 Jiacheng Lai
Jiacheng Lai Ziqiang Liu
Ziqiang Liu Yulei Zhao
Yulei Zhao Chengyuan Ma
Chengyuan Ma Haiyan Huang
Haiyan Huang- Department of Neurosurgery, The First Hospital of Jilin University, Changchun, China
I-BET151 is an inhibitor of bromodomain and extra-terminal domain (BET) proteins that selectively inhibits BET family members (BRD2, BRD3, BRD4, and BRDT). Over the past ten years, many studies have demonstrated the potential of I-BET151 in cancer treatment. Specifically, I-BET151 causes cell cycle arrest and inhibits tumor cell proliferation in some hematological malignancies and solid tumors, such as breast cancer, glioma, melanoma, neuroblastoma, and ovarian cancer. The anticancer activity of I-BET151 is related to its effects on NF-κB, Notch, and Hedgehog signal transduction pathway, tumor microenvironment (TME) and telomere elongation. Remarkably, the combination of I-BET151 with select anticancer drugs can partially alleviate the occurrence of drug resistance in chemotherapy. Especially, the combination of forskolin, ISX9, CHIR99021, I-BET151 and DAPT allows GBM cells to be reprogrammed into neurons, and this process does not experience an intermediate pluripotent state. The research on the anticancer mechanism of I-BET151 will lead to new treatment strategies for clinical cancer.
Introduction
Bromodomain and extra-terminal domain (BET) proteins function as epigenetic readers that mainly recognize acetylated lysine residues in chromatin proteins. The BET family consists of four members, among which BRD2, BRD3, and BRD4 are ubiquitously expressed, and BRDT is only expressed in the testis. Conserved structural components of these proteins include two characteristic bromine domains (BD1 and BD2) and an extra-terminal domain (ET), along with a C-terminal domain (CTD) found only in BRD4 and BRDT (1). BET proteins participate in the formation of multiple nuclear protein complexes and play an important role in regulating gene transcription, as well as DNA replication, damage, and repair (2).
The abnormal manifestations of BET family members, especially BRD2 and BRD4, occur in various cancer types. In nuclear protein in testis (NUT) midline carcinoma (NMC), BRD3 and BRD4 fuse with NUT and retain it in the nucleus, which interferes with the differentiation of epithelial cells and promotes cancer growth (3). In melanoma, glioma, ovarian cancer, and some other cancers, the overexpression of BRD2 and BRD4 is associated with poor prognosis, and their presence affects the pathways of nuclear factor-κB (NF-κB), Notch, and Hedgehog (Hh) signaling (4–6). The changes in the expression and distribution of BET family members in different cancer cells and even stem cells often promote the occurrence and development of cancer.
I-BET151 (Figure 1) is a new type of BET protein inhibitor with the chemical designation 7-(3,5-dimethyl-4-isoxazo1lyl)-8-(methyloxy)-1-[(1R)-1-(2-pyridinyl)ethyl]-1,3-dihydro-2H-imidazo[4,5-c]quinolin-2-one, and the molecular formula C23H21N5O3. In 2011, Dawson et al. developed and optimized I-BET151 as a BET inhibitor with good bioavailability and a prolonged terminal half-life. I-BET151 binds into BD1 acetyl-lysine recognition pocket and displaces BET proteins from nuclear chromatin. Of the 27 bromodomain proteins in the HL60 nuclear extract, the excess I-BET151 affects only BRD2, BRD3, BRD4, and BRD9. Among them, the effect of I-BET151 on BRD9 may be indirect because BRD9 and BRD4 form a complex.I-BET151 selectively inhibits leukemia mouse models and mixed-lineage leukemia (MLL) primary patient samples, and its half-life is significantly longer than that of similar BET inhibitors (JQ1, I-BET762) (7). Several recent studies have demonstrated the anticancer effects of I-BET151 on various solid tumors, apart from leukemia, which has attracted extensive attention (4–6). In this review, we will discuss the existing research on anticancer effects of I-BET151 and focus on the implications for cancer therapy.
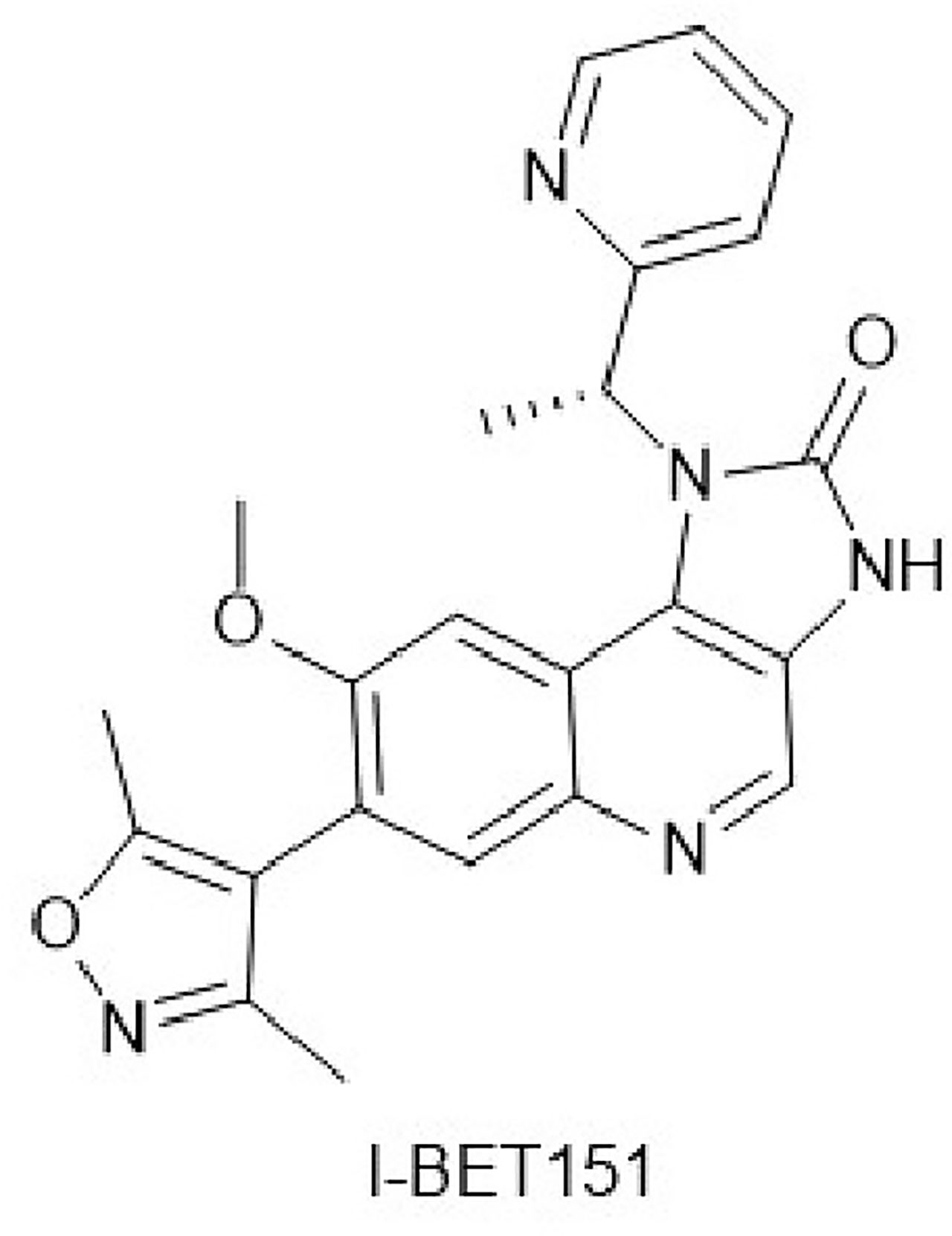
Figure 1 Chemical structure of I-BET151.
The Anticancer Mechanism of I-BET151
I-BET151 selectively inhibits members of the BET family, which affects intracellular signal transduction pathways, tumor microenvironment (TME), and telomere length mainly via the pathways for NF-κB, Notch, and Hh signaling (Figure 2).
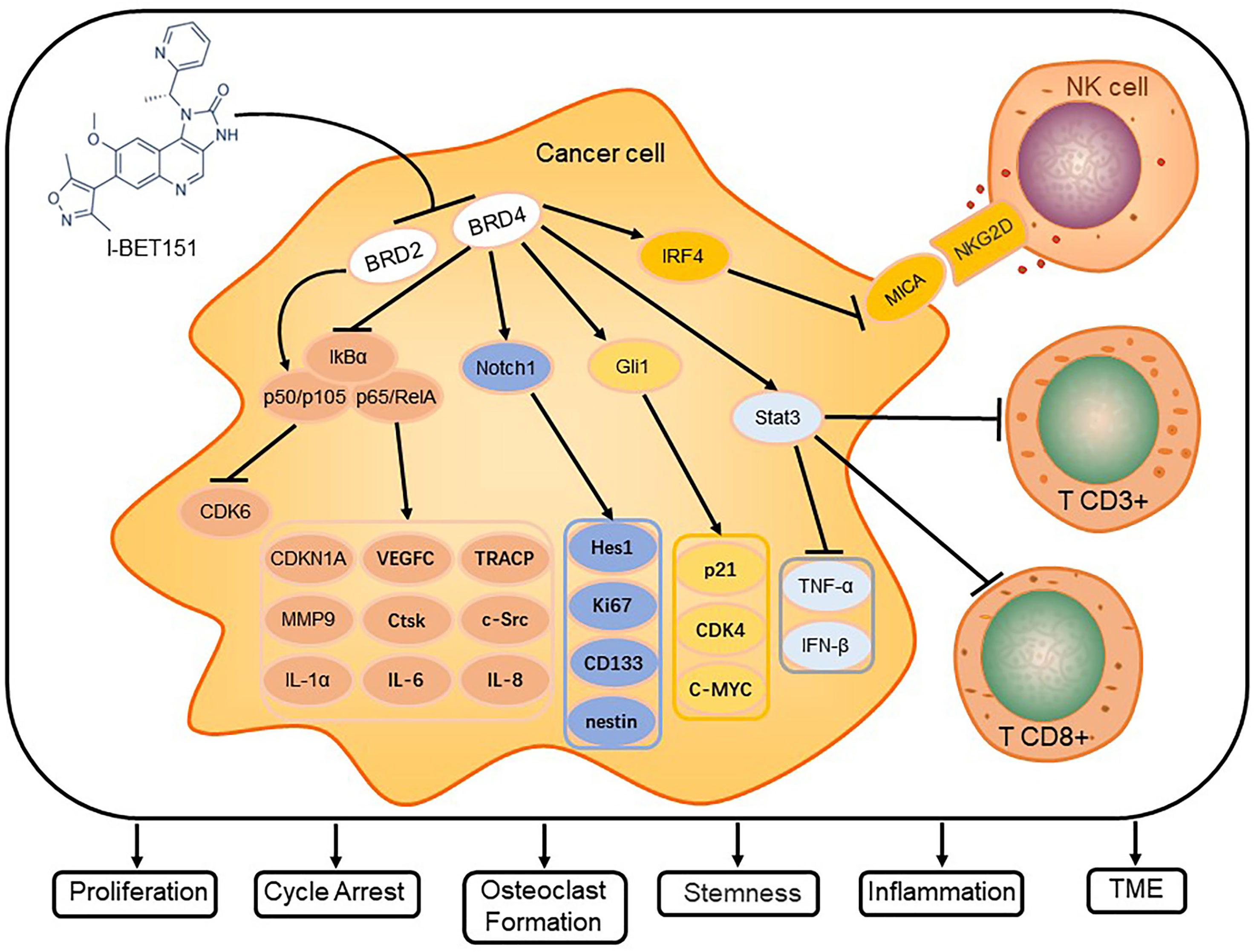
Figure 2 I-BET151 affects the signal transduction in cancer cells and modulates critical cellular processes. I-BET151 specifically inhibits BRD2 and BRD4, decreases the intracellular content of p50/p105, diminishes the degradation of IkB-α, prevents the dissociation of p50/p105 and p65/RelA from IkB-α and their transport into the nucleus, and decreases the activity of NF-B signal transduction. In addition, I-BET151 reduces the binding of BRD4 to the Notch1 and Gli1 promoter regions, inhibits the transcription of Notch1 and Gli1, and causes the target molecules of Notch and Hh signaling pathways to change. In the aspect of influencing tumor microenvironment, I-BET151 not only targets BRD4, which leads to the increase of MICA expression and promoting NK cell degranulation, but also inhibits Stat3 signal, which leads to more CD3+ and CD8+ cells in tumors. Eventually, I-BET151 leads to cell cycle arrest, inhibition of cancer cell proliferation, stemness of stem cells, inflammatory factor release, osteoclast formation and regulation of TME.
I-BET151 and Inhibition of NF-κB Signaling Pathways
As early as 1863, Rudolf Virchow linked inflammation with cancer. Subsequent studies suggested that inflammation may be an auxiliary factor in cancer (8). According to the global cancer attribution analysis in 2018, 2.2 million cancer cases were attributed to infections, and inflammation was the main component of these chronic infections (9). The seven members of the NF-κB family, RelA/p65, c-Rel, RelB, p100, p52, p105, and p50, are central mediators of inflammatory processes. Moreover, there is growing evidence that the NF-κB signaling pathway forms a critical connection between inflammation and cancer. Specifically, NF-κB can stimulate cancer cell proliferation, inhibit cancer cell apoptosis, and promote cancer-related migration and invasion in various cancers (10).
I-BET151 treatment reduces NF-κB activity in many melanoma cell lines, especially SK-Mel-28 and Mel-JD, and in primary cell lines with vemurafenib resistance, which is related to NF-κB overexpression. NF-κB activity inhibition by I-BET151 is mainly reflected in the reduction of p105 and p50, whereas RelA remains unchanged, which has also been confirmed in tumor-bearing animal models. Moreover, the expression of CDKN1A is increased while the CDK6 content is decreased, which indicates that I-BET151 is reducing cancer cell proliferation, resulting in cell cycle arrest. Furthermore, in melanoma, I-BET151 also inhibits the production of cytokines and chemokines, such as interleukin (IL)-1α, vascular endothelial growth factor C (VEGFC), IL-6, and IL-8, and its effect on NF-κB is mainly mediated via BRD2 (4).
Monocytes of patients with myeloma easily differentiate into osteoclasts because NF-κB signaling is activated in monocytes by the receptor agonist of NF-κB ligand (RANKL), leading to IκB-α degradation and RelA/P65 nuclear translocation, both of which promote osteoclast generation. I-BET151 specifically inhibits BRD4, thereby inhibiting RANKL-induced IκB-α degradation and p65 nuclear translocation. In isolated mononuclear cells from healthy donors and patients with multiple myeloma, I-BET151 inhibits NF-κB signaling pathways in monocytes in a dose-dependent manner and diminishes the expression of osteoclast-specific genes, such as TRACP, MMP9, Ctsk, and c-Src, all of which contributes to the inhibition of osteoclast formation. Moreover, BRD4 knockdown also enhances the effect of I-BET151 (11).
Thus, I-BET51 inhibits NF-κB signal by targeting different molecules (BRD2 or BRD4), which is caused by different cell types. However, abnormally activated NF-κB signaling may induce I-BET151 resistance in tumors, as demonstrated by triple-negative breast cancer (TNBC) and lymphoma cell line U937. In these and other similar cases, a select combination with other NF-κB pathway inhibitors can restore the susceptibility of tumor cells to I-BET151 (12, 13).
I-BET151 and Inhibition of Notch Signaling Pathways
The evolutionarily conserved Notch signaling pathway regulates cell fate during the development and the maintenance of tissue steady state; it also affects cell differentiation, proliferation, apoptosis, and epithelial-mesenchymal transition (EMT), as well as self-renewal and differentiation of stem cells (14–16). Notch signaling is related to both carcinogenesis and cancer suppression, depending on the context. In most studies, Notch appeared to be carcinogenic. However, some reports also indicate that the attenuation of Notch activity can induce certain types of brain cancer, breast cancer, ovarian cancer, small cell lung cancer, and hematologic malignancies (15, 17).
In the Notch signaling cascade, both the signal-inducing and -receiving cells interact with each other via ligand-receptor interactions. Mammals have four Notch receptors (Notch1-4) and five Delta-Serrate-Lag ligands (JAG1, JAG2, DLL1, DLL3, and DLL4) (18). Notch1 is activated via ligand-mediated cleavage by members of a disintegrin and metalloproteinase (ADAM) family and the γ-secretase complex (19). It promotes tumorigenesis in various tumor types and interferes with several signaling pathways, affecting cell proliferation, apoptosis, chemotherapy sensitivity, immune response, and self-renewal of cancer stem cells (20).
BRD4 binds to the proximal region of the TNBC Jagged1 promoter and affects migration and invasion of TNBC by regulating the Jagged1/Notch1 signaling pathway (21). Chromatin immunoprecipitation (ChIP) experiments demonstrated that BRD4 also has an affinity for the Notch1 promoter region. The inhibition and consumption of BRD4 downregulated Notch1 and suppressed stem cell marker‐related genes in glioma-initiating cells (GICs), which affected the self-renewal ability and tumorigenesis of these cells. Moreover, inhibiting Notch1 in BRD4 overexpressing cells, the self-renewal ability and proliferation of GICs are still inhibited. I-BET151 disrupted the effect of BRD4 on the Notch1 promoter by competing for acetylated histone binding sites. An immunohistochemistry analysis of intracranial orthotopic xenografts in female nude mice also found that the I-BET151 treatment suppressed the expression of Notch1, Hes1, Ki67, CD133, and nestin (6).
I-BET151 and Inhibition of Hh Signaling Pathways
The Hh pathway is evolutionarily conserved and necessary for normal embryo development. Specifically, the Hh gene family is involved in controlling the left-right asymmetry, the polarity of the central nervous system (CNS), body segments and limbs, organogenesis, chondrogenesis, and spermatogenesis (22, 23). A recent study found that abnormal Hh signal transduction can induce various cancers, including medulloblastoma, basal cell carcinoma, rhabdomyosarcoma, breast cancer, lung cancer, liver cancer, pancreatic cancer, gastric cancer, colon cancer, and prostate cancer (24).
In mammals, the core components of the Hh pathway include three Hh ligands (Sonic hedgehog, Indian hedgehog, and Desert hedgehog), the transmembrane receptor Patched (PTCH1), the G protein-coupled receptor-like transmembrane protein Smoothened (SMO), and three transcription factors (GLI1, GLI2, and GLI3). Moreover, the primary cilia are also involved in the signal transduction of the Hh pathway (24).
I-BET151 dose-dependently attenuates Hh signal transduction in Light2 cells, and its mechanism does not depend on inhibiting the SMO activity by binding, which is employed by most Hh inhibitors. An analysis of the expression levels of PTCH1, SMO, GLI2, and GLI1 after the treatment with I-BET151 demonstrates that the inhibitor significantly reduces the expression of GLI1 but has no effect on the SMO expression level. Furthermore, I-BET151 treatment reduces the expression levels of p21, CDK4, and C-MYC. In addition, the downregulation of BRD4 with siRNA also diminishes the GLI1 expression level. This indicates that I-BET151 inhibits GLI1 transcription by limiting the binding of BRD4 to the proximal regulatory region of the GLI1 locus. In a mouse model of Ptch1+/- derived medulloblastoma, I-BET151 treatment dose-dependently reduces the viability of isolated cancer stem cells, significantly suppresses the growth of medulloblastoma in vivo, and lowers the expression level of the Hh target gene GLI1 (25).
I-BET151 and Regulation of TME
TME is the cellular environment in which tumor cells are located, which is composed of a variety of cellular and non-cellular elements. Cells that TME involves include cancer-associated fibroblasts, natural killer (NK) cells, tumor-associated macrophages, tumor-associated neutrophils, tumor endothelial cells, pericytes, tumor-associated adipocytes, B lymphocytes or T lymphocytes. Non-cellular elements include blood vessels, lymphatic vessels, extracellular matrix, soluble molecules, and small organelles. TME is involved in tumor development, invasion, metastasis, recurrence, drug response, and maintenance of stem-like phenotype (26).
NK cells are the main effector cells in innate immunity, which kill cells by secreting granzymes and perforins. It interacts with extracellular matrix, cancer cells, stromal cells, and metabolites in TME to exert antitumor immunity (27). MHC class I polypeptide-related sequence A (MICA) is a natural killer group 2D ligand (NKG2DL) expressed by tumor cells. Natural killer group 2D (NKG2D) receptors activated on the surface of NK cells can bind to MICA to activate NK cells and kill tumor cells (28). I-BET151 targets BRD4 in multiple myeloma cells and inhibits the expression of C-MYC and IRF4, thereby improving the transcription and translation levels of MICA, promoting the degranulation of NK cells and inducing anti-tumor immune response (29). Multiple myeloma cells can secrete a variety of inflammatory cytokines, which interact with TME to induce osteoclast differentiation and inhibit osteoblast formation, thus promoting the development of multiple myeloma. I-BET151 inhibits the release of IL-1β, and IL-6 in peripheral blood mononuclear cells and myeloma cells by reducing BRD4-mediated activation of NF-κB (11). Furthermore, in melanoma, I-BET151 also inhibits the production of cytokines and chemokines, such as IL-1α, VEGFC, IL-6, and IL-8. This is also attributed to the inhibition of the BET family proteins by I-BET151 (4). In the ovarian cancer mouse model, I-BET151 treatment inhibits the Stat3 signaling pathway, induces more CD3+ and CD8+ cells in the tumor, increases TNF-α and IFN-β mRNA levels in the tumor and mouse spleen, and induces an anti-tumor immune response (30).
I-BET151 Prevents Telomere Elongation
Telomeres are composed of tandem repeats of the TTAGGG sequence motif. They are special chromatin structures that form the end of the chromosome. Over multiple rounds of cell division, telomeres gradually lose the TTAGGG tandem repeats and become shorter, which is a sign of aging in organisms. Telomere length is regulated by chromatin modification, telomere binding proteins, and telomerase (31). Importantly, the risk of cancer is increased by telomeres that are too long or too short (32).
Telomerase lengthens telomeres and keeps their length in a steady state. Most cancer cells modulate telomerase activity. Therefore, telomerase inhibitors represent a targeted strategy for cancer treatment (33). Interestingly, telomere extension induced by telomerase overexpression in 293T cells can be dose-dependently blocked by I-BET151. However, treatment of these 293T cells with the highest tolerated I-BET151 dose does not inhibit the telomerase activity, indicating that I-BET151 does not employ the same mechanism for blocking telomere elongation as conventional telomerase inhibitors. The results obtained with I-BET151 are similar to those observed with three known BRD4 inhibitors, suggesting that attenuation of telomere elongation by I-BET151 depends on the inhibition of BRD4. I-BET151 interferes with the binding of BRD4 to acetylated lysine residues by targeting the bromine domain (34). It is not completely clear how BRD4 coordinates telomere maintenance, but it is known that BRD4 selectively controls the expression of telomerase reverse transcriptase in the presence of cancer-related promoter mutations (35).
I-BET151 Is Effective Against Various Cancers
I-BET151 was first used for leukemia treatment, and later studies found that I-BET151 is also effective against various solid cancers, including breast cancer, glioma, and melanoma. Here, we summarize the anticancer activity of I-BET151 against various cancers (Table 1).
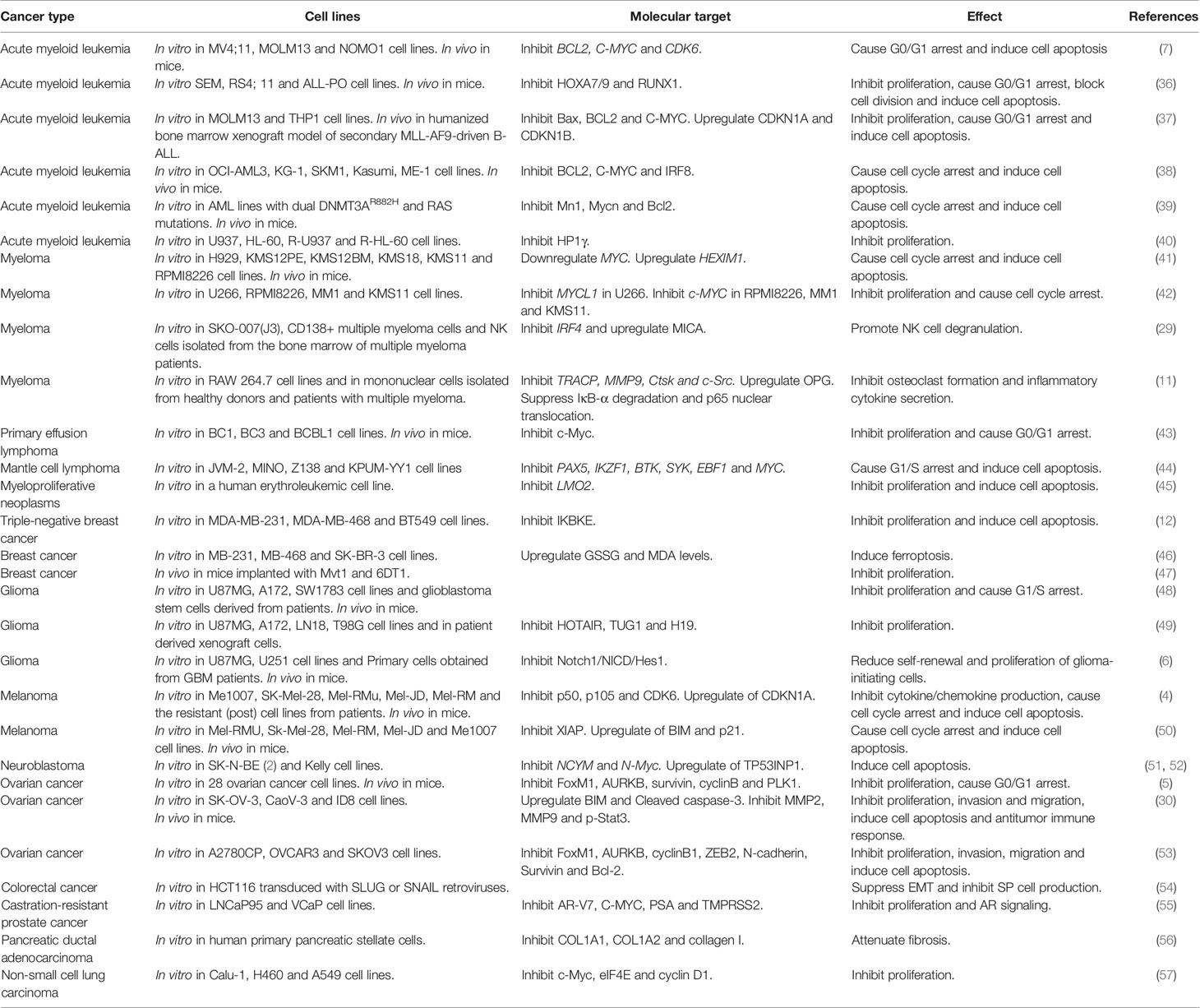
Table 1 Anticancer activity of I-BET151 against various cancer cells.
Hematological Malignancies
I-BET151 exerts anti-leukemia activity by decreasing the presence of BRD4 and CDK8 in the enhancer region and downregulating the genes related to super-enhancers (SEs) (58). Although I-BET151 treatment simultaneously dissociates BRD2, BRD3, and BRD4 from chromatin, BRD4 is the most susceptible BET protein. Specifically, in I-BET151-susceptible cell lines, the inhibitor mainly affects BRD4 and prolongs the suspension of RNA Pol II (59).
A special type of acute leukemia is caused by the translocation of the MLL gene encoding an MLL fusion protein, which can transform hematopoietic cells into leukemia stem cells, typically resulting in poor prognosis (60). I-BET151 inhibits the transcription of BCL2, C-MYC, and CDK6 by interfering with the chromatin recruitment of BRD3/4, which ensures efficacy in different MLL fusion cell lines and impairs the propagation of leukemia stem cells (7, 61). Administration of I-BET151 at 30 mg/kg in mouse models of MLL-AF9+ and MLL-AF4+ leukemia delays disease progression and significantly prolongs survival (7). Acute lymphocytic leukemia (ALL) in infants with MLL rearrangement is very invasive. In the preclinical mouse model of MLL-AF4+ infant acute lymphoblastic leukemia, I-BET151 downregulates the transcription of the BRD4, HOXA7/HOXA9, and RUNX1 gene network, which reduces the disease burden. In addition, I-BET151 increased the susceptibility of MLL-rearranged ALL cells to prednisolone in vitro, which provides a new treatment strategy for glucocorticoid-resistant ALL (36). In the MLL-AF9+ cell line, the HOXA gene is not downregulated by I-BET151. Comparative analysis of ChIP-seq data and RNA-seq data indicates that I-BET151 only targets less than 1/10 of MLL-AF9 directly targeted genes. Treatment with I-BET151 significantly delayed the progression of lymphocytic leukemia in NSG mice (37). The mice were implanted with ceramic scaffolds of human mesenchymal stem cells, which fully simulated the environment for human bone marrow, but it was not enough to completely eradicate leukemia cells (62). This suggests that the human bone marrow-like environment may have protective properties for leukemia cells.
I-BET151 is effective against a variety of acute myeloid leukemia (AML) subtypes (38). NPM1c AML is one of the most frequently reported subtypes, and its prognosis is related to synergistic mutations (63). However, regardless of the nature of the cooperative mutation, in vitro and in vivo analyses indicate that NPM1c AML is consistently susceptible to I-BET151 because the drug inhibits BRD4 rather than wild-type NPM1 (38). Somatic mutations in DNA methyltransferase 3A (DNMT3Amut) occur in a variety of hematological malignancies, including in AML and elderly individuals with clonal hematopoiesis, with hot-spot mutations at the Arg882 residue (DNMT3AR882mut) accounting for 50–60% among the identified DNMT3Amut in AML (64–67). I-BET151 causes the downregulation of DNMT3AR882H-related target genes by blocking BRD4; it also induces the upregulation of apoptosis-related genes and the downregulation of cell cycle progression genes. I-BET151 significantly delays the development of AML phenotypes, such as splenomegaly, increases the white blood cell count, and decreases the red blood cell count in an AML mouse model induced by two mutations, DNMT3A R882H and RAS G12D (39). The DNA methyltransferase inhibitor 5-azacytidine (AZA) is effective in myelodysplastic syndromes and AML (68). HP1γ is important in the survival of AZA drug-resistant cells, and I-BET151 can function as HP1γ inhibitor for the treatment of AZA drug-resistant hematological malignancies (40).
Critical mechanisms employed by BET inhibitors to fight multiple myeloma involve the inhibition of MYC transcription and MYC carcinogenesis (69), both of which are also caused by I-BET151 that exerts its inhibitory activity by attenuating the chromatin recruitment of CDK9 in a BRD2/3/4-dependent manner, which caused transcription inhibition of MYC and MYC carcinogenic programs. However, I-BET151 upregulates HEXIM1 transcription, which leads to cycle arrest and apoptosis of myeloma cells (41). The C-MYC- expressing myeloma cell lines are inhibited by I-BET51, which exerts its inhibitory activity by diminishing the c-MYC expression, but in U266 cells that do not express c-MYC, I-BET151 interferes with MYCL expression (42, 70). I-BET151 can also target the RANKL-NF-κB signaling pathway, inhibit the formation of osteoclasts, reduce the levels of osteoclast-specific genes TRACP, MMP9, Ctsk, and c-Src, and inhibit the secretion of inflammatory cytokines (11).
Primary exudative lymphoma (PEL) is an aggressive non-Hodgkin’s lymphoma, which is related to Kaposi’s sarcoma-associated herpesvirus (KSHV) infection. Non-PEL cell lines are much less susceptible to I-BET151 treatment than PEL cell lines, in which the drug downregulates the c-MYC level, inhibits lymphoma cell proliferation, and induces cell cycle arrest (43). Mantle cell lymphoma (MCL) is a refractory B-cell lymphoma caused by the translocation t (11, 14)(q13; Q32) (71). BRD4 directly regulates a series of genes related to the B cell receptor signaling pathway. I-BET151 promotes the G1/S cell cycle arrest and apoptosis in BRD4-induced MCL cells, which represents a new strategy for treating MCL disease (44).
The human erythroid leukemia (HEL) cell lines are susceptible to I-BET151, which functions as a JAK2 inhibitor and remains effective against JAK2 inhibitor-resistant HEL cells (45).
Breast Cancer
TNBC is the most aggressive breast cancer subtype, but I-BET151 can diminish NF-κB signaling by reducing IKBKE expression, which has a therapeutic effect on TNBC (12). High SIRT1 activity promotes DNA repair and cell cycle arrest and prevents various stress-induced apoptosis (72). I-BET151 increases the level of SIRT1 in MCF-7 and MDA-MB-231 cells, but it does not affect or even reduces the relative deacetylation activity of SIRT1 in the cells (73). I-BET151 is also known to induce ferroptosis in breast cancer cells (46). In mice implanted with highly metastatic breast cancer cell lines Mvt1 and 6DT1, I-BET151 inhibited the growth of primary tumors, but not the metastasis, which is related to the opposite effects of two BRD4 isoforms (47). Specifically, metastasis is diminished by the long BRD4 isoform but promoted by the short BRD4 isoform (74, 75).
Glioma
Gliomas have significantly higher BRD2 and BRD4 levels than control tissues, and the mRNA and expression levels of BRD4 are closely related to the tumor subtypes and the overall survival rate of the patients, indicating that I-BET151 can have a therapeutic effect on gliomas (6, 48). I-BET151 is known to inhibit the proliferation of U87MG cells, limit the cell cycle progression from G1 to S, and reduce the tumor size in U87MG xenografts (48). There is growing evidence that long non-coding RNA plays an important role in carcinogenesis and anticancer pathways (76–78). HOX transcribed antisense RNA (HOTAIR) is overexpressed in glioma and associated with the proliferation and periodic progression of this tumor. The anticancer effect of I-BET151 in glioma is achieved, at least in part, by downregulating HOTAIR (49). Notch signaling is involved in the self-renewal of glioma stem cells (GSCs) and the regulation of tumorigenesis. The direct association between BRD4 and the Notch1 promoter region contributes to transcriptional regulation. Therefore, I-BET151 can regulate the Notch signal transduction pathway by targeting BRD4, which affects the self-renewal of GSCs and tumorigenesis (6).
Melanoma
NF-kB is activated in melanoma (79). I-BET151 inhibits NF-kB activation in melanoma by targeting BRD2, causing cycle arrest, promoting apoptosis, and inhibiting the production of cytokines (e.g., IL6 and IL-8) and chemokines (e.g., CXCL10 and CCL5), which indicates that I-BET151 may have a therapeutic effect on melanoma (4). Another report shows that I-BET151 activates the BIM protein, a BH3-only pro-apoptotic protein family member, and the increase in BIM mediates caspase-dependent apoptosis, which is mainly related to the inhibition of BRD2. However, I-BET151-induced G1 arrest is associated with BRD4 inhibition and mediated by p21. The efficacy of I-BET151 is not identical across different melanoma cell lines; the NRAS mutant cell line (Mel-RM) and the NRAS/BRAF wild-type (Me1007) line are the most susceptible cell lines, whereas the NRAS mutant/BRAF wild-type (Mel-JD) line and the NRAS wild-type/BRAF mutant cell lines (SK-Mel-28, Mel-RMU) are relatively insensitive (50).
Neuroblastoma
Neuroblastoma is the most common extracranial solid tumor in children, accounting for 15% of the total tumor deaths in children (80). Statistical analysis of neuroblastoma specimens shows that low expression of nuclear protein 1 induced by tumor protein 53 (TP53INP1) in tumor tissues and high expression of N-Myc in neuroblastoma patients are closely related to poor prognosis. I-BET151 inhibits the transcription and expression of NCYM and N-Myc in neuroblastoma cells and significantly increases the mRNA and protein levels of TP53INP1, which promotes apoptosis of tumor cells (51, 52).
Ovarian Cancer
The expression of BRD4 is significantly higher in clinical ovarian cancer tissues than in non-malignant control tissues, whereas the levels of BRD2 and BRD3 do not significantly vary between malignant and non-malignant tissues. In addition, a pan-cancer analysis indicates that ovarian cancer is the most apparent tumor with BRD4 amplification. I-BET151 inhibits the viability of a wide range of ovarian tumor cells, including 28 epithelial ovarian cancer (EOC) cell lines that cover all histological types. This broad spectrum of activity is related to I-BET151-induced apoptosis mediated by mitochondria and the downregulation of the transcription and translation of FoxM1 and its transcription targets (5, 30). In addition, I-BET151 reduces the migration and invasion of EOC cells by inhibiting the Stat3 signaling pathway and downregulating ZEB2 and N-cadherin, which also inhibits tumor metastasis in the abdominal metastasis model of ovarian cancer (30, 53). Similar to the discovery in multiple myeloma, I-BET151 can also induce anticancer immunity in ovarian cancer (30).
Colorectal Cancer
Mutation or defect of succinate dehydrogenase B (SDHB) can lead to the loss of enzyme activity and expression, which can occur in various malignant tumors. However, colorectal cancer cells with SDHB knockout are highly susceptible to I-BET151 (81). EMT cells were generated from colorectal cancer tissue by SLUG or SNAIL retrovirus transduction, which also produced side population (SP) cells with low Hoechst 33342 staining and differentiation potential. However, I-BET151 not only inhibits the occurrence of EMT but also reduces the proportion of produced SP cells (54).
Prostatic Cancer
Androgen receptor (AR) is the main carcinogen in the development of prostate cancer. Second-generation antiandrogen therapy can enhance receptor signaling and improve the prognosis of castration-resistant prostate cancer (CRPC) (82–84). However, the expression of AR splice variants leads to drug resistance, including the AR splice variant 7 (AR-V7) (85, 86). Based on clinical prostate samples, BRD4 is associated with AR activity and patient survival. I-BET151 decreases AR-V7 and C-MYC expression levels and inhibits AR signaling, suggesting a new therapeutic strategy for patients with CRPC (55).
Pancreatic Ductal Adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC), as the most common pancreatic cancer type, is often associated with the development of a fibrotic reaction (87). Pancreatic stellate cells (PSCs) are the key regulators of fibrosis that produce only limited amounts of collagen I in the static state (88). However, unlike BRD2, BRD3, and the FOS-like 1 (FOSL1) protein, BRD4 promotes collagen I production in primary prostate cancer isolated from human PDAC. I-BET151 can effectively suppress the fibrotic reaction and collagen I production by inhibiting BRD4 and preventing BRD4-mediated blockage of FOSL1 (56).
Non-Small Cell Lung Carcinoma
Eukaryotic translation initiation factor 4E (eIF4E), a component of the translation initiation complex, is associated with cellular survival, EMT, and angiogenesis (89, 90). I-BET151 inhibits BRD4 and, therefore, downregulates eIF4E, causing dose-dependent inhibition of cell growth in non-small cell lung cancer (57).
Anticancer Efficacy of I-BET151 in Combination With Other Drugs
Drug resistance or insensitivity is a critical clinical issue associated with chemotherapy in cancer treatment. To overcome drug resistance and improve anticancer efficacy, an increasing number of experiments have been conducted to test I-BET151 in combination with other drugs (Table 2).
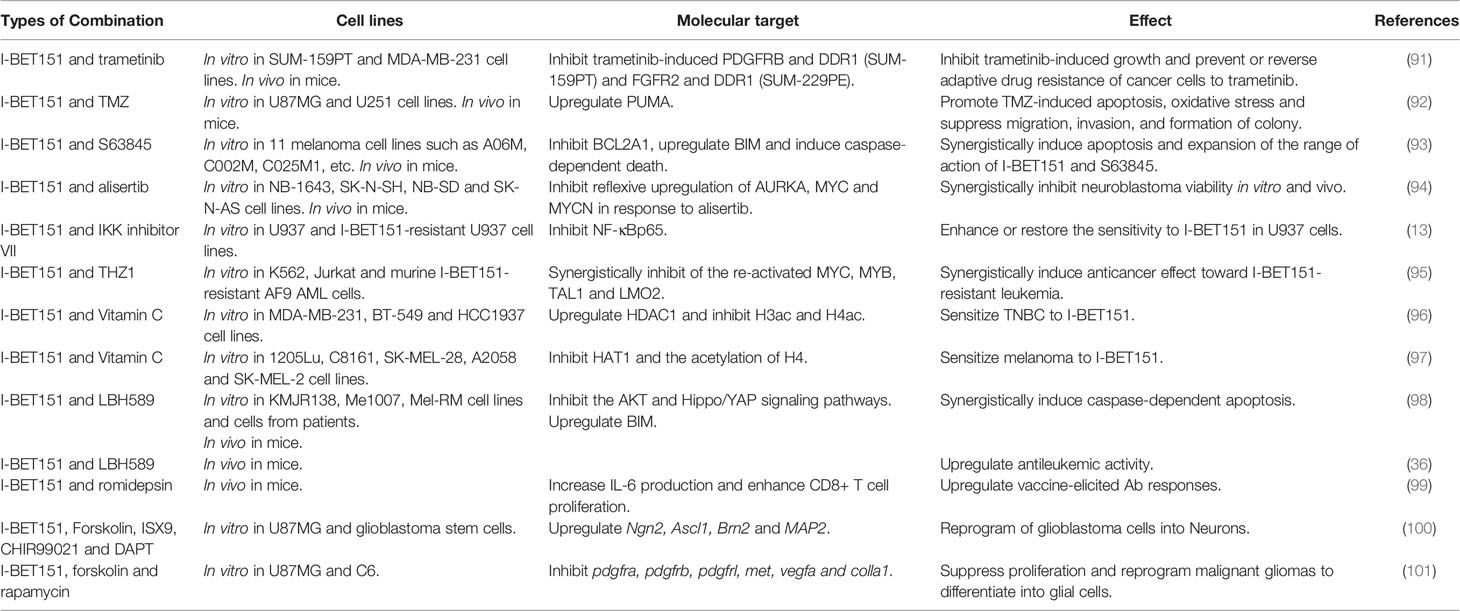
Table 2 Combination of I-BET151 and other drugs in cancer.
In TNBC patients who received trametinib for one week, an adaptive bypass reaction leading to trametinib resistance can be observed. The combination of I-BET151 and trametinib not only synergistically inhibits TNBC growth in vitro and in vivo but also prevents or reverses adaptive drug resistance of cancer cells to trametinib (91). In clinical practice, temozolomide (TMZ) is the main treatment for malignant glioma, but the drug resistance of glioma cells to TMZ will lead to treatment failure. I-BET151 can promote TMZ-mediated inhibition of glioma proliferation, invasion, and migration, enhance the oxidative stress induced by TMZ, and restore susceptibility of glioma cells to TMZ, all of which may be related to the I-BET151-induced expression of the p53-upregulated modulator of apoptosis (PUMA) (92). S63845 is a myeloid leukemia cell differentiation protein 1 (MCL1) inhibitor with therapeutic efficacy against a large number of melanoma cell lines, excluding a few that are not affected by MCL1 inhibition. The combination of this inhibitor with I-BET151 is superior to any single treatment, especially related to the caspase-dependent cell death induced by this combination (93). Neuroblastoma is mainly driven by MYC or MYCN. Inhibition of Aurora kinase A (AURKA) is an effective treatment, but treatment with an AURKA inhibitor (alisertib) often causes an upregulation of the transcription of AURKA, MYC, and MYCN. The combination of the AURKA inhibitor with I-BET151 significantly reduces the transcriptional upregulation and synergistically inhibits the tumor cell survival (94).
The mechanism of I-BET151 resistance of lymphoma cell line U937 is known to be related to the activation of NF-κB. The combination treatment with IKK inhibitor VII can inhibit the activation of NF-κBp65 protein in the nucleus of drug-resistant cells and enhance or restore the susceptibility of U937 cells to I-BET151 by targeting the NF-κB signaling pathway (13). MYC is significantly inhibited in I-BET151-susceptible cells, but it is not affected in I-BET151-resistant cells, indicating a functional compensation for MYC in I-BET151-resistant AML cells, which is related to enhancer remodeling. Plasmacytoma variant translocation 1 (PVT1) is a long non-coding RNA (lncRNA) that functions as an oncogene in many cancers and is known to promote MYC expression in I-BET151-resistant AML cells in a BRD4-independent manner (95, 102). Furthermore, CDK7 inhibitor (THZ1) inhibits MYC expression by interfering with RNA polymerase II activity of the PVT1 enhancer, which kills cancer cells in combination treatment with I-BET151 (95). Interestingly, vitamin C also improves the efficacy of I-BET151. In TNBC cells, the upregulation of histone deacetylase 1 (HDAC1) and the inhibition of histone H3 and H4 acetylation by vitamin C enhances the effect of I-BET151, as indicated by a lower half-maximal effective concentration (EC50), which allows a dose reduction of I-BET151 and concomitantly decreases the risk of side effects (96). However, vitamin C also diminishes the expression of histone acetyltransferase 1 (HAT1) and limits the acetylation of lysine 5 and lysine 12 on H4 without reducing the acetylation of H3 (97). These specific effects can be potentially related to the origin of the tumors from different tissue types, but they also indicate that the I-BET151 activity can be improved by mediating histone acetylation. Thus, combinations of I-BET151 and histone deacetylase (HDAC) inhibitors are increasingly used in patients. In melanoma, the combination of I-BET151 and HDAC inhibitor LBH589 effectively inhibits the AKT and Hippo/YAP signaling pathways, upregulates the BIM expression, synergistically induces caspase-dependent apoptosis of tumor cells, and significantly prolongs the survival time in a xenograft in vivo model (98). This combination also has a synergistic anti-leukemia effect in the preclinical mouse model of MLL-AF4+ infant ALL (36). In melanoma, the replacement of LBH589 with romidepsin for combination therapy with I-BET151 promotes apoptosis and changes the expression of IL-6/JAK/STAT-related genes, which increases the response frequency of CD8+ T cells in mice vaccinated with OVA+CpG tumor vaccine and improves the treatment efficacy and preventive protection of the vaccine (99).
Gliomas originate from glial precursor cells that can be reprogrammed to neurons with the help of nerve cell-specific transcription factors (103–105). Several small molecule combinations involving I-BET151 are known to treat malignant gliomas. The combination of cAMP enhancer Forskolin, ISX9, CHIR99021, and I-BET151, along with dual antiplatelet therapy (DAPT), can upregulate the expression levels of the Ngn2, Ascl1, Brn2, and MAP2 genes in U87 MG cells and reprograms the tumor cells to neuronal morphology without undergoing the intermediate pluripotent state, which can lead to the inhibition of U87MG cell growth and the formation of tumor-like spheroids (100). Another experiment also demonstrates that the combination treatment consisting of I-BET151, along with Forskolin and mammalian target of rapamycin (mTOR) inhibitor (rapamycin), can also reprogram malignant glioma cells into non-proliferative glial cells and strongly inhibit the proliferation of tumor cells. Although this combination is only effective in some glioma types, its inhibitory effect on glioma proliferation is stronger than that of TMZ, and it can still be used in TMZ resistant cells (101).
Conclusion
This review presents a discussion of the anticancer effects and mechanisms of I-BET151, which specifically targets BRD2 and BRD4, regulates the pathways of NF-κB, Notch, and Hh signal transduction, change TME and controls the telomere length. These I-BET151-mediated mechanisms cause the inhibition of proliferation, migration, and invasion of cancer cells, along with the induction of apoptosis. We also assessed the effects of I-BET151 used in combination with other drugs, and we describe different combination types that substantially increase the sensitivity of select chemotherapy drugs and achieve an improved therapeutic efficacy.
I-BET151 has a wide application prospect, and the most attractive one is its application in glioma. The combination of Forskolin, ISX9, CHIR99021, I-BET151 and DAPT can treat glioma by changing the differentiation state of cancer cells and reprogramming glioma cells into neurons. The process did not go through an intermediate pluripotent state, which means that the formed neurons are much less likely to become cancer cells again. This therapeutic strategy is expected to change the current treatment mode of glioma. For patients with small glioma and unobvious space-occupying effect, the use of this drug combination can promote tumor cell transformation and avoid the trauma caused by surgery. On the other hand, for patients with obvious space-occupying effects, surgical resection is required, and then the remaining tumor cells are converted into neurons by using the medicine combination, which can reduce the damage to healthy brain tissue caused by excessive surgical resection range. Moreover, compared with the emerging gene therapy, the side effects caused by drug therapy are easier to be found and solved, and the economic and technical costs required for treatment are relatively lower. This therapeutic strategy has provided new ideas for the clinical treatment of glioma and inspired the treatment of other types of cancer.
At present, there is no related clinical trials, which may be due to the short development time of I-BET151. However, I-BET151 is a valuable anticancer drug with a wide range of therapeutic effects based on preclinical experiments, which provides us with a new therapeutic strategy for clinical anticancer treatment.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author Contributions
JL wrote the article. ZL and YZ collected and organized data and figures. CM and HH revised the manuscript critically. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Sciences Foundation of Jilin Province (20180101158JC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kulikowski E, Rakai BD, Wong NCW. Inhibitors of Bromodomain and Extra-Terminal Proteins for Treating Multiple Human Diseases. Med Res Rev (2021) 41(1):223–45. doi: 10.1002/med.21730
2. Dawson MA, Kouzarides T, Huntly BJ. Targeting Epigenetic Readers in Cancer. N Engl J Med (2012) 367(7):647–57. doi: 10.1056/NEJMra1112635
3. French CA, Ramirez CL, Kolmakova J, Hickman TT, Cameron MJ, Thyne ME, et al. BRD-NUT Oncoproteins: A Family of Closely Related Nuclear Proteins That Block Epithelial Differentiation and Maintain the Growth of Carcinoma Cells. Oncogene (2008) 27(15):2237–42. doi: 10.1038/sj.onc.1210852
4. Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, et al. Control of NF-kB Activity in Human Melanoma by Bromodomain and Extra-Terminal Protein Inhibitor I-Bet151. Pigment Cell Melanoma Res (2014) 27(6):1126–37. doi: 10.1111/pcmr.12282
5. Zhang Z, Ma P, Jing Y, Yan Y, Cai MC, Zhang M, et al. BET Bromodomain Inhibition as a Therapeutic Strategy in Ovarian Cancer by Downregulating Foxm1. Theranostics (2016) 6(2):219–30. doi: 10.7150/thno.13178
6. Tao Z, Li X, Wang H, Chen G, Feng Z, Wu Y, et al. BRD4 Regulates Self-Renewal Ability and Tumorigenicity of Glioma-Initiating Cells by Enrichment in the Notch1 Promoter Region. Clin Transl Med (2020) 10(6):e181. doi: 10.1002/ctm2.181
7. Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Nature (2011) 478(7370):529–33. doi: 10.1038/nature10509
8. Balkwill F, Mantovani A. Inflammation and Cancer: Back to Virchow? Lancet (2001) 357(9255):539–45. doi: 10.1016/S0140-6736(00)04046-0
9. de Martel C, Georges D, Bray F, Ferlay J, Clifford GM. Global Burden of Cancer Attributable to Infections in 2018: A Worldwide Incidence Analysis. Lancet Glob Health (2020) 8(2):e180–90. doi: 10.1016/S2214-109X(19)30488-7
10. DiDonato JA, Mercurio F, Karin M. NF-kappaB and the Link Between Inflammation and Cancer. Immunol Rev (2012) 246(1):379–400. doi: 10.1111/j.1600-065X.2012.01099.x
11. Guo NH, Zheng JF, Zi FM, Cheng J. I-BET151 Suppresses Osteoclast Formation and Inflammatory Cytokines Secretion by Targetting BRD4 in Multiple Myeloma. Biosci Rep (2019) 39(5):BSR20181245. doi: 10.1042/BSR20181245
12. Qiao J, Chen Y, Mi Y, Jin H, Wang L, Huang T, et al. Macrophages Confer Resistance to BET Inhibition in Triple-Negative Breast Cancer by Upregulating IKBKE. Biochem Pharmacol (2020) 180:114126. doi: 10.1016/j.bcp.2020.114126
13. Hishiki K, Akiyama M, Kanegae Y, Ozaki K, Ohta M, Tsuchitani E, et al. NF-kappaB Signaling Activation via Increases in BRD2 and BRD4 Confers Resistance to the Bromodomain Inhibitor I-BET151 in U937 Cells. Leuk Res (2018) 74:57–63. doi: 10.1016/j.leukres.2018.09.016
14. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch Signaling: Cell Fate Control and Signal Integration in Development. Science (1999) 284(5415):770–6. doi: 10.1126/science.284.5415.770
15. Liu J, Sato C, Cerletti M, Wagers A. Notch Signaling in the Regulation of Stem Cell Self-Renewal and Differentiation. Curr Top Dev Biol (2010) 92:367–409. doi: 10.1016/S0070-2153(10)92012-7
16. Aster JC, Pear WS, Blacklow SC. The Varied Roles of Notch in Cancer. Annu Rev Pathol (2017) 12:245–75. doi: 10.1146/annurev-pathol-052016-100127
17. Nowell CS, Radtke F. Notch as a Tumour Suppressor. Nat Rev Cancer (2017) 17(3):145–59. doi: 10.1038/nrc.2016.145
18. Andersson ER, Sandberg R, Lendahl U. Notch Signaling: Simplicity in Design, Versatility in Function. Development (2011) 138(17):3593–612. doi: 10.1242/dev.063610
19. Rice MA, Hsu EC, Aslan M, Ghoochani A, Su A, Stoyanova T. Loss of Notch1 Activity Inhibits Prostate Cancer Growth and Metastasis and Sensitizes Prostate Cancer Cells to Antiandrogen Therapies. Mol Cancer Ther (2019) 18(7):1230–42. doi: 10.1158/1535-7163.MCT-18-0804
20. Gharaibeh L, Elmadany N, Alwosaibai K, Alshaer W. Notch1 in Cancer Therapy: Possible Clinical Implications and Challenges. Mol Pharmacol (2020) 98(5):559–76. doi: 10.1124/molpharm.120.000006
21. Andrieu G, Tran AH, Strissel KJ, Denis GV. BRD4 Regulates Breast Cancer Dissemination Through Jagged1/Notch1 Signaling. Cancer Res (2016) 76(22):6555–67. doi: 10.1158/0008-5472.CAN-16-0559
22. Hammerschmidt M, Brook A, McMahon AP. The World According to Hedgehog. Trends Genet (1997) 13(1):14–21. doi: 10.1016/s0168-9525(96)10051-2
23. Goodrich LV, Scott MP. Hedgehog and Patched in Neural Development and Disease. Neuron (1998) 21(6):1243–57. doi: 10.1016/s0896-6273(00)80645-5
24. Wu F, Zhang Y, Sun B, McMahon AP, Wang Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem Biol (2017) 24(3):252–80. doi: 10.1016/j.chembiol.2017.02.010
25. Long J, Li B, Rodriguez-Blanco J, Pastori C, Volmar CH, Wahlestedt C, et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-Driven Cancers. J Biol Chem (2014) 289(51):35494–502. doi: 10.1074/jbc.M114.595348
26. Hernandez-Camarero P, Lopez-Ruiz E, Marchal JA, Peran M. Cancer: A Mirrored Room Between Tumor Bulk and Tumor Microenvironment. J Exp Clin Cancer Res (2021) 40(1):217. doi: 10.1186/s13046-021-02022-5
27. Wu SY, Fu T, Jiang YZ, Shao ZM. Natural Killer Cells in Cancer Biology and Therapy. Mol Cancer (2020) 19(1):120. doi: 10.1186/s12943-020-01238-x
28. Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, et al. Natural Killer Group 2D Receptor and Its Ligands in Cancer Immune Escape. Mol Cancer (2019) 18(1):29. doi: 10.1186/s12943-019-0956-8
29. Abruzzese MP, Bilotta MT, Fionda C, Zingoni A, Soriani A, Vulpis E, et al. Inhibition of Bromodomain and Extra-Terminal (BET) Proteins Increases NKG2D Ligand MICA Expression and Sensitivity to NK Cell-Mediated Cytotoxicity in Multiple Myeloma Cells: Role of cMYC-IRF4-miR-125b Interplay. J Hematol Oncol (2016) 9(1):134. doi: 10.1186/s13045-016-0362-2
30. Liu A, Fan D, Wang Y. The BET Bromodomain Inhibitor I-BET151 Impairs Ovarian Cancer Metastasis and Improves Antitumor Immunity. Cell Tissue Res (2018) 374(3):577–85. doi: 10.1007/s00441-018-2906-y
31. Blasco MA. Telomeres and Human Disease: Ageing, Cancer and Beyond. Nat Rev Genet (2005) 6(8):611–22. doi: 10.1038/nrg1656
32. Aviv A, Anderson JJ, Shay JW. Mutations, Cancer and the Telomere Length Paradox. Trends Cancer (2017) 3(4):253–8. doi: 10.1016/j.trecan.2017.02.005
33. Greider CW. Telomerase Activation. One Step on the Road to Cancer? Trends Genet (1999) 15(3):109–12. doi: 10.1016/s0168-9525(98)01681-3
34. Wang S, Pike AM, Lee SS, Strong MA, Connelly CJ, Greider CW. BRD4 Inhibitors Block Telomere Elongation. Nucleic Acids Res (2017) 45(14):8403–10. doi: 10.1093/nar/gkx561
35. Donati B, Lorenzini E, Ciarrocchi A. BRD4 and Cancer: Going Beyond Transcriptional Regulation. Mol Cancer (2018) 17(1):164. doi: 10.1186/s12943-018-0915-9
36. Bardini M, Trentin L, Rizzo F, Vieri M, Savino AM, Garrido Castro P, et al. Antileukemic Efficacy of BET Inhibitor in a Preclinical Mouse Model of MLL-AF4(+) Infant ALL. Mol Cancer Ther (2018) 17(8):1705–16. doi: 10.1158/1535-7163.MCT-17-1123
37. Carretta M, Brouwers-Vos AZ, Bosman M, Horton SJ, Martens JHA, Vellenga E, et al. BRD3/4 Inhibition and FLT3-Ligand Deprivation Target Pathways That Are Essential for the Survival of Human MLL-AF9+ Leukemic Cells. PloS One (2017) 12(12):e0189102. doi: 10.1371/journal.pone.0189102
38. Dawson MA, Gudgin EJ, Horton SJ, Giotopoulos G, Meduri E, Robson S, et al. Recurrent Mutations, Including NPM1c, Activate a BRD4-Dependent Core Transcriptional Program in Acute Myeloid Leukemia. Leukemia (2014) 28(2):311–20. doi: 10.1038/leu.2013.338
39. Lu R, Wang J, Ren Z, Yin J, Wang Y, Cai L, et al. A Model System for Studying the DNMT3A Hotspot Mutation (DNMT3A(R882)) Demonstrates a Causal Relationship Between Its Dominant-Negative Effect and Leukemogenesis. Cancer Res (2019) 79(14):3583–94. doi: 10.1158/0008-5472.CAN-18-3275
40. Imanishi S, Umezu T, Kobayashi C, Ohta T, Ohyashiki K, Ohyashiki JH. Chromatin Regulation by HP1gamma Contributes to Survival of 5-Azacytidine-Resistant Cells. Front Pharmacol (2018) 9:1166. doi: 10.3389/fphar.2018.01166
41. Chaidos A, Caputo V, Gouvedenou K, Liu B, Marigo I, Chaudhry MS, et al. Potent Antimyeloma Activity of the Novel Bromodomain Inhibitors I-BET151 and I-Bet762. Blood (2014) 123(5):697–705. doi: 10.1182/blood-2013-01-478420
42. Suzuki K, Yamamoto K, Arakawa Y, Yamada H, Aiba K, Kitagawa M. Antimyeloma Activity of Bromodomain Inhibitors on the Human Myeloma Cell Line U266 by Downregulation of MYCL. Anticancer Drugs (2016) 27(8):756–65. doi: 10.1097/CAD.0000000000000389
43. Tolani B, Gopalakrishnan R, Punj V, Matta H, Chaudhary PM. Targeting Myc in KSHV-Associated Primary Effusion Lymphoma With BET Bromodomain Inhibitors. Oncogene (2014) 33(22):2928–37. doi: 10.1038/onc.2013.242
44. Tsukamoto T, Nakahata S, Sato R, Kanai A, Nakano M, Chinen Y, et al. BRD4-Regulated Molecular Targets in Mantle Cell Lymphoma: Insights Into Targeted Therapeutic Approach. Cancer Genomics Proteomics (2020) 17(1):77–89. doi: 10.21873/cgp.20169
45. Wyspianska BS, Bannister AJ, Barbieri I, Nangalia J, Godfrey A, Calero-Nieto FJ, et al. BET Protein Inhibition Shows Efficacy Against JAK2V617F-Driven Neoplasms. Leukemia (2014) 28(1):88–97. doi: 10.1038/leu.2013.234
46. Qiao J, Chen Y, Mi Y, Jin H, Huang T, Liu L, et al. NR5A2 Synergizes With NCOA3 to Induce Breast Cancer Resistance to BET Inhibitor by Upregulating NRF2 to Attenuate Ferroptosis. Biochem Biophys Res Commun (2020) 530(2):402–9. doi: 10.1016/j.bbrc.2020.05.069
47. Alsarraj J, Faraji F, Geiger TR, Mattaini KR, Williams M, Wu J, et al. BRD4 Short Isoform Interacts With RRP1B, SIPA1 and Components of the LINC Complex at the Inner Face of the Nuclear Membrane. PloS One (2013) 8(11):e80746. doi: 10.1371/journal.pone.0080746
48. Pastori C, Daniel M, Penas C, Volmar CH, Johnstone AL, Brothers SP, et al. BET Bromodomain Proteins Are Required for Glioblastoma Cell Proliferation. Epigenetics (2014) 9(4):611–20. doi: 10.4161/epi.27906
49. Pastori C, Kapranov P, Penas C, Peschansky V, Volmar CH, Sarkaria JN, et al. The Bromodomain Protein BRD4 Controls HOTAIR, a Long Noncoding RNA Essential for Glioblastoma Proliferation. Proc Natl Acad Sci USA (2015) 112(27):8326–31. doi: 10.1073/pnas.1424220112
50. Gallagher SJ, Mijatov B, Gunatilake D, Tiffen JC, Gowrishankar K, Jin L, et al. The Epigenetic Regulator I-BET151 Induces BIM-Dependent Apoptosis and Cell Cycle Arrest of Human Melanoma Cells. J Invest Dermatol (2014) 134(11):2795–805. doi: 10.1038/jid.2014.243
51. Shahbazi J, Scarlett CJ, Norris MD, Liu B, Haber M, Tee AE, et al. Histone Deacetylase 2 and N-Myc Reduce P53 Protein Phosphorylation at Serine 46 by Repressing Gene Transcription of Tumor Protein 53-Induced Nuclear Protein 1. Oncotarget (2014) 5(12):4257–68. doi: 10.18632/oncotarget.1991
52. Liu PY, Atmadibrata B, Mondal S, Tee AE, Liu T. NCYM Is Upregulated by Lncusmycn and Modulates N-Myc Expression. Int J Oncol (2016) 49(6):2464–70. doi: 10.3892/ijo.2016.3730
53. Momeny M, Eyvani H, Barghi F, Ghaffari SH, Javadikooshesh S, Hassanvand Jamadi R, et al. Inhibition of Bromodomain and Extraterminal Domain Reduces Growth and Invasive Characteristics of Chemoresistant Ovarian Carcinoma Cells. Anticancer Drugs (2018) 29(10):1011–20. doi: 10.1097/CAD.0000000000000681
54. Kato Y, Kondo S, Itakura T, Tokunaga M, Hatayama S, Katayama K, et al. SNAIL- and SLUG-Induced Side Population Phenotype of HCT116 Human Colorectal Cancer Cells and Its Regulation by BET Inhibitors. Biochem Biophys Res Commun (2020) 521(1):152–7. doi: 10.1016/j.bbrc.2019.10.094
55. Welti J, Sharp A, Yuan W, Dolling D, Nava Rodrigues D, Figueiredo I, et al. Targeting Bromodomain and Extra-Terminal (BET) Family Proteins in Castration-Resistant Prostate Cancer (CRPC). Clin Cancer Res (2018) 24(13):3149–62. doi: 10.1158/1078-0432.CCR-17-3571
56. Kumar K, DeCant BT, Grippo PJ, Hwang RF, Bentrem DJ, Ebine K, et al. BET Inhibitors Block Pancreatic Stellate Cell Collagen I Production and Attenuate Fibrosis in vivo. JCI Insight (2017) 2(3):e88032. doi: 10.1172/jci.insight.88032
57. Gao Z, Yuan T, Zhou X, Ni P, Sun G, Li P, et al. Targeting BRD4 Proteins Suppresses the Growth of NSCLC Through Downregulation of Eif4e Expression. Cancer Biol Ther (2018) 19(5):407–15. doi: 10.1080/15384047.2018.1423923
58. Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, et al. Mediator Kinase Inhibition Further Activates Super-Enhancer-Associated Genes in AML. Nature (2015) 526(7572):273–6. doi: 10.1038/nature14904
59. Khoueiry P, Ward Gahlawat A, Petretich M, Michon AM, Simola D, Lam E, et al. BRD4 Bimodal Binding at Promoters and Drug-Induced Displacement at Pol II Pause Sites Associates With I-BET Sensitivity. Epigenet Chromatin (2019) 12(1):39. doi: 10.1186/s13072-019-0286-5
60. Krivtsov AV, Armstrong SA. MLL Translocations, Histone Modifications and Leukaemia Stem-Cell Development. Nat Rev Cancer (2007) 7(11):823–33. doi: 10.1038/nrc2253
61. Shan X, Fung JJ, Kosaka A, Danet-Desnoyers G. Reproducibility Project: Cancer B. Replication Study: Inhibition of BET Recruitment to Chromatin as an Effective Treatment for MLL-Fusion Leukaemia. Elife (2017) 6:e25306. doi: 10.7554/eLife.25306
62. Sontakke P, Carretta M, Jaques J, Brouwers-Vos AZ, Lubbers-Aalders L, Yuan H, et al. Modeling BCR-ABL and MLL-AF9 Leukemia in a Human Bone Marrow-Like Scaffold-Based Xenograft Model. Leukemia (2016) 30(10):2064–73. doi: 10.1038/leu.2016.108
63. Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic Relevance of Integrated Genetic Profiling in Acute Myeloid Leukemia. N Engl J Med (2012) 366(12):1079–89. doi: 10.1056/NEJMoa1112304
64. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-Related Clonal Hematopoiesis Associated With Adverse Outcomes. N Engl J Med (2014) 371(26):2488–98. doi: 10.1056/NEJMoa1408617
65. Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, et al. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N Engl J Med (2013) 368(22):2059–74. doi: 10.1056/NEJMoa1301689
66. Yang L, Rau R, Goodell MA. DNMT3A in Haematological Malignancies. Nat Rev Cancer (2015) 15(3):152–65. doi: 10.1038/nrc3895
67. Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The Role of Mutations in Epigenetic Regulators in Myeloid Malignancies. Nat Rev Cancer (2012) 12(9):599–612. doi: 10.1038/nrc3343
68. Lund K, Cole JJ, VanderKraats ND, McBryan T, Pchelintsev NA, Clark W, et al. DNMT Inhibitors Reverse a Specific Signature of Aberrant Promoter DNA Methylation and Associated Gene Silencing in AML. Genome Biol (2014) 15(8):406. doi: 10.1186/s13059-014-0406-2
69. Holien T, Vatsveen TK, Hella H, Waage A, Sundan A. Addiction to C-MYC in Multiple Myeloma. Blood (2012) 120(12):2450–3. doi: 10.1182/blood-2011-08-371567
70. Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target C-Myc. Cell (2011) 146(6):904–17. doi: 10.1016/j.cell.2011.08.017
71. Maddocks K. Update on Mantle Cell Lymphoma. Blood (2018) 132(16):1647–56. doi: 10.1182/blood-2018-03-791392
72. Saunders LR, Verdin E. Sirtuins: Critical Regulators at the Crossroads Between Cancer and Aging. Oncogene (2007) 26(37):5489–504. doi: 10.1038/sj.onc.1210616
73. Tenhunen J, Kokkola T, Huovinen M, Rahnasto-Rilla M, Lahtela-Kakkonen M. Impact of Structurally Diverse BET Inhibitors on SIRT1. Gene (2020) 741:144558. doi: 10.1016/j.gene.2020.144558
74. Alsarraj J, Walker RC, Webster JD, Geiger TR, Crawford NP, Simpson RM, et al. Deletion of the Proline-Rich Region of the Murine Metastasis Susceptibility Gene Brd4 Promotes Epithelial-to-Mesenchymal Transition- and Stem Cell-Like Conversion. Cancer Res (2011) 71(8):3121–31. doi: 10.1158/0008-5472.CAN-10-4417
75. Crawford NP, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, et al. Bromodomain 4 Activation Predicts Breast Cancer Survival. Proc Natl Acad Sci USA (2008) 105(17):6380–5. doi: 10.1073/pnas.0710331105
76. Prensner JR, Chinnaiyan AM. The Emergence of lncRNAs in Cancer Biology. Cancer Discov (2011) 1(5):391–407. doi: 10.1158/2159-8290.CD-11-0209
77. Pastori C, Wahlestedt C. Involvement of Long Noncoding RNAs in Diseases Affecting the Central Nervous System. RNA Biol (2012) 9(6):860–70. doi: 10.4161/rna.20482
78. Gibb EA, Brown CJ, Lam WL. The Functional Role of Long Non-Coding RNA in Human Carcinomas. Mol Cancer (2011) 10:38. doi: 10.1186/1476-4598-10-38
79. Franco AV, Zhang XD, Van Berkel E, Sanders JE, Zhang XY, Thomas WD, et al. The Role of NF-Kappa B in TNF-Related Apoptosis-Inducing Ligand (TRAIL)-Induced Apoptosis of Melanoma Cells. J Immunol (2001) 166(9):5337–45. doi: 10.4049/jimmunol.166.9.5337
80. Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet (2007) 369(9579):2106–20. doi: 10.1016/s0140-6736(07)60983-0
81. Kitazawa S, Ebara S, Ando A, Baba Y, Satomi Y, Soga T, et al. Succinate Dehydrogenase B-Deficient Cancer Cells Are Highly Sensitive to Bromodomain and Extra-Terminal Inhibitors. Oncotarget (2017) 8(17):28922–38. doi: 10.18632/oncotarget.15959
82. Kumar A, Coleman I, Morrissey C, Zhang X, True LD, Gulati R, et al. Substantial Interindividual and Limited Intraindividual Genomic Diversity Among Tumors From Men With Metastatic Prostate Cancer. Nat Med (2016) 22(4):369–78. doi: 10.1038/nm.4053
83. Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in Metastatic Prostate Cancer Before Chemotherapy. N Engl J Med (2014) 371(5):424–33. doi: 10.1056/NEJMoa1405095
84. Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in Metastatic Prostate Cancer Without Previous Chemotherapy. N Engl J Med (2013) 368(2):138–48. doi: 10.1056/NEJMoa1209096
85. Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N Engl J Med (2014) 371(11):1028–38. doi: 10.1056/NEJMoa1315815
86. Guedes LB, Morais CL, Almutairi F, Haffner MC, Zheng Q, Isaacs JT, et al. Analytic Validation of RNA In Situ Hybridization (RISH) for AR and AR-V7 Expression in Human Prostate Cancer. Clin Cancer Res (2016) 22(18):4651–63. doi: 10.1158/1078-0432.CCR-16-0205
87. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res (2015) 21(15):3561–8. doi: 10.1158/1078-0432.CCR-14-1051
88. Apte MV, Wilson JS, Lugea A, Pandol SJ. A Starring Role for Stellate Cells in the Pancreatic Cancer Microenvironment. Gastroenterology (2013) 144(6):1210–9. doi: 10.1053/j.gastro.2012.11.037
89. Sonenberg N, Hinnebusch AG. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets. Cell (2009) 136(4):731–45. doi: 10.1016/j.cell.2009.01.042
90. Pelletier J, Graff J, Ruggero D, Sonenberg N. Targeting the Eif4f Translation Initiation Complex: A Critical Nexus for Cancer Development. Cancer Res (2015) 75(2):250–63. doi: 10.1158/0008-5472.CAN-14-2789
91. Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, et al. Enhancer Remodeling During Adaptive Bypass to MEK Inhibition Is Attenuated by Pharmacologic Targeting of the P-TEFb Complex. Cancer Discov (2017) 7(3):302–21. doi: 10.1158/2159-8290.CD-16-0653
92. Yao Z, Yang S, Zhao H, Yang H, Jiang X. BET Inhibitor I-BET151 Sensitizes GBM Cells to Temozolomide via PUMA Induction. Cancer Gene Ther (2020) 27(3-4):226–34. doi: 10.1038/s41417-018-0068-4
93. Tseng HY, Dreyer J, Emran AA, Gunatilake D, Pirozyan M, Cullinane C, et al. Co-Targeting Bromodomain and Extra-Terminal Proteins and MCL1 Induces Synergistic Cell Death in Melanoma. Int J Cancer (2020) 147(8):2176–89. doi: 10.1002/ijc.33000
94. Felgenhauer J, Tomino L, Selich-Anderson J, Bopp E, Shah N. Dual BRD4 and AURKA Inhibition Is Synergistic Against MYCN-Amplified and Nonamplified Neuroblastoma. Neoplasia (2018) 20(10):965–74. doi: 10.1016/j.neo.2018.08.002
95. Guo L, Li J, Zeng H, Guzman AG, Li T, Lee M, et al. A Combination Strategy Targeting Enhancer Plasticity Exerts Synergistic Lethality Against BETi-Resistant Leukemia Cells. Nat Commun (2020) 11(1):740. doi: 10.1038/s41467-020-14604-6
96. Mustafi S, Camarena V, Qureshi R, Yoon H, Volmar CH, Huff TC, et al. Vitamin C Supplementation Expands the Therapeutic Window of BETi for Triple Negative Breast Cancer. EBioMedicine (2019) 43:201–10. doi: 10.1016/j.ebiom.2019.04.006
97. Mustafi S, Camarena V, Volmar CH, Huff TC, Sant DW, Brothers SP, et al. Vitamin C Sensitizes Melanoma to BET Inhibitors. Cancer Res (2018) 78(2):572–83. doi: 10.1158/0008-5472.CAN-17-2040
98. Heinemann A, Cullinane C, De Paoli-Iseppi R, Wilmott JS, Gunatilake D, Madore J, et al. Combining BET and HDAC Inhibitors Synergistically Induces Apoptosis of Melanoma and Suppresses AKT and YAP Signaling. Oncotarget (2015) 6(25):21507–21. doi: 10.18632/oncotarget.4242
99. Badamchi-Zadeh A, Moynihan KD, Larocca RA, Aid M, Provine NM, Iampietro MJ, et al. Combined HDAC and BET Inhibition Enhances Melanoma Vaccine Immunogenicity and Efficacy. J Immunol (2018) 201(9):2744–52. doi: 10.4049/jimmunol.1800885
100. Lee C, Robinson M, Willerth SM. Direct Reprogramming of Glioblastoma Cells Into Neurons Using Small Molecules. ACS Chem Neurosci (2018) 9(12):3175–85. doi: 10.1021/acschemneuro.8b00365
101. Oh J, Kim Y, Baek D, Ha Y. Malignant Gliomas Can Be Converted to Nonproliferating Glial Cells by Treatment With a Combination of Small Molecules. Oncol Rep (2019) 41(1):361–8. doi: 10.3892/or.2018.6824
102. Tseng YY, Moriarity BS, Gong W, Akiyama R, Tiwari A, Kawakami H, et al. PVT1 Dependence in Cancer With MYC Copy-Number Increase. Nature (2014) 512(7512):82–6. doi: 10.1038/nature13311
103. Galvao RP, Kasina A, McNeill RS, Harbin JE, Foreman O, Verhaak RG, et al. Transformation of Quiescent Adult Oligodendrocyte Precursor Cells Into Malignant Glioma Through a Multistep Reactivation Process. Proc Natl Acad Sci USA (2014) 111(40):E4214–23. doi: 10.1073/pnas.1414389111
104. Su Z, Zang T, Liu ML, Wang LL, Niu W, Zhang CL. Reprogramming the Fate of Human Glioma Cells to Impede Brain Tumor Development. Cell Death Dis (2014) 5:e1463. doi: 10.1038/cddis.2014.425
Keywords: cancer, bromodomain and extra-terminal domain protein, I-BET151, signal transduction, drug combination
Citation: Lai J, Liu Z, Zhao Y, Ma C and Huang H (2021) Anticancer Effects of I-BET151, an Inhibitor of Bromodomain and Extra-Terminal Domain Proteins. Front. Oncol. 11:716830. doi: 10.3389/fonc.2021.716830
Received: 29 May 2021; Accepted: 17 August 2021;
Published: 02 September 2021.
Edited by:
Raquel Montenegro, Federal University of Ceara, BrazilReviewed by:
Samrein B. M. Ahmed, University of Sharjah, United Arab EmiratesDakang Xu, Shanghai Jiao Tong University, China
Copyright © 2021 Lai, Liu, Zhao, Ma and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haiyan Huang, aHVhbmdoeUBqbHUuZWR1LmNu; Chengyuan Ma, Y2hlbmd5dWFuQGpsdS5lZHUuY24=