 Yang Feng1
Yang Feng1 Jianping Cao
Jianping Cao Shuyu Zhang
Shuyu Zhang- 1School of Radiation Medicine and Protection, State Key Laboratory of Radiation Medicine, Soochow University, Suzhou, China
- 2China National Nuclear Corporation 416 Hospital (Second Affiliated Hospital of Chengdu Medical College), Chengdu, China
- 3West China Second University Hospital, Sichuan University, Chengdu, China
- 4West China School of Basic Medical Sciences and Forensic Medicine, Sichuan University, Chengdu, China
Ionizing radiation and radioactive materials have been widely used in industry, medicine, science and military. The efficacy of radiotherapy and adverse effects of normal tissues are closed related to cellular radiosensitivity. Molecular mechanisms underlying radiosensitivity are of significance to tumor cell radiosensitization as well as normal tissue radioprotection. 5,6,7,8-Tetrahydrobiopterin (BH4) is an essential cofactor for nitric oxide synthases (NOS) and aromatic amino acid hydroxylases, and its biosynthesis involves de novo biosynthesis and a pterin salvage pathway. In this review we overview the role of BH4 metabolism in modulating radiosensitivity. BH4 homeostasis determines the role of NOS, affecting the production of nitric oxide (NO) and oxygen free radicals. Under conditions of oxidative stress, such as UV-radiation and ionizing radiation, BH4 availability is diminished due to its oxidation, which subsequently leads to NOS uncoupling and generation of highly oxidative free radicals. On the other hand, BH4/NOS axis facilitates vascular normalization, a process by which antiangiogenic therapy corrects structural and functional flaws of tumor blood vessels, which enhances radiotherapy efficacy. Therefore, BH4/NOS axis may serve as an angel or a devil in regulating cellular radiosensitivity. Finally, we will address future perspectives, not only from the standpoint of perceived advances in treatment, but also from the potential mechanisms. These advances have demonstrated that it is possible to modulate cellular radiosensitivity through BH4 metabolism.
Radiation-Induced Injuries and ROS Generation
Ionizing radiation and radioactive materials have been widely used in industry, medicine, science and military. In addition, widespread application of nuclear technology may increase accidental or occupational radiation exposure, such as nuclear accidents, terrorist attacks, etc, which finally leads to radiation-induced injury or even mortality (1, 2). Radiosensitivity determines the injury severity or even survival exposed to ionizing radiation. Radiotherapy is an indispensable component of malignancy treatment, either alone or in combination with other treatments (3), which is applied to over 50% of all cancer patients (4). Although the accuracy of radiotherapy is improving, normal tissues are more or less damaged, resulting in toxicity, which may be a critical dose-limiting complication and affect the quality of life (5–7). Numerous approaches to modulate radiosensitivity, including increasing tumor response to radiotherapy and minimizing damage to normal tissues, have been reported in the past decade (8). Unfortunately, there has been a dearth of clinical treatments for radiation-induced injuries. Various compounds have been identified as potential radiation protection agents, such as free radical scavengers, antioxidants, cytokines, etc (9, 10). Amifostine is a FDA-approved radioprotector, an effective free radical scavenging agent (11), and has been extensively studied and used in clinical radiotherapy (12). It is worth noting that amifostine cannot protect all human organs from the toxic effects of ionizing radiation (13) and it has obvious side-effects, such as nausea and vomiting, which may cause its discontinuation during radiotherapy (14, 15). Thus, it is essential to uncover the mechanisms underlying cellular radiosensitivity and to innovate alternative agents with radioprotective or/and radiosensitization properties in clinical applications.
Ionizing radiation induces cellular damage through direct deposition of energy and indirect oxidative damage caused by excessive reactive oxygen species (ROS), which is the main toxic effects of ionizing radiation (16). Radiation-induced accumulation of ROS results in protein, lipid and DNA damage, leading to a series of pathophysiological changes and ultimately to acute and/or chronic damage (17). Microvascular injury is a distinctive feature of acute and chronic radiation injury. The dysfunction of vascular endothelium caused by ionizing radiation play a crucial role in the occurrence and development of radiation damage (18, 19). Radiation exposure can induce different degrees of functional and morphological changes in vascular endothelial cells, including apoptosis, loss of thrombus resistance and increased endothelial permeability. Endothelial nitric oxide synthase (eNOS) plays an important role in radiation injury. Radiation exposure impairs the function of eNOS and inhibits the production of endothelia nitric oxide (NO) (20). Collectively, strategies to prevent or ameliorate post-radiation endothelial dysfunction may improve the severity of radiation injury.
Functional Significance of BH4
5,6,7,8-tetrahydrobiopterin (BH4) is an essential cofactor for multiple enzymes, including three aromatic amino acid hydroxylases (phenylalanine hydroxylase, tyrosine hydroxylase and tryptophan hydroxylase) and nitric oxide synthases (NOSs) (Figure 1). Phenylalanine hydroxylase (PAH) is first enzyme recognized as a BH4-dependent enzyme (21). The activity of rat liver PAH is disrupted by ionizing radiation, thereby exerting a negative effect on BH4 activity (22, 23). Other aromatic amino acid hydroxylases, such as tyrosine hydroxylase (TH) and tryptophan hydroxylase (TPH), share common features with PAH with respect to the reaction mechanism (24), to BH4 (25) and substrate binding (26).
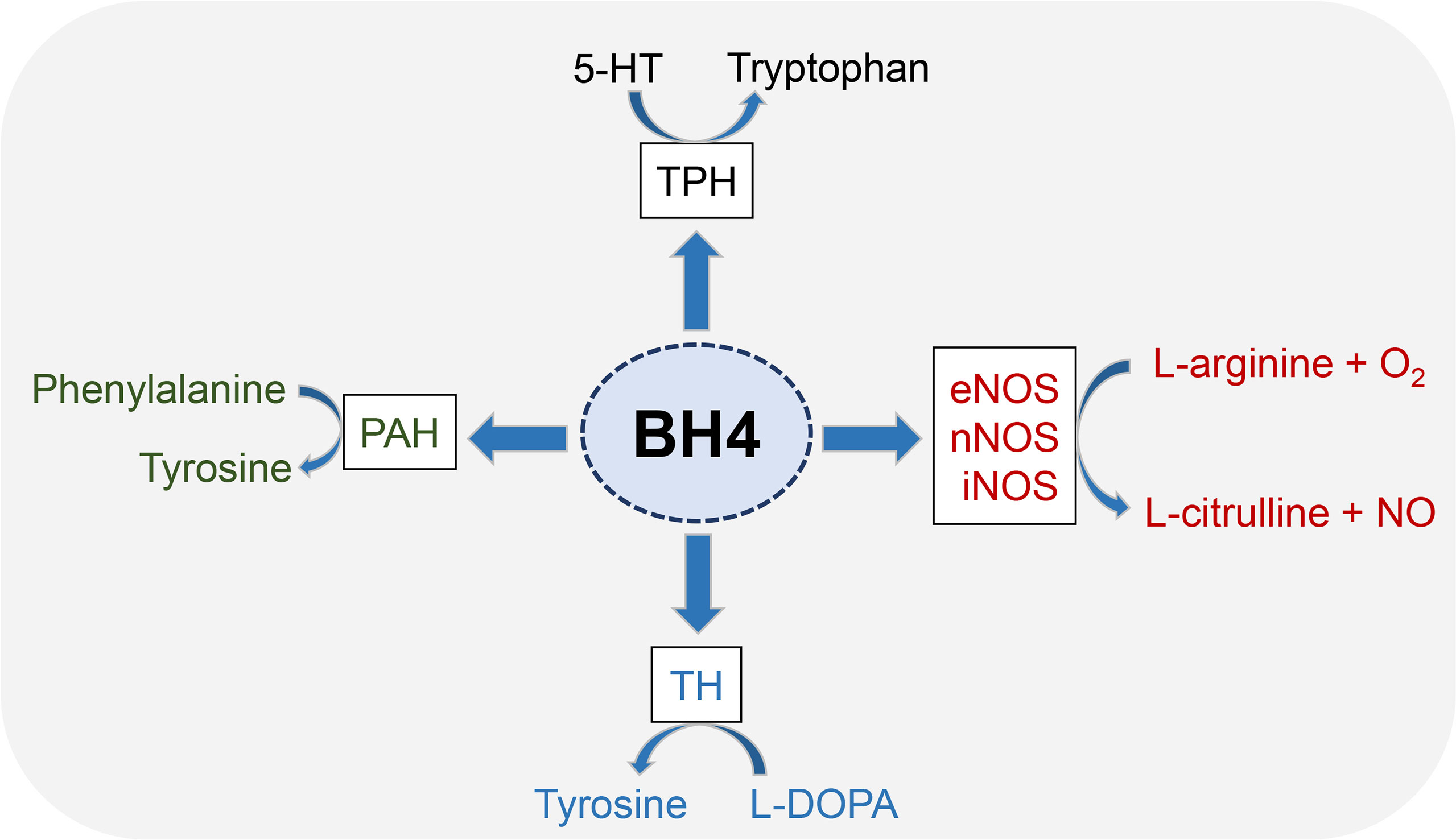
Figure 1 Enzyme cofactor activity of BH4. BH4 is an essential cofactor for multiple enzymes, including three aromatic amino acid hydroxylases (PAH, TH and TPH) and nitric oxide synthases (NOSs). All three NOSs need BH4 to produce NO. Generally, these enzymes combine the oxidation of L-arginine with the reduction of molecular oxygen to form NO and L-citrulline. BH4 is also the cofactor activity of three aromatic amino acid hydroxylases, which leads to the synthesis of neurotransmitters and prevents the accumulation of phenylalanine.
NOSs includes endothelial nitric oxide synthase (eNOS), neuronal nitric oxide synthase (nNOS) and induced nitric oxide synthase (iNOS) (27), wherein eNOS has been shown to play a key role in radiation damage and has been emerging as a therapeutic target (28). NOS catalyzes the conversion of L-arginine to L-citrulline and NO (29). NOS dimers consist of two identical monomers and each monomer consisting of a C-terminal reductase domain and an N-terminal oxygenase domain. The C-terminal binding flavin mononucleotide, flavin adenine dinucleotide and NADPH. The N-terminal binding sites containing heme, BH4, and L-arginine (20). BH4 has been proven to regulate NOS functions at a variety of levels. BH4 enhances NOS enzyme activity by increasing heme iron levels (30) and increases the affinity of NOS with its substrate (31). In addition, BH4 can promote the stability of NOS dimer structure, which is essential for NOS function (32).
Biosynthesis and Regulation of BH4
There are two distinct pathways for BH4 biosynthesis, including de novo pathway and salvage pathway (33) (Figure 2). The former refers to the synthesis of BH4 from guanosine triphosphate (GTP) through three enzymatic reactions; the latter refers to the process of converting sepiapterin as a substrate to BH2 and further reduction to BH4 (20). The relative contribution of the de novo and salvage pathways to the cellular availability of BH4 varies depending on the cell type.
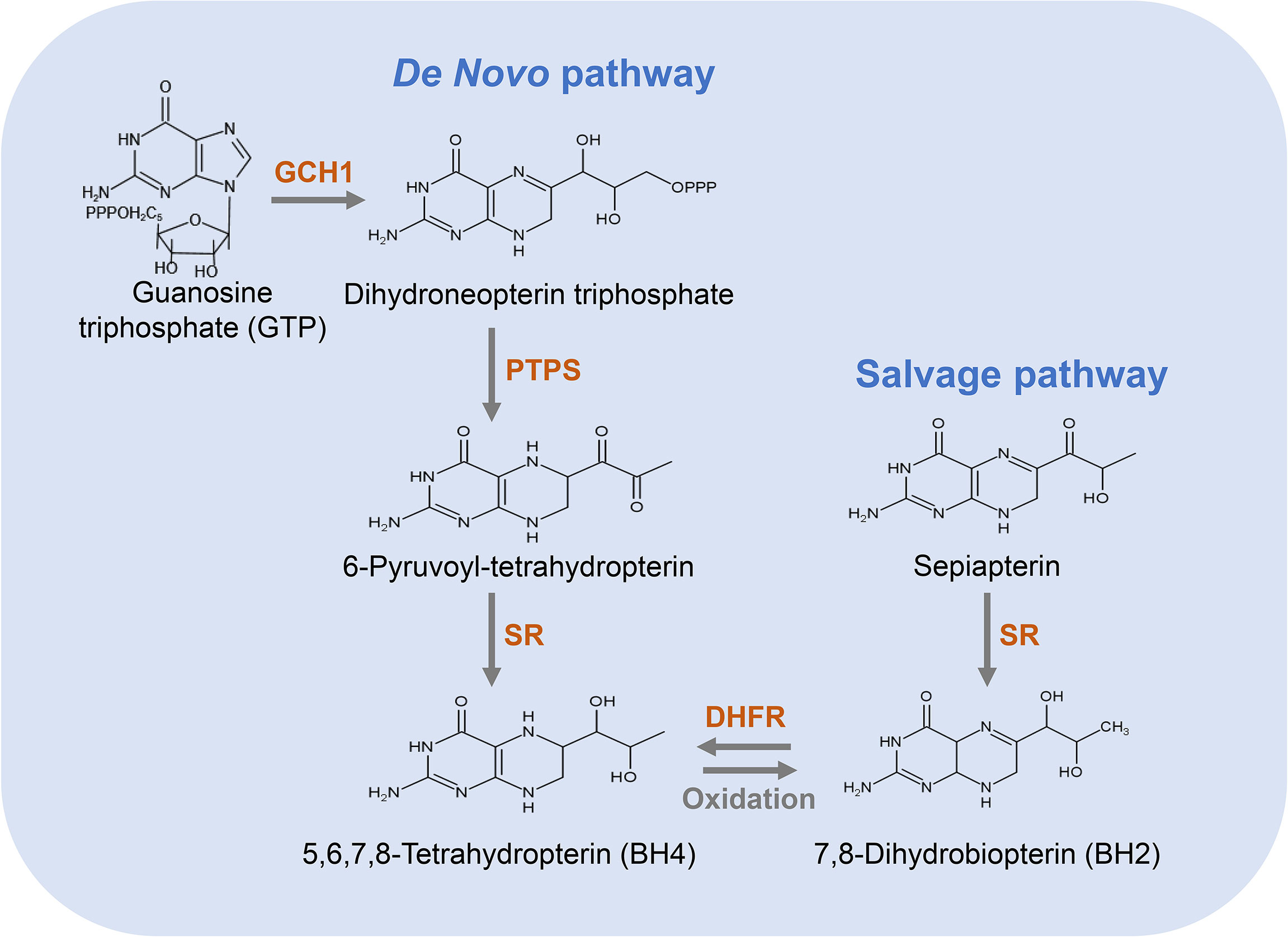
Figure 2 Pathways for the biosynthesis of BH4. The de novo pathway (left) is synthesized from GTP to BH4 in three steps. GCH1 is the rate-limiting enzyme in BH4 de novo biosynthesis. The salvage pathway (right) produces BH4 from its oxidized form, starting with sepiapterin in two steps, which is essential to convert exogenous sepiapterin into BH4.
De Novo Pathway of BH4 Synthesis
In the de novo pathway, BH4 is synthesized from GTP by three enzymes, namely GTP cyclohydrolase I (GCH1), 6-pyruvyl-tetrahydrobiopterin synthase (PTPS) and methotrexate reductase (SR). As shown in Figure 2, GCH1 is the rate-limiting enzyme in BH4 de novo biosynthesis, which catalyzes the formation of dihydroneopterin triphosphate (DNTP). The first step in the synthesis of BH4 is complicated and highly regulated at the level of transcription, translation and post-translation (34). Next, DNTP is converted into 6-pyruvyl-tetrahydrobiopterin by PTPS. Although the key rate-limiting enzyme in the de novo synthesis pathway of BH4 is GCH1 in most cells, PTPS has also been considered as a rate-limiting enzyme in certain types of cells, especially in human liver cells (35). After being stimulated by cytokines, LPS, hydrogen peroxide, insulin and other immune stimuli, GCH1 expression is induced and PTPS therefore becomes a rate-limiting enzyme (35). In the final step of this pathway, SR catalyzes the production of 6-pyruvyl-tetrahydrobiopterin to BH4. This step involves two consecutive NADPH-dependent reduction reactions. The side chain carboxyl of 6-pyruvyl-tetrahydrobiopterin is first reduced and rearranged to form the intermediate 6-lactanoyl-tetrahydrobiopterin, which is then reduced to BH4 on the second side chain carboxyl (35).
As a key enzyme for de novo pathway of BH4 synthesis, GCH1 activity is regulated by various factors, such as transcriptional factors, post-translational regulation, and activity regulatory proteins. In particular, we have demonstrated that AU-rich element RNA-binding factor 1 (AUF1) regulates GCH1 expression via its 3’UTR (36). Phosphorylation is a common post-translational modification in organisms (37). Post-translational regulation of GCH1 activity appears to occur by the protein phosphorylation. It has been reported that phosphorylation of GCH1 at serine 81 is critical in the activation of this enzyme because it not only improves its intrinsic activity and increases its protein expression level, but also reduces the feedback inhibition of its regulatory protein GTP cyclohydrolase I feedback regulator (GFRP) (38–40). The interaction between GCH1 and GFRP can either activate or inhibit GCH1 activity (41). In the presence of phenylalanine, GFRP interacts with GCH1 to activate the GCH1 activity (42, 43). In contrast, the binding of GCH1 with GFRP mediates the feedback inhibition of BH4 (42).
Salvage Pathway of BH4 Synthesis
The salvage pathway generates BH4 from its oxidized forms under the action of sepiapterin reductase (SR) and dihydrofolate reductase (DHFR) (Figure 2). SR is a homodimer composed of two subunits, which takes part not only in the de novo synthetic pathway of BH4 but also in the salvage biosynthetic pathway (44). Additionally, many non-pteridine derivatives, vicinal dicarbonyls, monoaldehydes and monoketones are sensitive as substrates of SR (44). It has been found that the regulation of endothelial BH4 content is mainly accomplished through salvage pathway (45, 46) and the decrease in SR leads to an impairment in endothelial BH4. Similarly, an increase in SR leads to an increase in BH4 level and NO production, and a reduction in oxygen radical production. DHFR is an enzyme necessary for the biosynthesis of folate in eukaryotic and prokaryotic cells (47). In addition to crucial roles in folate metabolism, DHFR can also reduce BH2 and thus regenerate BH4 (48). Previous studies have shown that DHFR plays a key role in determining BH4 homeostasis, NO bioavailability and NOS coupling in endothelial cells (49). When endothelial cells are stimulated via angiotensin II, DHFR expression is down-regulated, BH4 level is decreased, and NOS uncoupling is increased, which is restored by DHFR overexpression (45). Thus, DHFR is crucial in maintaining endothelial BH4 levels and NO bioavailability under oxidative stress.
The salvage pathway is very essential for the conversion of sepiapterin to BH4. Although sepiapterin is not a metabolite of mammals, it is a key exogenous substance that enhances BH4 levels in mammals (50). Thus, supplementation of cells with sepiapterin has been a common strategy to increase intracellular BH4 levels via the salvage pathway.
The Effect of Radiation on BH4 Metabolism and Possible Molecular Signaling Pathways
BH4 is reductive and easily oxidized to BH2 when damaged, such as UV-radiation and ionizing radiation. Oxidation of BH4 to BH2 and other oxidized biopterin species causes eNOS to produce higher superoxide levels instead of NO, a phenomenon commonly referred to eNOS, leading to increased oxidative stress (20, 51). Ionizing radiation oxidizes BH4. Engin et al. found that the urinary biopterin concentration is significantly higher in radiation-exposed hospital staff compared with the healthy subjects (52). BH4 plasma level is significantly lower in patients with abdominal radiotherapy one week after radiotherapy (53). Similarly, after daily exposure to 4 Gy, the plasma BH4 level of the rats decreases significantly, which is consistent with the downward trend of the plasma BH4 level of the patients receiving abdominal radiotherapy. Compared with wild-type mice, BH4 deficient mice show an increase in radiation-induced aortic peroxynitrite in lung tissues (54). Radiation-induced salivary gland dysfunction in mice is attributed to increased peroxynitrite (55). All these data indicate that ionizing radiation promotes the formation of peroxynitrite, which is likely to be the result of reduced BH4 availability after radiation (56).
GFRP overexpression increases the interaction between GFRP and GCH1, thereby negatively regulating the biosynthesis of BH4 and increasing the level of oxidative stress induced by ionizing radiation (54). The mRNA expressions of GFRP in lung and liver of wild- type mice increase after 8.5 Gy of total body Irradiation (TBI), suggesting that the inhibition of GCH1 activity mediated by GFRP may be a possible mechanism of BH4 inhibition after ionizing irradiation (54, 57).
iNOS activity is activated immediately after ionizing radiation (within 2 h) via NF-κB pathway (58), thereby inducing NO production, which may then interact with radiation-induced superoxide to form peroxynitrite (56). Peroxynitrite is prone to oxidize BH4, implying that the NF-κB pathway plays a key role in modulating the bioavailability of BH4 after ionizing radiation (59). Fascinatingly, coordinated activation of JAK-STAT pathway and NF-κB pathway may be involved in radiation-induced BH4 deficiency (56, 60).
Despite the finding that BH4 metabolism involves in NOS uncoupling and ROS production, the effect of radiation on BH4 metabolism may have other mechanisms. It has been reported that protein S-nitrosylation, an important post-translational modification (61, 62), requires NO participation. Dysregulated S-nitrosylation has been shown in multiple human diseases (63, 64). Microbiome-derived NO promotes extensive S-nitrosylation of the host proteome to regulate miRNAs, gene expression as well as host functions and physiology (65). BH4 production mediated by PTPS facilitates latent TGF-β binding protein 1 (LTBP1) S-nitrosylation, thereby suppressing TGF-β secretion and promoting tumor growth (66). Components of the ubiquitin-proteasome system are altered by BH4-dependent NO signaling via protein S-nitrosylation, which implicates the widespread impact of BH4 on downstream cellular signaling (67). Recent studies have delineated a previously unrecognized link between BH4 metabolism and ferroptosis (68), which is associated with radiotherapy (69). So dysregulated S-nitrosylation may also be responsible for the reduction of BH4 after irradiation.
BH4 and eNOS Function
NO is a potent endogenous vasodilator produced by NOS. eNOS produces NO which is an essential regulator of endothelial function, participating in various physiological events and is a key regulator of endothelial cell migration, survival and angiogenesis (70). The impaired NO production by eNOS is a main reason for endothelial dysfunction (71, 72). Reduced NO production in diabetic patients is associated with the pathogenesis of endothelial dysfunction (73, 74). Multiple studies have demonstrated that ionizing radiation inhibits the activity of eNOS and reduces the production of endothelial NO. Even years after radiotherapy, there is still endothelial dysfunction in the increased expression of pro-thrombotic and pro-inflammatory markers in irradiated blood vessels (75). Functional eNOS oxidizes L-arginine to L-citrulline and NO in the presence of BH4, which is an effective natural reducing agent (76). Suboptimal levels of BH4 due to the oxidation to BH2 via stimuli such as radiation exposure may lead to NOS uncoupling and the subsequent generation of highly oxidative radicals, including superoxide and peroxynitrite (77), which is the main mechanism of impaired vascular regulation (78). When BH4 is limited, activated NOS cannot catalyze the conversion of L-arginine to L-citrulline and NO, but can still accept electrons from NADPH and transfer electrons to another substrate O2, resulting in the production of O2- instead of NO. BH4 is oxidized by ONOO- to BH2 and then to biopterin (B). BH2 together with NOS causes ROS production instead of NO (20, 51). Thus, similar to BH4, BH2 has an affinity for the pterin-binding site, which makes it an efficient uncoupling agent for NOS (79). NOS activity is strictly regulated by plenty of biochemical pathways (80), including the availability of its cofactor BH4 (81). For example, compared with age-matched females, the higher oxidative stress of male spontaneously hypertensive rats leads to the relative lack of BH4, leading to the decrease of renal NOS activity and NO bioavailability (82–85).
BH4 Metabolism and Ionizing Radiation
BH4 Metabolism and Radiation-Induced Injuries
Since BH4 reduces ROS by regulating NOS product (86–88), BH4 has been shown to play a key role in the pathogenesis of multiple diseases characterized by increased oxidative stress, such as diabetes, arteriosclerosis, hypertension and radiation-induced injuries (48, 53, 89, 90). Stress-induced ROS production may reduce the availability of BH4, which may induce NOS uncoupling and increase the production of oxidative superoxide radicals. NOS can catalyze the production of NO and L-valine from L-arginine in the presence of sufficient BH4 (19, 20, 48). BH4 is likely to be involved in free radical production and may be related to the progression of radiogenic damage. So far, multiple studies have focused on the biochemical and mechanistic effects of BH4 in radiation-induced injuries and the radioprotective effect of BH4 has been confirmed (53, 90). It has been shown that radioprotection of BH4 through some mechanisms such as scavenging free radicals, promoting responses to DNA damage, and alleviating inflammatory responses, etc (53, 90).
BH4 has become a potential strategy for fibrosis and diastolic dysfunction, which are all related to ROS (91, 92). We have previously reported that GCH1 expression and BH4 levels in irradiated human skin and rat skin tissues are lower than that in the unirradiated counterparts, which impairs NO homeostasis and enhances ROS cascade (90). Oxidative stress-responsive transcriptional factor Nrf2 is able to transcriptionally activate GCH1, thereby restoring cellular BH4 level and attenuating procession of radiation-induced skin injury in vitro and in vivo (90).
BH4 treatment can decrease oxidative stress in irradiated cardiomyocytes, thereby reducing radiation damage and improving myocardial function (93). NO is insufficient after ionizing radiation, which is one of the key indicators of myocardial fibrosis. Patients with fibrotic diseases show low NO levels (94, 95). It has been reported that BH4 supplementation can restore NO and reduce animal myocardial fibrosis (96). BH4 can inhibit the decoupling of NOS and improve cardiac dysfunction (59, 79, 97–100). One month after the aortic arch narrowing of C57 mice, NOS decoupling and oxidative stress occur, exogenous administration of BH4 can improve myocardial function. When the coronary artery is severely narrowed, perfusion of BH4 can improve its diastolic function (101). GCH1 activity and BH4 level are decreased in irradiated mesenteric artery and endothelial cells. Administration of a GCH1 inhibitor DAHP significantly aggravates vascular injury and intestinal damage, while BH4 treatment can improve intestinal vascular injury and ischemia induced by ionizing radiation, and restore vascular function (53). Recent study has shown that the co-administration of Sildenafil (SD) and simvastatin (SV), NO donor/BH4 regulator, inhibits the cranial irradiation-induced oxidative stress, inflammation, NO-pathway dysregulation and neuronal apoptosis, indicating a neuroprotective effect role of SD/SV in irradiation-induced brain injury as a possible mechanism of its NO donor/BH4 regulatory activities (102).
GT3, a radioprotective vitamin E analog, can reduce radiation-induced oxidative/nitrosative stress (89). GT3 regulates the expression of GFRP and thus plays its radioprotective role in part by regulating the BH4 availability (89). A GFRP-overexpressing transgenic mice display reduced tissue BH4 and blood GSH levels, indicating a higher oxidative stress (54). Cheema et al. investigated liver metabolic changes following irradiation in control and GFRP overexpression mice (57). Compared with wild-type mice, GFRP transgenic mice show reduced glutathione levels and increased levels of glycocholic acid and N-arachidonic taurine after irradiation, suggesting the early occurrence of metabolic dysfunction. Thus, GFRP transgenic mice are susceptible to radiation stress and this sensitivity may lead to increased radiation-induced injuries (54).
Collectively, ionizing radiation oxidizes BH4, which results in NOS uncoupling and augmented radiation-induced secondary ROS, ultimately leading to radiation-induced injuries. While GCH1-mediated BH4 metabolism attenuates radiation-induced ROS production to improve radiation damage (Figure 3).
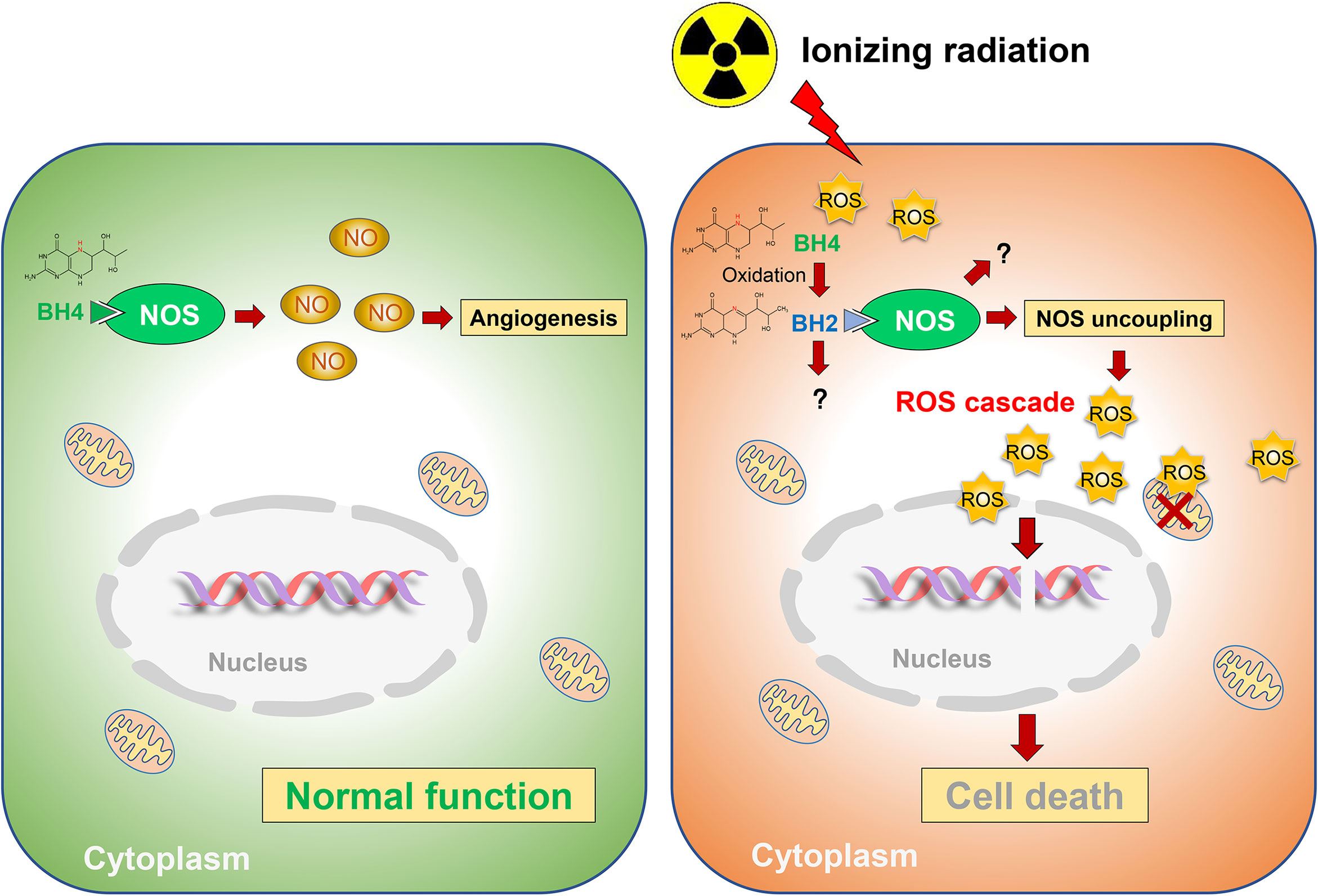
Figure 3 Schematic representation of BH4 metabolism in radiosensitivity. Radiation oxidates BH4, which results in NOS uncoupling and augmented radiation-induced secondary ROS, ultimately leading to radiation-induced injuries. While GCH1-mediated BH4 metabolism attenuated radiation-induced ROS production to improve radiation damage.
BH4 Metabolism and Cancer Radiosensitivity
We retrieved the expression of BH4 metabolic enzymes (GCH1, PTPS, SR and DHFR) in various tumors based on the Cancer Genome Atlas (TCGA) database (Figure 4) (44). According to the results, GCH1 exhibits relatively higher expression levels in the liver, endometrium and breast cancers than the tumor adjacent tissues. PTPS is overexpressed in lung, colon and endometrium cancers. The expression of the SPR gene is higher in liver cancer, colorectal cancer and invasive breast carcinoma. And DHFR gene is highly expressed in glioblastoma multiforme, invasive breast carcinoma, stomach adenocarcinoma and uterine corpus endometrial carcinoma. These different BH4 metabolic enzymes may be related to specific tissue functions. In a word, BH4 metabolic enzymes are generally overexpressed in tumor tissues than in corresponding normal tissues, which may be due to the higher ROS level in tumor cells (103). Therefore, BH4 metabolic enzymes are possible hallmarks and therapeutic targets.
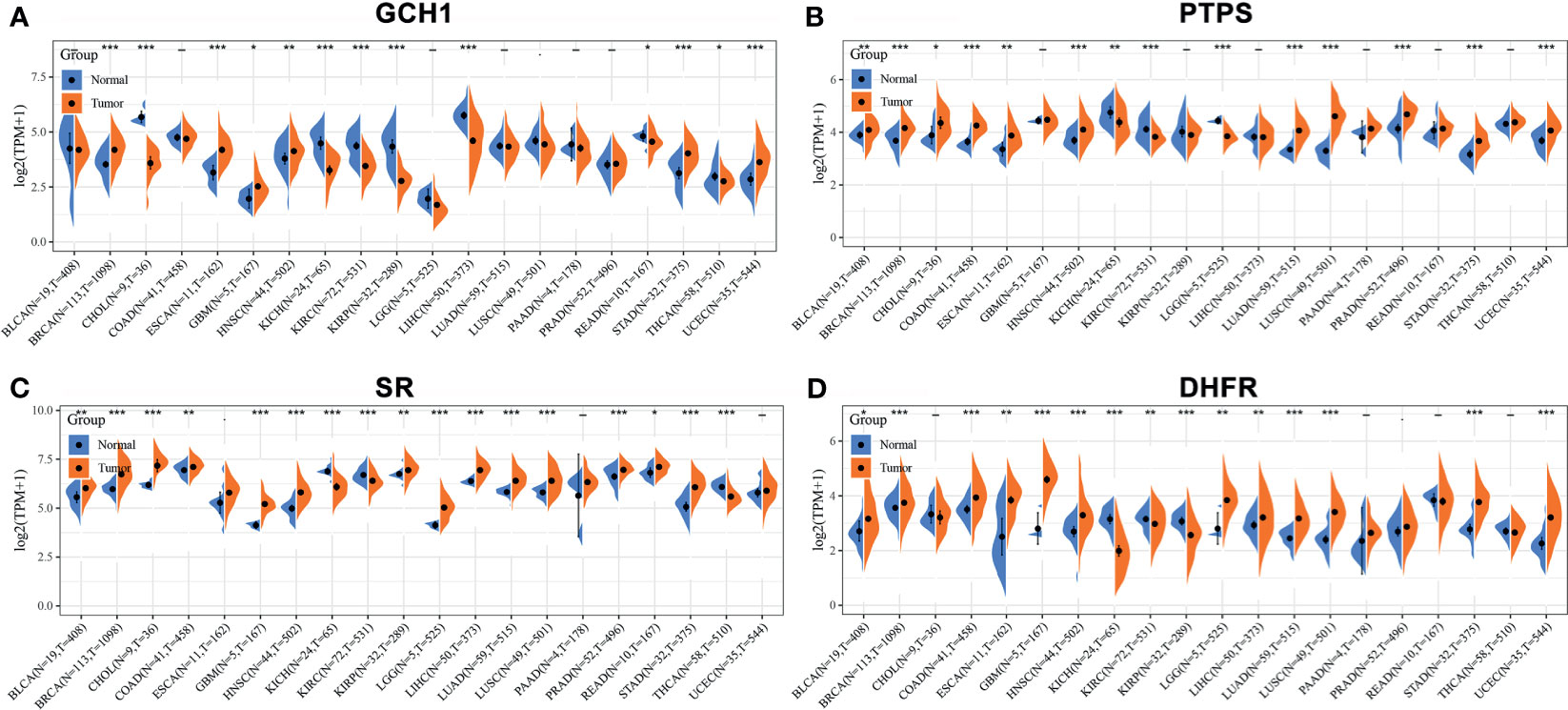
Figure 4 The expression of BH4 metabolic enzymes in human tumors. The comparison of (A) GCH1, (B) PTPS, (C) SR and (D) DHFR expression in in various tumor tissues and corresponding normal tissues. And BH4 metabolic enzymes generally overexpress in tumor tissues than corresponding normal tissues. *P < 0.05, **P < 0.01 and ***P < 0.001, compared with the normal tissues. Gene expression data are obtained from the Cancer Genome Atlas (TCGA) database (44). BLCA, Bladder urothelial carcinoma; BRCA, Breast invasive carcinoma; CHOL, Cholangiocarcinoma; COAD, Colon adenocarcinoma; ESCA, Esophageal carcinoma; GBM, Glioblastoma multiforme; HNSC, Head and Neck squamous cell carcinoma; KICH, Kidney Chromophobe; KIRC, Kidney renal clear cell carcinoma; KIRP, Kidney renal papillary cell carcinoma; LGG, Brain Lower Grade Glioma; LIHC, Liver hepatocellular carcinoma; LUAD, Lung adenocarcinoma; LUSC, Lung squamous cell carcinoma; PAAD, Pancreatic adenocarcinoma; PRAD, Prostate adenocarcinoma; READ, Rectum adenocarcinoma; STAD, Stomach adenocarcinoma; THCA, Thyroid carcinoma; UCEC, Uterine Corpus Endometrial Carcinoma.
In addition to the radioprotective effects of BH4 and its metabolites, some studies have shown that they can improve the therapeutic effect of radiotherapy. Therefore, BH4 and its metabolites are considered as radiosensitization targets in cancer radiotherapy. It has been reported that BH4/BH2 ratio in colorectal, breast and head and neck tumors is significantly lower than that in normal tissues (104). In mouse spontaneous breast cancer model, exogenous BH4 precursor sepiapterin increases BH4/BH2 ratio, which enhances the NOS activity and increases NO production. Sepiapterin finally leads to the transition from pro-inflammatory/pro-survival signals to anti-inflammatory/pro-apoptotic signals, thereby inhibiting spontaneous tumor growth (104). In addition, in a murine SCCVII tumor model, radiation-induced NO through increases eNOS activity mitigates tumor hypoxia and increases radiosensitivity (105). It is now clear that NO, which is associated with malignancy, may exhibit a dual activity: stimulating tumor growth and having the opposite anti-tumor effect (106), which depends on the concentration of NO (107, 108). At low concentrations, NO can inhibit apoptosis and cause mutations, which may lead to the formation of malignant growth loci. Conversely, high concentrations of NO seem to be harmful to malignant cells, especially when exposed to ionizing radiation (107–109). Kashiwagi et al. (110) demonstrated that NOS activity affects tumor blood vessels. Inhibition of NOS in glioma cells can improve oxygen delivery and a more normal phenotype (110). Whereas, vasculature normalization with antiangiogenics is short-lived. Treatment of mice with the NOS inhibitor L-NNA reduces tumor blood flow, resulting in delayed tumor growth, but quickly lost its effect (111). On the other hand, post ionizing radiation NOS inhibition delays tumor growth via Th1 immune polarization within the tumor microenvironment (112). A 6-day sepiapterin treatment in mice reduces tumor blood flow, delays tumor growth and improves animal survival, while tumor oxygenation continues to improve significantly after 10 days of sepiapterin treatment and improved tumor oxygenation is associated with increased tumor cell apoptosis (113). Pretreatment with sepiapterin not only enhances the killing of tumor by ionizing radiation, but also enhances the absorption of doxorubicin. Thus, as a vascular normalizing agent, sepiapterin can reduce tumor hypoxia, improve tumor %HbO2 and perfusion, and prevent cancer cells from acquiring aggressive phenotypes in the hypoxic microenvironment, ultimately leading to radiation-induced apoptosis, thereby enhancing tumor radio- and chemosensitivities (113, 114).
In the salvage pathway, DHFR exhibits a critical role in BH4 generation. Radiotherapy, however, tends to trigger DHFR amplification, thereby enhancing the activity of DHFR (115, 116). Enhanced DHFR activity promotes DNA replication in cervical cancer cells, leading to reduced therapeutic efficacy (117). The use of DHFR inhibitors, such as methotrexate (MTX) analogues as radiosensitizers is expected to improve the therapeutic effect (118, 119). Liang et al. (117) synthesized a series of 2,4-diaminopteridine analogues as DHFR inhibitors for radiosensitization. In particular, the combination of X-rays and a compound named 2a effectively suppresses cervical tumor growth and compound 2a has higher radiosensitization activity than MTX. Hence, if the DHFR activity is inhibited, the radiotherapy effect will be improved to varying degrees. The normal tissue or cancer types associated with BH4-mediated radiosensitivity are summarized in Figure 5.
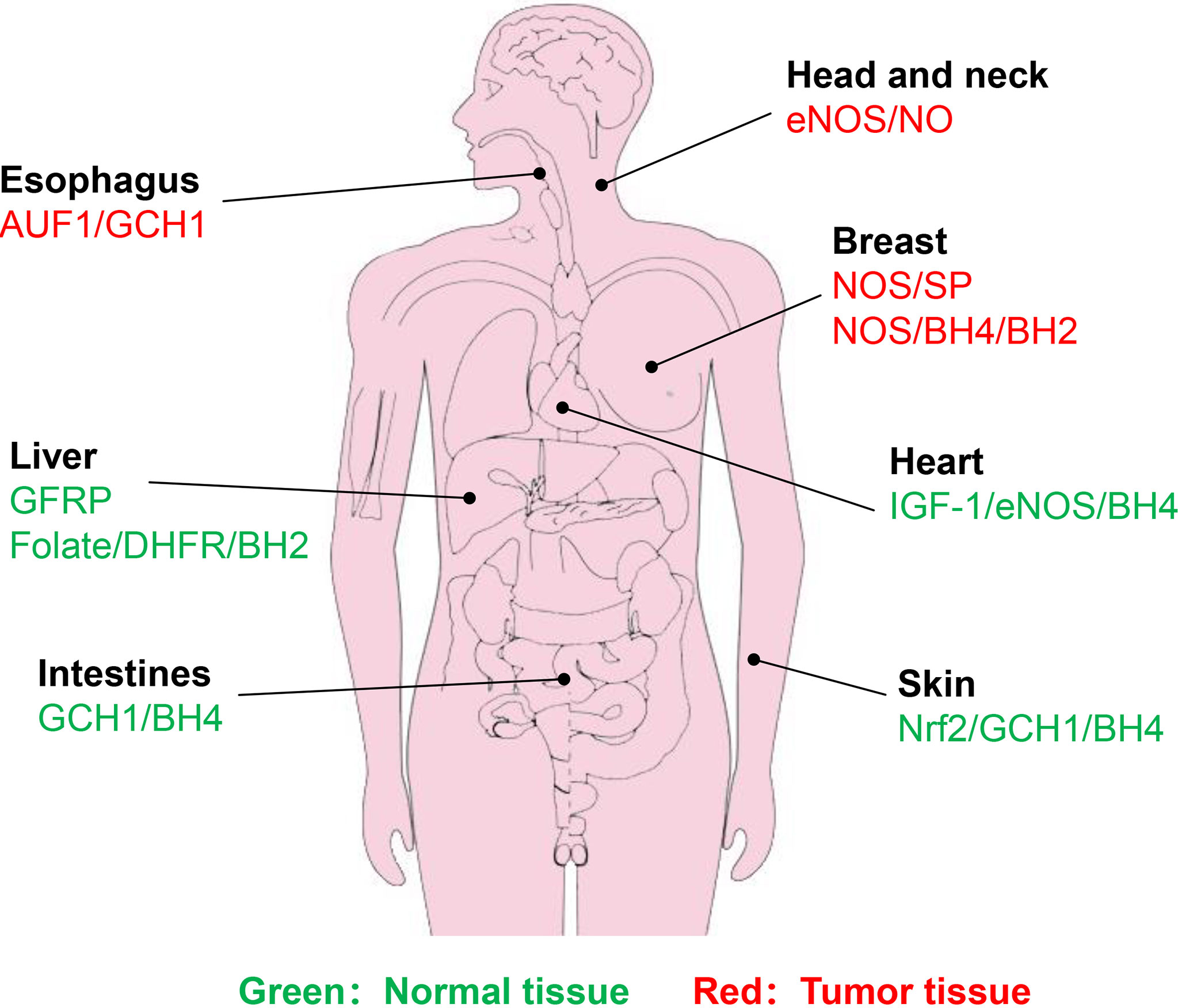
Figure 5 Identified normal tissues or cancer types associated with BH4-mdiated radiosensitivity. Green represents normal tissue, while red represents tumor tissue. Brief description of pathways associated with BH4 are shown.
Future Directions
Since BH4 is easily oxidated (20, 51, 120), novel approaches are needed to protect its integrity during delivery. Nanotechnology offers a new way to deliver drugs efficiently and specifically. It is reported that liposome formulations can improve the therapeutic effect of drugs with poor bioavailability (121). Liposomal BH4 has been used to reverse the loss of BH4 after ischemia-reperfusion injury (122, 123). Similarly, targeted delivery of BH4 nanocarriers can be used as a prophylactic treatment for atherosclerosis (124). Thus, it is possible to use novel approaches, such as liposomes and nanoparticles, to carry BH4 to enhance its stability and make its clinical application more promising. In addition, the design of ROS responsive nanomaterials based on the high ROS conditions at radiation-damaged sites provides a new approach for BH4 loading for radioprotection. Although it has been reported that vitamin C, folic acid, etc. can enhance the binding of BH4 to eNOS, thereby increasing the level of intracellular BH4 (48, 125), the clinical efficacy is compromised due to difficulties to combine with BH4. The rescue approach of regulating BH4 synthesis through its precursor sepiapterin may be another treatment strategy (126).
During cancer radiotherapy, the role of BH4 metabolism in cancer cell radiosensitivity yet to be determined. BH4 on one hand reduced radiogenic ROS, however, BH4 on the other hand normalizes vessels, which enhances radiotherapy efficacy. Given the rapid development of targeted therapies, specific radiosensitizers can be used for cancer radiotherapy. Tumor cells constantly interact with the surrounding microenvironment. Apart from the tumor cells, the tumor microenvironment includes a variety of cell types (endothelial cells, fibroblasts, immune cells, etc.) and extracellular components (cytokines, growth factors, hormones, extracellular matrix, etc.) (127). High expression of GCH1 in cancer-associated fibroblasts stimulates breast cancer cell proliferation and motility (128). As a critical T-cell regulator, BH4 can be manipulated to enhance immunity and inhibit tumor growth (129). The role of BH4 metabolism in tumor microenvironment is largely unknown. Further research on these mechanisms will accelerate the development of radiosensitizers based on BH4 metabolism.
Author Contributions
YF and YHF drafted the manuscript and figures. LG and PL collected literature. JC and SZ modified the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by the National Natural Science Foundation of China (U1967220, and 82073477), the Key Research & Development Program of Jiangsu Province (BE2020637), Post graduate Research & Practice Innovation Program of Jiangsu Province (No KYCX21_2969) and the Young Talent Project of China National Nuclear Corporation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mahmood J, Jelveh S, Calveley V, Zaidi A, Doctrow SR, Hill RP. Mitigation of Lung Injury After Accidental Exposure to Radiation. Radiat Res (2011) 176(6):770–80. doi: 10.1667/RR2562.1
2. Svendsen ER, Kolpakov IE, Stepanova YI, Vdovenko VY, Naboka MV, Mousseau TA, et al. 137Cesium Exposure and Spirometry Measures in Ukrainian Children Affected by the Chernobyl Nuclear Incident. Environ Health Perspect (2010) 118(5):720–5. doi: 10.1289/ehp.0901412
3. Tsoutsou PG, Koukourakis MI. Radiation Pneumonitis and Fibrosis: Mechanisms Underlying its Pathogenesis and Implications for Future Research. Int J Radiat Oncol Biol Phys (2006) 66(5):1281–93. doi: 10.1016/j.ijrobp.2006.08.058
4. Allen C, Her S, Jaffray DA. Radiotherapy for Cancer: Present and Future. Adv Drug Delivery Rev (2017) 109:1–2. doi: 10.1016/j.addr.2017.01.004
5. Abid SH, Malhotra V, Perry MC. Radiation-Induced and Chemotherapy-Induced Pulmonary Injury. Curr Opin Oncol (2001) 13(4):242–8. doi: 10.1097/00001622-200107000-00006
6. Kong F-M, Ten Haken R, Eisbruch A, Lawrence TS. Non-Small Cell Lung Cancer Therapy-Related Pulmonary Toxicity: An Update on Radiation Pneumonitis and Fibrosis. Semin Oncol (2005) 32(2 Suppl 3):S42–54. doi: 10.1053/j.seminoncol.2005.03.009
7. De Ruysscher D, Niedermann G, Burnet NG, Siva S, Lee AWM, Hegi-Johnson F. Radiotherapy Toxicity. Nat Rev Dis Primers (2019) 5(1):13. doi: 10.1038/s41572-019-0064-5
8. Liauw SL, Connell PP, Weichselbaum RR. New Paradigms and Future Challenges in Radiation Oncology: An Update of Biological Targets and Technology. Sci Transl Med (2013) 5(173):173sr172. doi: 10.1126/scitranslmed.3005148
9. Weiss JF, Landauer MR. Radioprotection by Antioxidants. Ann N Y Acad Sci (2000) 899:44–60. doi: 10.1111/j.1749-6632.2000.tb06175.x
10. Hosseinimehr SJ. Trends in the Development of Radioprotective Agents. Drug Discovery Today (2007) 12(19-20):794–805. doi: 10.1016/j.drudis.2007.07.017
11. Huang B, He T, Yao Q, Zhang L, Yao Y, Tang H, et al. Amifostine Suppresses the Side Effects of Radiation on BMSCs by Promoting Cell Proliferation and Reducing ROS Production. Stem Cells Int (2019) 2019:8749090. doi: 10.1155/2019/8749090
12. Rosenthal DI, Chambers MS, Weber RS, Eisbruch A. A Phase II Study to Assess the Efficacy of Amifostine for Submandibular/Sublingual Salivary Sparing During the Treatment of Head and Neck Cancer With Intensity Modulated Radiation Therapy for Parotid Salivary Sparing. Semin Oncol (2004) 31(6 Suppl 18):25–8. doi: 10.1053/j.seminoncol.2004.12.008
13. Singh VK, Fatanmi OO, Wise SY, Newman VL, Romaine PLP, Seed TM. The Potential of the Radioprotective Efficacy of Two Medical Countermeasures, Gamma Tocotrienol and Amifostine, By a CombinationPhrophylactic Modality. Radiat Prot Dosimetry (2016) 172(1-3):302–10. doi: 10.1093/rpd/ncw223
14. Thorstad WL, Chao KSC, Haughey B. Toxicity and Compliance of Subcutaneous Amifostine in Patients Undergoing Postoperative Intensity-Modulated Radiation Therapy for Head and Neck Cancer. Semin Oncol (2004) 31(6 Suppl 18):8–12. doi: 10.1053/j.seminoncol.2004.12.005
15. Rades D, Fehlauer F, Bajrovic A, Mahlmann B, Richter E, Alberti W. Serious Adverse Effects of Amifostine During Radiotherapy in Head and Neck Cancer Patients. Radiother Oncol (2004) 70(3):261–4. doi: 10.1016/j.radonc.2003.10.005
16. Mozdarani H. Biological Complexities in Radiation Carcinogenesis and Cancer Radiotherapy: Impact of New Biological Paradigms. Genes (Basel) (2012) 3(1):90–114. doi: 10.3390/genes3010090
17. Zhao W, Robbins MEC. Inflammation and Chronic Oxidative Stress in Radiation-Induced Late Normal Tissue Injury: Therapeutic Implications. Curr Med Chem (2009) 16(2):130–43. doi: 10.2174/092986709787002790
18. Wang J, Zheng H, Ou X, Fink LM, Hauer-Jensen M. Deficiency of Microvascular Thrombomodulin and Up-Regulation of Protease-Activated Receptor-1 in Irradiated Rat Intestine: Possible Link Between Endothelial Dysfunction and Chronic Radiation Fibrosis. Am J Pathol (2002) 160(6):2063–72. doi: 10.1016/S0002-9440(10)61156-X
19. Wang J, Boerma M, Fu Q, Hauer-Jensen M. Significance of Endothelial Dysfunction in the Pathogenesis of Early and Delayed Radiation Enteropathy. World J Gastroenterol (2007) 13(22):3047–55. doi: 10.3748/wjg.v13.i22.3047
20. Berbée M, Fu Q, Kumar KS, Hauer-Jensen M. Novel Strategies to Ameliorate Radiation Injury: A Possible Role for Tetrahydrobiopterin. Curr Drug Targets (2010) 11(11):1366–74. doi: 10.2174/1389450111009011366
21. Kaufman S. New Tetrahydrobiopterin-Dependent Systems. Annu Rev Nutr (1993) 13:261–86. doi: 10.1146/annurev.nu.13.070193.001401
22. Davis MD, Parniak MA, Kaufman S, Kempner E. The Role of Phenylalanine in Structure-Function Relationships of Phenylalanine Hydroxylase Revealed by Radiation Target Analysis. Proc Natl Acad Sci USA (1997) 94(2):491–5. doi: 10.1073/pnas.94.2.491
23. Davis MD, Parniak MA, Kaufman S, Kempner E. Structure-Function Relationships of Phenylalanine Hydroxylase Revealed by Radiation Target Analysis. Arch Biochem Biophys (1996) 325(2):235–41. doi: 10.1006/abbi.1996.0029
24. Pavon JA, Fitzpatrick PF. Insights Into the Catalytic Mechanisms of Phenylalanine and Tryptophan Hydroxylase From Kinetic Isotope Effects on Aromatic Hydroxylation. Biochemistry (2006) 45(36):11030–7. doi: 10.1021/bi0607554
25. Teigen K, Dao KK, McKinney JA, Gorren ACF, Mayer B, Frøystein NA, et al. Tetrahydrobiopterin Binding to Aromatic Amino Acid Hydroxylases. Ligand Recognition and Specificity. J Med Chem (2004) 47(24):5962–71. doi: 10.1021/jm0497646
26. Teigen K, McKinney JA, Haavik J, Martínez A. Selectivity and Affinity Determinants for Ligand Binding to the Aromatic Amino Acid Hydroxylases. Curr Med Chem (2007) 14(4):455–67. doi: 10.2174/092986707779941023
27. Kwon NS, Nathan CF, Stuehr DJ. Reduced Biopterin as a Cofactor in the Generation of Nitrogen Oxides by Murine Macrophages. J Biol Chem (1989) 264(34):20496–501. doi: 10.1016/S0021-9258(19)47089-0
28. Holler V, Buard V, Gaugler M-H, Guipaud O, Baudelin C, Sache A, et al. Pravastatin Limits Radiation-Induced Vascular Dysfunction in the Skin. J Invest Dermatol (2009) 129(5):1280–91. doi: 10.1038/jid.2008.360
29. Alderton WK, Cooper CE, Knowles RG. Nitric Oxide Synthases: Structure, Function and Inhibition. Biochem J (2001) 357(Pt 3):593–615. doi: 10.1042/bj3570593
30. Rodríguez-Crespo I, Moënne-Loccoz P, Loehr TM, Ortiz de Montellano PR. Endothelial Nitric Oxide Synthase: Modulations of the Distal Heme Site Produced by Progressive N-Terminal Deletions. Biochemistry (1997) 36(28):8530–8. doi: 10.1021/bi970192j
31. Klatt P, Schmid M, Leopold E, Schmidt K, Werner ER, Mayer B. The Pteridine Binding Site of Brain Nitric Oxide Synthase. Tetrahydrobiopterin Binding Kinetics, Specificity, and Allosteric Interaction With the Substrate Domain. J Biol Chem (1994) 269(19):13861–6. doi: 10.1016/S0021-9258(17)36726-1
32. Ghosh DK, Wu C, Pitters E, Moloney M, Werner ER, Mayer B, et al. Characterization of the Inducible Nitric Oxide Synthase Oxygenase Domain Identifies a 49 Amino Acid Segment Required for Subunit Dimerization and Tetrahydrobiopterin Interaction. Biochemistry (1997) 36(35):10609–19. doi: 10.1021/bi9702290
33. Thöny B, Auerbach G, Blau N. Tetrahydrobiopterin Biosynthesis, Regeneration and Functions. Biochem J (2000) 347 Pt 1(Pt 1):1–16. doi: 10.1042/bj3470001
34. Zhao Y, Zhu H, Zou M-H. Non-Covalent Interaction Between Polyubiquitin and GTP Cyclohydrolase 1 Dictates its Degradation. PloS One (2012) 7(9):e43306. doi: 10.1371/journal.pone.0043306
35. Longo N. Disorders of Biopterin Metabolism. J Inherit Metab Dis (2009) 32(3):333–42. doi: 10.1007/s10545-009-1067-2
36. Gao Y, Wang W, Cao J, Wang F, Geng Y, Cao J, et al. Upregulation of AUF1 is Involved in the Proliferation of Esophageal Squamous Cell Carcinoma Through GCH1. Int J Oncol (2016) 49(5):2001–10. doi: 10.3892/ijo.2016.3713
37. Vu LD, Gevaert K, De Smet I. Protein Language: Post-Translational Modifications Talking to Each Other. Trends Plant Sci (2018) 23(12):1068–80. doi: 10.1016/j.tplants.2018.09.004
38. Li L, Rezvan A, Salerno JC, Husain A, Kwon K, Jo H, et al. GTP Cyclohydrolase I Phosphorylation and Interaction With GTP Cyclohydrolase Feedback Regulatory Protein Provide Novel Regulation of Endothelial Tetrahydrobiopterin and Nitric Oxide. Circ Res (2010) 106(2):328–36. doi: 10.1161/CIRCRESAHA.109.210658
39. Widder JD, Chen W, Li L, Dikalov S, Thöny B, Hatakeyama K, et al. Regulation of Tetrahydrobiopterin Biosynthesis by Shear Stress. Circ Res (2007) 101(8):830–8. doi: 10.1161/CIRCRESAHA.107.153809
40. De Bono JP, Channon KM. Endothelial Cell Tetrahydrobiopterin: Going With the Flow. Circ Res (2007) 101(8):752–4. doi: 10.1161/CIRCRESAHA.107.162503
41. Ishii M, Shimizu S, Wajima T, Hagiwara T, Negoro T, Miyazaki A, et al. Reduction of GTP Cyclohydrolase I Feedback Regulating Protein Expression by Hydrogen Peroxide in Vascular Endothelial Cells. J Pharmacol Sci (2005) 97(2):299–302. doi: 10.1254/jphs.SC0040146
42. Maita N, Hatakeyama K, Okada K, Hakoshima T. Structural Basis of Biopterin-Induced Inhibition of GTP Cyclohydrolase I by GFRP, Its Feedback Regulatory Protein. J Biol Chem (2004) 279(49):51534–40. doi: 10.1074/jbc.M409440200
43. Gesierich A, Niroomand F, Tiefenbacher CP. Role of Human GTP Cyclohydrolase I and its Regulatory Protein in Tetrahydrobiopterin Metabolism. Basic Res Cardiol (2003) 98(2):69–75. doi: 10.1007/s00395-003-0394-y
44. Wu Y, Chen P, Sun L, Yuan S, Cheng Z, Lu L, et al. Sepiapterin Reductase: Characteristics and Role in Diseases. J Cell Mol Med (2020) 24(17):9495–506. doi: 10.1111/jcmm.15608
45. Chalupsky K, Cai H. Endothelial Dihydrofolate Reductase: Critical for Nitric Oxide Bioavailability and Role in Angiotensin II Uncoupling of Endothelial Nitric Oxide Synthase. Proc Natl Acad Sci USA (2005) 102(25):9056–61. doi: 10.1073/pnas.0409594102
46. Gao L, Chalupsky K, Stefani E, Cai H. Mechanistic Insights Into Folic Acid-Dependent Vascular Protection: Dihydrofolate Reductase (DHFR)-Mediated Reduction in Oxidant Stress in Endothelial Cells and Angiotensin II-Infused Mice: A Novel HPLC-Based Fluorescent Assay for DHFR Activity. J Mol Cell Cardiol (2009) 47(6):752–60. doi: 10.1016/j.yjmcc.2009.07.025
47. Raimondi MV, Randazzo O, La Franca M, Barone G, Vignoni E, Rossi D, et al. DHFR Inhibitors: Reading the Past for Discovering Novel Anticancer Agents. Molecules (2019) 24(6):1140. doi: 10.3390/molecules24061140
48. Bendall JK, Douglas G, McNeill E, Channon KM, Crabtree MJ. Tetrahydrobiopterin in Cardiovascular Health and Disease. Antioxid Redox Signal (2014) 20(18):3040–77. doi: 10.1089/ars.2013.5566
49. Crabtree MJ, Tatham AL, Hale AB, Alp NJ, Channon KM. Critical Role for Tetrahydrobiopterin Recycling by Dihydrofolate Reductase in Regulation of Endothelial Nitric-Oxide Synthase Coupling: Relative Importance of the De Novo Biopterin Synthesis Versus Salvage Pathways. J Biol Chem (2009) 284(41):28128–36. doi: 10.1074/jbc.M109.041483
50. Smith N, Longo N, Levert K, Hyland K, Blau N. Phase I Clinical Evaluation of CNSA-001 (Sepiapterin), a Novel Pharmacological Treatment for Phenylketonuria and Tetrahydrobiopterin Deficiencies, in Healthy Volunteers. Mol Genet Metab (2019) 126(4):406–12. doi: 10.1016/j.ymgme.2019.02.001
51. Crabtree MJ, Smith CL, Lam G, Goligorsky MS, Gross SS. Ratio of 5,6,7,8-Tetrahydrobiopterin to 7,8-Dihydrobiopterin in Endothelial Cells Determines Glucose-Elicited Changes in NO vs. Superoxide Production by eNOS. Am J Physiol Heart Circ Physiol (2008) 294(4):H1530–40. doi: 10.1152/ajpheart.00823.2007
52. Engin AB, Ergun MA, Yurtcu E, Kan D, Sahin G. Effect of Ionizing Radiation on the Pteridine Metabolic Pathway and Evaluation of Its Cytotoxicity in Exposed Hospital Staff. Mutat Res (2005) 585(1-2):184–92. doi: 10.1016/j.mrgentox.2005.05.005
53. Yan T, Zhang T, Mu W, Qi Y, Guo S, Hu N, et al. Ionizing Radiation Induces BH Deficiency by Downregulating GTP-Cyclohydrolase 1, a Novel Target for Preventing and Treating Radiation Enteritis. Biochem Pharmacol (2020) 180:114102. doi: 10.1016/j.bcp.2020.114102
54. Pathak R, Pawar SA, Fu Q, Gupta PK, Berbée M, Garg S, et al. Characterization of Transgenic Gfrp Knock-in Mice: Implications for Tetrahydrobiopterin in Modulation of Normal Tissue Radiation Responses. Antioxid Redox Signal (2014) 20(9):1436–46. doi: 10.1089/ars.2012.5025
55. Hanaue N, Takeda I, Kizu Y, Tonogi M, Yamane G-Y. Peroxynitrite Formation in Radiation-Induced Salivary Gland Dysfunction in Mice. BioMed Res (2007) 28(3):147–51. doi: 10.2220/biomedres.28.147
56. Pathak R, Cheema AK, Boca SM, Krager KJ, Hauer-Jensen M, Aykin-Burns N. Modulation of Radiation Response by the Tetrahydrobiopterin Pathway. Antioxidants (Basel) (2015) 4(1):68–81. doi: 10.3390/antiox4010068
57. Cheema AK, Pathak R, Zandkarimi F, Kaur P, Alkhalil L, Singh R, et al. Liver Metabolomics Reveals Increased Oxidative Stress and Fibrogenic Potential in Gfrp Transgenic Mice in Response to Ionizing Radiation. J Proteome Res (2014) 13(6):3065–74. doi: 10.1021/pr500278t
58. Ohta S, Matsuda S, Gunji M, Kamogawa A. The Role of Nitric Oxide in Radiation Damage. Biol Pharm Bull (2007) 30(6):1102–7. doi: 10.1248/bpb.30.1102
59. Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of Tetrahydrobiopterin Leads to Uncoupling of Endothelial Cell Nitric Oxide Synthase in Hypertension. J Clin Invest (2003) 111(8):1201–9. doi: 10.1172/JCI200314172
60. Schneider L, Pellegatta S, Favaro R, Pisati F, Roncaglia P, Testa G, et al. DNA Damage in Mammalian Neural Stem Cells Leads to Astrocytic Differentiation Mediated by BMP2 Signaling Through JAK-STAT. Stem Cell Rep (2013) 1(2):123–38. doi: 10.1016/j.stemcr.2013.06.004
61. Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, et al. S-Nitrosylated GAPDH Initiates Apoptotic Cell Death by Nuclear Translocation Following Siah1 Binding. Nat Cell Biol (2005) 7(7):665–74. doi: 10.1038/ncb1268
62. Hess DT, Matsumoto A, Kim S-O, Marshall HE, Stamler JS. Protein S-Nitrosylation: Purview and Parameters. Nat Rev Mol Cell Biol (2005) 6(2):150–66. doi: 10.1038/nrm1569
63. Foster MW, McMahon TJ, Stamler JS. S-Nitrosylation in Health and Disease. Trends Mol Med (2003) 9(4):160–8. doi: 10.1016/S1471-4914(03)00028-5
64. Cho D-H, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, et al. S-Nitrosylation of Drp1 Mediates Beta-Amyloid-Related Mitochondrial Fission and Neuronal Injury. Science (2009) 324(5923):102–5. doi: 10.1126/science.1171091
65. Seth P, Hsieh PN, Jamal S, Wang L, Gygi SP, Jain MK, et al. Regulation of MicroRNA Machinery and Development by Interspecies S-Nitrosylation. Cell (2019) 176(5):1014–25.e12. doi: 10.1016/j.cell.2019.01.037
66. Zhao Q, Zheng K, Ma C, Li J, Zhuo L, Huang W, et al. PTPS Facilitates Compartmentalized LTBP1 S-Nitrosylation and Promotes Tumor Growth Under Hypoxia. Mol Cell (2020) 77(1):95–107.e5. doi: 10.1016/j.molcel.2019.09.018
67. Bailey J, Davis S, Shaw A, Diotallevi M, Fischer R, Benson MA, et al. Tetrahydrobiopterin Modulates Ubiquitin Conjugation to UBC13/UBE2N and Proteasome Activity by S-Nitrosation. Sci Rep (2018) 8(1):14310. doi: 10.1038/s41598-018-32481-4
68. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis Through Lipid Remodeling. ACS Cent Sci (2020) 6(1):41–53. doi: 10.1021/acscentsci.9b01063
69. Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The Role of Ferroptosis in Ionizing Radiation-Induced Cell Death and Tumor Suppression. Cell Res (2020) 30(2):146–62. doi: 10.1038/s41422-019-0263-3
70. Suganya N, Bhakkiyalakshmi E, Sarada DVL, Ramkumar KM. Reversibility of Endothelial Dysfunction in Diabetes: Role of Polyphenols. Br J Nutr (2016) 116(2):223–46. doi: 10.1017/S0007114516001884
71. Napoli C, Ignarro LJ. Nitric Oxide and Pathogenic Mechanisms Involved in the Development of Vascular Diseases. Arch Pharm Res (2009) 32(8):1103–8. doi: 10.1007/s12272-009-1801-1
72. Naseem KM. The Role of Nitric Oxide in Cardiovascular Diseases. Mol Aspects Med (2005) 26(1-2):33–65. doi: 10.1016/j.mam.2004.09.003
73. Knapp M, Tu X, Wu R. ascular Endothelial Dysfunction, a Major Mediator in Diabetic Cardiomyopathy. Acta Pharmacol Sin (2019) 40(1):1–8. doi: 10.1038/s41401-018-0042-6
74. Taddei S, Ghiadoni L, Virdis A, Versari D, Salvetti A. Mechanisms of Endothelial Dysfunction: Clinical Significance and Preventive non-Pharmacological Therapeutic Strategies. Curr Pharm Des (2003) 9(29):2385–402. doi: 10.2174/1381612033453866
75. Preidl RHM, Möbius P, Weber M, Amann K, Neukam FW, Kesting M, et al. Long-Term Endothelial Dysfunction in Irradiated Vessels: An Immunohistochemical Analysis. Strahlenther Onkol (2019) 195(1):52–61. doi: 10.1007/s00066-018-1382-3
76. Heitzer T, Krohn K, Albers S, Meinertz T. Tetrahydrobiopterin Improves Endothelium-Dependent Vasodilation by Increasing Nitric Oxide Activity in Patients With Type II Diabetes Mellitus. Diabetologia (2000) 43(11):1435–8. doi: 10.1007/s001250051551
77. Förstermann U, Münzel T. Endothelial Nitric Oxide Synthase in Vascular Disease: From Marvel to Menace. Circulation (2006) 113(13):1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532
78. Williams IL, Wheatcroft SB, Shah AM, Kearney MT. Obesity, Atherosclerosis and the Vascular Endothelium: Mechanisms of Reduced Nitric Oxide Bioavailability in Obese Humans. Int J Obes Relat Metab Disord (2002) 26(6):754–64. doi: 10.1038/sj.ijo.0801995
79. Vásquez-Vivar J, Martásek P, Whitsett J, Joseph J, Kalyanaraman B. The Ratio Between Tetrahydrobiopterin and Oxidized Tetrahydrobiopterin Analogues Controls Superoxide Release From Endothelial Nitric Oxide Synthase: An EPR Spin Trapping Study. Biochem J (2002) 362(Pt 3):733–9. doi: 10.1042/bj3620733
80. Proskuryakov SY, Konoplyannikov AG, Skvortsov VG, Mandrugin AA, Fedoseev VM. Structure and Activity of NO Synthase Inhibitors Specific to the L-Arginine Binding Site. Biochem Mosc (2005) 70(1):8–23. doi: 10.1007/s10541-005-0002-x
81. Engin AB, Engin A. The Interactions Between Kynurenine, Folate, Methionine and Pteridine Pathways in Obesity. Adv Exp Med Biol (2017) 960:511–27. doi: 10.1007/978-3-319-48382-5_22
82. Gillis EE, Brinson KN, Rafikova O, Chen W, Musall JB, Harrison DG, et al. Oxidative Stress Induces BH Deficiency in Male, But Not Female, SHR. Biosci Rep (2018) 38(4):BSR20180111. doi: 10.1042/BSR20180111
83. Sasser JM, Brinson KN, Tipton AJ, Crislip GR, Sullivan JC. Blood Pressure, Sex, and Female Sex Hormones Influence Renal Inner Medullary Nitric Oxide Synthase Activity and Expression in Spontaneously Hypertensive Rats. J Am Heart Assoc (2015) 4(4):e001738. doi: 10.1161/JAHA.114.001738
84. Sullivan JC, Pardieck JL, Hyndman KA, Pollock JS. Renal NOS Activity, Expression, and Localization in Male and Female Spontaneously Hypertensive Rats. Am J Physiol Regul Integr Comp Physiol (2010) 298(1):R61–9. doi: 10.1152/ajpregu.00526.2009
85. Fortepiani LA, Reckelhoff JF. Role of Oxidative Stress in the Sex Differences in Blood Pressure in Spontaneously Hypertensive Rats. J Hypertens (2005) 23(4):801–5. doi: 10.1097/01.hjh.0000163149.05083.13
86. Nakamura K, Bindokas VP, Kowlessur D, Elas M, Milstien S, Marks JD, et al. Tetrahydrobiopterin Scavenges Superoxide in Dopaminergic Neurons. J Biol Chem (2001) 276(37):34402–7. doi: 10.1074/jbc.M103766200
87. Reif A, Fröhlich LG, Kotsonis P, Frey A, Bömmel HM, Wink DA, et al. Tetrahydrobiopterin Inhibits Monomerization and is Consumed During Catalysis in Neuronal NO Synthase. J Biol Chem (1999) 274(35):24921–9. doi: 10.1074/jbc.274.35.24921
88. Kotsonis P, Fröhlich LG, Shutenko ZV, Horejsi R, Pfleiderer W, Schmidt HH. Allosteric Regulation of Neuronal Nitric Oxide Synthase by Tetrahydrobiopterin and Suppression of Auto-Damaging Superoxide. Biochem J (2000) 346 Pt 3:767–76. doi: 10.1042/bj3460767
89. Berbee M, Fu Q, Boerma M, Pathak R, Zhou D, Kumar KS, et al. Reduction of Radiation-Induced Vascular Nitrosative Stress by the Vitamin E Analog γ-Tocotrienol: Evidence of a Role for Tetrahydrobiopterin. Int J Radiat Oncol Biol Phys (2011) 79(3):884–91. doi: 10.1016/j.ijrobp.2010.08.032
90. Xue J, Yu C, Sheng W, Zhu W, Luo J, Zhang Q, et al. The Nrf2/GCH1/BH4 Axis Ameliorates Radiation-Induced Skin Injury by Modulating the ROS Cascade. J Invest Dermatol (2017) 137(10):2059–68. doi: 10.1016/j.jid.2017.05.019
91. Cai S, Alp NJ, McDonald D, Smith I, Kay J, Canevari L, et al. GTP Cyclohydrolase I Gene Transfer Augments Intracellular Tetrahydrobiopterin in Human Endothelial Cells: Effects on Nitric Oxide Synthase Activity, Protein Levels and Dimerisation. Cardiovasc Res (2002) 55(4):838–49. doi: 10.1016/S0008-6363(02)00460-1
92. Cai S, Khoo J, Channon KM. Augmented BH4 by Gene Transfer Restores Nitric Oxide Synthase Function in Hyperglycemic Human Endothelial Cells. Cardiovasc Res (2005) 65(4):823–31. doi: 10.1016/j.cardiores.2004.10.040
93. Zhang Z-Y, Li Y, Li R, Zhang A-A, Shang B, Yu J, et al. Tetrahydrobiopterin Protects Against Radiation-Induced Growth Inhibition in H9c2 Cardiomyocytes. Chin Med J (2016) 129(22):2733–40. doi: 10.4103/0366-6999.193455
94. de Winter-de Groot KM, van Haren Noman S, Speleman L, Schilder AGM, van der Ent CK. Nasal Nitric Oxide Levels and Nasal Polyposis in Children and Adolescents With Cystic Fibrosis. JAMA Otolaryngol Head Neck Surg (2013) 139(9):931–6. doi: 10.1001/jamaoto.2013.4099
95. Michl RK, Hentschel J, Fischer C, Beck JF, Mainz JG. Reduced Nasal Nitric Oxide Production in Cystic Fibrosis Patients With Elevated Systemic Inflammation Markers. PloS One (2013) 8(11):e79141. doi: 10.1371/journal.pone.0079141
96. Moens AL, Takimoto E, Tocchetti CG, Chakir K, Bedja D, Cormaci G, et al. Reversal of Cardiac Hypertrophy and Fibrosis From Pressure Overload by Tetrahydrobiopterin: Efficacy of Recoupling Nitric Oxide Synthase as a Therapeutic Strategy. Circulation (2008) 117(20):2626–36. doi: 10.1161/CIRCULATIONAHA.107.737031
97. Moens AL, Kietadisorn R, Lin JY, Kass D. Targeting Endothelial and Myocardial Dysfunction With Tetrahydrobiopterin. J Mol Cell Cardiol (2011) 51(4):559–63. doi: 10.1016/j.yjmcc.2011.03.009
98. Shimizu S, Ishibashi M, Kumagai S, Wajima T, Hiroi T, Kurihara T, et al. Decreased Cardiac Mitochondrial Tetrahydrobiopterin in a Rat Model of Pressure Overload. Int J Mol Med (2013) 31(3):589–96. doi: 10.3892/ijmm.2013.1236
99. Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER. L-Ascorbic Acid Potentiates Endothelial Nitric Oxide Synthesis via a Chemical Stabilization of Tetrahydrobiopterin. J Biol Chem (2001) 276(1):40–7. doi: 10.1074/jbc.M004392200
100. Hattori Y, Nakanishi N, Akimoto K, Yoshida M, Kasai K. HMG-CoA Reductase Inhibitor Increases GTP Cyclohydrolase I mRNA and Tetrahydrobiopterin in Vascular Endothelial Cells. Arterioscler Thromb Vasc Biol (2003) 23(2):176–82. doi: 10.1161/01.ATV.0000054659.72231.A1
101. Maynard KI, Stewart-Lee AL, Milner P, Burnstock G. X-Irradiation Attenuates Relaxant Responses in the Rabbit Ear Artery. Br J Pharmacol (1992) 105(1):126–8. doi: 10.1111/j.1476-5381.1992.tb14222.x
102. Thabet NM, Rashed ER, Abdel-Rafei MK, Moustafa EM. Modulation of the Nitric Oxide/BH4 Pathway Protects Against Irradiation-Induced Neuronal Damage. Neurochem Res (2021) 46(7):1641–58. doi: 10.1007/s11064-021-03306-0
103. Hayes JD, Dinkova-Kostova AT, Tew KD. Oxidative Stress in Cancer. Cancer Cell (2020) 38(2):167–97. doi: 10.1016/j.ccell.2020.06.001
104. Rabender CS, Alam A, Sundaresan G, Cardnell RJ, Yakovlev VA, Mukhopadhyay ND, et al. The Role of Nitric Oxide Synthase Uncoupling in Tumor Progression. Mol Cancer Res (2015) 13(6):1034–43. doi: 10.1158/1541-7786.MCR-15-0057-T
105. Nagane M, Yasui H, Yamamori T, Zhao S, Kuge Y, Tamaki N, et al. Radiation-Induced Nitric Oxide Mitigates Tumor Hypoxia and Radioresistance in a Murine SCCVII Tumor Model. Biochem Biophys Res Commun (2013) 437(3):420–5. doi: 10.1016/j.bbrc.2013.06.093
106. Holotiuk VV, Kryzhanivska AY, Churpiy IK, Tataryn BB, Ivasiutyn DY. Role of Nitric Oxide in Pathogenesis of Tumor Growth and Its Possible Application in Cancer Treatment. Exp Oncol (2019) 41(3):210–5. doi: 10.32471/exp-oncology.2312-8852.vol-41-no-3.13515
107. Huerta S, Chilka S, Bonavida B. Nitric Oxide Donors: Novel Cancer Therapeutics (Review). Int J Oncol (2008) 33(5):909–27. doi: 10.3892/ijo_00000079
108. Weigert A, Brüne B. Nitric Oxide, Apoptosis and Macrophage Polarization During Tumor Progression. Nitric Oxide (2008) 19(2):95–102. doi: 10.1016/j.niox.2008.04.021
109. Vannini F, Kashfi K, Nath N. The Dual Role of iNOS in Cancer. Redox Biol (2015) 6:334–43. doi: 10.1016/j.redox.2015.08.009
110. Kashiwagi S, Tsukada K, Xu L, Miyazaki J, Kozin SV, Tyrrell JA, et al. Perivascular Nitric Oxide Gradients Normalize Tumor Vasculature. Nat Med (2008) 14(3):255–7. doi: 10.1038/nm1730
111. Cardnell RJG, Mikkelsen RB. Nitric Oxide Synthase Inhibition Enhances the Antitumor Effect of Radiation in the Treatment of Squamous Carcinoma Xenografts. PloS One (2011) 6(5):e20147. doi: 10.1371/journal.pone.0020147
112. Ridnour LA, Cheng RYS, Weiss JM, Kaur S, Soto-Pantoja DR, Basudhar D, et al. NOS Inhibition Modulates Immune Polarization and Improves Radiation-Induced Tumor Growth Delay. Cancer Res (2015) 75(14):2788–99. doi: 10.1158/0008-5472.CAN-14-3011
113. Rabender CS, Bruno N, Alam A, Sundaresan G, Zweit J, Mikkelsen RB. Sepiapterin Enhances Tumor Radio- and Chemosensitivities by Promoting Vascular Normalization. J Pharmacol Exp Ther (2018) 365(3):536–43. doi: 10.1124/jpet.117.245258
114. Viallard C, Larrivée B. Tumor Angiogenesis and Vascular Normalization: Alternative Therapeutic Targets. Angiogenesis (2017) 20(4):409–26. doi: 10.1007/s10456-017-9562-9
115. Lustig RA, DeMare PA, Kramer S. Adjuvant Methotrexate in the Radiotherapeutic Management of Advanced Tumors of the Head and Neck. Cancer (1976) 37(6):2703–8. doi: 10.1002/1097-0142(197606)37:6<2703::AID-CNCR2820370620>3.0.CO;2-H
116. Hahn P, Nevaldine B, Morgan WF. X-Ray Induction of Methotrexate Resistance Due to Dhfr Gene Amplification. Somat Cell Mol Genet (1990) 16(5):413–23. doi: 10.1007/BF01233191
117. Liang Y, Zeng D, You Y, Ma B, Li X, Chen T. Designing Dihydrofolate Reductase Inhibitors as X-Ray Radiosensitizers to Reverse Radioresistance of Cervical Cancer. ACS Med Chem Lett (2020) 11(7):1421–8. doi: 10.1021/acsmedchemlett.0c00105
118. Burns DD, Teppang KL, Lee RW, Lokensgard ME, Purse BW. Fluorescence Turn-On Sensing of DNA Duplex Formation by a Tricyclic Cytidine Analogue. J Am Chem Soc (2017) 139(4):1372–5. doi: 10.1021/jacs.6b10410
119. Cammarata M, Thyer R, Lombardo M, Anderson A, Wright D, Ellington A, et al. Characterization of Trimethoprim Resistant Dihydrofolate Reductase Mutants by Mass Spectrometry and Inhibition by Propargyl-Linked Antifolates. Chem Sci (2017) 8(5):4062–72. doi: 10.1039/C6SC05235E
120. Crabtree MJ, Hale AB, Channon KM. Dihydrofolate Reductase Protects Endothelial Nitric Oxide Synthase From Uncoupling in Tetrahydrobiopterin Deficiency. Free Radic Biol Med (2011) 50(11):1639–46. doi: 10.1016/j.freeradbiomed.2011.03.010
121. Mufamadi MS, Pillay V, Choonara YE, Du Toit LC, Modi G, Naidoo D, et al. A Review on Composite Liposomal Technologies for Specialized Drug Delivery. J Drug Deliv (2011) 2011:939851. doi: 10.1155/2011/939851
122. Dumitrescu C, Biondi R, Xia Y, Cardounel AJ, Druhan LJ, Ambrosio G, et al. Myocardial Ischemia Results in Tetrahydrobiopterin (BH4) Oxidation With Impaired Endothelial Function Ameliorated by BH4. Proc Natl Acad Sci USA (2007) 104(38):15081–6. doi: 10.1073/pnas.0702986104
123. Xie L, Talukder MAH, Sun J, Varadharaj S, Zweier JL. Liposomal Tetrahydrobiopterin Preserves eNOS Coupling in the Post-Ischemic Heart Conferring In Vivo Cardioprotection. J Mol Cell Cardiol (2015) 86:14–22. doi: 10.1016/j.yjmcc.2015.06.015
124. Hofmeister LH, Lee SH, Norlander AE, Montaniel KRC, Chen W, Harrison DG, et al. Phage-Display-Guided Nanocarrier Targeting to Atheroprone Vasculature. ACS Nano (2015) 9(4):4435–46. doi: 10.1021/acsnano.5b01048
125. Whitsett J, Rangel Filho A, Sethumadhavan S, Celinska J, Widlansky M, Vasquez-Vivar J. Human Endothelial Dihydrofolate Reductase Low Activity Limits Vascular Tetrahydrobiopterin Recycling. Free Radic Biol Med (2013) 63:143–50. doi: 10.1016/j.freeradbiomed.2013.04.035
126. Almudéver P, Milara J, De Diego A, Serrano-Mollar A, Xaubet A, Perez-Vizcaino F, et al. Role of Tetrahydrobiopterin in Pulmonary Vascular Remodelling Associated With Pulmonary Fibrosis. Thorax (2013) 68(10):938–48. doi: 10.1136/thoraxjnl-2013-203408
127. Hui L, Chen Y. Tumor Microenvironment: Sanctuary of the Devil. Cancer Lett (2015) 368(1):7–13. doi: 10.1016/j.canlet.2015.07.039
128. Chen L, Zeng X, Kleibeuker E, Buffa F, Barberis A, Leek RD, et al. Paracrine Effect of GTP Cyclohydrolase and Angiopoietin-1 Interaction in Stromal Fibroblasts on Tumor Tie2 Activation and Breast Cancer Growth. Oncotarget (2016) 7(8):9353–67. doi: 10.18632/oncotarget.6981
Keywords: radiation, nitric oxide synthase (NOS), nitric oxide (NO), 5, 6, 7, 8-tetrahydrobiopterin (BH4), radiosensitivity
Citation: Feng Y, Feng Y, Gu L, Liu P, Cao J and Zhang S (2021) The Critical Role of Tetrahydrobiopterin (BH4) Metabolism in Modulating Radiosensitivity: BH4/NOS Axis as an Angel or a Devil. Front. Oncol. 11:720632. doi: 10.3389/fonc.2021.720632
Received: 04 June 2021; Accepted: 12 August 2021;
Published: 27 August 2021.
Edited by:
Ronggui Cory Hu, Shanghai Institute of Biochemistry and Cell Biology (CAS), ChinaReviewed by:
Paola Maycotte, Instituto Mexicano del Seguro Social, MexicoWanessa Altei, Barretos Cancer Hospital, Brazil
Copyright © 2021 Feng, Feng, Gu, Liu, Cao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuyu Zhang, emhhbmcuc2h1eXVAaG90bWFpbC5jb20=; Jianping Cao, anBjYW9Ac3VkYS5lZHUuY24=