 Dev Karan
Dev Karan- Department of Pathology, Medical College of Wisconsin, Milwaukee, WI, United States
Endoplasmic reticulum (ER) stress is a cellular process in response to stress stimuli in protecting functional activities. However, sustained hyperactive ER stress influences tumor growth and development. Hepatocytes are enriched with ER and highly susceptible to ER perturbations and stress, which contribute to immunosuppression and the development of aggressive and drug-resistant hepatocellular carcinoma (HCC). ER stress-induced inflammation and tumor-derived chemokines influence the immune cell composition at the tumor site. Consequently, a decrease in the CCL23 chemokine in hepatic tumors is associated with poor survival of HCC patients and could be a mechanism hepatic tumor cells use to evade the immune system. This article describes the prospective role of CCL23 in alleviating ER stress and its impact on the HCC tumor microenvironment in promoting antitumor immunity. Moreover, approaches to reactivate CCL23 combined with immune checkpoint blockade or chemotherapy drugs may provide novel opportunities to target hepatocellular carcinoma.
Introduction
Hepatocellular carcinoma (HCC) is a basic form of liver malignancy and is the leading cause of liver cancer deaths worldwide. In 2021, approximately 43 000 new cases will be diagnosed, with an estimated 30 000 deaths in the United States (1). Like with any other cancer type, HCC development is a multistep process involving hepatic cell injury, chronic infection, fibrosis, cirrhosis, and finally, liver cancer. Understanding this process has helped identify new genetic/molecular drivers and develop treatment approaches. Thus far, the five-year survival expectancy rate for liver cancer is only ~18% and is further reduced to ~15% in black patients (1). Treatment with chemotherapy drugs has low efficacy due to the emergence of drug-resistance disease. For advanced HCC, systemic therapy with sorafenib, lenvatinib, or regorafenib delivers limited survival benefits (2, 3). Unfortunately, liver cancer incidence has increased rapidly (2 to 3% annually) in the United States, with a ~43% increase in HCC-related mortality over the past two decades. Besides, a projected surge in death rates of ~57.6% (6.6 to 10.4) from 2015 to 2035 is very alarming (2). Thus, understanding the biological process of HCC will help to create new and effective treatment opportunities.
As a vital organ, the liver tissue engages in essential physiological activities, adding new challenges to understanding liver oncogenesis. However, based on evolutionary history and the process of HCC development, the most common genetic and epigenetic alterations include mutations in the TERT promoter, TP53, β-catenin, AXIN1, ARID1A, ARID2, CDKN2A, and CCND1 genes (4–6). Here, we describe a novel perspective of a CC-chemokine ligand (CCL23) in context with HCC tumor development and its potential in mitigating the ER stress leading to remodeling the HCC tumor microenvironment in favor of enhanced antitumor immunity.
Functional Analysis of CCL23 in Cancer and Non-Cancerous Diseases
In general, chemokines contribute to a miscellany of biological activities during the process of HCC development, helping tumor cells evade the immune system and tumor angiogenesis, invasion, and metastasis (7). CCL23, also known as myeloid progenitor inhibitory factor-1, suppresses the production and release of polymorphonuclear leukocytes and hematopoietic progenitor cells from the bone marrow (8, 9). CCL23 is considered a relatively new chemokine, and its role in tumor cell progression, metastasis, and other disease conditions is not well characterized, instead primarily limited to correlated expression analysis. Several disease types – including acute myeloid leukemia, ischemic strokes, coronary atherosclerosis, chronic kidney disease, and systemic mastocytosis – have indicated expression of CCL23 correlated with disease progression (10–14). A few studies on cancer types have primarily demonstrated CCL23 gene expression in tissues without much attention to biological or molecular functions. For example, a high throughput genomic data analysis in ovarian cancer revealed CCL23 as one of the candidate genes associated with ovarian cancer (15). Colorectal studies displayed somewhat ambiguous observations. A protein-based array of 507 targets in six samples of colorectal patients revealed an upregulation of CCL23 in rectal cancer compared to non-rectal cancer (16). Conversely, inflammatory gene expression analysis in colorectal patients showed a reduced level of CCL23 in adenoma and adenocarcinoma than in normal mucosa (17). Furthermore, a meta-analysis of 1577 breast cancer patients from the Oncomine datasets revealed a negative correlation of CCL23 and its receptor CCR1 with metastasis-free survival. However, this association was limited to HER2+ breast cancer patients and did not persist in HER2- patients or other subtypes (18).
The mechanistic and functional studies on CCL23 are hampered because, as such, there is no defined murine CCL23. However, genomic analysis of CC chemokines suggested that the chemokine ligand CCL6 is the mouse homolog of the human CCL23 (19). Studies in a BCR-ABL-induced leukemia murine model showed that CCL6 was required for an interferon consensus sequence-binding protein (ICSBP)-induced, vaccine-like immunoprotective effect. Normal cells expressed high levels of CCL6 compared to leukemic cells, but IFN-α treatment could reactivate the expression of CCL6 in leukemic cells. In this mouse model, shRNA-directed CCL6 knockdown in mouse tumor cells abrogated the anti-leukemic survival response generated by ICSBP, suggesting the role of CCL6 as a potential immunomodulatory chemokine (20). Clinical extension of these studies revealed that chronic myeloid leukemia (CML) patients who responded to IFN-α treatment showed higher levels of CCL23 compared to non-responders. Similarly, increased CCL23 was observed in patients with multiple myeloma, melanoma, and renal cell carcinoma after exposure to IFN-α (20).
Mechanistic studies in mouse models further demonstrated that CCL6 promotes innate immunity via immune cell activation and serves as a chemoattractant for CD11b+, IFN-producing dendritic cells, NK cells, and CD4+ T cells (20, 21). Therefore, decreasing CCL23 expression in leukemic cells could be a mechanism for CML cells to escape from the immune system. In addition, an in vitro study on freshly prepared eosinophils from human PBMCs showed a slight increase in CCL23 mRNA following 6 hours of IFN-α treatment (22).
In a breast cancer mouse model, a protein array analysis in the lungs of 4T1 tumor-bearing mice showed a higher level of CCL6 along with IL-33, CCL12, CCL17, and MMP-9 than in normal mice. Treatment with Cordyceps sinensis (a Chinese herb) reduced 4T1 tumor cell metastasis to the lungs associated with a decrease in IL-33, CCL17, and MMP-9 (23). However, the level of CCL6 chemokine remains unaltered and warrant further investigation for its role in the anti-metastatic activity of cancer cells.
CCL23 in Hepatocellular Carcinoma
The Cancer Genome Atlas (TCGA) data sets have significantly helped in mining the genomic landscape of human diseases and in elucidating the role of the tumor microenvironment (TME) both from the perception of tumor cells and molecular subtyping of tumor-infiltrating lymphocytes (24). Nonetheless, TCGA data sets continue to offer many opportunities in identifying new molecular drivers, mutational analyses, gene signaling pathways, and diagnostic/prognostic biomarkers for various cancer types. We extracted the mRNA expression profile from a TCGA data set (TCGA Liver Cancer) for normal tissues and primary tumors to elucidate the potential role of CCL23 in association with the immunobiology of hepatocellular carcinoma (25). Analysis of the data set from the cohort TCGA Liver Cancer showed a significantly lower (p = 0.0001) expression of the CCL23 transcript in HCC compared to normal liver tissue (Figure 1A). Extension of this analysis to the Oncomine data set also revealed significant downregulation (-2.15 fold; p = 0.0004) of CCL23 in HCC compared to the normal liver (Figure 1B) (26). In support of these observations, we examined the Human Protein Atlas resources for CCL23 protein expression (v20.proteinatlas.org/ENSG00000274736-CCL23/pathology/liver+cancer). The immunohistochemistry (IHC) data revealed a low CCL23 protein level in HCC compared to the normal liver (Figure 1C). This data set comprises 12 HCC samples; CCL23 protein was not detectable in eight (8/12), showed low expression levels in three (3/12), and displayed a moderate level of CCL23 protein expression in one (1/12). In these small numbers of samples from the protein atlas data set, we also noticed that CCL23 protein expression in the normal liver of females was comparatively higher than males. These observations suggest a lower level of CCL23 in HCC at both the mRNA and protein levels than normal liver tissue.
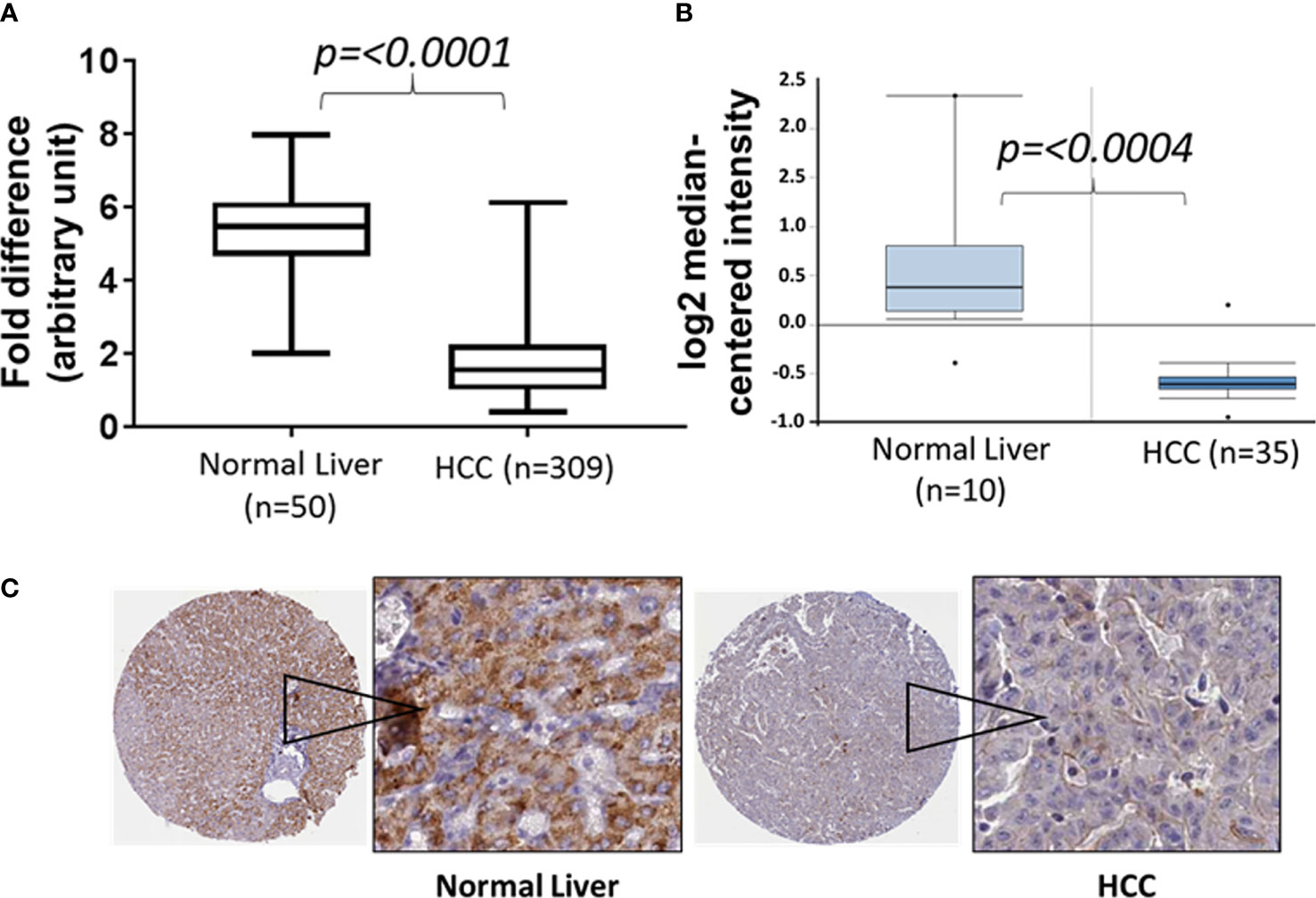
Figure 1 Box and Whisker plots of CCL23 transcript expression levels from two different data sets (A: TCGA liver cancer and B: Oncomine) and (C) the expression of CCL23 protein by immunohistochemistry data sets from the Human Protein Atlas (proteinatlas.org) in the normal liver and hepatocellular carcinoma (HCC). The expression level of CCL23 is significantly lower in HCC as compared to the normal liver.
We further selected mRNA (RNA-seq) liver cancer data sets from Kaplan Meier (KM) Plotter for the pan-cancer and examined the correlation between CCL23 mRNA and the survival probability (27). The analysis includes all stages, grades, both sex and race, and risk factors. The cutoff values used in the analysis are based on auto best cutoff values. KM Plotter analysis revealed a significantly poor (p = 6.4x10-10) prognosis in HCC patients having low levels of CCL23, with a median survival of 27.57 months vs. 82.87 months (Figure 2A). However, the expression of CCL23 receptor CCR1 revealed an opposite trend – high CCR1 expression was associated with shorter survival in HCC patients (Figure 2B). This seemingly conflicting observation for CCR1 is not surprising in light of the ambiguity regarding the functional activities of chemokine receptors. CCR1 serves as a receptor for multiple chemokines and may not necessarily follow a similar functional correlation as CCL23.
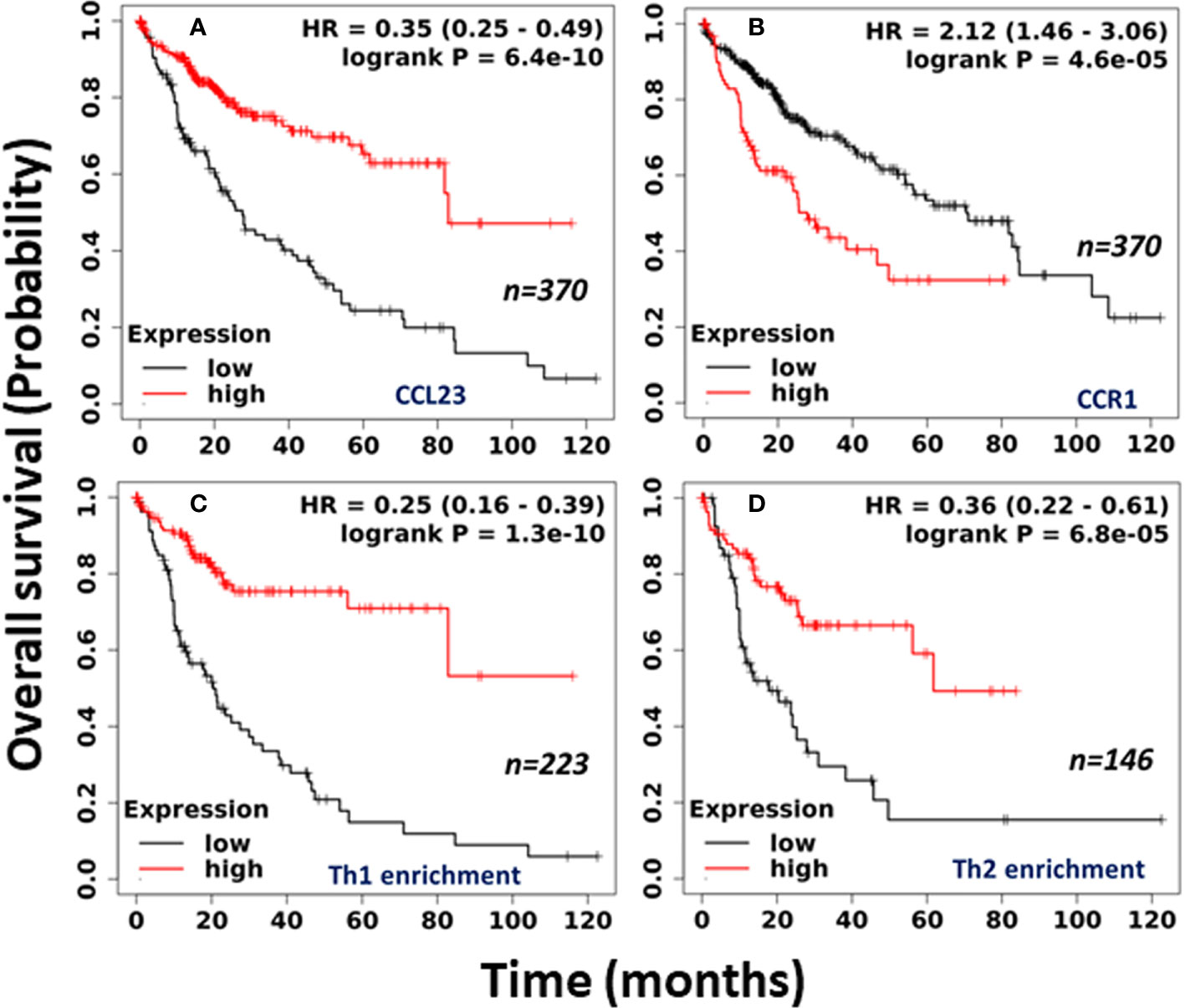
Figure 2 Clinical association of (A) CCL23; (B) CCR1; (C) Th1 cell enrichment; and (D) Th2 cell enrichment in human HCC tissues with overall survival. (Data source: Kaplan-Meier Plotter, Pan-cancer RNA-seq).
To understand the clinical significance between the low level of CCL23 and the infiltrated immune cells, we further examined the immune cell composition within the HCC tissues and its impact on overall survival. Surprisingly, in association with low levels of CCL23, enrichment of the human HCC tissues with type 1 T-helper (Th1) cells revealed significantly shorter (p = 1.3x10-10) median survival of 9.3 months vs. 56.17 months in HCC patients (Figure 2C). Enrichment with type 2 T-helper (Th2) cells also showed significantly reduced (p = 6.8x10-5) overall survival with low levels of CCL23, but the median survival was better than the Th1 cell enrichment (17.83 months vs. 61.73 months; Figure 2D). Furthermore, mechanistic studies on the role of T-helper cells showed that liver Th1 cells might drive the infiltration of myeloid-derived suppressor cells (MDSCs) in the inflamed TGF-β1 knockout mouse liver, leading to diminished survival (28).
On the other hand, Th2 cytokines IL-4 and IL-13 regulate CCL23 expression in CD14+ monocytes (29). Human neutrophils, which do not produce CCL23 in response to Th2 cytokines, express and release CCL23 upon stimulation with toll-like receptor agonists (Resiquimod and LPS) or TNF-α, suggesting the role of CCL23 in driving the recruitment of immune cells at the inflamed site in favor of a controlled immune response (30). Although the infiltration of Th1 immune cells in the HCC TME is linked with a favorable clinical response (31), in the absence of CCL23, Th1 cells may not exert cytolytic function. Also, the operational support of Th1 cells may be outweighed due to an anergic or exhausted cytotoxic immune cells ecosystem within the HCC tumor microenvironment due to a low level of CCL23. However, additional studies are essential to validate the infiltration of Th1 and Th2 immune cells in HCC tissue, predicting the survival benefit associated with the loss of CCL23.
Lu et al. performed bioinformatics analysis of CCL23 in human HCC tissues from multiple online databases (GEPIA, HCCDB, MetaScape, TIMER, TISIBD, and KM Plotter) and revealed low expression of CCL23 in all stages of HCC in association with poor prognosis (32). The gene ontology function and Kyoto gene and genome encyclopedia pathway of CCL23 co-expressed gene in HCC were enriched in immune cells and mainly associated with CD8+ T cells and macrophage activation (32). Jia-Jie et al. analyzed CCL23 expression in liver cancer and adjacent normal liver tissues from 196 cases of radical hepatectomy by real-time fluorescence quantitative-PCR and 82 HCC tissues with matched cancer and adjacent normal tissues by IHC. The multivariate Cox regression analysis revealed a significantly lower expression of CCL23 in liver cancer tissues compared to adjacent normal. The liver cancer patients with higher CCL23 expression showed better survival than those with low CCL23 (33).
Collectively, these observations suggest a critical role of CCL23 in HCC. The presence of CCL23 may help create a tumor-suppressive environment by recruiting leukocytes to the tumor site and postulate that the loss of CCL23 serves as a driver in the oncogenesis of hepatic tumor cells. Therefore, determining the onco-immunologic function of CCL23 may help to understand the process of HCC development better.
ER Stress and Immune Suppression in HCC
The endoplasmic reticulum is a vital cell organelle engaging in multiple physiological functions, including protein folding and transport of the synthesized proteins. During the process of tumor development, ER stress leads to the activation of the unfolded protein response (UPR), distressing cellular metabolic activities regulating various intracellular functions, including protein folding, calcium homeostasis, lipid metabolism, cell differentiation, and protein translocation (34, 35). UPR is an adaptive cellular mechanism to counteract the accumulated protein misfolding stress, ultimately evolving anti-apoptotic and drug-resistance machinery of tumor cells. In the absence of stress, ER chaperone glucose‐regulated protein 78 (GRP78) binds to UPR and sequesters the UPR sensors protein kinase R-like ER kinase (PERK), inositol‐requiring enzyme 1α (IRE1α), and transcription factor 6 (ATF6).
Various studies document that ER stress and UPR signaling pathways contribute to nearly all forms of acute and chronic liver diseases and promote HCC cell survival, proliferation, and angiogenesis (36–38). The expression levels of UPR signaling proteins IRE1α, XBP1s, PERK/ATF4, CHOP, and ATF6 are increased in HCC model systems and are associated with HCC growth and development (39–41). Interestingly, UPR kinetic studies in HCC revealed that IRE1α is activated during tumor initiation and the PERK pathway during tumor progression, while ATF6 is only moderately activated in developed tumors (39). HCC biopsies from human patients showed elevated XBP1 expression levels, whereas cancer cells deficient in XBP1 are less prone to developing solid tumors in nude mice (42). In an orthotopic mouse model of HCC, the PERK inhibitor significantly reduces the tumor burden by killing ER-stressed HCC cells (39). Similarly, liver-specific knockout IRE1α mice fed with a regular diet showed low diethylnitrosamine-induced HCC and suppressed HCC progression in mice fed with a high-fat diet. The tumor growth inhibition was associated with decreased hepatocyte proliferation, STAT3 activation, and reduced tumor-promoting inflammatory cytokines TNF-α and IL-6 (40).
An increased in GRP78 expression level is reported as a pro-survival factor for cells undergoing ER stress. In HCC tumors from patients treated with sorafenib, 73% showed high GRP78 expression, which was associated with the shortest progression-free survival (41). Elevated GRP78 is also proposed as a predictive biomarker in HCC patients treated with sorafenib (43). Moreover, GRP78 is linked with activation of the Wnt/catenin pathway in HCC (44). Higher GRP78 is positively correlated with Golgi protein 73 (GP73) and a high density of tumor-associated macrophages displaying CD206 expression leading to poor prognosis in HCC patients (45). The increased expression level of CD147 in HCC also serves as a UPR inducer, promoting ER stress and HCC cell survival, decreasing the efficacy of the chemotherapy drug adriamycin in animal studies (37, 46). Indeed, chemotherapy drug-induced cellular stress in cancer cells leading to adaptation, and drug-resistant cancer cells occur frequently in HCC.
Hepatocellular carcinoma is inflammation-associated cancer and usually progresses on the pretext of inflammation in the liver (47). The chronically inflamed tumor microenvironment is characterized by a high degree of ER stress, activating UPR signaling, and immune suppression within tumors (48, 49). Any of the UPR sensors can trigger NF-κB-inducing, tumor-promoting, pro-inflammatory cascade implicated in macrophage activation and TNF-α, IL-6, IL-1β, and IL-8 cytokines production (50–52). Similarly, vitamin D receptor deficiency leads to persistent UPR activation, promotes hepatic macrophage infiltration, and produces pro-inflammatory cytokines (53). In addition, inflammatory mediators can maintain and augment ER stress in the inflamed tissues, providing a feedback mechanism, attributing to the plasticity of macrophage polarization at the tumor site. Altogether, UPR is known to associate with cancer initiation, tumor cell quiescence and aggressiveness, EMT, angiogenesis, autophagy, and a metabolic switch in cancer cells to adapt to the challenging, stressful TME and supporting immunosuppression (37, 49, 54).
The degree of tumor-infiltrating lymphocytes in HCC is closely associated with tumor recurrence. High levels of MDSCs and T-regulatory cells in patients are reportedly related to aggressive HCC and low survival rates (55–57). Patients infected with the hepatitis-C or hepatitis-B virus accumulate T-regulatory cells in HCC tumor tissues involving TGF-β signaling (58). There are multiple mechanisms linked to the increased association of immune suppressor cells in HCC. However, emerging evidence suggests that ER stress promotes the escape of tumor cells from immune surveillance and hampers antitumor immunity through the regulation of immunosuppression (54, 59–61). UPR and the increased level of inflammatory cytokines by tumor cells could be transmitted into bone marrow-derived dendritic cells – such distressed DCs exhibit impaired antigen presentation and cross-priming CD8+ T cell, supporting enhanced tumor growth in mice (62). Increased ER stress-related proteins were also correlated with M2 macrophage recruitment and PD‐L1 expression in HCC tissues (63). Targeting MDSCs has been shown to enhance the therapeutic efficacy of sorafenib and immune checkpoint inhibitors in HCC murine models. Reversing the pro-tumor effects of MDSCs could be achieved by depleting MDSCs, blocking MDSC trafficking into TME (64, 65). CCL23 functionally contributes to the modulation of the immune response by promoting leucocyte trafficking as well as directing the migration of monocytes, macrophages, dendritic cells, and T lymphocytes to the sites of injury or infection (66, 67). Therefore, manipulation with CCL23 may help reprogram the HCC TME by targeting myeloid checkpoints to harness the power of antitumor immunity with immune checkpoint inhibitors or other forms of immunotherapy approaches.
CCL23-Directed ER Stress Mitigation
A high level of ER stress is closely linked to immune responses and activates pro-inflammatory cytokines/chemokines. For example, increased CXCL10 (IP10) following liver graft injury leads to ER stress-associated cisplatin-resistant HCC, and the neutralization of CXCL10 sensitizes cisplatin treatment, which suppresses tumor growth (68). A study in type 2 diabetic mouse model showed that inhibiting chemokine receptor 2 (receptor for CCL2) improves hepatic steatosis by reducing ER stress and inflammation (69). Additionally, visfatin-induced hepatic inflammation in a mouse model of methionine-choline-deficient diet showed increased CXCL2, CXCL8, and MCP-1 associated with ER stress upregulation (70). On the other hand, in a breast cancer study, tunicamycin-activated ER stress increases the endogenous level of CCL5 mRNA and protein and prevents MCF-7 cells migration (71). This study supports our observations that approaches to reactivate endogenous CCL23 may help repress hepatic tumor cell growth associated with reduced ER stress.
A bovine endometrial cell model showed that tunicamycin (an antibiotic)-induced ER stress inhibits epithelial cell proliferation (72). Addition of CCL23 rescued cell proliferation by activating PI3K/AKT and MAPK signaling while simultaneously reducing the expression levels of UPR-signaling proteins, including PERK, IRE1α, and ATF6. Moreover, CCL23 inhibited the expression of LPS-induced, pro-inflammatory cytokines TNF-α, IL-6, and IL-8 and restored intracellular Ca2+ levels required for optimal ER functioning. This observation corroborates with the HCC growth inhibition in liver-specific knockout IRE1α mice. In addition, CCL23 treatment restored the basal level of tunicamycin-induced downstream molecules (eIF2α and CHOP) and the ER chaperone protein (GRP78). A similar phenomenon of CCL23-induced ER stress alleviation was observed in porcine endometrial luminal epithelial cells (73). Thus, the CCL23 chemokine likely has a significant potential to alleviate ER stress by rescuing the UPR-sensors and reducing the pro-inflammatory cytokine profile, diminishing the immunosuppressive HCC tumor microenvironment and enhancing antitumor immunity.
Conclusions and Future Perspectives
ER stress is known to impair the immune cell function, promote MDSCs, T-regulatory cells, and M2 macrophages with an overall impact on immune evasion. Even though HCC is a deeply enriched site of immune cell engagement, the commonly used immune checkpoint inhibitor-based therapies showed limited success. ER stress or viral infection is associated with increased PD-L1 expression in HCC tissues, yet the PD-1 inhibitor nivolumab showed a ~15% response rate in HCC while the CTLA4 blockade showed a relatively lesser objective response (74–76).
This review accentuates the observations that low expression of CCL23 in HCC tissues is associated with the poor prognosis of HCC patients. Therefore, to understand the significance of CCL23 in HCC oncogenesis, we hypothesize two potential scenarios: 1) CCL23 might play a vital role in reducing ER stress and help recruit macrophages and dendritic cells, augmenting antitumor immunity; and 2) expression of CCL23 may serve as a tumor suppressor by inhibiting the invasive features (such as hepatic tumor cell invasion and migration). These molecular events will ultimately influence the TME and may rejuvenate immune cell functions by reducing the function and frequency of immune suppressor cells (Figure 3). Following CCL23-directed reprogramming of the TME, HCC may be amenable to immune-based therapies, including checkpoint blockade inhibitors to enhance the therapeutic efficacy. For example, since IFN-α induces antitumor immunity via CCL23 reactivation in humans, a combinatorial approach of IFN-α and checkpoint blockade inhibitors may provide a novel opportunity targeting hepatocellular carcinoma (Figure 3). These observations may inspire a new direction of research investigating the cross-talk between ER stress and immunomodulation in the HCC tumor microenvironment using CCL23 as a molecular target. Preclinical validations of this potential concept may provide much-needed information to improve clinical management of hepatocellular carcinoma.
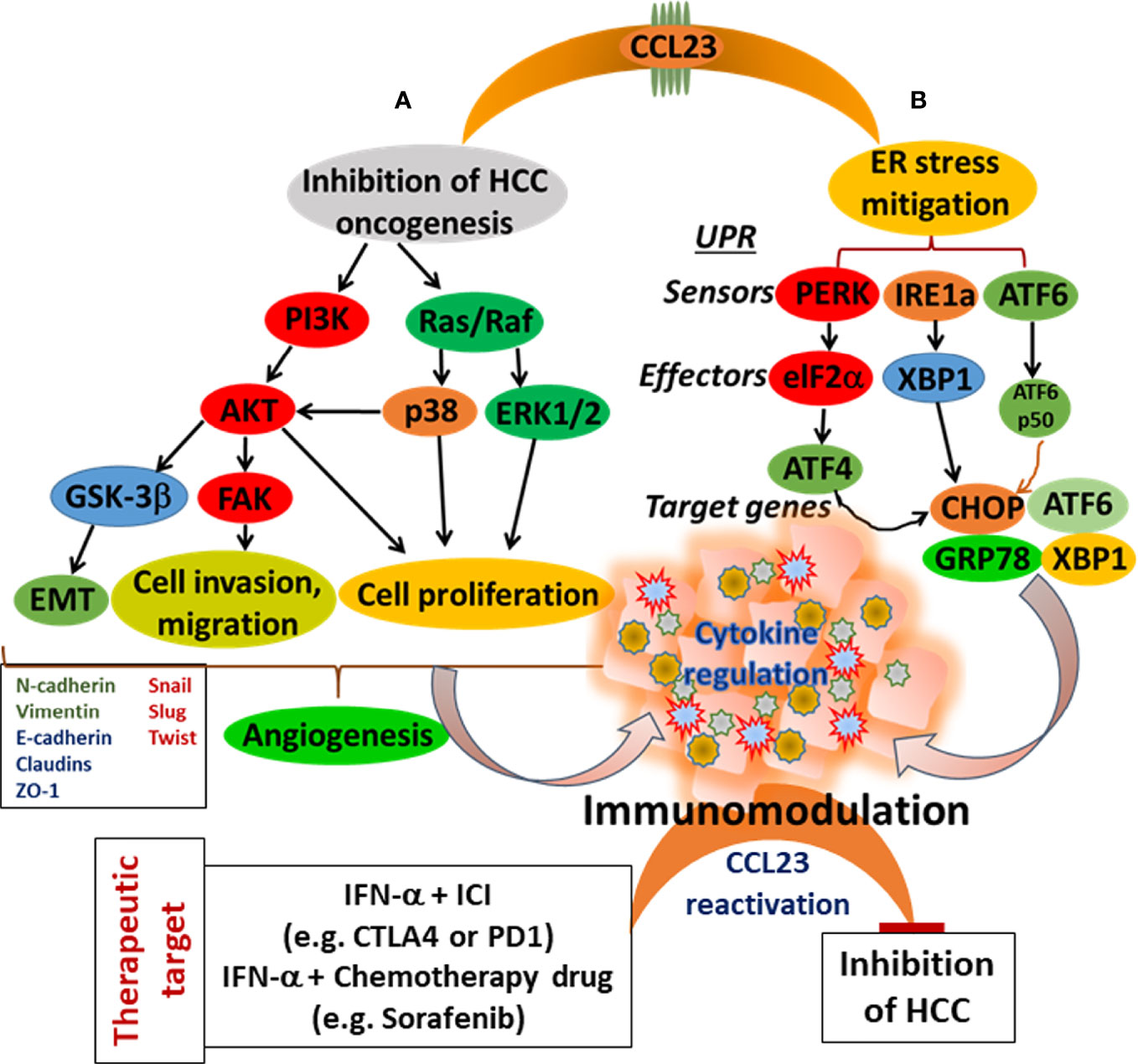
Figure 3 Schematic presentation of CCL23 functions for the modulation of the HCC tumor microenvironment. (A) Targeting cellular signaling cascade inhibiting HCC cell progression, and (B) Mitigating of ER stress. Both events lead to immunomodulation with a potential to enhance antitumor immunity.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The idea for this article emanated from the studies funded by the NIH/NCI (CA204786). We thank Dr. Seema Dubey for her help in preparing Figure 3 schemes.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654
2. Ma J, Jemal A, Fedewa SA, Islami F, Lichtenfeld JL, Wender RC, et al. The American Cancer Society 2035 Challenge Goal on Cancer Mortality Reduction. CA Cancer J Clin (2019) 69:351–62. doi: 10.3322/caac.21564
3. Forner A, Reig M, Bruix J. Hepatocellular Carcinoma. Lancet (2018) 391:1301–14. doi: 10.1016/S0140-6736(18)30010-2
4. Villanueva A. Hepatocellular Carcinoma. N Engl J Med (2019) 380:1450–62. doi: 10.1056/NEJMra1713263
5. Khemlina G, Ikeda S, Kurzrock R. The Biology of Hepatocellular Carcinoma: Implications for Genomic and Immune Therapies. Mol Cancer (2017) 16:149. doi: 10.1186/s12943-017-0712-x
6. Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, Villanueva A. Tumour Evolution in Hepatocellular Carcinoma. Nat Rev Gastroenterol Hepatol (2020) 17:139–52. doi: 10.1038/s41575-019-0229-4
7. Ehling J, Tacke F. Role of Chemokine Pathways in Hepatobiliary Cancer. Cancer Lett (2016) 379:173–83. doi: 10.1016/j.canlet.2015.06.017
8. Shih CH, van Eeden SF, Goto Y, Hogg JC. CCL23/myeloid Progenitor Inhibitory Factor-1 Inhibits Production and Release of Polymorphonuclear Leukocytes and Monocytes From the Bone Marrow. Exp Hematol (2005) 33:1101–8. doi: 10.1016/j.exphem.2005.06.028
9. Patel VP, Kreider BL, Li Y, Li H, Leung K, Salcedo T, et al. Molecular and Functional Characterization of Two Novel Human C-C Chemokines as Inhibitors of Two Distinct Classes of Myeloid Progenitors. J Exp Med (1997) 185:1163–72. doi: 10.1084/jem.185.7.1163
10. Castillo L, Rohatgi A, Ayers CR, Owens AW, Das SR, Khera A, et al. Associations of Four Circulating Chemokines With Multiple Atherosclerosis Phenotypes in a Large Population-Based Sample: Results From the Dallas Heart Study. J Interferon Cytokine Res (2010) 30:339–47. doi: 10.1089/jir.2009.0045
11. Pawlak K, Mysliwiec M, Pawlak D. Effect of Diabetes and Oxidative Stress on Plasma CCL23 Levels in Patients With Severe Chronic Kidney Disease. Pol Arch Med Wewn (2014) 124:459–66. doi: 10.20452/pamw.2405
12. Mayado A, Teodosio C, Garcia-Montero AC, Matito A, Rodriguez-Caballero A, Morgado JM, et al. Increased IL6 Plasma Levels in Indolent Systemic Mastocytosis Patients Are Associated With High Risk of Disease Progression. Leukemia (2016) 30:124–30. doi: 10.1038/leu.2015.176
13. Bonaventura A, Montecucco F. CCL23 Is a Promising Biomarker of Injury in Patients With Ischaemic Stroke. J Intern Med (2018) 283:476–8. doi: 10.1111/joim.12742
14. Simats A, Garcia-Berrocoso T, Penalba A, Giralt D, Llovera G, Jiang Y, et al. CCL23: A New CC Chemokine Involved in Human Brain Damage. J Intern Med (2018) 283:461–75. doi: 10.1111/joim.12738
15. Lu X, Lu J, Liao B, Li X, Qian X, Li K. Driver Pattern Identification Over the Gene Co-Expression of Drug Response in Ovarian Cancer by Integrating High Throughput Genomics Data. Sci Rep (2017) 7:16188. doi: 10.1038/s41598-017-16286-5
16. Myoshi H, Morishita A, Tani J, Sakamoto T, Fujita K, Katsura A, et al. Expression Profiles of 507 Proteins From a Biotin Label-Based Antibody Array in Human Colorectal Cancer. Oncol Rep (2014) 31:1277–81. doi: 10.3892/or.2013.2935
17. McLean MH, Murray GI, Stewart KN, Norrie G, Mayer C, Hold GL, et al. The Inflammatory Microenvironment in Colorectal Neoplasia. PLoS One (2011) 6:e15366. doi: 10.1371/journal.pone.0015366
18. Yan HH, Jiang J, Pang Y, Achyut BR, Lizardo M, Liang X, et al. CCL9 Induced by TGFbeta Signaling in Myeloid Cells Enhances Tumor Cell Survival in the Premetastatic Organ. Cancer Res (2015) 75:5283–98. doi: 10.1158/0008-5472.CAN-15-2282-T
19. Zlotnik A, Yoshie O. The Chemokine Superfamily Revisited. Immunity (2012) 36:705–16. doi: 10.1016/j.immuni.2012.05.008
20. Nardi V, Naveiras O, Azam M, Daley GQ. ICSBP-Mediated Immune Protection Against BCR-ABL-Induced Leukemia Requires the CCL6 and CCL9 Chemokines. Blood (2009) 113:3813–20. doi: 10.1182/blood-2008-07-167189
21. Coelho AL, Schaller MA, Benjamim CF, Orlofsky AZ, Hogaboam CM, Kunkel SL. The Chemokine CCL6 Promotes Innate Immunity via Immune Cell Activation and Recruitment. J Immunol (2007) 179:5474–82. doi: 10.4049/jimmunol.179.8.5474
22. Matsumoto K, Fukuda S, Hashimoto N, Saito H. Human Eosinophils Produce and Release a Novel Chemokine, CCL23, In Vitro. Int Arch Allergy Immunol (2011) 155(Suppl 1):34–9. doi: 10.1159/000327263
23. Cai H, Li J, Gu B, Xiao Y, Chen R, Liu X, et al. Extracts of Cordyceps Sinensis Inhibit Breast Cancer Cell Metastasis via Down-Regulation of Metastasis-Related Cytokines Expression. J Ethnopharmacol (2018) 214:106–12. doi: 10.1016/j.jep.2017.12.012
24. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity (2018) 48:812–30.e814. doi: 10.1016/j.immuni.2018.03.023
25. Goldman MJ, Craft B, Hastie M, Repecka K, McDade F, Kamath A, et al. Visualizing and Interpreting Cancer Genomics Data via the Xena Platform. Nat Biotechnol (2020) 38:675–8. doi: 10.1038/s41587-020-0546-8
26. Rhodes DR, Yu J, Shanker K. Deshpande N, Varambally R, Ghosh D, et al. ONCOMINE: A Cancer Microarray Database and Integrated Data-Mining Platform. Neoplasia (2004) 6:1–6. doi: 10.1016/s1476-5586(04)80047-2
27. Nagy A, Munkacsy G, Gyorffy B. Pancancer Survival Analysis of Cancer Hallmark Genes. Sci Rep (2021) 11:6047. doi: 10.1038/s41598-021-84787-5
28. Cripps JG, Wang J, Maria A, Blumenthal I, Gorham JD. Type 1 T Helper Cells Induce the Accumulation of Myeloid-Derived Suppressor Cells in the Inflamed Tgfb1 Knockout Mouse Liver. Hepatology (2010) 52:1350–9. doi: 10.1002/hep.23841
29. Novak H, Muller A, Harrer N, Gunther C, Carballido JM, Woisetschlager M. CCL23 Expression Is Induced by IL-4 in a STAT6-Dependent Fashion. J Immunol (2007) 178:4335–41. doi: 10.4049/jimmunol.178.7.4335
30. Arruda-Silva F, Bianchetto-Aguilera F, Gasperini S, Polletti S, Cosentino E, Tamassia N, et al. Human Neutrophils Produce CCL23 in Response to Various TLR-Agonists and TNFalpha. Front Cell Infect Microbiol (2017) 7:176. doi: 10.3389/fcimb.2017.00176
31. Foerster F, Hess M, Gerhold-Ay A, Marquardt JU, Becker D, Galle PR, et al. The Immune Contexture of Hepatocellular Carcinoma Predicts Clinical Outcome. Sci Rep (2018) 8:5351. doi: 10.1038/s41598-018-21937-2
32. Lu J, Yang Y, Yu P, Tao H, Lu X, Wang L, et al. Bioinformatics Analysis and Significance of Expression of CC Chemokine Ligand 23 (CCL23) in Hepatocellular Carcinoma. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2019) 35:903–9.
33. Hou J-J, Xia Q, Zhao Z-C, Wang J-Y. Expression of CCL23 in Hepatocellular Carcinoma and Its Effect on Prognosis. Chin Hepatol (2019) 24:368–72.
34. Kadowaki H, Nishitoh H. Signaling Pathways From the Endoplasmic Reticulum and Their Roles in Disease. Genes (Basel) (2013) 4:306–33. doi: 10.3390/genes4030306
35. Li A, Song NJ, Riesenberg BP, Li Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front Immunol (2019) 10:3154. doi: 10.3389/fimmu.2019.03154
36. Duvigneau JC, Luis A, Gorman AM, Samali A, Kaltenecker D, Moriggl R, et al. Crosstalk Between Inflammatory Mediators and Endoplasmic Reticulum Stress in Liver Diseases. Cytokine (2019) 124:154577. doi: 10.1016/j.cyto.2018.10.018
37. Tang J, Guo YS, Zhang Y, Yu XL, Li L, Huang W, et al. CD147 Induces UPR to Inhibit Apoptosis and Chemosensitivity by Increasing the Transcription of Bip in Hepatocellular Carcinoma. Cell Death Differ (2012) 19:1779–90. doi: 10.1038/cdd.2012.60
38. Guo B, Li Z. Endoplasmic Reticulum Stress in Hepatic Steatosis and Inflammatory Bowel Diseases. Front Genet (2014) 5:242. doi: 10.3389/fgene.2014.00242
39. Vandewynckel YP, Laukens D, Bogaerts E, Paridaens A, Van den Bussche A, Verhelst X, et al. Modulation of the Unfolded Protein Response Impedes Tumor Cell Adaptation to Proteotoxic Stress: A PERK for Hepatocellular Carcinoma Therapy. Hepatol Int (2015) 9:93–104. doi: 10.1007/s12072-014-9582-0
40. Wu Y, Shan B, Dai J, Xia Z, Cai J, Chen T, et al. Dual Role for Inositol-Requiring Enzyme 1alpha in Promoting the Development of Hepatocellular Carcinoma During Diet-Induced Obesity in Mice. Hepatology (2018) 68:533–46. doi: 10.1002/hep.29871
41. Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K, et al. Activation of the ATF6, XBP1 and Grp78 Genes in Human Hepatocellular Carcinoma: A Possible Involvement of the ER Stress Pathway in Hepatocarcinogenesis. J Hepatol (2003) 38:605–14. doi: 10.1016/S0168-8278(03)00029-1
42. Spiotto MT, Banh A, Papandreou I, Cao H, Galvez MG, Gurtner GC, et al. Imaging the Unfolded Protein Response in Primary Tumors Reveals Microenvironments With Metabolic Variations That Predict Tumor Growth. Cancer Res (2010) 70:78–88. doi: 10.1158/0008-5472.CAN-09-2747
43. Feng YH, Tung CL, Su YC, Tsao CJ, Wu TF. Proteomic Profile of Sorafenib Resistance in Hepatocellular Carcinoma; GRP78 Expression Is Associated With Inferior Response to Sorafenib. Cancer Genomics Proteomics (2019) 16:569–76. doi: 10.21873/cgp.20159
44. Xiong H, Xiao H, Luo C, Chen L, Liu X, Hu Z, et al. GRP78 Activates the Wnt/HOXB9 Pathway to Promote Invasion and Metastasis of Hepatocellular Carcinoma by Chaperoning LRP6. Exp Cell Res (2019) 383:111493. doi: 10.1016/j.yexcr.2019.07.006
45. Wei C, Yang X, Liu N, Geng J, Tai Y, Sun Z, et al. Tumor Microenvironment Regulation by the Endoplasmic Reticulum Stress Transmission Mediator Golgi Protein 73 in Mice. Hepatology (2019) 70:851–70. doi: 10.1002/hep.30549
46. Li Y, Xu J, Chen L, Zhong WD, Zhang Z, Mi L, et al. HAb18G (CD147), a Cancer-Associated Biomarker and Its Role in Cancer Detection. Histopathology (2009) 54:677–87. doi: 10.1111/j.1365-2559.2009.03280.x
47. Giannelli G, Rani B, Dituri F, Cao Y, Palasciano G. Moving Towards Personalised Therapy in Patients With Hepatocellular Carcinoma: The Role of the Microenvironment. Gut (2014) 63:1668–76. doi: 10.1136/gutjnl-2014-307323
48. Wang M, Kaufman RJ. The Impact of the Endoplasmic Reticulum Protein-Folding Environment on Cancer Development. Nat Rev Cancer (2014) 14:581–97. doi: 10.1038/nrc3800
49. Avril T, Vauleon E, Chevet E. Endoplasmic Reticulum Stress Signaling and Chemotherapy Resistance in Solid Cancers. Oncogenesis (2017) 6:e373. doi: 10.1038/oncsis.2017.72
50. Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P. ER Stress-Induced Inflammation: Does It Aid or Impede Disease Progression? Trends Mol Med (2012) 18:589–98. doi: 10.1016/j.molmed.2012.06.010
51. Malhi H, Kaufman RJ. Endoplasmic Reticulum Stress in Liver Disease. J Hepatol (2011) 54:795–809. doi: 10.1016/j.jhep.2010.11.005
52. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine Tumor Necrosis Factor Alpha Links Endoplasmic Reticulum Stress to the Membrane Death Receptor Pathway Through IRE1alpha-Mediated NF-kappaB Activation and Down-Regulation of TRAF2 Expression. Mol Cell Biol (2006) 26:3071–84. doi: 10.1128/MCB.26.8.3071-3084.2006
53. Zhou Y, Dong B, Kim KH, Choi S, Sun Z, Wu N, et al. Vitamin D Receptor Activation in Liver Macrophages Protects Against Hepatic Endoplasmic Reticulum Stress in Mice. Hepatology (2020) 71:1453–66. doi: 10.1002/hep.30887
54. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell (2017) 168:692–706. doi: 10.1016/j.cell.2016.12.004
55. Arihara F, Mizukoshi E, Kitahara M, Takata Y, Arai K, Yamashita T, et al. Increase in CD14+HLA-DR -/Low Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients and its Impact on Prognosis. Cancer Immunol Immunother (2013) 62:1421–30. doi: 10.1007/s00262-013-1447-1
56. Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R, et al. Delineation of an Immunosuppressive Gradient in Hepatocellular Carcinoma Using High-Dimensional Proteomic and Transcriptomic Analyses. Proc Natl Acad Sci U S A (2017) 114:E5900–9. doi: 10.1073/pnas.1706559114
57. Sun L, Xu G, Liao W, Yang H, Xu H, Du S, et al. Clinicopathologic and Prognostic Significance of Regulatory T Cells in Patients With Hepatocellular Carcinoma: A Meta-Analysis. Oncotarget (2017) 8:39658–72. doi: 10.18632/oncotarget.17340
58. Yang P, Li QJ, Feng Y, Zhang Y, Markowitz GJ, Ning S, et al. TGF-beta-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell (2012) 22:291–303. doi: 10.1016/j.ccr.2012.07.023
59. Lee BR, Chang SY, Hong EH, Kwon BE, Kim HM, Kim YJ, et al. Elevated Endoplasmic Reticulum Stress Reinforced Immunosuppression in the Tumor Microenvironment via Myeloid-Derived Suppressor Cells. Oncotarget (2014) 5:12331–45. doi: 10.18632/oncotarget.2589
60. Soto-Pantoja DR, Wilson AS, Clear KY, Westwood B, Triozzi PL, Cook KL. Unfolded Protein Response Signaling Impacts Macrophage Polarity to Modulate Breast Cancer Cell Clearance and Melanoma Immune Checkpoint Therapy Responsiveness. Oncotarget (2017) 8:80545–59. doi: 10.18632/oncotarget.19849
61. Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER Stress Sensor XBP1 Controls Anti-Tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell (2015) 161:1527–38. doi: 10.1016/j.cell.2015.05.025
62. Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M. Cell-Extrinsic Effects of Tumor ER Stress Imprint Myeloid Dendritic Cells and Impair CD8(+) T Cell Priming. PLoS One (2012) 7:e51845. doi: 10.1371/journal.pone.0051845
63. Liu J, Fan L, Yu H, Zhang J, He Y, Feng D, et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-Regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology (2019) 70:241–58. doi: 10.1002/hep.30607
64. Lu LC, Chang CJ, Hsu CH. Targeting Myeloid-Derived Suppressor Cells in the Treatment of Hepatocellular Carcinoma: Current State and Future Perspectives. J Hepatocell Carcinoma (2019) 6:71–84. doi: 10.2147/JHC.S159693
65. Wan S, Kuo N, Kryczek I, Zou W, Welling TH. Myeloid Cells in Hepatocellular Carcinoma. Hepatology (2015) 62:1304–12. doi: 10.1002/hep.27867
66. Kim J, Kim YS, Ko J. CK Beta 8/CCL23 Induces Cell Migration via the Gi/Go Protein/PLC/PKC Delta/NF-Kappa B and Is Involved in Inflammatory Responses. Life Sci (2010) 86:300–8. doi: 10.1016/j.lfs.2009.11.012
67. Nardelli B, Tiffany HL, Bong GW, Yourey PA, Morahan DK, Li Y, et al. Characterization of the Signal Transduction Pathway Activated in Human Monocytes and Dendritic Cells by MPIF-1, a Specific Ligand for CC Chemokine Receptor 1. J Immunol (1999) 162:435–44.
68. Geng W, Lo CM, Ng KT, Ling CC, Qi X, Li CX, et al. Interferon-Gamma Inducible Protein 10 (IP10) Induced Cisplatin Resistance of HCC After Liver Transplantation Through ER Stress Signaling Pathway. Oncotarget (2015) 6:28042–56. doi: 10.18632/oncotarget.4832
69. Kim HM, Lee ES, Lee BR, Yadav D, Kim YM, Ko HJ, et al. C-C Chemokine Receptor 2 Inhibitor Ameliorates Hepatic Steatosis by Improving ER Stress and Inflammation in a Type 2 Diabetic Mouse Model. PLoS One (2015) 10:e0120711. doi: 10.1371/journal.pone.0120711
70. Heo YJ, Choi SE, Lee N, Jeon JY, Han SJ, Kim DJ, et al. Visfatin Exacerbates Hepatic Inflammation and Fibrosis in a Methionine-Choline-Deficient Diet Mouse Model. J Gastroenterol Hepatol (2021) 36:2592–600. doi: 10.1111/jgh.15465
71. Zhang Y, Liao S, Fan W, Wei W, Wang C, Sun S. Tunicamycin-Induced ER Stress Regulates Chemokine CCL5 Expression and Secretion via STAT3 Followed by Decreased Transmigration of MCF-7 Breast Cancer Cells. Oncol Rep (2014) 32:2769–76. doi: 10.3892/or.2014.3479
72. Lim W, Bae H, Bazer FW, Song G. C-C Motif Chemokine Ligand 23 Abolishes ER Stress- and LPS-Induced Reduction in Proliferation of Bovine Endometrial Epithelial Cells. J Cell Physiol (2018) 233:3529–39. doi: 10.1002/jcp.26210
73. Jeong W, Bae H, Lim W, Bazer FW, Song G. Differential Expression and Functional Roles of Chemokine (C-C Motif) Ligand 23 and Its Receptor Chemokine (C-C Motif) Receptor Type 1 in the Uterine Endometrium During Early Pregnancy in Pigs. Dev Comp Immunol (2017) 76:316–25. doi: 10.1016/j.dci.2017.07.007
74. Peeraphatdit TB, Wang J, Odenwald MA, Hu S, Hart J, Charlton MR. Hepatotoxicity From Immune Checkpoint Inhibitors: A Systematic Review and Management Recommendation. Hepatology (2020) 72:315–29. doi: 10.1002/hep.31227
75. Liu Z, Lin Y, Zhang J, Zhang Y, Li Y, Liu Z, et al. Molecular Targeted and Immune Checkpoint Therapy for Advanced Hepatocellular Carcinoma. J Exp Clin Cancer Res (2019) 38:447. doi: 10.1186/s13046-019-1412-8
Keywords: tumor microenvironment, endoplasmic reticulum, hepatocellular carcinoma, antitumor immunity, chemokine ligand CCL23
Citation: Karan D (2021) CCL23 in Balancing the Act of Endoplasmic Reticulum Stress and Antitumor Immunity in Hepatocellular Carcinoma. Front. Oncol. 11:727583. doi: 10.3389/fonc.2021.727583
Received: 18 June 2021; Accepted: 17 September 2021;
Published: 04 October 2021.
Edited by:
Yun Dai, Peking University First Hospital, ChinaReviewed by:
Suraj Kadunganattil, Amala Cancer Research Centre, IndiaAntonella Argentiero, Istituto Nazionale dei Tumori (IRCCS), Italy
Copyright © 2021 Karan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dev Karan, ZGthcmFuQG1jdy5lZHU=