 Lin Tang
Lin Tang Rui Zhang
Rui Zhang Xiaoyu Zhang
Xiaoyu Zhang Li Yang
Li Yang- State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, and Collaborative Innovation Center for Biotherapy, Chengdu, China
In the past few decades, great progress has been made in the clinical application of dendritic cell (DC) vaccines loaded with personalized neoantigens. Personalized neoantigens are antigens arising from somatic mutations in cancers, with specificity to each patient. DC vaccines work based on the fundamental characteristics of DCs, which are professional antigen-presenting cells (APCs), responsible for the uptake, processing, and presentation of antigens to T cells to activate immune responses. Neoantigens can exert their antitumor effects only after they are taken up by APCs and presented to T cells. In recent years, neoantigen-based personalized tumor therapeutic vaccines have proven to be safe, immunogenic and feasible treatment strategies in patients with melanoma and glioblastoma that provide new hope in the treatment of cancer patients and a new approach to cure cancer. In addition, according to ClinicalTrials.gov, hundreds of registered DC vaccine trials are either completed or ongoing worldwide, of which 9 are in early phase I, 191 in phase I, 166 in phase II and 8 in phase III. Hundreds of clinical studies on therapeutic tumor vaccines globally have proven that DC vaccines are stable, reliable and very safe. However, in this process, many other factors still limit the effectiveness of the vaccine. This review will focus on the current research progress on personalized neoantigen-pulsed DC vaccines, their limitations and future research directions of DC vaccines loaded with neoantigens. This review aims to provide a better understanding of DCs biology and manipulation of activated DCs for DCs researchers to produce the next generation of highly efficient cancer vaccines for patients.
Introduction
Malignant tumors are still an acute threat for people worldwide and the incidence and mortality from cancer are still rapidly growing. GLOBOCAN showed an estimated 19.3 million new cases and 10 million cancer deaths worldwide in 2020; at the same time, an estimated 28.4 million new cancer cases are projected to occur in 2040 (1). Therefore, it is still difficult to find proper and effective ways to fight cancer.
After decades of effort, conventional methods and systems for treating cancer have been developed, including surgery, radiotherapy and chemotherapy alone or in combination. Surgery is the preferred treatment for most tumors; however, it is a traumatic and local treatment that easily leads to surgical complications. Although radiotherapy is the most suitable method for tumors in all parts of the body, the radiation dose that the body can withstand is limited, and normal cells are also damaged when tumor cells are destroyed. Although chemotherapy is successful for some tumors, such as testicular tumors, it can cause severe side effects, such as hair loss, anemia and organ damage, reducing the patients’ quality of life (2, 3). Because of the side effects of conventional treatments, cancer immunotherapy has been developed as a therapeutic method with better tumor targeting, safety, and a lower toxicity.
Cancer immunotherapy relies on the individual’s own immune system to recognize and control cancer progression to fight and cure cancer (4). At the same time, cancer immunotherapy has been developed to enhance the antitumor response of the immune system and reduce off-target effects and other serious side effects of other conventional therapies (5). There are five main types of cancer immunotherapy (6):
(i) Immune checkpoint inhibitors, in which the most extensive strategies involve the use of programmed death 1/programmed death ligand 1 blockade (PD1/PD-L1 blockade) and cytotoxic T lymphocyte-associated antigen-4 inhibition (CTLA-4 inhibition). Immune checkpoints are immunosuppressive pathways that regulate the immune response to maintain tolerance and protect the surrounding tissues. This property is used by tumor cells to escape the attack of immune cells, and immune checkpoint inhibitors can inhibit immune checkpoint activity and reactivate the immune response of T cells to the tumor to achieve an antitumor effect (7).
(ii) Cytokines, which contain three main types (interleukins, interferons, and granulocyte–macrophage colony-stimulating factor (GM-CSF)) (8), are the first class of approved immunotherapies for clinical use and have effects via stimulating immune cells directly (6, 9).
(iii) T cells and natural killer (NK) cells, in which T cells include engineered T cells and non-engineered T cells such as adoptive tumor infiltrating lymphocyte (TIL) and cultivated T cells. Engineered T cells contain chimeric antigen receptor T cells (CAR-T) and T cell receptor T cells (TCR-T), and CAR-T cells can trigger the death of tumor cells by recognizing the targeted antigens on tumor cells (10), and the antitumor activity of TCR-T cells is mainly stimulated by tumor-associated antigens presented by major histocompatibility complexes (MHCs) (11). NK cells also include engineered NK cells such as CAR-NK cells and many trials are under way.
(iv) Agonistic antibodies, which can specifically bind to receptors on the surface of T cells, triggering intracellular signaling pathways and inducing T cells to function as effectors to kill tumor cells (12).
(v) Cancer vaccines include those based on tumor cell lysates, nucleic acids, and peptides, which contain or can encode neoantigens (13). Neoantigen vaccines are an attractive type of cancer vaccine. In addition to being used separately as vaccines, DNA, RNA, peptide and tumor lysate can also be loaded onto DCs (Figure 1). Although DNA can be easily manipulated by molecular engineering, the successful use of the first generation of drug delivery platforms in humans is limited, and they tend to rely more on electroporation and it is also limited by its potential to integrate into the genome (14). On the other hand, there is no potential risk for RNA to integrate into the genome; however, it is still affected by RNase degradation although modification may prolong its half-life (14). In addition, synthetic long peptide (SLP) is easy to store, low toxicity and appropriate adjuvants are required (14). Therefore, when working with DC vaccines, the choice needs to be made between either loading with peptides, RNA, DNA or tumor lysate. Furthermore, DCs pulsed with neoantigens ex vivo to treat patients can effectively induce anti-tumor immune responses induced by activated T cells (13). Hundreds of research and clinical trials have been conducted or are underway since the first DC vaccine, sipuleucel-T, was approved for clinical use in 2010 (15). Although the safety of DC vaccines has been demonstrated in several clinical trials, several clinical trials have still failed due to the lack of clear efficacy (16, 17). The emergence of personalized neoantigens that were isolated, identified and selected from the patients’ tumors and their entry into the body after loading on DCs ex vivo can promote the efficient presentation of neoantigens by DCs to T cells to exert an anti-tumor role (16) (Figure 2). In this review, we summarize the progress and clinical application of personalized neoantigen-pulsed DC cancer vaccines.
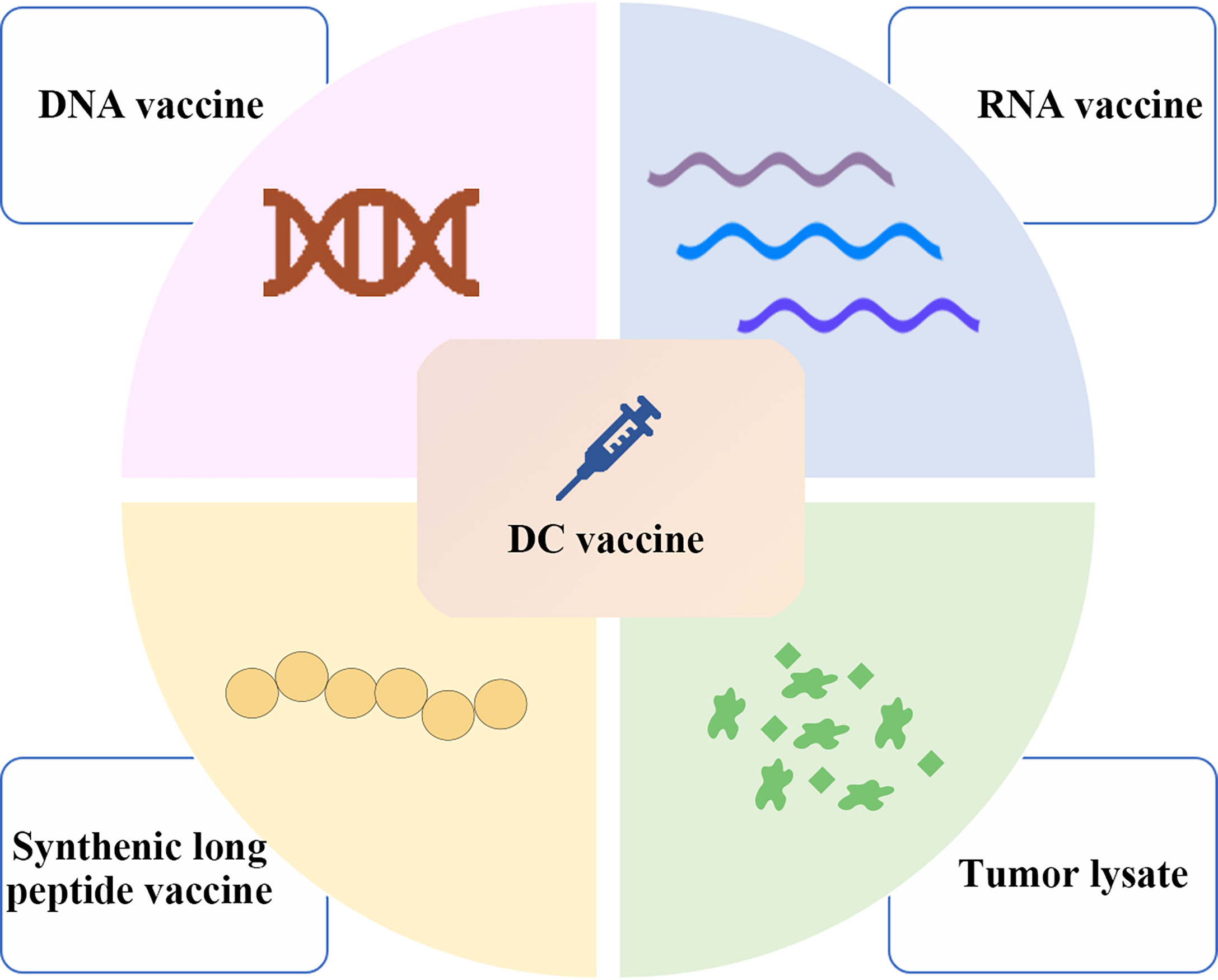
Figure 1 Major types of neoantigen vaccines in clinical research. Neoantigen vaccines mainly include nucleic acid vaccines consisting of DNA and RNA vaccines, synthetic long peptide vaccines and tumor lysate vaccines. In addition to being used separately as vaccines, these neoantigens formulations can also be loaded onto DCs. Therefore, when working with DC vaccines, still the choice needs to be made between loading with either peptides, RNA, DNA or tumor lysate.
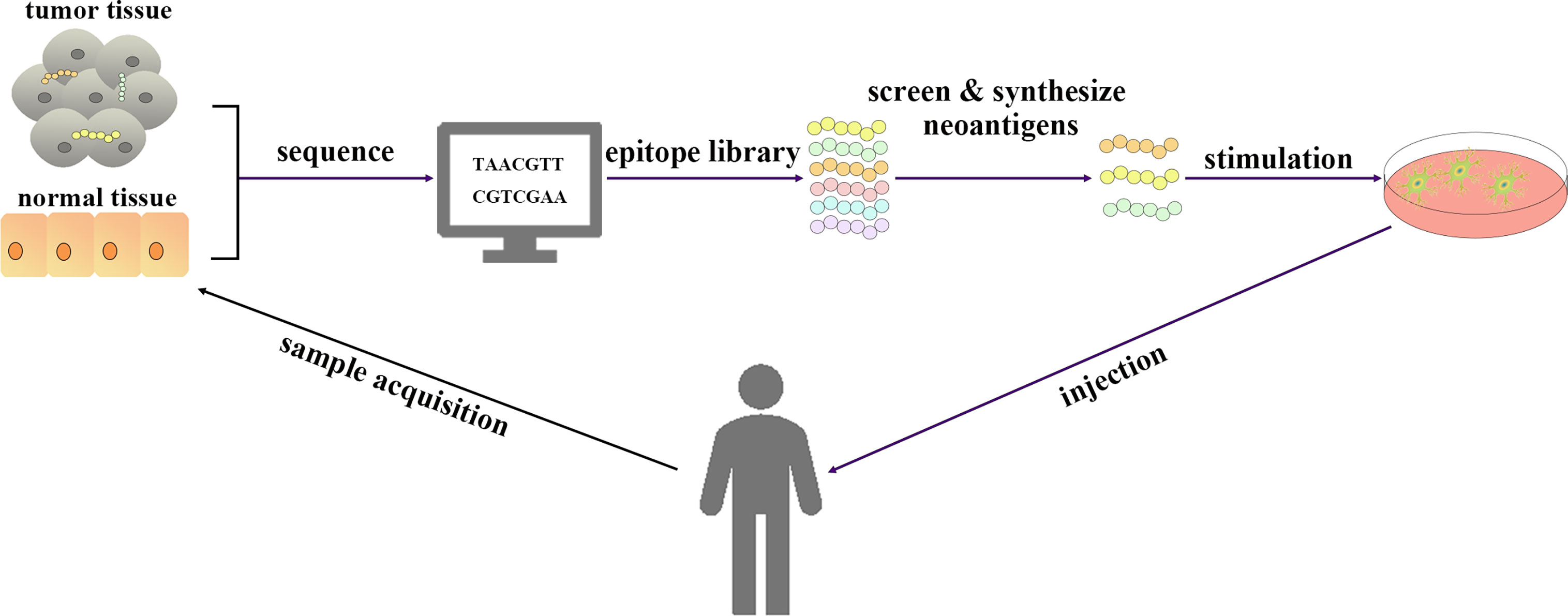
Figure 2 Schematic diagram of personalized neoantigen-pulsed DC vaccines. Tumor tissue and normal tissue of patients were sequenced. The epitope library is processed by bioinformatics methods, from which immunogenic neoantigens are screened and synthesized. The DC vaccines loaded with neoantigens are prepared by using DCs extracted from the peripheral blood of patients and injected into patients.
Personalized Neoantigens
Neoantigens are a series of peptides with tumor specificity that are present in proliferating tumor cells but not in normal tissues. Therefore, they are different from tumor-associated antigens (TAAs) mostly present in both normal and tumor tissues, which also include viral antigens (18, 19). At the same time, they are derived from viral proteins such as open reading frame-derived epitopes in the viral genome and tumor somatic nonsynonymous genetic alterations, including genomic variant level such as single nucleotide variants (SNVs), insertion-deletions, gene fusion, frame shift mutation and transcriptomic and proteomic variants (20–23). The change in peptide sequence and its spatial structure can result in a stronger affinity for major histocompatibility complexes (MHCs), and therefore, making it more likely to be recognized by T cells to induce antitumor immune responses (24). In general, neoantigens are divided into two subgroups: shared neoantigens and personalized neoantigens (25). Shared neoantigens are common in some tumor types and can be used broadly to treat patients who have the same tumor type and express these antigens; however, there are antigenic differences between different patients and different tumors, limiting the role of shared antigens (26, 27). Unlike shared neoantigens, personalized neoantigens are a class of antigens specific to individual patients and tumors. Since tumors of the same cancer type can vary greatly, personalized treatment with personalized neoantigens is a better way to ensure a response by each cancer type (25).
A series of completed or ongoing clinical trials on personalized tumor neoantigens is listed in Table 1 according to the data on ClinicalTrials.gov. In the trial conducted by Ott (28), they enrolled 10 patients, 8 of whom displayed a high degree of melanoma-related mutations as expected, and then 13-20 immunizing long peptides were synthesized for each patient. Finally, 6 patients completed the full series vaccinations. No disease recurrence was observed in 4 patients during a median follow-up period of 25 months (range 20-32 months) after vaccination. Two other patients suffered disease recurrence but also had a complete response after anti-PD1 therapy, which still shows that personalized neoantigen-based vaccines are safe and feasible and could be used in the clinic.
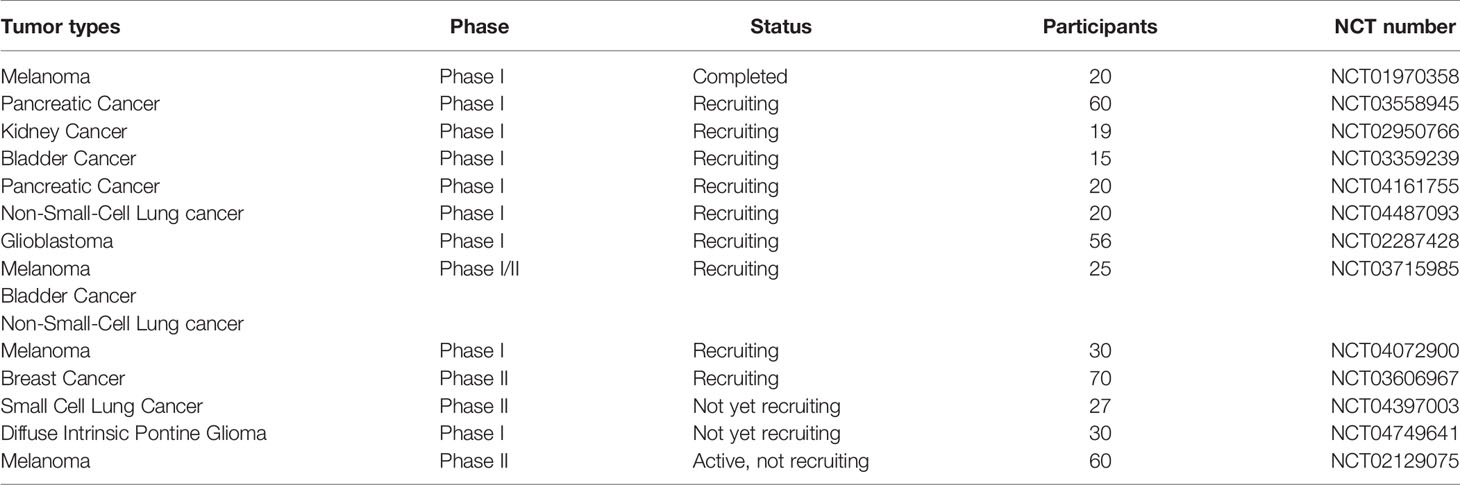
Table 1 The clinical application of personalized neoantigen vaccines.
Identification of Personalized Neoantigens
The identification of personalized neoantigens is an important part of tumor immunity therapy to allow personalized neoantigens to have an effect in each patient. The common approach is to compare DNA sequences in tumor tissues with those in normal tissues using high-throughput sequencing technologies (next-generation sequencing, NGS), which is rapid and efficient (29). However, many of the detected DNA mutations are not expressed as they are noncoding mutations or nonsense mutations, which poses new challenges to identify neoantigens (29). With the progress of sequencing technology, a more efficient and feasible sequencing technology with a lower false-negative rate was born: whole-exome sequencing technology (the exome is the protein-encoding part of the genome), which is currently widely used to identify personalized neoantigens (30). The mutant amino acid sequence that can be expressed needs to be translated and processed into short peptide fragments. These also need to be expressed on the cell surface in complex with MHC molecules to be recognized successfully by the immune system (19). Therefore, there are several crucial factors that determine whether a mutation can produce an effective personalized neoantigen: (i) whether the mutated DNA sequence can eventually be expressed and processed into short peptide fragments at the protein level; (ii) the ability of peptides to be presented and their affinity to MHC molecules; (iii) the affinity of the complex formed by the mutant peptides and MHC to TCR (31).
Because of lots of work involved in comparing high-throughput sequencing data, the development of computer simulation experiments or tools has effectively promoted the identification of personalized tumor neoantigens. On the one hand, for different neoantigen sources, there are corresponding computational tools. For single nucleotide variants (SNVs), small insertions and deletions (INDELs), or gene fusion at the genomic variant level, pVAC-Seq, TSNAD, CloudNeo, Tlminer, MuPeXI, Neopepsee, and INTEGRATE-Neo are usually utilized (32–37). For alternative transcript splicing at the transcriptomic variant level, NeoantigenR is widely used (38). On the other hand, these mutations need to be ranked according to their affinity with individual autologous MHC molecules; for this, the tools NetMHC, SMMPMBEC and SMM, among others, are used (39–41). Of the MHC molecule, MHC-I is directly related to neoantigen presentation on tumor cells, and the methods of using the MHC-I molecule to predict neoantigens are relatively mature at present, while CD4+ T cells recognize predicted neoantigens presented by MHC-II molecules. Compared to MHC-I molecules, where the peptide-binding groove is closed at both ends, the binding groove of MHC-II molecules is open at both ends and can deliver longer peptides (11-20 amino acids) (42). However, there is currently a lack of robust and rich databases and effective tools for assessing the interactions between MHC-II molecules and peptides compared with what is available for MHC-I (42). The further development of bioinformatics resources and the use of other cross-disciplinary methods are expected to improve neoantigen identification.
Personalized Neoantigens Manufacturing
Personalized neoantigens are a unique class of neoantigens specifically prepared for each patient; therefore, a rapid, simple, and mature system for the synthesis of personalized neoantigens is needed, as this is the first step of manufacturing neoantigens (43). In addition, the formulation of neoantigens such as buffering agents and surfactants are another crucial factor due to the different compositions and properties of each personalized neoantigen and these other components play important roles in ensuring the solubility and stability of neoantigens (44). The next step is purification. Many systems are used to purify neoantigens such as RP-HPLC and flash-like systems. With the development of new technologies, an increasing number of manual operations have been replaced by automated processes such as auto-sampling systems and ultra-performance liquid chromatography (UPLC) (45), saving time while enhancing the productivity and quality. The last step is lyophilization to make the newly prepared neoantigens easier to transport and store until used. Similarly, as technology advances, the processing of personalized neoantigens will become faster and more efficient, saving the patient precious time and increasing the effectiveness of cures.
Personalized Neoantigens-Pulsed DC Vaccines
Definition and Types of DCs
Immune cells, which include B cells, T cells, natural killer cells (NK cells) derived from lymphoid stem cells and neutrophils, eosinophils, basophils, and monocytes derived from myeloid progenitor, produce an immune response to resist the invasion of bacteria and viruses, kill tumor cells and maintain the human body’s immune balance. DCs, often differentiated from monocytes, are professional antigen-presenting cells and are responsible for efficient uptake, processing and presentation of antigens, which can not only teach naive T cells to become antigen-specific cytotoxic T cells (CTLs) through antigen presentation but also allow for interaction with other immune cells in the body, such as NK cells, T cells and B cells, activating the immune system to recognize and kill tumors (46).
In humans, committed DC precursors (CDPs) in bone marrow are divided into two major subsets of DCs, plasmacytoid DCs (pDCs) and conventional DCs (cDCs), which include two major categories-cDC1 and cDC2 based on their phenotype (47). These cells circulate in the blood and continue to enter the lymphoid organs and peripheral tissues as a supplement to DCs. For pDCs, surface markers mainly include CD123, CD303, CD304, and CD45RA, and they specifically secrete type I interferons (IFN-I) while presenting antigens to T cells and activating T cells (48, 49). For cDC1, surface markers mainly include Cleca9A, XCR1, and CD141, and cDC1 have the ability to cross-present and induce cytotoxic T cell immune responses and can also significantly stimulate the immune response of allogeneic or autologous CD4+ T cells (48, 49). For cDC2, surface markers mainly include CD1c, CD1a, and CD103, which can present soluble antigens but rarely present antigens derived from necrotic cells (48–50). In conclusion, different DCs play different physiological functions in the body, promoting the important role of DCs in immune regulation.
DC Vaccines
The principle of preparing a DC vaccine is simple. The precursor cells of DCs in patients are isolated and cultured in vitro, loaded with tumor antigens, and then transferred back into patients. Then, the antitumor effect can be exerted by specific antitumor T cells stimulated by DCs. After nearly 10 years of effort in the field of DC vaccines, in 2000, DC-based immunotherapy was used for the first time in a patient with a primary intracranial tumor. The patient received 3 treatments with an allogeneic MHC class I glioblastoma peptide-pulsed DC vaccine. The trial showed that the DC vaccine was tolerated, and the patient received a positive immune response. However, no objective clinical response was observed (51). In 2010, the United States FDA approved Sipuleucel-T as the first therapeutic DC vaccine for prostate cancer. Sipuleucel-T consists of peripheral blood mononuclear cells (PBMCs), which include APCs, activated ex vivo by PA2024, a recombinant protein including mainly prostate-specific antigen and prostatic acid phosphatase (15).
Philip W. Kantoff’s group divided 512 patients into two groups at a ratio of 2:1 to receive treatment with Sipuleucel-T and placebo every two weeks by intravenous injection, for 3 treatments in total (52). The results showed that the 36-month survival was 31.7% in the Sipuleucel-T group and 23% in the placebo group. The median survival duration in the Sipuleucel-T group was 25.8 months, an increase of 4.1 months compared with 21.7 months in the placebo group. This revealed that the drug could significantly prolong the survival period, suggesting that the DC vaccine can give patients a survival benefit. Another clinical trial on glioblastoma also showed superior efficacy of a DC vaccine. ICT-107 is an autologous DC vaccine pulsed with 6 different peptides targeting glioblastoma. In a prior phase I study, 21 patients with glioblastoma administered ICT-107 showed good tolerance and in 16 newly diagnosed patients, 6 patients did not show tumor recurrence, which showed that this DC vaccine was well tolerated and possessed antitumor activity (53).
In the following phase IIb trial conducted by Patrick Y. Wen, among HLA-A2+ patients with a matriculated MGMT promoter, progression-free survival (PFS) in the ICT-107 group (24.1 months) was significantly higher than that in the control group (8.5 months) and the patients in the ICT-107 group showed improved immune responses (54). Although many trials have focused on DC vaccines in recent years (Table 2), the basis of DC vaccines is the selection of immunogenic antigens to activate the immune system effectively in addition to the maturation of DCs. Because the antigens in each patient’s tumor are highly specific, DCs loaded with personalized neoantigens for fusion into therapeutic tumor vaccines are another attractive strategy.
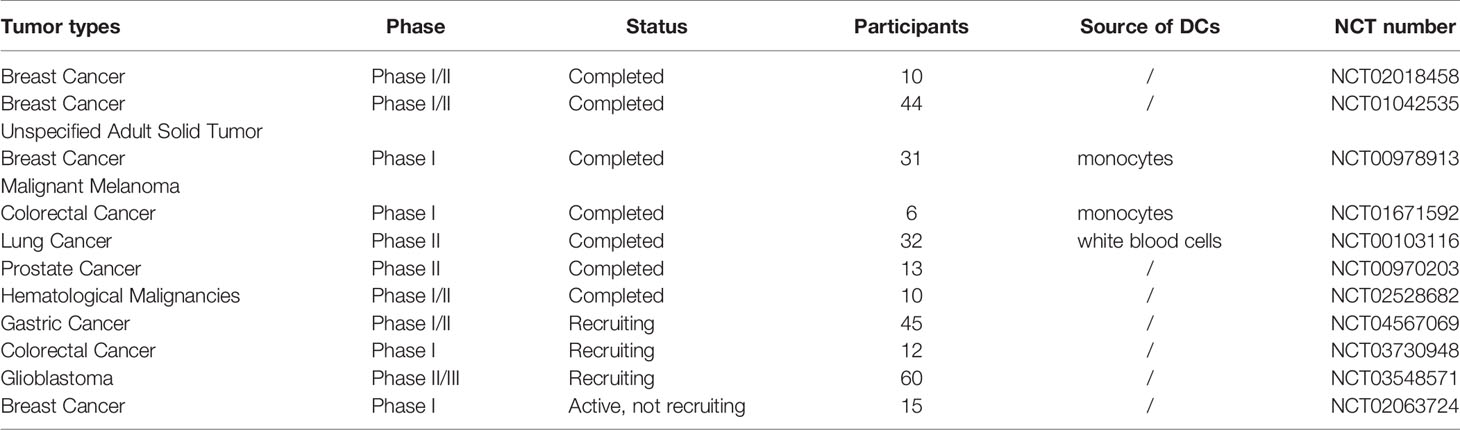
Table 2 The clinical application of DC vaccines.
Clinical Trial Progress of Personalized Neoantigen-Pulsed DC Vaccines
Tumor vaccines that rely on neoantigens alone cannot completely eliminate malignant tumors (55). The reason for this is not the neoantigen itself but more because most of the trials used neoantigens to solve the problem of the weak antigenicity of tumor cells but did not solve the problem of immune cell functional defects in cancer patients. Patients with malignant tumors usually have a low level of immune function, and it is difficult to initiate the antitumor immune response in vivo. One of the main reasons is that the function of antigen-presenting cells in patients is inhibited, and antigen-activated T cells cannot be effectively presented. Therefore, to achieve good clinical efficacy, immunotherapy should not only solve the problems related to antigens but also the problems of immunosuppression in tumor patients. In other words, when many tumor-specific antigens are injected into the body, it is necessary to ensure that they are efficiently taken up and presented by the body’s antigen-presenting cells and that a sufficient number of effector T cells are activated.
In 2015, the first personalized neoantigen-loaded DC vaccine began testing in a phase I clinical trial (56). They enrolled 3 melanoma patients with stage III resected cutaneous melanoma and treated them with ipilimumab. Then, they identified somatic mutations from their own surgically excised tumors by whole-exome sequencing and computer-simulated epitope prediction to screen for suitable neoantigens. Furthermore, 7 neoantigens selected from each patient were loaded with DCs isolated from PBMCs, cultured ex vivo, and then intravenously injected into the patients for a total of three treatments. After the treatments, an enhanced immune response triggered by T cells was observed, while three patients were all surviving and no autoimmune adverse reactions were observed, which showed that DC vaccines pulsed with personalized neoantigens was safe and reliable.
In another trial conducted by Ding’s group in 2020, they demonstrated for the first time the activity of a personalized neoantigen-pulsed DC vaccine in patients with advanced NSCLC (57). In their study, they enrolled 12 patients with advanced lung cancer and 13-30 peptide-based personalized neoantigens were isolated and identified from each patient’s tumor tissue. At the same time, PBMCs were derived from each patient, DCs were separated, then DCs were pulsed with the corresponding selected neoantigens to form a personalized neoantigen-pulsed DC vaccine to treat patients. Their study showed a 25% objective response rate, while a 75% disease control rate was observed after treatment of a personalized neoantigen-pulsed DC vaccine. In addition, only low-grade and transient side effects were observed, which also demonstrated that the vaccine was safe and able to induce specific T cell immune response. In particular, a patient with metastatic lung cancer whose main metastases were in bone, pelvis, and inferior vena cava lymph nodes failed to show a tumor response after three treatments. Then, he received personalized neoantigen-pulsed DC vaccine treatment, and after 5 doses of this vaccine, almost no metastatic lymph nodes and shrinking pelvic lesions were observed. There was a 29% reduction in overall tumor lesions, which showed a good therapeutic effect of the vaccine.
In Sarivalasis’s paper published in 2019, they present another phase I/II trial that uses personalized peptides, including tumor-specific neoantigens and TAAs derived from patients, and pulses them into DCs isolated from autologous monocytes (58). They will acquire the tumor specimens from each patient for NGS analysis. Then they will analyze the data to generate personalized databases, and up to 10 will be selected per patient by verifying the immune response of the candidate peptides to T cells isolated from the patient. This trial will investigate the feasibility and safety of a personalized neoantigen-loaded DC vaccine in patients with ovarian cancer and evaluate overall survival (OS) progression time and disease-free survival at 12, 24, and 36 months. This trial is the first of its kind to test a personalized neoantigen-pulsed DC vaccine in ovarian cancer patients. We look forward to its expected efficacy in a clinical trial, providing additional strong evidence of the efficacy and safety of a personalized neoantigen-pulsed DC vaccine and bringing benefits to patients.
In addition, according to ClinicalTrials.gov, there are several clinical trials around personalized neoantigen-loaded DC vaccines under way that are in phase I (Table 3). Although these trials are underway, the fact that they have been carried out only in the last decade shows that they are still forward-looking and innovative.
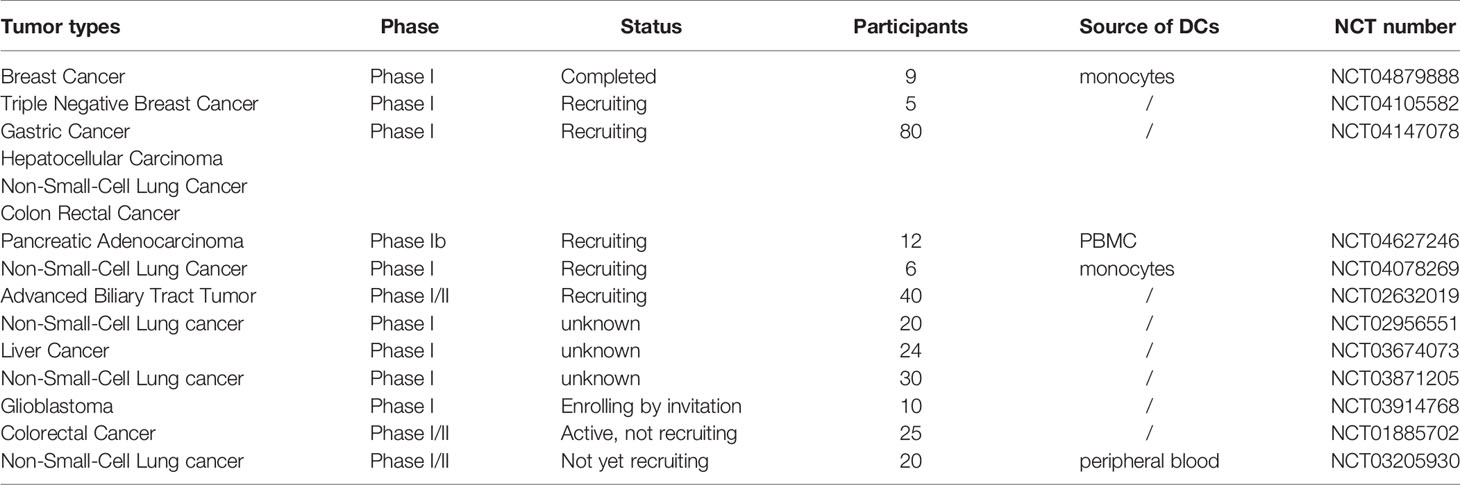
Table 3 The clinical application of personalized neoantigen-pulsed DC vaccines.
Personalized Neoantigen-Pulsed DC Vaccines in Combination With Other Therapies
The combination of personalized neoantigen-pulsed DC vaccines with other strategies, such as chemotherapy and immune checkpoint inhibitors, is another attractive approach to enhance the tumor therapeutic vaccine efficacy. Chemotherapy is considered an immunotherapy partner to improve immunotherapy efficacy by enhancing antigen production and presentation, and inducing T cell immune response, although it still has several side effects (59). In a trial conducted by Batich and colleagues (60), cytomegalovirus antigen pp65 was found to be present in glioma cells instead of surrounding normal tissues. Then, they used a pp65-pulsed DC vaccine combined with dose-intensified temozolomide, which is a chemotherapeutic drug to treat glioma. As expected, the median PFS was 25.3 months and OS was 41.1 months, and both were much higher than the statistical median survival of patients (less than 15 months) with newly diagnosed glioma.
Immune checkpoints exert strong immunosuppressive effects to block the antitumor immune response; thus, neoantigen vaccines combined with immune checkpoint inhibitors, which involve mainly specific monoclonal antibodies such as anti-PD-1, anti-PD-L1, and anti-CTLA-4 antibodies, are thought to generate strong a T cell immune response to kill tumors (7, 61, 62). In Sahin’s trial, neoantigen-specific T cells were PD1+ and after the neoantigen vaccine, PD-L1 upregulation was observed (63). Then, anti-PD1 treatment was applied after the neoantigen vaccine, and a complete response to the neoantigen vaccine was observed. In a phase I trial, the combination of MART-1 peptide-pulsed DCs and tremelimumab, an anti-CTLA-4 antibody, was used for 16 patients with melanoma and they acquired a higher durable objective tumor response rate than treatment alone (64). In addition, a trial conducted by Ding showed that an enlarged tumor was still observed in a patient with lung cancer after treatment with a personalized neoantigen-pulsed DC vaccine (57). When nivolumab, an anti-PD-1 antibody, was combined with this DC vaccine, the patient’s tumor became cavitated, which demonstrated the superiority of combination therapy for cancer.
Factors That Limit the Effectiveness of Personalized Neoantigen-Pulsed DC Vaccines
Although the current clinical application shows efficacy, personalized tumor neoantigen-pulsed DC vaccines are still limited in several aspects. (i) The selection of neoantigens: The sequencing and screening of tumor neoantigens requires individual detection and analysis for each patient’s tumor, which is a complex and time-consuming process. Additionally, the manufacture of neoantigens requires a better manufacturing conditions to ensure the consistency of neoantigens (65). As a result, the development and wide application of advanced technology are urgently needed. It is believed that the time and production cost of this process will be greatly reduced in the near future. (ii) The source and maturation conditions of DCs: DCs applied in personalized neoantigen-pulsed DC vaccines are also individualized, and it is necessary to extract DCs from each patient for separate culture. In addition, mature DCs are needed to enhance antigen processing, presentation and stimulate B and T cells. Antigens, cytokines such as GM-CSF and other factors such as LPS could stimulate DCs maturation. This process still has problems such as the intensive labor required for the ex vivo culture process and the skill required for inducing DC maturation. Thus, in future studies, efforts are needed to optimize ex vivo culture while inducing mature and high-quality DCs (66–69). (iii) The efficiency of DC migration: DCs injected back into patients should migrate to the lymphoid organs to stimulate T cells to achieve effective immune responses, and some proinflammatory cytokines, such as prostaglandin E2 (PGE2), could promote the migration of DCs to some extent (70–72). However, selective migration of DCs and their residence in nonlymphoid and lymphoid organs are tightly regulated events. The molecular control mechanisms need to be elucidated in future studies to lay the foundation for improving the stimulation conditions of DC vaccines in clinical trials.
Conclusion
Personalized tumor neoantigens are highly specific to individuals, and tumor vaccines targeting neoantigens can effectively induce T cells to produce a strong immune response against tumors. However, the key to the effectiveness of personalized tumor neoantigens is that they can be efficiently taken up and processed by APCs and delivered to T cells to induce an antitumor immune response. However, the function of antigen-presenting cells in patients with malignant tumors is usually inhibited. Therefore, the treatment of patients with DC vaccines loaded with neoantigens can specifically target the tumor and ensure that DCs can exert their efficacy to the maximum extent. An increasing number of research and clinical trials are currently underway, promising to offer new hope to patients with solid tumors.
Author Contributions
LY and LT conceptualized the study. LT and RZ finished the manuscript and figures, and XZ helped LT collect the related paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82073366), West China Hospital, Sichuan University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 0:1–41. doi: 10.3322/caac.21660
2. Airley R. Cancer Chemotherapy: Basic Science to the Clinic. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA (2009).
3. Santhosh S, Kumar P, Ramprasad V. Chaudhuri. Evolution of Targeted Therapies in Cancer: Opportunities and Challenges in the Clinic. Future Oncol (2015) 11(2):279–93. doi: 10.2217/fon.14.198
4. Yang YP. Cancer Immunotherapy: Harnessing the Immune System to Battle Cancer. J Clin Invest (2015) 125(9):3335–7. doi: 10.1172/JCI83871
5. Rosenberg SA. IL-2: The First Effective Immunotherapy for Human Cancer. J Immunol (2014) 192:5451–8. doi: 10.4049/jimmunol.1490019
6. Riley RS, June CH, Langer R, Mitchell MJ. Delivery Technologies for Cancer Immunotherapy. Nat Rev Drug Discov (2019) 18(3):175–96. doi: 10.1038/s41573-018-0006-z
7. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
8. Lee S, Margolin K. Cytokines in Cancer Immunotherapy. Cancers (Basel) (2011) 3(4):3856–93. doi: 10.3390/cancers3043856
9. Ahmed S, Rai K. Interferon in the Treatment of Hairy- Cell Leukemia. Best Pract Res. Clin Haematol (2003) 16:69–81. doi: 10.1016/S1521-6926(02)00084-1
10. Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell (2017) 168(4):724–40. doi: 10.1016/j.cell.2017.01.016
11. Cohen M. Reiter Y. T-Cell Receptor-Like Antibodies: Targeting the Intracellular Proteome Therapeutic Potential and Clinical Applications. Antibodies (2013) 2(3):517–34. doi: 10.3390/antib2030517
12. Peggs KS, Quezada SA, Allison JP. Cancer Immunotherapy: Co-Stimulatory Agonists and Co-Inhibitory Antagonists. Clin Ex Immunol (2009) 157(1):9–19. doi: 10.1111/j.1365-2249.2009.03912.x
13. Guo CQ, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic Cancer Vaccines; Past, Present and Future. Adv Cancer Res (2013) 119:421–75. doi: 10.1016/B978-0-12-407190-2.00007-1
14. Li L, Goedegebuure SP, Gillanders WE. Preclinical and Clinical Development of Neoantigen Vaccines. Ann Oncol (2017) 28:xii11–7. doi: 10.1093/annonc/mdx681
15. Goldman B, DeFrancesco L. The Cancer Vaccine Roller Coaster. Nat Biotechnol (2009) 27(2):129–39. doi: 10.1038/nbt0209-129
16. Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P. Integrating Next-Generation Dendritic Cell Vaccines Into the Current Cancer Immunotherapy Landscape. Trends Immunol (2017) 38(8):577–93. doi: 10.1016/j.it.2017.05.006
17. Rosenberg SA, Yang JC, Restifo NP. Cancer Immunotherapy: Moving Beyond Current Vaccines. Nat Med (2004) 10:909–15. doi: 10.1038/nm1100
18. Liu XS, Mardis ER. Applications of Immunogenomics to Cancer. Cell (2017) 168:600–12. doi: 10.1016/j.cell.2017.01.014
19. Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination With Ipilimumab in Small-Cell Lung Cancer. Cancer Cell (2018) 33(5):853–61. doi: 10.1016/j.ccell.2018.04.001
20. Ward JP, Gubin MM, Schreiber RD. The Role of Neoantigens in Naturally Occurring and Therapeutically Induced Immune Responses to Cancer. Adv Immunol (2016) 130:25–74. doi: 10.1016/bs.ai.2016.01.001
21. Schumacher TN, Hacohen N. Neoantigens Encoded in the Cancer Genome. Curr Opin Immunol (2016) 41:98–103. doi: 10.1016/j.coi.2016.07.005
22. Katsnelson A. Mutations as Munitions: Neoantigen Vaccines Get a Closer Look as Cancer Treatment. Nat Med (2016) 22(2):122–4. doi: 10.1038/nm0216-122
23. Capietto AH, Jhunjhunwala S, Delamarre L. Characterizing Neoantigens for Personalized Cancer Immunotherapy. Curr Opin Immunol (2017) 46:58–65. doi: 10.1016/j.coi.2017.04.007
24. Chen DS, Mellman I. Elements of Cancer Immunity and the Cancer-Immune Set Point. Nature (2017) 541(7637):321–30. doi: 10.1038/nature21349
25. Aldous AR, Dong JZ. Personalized Neoantigen Vaccines: A New Approach to Cancer Immunotherapy. Bioorg Med Chem (2017) 26(10):2842–9. doi: 10.1016/j.bmc.2017.10.021. S0968089617312208.
26. Mullard A. The Cancer Vaccine Resurgence. Nat Rev Drug Discov (2016) 15(10):663–5. doi: 10.1038/nrd.2016.201
27. Romero P, Banchereau J, Bhardwaj N, Cockett M, Disis ML, Dranoff G, et al. The Human Vaccines Project: A Roadmap for Cancer Vaccine Development. Sci Transl Med (2016) 8(334):3349. doi: 10.1126/scitranslmed.aaf0685
28. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An Immunogenic Personal Neoantigen Vaccine for Patients With Melanoma. Nature (2017) 547(7662):217–21. doi: 10.1038/nature22991
29. Li L, Goedegebuure P, Mardis ER, Mardis ER, Ellis MJC, Zhang X, et al. Cancer Genome Sequencing and Its Implications for Personalized Cancer Vaccines. Cancers (Basel) (2011) 3(4):4191–211. doi: 10.3390/cancers3044191
30. van Buuren MM, Calis JJ, Schumacher TN. High Sensitivity of Cancer Exome-Based CD8 T Cell Neo-Antigen Identification. Oncoimmunology (2014) 3:e28836. doi: 10.4161/onci.28836
31. Schumacher TN, Schreiber RD. Neoantigens in Cancer Immunotherapy. Science (2015) 348(6230):69–74. doi: 10.1126/science.aaa4971
32. Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, et al. pVAC-Seq: A Genome-Guided In Silico Approach to Identifying Tumor Neoantigens. Genome Med (2016) 8(1):11. doi: 10.1186/s13073-016-0264-5
33. Zhou Z, Lyu X, Wu J, Yang X, Wu S, Zhou J, et al. TSNAD: An Integrated Software for Cancer Somatic Mutation and Tumour-Specific Neoantigen Detection. R Soc Open Sci (2017) 4:170050. doi: 10.1098/rsos.170050
34. Bais P, Namburi S, Gatti DM, Zhang X, Chuang JH. CloudNeo: A Cloud Pipeline for Identifying Patient-Specific Tumor Neoantigens. Bioinformatics (2017) 33:3110–2. doi: 10.1093/bioinformatics/btx375
35. Tappeiner E, Finotello F, Charoentong P, Mayer C, Rieder D, Trajanoski Z. TIminer: NGS Data Mining Pipeline for Cancer Immunology and Immunotherapy. Bioinformatics (2017) 33:3140–1. doi: 10.1093/bioinformatics/btx377
36. Bjerregaard AM, Nielsen M, Hadrup SR, Szallasi Z, Eklund AC. MuPeXI: Prediction of Neo-Epitopes From Tumor Sequencing Data. Cancer Immunol Immunother (2017) 66:1123–30. doi: 10.1007/s00262-017-2001-3
37. Kim S, Kim HS, Kim E, Lee MG, Shin EC, Paik S, et al. Neopepsee: Accurate Genome-Level Prediction of Neoantigens by Harnessing Sequence and Amino Acid Immunogenicity Information. Ann Oncol (2018) 29(4):1030–6. doi: 10.1093/annonc/mdy022
38. Zhang J, Mardis ER, Maher CA. INTEGRATE-Neo: A Pipeline for Personalized Gene Fusion Neoantigen Discovery. Bioinformatics (2017) 33:555–7. doi: 10.1093/bioinformatics/btw674
39. Buus S, Lauemoller SL, Worning P, Kesmir C, Frimurer T, Corbet S, et al. Sensitive Quantitative Predictions of Peptide-MHC Binding by a ‘Query by Committee’ Artificial Neural Network Approach. Tissue Antigens (2010) 62(5):378–84. doi: 10.1034/j.1399-0039.2003.00112.x
40. Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an Amino Acid Similarity Matrix for Peptide: MHC Binding and its Application as a Bayesian Prior. BMC Bioinf (2009) 10:394. doi: 10.1186/1471-2105-10-394
41. Peters B, Sette A. Generating Quantitative Models Describing the Sequence Specificity of Biological Processes With the Stabilized Matrix Method. BMC Bioinf (2005) 6:132. doi: 10.1186/1471-2105-6-132
42. Nielsen M, Lund O, Buus S, Lundegaard C. MHC Class II Epitope Predictive Algorithms. Immunology (2010) 130(3):319–28. doi: 10.1111/j.1365-2567.2010.03268.x
43. Fritsch EF, Hacohen N, Wu CJ. Personal Neoantigen Cancer Vaccines: The Momentum Builds. Onco Immunol (2014) 3(6):e29311. doi: 10.4161/onci.29311
44. Uhlig T, Kyprianou T, Martinelli FG, Oppici CA, Heiligers D, Hills D, et al. The Emergence of Peptides in the Pharmaceutical Business: From Exploration to Exploitation. EuPA Open Proteomics (2014) 4:58–69. doi: 10.1016/j.euprot.2014.05.003
45. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, et al. Non-Invasive Analysis of Acquired Resistance to Cancer Therapy by Sequencing of Plasma DNA. Nature (2013) 497:108–12. doi: 10.1038/nature12065
46. Steinman RM, Cohn ZA. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice. I. Morphology, Quantitation, Tissue Distribution. J Exp Med (1973) 137:1142–62. doi: 10.1084/jem.139.2.380
47. Shortman K, Sathe P, Vremec D, Naik S, Keeffe MO. Plasmacytoid Dendritic Cell Development. Adv Immunol (2013) 120:105–26. doi: 10.1016/B978-0-12-417028-5.00004-1
48. Macri C, Pang ES, Patton T, Keeffe MO. Dendritic Cell Subsets. Semin Cell Dev Biol (2018) 84:11–21. doi: 10.1016/j.semcdb.2017.12.009
49. Keeffe MO, Mok WH, Radford KJ. Human Dendritic Cell Subsets and Function in Health and Disease. Cell Mol Life Sci (2015) 72(22):4309–25. doi: 10.1007/s00018-015-2005-0
50. Chiang MC, Tullett KM, Lee YS, Idris A, Ding Y, McDonald KJ, et al. Differential Uptake and Cross-Presentation of Soluble and Necrotic Cell Antigen by Human DC Subsets. Eur J Immunol (2016) 46(2):329–39. doi: 10.1002/eji.201546023
51. Liau LM, Black KL, Martin NA, Sykes SN, Bronstein JM, Steele LJ, et al. Treatment of a Glioblastoma Patient by Vaccination With Autologous Dendritic Cells Pulsed With Allogeneic Major Histocompatibility Complex Class I–matched Tumor Peptides. Neurosurg Focus (2000) 9(6):8. doi: 10.3171/foc.2000.9.6.9
52. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N Engl J Med (2010) 363(5):411–22. doi: 10.1056/NEJMoa1001294
53. Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, et al. Phase I Trial of a Multi-Epitope-Pulsed Dendritic Cell Vaccine for Patients With Newly Diagnosed Glioblastoma. Cancer Immunol Immunother (2013) 62(1):125–35. doi: 10.1007/s00262-012-1319-0
54. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients With Glioblastoma. Clin Cancer Res (2019) 25(19):5799–807. doi: 10.1158/1078-0432.CCR-19-0261
55. Melief CJ, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic Cancer Vaccines. J Clin Invest (2015) 125(9):3401–12. doi: 10.1172/JCI80009
56. Carreno BM, Magrini V, Hapak MB, Kaabinejadian S, Hundal J, Petti AA, et al. A Dendritic Cell Vaccine Increases the Breadth and Diversity of Melanoma Neoantigen-Specific T Cells. Science (2015) 348(6236):803–8. doi: 10.1126/science.aaa3828
57. Ding ZY, Li Q, Zhang R, Xie L, Shu Y, Gao S, et al. Personalized Neoantigen Pulsed Dendritic Cell Vaccine for Advanced Lung Cancer. Sig Transduct Target Ther (2021) 6:26. doi: 10.1038/s41392-020-00448-5
58. Sarivalasis A, Boudousquié C, Balint K, Stevenson BJ, Gannon PO, Iancu EM, et al. A Phase I/II Trial Comparing Autologous Dendritic Cell Vaccine Pulsed Either With Personalized Peptides (PEP-DC) or With Tumor Lysate (OC-DC) in Patients With Advanced High-Grade Ovarian Serous Carcinoma. J Transl Med (2019) 17(1):391. doi: 10.1186/s12967-019-02133-w
59. Lake RA, Robinson BW. Immunotherapy and Chemotherapy–A Practical Partnership. Nat Rev Cancer (2005) 5:397–405. doi: 10.1038/nrc1613
60. Batich KA, Reap EA, Archer GE, Perez LS, Nair SK, Schmittling RJ, et al. Long-Term Survival in Glioblastoma With Cytomegalovirus Pp65-Targeted Vaccination. Clin Cancer Res (2017) 23(8):1898–909. doi: 10.1158/1078-0432.CCR-16-2057
61. Hodi FS, O’Day SJ, Mcdermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival With Ipilimumab in Patients With Metastatic Melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
62. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
63. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity Against Cancer. Nature (2017) 547(7662):222–6. doi: 10.1038/nature23003
64. Plaen ED, Lurquin C, Pel AV, Mariame B, Szikora JP, Wolfel T, et al. Immunogenic (Tum-) Variants of Mouse Tumor P815: Cloning of the Gene of Tum- Antigen P91A and Identification of the Tum- Mutation. Proc Natl Acad Sci (1988) 85(7):2274–8. doi: 10.1073/pnas.85.7.2274
65. Ophir E, Bobisse S, Coukos G, Harari A, Kandalaft LE. Personalized Approaches to Active Immunotherapy in Cancer. Biochem Biophys Acta (2016) 1865(1):72–82. doi: 10.1016/j.bbcan.2015.07.004
66. Kalinski P, Okada H. Polarized Dendritic Cells as Cancer Vaccines: Directing Effector-Type T Cells to Tumors. Semin Immunol (2010) 22(3):173–82. doi: 10.1016/j.smim.2010.03.002
67. Schreibelt G, Benitez Ribas D, Schuurhuis D, Lambeck AJA, van Hout-Kuijer M, Schaft N, et al. Commonly Used Prophylactic Vaccines as an Alternative for Synthetically Produced TLR Ligands to Mature Monocyte-Derived Dendritic Cells. Blood (2010) 116(4):564–74. doi: 10.1182/blood-2009-11-251884
68. Engell-Noerregaard L, Hansen TH, Andersen MH, Straten PT, Svane IM. Review of Clinical Studies on Dendritic Cell-Based Vaccination of Patients With Malignant Melanoma: Assessment of Correlation Between Clinical Response and Vaccine Parameters. Cancer Immunol Immunother (2009) 58(1):1–14. doi: 10.1007/s00262-008-0568-4
69. Wimmers F, Schreibelt G, Skold AE, Figdor CG, De Vries IJM. Paradigm Shift in Dendritic Cell-Based Immunotherapy: From In Vitro Generated Monocyte-Derived DCs to Naturally Circulating DC Subsets. Front Immunol (2014) 5:165. doi: 10.3389/fimmu.2014.00165
70. Breton G, Lee J, Zhou YJ, Schreiber JJ, Keler T, Puhr S, et al. Circulating Precursors of Human CD1c+ and CD141+ Dendritic Cells. J Exp Med (2015) 212(3):401–13. doi: 10.1084/jem.20141441
71. Lee AW, Truong T, Bickham K, Fonteneau JF, Larsson M, Silva ID, et al. A Clinical Grade Cocktail of Cytokines and PGE2 Results in Uniform Maturation of Human Monocyte-Derived Dendritic Cells: Implications for Immunotherapy. Vaccine (2002) 20(4):A8–A22. doi: 10.1016/S0264-410X(02)00382-1
Keywords: personalized neoantigen, DC vaccine, tumor, immunotherapy, clinical applications
Citation: Tang L, Zhang R, Zhang X and Yang L (2021) Personalized Neoantigen-Pulsed DC Vaccines: Advances in Clinical Applications. Front. Oncol. 11:701777. doi: 10.3389/fonc.2021.701777
Received: 28 April 2021; Accepted: 12 July 2021;
Published: 26 July 2021.
Edited by:
Min Cheng, Weifang Medical University, ChinaReviewed by:
Krithika Kodumudi, Moffitt Cancer Center, United StatesAn Maria Theophiel Van Nuffel, Anticancer Fund, Belgium
Copyright © 2021 Tang, Zhang, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Yang, eWwudHJhY3k3M0BnbWFpbC5jb20=
†These authors have contributed equally to this work