensively studied in PDAC and their biological importance during the development and progression of PDAC has been uncovered. In this review, we summarize the biological significance of a dysregulated Hippo signaling pathway and activated YAP/TAZ in PDAC, and propose a role for YAP/TAZ as a therapeutic target.
Biological Role of Hippo Signaling Pathway During Normal Pancreas Development
The mammalian pancreas is a dual-function organ that is critical for the regulation of basic metabolism. In the mouse, development of the pancreas is divided into two stages, commonly denoted as the primary and secondary transitions (33). In a report using pancreatic MST1/2 double knockout (DKO) mice, George et al. (34) found that YAP is broadly expressed throughout the pancreatic and duodenal homeobox 1 (Pdx1)-positive embryonic day (E)12.5 mouse pancreas (primary transition) (34) (Figure 2). YAP expression then gradually becomes limited to prospective ductal and acinar regions at E16.5 (secondary transition). At E16.5, the productal cells show high YAP expression in the nucleus, whereas acinus-fated cells display expression mainly within the cytoplasm. Strikingly, prospective endocrine cells are negative for YAP expression. Pancreas development at E12.5 is characterized by compartmentalization, whereas E16.5 is characterized by massive cell proliferation and differentiation throughout the pancreas epithelium. In the adult mouse pancreas at 6 weeks (34), YAP expression is markedly decreased and strong expression is largely confined to ductal and terminal-duct centroacinar cells, unlike in the embryonic pancreas. YAP expression in acinar cells displays a weak cytoplasmic staining pattern, and is undetectable within islets. YAP expression in the adult human pancreas mirrors that in the mouse. On the other hand, phosphorylated-MST1/2 expression (indicative of active Hippo signaling) is broadly detectable in adult human pancreas, and islet cells display strong expression of phosphorylated MST1/2 (34). In another report using pancreatic MST1/2 DKO mice by Gao et al. (35), nuclear YAP staining was observed in the “trunk” regions at E15.5, and was almost undetectable at birth. Thereafter, YAP expression is weak and confined mainly to the ductal compartment at postnatal day (P)7 and later stages. MST1/2 mRNA levels are highest at E15.5 and lowest at birth; MST1 mRNA expression reappears at P7 and later stages. Interestingly, YAP expression is decreased and absent during the late embryonic and perinatal periods, raising the possibility that YAP must be silenced for proper pancreas differentiation. Such sequential changes in YAP expression have a crucial role for proper pancreas development (34, 35).
Figure 2
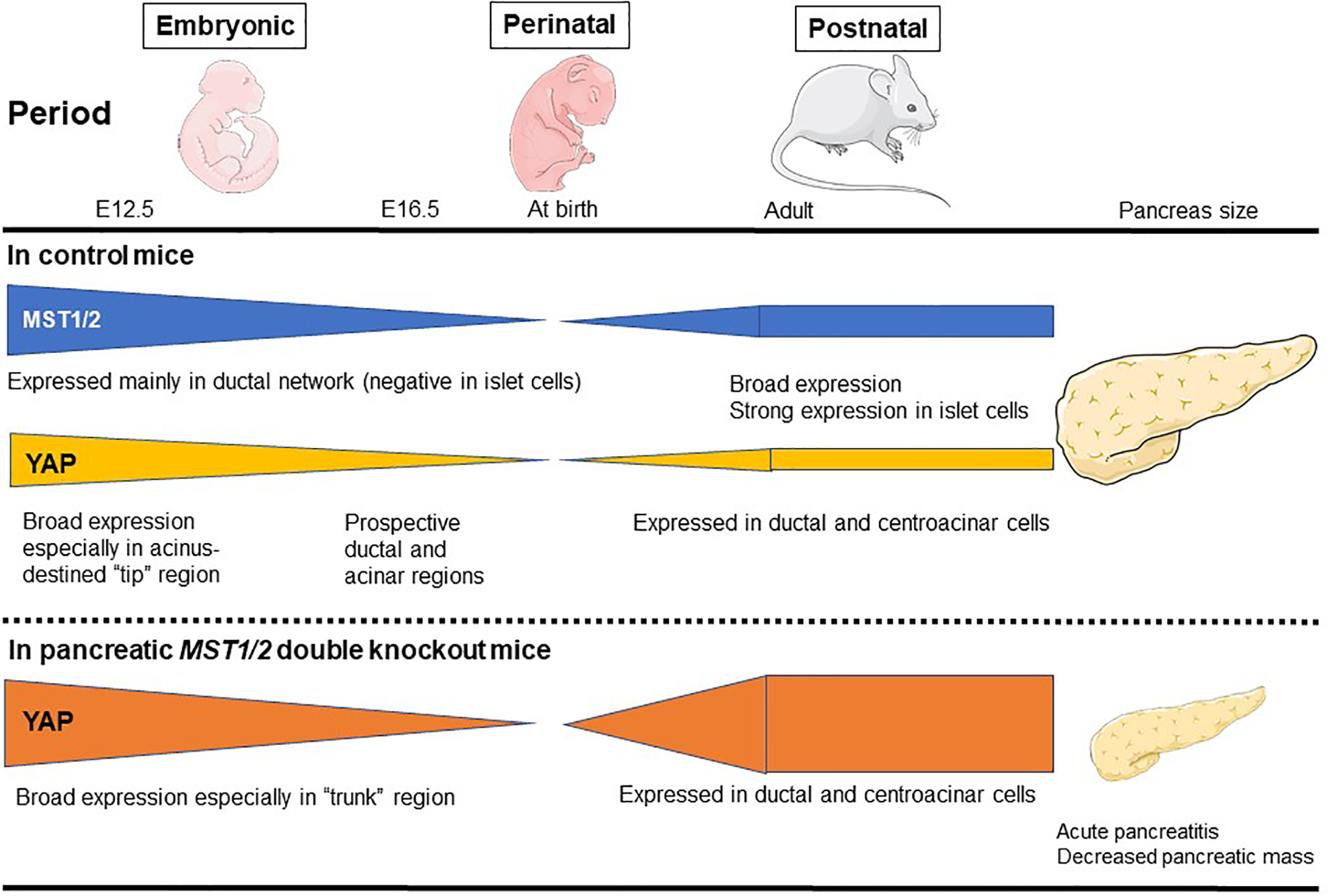
MST1/2 and YAP expression during normal pancreas development. MST1/2 mRNA levels are highest during the embryonic phase and lowest at birth. MST1 mRNA expression reappears at postnatal day 7 and later stages. Phosphorylated MST1/2 expression (active Hippo signaling) is broadly detectable in adult human pancreas, and islet cells display strong expression of phosphorylated MST1/2. YAP is broadly expressed throughout the pancreatic and duodenal E12.5 mouse pancreas (primary transition), but then gradually becomes restricted to prospective ductal and acinar regions at E16.5 (secondary transition). In the adult mouse pancreas, YAP expression is markedly decreased and the strong expression is largely confined to ductal and terminal-duct centroacinar cells, unlike in the embryonic pancreas. In pancreatic MST1/2 double knockout (DKO) mice, nuclear YAP staining is observed in the “trunk” regions during the embryonic phase, but is almost undetectable at birth. Thereafter, YAP expression is weak and confined mainly to the ductal compartment at birth and later postnatal stages. In pancreatic MST1/2 DKO mice, abundant YAP expression was observed in abnormally numerous duct-like structures during the postnatal phase. YAP remains undetectable within endocrine cells even in the absence of MST1/2. Pancreatic MST1/2 DKO mice show the histologic features of acute pancreatitis and decreased size (an approximately 2-fold decrease in pancreas mass).
Indeed, in pancreatic MST1/2 DKO mice, abundant YAP expression was observed in the abnormally numerous duct-like structures from P7 to P14 (35). Furthermore, the duct-like cells in MST1/2 DKO mice originated from acinar cells. In the absence of MST1/2, acinar cells differentiate normally but fail to maintain their differentiated state and de-differentiate or trans-differentiate into a duct-like state (35). On the other hand, MST1/2 deletion does not affect perinatal YAP expression, suggesting that perinatal YAP repression occurs via an MST1/2-independent mechanism (35).
In addition, pancreatic MST1/2 DKO mice show the histologic features of acute pancreatitis. While no discernible difference is observed between control and pancreatic Mst1/2 DKO mice at E12.5 (primary transition), a dramatic reduction in the overall expression of amylase is found in the pancreatic MST1/2 DKO mice at E16.5 (secondary transition), suggesting a defect in exocrine differentiation (34). In these mice, acini fail to form the classic rosette-like structure (34, 35). Robust immune cell infiltration and TUNEL-positive cell death are also detectable with a pancreatitis-like phenotype (35). These findings further suggest that Hippo signaling becomes active during the secondary transition where it regulates acinar cell proliferation and differentiation.
By 6 weeks, the majority of pancreatic cells are not proliferating in mice (34). In contrast, one-third of amylase-positive acinar cells and cytokeratin 19-positive ductal cells display sustained cell proliferation with BrdU incorporation in the pancreatic MST1/2 DKO mice (34). Thus, MST1/2 play a role as suppressors of proliferation in the mammalian pancreas.
On the other hand, for endocrine cells, YAP is not expressed in glucagon- or insulin-expressing cells at E12.5 and E16.5, respectively (34). Even at P30, YAP is not detectable in the β-cells (35). YAP remains undetectable within endocrine cells even in the absence of MST1/2 (34). Additionally, islet cells are largely Ki-67-negative, in agreement with undetectable YAP expression in both control and pancreatic MST1/2 DKO mice (34). The ratio of insulin-positive to glucagon-positive cells is not different between control and MST1/2 DKO mice (34), and blood glucose level also shows no significant difference between them (34, 35). On the other hand, complete loss of YAP in Yapflox/flox:p48-Cre mice also has no effect on blood glucose level (36). Hippo signaling does not play a crucial role in the pancreatic endocrine compartment.
As a consequence of the above features, the pancreas in pancreatic MST1/2 DKO mice is smaller (approximately 2-fold decrease in pancreas mass), displaying a pale white color and atrophy (34, 35). While Hippo deficiency in liver results in liver hypertrophy (37, 38), the Hippo-deficient pancreas is reduced in size (34, 35). Thus, pancreas mass and tissue architecture are greatly disrupted in the absence of MST1/2. YAP plays a crucial role downstream of MST1/2 during pancreas development, and dysregulation of Hippo signaling may contribute to human pancreatic disease phenotypes.
Biological Role of the Hippo Signaling Pathway in Pancreatic Cancer Development—Lessons From Genetically Engineered Mouse Models
The genetic landscape of PDAC is characterized by four frequently mutated genes: KRAS, TP53, CDKN2A (p16), and SMAD4 (39). The four predominant gene mutations appear to occur sequentially as PanIN progresses. KRAS mutations can be found even in normal pancreas and in PanIN1. In PDAC, KRAS mutation ranges from 88% to 100% in several studies (40–45). Although the initial step in PDAC development remains to be elucidated, oncogenic KRAS mutation is a key event, as evidenced by its presence in PanIN lesions (46, 47) and the development of PanIN lesions in oncogenic KRAS-driven genetically engineered mouse models (GEMMs) (48, 49). The oncogenic KRAS mutation perturbs the constitutively activated RAS protein, and results in the dysregulated activation of proliferation and survival pathways. GEMMs have provided several insights into the development of PDAC (50–53). Although oncogenic KRAS mutations are recognized early events in PDAC development, they are not entirely sufficient for the development of fully invasive PDAC. Indeed, only 5–10% of animals in GEMMs with mutated KRAS (without additional genetic alterations) develop frank PDAC, and do so very late (usually after 9 months) (50). PDAC development can be enhanced by the existence of another mutation (e.g., TP53) (51, 54). Pancreatic inflammation by administration of cerulein accelerates the formation of PanINs and PDAC in KRASG12V mice (55). In addition to the role of oncogenic KRAS in development of PDAC, KRAS mutations have also been shown to be important for PDAC maintenance (56, 57). Interestingly, inactivation of KRASG12D in confirmed precursor lesions and during progression to PDAC leads to tumor regression of those lesions, showing that KRASG12D is required for tumor cell maintenance (56, 58) (Figure 3). In an analysis of KRAS mutation type, codon G12D mutation was the most frequent (48%), followed by G12V (31%) and G12R (21%) (45).
Figure 3
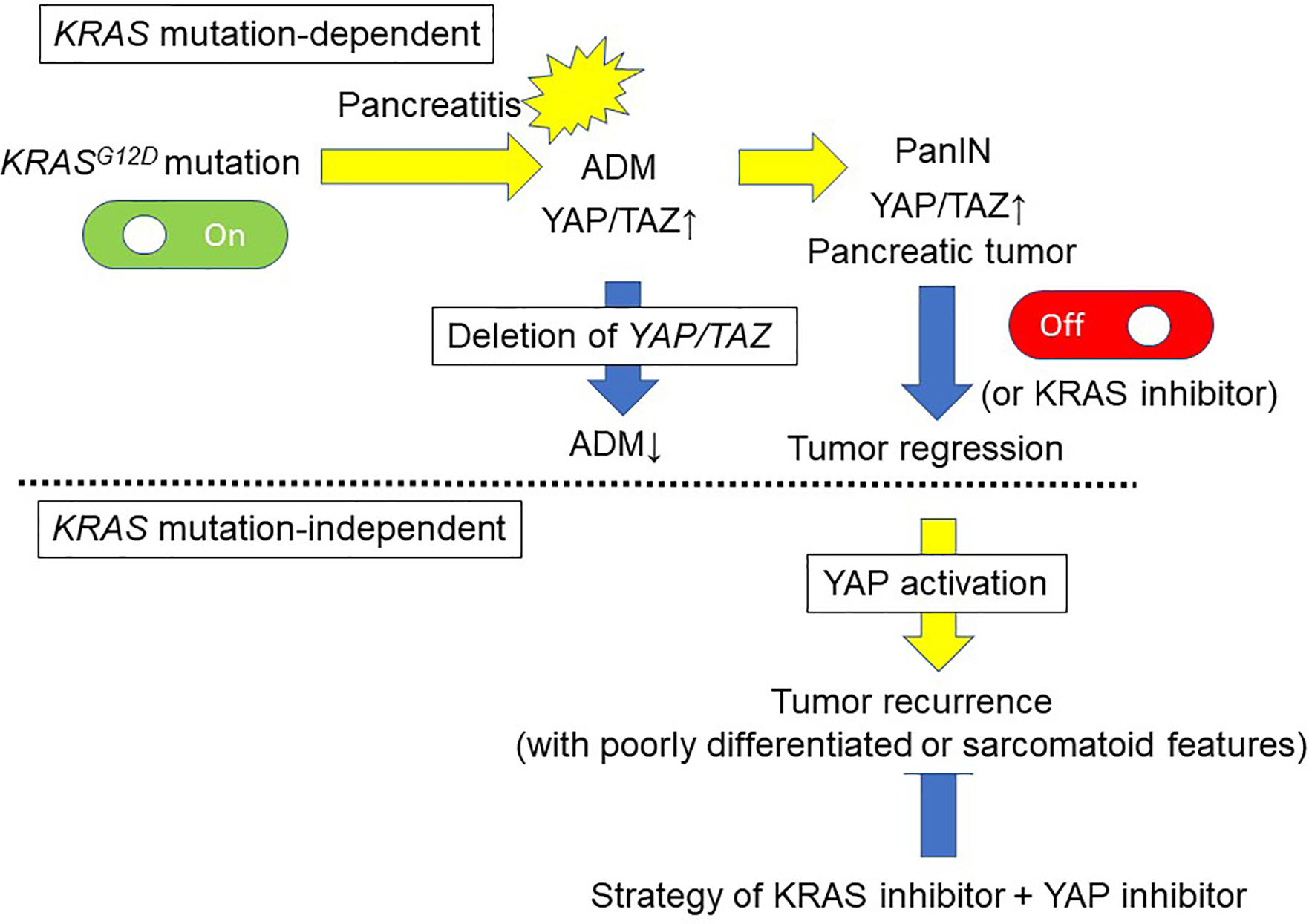
Pancreatic tumor development and maintenance by KRAS-dependent and KRAS-independent mechanisms via YAP activation. Oncogenic KRAS mutation is a key event of the development of PanIN lesions. Acinar-to-ductal metaplasia (ADM) caused by pancreatitis is an initiating step in pancreatic tumor development. The pancreatic tumor mouse model LSL-KRASG12D; Pdx1-Cre displays increased ADM lesions and development of PanIN in response to cerulein, accompanied by YAP/TAZ expression. On the other hand, deletion of YAP/TAZ reduces the ability of KRASG12D mutant mice to develop ADM in response to cerulein, and these mice are free of PanIN lesions even after cerulein-induced pancreatitis. Inactivation of KRASG12D in established tumor lesions can lead to tumor regression. Although KRASG12D extinction induces regression of pancreatic tumors, 70% of the mice develop relapsed tumors via oncogenic KRAS-independent mechanisms involving the YAP1 oncogene. An anti-YAP1 therapeutic strategy with KRAS-targeting agents may be required for elective tumors.
Acinar-to-ductal metaplasia (ADM) caused by pancreatitis is an initiating step in pancreatic tumor development (55). Cerulein treatment reduces phosphorylation of LATS1, and increases YAP/TAZ protein levels accompanied by strong nuclear localization (59). Following cerulein treatment, cytokeratin 19 (duct cell marker) expression is also increased, consistent with acinar-to-ductal reprogramming (59). Thus, YAP/TAZ activity is accelerated in the injured pancreas, particularly in the subset of cells undergoing ADM (Figure 3).
Recent studies have demonstrated that YAP/TAZ play a crucial role in the induction of ADM, an initial step in the progression to PDAC, in GEMMs (36, 59). The pancreatic tumor mouse model LSL-KRASG12D;Pdx1-Cre displays the whole spectrum of preneoplastic lesions (50). In these mice, increased ADM lesions and development of PanIN with strong YAP/TAZ expression are detectable, and YAP/TAZ levels are elevated in pancreatic protein lysates (59). Deletion of YAP/TAZ significantly reduced the ability of KRASG12D mice to induce ADM in response to cerulein, and these mice (KRASG12D;YAP1fl/fl;TAZfl/fl) were free of PanIN lesions at 3 months after the transient induction of pancreatitis by cerulein, similar to control mice (59). Thus, YAP/TAZ are required for KRASG12D-induced ADM in response to pancreatitis in vivo (Figure 3). Deletion of YAP/TAZ in the KRASG12D mice reduced Ras activation even after cerulein treatment (59). In contrast, ectopic YAP/TAZ activation in acinar cells by adenoviral vectors converted the infected acinar cells to duct cell morphology (59). Overexpression of constitutively active YAP1 in primary acinar cells also enhances Ras activity (59). YAP/TAZ are necessary and sufficient for ADM induction (59). Acinar cell-specific YAP/TAZ signaling may be essential for oncogenic KRASG12D-induced PanIN formation in the context of pancreatitis.
Zhang et al. (36) genetically engineered KRASG12D/+:TP53R172H/+:YAPflox/flox:p48-Cre mice to determine whether YAP is involved in PDAC development. In their study, KRASG12D/+:p48-Cre or KRASG12D/+:TP53R172H/+:p48-Cre mice with one or two intact YAP alleles developed ADM and early PanINs from 4 to 8 weeks of age, respectively (36). These ADM and early PanINs progressed through late-stage PanINs and eventually to invasive PDAC by 2 to 4 months in KRASG12D/+:TP53R172H/+:p48-Cre mice, or from 6 months to 2 years in KRASG12D/+:p48-Cre or KRASG12D/+ mice (36). In contrast, when these mice underwent homozygous YAP deletion (KRASG12D/+:YAPflox/flox:p48-Cre and KRASG12D/+:TP53R172H/+:YAPflox/flox:p48-Cre), they entirely lacked any late-stage PanINs or PDAC (36).
Zhang et al. generated p48-Cre;LSL-KRASG12D;FBXW7fl/fl mice to examine whether loss of the tumor suppressor FBXW7 might be an additional gene alteration in the development of PDAC (60). They found that the mice displayed accelerated tumorigenesis: PDACs were detectable by P14 and all mice yielded PDACs by P40 PDAC in p48-Cre;LSL-KRASG12D;FBXW7fl/fl mice was preceded by earlier onset of ADM and PanIN lesions, and accompanied by chromosomal instability and the accumulation of YAP (60). Furthermore, in a pancreatic cell line established from p48-Cre;LSL-KRASG12D;FBXW7fl/fl mice and in FBXW7-deficient human pancreatic cancer cells, down-regulation of YAP attenuated cell growth. Thus, deletion of the tumor suppressor FBXW7 accelerates KRAS-driven pancreatic tumorigenesis with YAP expression (60).
Kapoor et al. examined the mechanism of KRASG12D-independent PDAC recurrence using a doxycycline-inducible KRASG12D transgene and conditional p53 null alleles (p48Cre; tetO_LSL-KRASG12D; ROSA_rtTA; p53L/+, designated iKras) (61). In their investigation, KRASG12D extinction by doxycycline withdrawal induced complete regression of pancreatic tumors at three weeks, as determined by MRI imaging. However, 70% of the mice developed relapsed tumors between 9 and 47 weeks after doxycycline withdrawal, with a median survival of 36.6 weeks compared to 15.4 weeks for iKras mice with continued doxycycline treatment (61). Kapoor et al. revealed oncogenic KRAS-independent bypass mechanisms involving the YAP1 oncogene in KRASG12D-independent PDAC recurrence, and emphasized the potential importance of an anti-YAP1 therapeutic strategy for elective tumors in the clinical setting with agents that targeted KRAS and its signaling pathways (61) (Figure 2). Shao et al. reported similar mechanisms in KRAS-driven lung cancer (62). These findings suggest that murine PDAC cells can survive in the absence of oncogenic KRAS signaling and acquire alternative mechanisms to foster their own growth (61, 63). The activity of the transcriptional co-activator YAP plays a critical role in the promotion and maintenance of PDAC by operating as a key downstream target of KRAS signaling. YAP/TAZ amplification frequency ranged from 0% to 19% in 9,125 tumor samples among 33 cancer types from The Cancer Genome Atlas (64). Among these 33, the top six cancer types with the highest amplification frequency of YAP/TAZ included all five squamous cell-involved cancers (lung squamous cell carcinoma, esophageal squamous cell carcinoma, head and neck squamous cell carcinomas, and bladder urothelial carcinoma), whereas the frequency in PDAC was about 2% and 14th among 33 cancer types (64). Overall, YAP has emerged as a central node of transcriptional convergence in growth-promoting signaling in PDAC cells by both KRAS-dependent and KRAS-independent bypass mechanisms. (Figure 3). Collectively, these observations indicate that YAP/TAZ also play a crucial role in the development and recurrence of PDACs.
Clinical Impact of YAP/TAZ Expression in PDAC Patients
The clinical function of YAP as a prognostic marker has been investigated in several studies (Table 1), which have indicated that YAP and/or TAZ are overexpressed in tumor samples from patients with PDACs (61, 65–68). It has been found that nuclear overexpression of YAP is an independent prognostic marker for poor survival and is associated with liver metastasis (68). Furthermore, using public mRNA expression data, YAP was confirmed to be correlated with poor survival (69, 70). The 5-year survival rate was 0% in patients with high YAP mRNA expression compared to 32% in those with low expression. Furthermore, multiple YAP/TEAD-regulated genes were associated with poor prognosis, such as transforming growth factor alpha, heparin-binding EGF-like growth factor, integrin subunit alpha 2, P2Y2 receptor, G protein-coupled receptor 87, and mucin 1. On the other hand, YAP-inhibitory pathway components were associated with a favorable prognosis, such as STE20-related kinase adaptor/liver kinase B1, protein kinase A/large tumor suppressor, and tuberous sclerosis complex/mammalian target of rapamycin complex 1.
Table 1
| Reference | Number | Target | Location | Positive ratio | Outcomes |
|---|---|---|---|---|---|
| Diep et al. (65) | |||||
| 64 | YAP1 | Primary | 77% | Not available | |
| Yang et al. (66) | |||||
| 38 | YAP | Primary | 61% | Not available | |
| 25 | YAP | Metastatic site | 72% | Not available | |
| Xie et al. (67) | |||||
| 57 | TAZ | Primary | 82%* | Not available | |
| Salcedo Allende et al. (68) | |||||
| 64 | YAP1 | Primary | 90.62% | Poor OS | |
| Rozengurt et al. (69)** | |||||
| 176 | YAP mRNA | Primary | 20% | Poor OS | |
| Zhou et al. (70)*** | |||||
| 176 | YAP1 mRNA | Primary | 50% | Poor OS | |
YAP/TAZ expression and functional relevance in human pancreatic cancers.
*Weak, moderate, and strong expression of TAZ were identified as positive.
**A published interactive open-access database (www.proteinatlas.org/pathology) was used.
***The cohort of The Cancer Genome Atlas (TCGA) was used.
OS, overall survival.
Biological Role of YAP/TAZ in Cancer Cells
There is accumulating evidence that YAP and TAZ promote proliferation and growth of PDAC cells. Treatment of PDAC cells with YAP-targeting small interfering RNA oligonucleotides significantly reduced tumor growth (65). It has been reported that eukaryotic translation initiation factor 5A–pseudopodium-enriched atypical kinase 1 signaling regulates YAP and TAZ expression and pancreatic cancer cell growth (71). Disrupting this signaling in pancreatic cancer cells inhibited YAP/TAZ protein expression, reducing the expression of stem cell-associated transcription factors and tumor sphere growth (71).
In human PDAC cells, YAP functions as a downstream effector of the crosstalk between insulin/IGF-1 receptor and G protein-coupled receptor systems (72). Stimulation with insulin and the G protein-coupled receptor agonist neurotensin induced rapid YAP nuclear import and markedly augmented the mRNA levels of YAP/TEAD-regulated genes, including CTGF and Cyr61. The growth-promoting agonists regulated YAP activity via PI3K and protein kinase D in PANC-1 and MiaPaCa-2 (72), human cell lines that correspond to the squamous/quasi mesenchymal/basal-like sub-type of PDAC. It is of great interest that YAP function has been associated with this PDAC sub-type, considered the most clinically aggressive form.
The epithelial-to-mesenchymal transition (EMT) is a developmental regulatory program defined by the phenotypical transition from an epithelial to a mesenchymal cell state. The EMT is an essential step for metastasis and confers resistance to therapy (73). Active YAP promotes pancreatic cancer cell motility, invasion, and tumorigenesis in a mitotic phosphorylation-dependent manner and contributes to the EMT in pancreatic cancer cells by several mechanisms, including hyperactivation of AKT signaling (66, 67, 74, 75). YAP/TAZ also interact with nuclear factors such as ZEB1 (29) and SMADs (76, 77), both of which are important EMT regulators. TGF-β is a well-known EMT inducer in cancer cells. TGF-β enhances YAP nuclear localization and stabilizes YAP activity, and TGF-β-induced EMT and YAP activity are both blocked by inhibition of AKT signaling in PDAC cells (78). Xie et al. (67) focused on TAZ activation in pancreatic cancer cells and examined its functional roles in the EMT. Aberrant expression and activation of TAZ in pancreatic cancer cells promoted the EMT via down-regulation of E-cadherin and up-regulation of vimentin expression. In contrast, depletion of TAZ in pancreatic cancer cells suppressed the EMT phenotype (67).
PDAC is characterized by a high degree of chemoresistance. Gemcitabine has been the standard chemotherapeutic agent in PDAC since 1997 (79). Several mechanisms of YAP-induced chemoresistance have been proposed. One mechanism suggests that YAP overexpression induces the EMT in pancreatic cancer cells by activating the AKT cascade, which can cause resistance to gemcitabine (74). Another proposed mechanism involves microRNA, since microRNA 181c was overexpressed in PDAC samples and correlated with poor prognosis. microRNA 181c directly repressed MST1, LATS2, salvador homolog 1, and MOB kinase activator 1, leading to YAP and TAZ activation, and gemcitabine resistance in vitro and in vivo (80). Isoprenylcysteine carboxylmethyltransferase (ICMT) is the catalytic enzymes in the three step prenylation processing that posttranslationally modifies substrate proteins including RAS isoforms. Suppression of ICMT inhibits cancer stem cell self-renewal and chemoresistance of mutant KRAS pancreatic cancer cells with TAZ protein degradation (81). On the other hand, expression of constitutively active KRASG12V restores TAZ protein level and the self-renewal ability of pancreatic cancer cells. Thus, mutant KRAS plays a major role in TAZ expression and cancer stem self-renewal in pancreatic cancer cells, and ICMT has potential as a pharmacological target in the treatment of mutant KRAS pancreatic cancer cells (81).
Biological Role of YAP/TAZ in the Tumor Microenvironment
An important feature of human and murine PDAC is an extensive desmoplastic stroma (82) that increases the stiffness of the extracellular matrix (ECM) surrounding epithelial cancer cells (83). The Hippo/YAP pathway has been recognized to play a critical role in mechano-transduction (84, 85) and in sensing ECM stiffness (86). High stiffness leads to inhibition of the Hippo tumor suppressive pathway while enhancing the activity of YAP/TAZ. The stroma contains cancer-associated fibroblasts (CAFs), immune cells, endothelial cells, and the ECM. Pancreatic stellate cells are resident mesenchymal cells of the pancreas that represent the major source of CAFs. It has been found that YAP and TAZ are expressed at high levels in activated pancreatic stellate cells in PDAC, as well as in chronic pancreatitis (87).
Transglutaminase 2 secreted by pancreatic cancer cells promotes cross-linking of collagen, which activates CAFs and stimulates their proliferation, and results in higher collagen production by CAFs and further stiffening of the stroma. In turn, such a stiff tumor microenvironment conveys mechanical signals to cancer cells, leading to activation of YAP/TAZ and tumor progression (88). Environmental stimuli, including obesity and metabolic syndrome, also enhance the promotion of invasive PDAC (89, 90).
PDAC is characterized by a profound inflammatory reaction and an immunosuppressive state (91). Pancreatic tumors are associated with immune dysfunction, partly mediated by the recruitment of immunosuppressive cells, such as tumor-associated macrophages and myeloid-derived suppressor cells (92, 93). These cells are recruited to the tumor microenvironment and can inhibit T-cell activity. YAP has been identified as a critical regulator of the immunosuppressive microenvironment in PDAC. YAP inactivation prevented recruitment of myeloid-derived suppressor cells while in turn supporting infiltration of antigen-presenting macrophages and T-cell activation, thereby promoting apoptosis of tumor cells (30). Although T-cell activity is critical for tumor immunity, T-cell fate is governed by Hippo signaling (94–96). Geng et al. reported that TAZ induces Th17 cell differentiation and suppresses the differentiation of immunosuppressive regulatory T-cells (95). Ni et al. reported that immunosuppressive activity of regulatory T-cells was dependent on YAP expression in melanoma, and the anti-tumor immunity was enhanced in the absence of YAP (97). In hepatocellular carcinoma, YAP mediates the migration of macrophages in vitro and in vivo (98). Thus, YAP/TAZ are capable of regulating the biological activity and function of T-cells and macrophages, which is crucial for tumor immunity. They thereby participate in immune escape by suppressing normal immunological activity.
Whole-genome and whole-exome sequencing of PDACs have revealed a mean mutation load of 1.8 and 1.1 mutations per megabase, respectively, and only 5% of PDACs displayed a hypermutated phenotype (99). The identification of hypermutated PDACs is important because these tumors are sensitive to immunotherapy (99). Furthermore, the prevalence of microsatellite instability was found to be around 5% in many solid tumors, while in PDAC it was only 2% (100). These DNA mismatch repair (MMR)-deficient tumors carried high neo-antigen load and displayed considerably improved responses to programmed cell death 1 blockade (100). These authors reported that solid tumors with MMR deficiency are responsive to immune checkpoint blockade with pembrolizumab. Pembrolizumab has subsequently been approved by the FDA for solid tumors with MMR deficiency, regardless of tissue of origin (101). Furthermore, the clinical benefit of pembrolizumab was confirmed in patients with microsatellite instability high MMR-deficient non-colorectal cancers including pancreatic cancer (102). Thus, immunotherapy is a rapidly progressing field in cancer treatment. Among the immunotherapy modalities, immune checkpoint inhibition has displayed considerable success in several solid tumors, but there is still no significant benefit in PDAC.
In cancers other than PDAC, there is accumulating evidence that YAP/TAZ play a pivotal role in PD-L1 expression. Overexpression of constitutively active YAP or TAZ by the deletion of MST1/2 or LATS1/2 enhances PD-L1 expression in breast and lung cancer cell lines (103). Furthermore, PD-L1 expression is also induced by YAP in BRAF inhibitor-resistant melanoma, and the relationship between YAP and PD-L1 expression was validated in human clinical melanoma tissues (104). In human non-small cell lung cancer, YAP regulated PD-L1 at the transcriptional level, suggesting that YAP has potential as an immunotherapeutic target (105). Lee et al. found that YAP regulates PD-L1 by directly binding to the PD-L1 promoter and that YAP/PD-L1 signaling modulated tumor cell proliferation and migration in EGFR–TKI-resistant lung adenocarcinoma, and also that YAP down-regulation inhibited PD-L1 expression (106). It is worth further exploring the role of YAP/TAZ in tumor immunotherapy. Thus, targeting YAP/TAZ may be an alternative approach for combination with immunotherapy in cancer cells and the tumor microenvironment.
Therapeutic Targeting of the Hippo Signaling Pathway in PDAC
According to the above collected evidence, it is reasonable to develop drugs that target YAP and TAZ activities in PDAC. As indicated above, tumor cells with YAP activation can evade the requirement for KRAS mutant expression in PDAC (63). YAP is a key element not only downstream of Ras but also as an alternative route to bypass the need of this oncogene for tumor relapse. Recently, the KRASG12C inhibitor Sotorasib is effectively developed for solid cancers (107). In 11 PDAC patients with KRASG12C mutation, one patient had a confirmed partial response, 9 had stable disease, and one had progressive disease in response to Sotorasib (107). Even if Ras could be effectively inhibited by this new therapy, YAP amplification offers a potential pathway to induce tumor recurrence. Recent studies suggest novel approaches to inhibit YAP/TAZ activity with drug repositioning in clinical use, including statins. Statins, which have been used to treat dyslipidemia and prevent heart diseases, selectively inhibit 3-hydroxy-methylglutaryl (HMG) CoA reductase (108), the rate-limiting enzyme in the generation of mevalonate. Accelerated mevalonate biosynthesis through mutant p53 (109–111) and AKT/mTORC1 (111) has been reported in cancers. The mevalonate pathway plays an important role in the generation of lipids and lipid intermediates, including farnesyl pyrophosphate, geranylgeranyl pyrophosphate, and cholesterol. In preclinical studies (112, 113), statins delayed the progression of PDAC in mice harboring KRASG12D mutation. Statins were identified as potential YAP inhibitors by screens of molecules that changed the nuclear/cytoplasmic distribution of YAP (114). In our previous study, statin treatment suppressed cancer cell growth via TAZ down-regulation in hepatocellular carcinoma (115). Several epidemiological studies have indicated that statin use correlates with favorable oncologic effects in PDAC (116–124), especially in males (119, 120). A large study demonstrated that statins were associated with a significantly reduced PDAC risk (by 34%), with a stronger effect in males (119). The beneficial effects of statins depend on the type of statins used, with several reports showing positive associations with lipophilic (and not hydrophilic) statins and reduced cancer risk (125–128). On the other hand, Hamada et al. reported that regular statin use was not associated with pancreatic cancer risk in two large prospective cohort studies in the U.S (129). Nevertheless, Hamada et al. also reported increased survival in PDAC patients with regular pre-diagnosis use of statins (130). Recently, a meta-analysis of PDAC risk that included more than 3 million participants and 170,000 PDAC patients has been published (131). This study indicates a significant decrease in PDAC risk with statin use, thus reinforcing the conclusion that statin administration is associated with beneficial effects in PDAC patients. In addition to their potential efficacy in primary prevention and interception, statins may improve the outcome for patients after surgical removal of their primary PDAC (116, 117, 132), indicating a possible role for statins in the prevention of PDAC recurrence. Collectively, accumulating evidence from epidemiological and preclinical studies indicates a protective effect of statins in PDAC. Of the evaluated treatments in PDAC, verteporfin (133, 134) has a direct effect on Hippo signaling by inhibiting YAP–TEAD interactions. Erlotinib (135), FG-3019 (CTGF antagonism) (136), BIS 1 (135), LY3009120 (133) and ICMT small molecule inhibitor (81) indirectly affect YAP and/or TAZ signaling. Although the mechanism is not fully clarified for several natural substances, curcumin (32, 137), resveratrol (138), Stichopus japonicus acidic mucopolysaccharide (139), and pseudolaric acid B (140) have been reported to target YAP/TAZ signaling. Also, in our previous study, curcumin, a major component of turmeric and an old Indian spice, successfully suppressed TAZ/YAP expression and exerted anticancer effects in hepatocellular carcinoma cell lines (32). In the future, direct or indirect pharmacological modulation of YAP/TAZ expression may become promising approaches to fight this deadly disease.
Conclusions
The Hippo pathway is an evolutionarily conserved signaling pathway in mammals, and YAP and TAZ are key downstream regulators in the Hippo pathway that play a crucial role in the development of the normal pancreas and of PDAC in GEMMs. Furthermore, YAP and TAZ play a crucial role in the development of PDAC by both KRAS-dependent and KRAS-independent bypass mechanisms. Also in PDAC progression, aberrant transcriptional activity of YAP and TAZ has a pivotal role in malignant behavior, including cell growth, EMT, and drug resistance. Besides, YAP/TAZ play a tumor-promoting role in the tumor microenvironment. PDAC features an extensive desmoplastic stroma, and the stroma contains CAFs and immune cells. YAP promotes CAF activation and subsequent fibroinflammatory responses, and the resultant high stiffness enhances the malignant behavior of PDAC with high activity of YAP/TAZ. In addition, YAP acts as a critical regulator of the immunosuppressive microenvironment in PDAC. YAP/TAZ have potential as a therapeutic target not only for cancer cells, but also for the tumor microenvironment in PDAC. Thus, accumulating evidence supports the biological importance of YAP/TAZ in the development and progression of PDACs, and the regulation of YAP/TAZ signaling is increasingly recognized as a therapeutic target. In the near future, direct or indirect pharmacological modulation of YAP/TAZ may become promising therapeutic approaches in PDACs. On the other hand, complete deletion of YAP in knockout mice induced embryonic lethality at E8.5 due to severe developmental defects (141). Although TAZ knockout mice show only partial lethality, with 20% of the mice remaining viable, the survivors develop renal cysts and lung emphysema (142–144). Since YAP/TAZ has so many important physiological functions, as evidenced by YAP-null and TAZ-null mice, careful targeting of the YAP/TAZ signaling pathway to minimize systemic effects is clearly a highly desirable goal in PDAC treatment. Anti-YAP/TAZ strategies to selectively block aberrant YAP/TAZ signal activation are attractive and rational. Biomarker analysis to identify aberrant YAP/TAZ signal activation may therefore be the next step to establish an efficient therapeutic approach.
Funding
This work was supported by a Grant-in-Aid for Scientists (C); the Ministry of Education, Culture, Sports, Science, and Technology of Japan, No. 19K09177 (to HH); the Shinnihon Foundation of Advanced Medical Treatment Research (HH), the Takeda Science Foundation, Japan (to HH), and Public Trust Surgery Research Fund (to HH).
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
HH and HB conducted the topic investigated in this paper. NU, LS, KM, HS, and YS assisted in the useful discussions and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ADM, Acinar-to-ductal metaplasia; CAFs, cancer-associated fibroblasts; CTGF, connective tissue growth factor, CYR61, cysteine-rich angiogenic inducer 61; DKO, double knockout; ECM, extracellular matrix; EMT, epithelial-to-mesenchymal transition; FBXW7, F-box and WD repeat domain containing 7; FOLFIRINOX, 5-fluorouracil, folinic acid, irinotecan, and oxaliplatin; GEMM, genetically engineered mouse model; GnP, gemcitabine plus nab-paclitaxel; ICMT, Isoprenylcysteine carboxylmethyltransferase; LATS1/2, large tumor suppressor 1/2; MST1/2, mammalian sterile 20-like kinase 1/2; MMR, mismatch repair; PDAC, pancreatic ductal adenocarcinoma; TAZ, transcriptional co-activator with PDZ-binding motif; TEAD, TEA domain transcriptional factor; PD-L1, programmed cell death-ligand 1; Pdx-1, pancreatic and duodenal homeobox 1; YAP, Yes-associated protein.
References
1
Siegel RL Miller KD Jemal A . Cancer Statistics, 2019. CA Cancer J Clin (2019) 69(1):7–34. doi: 10.3322/caac.21551
2
Notta F Chan-Seng-Yue M Lemire M Li Y Wilson GW Connor AA et al . A Renewed Model of Pancreatic Cancer Evolution Based on Genomic Rearrangement Patterns. Nature (2016) 538(7625):378–82. doi: 10.1038/nature19823
3
Conroy T Desseigne F Ychou M Bouche O Guimbaud R Becouarn Y et al . FOLFIRINOX Versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med (2011) 364(19):1817–25. doi: 10.1056/NEJMoa1011923
4
Von Hoff DD Ervin T Arena FP Chiorean EG Infante J Moore M et al . Increased Survival in Pancreatic Cancer With Nab-Paclitaxel Plus Gemcitabine. N Engl J Med (2013) 369(18):1691–703. doi: 10.1056/NEJMoa1304369
5
Nishio K Kimura K Amano R Yamazoe S Ohrira G Nakata B et al . Preoperative Predictors for Early Recurrence of Resectable Pancreatic Cancer. World J Surg Oncol (2017) 15(1):16. doi: 10.1186/s12957-016-1078-z
6
Chand S O’Hayer K Blanco FF Winter JM Brody JR . The Landscape of Pancreatic Cancer Therapeutic Resistance Mechanisms. Int J Biol Sci (2016) 12(3):273–82. doi: 10.7150/ijbs.14951
7
Hayashi H Higashi T Miyata T Yamashita Y-i Baba H . Recent Advances in Precision Medicine for Pancreatic Ductal Adenocarcinoma. Ann Gastroenterol Surg (2021). doi: 10.1002/ags3.12436 [in press]
8
Harvey KF Pfleger CM Hariharan IK . The Drosophila Mst Ortholog, Hippo, Restricts Growth and Cell Proliferation and Promotes Apoptosis. Cell (2003) 114(4):457–67. doi: 10.1016/S0092-8674(03)00557-9
9
Wu S Huang J Dong J Pan D . Hippo Encodes a Ste-20 Family Protein Kinase That Restricts Cell Proliferation and Promotes Apoptosis in Conjunction With Salvador and Warts. Cell (2003) 114(4):445–56. doi: 10.1016/S0092-8674(03)00549-X
10
Huang J Wu S Barrera J Matthews K Pan D . The Hippo Signaling Pathway Coordinately Regulates Cell Proliferation and Apoptosis by Inactivating Yorkie, the Drosophila Homolog of YAP. Cell (2005) 122(3):421–34. doi: 10.1016/j.cell.2005.06.007
11
Maugeri-Sacca M De Maria R . The Hippo Pathway in Normal Development and Cancer. Pharmacol Ther (2018) 186:60–72. doi: 10.1016/j.pharmthera.2017.12.011
12
Dupont S Morsut L Aragona M Enzo E Giulitti S Cordenonsi M et al . Role of YAP/TAZ in Mechanotransduction. Nature (2011) 474(7350):179–83. doi: 10.1038/nature10137
13
Dupont S . Role of YAP/TAZ in Cell-Matrix Adhesion-Mediated Signalling and Mechanotransduction. Exp Cell Res (2016) 343(1):42–53. doi: 10.1016/j.yexcr.2015.10.034
14
Varelas X Samavarchi-Tehrani P Narimatsu M Weiss A Cockburn K Larsen BG et al . The Crumbs Complex Couples Cell Density Sensing to Hippo-Dependent Control of the TGF-Beta-SMAD Pathway. Dev Cell (2010) 19(6):831–44. doi: 10.1016/j.devcel.2010.11.012
15
Fujii M Toyoda T Nakanishi H Yatabe Y Sato A Matsudaira Y et al . TGF-Beta Synergizes With Defects in the Hippo Pathway to Stimulate Human Malignant Mesothelioma Growth. J Exp Med (2012) 209(3):479–94. doi: 10.1084/jem.20111653
16
Azzolin L Zanconato F Bresolin S Forcato M Basso G Bicciato S et al . Role of TAZ as Mediator of Wnt Signaling. Cell (2012) 151(7):1443–56. doi: 10.1016/j.cell.2012.11.027
17
Varelas X Miller BW Sopko R Song S Gregorieff A Fellouse FA et al . The Hippo Pathway Regulates Wnt/beta-Catenin Signaling. Dev Cell (2010) 18(4):579–91. doi: 10.1016/j.devcel.2010.03.007
18
Fernandez LA Northcott PA Dalton J Fraga C Ellison D Angers S et al . YAP1 is Amplified and Up-Regulated in Hedgehog-Associated Medulloblastomas and Mediates Sonic Hedgehog-Driven Neural Precursor Proliferation. Genes Dev (2009) 23(23):2729–41. doi: 10.1101/gad.1824509
19
Lin YT Ding JY Li MY Yeh TS Wang TW Yu JY . YAP Regulates Neuronal Differentiation Through Sonic Hedgehog Signaling Pathway. Exp Cell Res (2012) 318(15):1877–88. doi: 10.1016/j.yexcr.2012.05.005
20
Chen HJ Wang CM Wang TW Liaw GJ Hsu TH Lin TH et al . The Hippo Pathway Controls Polar Cell Fate Through Notch Signaling During Drosophila Oogenesis. Dev Biol (2011) 357(2):370–9. doi: 10.1016/j.ydbio.2011.07.003
21
Yu J Poulton J Huang YC Deng WM . The Hippo Pathway Promotes Notch Signaling in Regulation of Cell Differentiation, Proliferation, and Oocyte Polarity. PLoS One (2008) 3(3):e1761. doi: 10.1371/journal.pone.0001761
22
Lai ZC Wei X Shimizu T Ramos E Rohrbaugh M Nikolaidis N et al . Control of Cell Proliferation and Apoptosis by Mob as Tumor Suppressor, Mats. Cell (2005) 120(5):675–85. doi: 10.1016/j.cell.2004.12.036
23
He M Zhou Z Shah AA Hong Y Chen Q Wan Y . New Insights Into Posttranslational Modifications of Hippo Pathway in Carcinogenesis and Therapeutics. Cell Div (2016) 11:4. doi: 10.1186/s13008-016-0013-6
24
Zhao B Li L Tumaneng K Wang CY Guan KL . A Coordinated Phosphorylation by Lats and CK1 Regulates YAP Stability Through SCF(beta-TRCP). Genes Dev (2010) 24(1):72–85. doi: 10.1101/gad.1843810
25
Lei QY Zhang H Zhao B Zha ZY Bai F Pei XH et al . TAZ Promotes Cell Proliferation and Epithelial-Mesenchymal Transition and Is Inhibited by the Hippo Pathway. Mol Cell Biol (2008) 28(7):2426–36. doi: 10.1128/MCB.01874-07
26
Yu FX Zhao B Guan KL . Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell (2015) 163(4):811–28. doi: 10.1016/j.cell.2015.10.044
27
Poma AM Torregrossa L Bruno R Basolo F Fontanini G . Hippo Pathway Affects Survival of Cancer Patients: Extensive Analysis of TCGA Data and Review of Literature. Sci Rep (2018) 8(1):10623. doi: 10.1038/s41598-018-28928-3
28
Moroishi T Hansen CG Guan KL . The Emerging Roles of YAP and TAZ in Cancer. Nat Rev Cancer (2015) 15(2):73–9. doi: 10.1038/nrc3876
29
Lehmann W Mossmann D Kleemann J Mock K Meisinger C Brummer T et al . ZEB1 Turns Into a Transcriptional Activator by Interacting With YAP1 in Aggressive Cancer Types. Nat Commun (2016) 7:10498. doi: 10.1038/ncomms10498
30
Murakami S Shahbazian D Surana R Zhang W Chen H Graham GT et al . Yes-Associated Protein Mediates Immune Reprogramming in Pancreatic Ductal Adenocarcinoma. Oncogene (2017) 36(9):1232–44. doi: 10.1038/onc.2016.288
31
Plouffe SW Lin KC Moore JL 3rd Tan FE Ma S Ye Z et al . The Hippo Pathway Effector Proteins YAP and TAZ Have Both Distinct and Overlapping Functions in the Cell. J Biol Chem (2018) 293(28):11230–40. doi: 10.1074/jbc.RA118.002715
32
Hayashi H Higashi T Yokoyama N Kaida T Sakamoto K Fukushima Y et al . An Imbalance in TAZ and YAP Expression in Hepatocellular Carcinoma Confers Cancer Stem Cell-Like Behaviors Contributing to Disease Progression. Cancer Res (2015) 75(22):4985–97. doi: 10.1158/0008-5472.CAN-15-0291
33
Pictet RL Clark WR Williams RH Rutter WJ . An Ultrastructural Analysis of the Developing Embryonic Pancreas. Dev Biol (1972) 29(4):436–67. doi: 10.1016/0012-1606(72)90083-8
34
George NM Day CE Boerner BP Johnson RL Sarvetnick NE . Hippo Signaling Regulates Pancreas Development Through Inactivation of Yap. Mol Cell Biol (2012) 32(24):5116–28. doi: 10.1128/MCB.01034-12
35
Gao T Zhou D Yang C Singh T Penzo-Mendez A Maddipati R et al . Hippo Signaling Regulates Differentiation and Maintenance in the Exocrine Pancreas. Gastroenterology (2013) 144(7):1543–53.e1. doi: 10.1053/j.gastro.2013.02.037
36
Zhang W Nandakumar N Shi Y Manzano M Smith A Graham G et al . Downstream of Mutant KRAS, the Transcription Regulator YAP Is Essential for Neoplastic Progression to Pancreatic Ductal Adenocarcinoma. Sci Signal (2014) 7(324):ra42. doi: 10.1126/scisignal.2005049
37
Dong J Feldmann G Huang J Wu S Zhang N Comerford SA et al . Elucidation of a Universal Size-Control Mechanism in Drosophila and Mammals. Cell (2007) 130(6):1120–33. doi: 10.1016/j.cell.2007.07.019
38
Lu L Li Y Kim SM Bossuyt W Liu P Qiu Q et al . Hippo Signaling is a Potent In Vivo Growth and Tumor Suppressor Pathway in the Mammalian Liver. Proc Natl Acad Sci USA (2010) 107(4):1437–42. doi: 10.1073/pnas.0911427107
39
Aung KL Fischer SE Denroche RE Jang GH Dodd A Creighton S et al . Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results From the COMPASS Trial. Clin Cancer Res (2018) 24(6):1344–54. doi: 10.1158/1078-0432.CCR-17-2994
40
Jones S Zhang X Parsons DW Lin JC Leary RJ Angenendt P et al . Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science (2008) 321(5897):1801–6. doi: 10.1126/science.1164368
41
Biankin AV Waddell N Kassahn KS Gingras MC Muthuswamy LB Johns AL et al . Pancreatic Cancer Genomes Reveal Aberrations in Axon Guidance Pathway Genes. Nature (2012) 491(7424):399–405. doi: 10.1038/nature11547
42
Waddell N Pajic M Patch AM Chang DK Kassahn KS Bailey P et al . Whole Genomes Redefine the Mutational Landscape of Pancreatic Cancer. Nature (2015) 518(7540):495–501. doi: 10.1038/nature14169
43
Witkiewicz AK McMillan EA Balaji U Baek G Lin WC Mansour J et al . Whole-Exome Sequencing of Pancreatic Cancer Defines Genetic Diversity and Therapeutic Targets. Nat Commun (2015) 6:6744. doi: 10.1038/ncomms7744
44
Sausen M Phallen J Adleff V Jones S Leary RJ Barrett MT et al . Clinical Implications of Genomic Alterations in the Tumour and Circulation of Pancreatic Cancer Patients. Nat Commun (2015) 6:7686. doi: 10.1158/1538-7445.AM2015-619
45
Cancer Genome Atlas Research Network Electronic address aadhe . Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell (2017) 32(2):185–203.e13. doi: 10.1016/j.ccell.2017.07.007
46
Murphy SJ Hart SN Lima JF Kipp BR Klebig M Winters JL et al . Genetic Alterations Associated With Progression From Pancreatic Intraepithelial Neoplasia to Invasive Pancreatic Tumor. Gastroenterology (2013) 145(5):1098–109.e1. doi: 10.1053/j.gastro.2013.07.049
47
Kanda M Matthaei H Wu J Hong SM Yu J Borges M et al . Presence of Somatic Mutations in Most Early-Stage Pancreatic Intraepithelial Neoplasia. Gastroenterology (2012) 142(4):730–3.e9. doi: 10.1053/j.gastro.2011.12.042
48
Hezel AF Kimmelman AC Stanger BZ Bardeesy N Depinho RA . Genetics and Biology of Pancreatic Ductal Adenocarcinoma. Genes Dev (2006) 20(10):1218–49. doi: 10.1101/gad.1415606
49
Perez-Mancera PA Guerra C Barbacid M Tuveson DA . What We Have Learned About Pancreatic Cancer From Mouse Models. Gastroenterology (2012) 142(5):1079–92. doi: 10.1053/j.gastro.2012.03.002
50
Hingorani SR Petricoin EF Maitra A Rajapakse V King C Jacobetz MA et al . Preinvasive and Invasive Ductal Pancreatic Cancer and Its Early Detection in the Mouse. Cancer Cell (2003) 4(6):437–50. doi: 10.1016/S1535-6108(03)00309-X
51
Hingorani SR Wang L Multani AS Combs C Deramaudt TB Hruban RH et al . Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell (2005) 7(5):469–83. doi: 10.1016/j.ccr.2005.04.023
52
Hruban RH Adsay NV Albores-Saavedra J Anver MR Biankin AV Boivin GP et al . Pathology of Genetically Engineered Mouse Models of Pancreatic Exocrine Cancer: Consensus Report and Recommendations. Cancer Res (2006) 66(1):95–106. doi: 10.1158/0008-5472.CAN-05-2168
53
Hruban RH Rustgi AK Brentnall TA Tempero MA Wright CV Tuveson DA . Pancreatic Cancer in Mice and Man: The Penn Workshop 2004. Cancer Res (2006) 66(1):14–7. doi: 10.1158/0008-5472.CAN-05-3914
54
Aguirre AJ Bardeesy N Sinha M Lopez L Tuveson DA Horner J et al . Activated Kras and Ink4a/Arf Deficiency Cooperate to Produce Metastatic Pancreatic Ductal Adenocarcinoma. Genes Dev (2003) 17(24):3112–26. doi: 10.1101/gad.1158703
55
Guerra C Schuhmacher AJ Canamero M Grippo PJ Verdaguer L Perez-Gallego L et al . Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell (2007) 11(3):291–302. doi: 10.1016/j.ccr.2007.01.012
56
Collins MA Bednar F Zhang Y Brisset JC Galban S Galban CJ et al . Oncogenic Kras Is Required for Both the Initiation and Maintenance of Pancreatic Cancer in Mice. J Clin Invest (2012) 122(2):639–53. doi: 10.1172/JCI59227
57
di Magliano MP Logsdon CD . Roles for KRAS in Pancreatic Tumor Development and Progression. Gastroenterology (2013) 144(6):1220–9. doi: 10.1053/j.gastro.2013.01.071
58
Ying H Kimmelman AC Lyssiotis CA Hua S Chu GC Fletcher-Sananikone E et al . Oncogenic Kras Maintains Pancreatic Tumors Through Regulation of Anabolic Glucose Metabolism. Cell (2012) 149(3):656–70. doi: 10.1016/j.cell.2012.01.058
59
Gruber R Panayiotou R Nye E Spencer-Dene B Stamp G Behrens A . YAP1 and TAZ Control Pancreatic Cancer Initiation in Mice by Direct Up-Regulation of JAK-STAT3 Signaling. Gastroenterology (2016) 151(3):526–39. doi: 10.1053/j.gastro.2016.05.006
60
Zhang Q Zhang Y Parsels JD Lohse I Lawrence TS Pasca di Magliano M et al . Fbxw7 Deletion Accelerates Kras(G12D)-Driven Pancreatic Tumorigenesis via Yap Accumulation. Neoplasia (2016) 18(11):666–73. doi: 10.1016/j.neo.2016.08.009
61
Kapoor A Yao W Ying H Hua S Liewen A Wang Q et al . Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell (2014) 158(1):185–97. doi: 10.1016/j.cell.2014.06.003
62
Shao DD Xue W Krall EB Bhutkar A Piccioni F Wang X et al . KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell (2014) 158(1):171–84. doi: 10.1016/j.cell.2014.06.004
63
Ying H Dey P Yao W Kimmelman AC Draetta GF Maitra A et al . Genetics and Biology of Pancreatic Ductal Adenocarcinoma. Genes Dev (2016) 30(4):355–85. doi: 10.1101/gad.275776.115
64
Wang Y Xu X Maglic D Dill MT Mojumdar K Ng PK et al . Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep (2018) 25(5):1304–17.e5. doi: 10.1016/j.celrep.2018.10.001
65
Diep CH Zucker KM Hostetter G Watanabe A Hu C Munoz RM et al . Down-Regulation of Yes Associated Protein 1 Expression Reduces Cell Proliferation and Clonogenicity of Pancreatic Cancer Cells. PLoS One (2012) 7(3):e32783. doi: 10.1371/journal.pone.0032783
66
Yang S Zhang L Purohit V Shukla SK Chen X Yu F et al . Active YAP Promotes Pancreatic Cancer Cell Motility, Invasion and Tumorigenesis in a Mitotic Phosphorylation-Dependent Manner Through LPAR3. Oncotarget (2015) 6(34):36019–31. doi: 10.18632/oncotarget.5935
67
Xie D Cui J Xia T Jia Z Wang L Wei W et al . Hippo Transducer TAZ Promotes Epithelial Mesenchymal Transition and Supports Pancreatic Cancer Progression. Oncotarget (2015) 6(34):35949–63. doi: 10.18632/oncotarget.5772
68
Salcedo Allende MT Zeron-Medina J Hernandez J Macarulla T Balsells J Merino X et al . Overexpression of Yes Associated Protein 1, an Independent Prognostic Marker in Patients With Pancreatic Ductal Adenocarcinoma, Correlated With Liver Metastasis and Poor Prognosis. Pancreas (2017) 46(7):913–20. doi: 10.1097/MPA.0000000000000867
69
Rozengurt E Sinnett-Smith J Eibl G . Yes-Associated Protein (YAP) in Pancreatic Cancer: At the Epicenter of a Targetable Signaling Network Associated With Patient Survival. Signal Transduct Target Ther (2018) 3:11. doi: 10.1038/s41392-017-0005-2
70
Zhou Q Bauden M Andersson R Hu D Marko-Varga G Xu J et al . YAP1 is an Independent Prognostic Marker in Pancreatic Cancer and Associated With Extracellular Matrix Remodeling. J Transl Med (2020) 18(1):77. doi: 10.1186/s12967-020-02254-7
71
Strnadel J Choi S Fujimura K Wang H Zhang W Wyse M et al . Eif5a-PEAK1 Signaling Regulates YAP1/TAZ Protein Expression and Pancreatic Cancer Cell Growth. Cancer Res (2017) 77(8):1997–2007. doi: 10.1158/0008-5472.CAN-16-2594
72
Hao F Xu Q Zhao Y Stevens JV Young SH Sinnett-Smith J et al . Insulin Receptor and GPCR Crosstalk Stimulates YAP via PI3K and PKD in Pancreatic Cancer Cells. Mol Cancer Res (2017) 15(7):929–41. doi: 10.1158/1541-7786.MCR-17-0023
73
Tsai JH Yang J . Epithelial-Mesenchymal Plasticity in Carcinoma Metastasis. Genes Dev (2013) 27(20):2192–206. doi: 10.1101/gad.225334.113
74
Yuan Y Li D Li H Wang L Tian G Dong Y . YAP Overexpression Promotes the Epithelial-Mesenchymal Transition and Chemoresistance in Pancreatic Cancer Cells. Mol Med Rep (2016) 13(1):237–42. doi: 10.3892/mmr.2015.4550
75
Wei H Xu Z Liu F Wang F Wang X Sun X et al . Hypoxia Induces Oncogene Yes-Associated Protein 1 Nuclear Translocation to Promote Pancreatic Ductal Adenocarcinoma Invasion via Epithelial-Mesenchymal Transition. Tumour Biol (2017) 39(5):1010428317691684. doi: 10.1177/1010428317691684
76
Narimatsu M Labibi B Wrana JL Attisano L . Analysis of Hippo and TGFbeta Signaling in Polarizing Epithelial Cells and Mouse Embryos. Differentiation (2016) 91(4-5):109–18. doi: 10.1016/j.diff.2016.01.003
77
Pefani DE Pankova D Abraham AG Grawenda AM Vlahov N Scrace S et al . TGF-Beta Targets the Hippo Pathway Scaffold RASSF1A to Facilitate YAP/SMAD2 Nuclear Translocation. Mol Cell (2016) 63(1):156–66. doi: 10.1016/j.molcel.2016.05.012
78
Gao C Quan MY Chen QJ Yang R Wu Y Liu JY et al . Yap1-2 Isoform Is the Primary Mediator in TGF-Beta1 Induced EMT in Pancreatic Cancer. Front Oncol (2021) 11:649290. doi: 10.3389/fonc.2021.649290
79
Burris HA 3rd Moore MJ Andersen J Green MR Rothenberg ML Modiano MR et al . Improvements in Survival and Clinical Benefit With Gemcitabine as First-Line Therapy for Patients With Advanced Pancreas Cancer: A Randomized Trial. J Clin Oncol (1997) 15(6):2403–13. doi: 10.1200/JCO.1997.15.6.2403
80
Chen M Wang M Xu S Guo X Jiang J . Upregulation of miR-181c Contributes to Chemoresistance in Pancreatic Cancer by Inactivating the Hippo Signaling Pathway. Oncotarget (2015) 6(42):44466–79. doi: 10.18632/oncotarget.6298
81
Chai TF Manu KA Casey PJ Wang M . Isoprenylcysteine Carboxylmethyltransferase Is Required for the Impact of Mutant KRAS on TAZ Protein Level and Cancer Cell Self-Renewal. Oncogene (2020) 39(31):5373–89. doi: 10.1038/s41388-020-1364-7
82
Suklabaidya S Dash P Das B Suresh V Sasmal PK Senapati S . Experimental Models of Pancreatic Cancer Desmoplasia. Lab Invest (2018) 98(1):27–40. doi: 10.1038/labinvest.2017.127
83
Ren B Liu X Suriawinata AA . Pancreatic Ductal Adenocarcinoma and Its Precursor Lesions: Histopathology, Cytopathology, and Molecular Pathology. Am J Pathol (2019) 189(1):9–21. doi: 10.1016/j.ajpath.2018.10.004
84
Panciera T Azzolin L Cordenonsi M Piccolo S . Mechanobiology of YAP and TAZ in Physiology and Disease. Nat Rev Mol Cell Biol (2017) 18(12):758–70. doi: 10.1038/nrm.2017.87
85
Low BC Pan CQ Shivashankar GV Bershadsky A Sudol M Sheetz M . YAP/TAZ as Mechanosensors and Mechanotransducers in Regulating Organ Size and Tumor Growth. FEBS Lett (2014) 588(16):2663–70. doi: 10.1016/j.febslet.2014.04.012
86
Ibar C Irvine KD . Rapping About Mechanotransduction. Dev Cell (2018) 46(6):678–9. doi: 10.1016/j.devcel.2018.09.007
87
Morvaridi S Dhall D Greene MI Pandol SJ Wang Q . Role of YAP and TAZ in Pancreatic Ductal Adenocarcinoma and in Stellate Cells Associated With Cancer and Chronic Pancreatitis. Sci Rep (2015) 5:16759. doi: 10.1038/srep16759
88
Lee J Condello S Yakubov B Emerson R Caperell-Grant A Hitomi K et al . Tissue Transglutaminase Mediated Tumor-Stroma Interaction Promotes Pancreatic Cancer Progression. Clin Cancer Res (2015) 21(19):4482–93. doi: 10.1158/1078-0432.CCR-15-0226
89
Xu M Jung X Hines OJ Eibl G Chen Y . Obesity and Pancreatic Cancer: Overview of Epidemiology and Potential Prevention by Weight Loss. Pancreas (2018) 47(2):158–62. doi: 10.1097/MPA.0000000000000974
90
Abbruzzese JL Andersen DK Borrebaeck CAK Chari ST Costello E Cruz-Monserrate Z et al . The Interface of Pancreatic Cancer With Diabetes, Obesity, and Inflammation: Research Gaps and Opportunities: Summary of a National Institute of Diabetes and Digestive and Kidney Diseases Workshop. Pancreas (2018) 47(5):516–25. doi: 10.1097/MPA.0000000000001037
91
von Bernstorff W Voss M Freichel S Schmid A Vogel I Johnk C et al . Systemic and Local Immunosuppression in Pancreatic Cancer Patients. Clin Cancer Res (2001) 7(3 Suppl):925s–32s.
92
Mace TA Ameen Z Collins A Wojcik S Mair M Young GS et al . Pancreatic Cancer-Associated Stellate Cells Promote Differentiation of Myeloid-Derived Suppressor Cells in a STAT3-Dependent Manner. Cancer Res (2013) 73(10):3007–18. doi: 10.1158/0008-5472.CAN-12-4601
93
Stromnes IM Brockenbrough JS Izeradjene K Carlson MA Cuevas C Simmons RM et al . Targeted Depletion of an MDSC Subset Unmasks Pancreatic Ductal Adenocarcinoma to Adaptive Immunity. Gut (2014) 63(11):1769–81. doi: 10.1136/gutjnl-2013-306271
94
Thaventhiran JE Hoffmann A Magiera L de la Roche M Lingel H Brunner-Weinzierl M et al . Activation of the Hippo Pathway by CTLA-4 Regulates the Expression of Blimp-1 in the CD8+ T Cell. Proc Natl Acad Sci USA (2012) 109(33):E2223–9. doi: 10.1073/pnas.1209115109
95
Geng J Yu S Zhao H Sun X Li X Wang P et al . The Transcriptional Coactivator TAZ Regulates Reciprocal Differentiation of TH17 Cells and Treg Cells. Nat Immunol (2017) 18(7):800–12. doi: 10.1038/ni.3748
96
Onuora S . Immunology: Hippo Signalling Influences T Cell Fate. Nat Rev Rheumatol (2017) 13(7):389. doi: 10.1038/nrrheum.2017.82
97
Ni X Tao J Barbi J Chen Q Park BV Li Z et al . YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov (2018) 8(8):1026–43. doi: 10.1158/2159-8290.CD-17-1124
98
Zhang YL Li Q Yang XM Fang F Li J Wang YH et al . SPON2 Promotes M1-Like Macrophage Recruitment and Inhibits Hepatocellular Carcinoma Metastasis by Distinct Integrin-Rho GTPase-Hippo Pathways. Cancer Res (2018) 78(9):2305–17. doi: 10.1158/0008-5472.CAN-17-2867
99
Humphris JL Patch AM Nones K Bailey PJ Johns AL McKay S et al . Hypermutation In Pancreatic Cancer. Gastroenterology (2017) 152(1):68–74.e2. doi: 10.1053/j.gastro.2016.09.060
100
Le DT Durham JN Smith KN Wang H Bartlett BR Aulakh LK et al . Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
101
Marcus L Lemery SJ Keegan P Pazdur R . FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin Cancer Res (2019) 25(13):3753–8. doi: 10.1158/1078-0432.CCR-18-4070
102
Marabelle A Le DT Ascierto PA Di Giacomo AM De Jesus-Acosta A Delord JP et al . Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol (2020) 38(1):1–10. doi: 10.1200/JCO.19.02105
103
Janse van Rensburg HJ Azad T Ling M Hao Y Snetsinger B Khanal P et al . The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer Through PD-L1. Cancer Res (2018) 78(6):1457–70. doi: 10.1158/0008-5472.CAN-17-3139
104
Kim MH Kim CG Kim SK Shin SJ Choe EA Park SH et al . YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol Res (2018) 6(3):255–66. doi: 10.1158/2326-6066.CIR-17-0320
105
Miao J Hsu PC Yang YL Xu Z Dai Y Wang Y et al . YAP Regulates PD-L1 Expression in Human NSCLC Cells. Oncotarget (2017) 8(70):114576–87. doi: 10.18632/oncotarget.23051
106
Lee BS Park DI Lee DH Lee JE Yeo MK Park YH et al . Hippo Effector YAP Directly Regulates the Expression of PD-L1 Transcripts in EGFR-TKI-Resistant Lung Adenocarcinoma. Biochem Biophys Res Commun (2017) 491(2):493–9. doi: 10.1016/j.bbrc.2017.07.007
107
Hong DS Fakih MG Strickler JH Desai J Durm GA Shapiro GI et al . KRAS(G12C) Inhibition With Sotorasib in Advanced Solid Tumors. N Engl J Med (2020) 383(13):1207–17. doi: 10.1056/NEJMoa1917239
108
Istvan ES Deisenhofer J . Structural Mechanism for Statin Inhibition of HMG-CoA Reductase. Science (2001) 292(5519):1160–4. doi: 10.1126/science.1059344
109
Freed-Pastor WA Mizuno H Zhao X Langerod A Moon SH Rodriguez-Barrueco R et al . Mutant P53 Disrupts Mammary Tissue Architecture via the Mevalonate Pathway. Cell (2012) 148(1-2):244–58. doi: 10.1016/j.cell.2011.12.017
110
Clendening JW Pandyra A Boutros PC El Ghamrasni S Khosravi F Trentin GA et al . Dysregulation of the Mevalonate Pathway Promotes Transformation. Proc Natl Acad Sci USA (2010) 107(34):15051–6. doi: 10.1073/pnas.0910258107
111
Kuzu OF Noory MA Robertson GP . The Role of Cholesterol in Cancer. Cancer Res (2016) 76(8):2063–70. doi: 10.1158/0008-5472.CAN-15-2613
112
Mohammed A Qian L Janakiram NB Lightfoot S Steele VE Rao CV . Atorvastatin Delays Progression of Pancreatic Lesions to Carcinoma by Regulating PI3/AKT Signaling in p48Cre/+ LSL-KrasG12D/+ Mice. Int J Cancer (2012) 131(8):1951–62. doi: 10.1002/ijc.27456
113
Fendrich V Sparn M Lauth M Knoop R Plassmeier L Bartsch DK et al . Simvastatin Delay Progression of Pancreatic Intraepithelial Neoplasia and Cancer Formation in a Genetically Engineered Mouse Model of Pancreatic Cancer. Pancreatology (2013) 13(5):502–7. doi: 10.1016/j.pan.2013.08.002
114
Sorrentino G Ruggeri N Specchia V Cordenonsi M Mano M Dupont S et al . Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat Cell Biol (2014) 16(4):357–66. doi: 10.1038/ncb2936
115
Higashi T Hayashi H Kitano Y Yamamura K Kaida T Arima K et al . Statin Attenuates Cell Proliferative Ability via TAZ (WWTR1) in Hepatocellular Carcinoma. Med Oncol (2016) 33(11):123. doi: 10.1007/s12032-016-0845-6
116
Jeon CY Pandol SJ Wu B Cook-Wiens G Gottlieb RA Merz CN et al . The Association of Statin Use After Cancer Diagnosis With Survival in Pancreatic Cancer Patients: A SEER-Medicare Analysis. PLoS One (2015) 10(4):e0121783. doi: 10.1371/journal.pone.0121783
117
Wu BU Chang J Jeon CY Pandol SJ Huang B Ngor EW et al . Impact of Statin Use on Survival in Patients Undergoing Resection for Early-Stage Pancreatic Cancer. Am J Gastroenterol (2015) 110(8):1233–9. doi: 10.1038/ajg.2015.217
118
Chen MJ Tsan YT Liou JM Lee YC Wu MS Chiu HM et al . Statins and the Risk of Pancreatic Cancer in Type 2 Diabetic Patients–A Population-Based Cohort Study. Int J Cancer (2016) 138(3):594–603. doi: 10.1002/ijc.29813
119
Walker EJ Ko AH Holly EA Bracci PM . Statin Use and Risk of Pancreatic Cancer: Results From a Large, Clinic-Based Case-Control Study. Cancer (2015) 121(8):1287–94. doi: 10.1002/cncr.29256
120
Carey FJ Little MW Pugh TF Ndokera R Ing H Clark A et al . The Differential Effects of Statins on the Risk of Developing Pancreatic Cancer: A Case-Control Study in Two Centres in the United Kingdom. Dig Dis Sci (2013) 58(11):3308–12. doi: 10.1007/s10620-013-2778-7
121
JY E Lu SE Lin Y Graber JM Rotter D Zhang L et al . Differential and Joint Effects of Metformin and Statins on Overall Survival of Elderly Patients With Pancreatic Adenocarcinoma: A Large Population-Based Study. Cancer Epidemiol Biomarkers Prev (2017) 26(8):1225–32. doi: 10.1158/1055-9965.EPI-17-0227
122
Iarrobino NA Gill B Bernard ME Mishra MV Champ CE . Targeting Tumor Metabolism With Statins During Treatment for Advanced-Stage Pancreatic Cancer. Am J Clin Oncol (2018) 41(11):1125–31. doi: 10.1097/COC.0000000000000433
123
Jian-Yu E Graber JM Lu SE Lin Y Lu-Yao G Tan XL . Effect of Metformin and Statin Use on Survival in Pancreatic Cancer Patients: A Systematic Literature Review and Meta-Analysis. Curr Med Chem (2018) 25(22):2595–607. doi: 10.2174/0929867324666170412145232
124
Bang UC Watanabe T Bendtsen F . The Relationship Between the Use of Statins and Mortality, Severity, and Pancreatic Cancer in Danish Patients With Chronic Pancreatitis. Eur J Gastroenterol Hepatol (2018) 30(3):346–51. doi: 10.1097/MEG.0000000000001060
125
Liu Y Tang W Wang J Xie L Li T He Y et al . Association Between Statin Use and Colorectal Cancer Risk: A Meta-Analysis of 42 Studies. Cancer Causes Control (2014) 25(2):237–49. doi: 10.1007/s10552-013-0326-6
126
Manthravadi S Shrestha A Madhusudhana S . Impact of Statin Use on Cancer Recurrence and Mortality in Breast Cancer: A Systematic Review and Meta-Analysis. Int J Cancer (2016) 139(6):1281–8. doi: 10.1002/ijc.30185
127
Anothaisintawee T Udomsubpayakul U McEvoy M Lerdsitthichai P Attia J Thakkinstian A . Effect of Lipophilic and Hydrophilic Statins on Breast Cancer Risk in Thai Women: A Cross-Sectional Study. J Cancer (2016) 7(9):1163–8. doi: 10.7150/jca.14941
128
Liu B Yi Z Guan X Zeng YX Ma F . The Relationship Between Statins and Breast Cancer Prognosis Varies by Statin Type and Exposure Time: A Meta-Analysis. Breast Cancer Res Treat (2017) 164(1):1–11. doi: 10.1007/s10549-017-4246-0
129
Hamada T Khalaf N Yuan C Babic A Morales-Oyarvide V Qian ZR et al . Statin Use and Pancreatic Cancer Risk in Two Prospective Cohort Studies. J Gastroenterol (2018) 53(8):959–66. doi: 10.1007/s00535-018-1430-x
130
Hamada T Khalaf N Yuan C Morales-Oyarvide V Babic A Nowak JA et al . Prediagnosis Use of Statins Associates With Increased Survival Times of Patients With Pancreatic Cancer. Clin Gastroenterol Hepatol (2018) 16(8):1300–6.e3. doi: 10.1016/j.cgh.2018.02.022
131
Zhang Y Liang M Sun C Qu G Shi T Min M et al . Statin Use and Risk of Pancreatic Cancer: An Updated Meta-Analysis of 26 Studies. Pancreas (2019) 48(2):142–50. doi: 10.1097/MPA.0000000000001226
132
Lee HS Lee SH Lee HJ Chung MJ Park JY Park SW et al . Statin Use and Its Impact on Survival in Pancreatic Cancer Patients. Med (Baltimore) (2016) 95(19):e3607. doi: 10.1097/MD.0000000000003607
133
Zhao X Wang X Fang L Lan C Zheng X Wang Y et al . A Combinatorial Strategy Using YAP and Pan-RAF Inhibitors for Treating KRAS-Mutant Pancreatic Cancer. Cancer Lett (2017) 402:61–70. doi: 10.1016/j.canlet.2017.05.015
134
Wei H Wang F Wang Y Li T Xiu P Zhong J et al . Verteporfin Suppresses Cell Survival, Angiogenesis and Vasculogenic Mimicry of Pancreatic Ductal Adenocarcinoma via Disrupting the YAP-TEAD Complex. Cancer Sci (2017) 108(3):478–87. doi: 10.1111/cas.13138
135
Thongon N Castiglioni I Zucal C Latorre E D’Agostino V Bauer I et al . The GSK3beta Inhibitor BIS I Reverts YAP-Dependent EMT Signature in PDAC Cell Lines by Decreasing SMADs Expression Level. Oncotarget (2016) 7(18):26551–66. doi: 10.18632/oncotarget.8437
136
Neesse A Frese KK Bapiro TE Nakagawa T Sternlicht MD Seeley TW et al . CTGF Antagonism With mAb FG-3019 Enhances Chemotherapy Response Without Increasing Drug Delivery in Murine Ductal Pancreas Cancer. Proc Natl Acad Sci USA (2013) 110(30):12325–30. doi: 10.1073/pnas.1300415110
137
Zhou X Su J Feng S Wang L Yin X Yan J et al . Antitumor Activity of Curcumin is Involved in Down-Regulation of YAP/TAZ Expression in Pancreatic Cancer Cells. Oncotarget (2016) 7(48):79076–88. doi: 10.18632/oncotarget.12596
138
Jiang Z Chen X Chen K Sun L Gao L Zhou C et al . YAP Inhibition by Resveratrol via Activation of AMPK Enhances the Sensitivity of Pancreatic Cancer Cells to Gemcitabine. Nutrients (2016) 8(10):546. doi: 10.3390/nu8100546
139
Li X Liu Y Zhang C Niu Q Wang H Che C et al . Stiehopus Japonieus Acidic Mucopolysaccharide Inhibits the Proliferation of Pancreatic Cancer SW1990 Cells Through Hippo-YAP Pathway. Oncotarget (2017) 8(10):16356–66. doi: 10.18632/oncotarget.14633
140
Li X Zhao X Song W Tian Z Yang L Niu Q et al . Pseudolaric Acid B Inhibits Proliferation, Invasion and Epithelial-To-Mesenchymal Transition in Human Pancreatic Cancer Cell. Yonsei Med J (2018) 59(1):20–7. doi: 10.3349/ymj.2018.59.1.20
141
Morin-Kensicki EM Boone BN Howell M Stonebraker JR Teed J Alb JG et al . Defects in Yolk Sac Vasculogenesis, Chorioallantoic Fusion, and Embryonic Axis Elongation in Mice With Targeted Disruption of Yap65. Mol Cell Biol (2006) 26(1):77–87. doi: 10.1128/MCB.26.1.77-87.2006
142
Makita R Uchijima Y Nishiyama K Amano T Chen Q Takeuchi T et al . Multiple Renal Cysts, Urinary Concentration Defects, and Pulmonary Emphysematous Changes in Mice Lacking TAZ. Am J Physiol Renal Physiol (2008) 294(3):F542–53. doi: 10.1152/ajprenal.00201.2007
143
Hossain Z Ali SM Ko HL Xu J Ng CP Guo K et al . Glomerulocystic Kidney Disease in Mice With a Targeted Inactivation of Wwtr1. Proc Natl Acad Sci USA (2007) 104(5):1631–6. doi: 10.1073/pnas.0605266104
144
Tian Y Kolb R Hong JH Carroll J Li D You J et al . TAZ Promotes PC2 Degradation Through a SCFbeta-Trcp E3 Ligase Complex. Mol Cell Biol (2007) 27(18):6383–95. doi: 10.1128/MCB.00254-07
Summary
Keywords
pancreatic cancer, Hippo signaling pathway, Yes-associated protein, TAZ, KRAS mutation, pancreatic ductal adenocarcinoma
Citation
Hayashi H, Uemura N, Zhao L, Matsumura K, Sato H, Shiraishi Y and Baba H (2021) Biological Significance of YAP/TAZ in Pancreatic Ductal Adenocarcinoma. Front. Oncol. 11:700315. doi: 10.3389/fonc.2021.700315
Received
26 April 2021
Accepted
12 July 2021
Published
29 July 2021
Volume
11 - 2021
Edited by
Kanjoormana Aryan Manu, Amala Cancer Research Centre, India
Reviewed by
Bowen Zhu, Genome Institute of Singapore, Singapore; Jun Zhang, China Medical University, China
Updates
Copyright
© 2021 Hayashi, Uemura, Zhao, Matsumura, Sato, Shiraishi and Baba.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiromitsu Hayashi, hhayasi@kumamoto-u.ac.jp
This article was submitted to Gastrointestinal Cancers, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.