 Panagiotis T. Diamantopoulos
Panagiotis T. Diamantopoulos Nora-Athina Viniou
Nora-Athina Viniou- First Department of Internal Medicine, Laikon General Hospital, National and Kapodistrian University of Athens, Athens, Greece
Atypical chronic myelogenous leukemia (aCML), BCR/ABL1 negative is a rare myelodysplastic/myeloproliferative neoplasm, usually manifested with hyperleukocytosis without monocytosis or basophilia, organomegaly, and marked dysgranulopoiesis. In this review, we will discuss the classification and diagnostic criteria of aCML, as these have been formulated during the past 30 years, with a focus on the recent advances in the molecular characterization of the disease. Although this entity does not have a definitive molecular profile, its molecular characterization has contributed to a better understanding and more accurate classification and diagnosis of aCML. At the same time, it has facilitated the identification of adverse prognostic factors and the stratification of patients according to their risk for leukemic transformation. What is more, the molecular characterization of the disease has expanded our therapeutic choices, thoroughly presented and analyzed in this review article.
Introduction
Atypical chronic myelogenous leukemia (aCML), BCR/ABL1 negative is a rare disorder classified into the category of myelodysplastic/myeloproliferative neoplasms (MDS/MPN), according to the 2016 revision of the World Health organization (WHO) classification of myeloid neoplasms and acute leukemia (1). It is, by definition, a BCR-ABL1-negative clonal disorder sharing myelodysplastic and myeloproliferative features. Due to its rarity, the diagnostic criteria for aCML have been changing since its first description while there are no established standards of care for patients with this condition; hence we will focus on the diagnostic criteria of the disease, new molecular characteristics that have emerged during the last few years, some of which have already been incorporated in the latest diagnostic criteria of the WHO, the proposed risk stratification systems, and the available treatment approaches for aCML.
Epidemiology, Clinical and Laboratory Characteristics
The incidence of aCML is low, a fact leading to limited knowledge about this disease entity. Its true incidence is largely unknown, since it is usually estimated in comparison to that of (BCR-ABL1-positive) CML at about one to two cases per 100 cases of CML (2). The cases reported so far concern adults. Patients usually present in the seventh or eighth decade of life and there is an apparent male predominance (1:1 to 4:1 in several case series).
The patients usually present with organomegaly and hyperleukocytosis. Splenomegaly has been reported in 50% to 75% of the patients while in one patient series, hepatomegaly has been reported to be present in 49% of the patients. The median white blood cell (WBC) count at presentation has been reported to fluctuate between 23x109/L to 97x109/L in several case series. The patients are usually moderately to severely anemic [median hemoglobin (Hb) level, 8.6 g/dL to 11.7 g/dL], while about two thirds of them are transfusion dependent. The platelet count may be decreased or normal but can also be found increased in several cases (3). Monocyte and basophile counts are usually within normal range while immature myeloid precursors (promyelocytes, myelocytes, metamyelocytes) are usually present in the peripheral blood smear (10% to 20%), but peripheral blasts are generally absent or low. The neutrophils are highly dysplastic in most cases with typical pseudo-Pelger-Huet changes and hypogranularity of the cytoplasm while dyserythropoiesis and dysmegakaryopoiesis may also be present (3). The bone marrow is hypercellular, with an increased myeloid-to-erythroid ratio (usually up to 10:1) while marrow fibrosis may be present. By definition, blood or bone marrow blasts are less than 20%..
Classification and Diagnostic Criteria
In the late 80s, the first reports of the disease came to light, with the scientific community wondering whether a Philadelphia-negative CML exists (4). This lead to the identification of “masked” or “hidden” cases of BCR/ABL1 translocation (5–8), and to the understanding that “CML-like” changes can be found without the diagnostic hallmark of CML. At the same time, cases with features compatible with chronic neutrophilic leukemia (CNL) (9) or chronic myelomonocytic leukemia (10, 11) but atypical presentation were identified, leading to the initial descriptions of the clinical and laboratory characteristics of the disease that eventually led to its classification in the group of chronic myeloid leukemias (12). The French-American-British (FAB) Cooperative Leukaemia Group also classified the disease in 1994 as a subtype of chronic myeloid leukemia with distinct features such as the low basophil and monocyte count, granulocytic dysplasia, presence of immature granulocytes up to 10%-20% of WBC, blasts <2%, and the absence of Philadelphia (Ph) chromosome and the BCR-ABL1 fusion gene (13). The WHO 2001 classification had minimal differences from the original FAB classification, mainly referring to the marked multilineage dysplasia and the blast percentage in the bone marrow at <20%. In 2008, the new WHO classification added the threshold of 13x109/L to define leukocytosis while it also proposed the limit of 10% of total WBC for monocytes. Moreover, it limited dysplasia to dysgranulopoiesis and for the first time it required the absence of other rearrangements besides that of BCR-ABL1 (2). Finally, the latest WHO 2016 classification (1) emphasized molecular changes in newly described genes (ETNK1, SETBP1) and the absence of rearrangement of PDGFRA, PDGFRB, FGFR1, and PCM1-JAK2. Moreover, for the first time, it was added as an exclusion criterion that WHO criteria for other myeloproliferative neoplasms should not be met. Finally, it is the first time that marrow cellularity is commented on. Nevertheless, there is no quantification of bone marrow cellularity and dysplasia in any of the available diagnostic criteria as is usually the case in other disease entities. Although a major feature of aCML, prominent dysgranulopoiesis is not further specified qualitatively and, especially, quantitatively. Granulocytic dysplasia includes pseudo-Pelger-Huët cells, other types of abnormal segmentation of the nucleus or abnormal chromatin clumping, and reduced cytoplasmic granularity. Although, dysgranulopoiesis is the main morphologic feature of the disorder, dyserythropoiesis and, especially, dysmegakaryopoiesis are not rare, although more subtle. These defining morphologic features help distinguish aCML from other entities such as CML and CNL where dysplastic features are minimal or absent (14). A comprehensive list of the diagnostic criteria for aCML as these were shaped during the last 35 years is presented in Table 1.

Table 1 Diagnostic criteria of atypical chronic myeloid leukemia, BCR/ABL1 negative.
In conclusion, the defining characteristics of aCML are granulocytic proliferation with marked dysgranulopoiesis, along with minimal or absent monocytosis and absence of basophilia, without BCR/ABL1 translocation or rearrangements of genes that define other hematologic neoplasms. Because of the rarity of aCML, it remains virtually an exclusion diagnosis that should be made when other MDS and MPN can be safely ruled out. In recent years, the molecular profile of the disease is being increasingly outlined through the accumulation of data on the detection of recurrent molecular abnormalities that may contribute to the differential diagnosis of aCML. These molecular changes may be incorporated in the diagnostic criteria for the disease in the future.
The cytogenetic abnormalities in aCML, apart from the lack of the Philadelphia chromosome, are not disease-specific and include trisomy 8, deletion Y, deletion 20q, isochromosome 17(q), and other cytogenetic changes along with complex karyotype that are usually found in MDS. Nevertheless, about 50% to 80% of the cases have normal karyotypes (15, 16).
Although the presence of several molecular changes can help to rule out the diagnosis of aCML, there is no definitive molecular profile of the disease. During the last few years, recurrent mutations have been increasingly identified in patients with aCML, but the percentage of patients bearing those mutations vary significantly among different studies. Frequently mutated genes (i.e. >20% of cases) are SETBP1, ASXL1, NRAS/KRAS, SRSF2, and TET2 while a variety of genes, including CSF3R, CBL, EZH2, ETNK1, U2AF1 and others (14, 15, 17–22) are found mutated in a lower frequency (<10% of cases). The genes found recurrently mutated in aCML, along with their chromosomal location, normal function, affected exons, mutation types, and clinical implications are listed in Table 2 (23). Moreover, an algorithm for the diagnosis of aCML incorporating molecular data has been proposed and can be found in Figure 1. Since some of the mutated genes found in patients with aCML may be targetable, below we will discuss frequently mutated genes that may affect treatment decision. A list of actionable and non-actionable mutations along with potential targeted therapies can be found in Table 3.
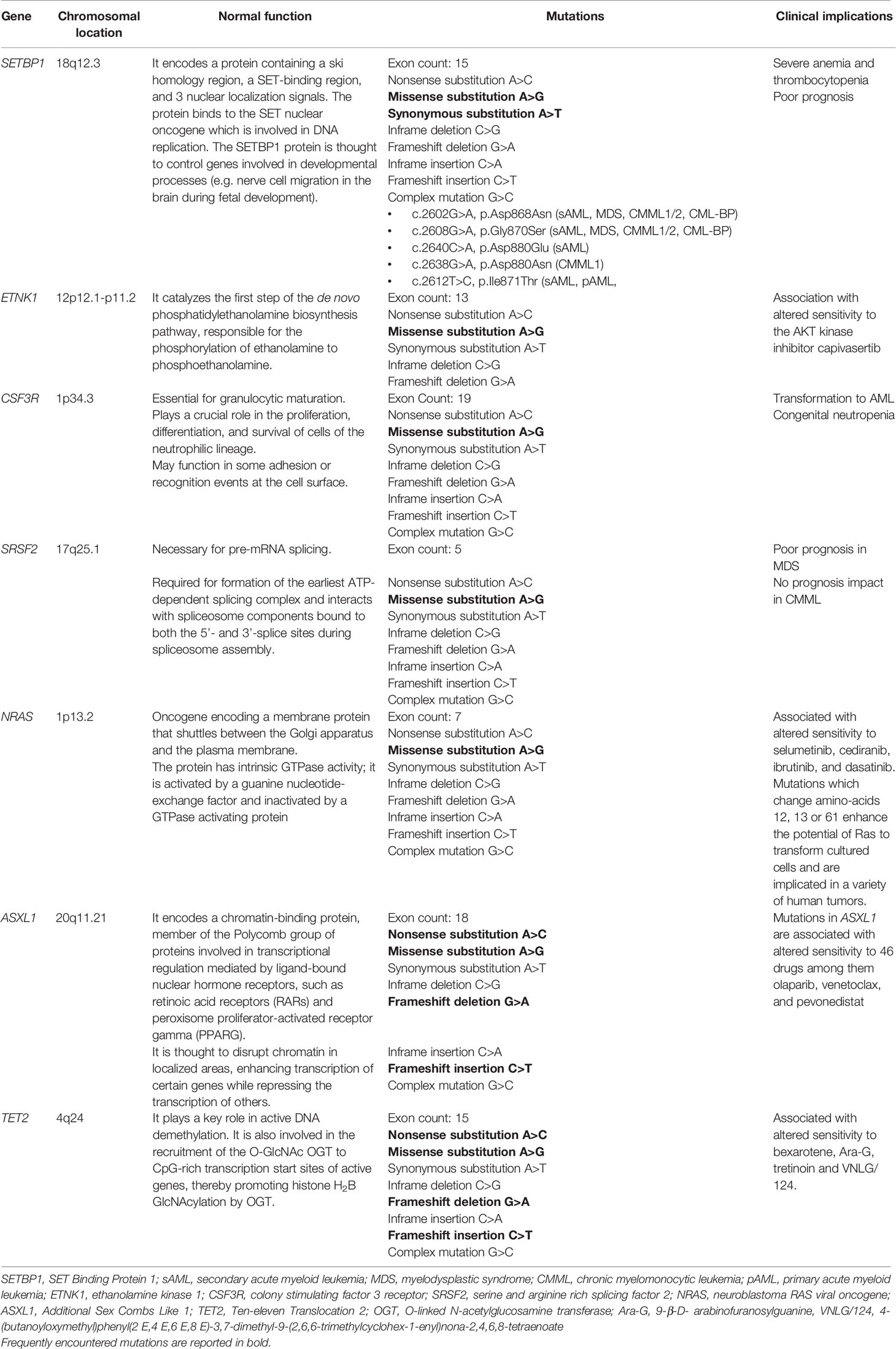
Table 2 Recurrently mutated genes in aCML.
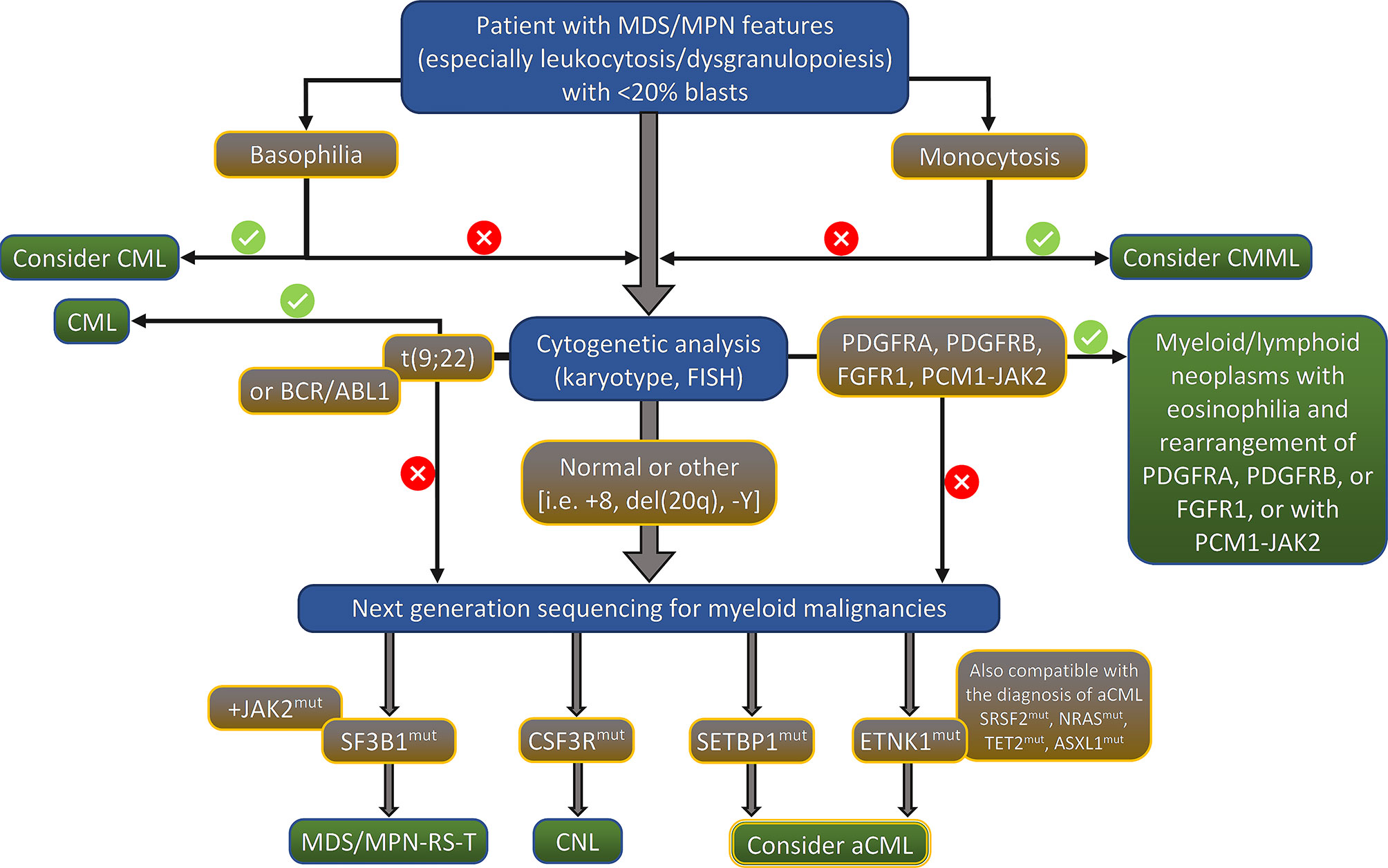
Figure 1 Proposed algorithm for the diagnosis of aCML. MDS/MPN, myelodysplastic syndrome/myeloproliferative neoplasm; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; FISH, fluorescent in situ hybridization; RS, ring sideroblasts; T, thrombocytosis; CNL, chronic neutrophilic leukemia; aCML, atypical chronic myelogenous leukemia, BCR/ABL1 negative.

Table 3 Actionable and non-actionable mutations in aCML.
SET Binding Protein 1 (SETBP1)
The most frequently identified mutated gene in aCML, in up to one third of cases, is SETBP1 (15, 20, 21, 30). SETBP1 binds to SET which inhibits the tumor suppressor PP2A. This binding protects SET from cleavage, thus repressing PP2A activity. Mutations of SETBP1 lead to ubiquitination and, thus, degradation of the protein, increasing SET expression, and, consequently, cellular proliferation through inhibition of PP2A. SETPB1 mutations have been correlated with more pronounced dysplasia, higher WBC counts, more severe anemia and thrombocytopenia, as well as a worse prognosis (20, 21). Moreover, SETBP1 mutations have been associated with ASXL1 and CBL mutations while they have been found to be mutually exclusive with JAK2 and TET2 mutations (21). SETBP1 mutations have been incorporated as a supportive criterion for aCML in the WHO 2016 diagnostic criteria.
Ethanolamine Kinase 1 (ETNK1)
ETNK1 is responsible for the phosphorylation of ethanolamine to phosphoethanolamine as part of the Kennedy pathway, which is the main metabolic route for the synthesis of phosphatidylethanolamine and phosphatidylcholine (31). The presence of phosphatidylethanolamine is crucial for cytokinesis and cells lacking phosphatidylethanolamine are unable to complete the mitotic process while reduced intracellular phosphoethanolamine causes hyperactivation of the mitochondria, ROS production, and Histone H2AX phosphorylation, ultimately leading to DNA damage. Recurrent somatic mutations of ETNK1 have been found in about 13% of patients with aCML (32). A recent study on 43 aCML samples identified ETNK1 mutations in 16.2% of the analyzed samples while it also suggested that whereas ETNK1 mutations are an early event in the clonal evolution history of the disease, SETBP1 mutations are usually a late event (33). Although the presence of ETNK1 mutations has been considered as a relatively specific finding for aCML, leading to their inclusion in the 2016 WHO classification as a support criterion for the diagnosis of the disease, they have also been found in chronic myelomonocytic leukemia (CMML) and systemic mastocytosis with eosinophilia (34) while they were recently described in diffuse large B-cell lymphoma (35). Nevertheless, the interest in ETNK1 mutations remains high due to the fact that phosphoethanolamine, the metabolic product of ETNK1, has been shown to negatively control mitochondrial activity, thus restoring a normal phenotype (24).
Colony Stimulating Factor 3 Receptor (CSF3R)
Although, in an early study, CSF3R mutations had been reported in 59% of patients with aCML and CNL (25, 36), further studies did not confirm this high frequency, and it is now believed that CSF3R-mutated aCML is in fact rare (19, 37). Nevertheless, these mutations have been proposed to activate either the JAK-STAT pathway (CSF3R membrane proximal mutation), or the SRC-kinase signaling pathway (CSF3R truncating mutation), thus being targetable with ruxolitinib or dasatinib, respectively (38). Moreover, due to the extremely high frequency of CSF3R mutations in CNL (39), these mutations constitute a criterion for the diagnosis of CNL and, along with other morphological and clinical features, help distinguish CNL from aCML In fact, the identification of a CSF3R mutation in a patient with hepatosplenomegaly, hyperleukocytosis with prominence of neutrophils, low percentage of peripheral blood immature granulocytes and myeloblasts, along with minimal or absent dysgranulopoiesis in the bone marrow strongly favors the diagnosis of CNL over aCML.
Serine and Arginine Rich Splicing Factor 2 (SRSF2)
SRSF2 mutations have been reported in 12%, 13.5%, and 40% of aCML cases in three studies (15, 33, 37). In the study by Patnaik et al., the presence of SRSF2 mutations in three of the examined cases did not affect OS, while one patient had a concomitant TET2 mutation. On the other hand, in the study by Meggendorfer et al., it was shown that SRSF2 mutations tended to coexist with SETBP1 and ASXL1 mutations.
RAS Mutations
NRAS mutations resulting is constitutive activation of the MAPK pathway, thus, promoting malignant cell survival and proliferation, have been detected in up to one third of patients with aCML (19, 40). Moreover, it has been shown that enhanced MAPK signaling is crucial to leukemogenesis by CSF3R mutants in CNL (41).
Other Gene Mutations
Mutations of ASXL1 and TET2 are found in over 15% of cases with aCML. In one study, TET2 mutations were correlated with lower OS along with advanced age and low hemoglobin level. On the other hand, ASXL1 is considered to be the most commonly mutated gene in aCML (28%), but it has also been shown not to affect overall survival (OS) despite the detrimental effect on survival in CMML, CNL, and PMF (37). Mutations of ASXL1 have been found to accompany spliceosome gene mutations in as high as 65% of cases (21), a fact supporting the hypothesis of serial accumulation of mutations in the malignant clone, that seems to apply to all MDS/MPNs (42). It should be noted though, that the correlation of OS with the mutational status has been based in small patient series and should, therefore, be considered with caution.
Treatment Approaches for aCML
As already mentioned, there is no standard of care for the management of patients with aCML. The rarity of the disease has led to its misclassification, resulting in the lack of a universally accepted risk stratification system that would lead to the formulation of recommendations for risk-based treatment strategies. Treatment choices are based on the results of small trials and patient series while some classical therapeutic options have never actually been studied and their use is only based on their efficacy in other MDS/MPN. Recently, the molecular profile of the disease has led to the emergence of new treatment options based on targetable mutations, but their use is mostly based on preclinical data or case reports. Most patients require some kind of treatment since their initial presentation, due to the accelerating leukocytosis and the deteriorating anemia, splenomegaly, and constitutional symptoms, thus a wait-and-watch strategy is rarely advisable.
As already mentioned, there is a lack of risk stratification systems for aCML, mainly due to the rarity of the condition and to the absence of large studies evaluating prognosis. Nevertheless, there have been some efforts to identify adverse prognostic factors and to stratify the patients according to their risk for AML transformation and death. In one of the largest studies in aCML, the authors analyzed the prognostic characteristics of 76 treatment-naïve patients with BCR/ABL1 negative CML. Multivariate analysis identified age >65 years, hemoglobin level <10 g/dL, and WBC>50x109/L as independent prognostic factors of poor survival (43). In the same study it was shown that treatment did not significantly affect survival. Moreover, a simple scoring system assigning one point to each one of the three above-mentioned independent prognostic factors was designed to stratify patients according to their expected survival. The system stratified the patients into two risk-groups (low risk with 0-1 point and high risk with >1 points), with a corresponding median survival of 38 months versus 9 months (p<0.01). Although this risk stratification system has not been widely used, the value of its prognostic parameters has been confirmed in more recent studies. Thus, in a study of 55 patients with aCML (44) multivariate analysis associated shorter survival with age >65 years, female sex, WBC>50x109/L, and presence of circulating immature precursors while the hemoglobin level did not retain its statistical significance. The authors also evaluated the risk of leukemic transformation that was found to be higher in patients with palpable hepatosplenomegaly, monocytosis, bone marrow blasts >5%, marked dyserythropoiesis, and transfusion dependence. Moreover, they tried to validate the prognostic scoring system by Onida et al. and reported that, in their cohort, it could not identify poor survival but identified patients at higher risk for AML transformation. A smaller study reported longer survival rates in patients with normal platelet counts and hemoglobin level >10 g/dL (45) while in a study of 65 patients, WBC>50x109/L, and a high blood immature myeloid cell count and bone marrow blast count (as continuous variables) were correlated with lower OS in univariate analysis. However, no multivariate analysis was carried out (19). Finally, in the most recent study in aCML, age >67 years, hemoglobin level <10 g/dL, and TET2 mutations were correlated with lower survival rates in multivariate analysis (37). The authors also provided a two-group prognostic model based on the above parameters, with a median OS of 18 and seven months, respectively. Table 4 summarizes the proposed prognostic factors for aCML by the above referenced studies.
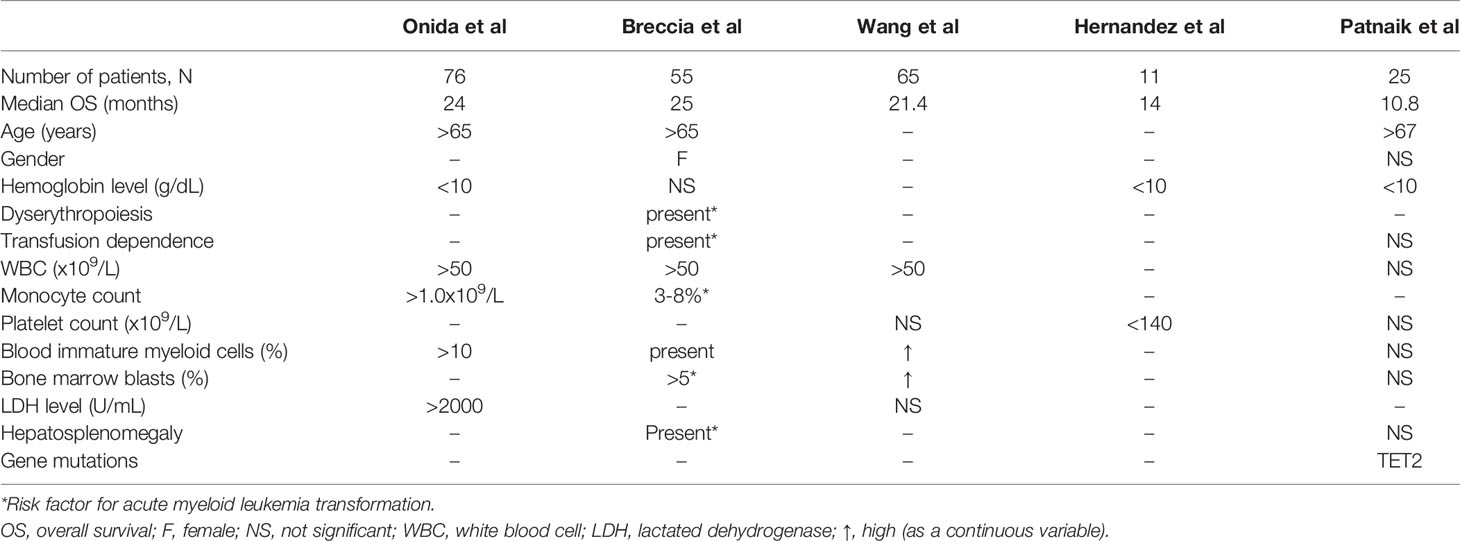
Table 4 Factors correlated with lower overall survival in patients with atypical chronic myelogenous leukemia, BCR/ABL1 negative.
In conclusion, most of the studies reporting on prognostic factors in aCML agree that advanced age, anemia, hyperleukocytosis, and presence of immature myeloid cells in the peripheral blood adversely affect OS. It is becoming more and more obvious that the underlying molecular mechanisms may be well correlated with prognosis; thus, further analysis of the molecular footprint of the disease will allow the design of more accurate risk stratification systems that will define the treatment needs of each patient. The available treatment options for patients with aCML are presented in detail in the following paragraphs.
Allogeneic Hematopoietic Stem Cell Transplantation (Allo-HSCT)
Allo-HSCT should be considered in all eligible patients, given the unfavorable prognosis of aCML. Nevertheless, the age of the patients is usually a prohibitive factor while at the same time there is limited reliable data on the efficacy of allo-HSCT in patients with aCML. Several small studies of retrospective nature have been published. In a study of nine transplanted patients (four from HLA-identical siblings, four from HLA-compatible unrelated donors and one from a twin sibling) followed for a median period of 55 months, all nine achieved complete chimerism and remained in complete remission (CR) while, with the exception of one patient who died from cerebral toxoplasmosis, the remaining eight were alive at the time of the analysis (46). In another study of seven patients with aCML treated with allo-HSCT, after a follow-up period of 17.5 months, only two were alive; one had died due to aCML, and the remaining four due to transplantation related complications (47). In an analysis of 42 cases reported to the European Society for Blood and Marrow Transplantation (EBMT) registry, donors were HLA-identical siblings in 64% and matched unrelated in 36% of the cases. A CR was observed in 87% of patients and the 5-year relapse-free survival was 36%. Younger patients with low EBMT risk scores were found to be the best candidates for allo-HSCT (43). In a recent study of 14 patients with aCML treated with allo-HSCT, 13/14 had received first-line therapy (10/14 with hydroxyurea) with variable responses, 8/13 had received second-line therapy before transplant, and 4/8 third-line therapy while at least two had progressed to acute myelogenous leukemia (AML) before allo-HSCT. Five, seven, and two patients received an allograft from HLA-matched related, unrelated marrow, and unrelated cord blood donors, respectively. A myeloablative regimen was used in 11/14 patients. Among patients with neutrophil engraftment, 9/13 achieved a CR and the 1-year OS was 54.4%, being higher in univariate analysis in patients with related donors, <5% myeloblasts in the peripheral blood, and a Karnofsky performance status of ≤80% (48).
Questions on the donor source, the correct timing (i.e. upfront transplantation versus transplantation after initial treatment to reduce the disease burden), the intensity of the conditioning regimen, the impact of previously administered treatments, and the possible impact of the molecular profile of the patients still remain unanswered. Nevertheless, the advanced age of most patients with aCML probably dictates the use of reduced intensity conditioning regimens while the molecular profile of the patients may be useful for the monitoring of minimal residual disease (49) and as a guide for post-transplant maintenance in cases with targetable mutations.
Hypomethylating Agents (HMAs)
The use of HMAs in patients with aCML should be a case-by-case approach since patients with striking myeloproliferative features are less likely to respond to HMAs. Although data on the efficacy and safety of HMAs in aCML is limited, the rationale for their use is sound, based on their established activity in CMML and other MDS/MPN. Early data on the use of decitabine in seven patients with aCML and a median age of 67 years showed an OS rate of only 13 months and a 14% 2-year survival rate (50). In a study of 76 patients with aCML treated with several regimens (5 patients treated with an HMA), OS was not affected by the treatment choice, but no information is specifically provided for the prognosis of patients treated with HMAs (51). Decitabine has also been administered in four patients with aCML and after a median of 2.5 cycles of treatment and a median follow-up of 13 months, three patients were still alive (one of them eventually treated with allo-HSCT) (52). Finally, in a report of five patients with aCML treated with HMAs, the best achieved response was stable disease (37), while a few case reports give mixed results on the efficacy of HMAs. Thus, a general rule would be that the use of HMAs in patients with aCML should be restricted in elderly patients with prominent dysplastic and more subtle myeloproliferative features, or as a bridge to a transplant.
Cytoreduction and Intensive Chemotherapy
Historically, hydroxyurea has been the mainstay of treatment for patients with aCML and other MDS/MPNs. In recent reports, it remains the most widely used factor for the management of myeloproliferation in aCML and it can effectively control leukocytosis and splenomegaly, but responses are typically short-lived. In an early study on 11 patients with aCML, nine were treated with hydroxyurea achieving a partial remission (8). In a study of 55 patients with aCML, 48 (87.3%) patients had been treated with hydroxyurea, but the effect of treatment on OS was not further discussed by the authors (44). Furthermore, in a study on the characteristics and outcome of 76 patients with aCML, the authors stated that 53% of the patients had already been treated with hydroxyurea or busulfan at the time of their referral to the study center while nine of them were also treated with hydroxyurea after their referral. Although there was no specific mention of the outcome per regimen, the authors reported that there was no significant difference in OS between treated and untreated patients (43). Finally, in a more recent study of 25 patients, hydroxyurea had been administered in 15 (60.0%) patients, but treatment outcomes were not evaluated (37). Busulfan and low-dose cytarabine have also been used in the treatment of patients with aCM while intensive chemotherapy is not a standard treatment option for patients with aCML and should be reserved for patients with leukemic transformation.
Ruxolitinib
Activating mutations in CSF3R have been found to drive myeloproliferative disorders resembling aCML and CNL in mouse models, where these mutations are sensitive to JAK inhibition that may effectively reduce the WBC count and the spleen size (53). Preliminary data has shown that treatment of aCML/CNL cells carrying activating mutations of CSF3R with ruxolitinib resulted in inhibition of cell growth while a patient with CNL bearing the CSF3R T618I mutation was effectively treated with ruxolitinib (25). Although the experience with ruxolitinib was limited to case reports, a recent phase II study was conducted to investigate the hematologic response to ruxolitinib in patients with aCML/CNL. The study included 23 patients with aCML, six of whom carried an activating mutation of CSF3R (T618I, T640N, or T615A). Response (PR and CR) was reported in only two (8.7%) patients while no serious adverse events attributed to ruxolitinib were observed (54). The results of this trial reduced the initial enthusiasm on the efficacy of targeting CSF3R mutations with ruxolotinib in aCML, especially since these mutations proved to be rather rare in this condition. The use of ruxolitinib as a bridge to transplant, though, is still a valid choice in an effort to reduce the WBC count and splenomegaly following the paradigm of myelofibrosis although clinical trials are still lacking.
Dasatinib
CSF3R truncation mutations have been shown to activate the SRC family pathway offering an option of targeted therapy with dasatinib. This SRC family kinase inhibitor used in CML has been proposed to potentially have therapeutic value in aCML with CSF3R truncation mutations since in vitro studies of cell lines with such mutations have shown sensitivity of the cells to the drug (18, 25). However, these preclinical data have not been supported by in vivo reports to date.
Trametinib
The mitogen-activated protein kinase kinase (MEK) inhibitor trametinib, approved for the treatment of advanced or metastatic melanoma, has been proven active against NRAS-mutated AML in human cell lines and murine models (55). Moreover, it has been shown that MEK inhibition with trametinib is sufficient to suppress CNL induced by CSF3R mutations, highlighting a MAPK-dependent mechanism of CSF3R-induced pathogenesis that is valid at least in CNL (41). A case report of a patient with aCML responding to trametinib (56) and a phase I/II nonrandomized study showing clinical activity of trametinib in several RAS-mutated myeloid malignancies (27) have confirmed these preclinical data although the beneficial effect of the drug in reducing the blast count was not translated into a survival benefit. Further studies with trametinib as monotherapy or in combination with other targeted therapies are needed to test its efficacy in aCML.
Other Agents
Interferon has been used in aCML with mixed results. In an early study of 14 patients, interferon-α was administered to seven patients following treatment with hydroxyurea, and CR was achieved in five (57). In another study on the characteristics and outcome of patients with aCML, interferon-α and interferon-γ were used as single-agent therapy in 17 patients with aCML; nevertheless, response rates were not presented (43). Moreover, among 11 patients with aCML, six were treated with interferon-α, two of whom achieved a CR lasting for 9 and 40 months (8). In another early study, out of 14 patients treated with interferon-α, six (43%) of them responded (five with a CR) with a duration of response ranging from 1 to >100 months (58). Finally, among five patients with aCML treated with peg-interferon-α-2b in the context of a phase II study, two responded to treatment achieving a CR after three months of treatment (59). The above-referenced results show that the use of interferon-α and its pegylated formulations should be further investigated.
Supportive and Palliative Treatment
Supportive treatment should be offered to all patients in the form of erythropoiesis-stimulating agents (ESAs) and red blood cell transfusions. The use of ESAs in terms of efficacy and safety has never been specifically studied for aCML. Thus, factors predicting response to ESAs have not been established. Moreover, heavily transfused patients should probably also receive iron chelation therapy, depending on their estimated prognosis, but this strategy is not supported by any published data. Finally, the use of corticosteroids, danazol, and thalidomide or lenalidomide to treat anemia has never been studied or reported in patients of aCML. Splenectomy or splenic irradiation have been scarcely used in the past as palliative measures in patients with aCML, with no disease improvement (8, 50, 60). Thus, they should be avoided since any possible benefit is cancelled out by potentially severe complications, such as bleeding, thrombosis, infection, and potential acceleration of leukocytosis and/or hepatomegaly (22).
Conclusions on Treatment Approach
Based on the available data, the general principals of the management of patients with aCML can be summarized into the following.
a. Allo-HSCT should be offered to all eligible patients.
b. Myeloid mutation panel testing should be performed to all patients to detect potentially targetable mutations.
c. Although clinical trials focusing on patients with aCML are rare, inclusion in a clinical trial should be an early choice, especially for patients without targetable mutations.
d. In patients not eligible for allo-HSCT, the treatment should focus on addressing the patient’s major clinical issues (constitutional symptoms, anemia, organomegaly) and potentially on decreasing the possibility of progression to AML although supporting data about the latter is lacking.
e. Treatment strategies applicable to patients with MDS or MPN can be selected on a case-by-case approach.
f. Given the poor prognosis of aCML, initiation of treatment is generally favored over a watch-and-wait strategy.
The prognosis of patients with aCML remains poor and cases with long survival are the exception to the rule. Further identification of the molecular footprint of the disease may allow for the emergence of new targeted treatment choices that may reduce the risk for AML transformation and prolong survival.
Author Contributions
PD has drafted the manuscript and N-AV has critically revised the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Ms Evita Alexopoulos for copyediting the final manuscript.
References
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
2. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 Revision of the World Health Organization (WHO) Classification of Myeloid Neoplasms and Acute Leukemia: Rationale and Important Changes. Blood (2009) 114(5):937–51. doi: 10.1182/blood-2009-03-209262
3. Drozd-Sokołowska JE, Waszczuk-Gajda A, Mądry K, Dwilewicz-Trojaczek J. Atypical Chronic Myeloid Leukaemia - A Rare Subtype of Myelodysplastic/Myeloproliferative Neoplasm. Contemp Oncol (Pozn) (2018) 22(1):14–9. doi: 10.5114/wo.2018.74388
4. Iavorkovskiĭ LI, Iavorkovskiĭ LL. [Does Ph’-Negative Chronic Myeloleukemia Exist]? . Gematol Transfuziol (1989) 34(11):3–10.
5. van der Plas DC, Hermans AB, Soekarman D, Smit EM, de Klein A, Smadja N, et al. Cytogenetic and Molecular Analysis in Philadelphia Negative CML. Blood (1989) 73(4):1038–44. doi: 10.1182/blood.V73.4.1038.1038
6. Sessarego M, Mareni C, Vimercati R, Defferrari R, Origone P, Damasio E, et al. Translocation T(9;9)(P13;Q34) in Philadelphia-Negative Chronic Myeloid Leukemia With Breakpoint Cluster Region Rearrangement. Cancer Genet Cytogenet (1989) 43(1):51–6. doi: 10.1016/0165-4608(89)90126-X
7. Selleri L, Emilia G, Luppi M, Temperani P, Zucchini P, Tagliafico E, et al. Chronic Myelogenous Leukemia With Typical Clinical and Morphological Features can be Philadelphia Chromosome Negative and “Bcr Negative”. Hematol Pathol (1990) 4(2):67–77.
8. Kurzrock R, Kantarjian HM, Shtalrid M, Gutterman JU, Talpaz M. Philadelphia Chromosome-Negative Chronic Myelogenous Leukemia Without Breakpoint Cluster Region Rearrangement: A Chronic Myeloid Leukemia With a Distinct Clinical Course. Blood (1990) 75(2):445–52. doi: 10.1182/blood.V75.2.445.445
9. Zoumbos NC, Symeonidis A, Kourakli-Symeonidis A. Chronic Neutrophilic Leukemia With Dysplastic Features. A New Variant of the Myelodysplastic Syndromes. Acta Haematol (1989) 82(3):156–60. doi: 10.1159/000205367
10. Kantarjian HM, Kurzrock R, Talpaz M. Philadelphia Chromosome-Negative Chronic Myelogenous Leukemia and Chronic Myelomonocytic Leukemia. Hematol Oncol Clin North Am (1990) 4(2):389–404. doi: 10.1016/S0889-8588(18)30494-5
11. Michaux JL, Martiat P. Chronic Myelomonocytic Leukaemia (CMML)–a Myelodysplastic or Myeloproliferative Syndrome? Leuk Lymphoma (1993) 9(1-2):35–41. doi: 10.3109/10428199309148501
12. Shepherd PC, Ganesan TS, Galton DA. Haematological Classification of the Chronic Myeloid Leukaemias. Baillieres Clin Haematol (1987) 1(4):887–906. doi: 10.1016/S0950-3536(87)80031-8
13. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick H, et al. The Chronic Myeloid Leukaemias: Guidelines for Distinguishing Chronic Granulocytic, Atypical Chronic Myeloid, and Chronic Myelomonocytic Leukaemia. Proposals by the French-American-British Cooperative Leukaemia Group. Br J Haematol (1994) 87(4):746–54. doi: 10.1111/j.1365-2141.1994.tb06734.x
14. Dao KH, Tyner JW. What’s Different About Atypical CML and Chronic Neutrophilic Leukemia? Hematol Am Soc Hematol Educ Program (2015) 2015:264–71. doi: 10.1182/asheducation-2015.1.264
15. Meggendorfer M, Haferlach T, Alpermann T, Jeromin S, Haferlach C, Kern W, et al. Specific Molecular Mutation Patterns Delineate Chronic Neutrophilic Leukemia, Atypical Chronic Myeloid Leukemia, and Chronic Myelomonocytic Leukemia. Haematologica (2014) 99(12):e244–6. doi: 10.3324/haematol.2014.113159
16. Sadigh S, Hasserjian RP, Hobbs G. Distinguishing Atypical Chronic Myeloid Leukemia From Other Philadelphia-Negative Chronic Myeloproliferative Neoplasms. Curr Opin Hematol (2020) 27(2):122–7. doi: 10.1097/MOH.0000000000000565
17. Gotlib J, Maxson JE, George TI, Tyner JW. The New Genetics of Chronic Neutrophilic Leukemia and Atypical CML: Implications for Diagnosis and Treatment. Blood (2013) 122(10):1707–11. doi: 10.1182/blood-2013-05-500959
18. Zoi K, Cross NC. Molecular Pathogenesis of Atypical CML, CMML and MDS/MPN-Unclassifiable. Int J Hematol (2015) 101(3):229–42. doi: 10.1007/s12185-014-1670-3
19. Wang SA, Hasserjian RP, Fox PS, Rogers HJ, Geyer JT, Chabot-Richards D, et al. Atypical Chronic Myeloid Leukemia Is Clinically Distinct From Unclassifiable Myelodysplastic/Myeloproliferative Neoplasms. Blood (2014) 123(17):2645–51. doi: 10.1182/blood-2014-02-553800
20. Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 Mutations in Atypical Chronic Myeloid Leukemia. Nat Genet (2013) 45(1):18–24. doi: 10.1038/ng.2495
21. Meggendorfer M, Bacher U, Alpermann T, Haferlach C, Kern W, Gambacorti-Passerini C, et al. SETBP1 Mutations Occur in 9% of MDS/MPN and in 4% of MPN Cases and Are Strongly Associated With Atypical CML, Monosomy 7, Isochromosome I(17)(Q10), ASXL1 and CBL Mutations. Leukemia (2013) 27(9):1852–60. doi: 10.1038/leu.2013.133
22. Gotlib J. How I Treat Atypical Chronic Myeloid Leukemia. Blood (2017) 129(7):838–45. doi: 10.1182/blood-2016-08-693630
23. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Research (2019) 47: (D1):D941–7. doi: 10.1093/nar/gky1015
24. Fontana D, Mauri M, Renso R, Docci M, Crespiatico I, Røst LM, et al. ETNK1 Mutations Induce a Mutator Phenotype That can be Reverted With Phosphoethanolamine. Nat Commun (2020) 11(1):5938. doi: 10.1038/s41467-020-19721-w
25. Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R Mutations in Chronic Neutrophilic Leukemia and Atypical CML. N Engl J Med (2013) 368(19):1781–90. doi: 10.1056/NEJMoa1214514
26. Yoda A, Morishita D, Mizutani A, Satoh Y, Ochi Y, Nannya Y, et al. CTX-712, a Novel Clk Inhibitor Targeting Myeloid Neoplasms With SRSF2 Mutation. Blood (2019) 134(Supplement_1):404.
27. Borthakur G, Popplewell L, Boyiadzis M, Foran J, Platzbecker U, Vey N, et al. Activity of the Oral Mitogen-Activated Protein Kinase Kinase Inhibitor Trametinib in RAS-Mutant Relapsed or Refractory Myeloid Malignancies. Cancer (2016) 122(12):1871–9. doi: 10.1002/cncr.29986
28. Wang L, Birch NW, Zhao Z, Nestler CM, Kazmer A, Shilati A, et al. Epigenetic Targeted Therapy of Stabilized BAP1 in ASXL1 Gain-of-Function Mutated Leukemia. Nat Cancer (2021) 2:515–26. doi: 10.1038/s43018-021-00199-4
29. Guan Y, Tiwari AD, Phillips JG, Hasipek M, Grabowski DR, Pagliura S, et al. A Therapeutic Strategy for Preferential Targeting of TET2 Mutant and TET-Dioxygenase Deficient Cells in Myeloid Neoplasms. Blood Cancer Discovery (2021) 2(2):146–61. doi: 10.1158/2643-3230.BCD-20-0173
30. Makishima H, Yoshida K, Nguyen N, Przychodzen B, Sanada M, Okuno Y, et al. Somatic SETBP1 Mutations in Myeloid Malignancies. Nat Genet (2013) 45(8):942–6. doi: 10.1038/ng.2696
31. Kennedy EP, Weiss SB. The Function of Cytidine Coenzymes in the Biosynthesis of Phospholipides. J Biol Chem (1956) 222(1):193–214. doi: 10.1016/S0021-9258(19)50785-2
32. Gambacorti-Passerini CB, Donadoni C, Parmiani A, Pirola A, Redaelli S, Signore G, et al. Recurrent ETNK1 Mutations in Atypical Chronic Myeloid Leukemia. Blood (2015) 125(3):499–503. doi: 10.1182/blood-2014-06-579466
33. Fontana D, Ramazzotti D, Aroldi A, Redaelli S, Magistroni V, Pirola A, et al. Integrated Genomic, Functional, and Prognostic Characterization of Atypical Chronic Myeloid Leukemia. Hemasphere (2020) 4(6):e497. doi: 10.1097/HS9.0000000000000497
34. Lasho TL, Finke CM, Zblewski D, Patnaik M, Ketterling RP, Chen D, et al. Novel Recurrent Mutations in Ethanolamine Kinase 1 (ETNK1) Gene in Systemic Mastocytosis With Eosinophilia and Chronic Myelomonocytic Leukemia. Blood Cancer J (2015) 5(1):e275. doi: 10.1038/bcj.2014.94
35. Zhou XA, Louissaint A Jr., Wenzel A, Yang J, Martinez-Escala ME, Moy AP, et al. Genomic Analyses Identify Recurrent Alterations in Immune Evasion Genes in Diffuse Large B-Cell Lymphoma, Leg Type. J Invest Dermatol (2018) 138(11):2365–76. doi: 10.1016/j.jid.2018.04.038
36. Langabeer SE, McCarron SL, Haslam K, O’Donovan MT, Conneally E. The CSF3R T618I Mutation as a Disease-Specific Marker of Atypical CML Post Allo-SCT. Bone Marrow Transplant (2014) 49(6):843–4. doi: 10.1038/bmt.2014.35
37. Patnaik MM, Barraco D, Lasho TL, Finke CM, Reichard K, Hoversten KP, et al. Targeted Next Generation Sequencing and Identification of Risk Factors in World Health Organization Defined Atypical Chronic Myeloid Leukemia. Am J Hematol (2017) 92(6):542–8. doi: 10.1002/ajh.24722
38. Schwartz LC, Mascarenhas J. Current and Evolving Understanding of Atypical Chronic Myeloid Leukemia. Blood Rev (2019) 33:74–81. doi: 10.1016/j.blre.2018.07.004
39. Pardanani A, Lasho TL, Laborde RR, Elliott M, Hanson CA, Knudson RA, et al. CSF3R T618I Is a Highly Prevalent and Specific Mutation in Chronic Neutrophilic Leukemia. Leukemia (2013) 27(9):1870–3. doi: 10.1038/leu.2013.122
40. Patnaik MM, Lasho TL. Genomics of Myelodysplastic Syndrome/Myeloproliferative Neoplasm Overlap Syndromes. Hematol Am Soc Hematol Educ Program (2020) 2020(1):450–9. doi: 10.1182/hematology.2020000130
41. Rohrabaugh S, Kesarwani M, Kincaid Z, Huber E, Leddonne J, Siddiqui Z, et al. Enhanced MAPK Signaling Is Essential for CSF3R-Induced Leukemia. Leukemia (2017) 31(8):1770–8. doi: 10.1038/leu.2016.376
42. Deininger MWN, Tyner JW, Solary E. Turning the Tide in Myelodysplastic/Myeloproliferative Neoplasms. Nat Rev Cancer (2017) 17(7):425–40. doi: 10.1038/nrc.2017.40
43. Onida F, de Wreede LC, van Biezen A, Eikema DJ, Byrne JL, Iori AP, et al. Allogeneic Stem Cell Transplantation in Patients With Atypical Chronic Myeloid Leukaemia: A Retrospective Study From the Chronic Malignancies Working Party of the European Society for Blood and Marrow Transplantation. Br J Haematol (2017) 177(5):759–65. doi: 10.1111/bjh.14619
44. Breccia M, Biondo F, Latagliata R, Carmosino I, Mandelli F, Alimena G. Identification of Risk Factors in Atypical Chronic Myeloid Leukemia. Haematologica (2006) 91(11):1566–8.
45. Hernández JM, del Cañizo MC, Cuneo A, García JL, Gutiérrez NC, González M, et al. Clinical, Hematological and Cytogenetic Characteristics of Atypical Chronic Myeloid Leukemia. Ann Oncol (2000) 11(4):441–4. doi: 10.1023/A:1008393002748
46. Koldehoff M, Beelen DW, Trenschel R, Steckel NK, Peceny R, Ditschkowski M, et al. Outcome of Hematopoietic Stem Cell Transplantation in Patients With Atypical Chronic Myeloid Leukemia. Bone Marrow Transplant (2004) 34(12):1047–50. doi: 10.1038/sj.bmt.1704686
47. Mittal P, Saliba RM, Giralt SA, Shahjahan M, Cohen AI, Karandish S, et al. Allogeneic Transplantation: A Therapeutic Option for Myelofibrosis, Chronic Myelomonocytic Leukemia and Philadelphia-Negative/BCR-ABL-Negative Chronic Myelogenous Leukemia. Bone Marrow Transplant (2004) 33(10):1005–9. doi: 10.1038/sj.bmt.1704472
48. Itonaga H, Ota S, Ikeda T, Taji H, Amano I, Hasegawa Y, et al. Allogeneic Hematopoietic Stem Cell Transplantation for the Treatment of BCR-ABL1-Negative Atypical Chronic Myeloid Leukemia and Chronic Neutrophil Leukemia: A Retrospective Nationwide Study in Japan. Leuk Res (2018) 75:50–7. doi: 10.1016/j.leukres.2018.11.003
49. Langabeer SE, Comerford CM, Quinn J, Murphy PT. Molecular Profiling and Targeted Inhibitor Therapy in Atypical Chronic Myeloid Leukaemia in Blast Crisis. J Clin Pathol (2017) 70(12):1089. doi: 10.1136/jclinpath-2017-204621
50. Kantarjian HM, O’Brien S, Cortes J, Giles FJ, Faderl S, Issa JP, et al. Results of Decitabine (5-Aza-2’deoxycytidine) Therapy in 130 Patients With Chronic Myelogenous Leukemia. Cancer (2003) 98(3):522–8. doi: 10.1002/cncr.11543
51. Onida F, Ball G, Kantarjian HM, Smith TL, Glassman A, Albitar M, et al. Characteristics and Outcome of Patients With Philadelphia Chromosome Negative, Bcr/Abl Negative Chronic Myelogenous Leukemia. Cancer (2002) 95(8):1673–84. doi: 10.1002/cncr.10832
52. Tong X, Li J, Zhou Z, Zheng D, Liu J, Su C. Efficacy and Side-Effects of Decitabine in Treatment of Atypical Chronic Myeloid Leukemia. Leuk Lymphoma (2015) 56(6):1911–3. doi: 10.3109/10428194.2014.986477
53. Fleischman AG, Maxson JE, Luty SB, Agarwal A, Royer LR, Abel ML, et al. The CSF3R T618I Mutation Causes a Lethal Neutrophilic Neoplasia in Mice That Is Responsive to Therapeutic JAK Inhibition. Blood (2013) 122(22):3628–31. doi: 10.1182/blood-2013-06-509976
54. Dao KT, Gotlib J, Deininger MMN, Oh ST, Cortes JE, Collins RH Jr., et al. Efficacy of Ruxolitinib in Patients With Chronic Neutrophilic Leukemia and Atypical Chronic Myeloid Leukemia. J Clin Oncol (2020) 38(10):1006–18. doi: 10.1200/JCO.19.00895
55. Burgess MR, Hwang E, Firestone AJ, Huang T, Xu J, Zuber J, et al. Preclinical Efficacy of MEK Inhibition in Nras-Mutant AML. Blood (2014) 124(26):3947–55. doi: 10.1182/blood-2014-05-574582
56. Khanna V, Pierce ST, Dao KH, Tognon CE, Hunt DE, Junio B, et al. Durable Disease Control With MEK Inhibition in a Patient With NRAS-Mutated Atypical Chronic Myeloid Leukemia. Cureus (2015) 7(12):e414. doi: 10.7759/cureus.414
57. Costello R, Lafage M, Toiron Y, Brunel V, Sainty D, Arnoulet C, et al. Philadelphia Chromosome-Negative Chronic Myeloid Leukaemia: A Report of 14 New Cases. Br J Haematol (1995) 90(2):346–52. doi: 10.1111/j.1365-2141.1995.tb05157.x
58. Kurzrock R, Bueso-Ramos CE, Kantarjian H, Freireich E, Tucker SL, Siciliano M, et al. BCR Rearrangement-Negative Chronic Myelogenous Leukemia Revisited. J Clin Oncol (2001) 19(11):2915–26. doi: 10.1200/JCO.2001.19.11.2915
59. Jabbour E, Kantarjian H, Cortes J, Thomas D, Garcia-Manero G, Ferrajoli A, et al. PEG-IFN-Alpha-2b Therapy in BCR-ABL-Negative Myeloproliferative Disorders: Final Result of a Phase 2 Study. Cancer (2007) 110(9):2012–8. doi: 10.1002/cncr.23018
Keywords: atypical chronic myelogenous leukemia, myelodysplastic syndrome/myeloproliferative neoplasm, diagnostic criteria, treatment, molecular characterization
Citation: Diamantopoulos PT and Viniou N-A (2021) Atypical Chronic Myelogenous Leukemia, BCR-ABL1 Negative: Diagnostic Criteria and Treatment Approaches. Front. Oncol. 11:722507. doi: 10.3389/fonc.2021.722507
Received: 08 June 2021; Accepted: 02 November 2021;
Published: 17 November 2021.
Edited by:
Argiris Symeonidis, University of Patras, GreeceReviewed by:
Deepshi Thakral, All India Institute of Medical Sciences, IndiaNicholas Cross, University of Southampton, United Kingdom
Copyright © 2021 Diamantopoulos and Viniou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiotis T. Diamantopoulos, cGFuZGlhbWFudG9wb3Vsb3NAZ21haWwuY29t