 Lu Shen
Lu Shen Shizhen Zhang
Shizhen Zhang Kaiyue Wang
Kaiyue Wang Xiaochen Wang
Xiaochen Wang- Department of Breast Surgery and Oncology, Key Laboratory of Cancer Prevention and Intervention, Ministry of Education, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Background: About 5%–10% of the breast cancer cases have a hereditary background, and this subset is referred to as familial breast cancer (FBC). In this review, we summarize the susceptibility genes and genetic syndromes associated with FBC and discuss the FBC screening and high-risk patient consulting strategies for the Chinese population.
Methods: We searched the PubMed database for articles published between January 2000 and August 2021. Finally, 380 pieces of literature addressing the genes and genetic syndromes related to FBC were included and reviewed.
Results: We identified 16 FBC-related genes and divided them into three types (high-, medium-, and low-penetrance) of genes according to their relative risk ratios. In addition, six genetic syndromes were found to be associated with FBC. We then summarized the currently available screening strategies for FBC and discussed those available for high-risk Chinese populations.
Conclusion: Multiple gene mutations and genetic disorders are closely related to FBC. The National Comprehensive Cancer Network (NCCN) guidelines recommend corresponding screening strategies for these genetic diseases. However, such guidelines for the Chinese population are still lacking. For screening high-risk groups in the Chinese population, genetic testing is recommended after genetic counseling.
1 Introduction
As reported by the 2020 Cancer Statistics, the most common type of cancer diagnosed is breast cancer (BC), with approximately 2.26 million new cases worldwide in 2020. In China, BC is the fourth most commonly diagnosed malignancy (approximately 420,000 patients in 2020), after lung cancer, colorectal cancer, and stomach cancer. Notably, BC is the most commonly diagnosed cancer and the leading cause of death among women (1). BCs with a hereditary background are termed familial breast cancers (FBCs) and receive significant focus because they make up about 5%–7% of the BCs (2, 3). Many susceptibility genes, such as BRCA1/2, have been found to be related to FBC (4). Moreover, several genetic syndromes, such as hereditary breast and ovarian cancer (HBOC) syndrome, have also been associated with FBC (5). Due to the substantial heterogeneity among patients with BC, the prevalence and genetic susceptibility of BC in different races or regions vary depending on the type of disease. At present, several disease-related gene mutations have been confirmed in FBCs, and some exist specifically in the Chinese population.
In this review, we aim to summarize the FBC-related susceptibility genes and syndromes, introduce risk assessment models and explore screening methods, such as genetic counseling, for Chinese individuals with a potentially high risk of FBC.
2 Methods
We searched the literature published in PubMed between January 2000 and August 2021 by searching the terms “familial breast cancer,” “family breast cancer,” and “gene” in the title or abstract. Subsequently, 380 studies addressing genetic mutations in familial breast cancer were identified. Among those, we excluded irrelevant articles and classified the remaining articles (Figure 1). After analyzing the selected articles, we selected the most relevant genes and searched the PubMed database to acquire pertinent information.
Figure 1 Literature screening steps.
3 Results
3.1 Familial Breast Cancer (FBC)
Pathologically, BCs are classified into four subtypes: Luminal A, Luminal B, HER2-positive, and triple-negative BC. The treatment and prognosis of each subtype differ (6). Epidemiologically, BCs are mainly divided into three categories: 1) sporadic breast cancer (SBC), 2) hereditary breast cancer (HBC), and 3) FBC (Figure 2) (7). HBC differs from FBC and refers explicitly to patients with BC with definite genetic factors, accounting for approximately 5%–10% of the BCs. About 10%–15% of the HBCs have a positive family history (FH) (8, 9). Meanwhile, FBC is a subset of BC within a family, where the underlying genetic cause is not entirely known. The incidence of BC within a family is mainly due to genetic factors and partly due to environmental factors (10, 11). Environmental factors come from both shared and non-shared environments. The shared environment includes eating habits and lifestyles. The non-shared environment includes age at menarche, age at first full-term pregnancy, and age at menopause (10).. According to the prediction model proposed by Lichtenstein et al., the shared environmental factors contributed 6%, and non-shared environmental factors contributed 67% of the risk for developing BC (12). In subsequent studies, the discovery of low-penetrance genes reduced the influence rate of environmental factors to 27% (11). Recent studies have focused on the contribution of gene-environment interactions in familial aggregation (13). Nevertheless, environmental factors are still a major cause of FBC in various members of a family.
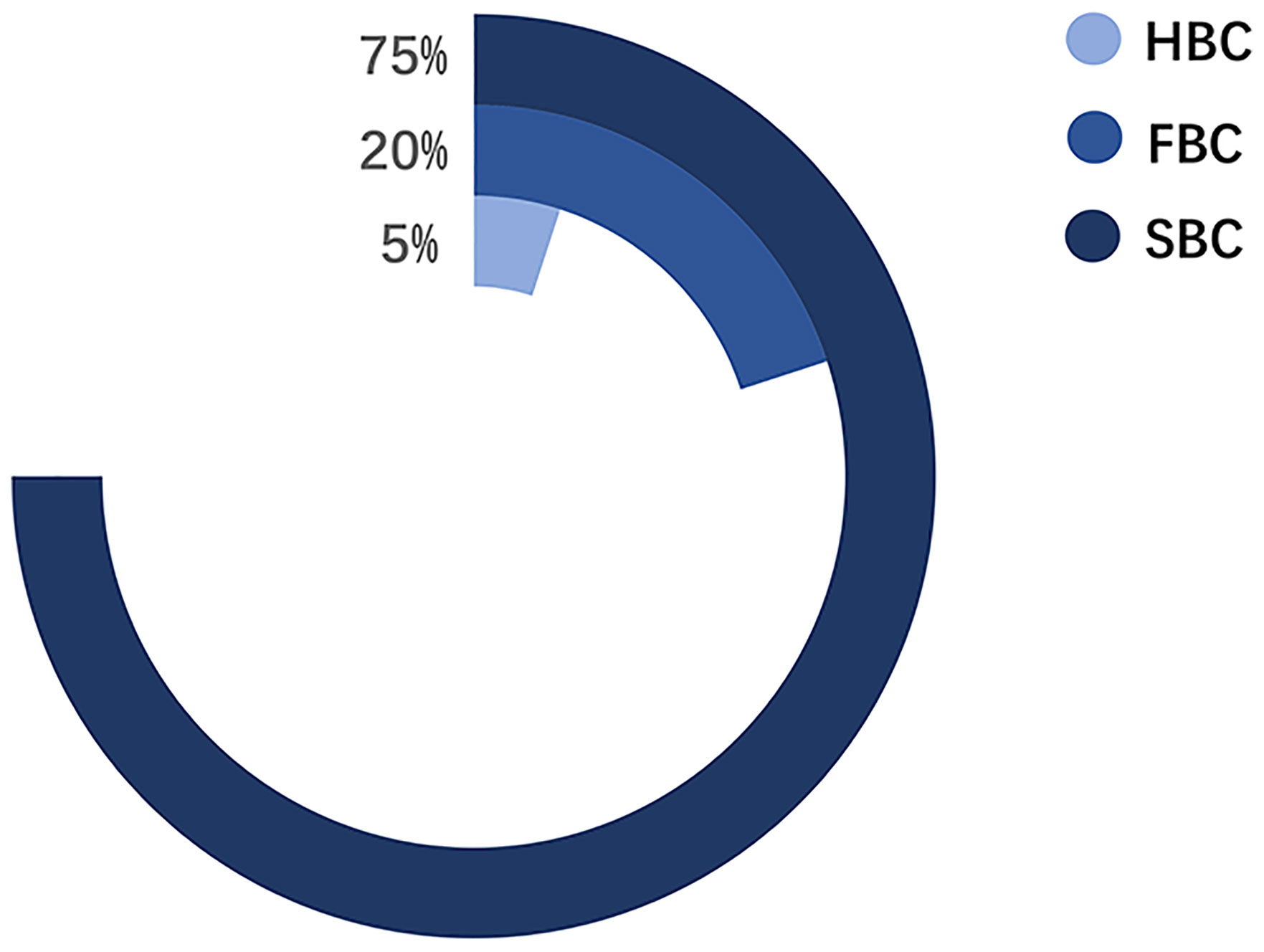
Figure 2 The proportion of three categories in BC.
The first typical case of FBC was reported by Broca in 1866. In that case, ten females were diagnosed with BC among the thirty-eight family members, which strongly suggested that the family members carried specific BC susceptibility genes or were exposed to the same environmental factors contributing to BC. In 1979, Lynch defined FBC with clinical characteristics of earlier onset age, two or more first-degree relatives with a history of BC, a higher incidence of bilateral BC, and multicentric cancer (14). In 1988, Phipps revised the definition of FBC to include early age of onset, excess bilaterality, specific tumor association with colon and ovary, and vertical transmission (15). Presently, the criteria for FBC diagnosis include (1) in addition to the first patient (proband) in the family, there are more than three BCs in first-degree relatives or: (2) in addition to the proband in a family, there are more than 2 BCs in first-degree relatives, and at least one of them meets one of the following conditions: age less than 40 years at the time of onset, simultaneous or heterochronous bilateral BC, or simultaneous or heterochronous non-breast repeat cancer (16).
The definitions of FBC and HBC are unclear and partially overlapping. Many studies have not distinguished between FBC and HBC in terms of clinical features and prognosis (17). One important reason for distinguishing FBC from other types of BCs is that it allows us to target the high-risk population and predict the risk of BC based on FH. As early as 1972, Anderson reported that first-degree relatives of patients with BC have a 2–3 times higher risk of developing BC than those with no FH of BC; if the patient has bilateral BC, the risk is about five times higher for the relatives and if the patient has premenopausal bilateral BC, the risk increases to 9 times higher for relatives (18). Recent studies have found that as the number of first-degree relatives with BC increases, the probability that women 20–80 years old with an FH of BC may develop BC increases correspondingly (19). Interestingly, a positive FH of BC appears to increase the patient’s lifetime risk of FBC because FH may affect their BC screening behaviors (20). With the combination of molecular genetic techniques and knowledge of FH of BC, genetic counselors can provide more precise disease risk prediction, which is critical for the prevention and treatment of FBC.
3.2 Gene Mutations Related to FBC
Multiple genes have been identified to be associated with FBC. For example, BRCA1/2 gene mutations account for 5% of the BC mutant genes and can lead to 16% to 25% of the FBC cases (21, 22). Moreover, mutant genes associated with genetic syndromes, such as TP53, PTEN, STK11, and CDH1, account for 5% of the FBC risk. Moderate mutations in penetrance genes, such as ATM gene mutations, also account for approximately 5% of the risk of FBC. More than 180 low-sensitivity genes account for approximately 18% of the FBC risk (23). However, the remaining FBCs showed no mutations in any of these genes and were therefore classified as BRCAX (BRCA1/2-negative, high-risk) BCs (Figure 3). This type of FBC may carry one or multiple unidentified genetic mutations (22, 24).
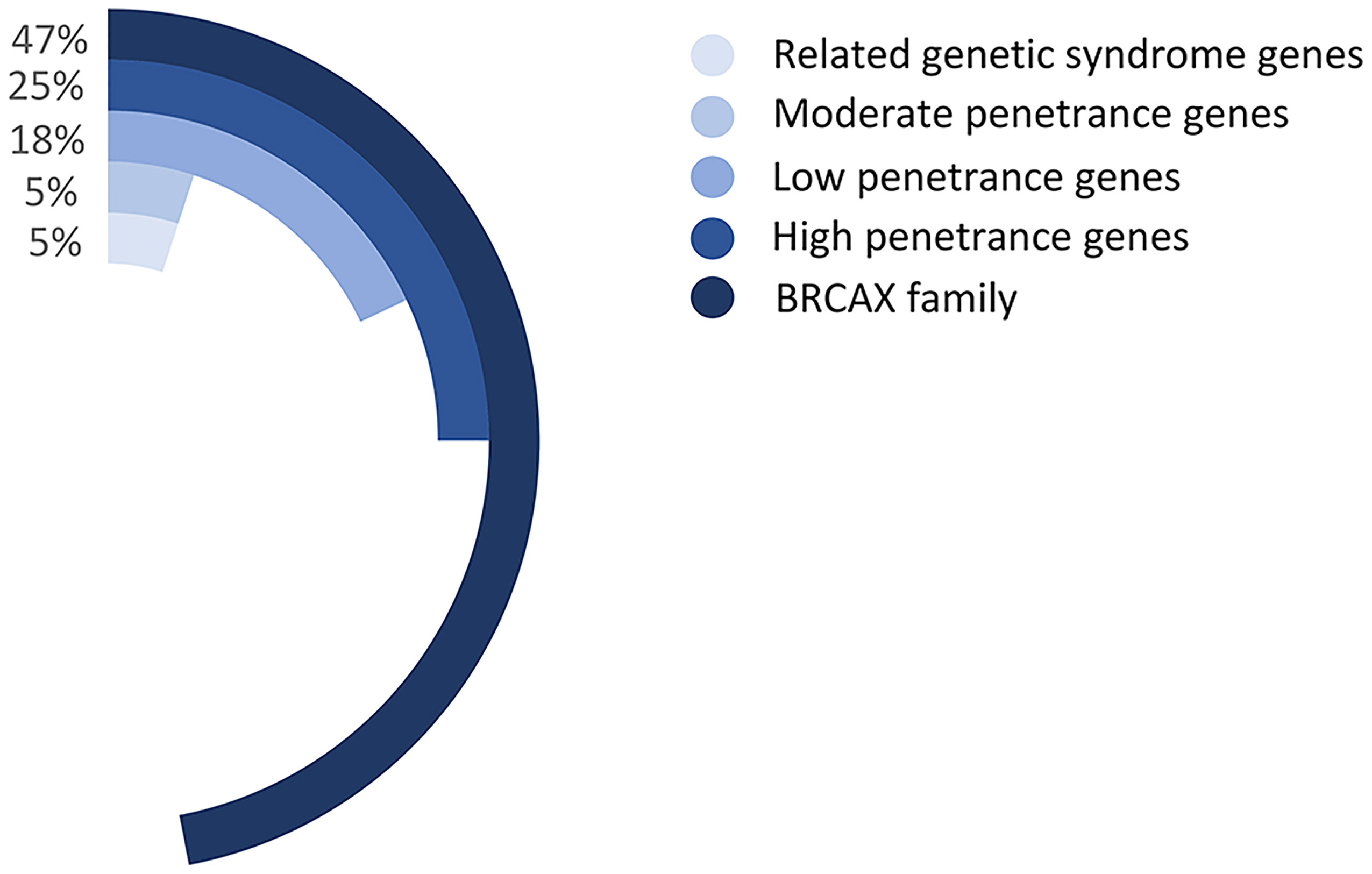
Figure 3 The proportion of various gene mutations in FBCs. Related genetic syndrome genes refer to mutated genes found in genetic syndromes related to BC, such as HBOC. Related genetic syndrome genes accounts for about 5% of whole mutation genes. Low-, moderate-, and high- penetrance genes are classified as BC mutation genes based on lifetime risk of disease. The BRCAX family accounted for the largest proportion, reaching 47% of whole mutation genes. Patients belong to The BRCAX family have not found any currently known disease-causing mutation genes.
The gene penetration rate, which refers to the estimated risk of a specific disease in the presence of this genotype of a particular gene, is commonly used as a genetic biomarker to predict cancer risk. Relative risk (RR) represents the risk of obtaining a disease compared with the general population’s risk. In general, BC susceptibility genes are associated with different risk levels and are roughly divided into high- (RR ≥5.0), moderate- (1.5≤ RR <5.0), and low-penetrance (1.0≤ RR <1.5) alleles (25, 26) (Table 1).
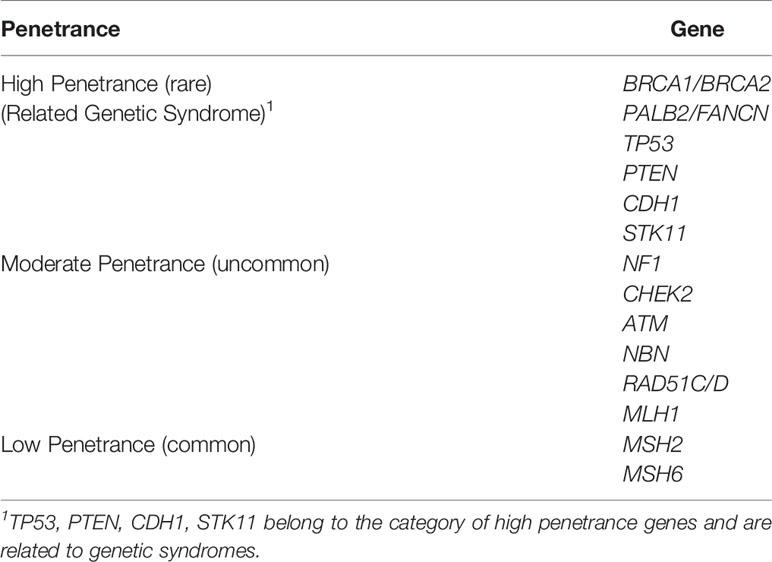
Table 1 Penetrance of genes in BC.
People with high-penetrance alleles usually have a lifetime risk of developing BC of more than 50%, those with moderate-penetrance have a lifetime risk greater than 20%, and those with low-penetrance alleles have a lifetime risk of 10%–20% (27–29). Thus, discovering the mutant genes highly related to BC is vital for disease screening and prediction.
3.3 Genes With High Penetrance
3.3.1 BRCA1 and BRCA2
In 1990, chromosome 17q21.2 was identified to be related to FBC (30), and the loss of 17q heterozygosity is frequently detected in familial breast and ovarian tumors (31). Using positional cloning, the BC susceptibility gene BRCA1 was found to be located on chromosome 17 at the 17q21 position (32). In 1995, the BRCA2 gene was identified on chromosome 13q12.36 (7). BRCA1 contains 24 exons and encodes a protein of 1,863 amino acids. The exon contains three mutation domains: a central N-terminal RING fingerprint domain (exons 2–7), a C-terminal BRCT domain (exons 16–24), and exons 11–13. The N-terminal RING fingerprint domain of BRCA1 binds to BRCA1-associated RING domain protein 1 (BARD1) (33), and the C-terminal BRCT domain binds to the phosphorylated protein (34, 35). BRCA2 contains 27 exons and encodes a protein of 3,418 amino acids. The N-terminal of BRCA2 contains the transcriptional activation domain, the middle section includes eight conserved motifs called BRC repeats that bind to RAD51, and the C-terminal contains the DNA-binding domain, two nuclear localization signals, and one TR2 (C-terminal RAD51 binding site) (36). BRCA1 and BRCA2 participate in RAD51-mediated homologous recombination (HR) for DNA repair. In the case of DNA double-strand damage, BRCA1 can be accurately located and phosphorylated at the damage site, and BRCA2 forms a complex with RAD51 to move it from the site of synthesis to the site of DNA damage processing (37). Furthermore, PALB2 acts as a bridge between BRCA1 and BRCA2. Its N-terminal coiled-coil motif binds to the coiled-coil motif encoded by exon 11 of BRCA1, and its C-terminal WD-40 repeats bind to the N-terminal of BRCA2 to form the BRCA1/PALB2/BRCA2 complex (Figures 4, 5). This complex is critical for HR after DNA double-strand breaks (38). In the absence of BRCA1 and BRCA2, HR is suppressed. DNA damage repair is alternatively carried out through the non-homologous end-joining pathway, which is more error-prone and leads to genome instability (7, 39). As a result, BRCA1 and BRCA2 play an essential role in maintaining genome integrity (40).
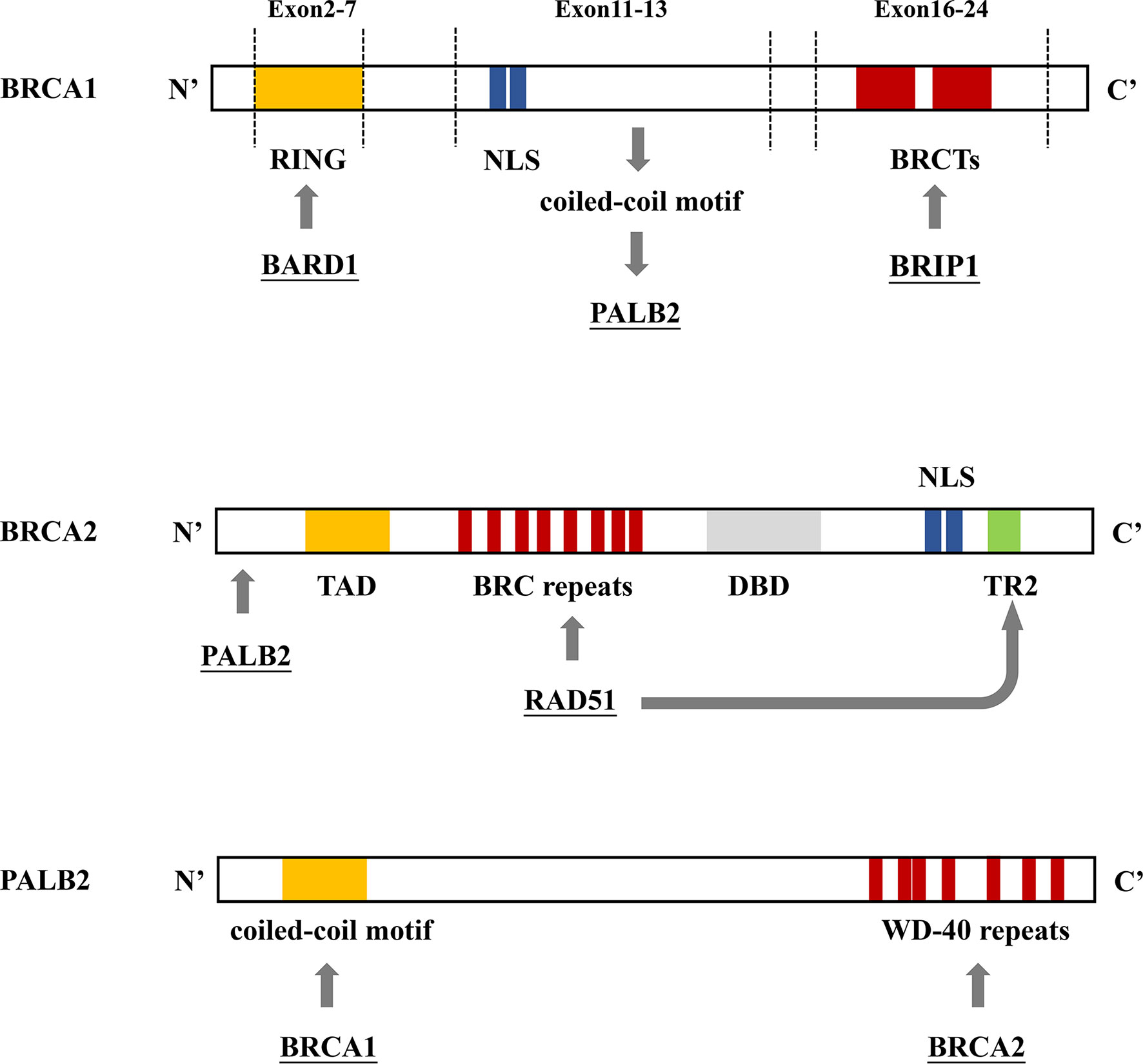
Figure 4 BRCA1/2 and PALB2 gene binding sites. (A) BRCA1 contains 3 mutation domains: a central N-terminal RING fingerprint domain (exons 2–7) that binds to BARD, two nuclear localization signals (NLSs) (exons 11–13) that import BRCA1 into the nucleus, and a C-terminal BRCT domain (exons 16–24) that interact with BRIP1. (B) The N-terminal region of BRCA2 interacts with PALB2. The N-terminal contains the topologically associating domain (TAD). Eight RAD51 binding sites (BRC repeats) located on the central part and TR2 located on the C-terminal bind to RAD51 to promote the RAD51-mediated DNA strand exchange process. The C-terminal contains the DNA binding domain (DBD), which includes 3 oligonucleotide/oligosaccharide-binding folds that bind to double-stranded DNA, two NLSs. (C) The N-terminal coiled-coil motif of PALB2 binds to BRCA1; the C-terminal WD-40 repeats bind to BRCA2, forming the BRCA1/PALB2/BRCA2 complex.
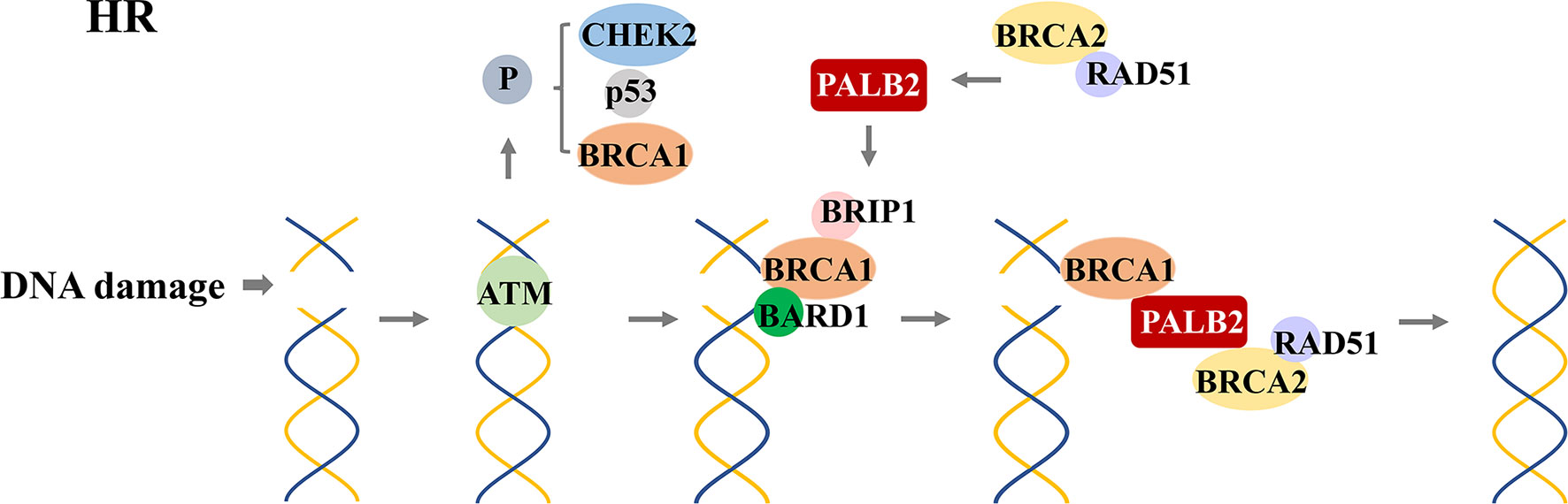
Figure 5 RAD51-mediated homologous recombination (HR) for DNA repair. After detecting DNA double-strand damage, ATM is recruited and activated, leading to the phosphorylation of downstream effectors, including BRCA1, p53, and CHEK2. After phosphorylation, BRCA1 and BRAD1 form a heterodimer; BRIP1 interacts with BRCT repeats; which constitute a scaffold to recruit BRCA2, PALB2, and RAD51 to form a complex. This complex locates the DNA damage site and promotes the HR process.
As tumor suppressor genes, mutations of BRCA1 (MIM 113705) and BRCA2 (MIM 600185) are closely related to the development of BC (41, 42). Carriers of these mutations have a 10–20 times higher risk of developing BC than those without BRCA mutations (43). Approximately 16%–25% of the FBC cases harbor harmful variants of BRCA1 and BRCA2 (44, 45). Although many genetic variants of BRCA1 and BRCA2 have been recorded, approximately 53%–55% of the variants occur in only one family. The most common BRCA1 variants were 185delAG (16.5%), 5382insC (8.8%), and missense variant C61G (1.8%). Meanwhile, the most frequently reported BRCA2 variants include 6174delT (9.6%), K3326X (2.6%), 3036del4 (0.9%), and 6503delTT (0.8%) (46). Notably, the mutation spectrum of BRCA1/BRCA2 varies significantly depending on geographic origin or ethnicity (47). For example, in China, the mutation rate of BRCA2 in FBC was higher than that of BRCA1 in the Shandong Province (48). However, other studies including different Chinese regions, such as Shanghai and the Henan Province, showed that the mutation rate of BRCA1 in FBC is higher than that of BRCA2 (49). The variant hotspots of BRCA1 in the Henan cohort were A3113G and A3780G, which was first reported in this population (50). Moreover, in the participants from Shanghai, two other new splice site variants in the BRCA1 gene (IVS17-1G>T, IVS21+1G>C) were discovered (51). BRCA2 gene mutations dominated in FBCs of the eastern Shandong population, and three BRCA2 gene variants, 2001delTTAT, 4099C to T, and 5873C to A, were discovered for the first time in this population (52). A new BRCA1 missense variant, c.5191C>A, was identified in the Taiwanese population, but whether it is pathogenic remains inconclusive (53). Moreover, the recurrent variant of BRCA1, 1100delAT, was found in the Shanghai, Jinan, Qingdao, and Shenyang populations (54), while the BRCA1 c.470_471delCT and c.981_982delAT variants were considered to be recurrent variants in the Hong Kong population (55). Additionally, racial differences have an impact on gene mutations. For example, a Singapore study that studied individuals with a personal or FH of familial breast/ovarian cancers (FBOCs) with the BRCA1 c.442-22_442-13del variant, found that this variant was more common in patients of Chinese origin. The study also implied that the BRCA1 c.442-22_442-13del variant could be a founding variant in Chinese individuals of ancient southern Han descent (56).
Recently, a study including 21,216 unselected patients with BC and 6,434 healthy controls from 19 medical centers throughout 11 Chinese provinces identified 1,958 BRAC1/2 variants through panel-based sequencing, of which 532 (27.2%) were pathogenic variants, and 858 (43.8%) were pathogenic variants of uncertain significance. The remaining 568 variants (29.0%) were benign. A total of 268 mutations in the BRCA1 gene and 242 mutations in BRCA2 were found in Asian patients with BC, most of which were meaningless mutations. Among these variants, researchers found 13 types of high-frequency lesions: p.Cys328fs, p.Asn704fs, p.Ser1862fs, and p.Ile1845fs in BRCA1; p.Ala938fs, p.Gln1037*, p.Ser1722fs, p. Tyr1894*, p.Leu1908fs, p.Glu2198fs, p.Ser2378*, p.Pro2802fs, and p.Thr3033fs in BRCA2. Eight of these variants have not been reported as high-frequency variants in Caucasians (57). Of note, single nucleotide polymorphisms (SNPs), a single nucleotide substitution at a specific position in the genome, may also contribute to BRCA1 changes. For example, two pathogenic SNPs were found on the 11th exon of BRCA1, which may be related to early-onset BC in the Chinese population (58).
Although various BRCA1/BRCA2 mutations are currently being investigated, variants of unknown significance that occur in BRCA1/BRCA2 account for 12%–13% of the cases, the functions of which are still unclear. Therefore, more research is required to determine the clinical importance of variants of unknown significance in BC (59).
3.3.2 TP53
The tumor suppressor gene TP53, located on chromosome 17p13.1, is the most commonly mutated gene in patients with cancer (60). The TP53 gene encodes the cellular tumor antigen p53, an intracellular transcription factor that controls multiple tumor suppressive pathways (61, 62). TP53 is defined as the “guardian of the genome” because of its role in conserving stability by preventing genetic mutations. The germline loss of TP53 can quickly lead to the formation of spontaneous cancers (63, 64). Moreover, cancers with wild-type TP53 predict a good prognosis (65), while those with mutant TP53 predict a worse prognosis (66–68).
Notably, about 30% of the BCs have TP53 mutations (60), most occurring in exons 5–8 (69). It has been reported that about 5% of the patients with BC with a positive FH and wild-type BRCA1 and BRCA2 carried a mutation in either CHEK2 or TP53 (70). In patients with Li-Fraumeni syndrome (LFS) with TP53 mutations, the risk of developing BC under 45 years of age is 18–60 times higher than that of the general population (71). One study of 150 patients with familial and early-onset BC revealed that the deletion variant of TP53 (643_660del18) appeared to have occurred only in the Chinese population (72).
3.3.3 Phosphatase and Tensin Homolog (PTEN)
The PTEN gene is located on chromosome 10q23.31, which encodes phosphatidylinositol 3,4,5-triphosphate 3-phosphatase. This has lipid phosphatase activity and works antagonistically to the PI3K signaling pathway. It also negatively regulates the mitogen-activated protein kinase (MAPK) pathway through its protein phosphatase activity (73). PTEN mutation is also associated with Cowden syndrome, an autosomal dominant genetic disease that increases the lifetime risk of BC. In PTEN variant carriers, the lifetime risk of BC is estimated to be 67%–85% (74).
3.3.4 PALB2/FANCN
PALB2 is located on chromosome 16p12.2 and encodes a protein that interacts with BRCA2 as a functional partner. Thus, PALB2 affects the nuclear localization and stability of BRCA2 and can also act as a bridge between BRCA1 and BRCA2 (38, 75). The biallelic PALB2 mutation causes a new Fanconi anemia subtype, FA-N, also called FANCN (76, 77). Recent studies have found that germline mutations of PALB2 exist in families with BC, indicating that PALB2 may be a tumor suppressor for FBC (78, 79). Moreover, individuals with PALB2 mutations have a 2.3 times higher risk of developing BC (43). One study conducted in Finland showed that PALB2 c.1592delT is a founder variant, which causes truncated protein products with functional defects that cannot support BRCA2 to complete DNA repair. As such, females with this PALB2 mutation have a four times higher risk of developing BC (80). By age 50, the cumulative risk of BC in women with such mutations is estimated to be 14%, and by age 70, it increases to 35% (81). Zhang et al. also identified three harmful variants (c.3271delC, c.103C>T, and c.3035C>T) of PALB2 in 305 cases of FBC in China, and the mutation rates were all 0.33% (82). In addition, studies have found that women with PALB2 mutations from families with a history of BC have a greater risk of BC than those with no FH of BC (83).
3.3.5 E-Cadherin (CDH1)
The CDH1 gene is located on chromosome 16q22.1 and encodes the tumor suppressor, E-cadherin (a transmembrane calcium-dependent protein involved in cell-cell adhesion) (84–87). CDH1 mutation is closely related to lobular breast cancer, and the lifetime risk of developing BC in those with a CDH1 mutation is approximately 39%. Moreover, somatic and epigenetic changes in the CDH1 gene are frequently detected in sporadic tumors, including BC, and are associated with worse survival rates (88, 89). Pathogenic mutations of CDH1 are also the leading cause of hereditary diffuse gastric cancer (HDGC) (90), whose first clinical manifestation could be lobular breast cancer (91).
3.3.6 Serine/Threonine Kinase 11/Liver Kinase B1 (STK11/LKB1)
The STK11 gene is located on chromosome 19p13.3 and encodes a serine/threonine kinase that regulates many physiological processes, including energy metabolism and cell polarity (92). Most importantly, it is estimated that 32%–54% of the STK11 gene mutation carriers under 60 years of age have a high risk of developing BC during their lifetime. The risk of developing BC for these carriers is 8% at the age of 40, increasing to 32% at the age of 60. Compared with the general population, the risk of BC in this subpopulation is seven times higher (93).
3.4 Genes With Moderate Penetrance
3.4.1 Neurofibromatosis Type 1 (NF1)
The NF1 gene is located on chromosome 17q11.2 and encodes neurofibromin, a cytoplasmic protein that regulates multiple critical signaling pathways, such as the Ras-cAMP pathway (94). Neurofibromin can increase the guanosine triphosphate (GTP) hydrolytic rate of Ras and, thus, plays a tumor-suppressive role by reducing Ras activity (95). Females under the age of 50 with NF1 gene mutations have an up to five times higher risk of BC morbidity and mortality (96), and the risk was high among women under 40 years of age (97).
3.4.2 Checkpoint Kinase 2 (CHEK2)
Cell cycle checkpoint kinase 2 (CHEK2) is involved in DNA damage and replication checkpoint responses and has been widely considered a BC-sensitive factor (98). The CHEK2 gene is located on chromosome 22 and is critical for cell cycle regulation. The pluripotent kinase CHEK2 is important for DNA damage response by causing cell cycle arrest or apoptosis. CHEK2 phosphorylates the TP53 tumor suppressor protein and prevents its degradation during DNA damage, leading to G1 cell cycle arrest (99). In addition, CHEK2 can induce cell apoptosis independent of TP53 by phosphorylating the tumor suppressor, promyelocytic leukemia protein (100). CHEK2.1100delC is a protein truncation variant first found in a family with LFS (101). Moreover, CHEK2.1100delC can be detected in 5.1% of the non-carriers of the BRCA1 or BRCA2 mutations with FH in northern Europe (102), and such mutation in females can increase the risk of developing BC by 2–3 times, and by 10 times in males (103, 104). A study based on a Chinese population with the CHEK2 mutation in 74 patients with BC with FH and 50 control subjects identified that the missense variant of CHEK2 in these cases was 1111C>T (His371Tyr) instead of CHEK2.1100delC, which implies that the CHEK2.1100delC variant was relatively rare in the Chinese population and that CHEK2 c.1111C>T mutation might be related to the genetic susceptibility of BC (105). Another study screened the CHEK2 coding sequence of 118 cases of Chinese FBC negative for BRCA1 and BRCA2 mutations and confirmed that the incidence of CHEK2 c.1111C>T in FBC was higher than that in non-selective BC (4.24% vs. 1.76%) (106).
3.4.3 Ataxia Telangiectasia Mutated (ATM)
The ATM gene is located on chromosome 11q, which encodes a phosphatase essential for substrate phosphorylation involved in DNA repair and cell cycle regulation (107). The ATM gene mutation rate in the general population is about 1%, and ATM heterozygotes have an increased risk of developing BC (108), especially women over 50 years of age (109). The lifetime risk of developing BC in patients with ATM monoallelic mutations is about 17%–52% (110, 111). A recent meta-analysis showed that the fully deleterious variants of ATM could cause a BC risk 2–4 times higher than that of the general population (112).
3.4.4 Nibrin (NBN)
The NBN gene is located on chromosome 8q21.3 and encodes the Nibrin protein, responsible for interaction with DNA repair proteins involved in DNA double-strand break signaling (113, 114). Carriers of NBN monoallelic mutations have a significantly increased risk of BC, with an estimated odds ratio of 3.1 (115). Moreover, the truncated c.657del5 variant of NBN is also regarded as a high-risk factor for BC (116).
3.4.5 RAD51C and RAD51D
Proteins encoded by the RAD51 gene are important for DNA double-strand break repair. Seven RAD51 paralogs have been identified in mammals, including RAD51, RAD51B, RAD51C, RAD51D, XRCC2, XRCC3, and DMC1 (117). Based on the present knowledge, mutations in RAD51C and RAD51D are closely related to carcinogenesis (118, 119). For example, RAD51C and RAD51D gene mutations can be detected in patients with FBOC (120). One study from China conducted a genetic analysis of 273 patients with BRCA1/2-negative FBC and identified four previously unknown amino acid substitution variants in the RAD51C gene, the 4C>G (R2G) located in exon 1, 635G>A (R212H), and 644A>G (D215G) in exon 4, and 882G>C (Q294H) in exon 6. The R212H variant was unlikely to be pathogenic, as it only existed in healthy individuals. However, the R2G and D215G variants were suggested to be pathogenic to Chinese women after analysis using the SIFT, PolyPhen, and PMut algorithms (121). Moreover, a recent study showed that the protein-truncating variants in RAD51C and RAD51D are related to FBC. The estimated relative risks of BC associated with RAD51C and RAD51D mutations were 1.99 and 1.83, respectively. Therefore, they were classified into the moderate-risk category based on the current National Institute for Health and Care Excellence guidelines (57, 122).
3.5 Genes With Low Penetrance
3.5.1 MLH1, MSH2, MSH6, and PMS2
MLH1, MSH2, MSH6, and PMS2 encode DNA mismatch repair (MMR) proteins responsible for DNA mismatch repair (123, 124). Mutations in these genes can cause Lynch syndrome. Several studies have found that MMR gene mutations frequently exist in patients with BC (125), but the association between Lynch syndrome and BC is unclear (126).
With the widespread application of high-throughput sequencing, a large number of genes related to BC risk have been identified, such as BAP1, PPM1D, and ABRAXAS1 (127, 128). However, their exact connection with BC and its penetrance remains unclear.
3.6 Gene Mutations in Chinese Patients with BC
Gene mutations related to BC are thought to vary among patients from different regions and races. Specifically, several gene mutations occurred mainly in Chinese populations (Table 2).
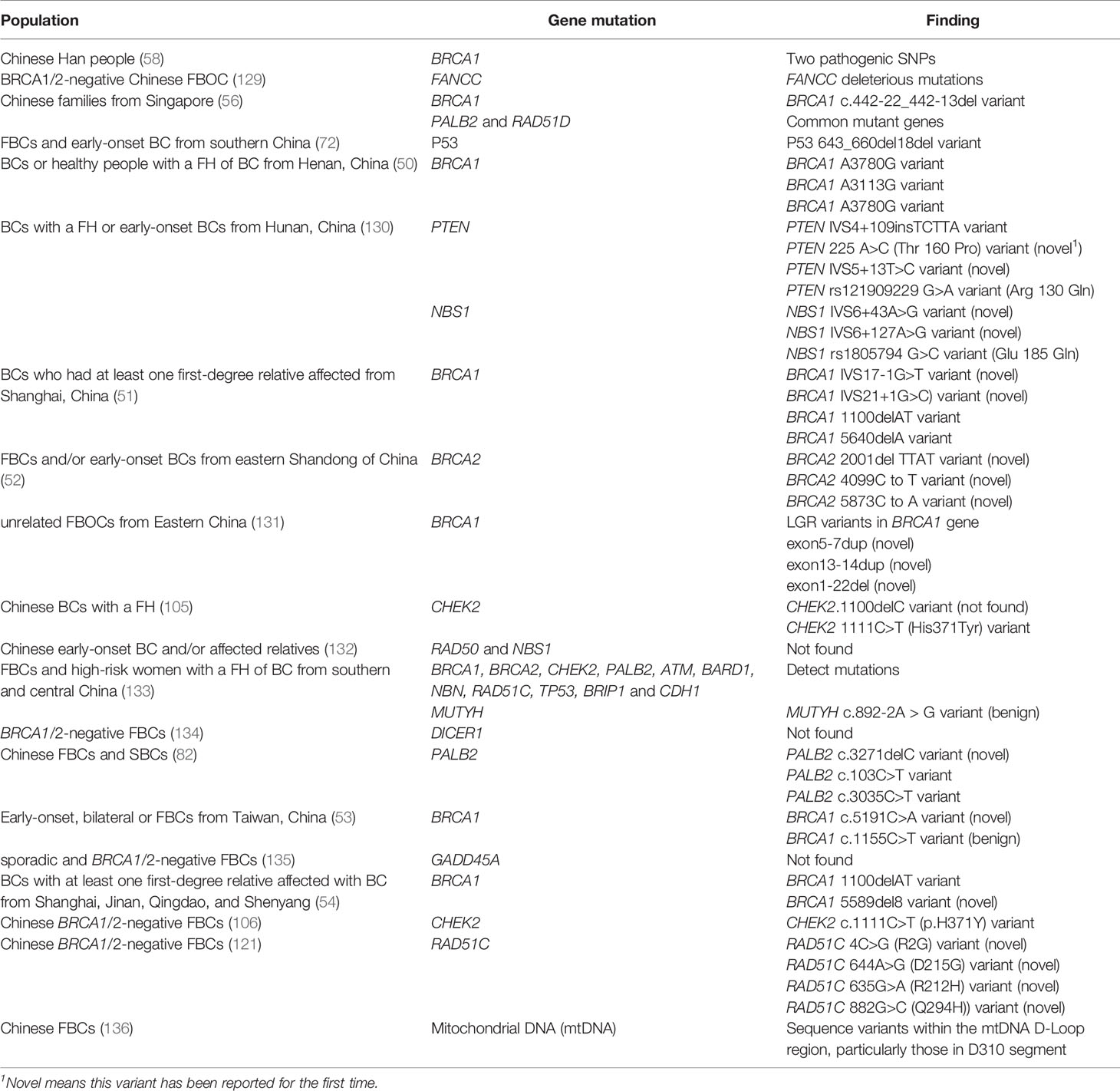
Table 2 Gene mutations in the Chinese population.
Generally, most mutations in BRCA1/2 genes exist in Chinese populations; however, ATM, CHEK2, PALB2, and BRIP1 have more pathogenic mutations among non- BRCA1/2 carriers (137).. One study conducted a 27-gene panel analysis of 120 patients with BC and 120 high-risk women with first-degree or second-degree relatives. The study identified that 12 genes contained harmful mutations in the Chinese population, including BRCA1, BRCA2, MUTYH, CHEK, PALB2, ATM, BARD1, NBN, RAD51C, TP53, BRIP1, and CDH1 (133).
One study conducted in Hunan, China, first reported two variants in the PTEN gene (225A> C (T160P) and IVS5 +13T> C) and two variants in the NBS1 gene (IVS6 + 43A> G and IVS6 + 127A> G) in familial and early-onset BC in the study population (130). FANCC, which belongs to the Fanconi anemia complementation group, has been reported as a susceptibility gene for BC. It was found that there were harmful variants of FANCC in patients with FBOC in China, but its penetration and spectrum require further study (129). Other genes, such as DICER1 (134), and growth arrest and DNA damage-induced 45 alpha (GADD45A) (135) have been reported as candidate susceptibility genes for FBC, but no mutations in these genes have yet been discovered to be harmful among the Chinese population.
3.7 BC-Related Genetic Syndromes
The above-mentioned BC susceptibility genes are active in several genetic syndromes related to BC. Due to the existence of mutant genes, patients with a family history of these genetic syndromes have a higher risk of BC than the general population. These BCs show familial aggregation, i.e., FBC.
Next, we introduce the genetic syndromes that have been confirmed to be associated with an increased risk of BC (Table 3).

Table 3 BC-related Genetic Syndromes.
3.7.1 Hereditary Breast and Ovarian Cancer (HBOC)
HBOC is an inherited disorder that was first reported in the 1970s (144). The clinical characteristics of HBOC include young age of onset, multiple family members with BC or OC, or both, or one family member with both BC and OC, or bilateral BC (145). The most common genetic changes associated with HBOC are mutations in the BRCA1 and BRCA2 genes (30). Approximately 60% of the typical families with HBOC harbor BRCA mutations (70). In China, this proportion was approximately 15.8% (146). Finally, because BRCA gene mutations are common in FBC, some patients with HBOC exhibit similar symptoms as patients with FBC.
3.7.2 Li-Fraumeni Syndrome (LFS)
LFS was first discovered in 1969 as an autosomal dominant malignant tumor syndrome caused by a germline mutation in the TP53 tumor suppressor gene. The TP53 gene mutation can be detected in approximately 50%–70% of the LFS cases, which is much higher than its prevalence of 1% in BC cases (147, 148). Furthermore, patients with LFS may suffer from multiple cancers, including BC, brain tumors, soft tissue sarcoma, leukemia, osteosarcoma, adrenal cortical malignancies, and broncho-alveolar lung cancer. The lifetime risk of developing BC in patients with LFS is estimated to be 25%–79% (149, 150). Some patients with BC of LFS show familial aggregation. Therefore, the NCCN guidelines recommend that women with TP53 pathogenic variant/likely pathogenic variant undergo a clinical breast examination every 6–12 months beginning at the age of 20 and a breast magnetic resonance imaging (MRI) with contrast screening every year from 20–75 years (151).
3.7.3 Hereditary Diffuse Gastric Cancer (HDGC)
HDGC is an autosomal dominant genetic disease caused by a CDH1 mutation. Patients with HDGC are susceptible to lobular breast cancer (LBC). The cumulative risk of LBC in women with CDH1 mutations is estimated to be 39%–52% by age 80 (143, 152, 153).
3.7.4 Ataxia Telangiectasia
Ataxia telangiectasia is an autosomal recessive genetic disease that is closely related to ATM mutations. The clinical manifestations include eyelid telangiectasia, cerebellar ataxia, and immunodeficiency (140). Some studies have shown that heterozygous carriers of ATM mutations have an increased risk of BC. A meta-analysis including three cohort studies of relatives of patients with ataxia telangiectasia estimated that the relative risk of BC in patients with ataxia telangiectasia is approximately 18% at 80 years of age (112). Moreover, if one has a history of radiation exposure, the risk increases further (154).
3.7.5 Cowden Syndrome
Cowden syndrome is an autosomal dominant genetic disease, which is clinically characterized by hamartoma-like lesions, pathognomonic skin lesions, benign breast disease, early-onset BC, and thyroid cancer. This disease is closely related to the PTEN/MMAC1/TEP1 gene mutations. Furthermore, approximately 75% of the female patients with Cowden syndrome harbor multiple benign breast lesions, such as fibroadenoma, cystic lesions, and ductal hyperplasia (155). A French study estimated that the cumulative BC risk in patients with Cowden syndrome was between 25 and 85%, and the cumulative incidence of BC by the age of 70 was 77% (95% CI: 59%–91%) (156).
3.7.6 Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome is a rare autosomal dominant genetic disease caused by mutations in the STK11 gene. The clinical manifestations of Peutz-Jeghers syndrome include hamartoma-like polyps in the gastrointestinal tract, melanin deposition in the skin and mucous membranes, pancreatic cancer, and mucocutaneous periorificial lentiginosis. The lifetime risk of patients with Peutz-Jeghers syndrome developing BC is 24%–54%, and the average age of onset is approximately 39 years (157, 158). The NCCN guidelines recommend that carriers of STK11 gene mutations undergo clinical breast examinations every 6 months and annual mammography and breast MRI examinations from the age of 25 (159).
3.8 Screening Strategies for FBC
FBC is characterized by familial aggregation, and family members of patients with FBC have a higher lifetime risk of disease than the general population (8). Generally, the screening processes for high-risk groups of FBC are as follows: professional genetic counselors use screening tools to identify potentially diseased members of the family; women who have a positive screening result receive genetic counseling to decide whether to perform advanced genetic counseling or BRCA genetic testing; and finally, early monitoring and physical examination of these high-risk groups are carried out to achieve early detection and treatment (160). This set of procedures is utilized in some countries (161); however, due to the differing FBC gene mutations among ethnic and geographical groups, when applied to the Chinese population, this procedure needs to be modified to meet the differing needs of this specific population (Figure 6).
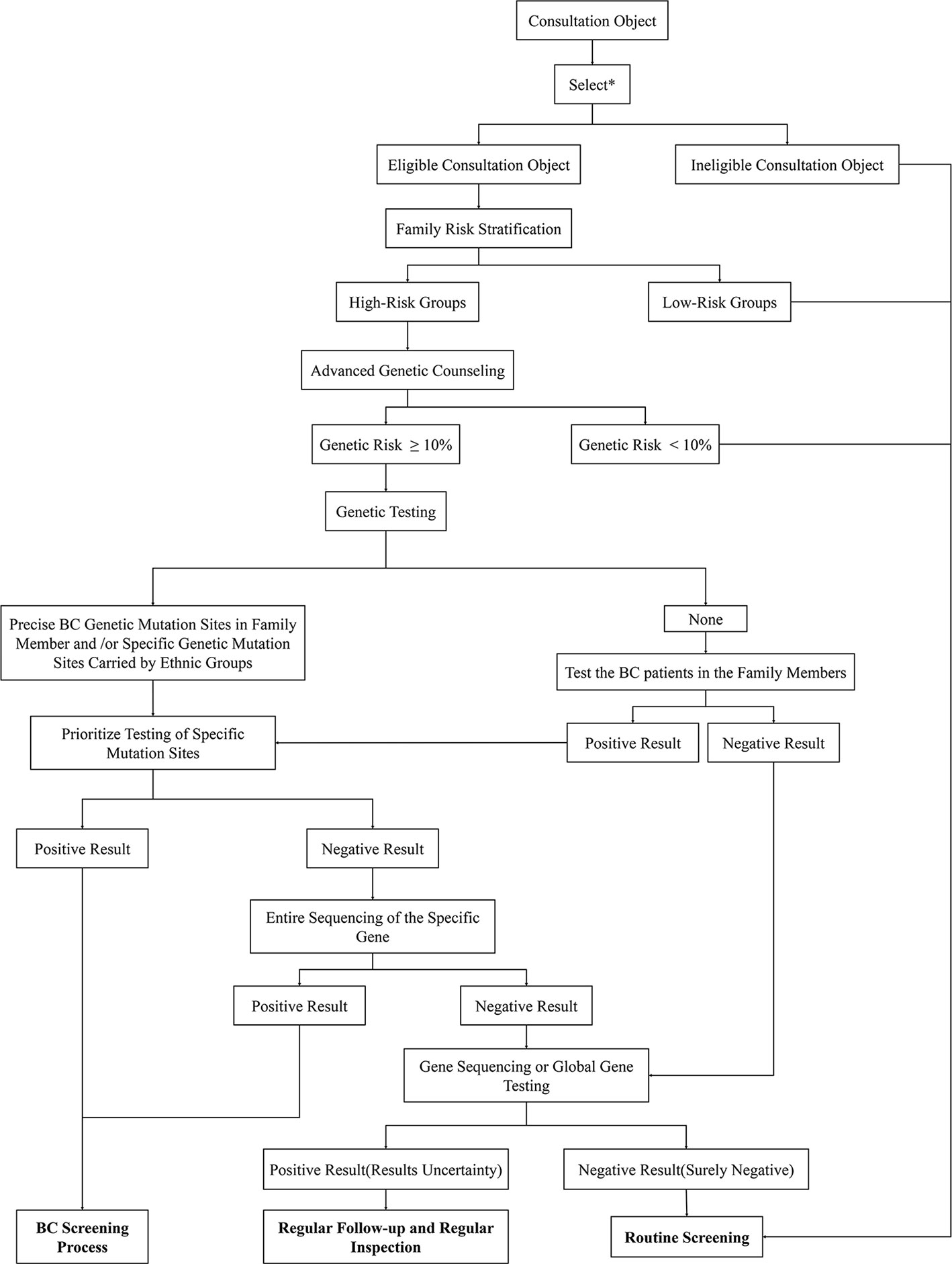
Figure 6 Genetic counseling strategies for high-risk BCs in China. First, identify eligible consultation participants through the listed criteria. Then use risk stratification tools to screen high-risk groups from the selected subjects. The lifetime risk of BC of these participants will be calculated through the risk assessment model in preliminary genetic counseling. High-risk subjects with a disease risk greater than 10% will be further subjected to follow-up genetic testing. Genetic testing gives priority to the detection of high-risk mutant gene sites in individuals of a family or Chinese populations. If obvious abnormalities are found, then specific mutation sites can be considered, which should undergo the BC screening process; if there are no abnormal findings, the entire sequence can be considered. In addition, the positive subjects receive BC screening while the negative participants receive whole genome sequencing. The final genetic test results need to be compared with the genetic test results of patients with BC in the family. If the mutant gene is the same, the consultation subject is confirmed to be in the high-risk group; if the results are inconsistent, the subject will not be listed as high-risk for the time being. For those whose family members have not been tested for genetic mutations, the first step is to detect the mutated gene sites in patients with BC. If the family member cannot be tested or the test result is negative, whole-genome sequencing can be performed. If there are clear mutation sites in the family members with BC, priority will be given to monitoring these gene sites. The subsequent detection steps are the same as those described above. Result analysis: 1. Positive Result (The consultation participant showed clear mutation genes that were consistent with those of a family member or ethnic group): Corresponding measures can be taken to actively intervene in the clinic; 2. Uncertainty negative (no obvious abnormality was found in the gene testing, but whole genome sequencing was not performed due to other factors): regular clinical follow-up; 3. Results Uncertainty (the patient found clear mutation genes but inconsistent with family): Regular clinical follow-up; 4. Surely negative (no abnormality was found after all inspections were completed): Routine examination. *The selection criteria are shown in Figure 7.
3.8.1 Genetic Counseling
Genetic counseling is a counseling process for relatives who have genetic diseases or are at risk of infection to provide disease occurrence and early detection or prophylactic intervention methods. Genetic counseling can prevent genetic diseases and provide insight regarding reproductive options and should be conducted by a well-trained professional counselor. The professional counselor conducts family investigation and analysis by evaluating the proband in the family and estimating the possibility of the disease in the offspring.
Genetic counseling for healthy family members of FBC is an important step in the screening process for high-risk populations and should be performed before blood is drawn to collect DNA to identify pathogenic mutations. However, not all individuals are required to receive genetic counseling. Eligible participants should meet one of the following criteria: 1. Personal medical history with genetic risk of i) early-onset BC (≤35 years old); ii) both BC and OC; iii) simultaneous cancers other than BC and OC; and 2. Significant FH of BC/OC: i) BC ≤50 years old; ii) bilateral BC; iii) at least three family members with ovarian, peritoneal, or tubal cancer; iv) at least one male family member with BC; v) multiple cases of BC in the family; vi) at least one primary cancer patient in the family with BRCA-related diseases; and vii) Nordic Jewish descent (Figure 7) (4, 160).
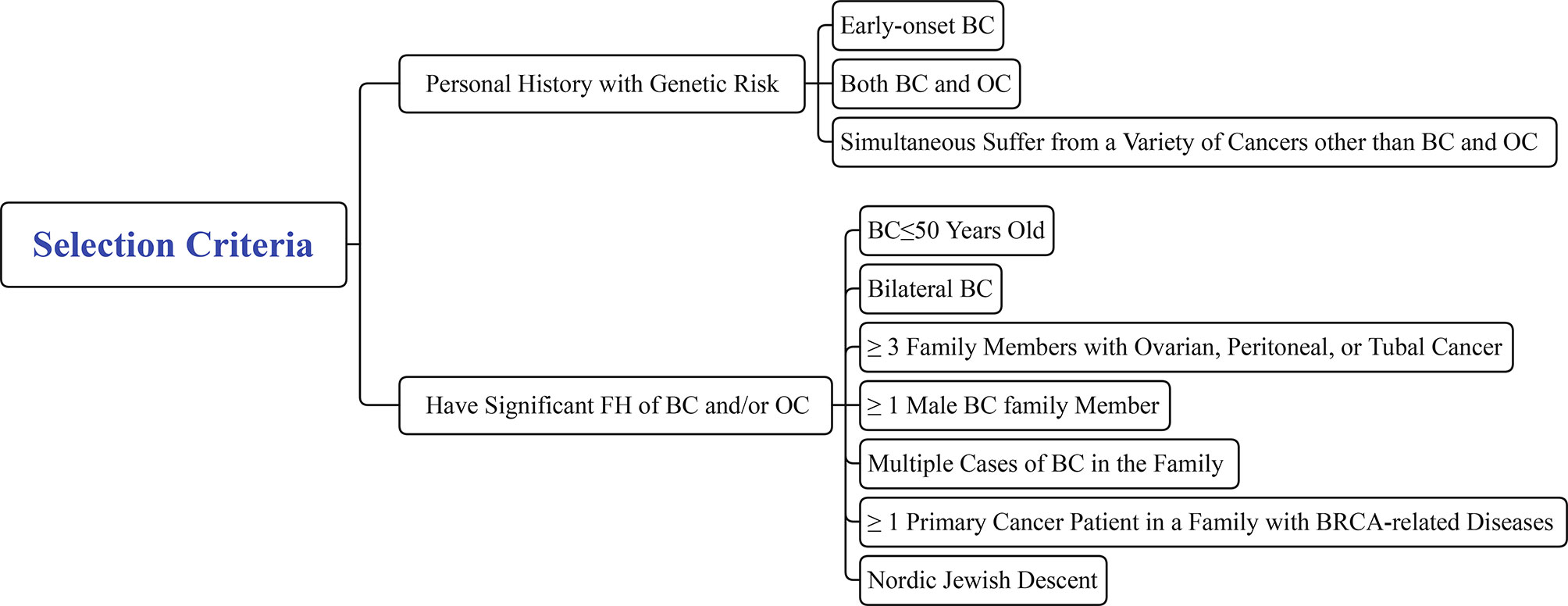
Figure 7 Selection criteria for consulting participants.
During genetic counseling, genetic counselors can use family risk stratification tools, such as the Manchester Scoring System (MSS), Family History Screen-7, Pedigree Assessment Tool, Referral Screening Tool, and Ontario Family History Assessment Tool (Ontario-FHAT) to distinguish the participants who need follow-up consultation. Healthy populations with a high risk of BC need advanced genetic counseling or BRCA gene mutation testing (162, 163). For others with a low risk of BC, routine screening strategies are recommended (161). Following the BC screening strategy for the Chinese population (2021 version), the low risk population undergoes X-ray or B-ultrasound examinations every 1–2 years, while high-risk groups undergo annual X-ray and B-ultrasound examinations and breast MRI when necessary. Patients that fall between low and high risk undergo X-ray and B-ultrasound examinations every 1–2 years (164).
3.8.2 Genetic Testing
Genetic testing is conducive to the early detection of high-risk groups of BC, providing preventive measures, and improving prognosis (165). Currently, only two genes (BRCA1 and BRCA2) are routinely used for BC genetic testing, and the test subjects are usually limited to women with an FH of BC and OC. However, given that genetic testing is relatively expensive and may cause adverse socio-psychological effects, testing should target individuals that most likely have gene mutations (166, 167). For example, BRCA1/2 testing is only provided to individuals with a mutation probability greater than or equal to 10% in many regions such as British (168). Therefore, advanced genetic counseling or models should be used to predict the likelihood of BRCA1 and BRCA2 mutations before performing genetic testing. When a patient qualifies as a test participant, they should fully understand the advantages and disadvantages of genetic testing and provide fully informed and written consent before genetic testing is possible (5).
It is generally recommended that female test subjects younger than 18 years of age undergo BRCA genetic testing. The focus of mutation analysis depends on the FH. If there are family members with BC with precise genetic mutation sites or ethnic groups carrying specific genetic mutation sites, these sites can be tested first (169). If the test subject does not have these characteristics, the entire specific gene sequence must be tested to exclude other site mutations. It is worth noting that women who are clinically negative for BRCA mutations are also at risk for BC (170). Walsh et al. found that among high-risk American families for BC and whose BRCA1 and BRCA2 gene test results are negative, approximately 12% still carry genetic mutations in BRCA1, BRCA2, CHEK2, TP53, and PTEN (70). These false negatives can easily lead to a missed diagnosis in high-risk groups. In addition, most of the gene mutations in FBC are unrelated to BRCA1, such as ATM, CHEK2, and BARD1, in the homologous recombination pathway (171). Further studies have shown that the truncated variants of ATM and CHEK2 are more closely associated with estrogen receptor (ER)-positive BC, while BARD1, BRCA1, BRCA2, PALB2, RAD51C, and RAD51D are more closely related to ER-negative BC (29). Therefore, it is believed that both sequencing and global screening for rearrangements should be performed for women with high risk for BC who are BRCA1/2 negative. For families with only negative BRCA1/2 sequencing test results, multiplex ligation-dependent probe amplification testing can be performed.
If the result of genetic testing is positive, that indicates the detected mutant gene is the same as the mutant gene of the BC patient in the family; therefore, the test subject needs to continue the screening process. Subsequent BC screening includes regular breast self-examination and imaging examinations. If the BC patient is not a mutation carrier, the test subject will temporarily not be listed as a high-risk patient. Those participants whose tests are all negative will temporarily not be classified as high-risk groups, and the general population screening process is recommended (161).
With the development of genetic testing, many new testing methods have emerged, such as multigene panel testing, which can detect multiple gene loci simultaneously. In a study of 35,000 women with BC, the 25-gene panel showed mutations most commonly in BRCA1, BRCA2, CHEK2, ATM, and PALB2 (172). Moreover, NCCN Guidelines 2017 stated that if more than one gene can explain hereditary cancer, polygenic testing may be required to be more effective and cost-effective. A recent study also demonstrated that population-based multigene panel testing is more cost-effective than individual BRCA1/BRCA2 testing (173). Moreover, a current study in China showed that multigene panel testing could increase the mutation detection rate in patients with a high risk of BC (137). Thus, although multigene panel testing still has shortcomings for practical applications (174), it may become an important technology in future FBC screening strategies.
From a technical point of view, it is practical to accurately detect BRCA1/2 mutations and other susceptible gene mutations in China. However, there is currently no uniform standard for the hot spots of FBC mutant genes in the Chinese population. Moreover, there is no uniform standard for genetic testing. The shortage of professionals to provide genetic counseling results in the imperfect prevention of uncertain genetic mutation carriers. Therefore, establishing an entirely professional team and standardized genetic counseling processes for FBC prevention and treatment are urgently needed.
3.8.3 Predictive Model
Only 5%–10% of the BC susceptibility gene carriers will eventually develop BC due to gene variations. Predicting the risk of BC in individuals with FH is helpful for the clinical prevention and treatment of FBC. At present, many clinical models that combine the patient’s personal history, FH, and other factors to assess the risk of BC have been proposed (175), although no standardized prediction method has been widely accepted.
3.8.4 Gail Model (1989)
The Gail model is a statistical analysis model based on case-control data, which integrates the risk factors of BC, including BC history, age, age at menarche, age at first birth, number of first-degree relatives with BC, breast biopsy results, and race. Gail-1 in 1989 was initially used to predict the risk of invasive BC and carcinoma in situ in white women who underwent mammography each year (176). Gail et al. modified Gail-2 in 1990 to improve its predictive power (175). Since then, the Gail model has been validated in many different populations (white, African American, Hispanic, Asian American, American Indian, and Alaska Natives).
3.8.5 Claus Model (1994)
The Claus model estimates the lifetime risk of BC based on the FH of first- and second-degree relatives with BC and OC (177). The reference parameters included the age of onset of the paternal and maternal lines and information on cancer history (178).
3.8.6 Penn Model (1997)
The Couch (Penn I) model was initially used to predict the probability of BRCA1 mutations in 169 families with BC. The Penn II model is based on logistic regression analysis and integrates specific clinical features (such as bilateral BC) to predict the possibility of BRCA1/2 mutations in individuals or families. Studies have shown that the prediction accuracy of the Penn II model is higher than that of the Penn I and Myriad II models (179).
3.8.7 BRCAPRO Model (1998)
The BRCAPRO model is based on the Bayes theorem, the prevalence of BC and OC in first- and second-degree relatives, and the age of onset of the disease in family members to screen BRCA1/2 gene mutation carriers. The model was developed into a computer software in 2002 (180, 181). At present, the model is continuously updated, and its feasibility has been confirmed (182).
3.8.8 Myriad Model (2002)
The Myriad model is based on 7,461 samples tested for BRCA1/2 mutations and 2,539 samples of detected mutations in three descendants of Ashkenazi Jewish ancestry (including FH, age of onset of FBOC, and presence of invasive cancer) to establish a model to predict the possibility of carrying mutations. The model has been publicly released at present, and the sample size is being expanded for research and updates (183).
3.8.9 BOADICEA Model (2004)
The BOADICEA model was developed based on the complex isolation analysis of the occurrence of BC and OC in the combined data of 1,484 BC cases and 156 multi-case families (184). This model not only allows for simultaneous effects of both BRCA1 and BRCA2 but also allows for the effects of genetic modifiers and the multiplier effect of low penetrance genes on BC risk (185).
3.8.10 Manchester Scoring System (2004)
Evans et al. used whole-gene screening technology to screen the DNA samples of 422 non-Jewish patients with a history of BC/OC for BRCA1 mutations and performed BRCA2 screening for 318 subsets. After combining the screening results and FH, a simple scoring system, the Manchester scoring system, was designed to predict pathogenic mutations (186). In 2009 and 2017, MSS2 and MSS3, respectively, were released, where the pathological weight was considered in the scoring system, and the corresponding score was adjusted (187, 188). For subjects in Northwest England, a total MSS3 score of 15–19 is equal to the 10% threshold, and a score of 20 points is equal to the 20% threshold, but the threshold may need to be adjusted when applied to other populations.
4 Discussion
BC is currently the most common cancer among women worldwide. The incidence of BC in 2020 is the highest while the mortality rate is only fifth in the world, highlighting the need to prevent and treat BC. Compared with SBC, the cause of FBC seems to be clearer. Many studies have discovered multiple gene mutations related to FBC, such as BRCA1, ATM, and CHEK2. Scholars divide these gene mutations into three categories based on the RR. As the research progressed, some genes that were originally considered to be moderate risk were reclassified into the high penetrance category, such as TP53. Some of these gene mutations also exist in the Chinese BC population, while some specific gene variants, such as PALB2 c.3271delC, are relatively unique to the Chinese population. In summary, we introduced 16 genes that have been identified to be closely associated with FBC. Accordingly, we described these genes in detail based on their different trajectories. Moreover, genetic syndromes associated with FBC have also been discussed with the corresponding risk of disease, such as HBOC, LFS, and HDGC. The NCCN guidelines also recommend corresponding screening strategies for these genetic diseases. However, such guidelines for the Chinese population have not yet been released. Considering the heterogeneity of BC, we listed the hotspot mutation genes and their loci in the Chinese FBC population and then proposed a screening flowchart for high-risk Chinese populations for FBC based on existing screening strategies.
At present, there is no standardized process for the diagnosis and treatment of FBC in China. High-risk groups can be screened out only through FH screening. After genetic counseling, some people are selected for corresponding genetic testing based on economic conditions as well as the counseling results, and the genetic testing results are combined to provide complementary preventive treatments. Although many predictive models have been established to predict disease risk, and some of them seem to be applicable to the Chinese population, there is currently no professional model designed specifically for the Chinese population. The lack of suitable predictive models and professional genetic counseling has led to the lack of standardization of early screening of FBC high-risk populations in China. An FBC screening strategy based on the risk of mutant genes specifically for the Chinese population is therefore urgently needed. This strategy can help in the early identification of high-risk patients among the families of patients with BC with FH in the clinic and provide a framework for subsequent in-depth therapeutic interventions and research. Due to the vast territory of China, the characteristics of BC in different areas or ethnic groups vary. It is difficult to collect the BC characteristics of the entire country, and further studies that can provide more objective evidence are required.
This study has several limitations. First, it lacks a systematic quantitative analysis, and there is a certain selection bias in the selection of the cited documents in this review, which may lead to bias in the research results and conclusions. In addition, the study of the Chinese population only selected populations in a particular region. Whether the results apply to the entire Chinese population remains to be confirmed.
In conclusion, early detection of FBC is a critical step in its treatment. FH, related mutant genes, and genetic syndromes provide a solid foundation for genetic counseling. For the Chinese population, different screening strategies need to be adopted based on unique genetic information.
Author Contributions
LS wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (grant no. Y19H160283).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
LS especially thanked her mentor, XW, who gave constructive suggestions in the conception and writing of the review.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 686:394–424. doi: 10.3322/caac.21492
2. Rosen EM, Fan S, Pestell RG, Goldberg ID. BRCA1 Gene in Breast Cancer. J Cell Physiol (2003) 1961:19–41. doi: 10.1002/jcp.10257
3. Ripperger T, Gadzicki D, Meindl A, Schlegelberger B. Breast Cancer Susceptibility: Current Knowledge and Implications for Genetic Counselling. Eur J Hum Genet (2009) 176:722–31. doi: 10.1038/ejhg.2008.212
4. Arpino G, Pensabene M, Condello C, Ruocco R, Cerillo I, Lauria R, et al. Tumor Characteristics and Prognosis in Familial Breast Cancer. BMC Cancer (2016) 16:924. doi: 10.1186/s12885-016-2962-1
5. Lynch HT, Silva E, Snyder C, Lynch JF. Hereditary Breast Cancer: Part I. Diagnosing Hereditary Breast Cancer Syndromes. Breast J (2008) 141:3–13. doi: 10.1111/j.1524-4741.2007.00515.x
6. Tsang JYS, Tse GM. Molecular Classification of Breast Cancer. Adv Anat Pathol (2020) 271:27–35. doi: 10.1097/pap.0000000000000232
7. Prado A, Andrades P, Parada F. Recent Developments in the Ability to Predict and Modify Breast Cancer Risk. J Plast Reconstr Aesthet Surg (2010) 6310:1581–7. doi: 10.1016/j.bjps.2009.06.034
8. Margolin S, Johansson H, Rutqvist LE, Lindblom A, Fornander T. Family History, and Impact on Clinical Presentation and Prognosis, in a Population-Based Breast Cancer Cohort From the Stockholm County. Fam Cancer (2006) 54:309–21. doi: 10.1007/s10689-006-7851-3
9. Honrado E, Benítez J, Palacios J. The Molecular Pathology of Hereditary Breast Cancer: Genetic Testing and Therapeutic Implications. Mod Pathol (2005) 1810:1305–20. doi: 10.1038/modpathol.3800453
10. Hemminki K, Granström C. Familial Breast Cancer: Scope for More Susceptibility Genes? Breast Cancer Res Treat (2003) 821:17–22. doi: 10.1023/B:BREA.0000003871.38587.8b
11. Couto E, Hemminki K. Estimates of Heritable and Environmental Components of Familial Breast Cancer Using Family History Information. Br J Cancer (2007) 9611:1740–2. doi: 10.1038/sj.bjc.6603753
12. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and Heritable Factors in the Causation of Cancer–Analyses of Cohorts of Twins From Sweden, Denmark, and Finland. N Engl J Med (2000) 3432:78–85. doi: 10.1056/nejm200007133430201
13. Huang Y, Ballinger DG, Dai JY, Peters U, Hinds DA, Cox DR, et al. Genetic Variants in the MRPS30 Region and Postmenopausal Breast Cancer Risk. Genome Med (2011) 36:42. doi: 10.1186/gm258
14. Lynch HT, Lynch PM, Albano WA, Edney J, Organ CH, Lynch JF. Hereditary Cancer: Ascertainment and Management. CA Cancer J Clin (1979) 294:216–32. doi: 10.3322/canjclin.29.4.216
15. Phipps RF, Perry PM. Familial Breast Cancer. Postgrad Med J (1988) 64757:847–9. doi: 10.1136/pgmj.64.757.847
16. Fukutomi T. Hereditary/familial Breast Cancer. Nihon rinsho Japanese J Clin Med (2007) 65 Suppl6:245–8.
17. Lee JY, Kim J, Kim SW, Park SK, Ahn SH, Lee MH, et al. BRCA1/2-Negative, High-Risk Breast Cancers (BRCAX) for Asian Women: Genetic Susceptibility Loci and Their Potential Impacts. Sci Rep (2018) 81:15263. doi: 10.1038/s41598-018-31859-8
19. Beral V, Bull D, Doll R, Peto R, Reeves G, Skegg G, et al. Familial Breast Cancer: Collaborative Reanalysis of Individual Data From 52 Epidemiological Studies Including 58,209 Women With Breast Cancer and 101,986 Women Without the Disease. Lancet (2001) 3589291:1389–99. doi: 10.1016/s0140-6736(01)06524-2
20. Bertoni N, de Souza MC, Crocamo S, Szklo M, de Almeida LM. Is a Family History of the Breast Cancer Related to Women’s Cancer Prevention Behaviors? Int J Behav Med (2019) 261:85–90. doi: 10.1007/s12529-018-9737-9
21. Skol AD, Sasaki MM, Onel K. The Genetics of Breast Cancer Risk in the Post-Genome Era: Thoughts on Study Design to Move Past BRCA and Towards Clinical Relevance. Breast Cancer Res (2016) 181:99. doi: 10.1186/s13058-016-0759-4
22. Stratton MR, Rahman N. The Emerging Landscape of Breast Cancer Susceptibility. Nat Genet (2008) 401:17–22. doi: 10.1038/ng.2007.53
23. Michailidou K, Lindström S, Dennis J, Beesley J, Hui S, Kar S, et al. Association Analysis Identifies 65 New Breast Cancer Risk Loci. Nature (2017) 5517678:92–4. doi: 10.1038/nature24284
24. Melchor L, Benítez J. The Complex Genetic Landscape of Familial Breast Cancer. Hum Genet (2013) 1328:845–63. doi: 10.1007/s00439-013-1299-y
25. Foulkes WD. Inherited Susceptibility to Common Cancers. N Engl J Med (2008) 35920:2143–53. doi: 10.1056/NEJMra0802968
26. Sud A, Kinnersley B, Houlston RS. Genome-Wide Association Studies of Cancer: Current Insights and Future Perspectives. Nat Rev Cancer (2017) 1711:692–704. doi: 10.1038/nrc.2017.82
27. Ghoussaini M, Pharoah PDP, Easton DF. Inherited Genetic Susceptibility to Breast Cancer: The Beginning of the End or the End of the Beginning? Am J Pathol (2013) 1834:1038–51. doi: 10.1016/j.ajpath.2013.07.003
28. Yip CH, Evans DG, Agarwal G, Buccimazza I, Kwong A, Morant R, et al. Global Disparities in Breast Cancer Genetics Testing, Counselling and Management. World J Surg (2019) 435:1264–70. doi: 10.1007/s00268-018-04897-6
29. Dorling L, Carvalho S, Allen J, González-Neira A, Luccarini C, Wahlström C, et al. Breast Cancer Risk Genes - Association Analysis in More Than 113,000 Women. N Engl J Med (2021) 3845:428–39. doi: 10.1056/NEJMoa1913948
30. Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, et al. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science (1990) 2504988:1684–9. doi: 10.1126/science.2270482
31. Smith SA, Easton DF, Evans DG, Ponder BA. Allele Losses in the Region 17q12-21 in Familial Breast and Ovarian Cancer Involve the Wild-Type Chromosome. Nat Genet (1992) 22:128–31. doi: 10.1038/ng1092-128
32. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A Strong Candidate for the Breast and Ovarian Cancer Susceptibility Gene BRCA1. Science (1994) 2665182:66–71. doi: 10.1126/science.7545954
33. Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, et al. The RING Heterodimer BRCA1-BARD1 Is a Ubiquitin Ligase Inactivated by a Breast Cancer-Derived Mutation. J Biol Chem (2001) 27618:14537–40. doi: 10.1074/jbc.C000881200
34. Yu X, Chini CC, He M, Mer G, Chen J. The BRCT Domain Is a Phospho-Protein Binding Domain. Science (2003) 3025645:639–42. doi: 10.1126/science.1088753
35. Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT Repeats as Phosphopeptide-Binding Modules Involved in Protein Targeting. Science (2003) 3025645:636–9. doi: 10.1126/science.1088877
36. Chatterjee G, Jimenez-Sainz J, Presti T, Nguyen T, Jensen RB. Distinct Binding of BRCA2 BRC Repeats to RAD51 Generates Differential DNA Damage Sensitivity. Nucleic Acids Res (2016) 4411:5256–70. doi: 10.1093/nar/gkw242
37. Davies AA, Masson JY, McIlwraith MJ, Stasiak AZ, Stasiak A, Venkitaraman AR, et al. Role of BRCA2 in Control of the RAD51 Recombination and DNA Repair Protein. Mol Cell (2001) 72:273–82. doi: 10.1016/s1097-2765(01)00175-7
38. Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 Links BRCA1 and BRCA2 in the DNA-Damage Response. Curr Biol (2009) 196:524–9. doi: 10.1016/j.cub.2009.02.018
39. Smith TM, Lee MK, Szabo CI, Jerome N, McEuen M, Taylor M, et al. Complete Genomic Sequence and Analysis of 117 Kb of Human DNA Containing the Gene BRCA1. Genome Res (1996) 611:1029–49. doi: 10.1101/gr.6.11.1029
40. Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as Regulators of DNA Repair, Transcription, and Cell Cycle in Response to DNA Damage. Cancer Sci (2004) 9511:866–71. doi: 10.1111/j.1349-7006.2004.tb02195.x
41. Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, et al. Localization of a Breast Cancer Susceptibility Gene, BRCA2, to Chromosome 13q12-13. Science (1994) 2655181:2088–90. doi: 10.1126/science.8091231
42. Tavtigian SV, Simard J, Rommens J, Couch F, Shattuck-Eidens D, Neuhausen S, et al. The Complete BRCA2 Gene and Mutations in Chromosome 13q-Linked Kindreds. Nat Genet (1996) 123:333–7. doi: 10.1038/ng0396-333
43. Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, et al. Truncating Mutations in the Fanconi Anemia J Gene BRIP1 Are Low-Penetrance Breast Cancer Susceptibility Alleles. Nat Genet (2006) 3811:1239–41. doi: 10.1038/ng1902
44. Fackenthal JD, Olopade OI. Breast Cancer Risk Associated With BRCA1 and BRCA2 in Diverse Populations. Nat Rev Cancer (2007) 712:937–48. doi: 10.1038/nrc2054
45. Couch FJ, Nathanson KL, Offit K. Two Decades After BRCA: Setting Paradigms in Personalized Cancer Care and Prevention. Science (2014) 3436178:1466–70. doi: 10.1126/science.1251827
46. Wang F, Fang Q, Ge Z, Yu N, Xu S, Fan X. Common BRCA1 and BRCA2 Mutations in Breast Cancer Families: A Meta-Analysis From Systematic Review. Mol Biol Rep (2012) 393:2109–18. doi: 10.1007/s11033-011-0958-0
47. Jara L, Morales S, de Mayo T, Gonzalez-Hormazabal P, Carrasco V, Godoy R. Mutations in BRCA1, BRCA2 and Other Breast and Ovarian Cancer Susceptibility Genes in Central and South American Populations. Biol Res (2017) 501:35. doi: 10.1186/s40659-017-0139-2
48. Kwong A, Shin VY, Ho JCW, Kang E, Nakamura S, Teo S-H, et al. Comprehensive Spectrum of BRCA1 and BRCA2 Deleterious Mutations in Breast Cancer in Asian Countries. J Med Genet (2016) 531:15–23. doi: 10.1136/jmedgenet-2015-103132
49. Zhu X, Tian T, Ruan M, Rao J, Yang W, Cai X, et al. Expression of DNA Damage Response Proteins and Associations With Clinicopathologic Characteristics in Chinese Familial Breast Cancer Patients With BRCA1/2 Mutations. J Breast Cancer (2018) 213:297–305. doi: 10.4048/jbc.2018.21.e38
50. Wang L, Zhou S, Xie J, Gao H, Wang F, Zhou J, et al. BRCA1 Germline Mutations Dominate Familial Breast Cancer Patients in Henan China. J Thorac Dis (2017) 912:5295–99. doi: 10.21037/jtd.2017.11.71
51. Song CG, Hu Z, Yuan WT, Di GH, Shen ZZ, Huang W, et al. BRCA1 and BRCA2 Gene Mutations of Familial Breast Cancer From Shanghai in China. Zhonghua Yi Xue Yi Chuan Xue Za Zhi (2006) 231:27–31.
52. Ma ZL, Cao MZ, Li WF. Analysis of BRCA2 Gene Mutations Among Familial and/or Early-Onset Breast Cancer Patients in Eastern Shandong of China. Zhonghua Yi Xue Yi Chuan Xue Za Zhi (2008) 252:195–8.
53. Kuo WH, Lin PH, Huang AC, Chien YH, Liu TP, Lu YS, et al. Multimodel Assessment of BRCA1 Mutations in Taiwanese (Ethnic Chinese) Women With Early-Onset, Bilateral or Familial Breast Cancer. J Hum Genet (2012) 572:130–8. doi: 10.1038/jhg.2011.142
54. Li WF, Hu Z, Zhang B, Cao MZ, Wang YS, Liu XY, et al. BRCA1 1100delat Is a Recurrent Mutation in Chinese Women With Familial Breast Cancer. Zhonghua Yi Xue Za Zhi (2007) 872:76–80.
55. Kwong A, Ng EKO, Wong CLP, Law FBF, Au T, Wong HN, et al. Identification of BRCA1/2 Founder Mutations in Southern Chinese Breast Cancer Patients Using Gene Sequencing and High Resolution DNA Melting Analysis. PloS One (2012) 79:e43994. doi: 10.1371/journal.pone.0043994
56. Shaw T, Chan SH, Teo JX, Chong ST, Li ST, Courtney E, et al. Investigation Into the Origins of an Ancient BRCA1 Founder Mutation Identified Among Chinese Families in Singapore. Int J Cancer (2021) 1483:637–45. doi: 10.1002/ijc.33241
57. Liu Y, Wang H, Wang X, Liu J, Li J, Wang X, et al. Prevalence and Reclassification of BRCA1 and BRCA2 Variants in a Large, Unselected Chinese Han Breast Cancer Cohort. J Hematol Oncol (2021) 141:18. doi: 10.1186/s13045-020-01010-0
58. Zhou YZ, Sun Q, Lin SQ, Wang J, Liu B, Li JX, et al. Germline Mutations in the BRCA1 and BRCA2 Genes From Breast Cancer Families in China Han People. Zhonghua Yi Xue Za Zhi (2004) 844:294–8.
59. Findlay GM, Daza R, Martin BK, Zhang MD, Leith AP, Gasperini M, et al. Accurate Functional Classification of Thousands of BRCA1 Variants With Saturation Genome Editing. Eur J Hum Genet (2019) 27:828–28. doi: 10.1101/294520
60. Perou CM, Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, et al. Comprehensive Molecular Portraits of Human Breast Tumours. Nature (2012) 4907418:61–70. doi: 10.1038/nature11412
61. Harris CC. Structure and Function of the P53 Tumor Suppressor Gene: Clues for Rational Cancer Therapeutic Strategies. J Natl Cancer Inst (1996) 8820:1442–55. doi: 10.1093/jnci/88.20.1442
62. Riley T, Sontag E, Chen P, Levine A. Transcriptional Control of Human P53-Regulated Genes. Nat Rev Mol Cell Biol (2008) 95:402–12. doi: 10.1038/nrm2395
63. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, et al. Mice Deficient for P53 Are Developmentally Normal But Susceptible to Spontaneous Tumours. Nature (1992) 3566366:215–21. doi: 10.1038/356215a0
64. Kuperwasser C, Hurlbut GD, Kittrell FS, Dickinson ES, Laucirica R, Medina D, et al. Development of Spontaneous Mammary Tumors in BALB/c P53 Heterozygous Mice. A Model for Li-Fraumeni Syndrome. Am J Pathol (2000) 1576:2151–9. doi: 10.1016/s0002-9440(10)64853-5
65. Kastenhuber ER, Lowe SW. Putting P53 in Context. Cell (2017) 1706:1062–78. doi: 10.1016/j.cell.2017.08.028
66. Bergh J, Norberg T, Sjögren S, Lindgren A, Holmberg L. Complete Sequencing of the P53 Gene Provides Prognostic Information in Breast Cancer Patients, Particularly in Relation to Adjuvant Systemic Therapy and Radiotherapy. Nat Med (1995) 110:1029–34. doi: 10.1038/nm1095-1029
67. Blaszyk H, Hartmann A, Cunningham JM, Schaid D, Wold LE, Kovach JS, et al. A Prospective Trial of Midwest Breast Cancer Patients: A P53 Gene Mutation Is the Most Important Predictor of Adverse Outcome. Int J Cancer (2000) 891:32–8. doi: 10.1002/(sici)1097-0215(20000120)89:1<32::aid-ijc6>3.0.co;2-g
68. Shahbandi A, Nguyen HD, Jackson JG. TP53 Mutations and Outcomes in Breast Cancer: Reading Beyond the Headlines. Trends Cancer (2020) 62:98–110. doi: 10.1016/j.trecan.2020.01.007
69. Hollstein M, Hergenhahn M, Yang Q, Bartsch H, Wang ZQ, Hainaut P. New Approaches to Understanding P53 Gene Tumor Mutation Spectra. Mutat Res (1999) 4312:199–209. doi: 10.1016/s0027-5107(99)00162-1
70. Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, et al. Spectrum of Mutations in BRCA1, BRCA2, CHEK2, and TP53 in Families at High Risk of Breast Cancer. Jama (2006) 29512:1379–88. doi: 10.1001/jama.295.12.1379
71. Malkin D. Germline P53 Mutations and Heritable Cancer. Annu Rev Genet (1994) 28:443–65. doi: 10.1146/annurev.ge.28.120194.002303
72. Hu X, Ouyang H, Wang H, Li H, Chen F, Dai X, et al. P53 Gene Mutations of Familial Breast Cancer and Early-Onset Breast Cancer in Part Population of Southern China. Zhong Nan Da Xue Xue Bao Yi Xue Ban (2017) 425:493–500. doi: 10.11817/j.issn.1672-7347.2017.05.002
73. Pezzolesi MG, Zbuk KM, Waite KA, Eng C. Comparative Genomic and Functional Analyses Reveal a Novel Cis-Acting PTEN Regulatory Element as a Highly Conserved Functional E-Box Motif Deleted in Cowden Syndrome. Hum Mol Genet (2007) 169:1058–71. doi: 10.1093/hmg/ddm053
74. Ngeow J, Sesock K, Eng C. Breast Cancer Risk and Clinical Implications for Germline PTEN Mutation Carriers. Breast Cancer Res Treat (2017) 1651:1–8. doi: 10.1007/s10549-015-3665-z
75. Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, et al. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol Cell (2006) 226:719–29. doi: 10.1016/j.molcel.2006.05.022
76. Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic Mutations in PALB2 Cause Fanconi Anemia Subtype FA-N and Predispose to Childhood Cancer. Nat Genet (2007) 392:162–4. doi: 10.1038/ng1947
77. Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, et al. Fanconi Anemia Is Associated With a Defect in the BRCA2 Partner PALB2. Nat Genet (2007) 392:159–61. doi: 10.1038/ng1942
78. Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li G, et al. Analysis of PALB2/FANCN-Associated Breast Cancer Families. Proc Natl Acad Sci USA (2007) 10416:6788–93. doi: 10.1073/pnas.0701724104
79. Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, et al. PALB2, Which Encodes a BRCA2-Interacting Protein, Is a Breast Cancer Susceptibility Gene. Nat Genet (2007) 392:165–7. doi: 10.1038/ng1959
80. Erkko H, Xia B, Nikkilä J, Schleutker J, Syrjäkoski K, Mannermaa A, et al. A Recurrent Mutation in PALB2 in Finnish Cancer Families. Nature (2007) 4467133:316–9. doi: 10.1038/nature05609
81. Sopik V, Narod SA. Breast-Cancer Risk in Families With Mutations in PALB2. N Engl J Med (2014) 37117:1650. doi: 10.1056/NEJMc1410673#SA1
82. Zhang K, Zhou J, Zhu X, Luo M, Xu C, Yu J, et al. Germline Mutations of PALB2 Gene in a Sequential Series of Chinese Patients With Breast Cancer. Breast Cancer Res Treat (2017) 1663:865–73. doi: 10.1007/s10549-017-4425-z
83. Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkäs K, Roberts J, et al. Breast-Cancer Risk in Families With Mutations in PALB2. N Engl J Med (2014) 3716:497–506. doi: 10.1056/NEJMoa1400382
84. van Roy F, Berx G. The Cell-Cell Adhesion Molecule E-Cadherin. Cell Mol Life Sci (2008) 6523:3756–88. doi: 10.1007/s00018-008-8281-1
85. Christofori G, Semb H. The Role of the Cell-Adhesion Molecule E-Cadherin as a Tumour-Suppressor Gene. Trends Biochem Sci (1999) 242:73–6. doi: 10.1016/s0968-0004(98)01343-7
86. Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, et al. E-Cadherin-Mediated Cell-Cell Adhesion Prevents Invasiveness of Human Carcinoma Cells. J Cell Biol (1991) 1131:173–85. doi: 10.1083/jcb.113.1.173
87. Vleminckx K, Vakaet L Jr., Mareel M, Fiers W, van Roy F. Genetic Manipulation of E-Cadherin Expression by Epithelial Tumor Cells Reveals an Invasion Suppressor Role. Cell (1991) 661:107–19. doi: 10.1016/0092-8674(91)90143-m
88. Berx G, van Roy F. Involvement of Members of the Cadherin Superfamily in Cancer. Cold Spring Harb Perspect Biol (2009) 16:a003129. doi: 10.1101/cshperspect.a003129
89. Corso G, Carvalho J, Marrelli D, Vindigni C, Carvalho B, Seruca R, et al. Somatic Mutations and Deletions of the E-Cadherin Gene Predict Poor Survival of Patients With Gastric Cancer. J Clin Oncol (2013) 317:868–75. doi: 10.1200/jco.2012.44.4612
90. Hansford S, Kaurah P, Li-Chang H, Woo M, Senz J, Pinheiro H, et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol (2015) 11:23–32. doi: 10.1001/jamaoncol.2014.168
91. Benusiglio PR, Malka D, Rouleau E, De Pauw A, Buecher B, Noguès C, et al. CDH1 Germline Mutations and the Hereditary Diffuse Gastric and Lobular Breast Cancer Syndrome: A Multicentre Study. J Med Genet (2013) 507:486–9. doi: 10.1136/jmedgenet-2012-101472
92. Xu X, Jin D, Durgan J, Hall A. LKB1 Controls Human Bronchial Epithelial Morphogenesis Through P114rhogef-Dependent RhoA Activation. Mol Cell Biol (2013) 3314:2671–82. doi: 10.1128/mcb.00154-13
93. Lim W, Olschwang S, Keller JJ, Westerman AM, Menko FH, Boardman LA, et al. Relative Frequency and Morphology of Cancers in STK11 Mutation Carriers. Gastroenterology (2004) 1267:1788–94. doi: 10.1053/j.gastro.2004.03.014
94. Trovó-Marqui AB, Tajara EH. Neurofibromin: A General Outlook. Clin Genet (2006) 701:1–13. doi: 10.1111/j.1399-0004.2006.00639.x
95. Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant Regulation of Ras Proteins in Malignant Tumour Cells From Type 1 Neurofibromatosis Patients. Nature (1992) 3566371:713–5. doi: 10.1038/356713a0
96. Evans DG, O’Hara C, Wilding A, Ingham SL, Howard E, Dawson J, et al. Mortality in Neurofibromatosis 1: In North West England: An Assessment of Actuarial Survival in a Region of the UK Since 1989. Eur J Hum Genet (2011) 1911:1187–91. doi: 10.1038/ejhg.2011.113
97. Uusitalo E, Leppävirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, et al. Incidence and Mortality of Neurofibromatosis: A Total Population Study in Finland. J Invest Dermatol (2015) 1353:904–06. doi: 10.1038/jid.2014.465
98. Cui J, Antoniou AC, Dite GS, Southey MC, Venter DJ, Easton DF, et al. After BRCA1 and BRCA2-What Next? Multifactorial Segregation Analyses of Three-Generation, Population-Based Australian Families Affected by Female Breast Cancer. Am J Hum Genet (2001) 682:420–31. doi: 10.1086/318187
99. Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 Functions as a DNA Damage Checkpoint in G(1) by Stabilizing P53. Genes Dev (2000) 143:278–88. doi: 10.1101/gad.14.3.278
100. Yang S, Kuo C, Bisi JE, Kim MK. PML-Dependent Apoptosis After DNA Damage Is Regulated by the Checkpoint Kinase Hcds1/Chk2. Nat Cell Biol (2002) 411:865–70. doi: 10.1038/ncb869
101. Bell DW, Varley JM, Szydlo TE, Kang DH, Wahrer DC, Shannon KE, et al. Heterozygous Germ Line Hchk2 Mutations in Li-Fraumeni Syndrome. Science (1999) 2865449:2528–31. doi: 10.1126/science.286.5449.2528
102. Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, et al. Low-Penetrance Susceptibility to Breast Cancer Due to CHEK2(*)1100delC in Noncarriers of BRCA1 or BRCA2 Mutations. Nat Genet (2002) 311:55–9. doi: 10.1038/ng879
103. Douglas E, Lesley G, Deborah H, Alison D, Louise T, Caroline B, et al. CHEK2*1100delC and Susceptibility to Breast Cancer: A Collaborative Analysis Involving 10,860 Breast Cancer Cases and 9,065 Controls From 10 Studies. Am J Hum Genet (2004) 746:1175–82. doi: 10.1086/421251
104. Bernstein JL, Teraoka SN, John EM, Andrulis IL, Knight JA, Lapinski R, et al. The CHEK2*1100delC Allelic Variant and Risk of Breast Cancer: Screening Results From the Breast Cancer Family Registry. Cancer Epidemiol Biomarkers Prev (2006) 152:348–52. doi: 10.1158/1055-9965.Epi-05-0557
105. Chen W, Yurong S, Liansheng N. Breast Cancer Low-Penetrance Allele 1100delc in the CHEK2 Gene: Not Present in the Chinese Familial Breast Cancer Population. Adv Ther (2008) 255:496–501. doi: 10.1007/s12325-008-0057-3
106. Liu Y, Liao J, Xu Y, Chen W, Liu D, Ouyang T, et al. A Recurrent CHEK2 P.H371Y Mutation Is Associated With Breast Cancer Risk in Chinese Women. Hum Mutat (2011) 329:1000–3. doi: 10.1002/humu.21538
107. Zaki-Dizaji M, Akrami SM, Abolhassani H, Rezaei N, Aghamohammadi A. Ataxia Telangiectasia Syndrome: Moonlighting ATM. Expert Rev Clin Immunol (2017) 1312:1155–72. doi: 10.1080/1744666x.2017.1392856
108. Swift M, Reitnauer PJ, Morrell D, Chase CL. Breast and Other Cancers in Families With Ataxia-Telangiectasia. N Engl J Med (1987) 31621:1289–94. doi: 10.1056/nejm198705213162101
109. Kelsey KT, Wiencke JK. Growing Pains for the Environmental Genetics of Breast Cancer: Observations on a Study of the Glutathione S-Transferases. J Natl Cancer Inst (1998) 907:484–5. doi: 10.1093/jnci/90.7.484
110. Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, et al. Cancer Risks and Mortality in Heterozygous ATM Mutation Carriers. J Natl Cancer Inst (2005) 9711:813–22. doi: 10.1093/jnci/dji141
111. van Os NJ, Roeleveld N, Weemaes CM, Jongmans MC, Janssens GO, Taylor AM, et al. Health Risks for Ataxia-Telangiectasia Mutated Heterozygotes: A Systematic Review, Meta-Analysis and Evidence-Based Guideline. Clin Genet (2016) 902:105–17. doi: 10.1111/cge.12710
112. Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N Engl J Med (2015) 37223:2243–57. doi: 10.1056/NEJMsr1501341
113. Rupnik A, Grenon M, Lowndes N. The MRN Complex. Curr Biol (2008) 1811:R455–7. doi: 10.1016/j.cub.2008.03.040
114. Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, et al. Nibrin, a Novel DNA Double-Strand Break Repair Protein, Is Mutated in Nijmegen Breakage Syndrome. Cell (1998) 933:467–76. doi: 10.1016/s0092-8674(00)81174-5
115. Bogdanova N, Feshchenko S, Schürmann P, Waltes R, Wieland B, Hillemanns P, et al. Nijmegen Breakage Syndrome Mutations and Risk of Breast Cancer. Int J Cancer (2008) 1224:802–6. doi: 10.1002/ijc.23168
116. Ciara E, Piekutowska-Abramczuk D, Popowska E, Grajkowska W, Barszcz S, Perek D, et al. Heterozygous Germ-Line Mutations in the NBN Gene Predispose to Medulloblastoma in Pediatric Patients. Acta Neuropathol (2010) 1193:325–34. doi: 10.1007/s00401-009-0608-y
117. Kawabata M, Kawabata T, Nishibori M. Role of Reca/RAD51 Family Proteins in Mammals. Acta Med Okayama (2005) 591:1–9. doi: 10.18926/amo/31987
118. Loveday C, Turnbull C, Ruark E, Xicola RM, Ramsay E, Hughes D, et al. Germline RAD51C Mutations Confer Susceptibility to Ovarian Cancer. Nat Genet (2012) 445:475–6; author reply 76. doi: 10.1038/ng.2224
119. Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, et al. Germline Mutations in RAD51D Confer Susceptibility to Ovarian Cancer. Nat Genet (2011) 439:879–82. doi: 10.1038/ng.893
120. Antoniou AC, Beesley J, McGuffog L, Sinilnikova OM, Healey S, Neuhausen SL, et al. Common Breast Cancer Susceptibility Alleles and the Risk of Breast Cancer for BRCA1 and BRCA2 Mutation Carriers: Implications for Risk Prediction. Cancer Res (2010) 7023:9742–54. doi: 10.1158/0008-5472.Can-10-1907
121. Pang Z, Yao L, Zhang J, Ouyang T, Li J, Wang T, et al. RAD51C Germline Mutations in Chinese Women With Familial Breast Cancer. Breast Cancer Res Treat (2011) 1293:1019–20. doi: 10.1007/s10549-011-1574-3
122. Yang X, Song H, Leslie G, Engel C, Hahnen E, Auber B, et al. Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. J Natl Cancer Inst (2020) 11212:1242–50. doi: 10.1093/jnci/djaa030
123. Chung DC, Rustgi AK. DNA Mismatch Repair and Cancer. Gastroenterology (1995) 1095:1685–99. doi: 10.1016/0016-5085(95)90660-6
124. Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA Mismatch Repair: Functions and Mechanisms. Chem Rev (2006) 1062:302–23. doi: 10.1021/cr0404794
125. Tedaldi G, Tebaldi M, Zampiga V, Danesi R, Arcangeli V, Ravegnani M, et al. Multiple-Gene Panel Analysis in a Case Series of 255 Women With Hereditary Breast and Ovarian Cancer. Oncotarget (2017) 829:47064–75. doi: 10.18632/oncotarget.16791
126. Walsh MD, Buchanan DD, Cummings MC, Pearson SA, Arnold ST, Clendenning M, et al. Lynch Syndrome-Associated Breast Cancers: Clinicopathologic Characteristics of a Case Series From the Colon Cancer Family Registry. Clin Cancer Res (2010) 167:2214–24. doi: 10.1158/1078-0432.Ccr-09-3058
127. Cunniff C, Bassetti JA, Ellis NA. Bloom’s Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition. Mol Syndromol (2017) 81:4–23. doi: 10.1159/000452082
128. Shahi RB, De Brakeleer S, Caljon B, Pauwels I, Bonduelle M, Joris S, et al. Identification of Candidate Cancer Predisposing Variants by Performing Whole-Exome Sequencing on Index Patients From BRCA1 and BRCA2-Negative Breast Cancer Families. BMC Cancer (2019) 191:313. doi: 10.1186/s12885-019-5494-7
129. Pan ZW, Wang XJ, Chen T, Ding XW, Jiang X, Gao Y, et al. Deleterious Mutations in DNA Repair Gene FANCC Exist in BRCA1/2-Negative Chinese Familial Breast and/or Ovarian Cancer Patients. Front Oncol (2019) 9:169. doi: 10.3389/fonc.2019.00169
130. Wu Y, Jiang B, Dai X, Hu X, Wang S, Jiang P, et al. PTEN and NBS1 Gene Mutations in Familial Breast Cancer and Early-Onset Breast Cancer From Hunan Province in China. Zhong Nan Da Xue Xue Bao Yi Xue Ban (2016) 412:121–6. doi: 10.11817/j.issn.1672-7347.2016.02.002
131. Cao WM, Zheng YB, Gao Y, Ding XW, Sun Y, Huang Y, et al. Comprehensive Mutation Detection of BRCA1/2 Genes Reveals Large Genomic Rearrangements Contribute to Hereditary Breast and Ovarian Cancer in Chinese Women. BMC Cancer (2019) 191:551. doi: 10.1186/s12885-019-5765-3
132. He M, Di GH, Cao AY, Hu Z, Jin W, Shen ZZ, et al. RAD50 and NBS1 Are Not Likely to be Susceptibility Genes in Chinese Non-BRCA1/2 Hereditary Breast Cancer. Breast Cancer Res Treat (2012) 1331:111–6. doi: 10.1007/s10549-011-1700-2
133. Jian W, Shao K, Qin Q, Wang X, Song S, Wang X. Clinical and Genetic Characterization of Hereditary Breast Cancer in a Chinese Population. Hered Cancer Clin Pract (2017) 15:19. doi: 10.1186/s13053-017-0079-4
134. Cao WM, Gao Y, Yang HJ, Xie SN, Meng XL, Pan ZW, et al. Germline Mutations of DICER1 in Chinese Women With BRCA1/BRCA2-Negative Familial Breast Cancer. Genet Mol Res (2014) 134:10754–60. doi: 10.4238/2014.December.18.16
135. Yu KD, Di GH, Li WF, Rao NY, Fan L, Yuan WT, et al. Genetic Contribution of GADD45A to Susceptibility to Sporadic and Non-BRCA1/2 Familial Breast Cancers: A Systematic Evaluation in Chinese Populations. Breast Cancer Res Treat (2010) 1211:157–67. doi: 10.1007/s10549-009-0516-9
136. Yu M, Shi Y, Zhang F, Zhou Y, Yang Y, Wei X, et al. Sequence Variations of Mitochondrial DNA D-Loop Region Are Highly Frequent Events in Familial Breast Cancer. J BioMed Sci (2008) 154:535–43. doi: 10.1007/s11373-007-9229-4
137. Wang J, Li W, Shi Y, Huang Y, Sun T, Tang L, et al. Germline Mutation Landscape of Chinese Patients With Familial Breast/Ovarian Cancer in a Panel of 22 Susceptibility Genes. Cancer Med (2019) 85:2074–84. doi: 10.1002/cam4.2093
138. Chen S, Parmigiani G. Meta-Analysis of BRCA1 and BRCA2 Penetrance. J Clin Oncol (2007) 2511:1329–33. doi: 10.1200/jco.2006.09.1066
139. Bougeard G, Renaux-Petel M, Flaman JM, Charbonnier C, Fermey P, Belotti M, et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J Clin Oncol (2015) 3321:2345–52. doi: 10.1200/jco.2014.59.5728
140. Ding SL, Sheu LF, Yu JC, Yang TL, Chen BF, Leu FJ, et al. Abnormality of the DNA Double-Strand-Break Checkpoint/Repair Genes, ATM, BRCA1 and TP53, in Breast Cancer Is Related to Tumour Grade. Br J Cancer (2004) 9010:1995–2001. doi: 10.1038/sj.bjc.6601804
141. Macken WL, Tischkowitz M, Lachlan KL. PTEN Hamartoma Tumor Syndrome in Childhood: A Review of the Clinical Literature. Am J Med Genet C Semin Med Genet (2019) 1814:591–610. doi: 10.1002/ajmg.c.31743
142. McGarrity TJ, Amos CI, Baker MJ, Adam MP, Ardinger HH, Pagon RA, et al. Peutz-Jeghers Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, editors. GeneReviews(®). (1993) Seattle (WA: University of Washington, Seattle.
143. van der Post RS, Vogelaar IP, Carneiro F, Guilford P, Huntsman D, Hoogerbrugge N, et al. Hereditary Diffuse Gastric Cancer: Updated Clinical Guidelines With an Emphasis on Germline CDH1 Mutation Carriers. J Med Genet (2015) 526:361–74. doi: 10.1136/jmedgenet-2015-103094
144. Lynch HT, Krush AJ, Lemon HM, Kaplan AR, Condit PT, Bottomley RH. Tumor Variation in Families With Breast Cancer. Jama (1972) 22213:1631–5. doi: 10.1001/jama.222.13.1631
145. ACOG Practice Bulletin No. 103. Hereditary Breast and Ovarian Cancer Syndrome. Obstet Gynecol (2009) 1134:957–66. doi: 10.1097/AOG.0b013e3181a106d4
146. Kim YC, Zhao L, Zhang H, Huang Y, Cui J, Xiao F, et al. Prevalence and Spectrum of BRCA Germline Variants in Mainland Chinese Familial Breast and Ovarian Cancer Patients. Oncotarget (2016) 78:9600–12. doi: 10.18632/oncotarget.7144
147. Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ, et al. Cancer Phenotype Correlates With Constitutional TP53 Genotype in Families With the Li-Fraumeni Syndrome. Oncogene (1998) 179:1061–8. doi: 10.1038/sj.onc.1202033
148. Malkin D, Li F, Strong L, Fraumeni J, Nelson C, Kim D, et al. Germ Line P53 Mutations in a Familial Syndrome of Breast Cancer, Sarcomas, and Other Neoplasms. Science (1990) 2504985:1233–38. doi: 10.1126/science.1978757
149. Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, et al. Relative Frequency and Morphology of Cancers in Carriers of Germline TP53 Mutations. Oncogene (2001) 2034:4621–8. doi: 10.1038/sj.onc.1204621
150. Masciari S, Dillon DA, Rath M, Robson M, Weitzel JN, Balmana J, et al. Breast Cancer Phenotype in Women With TP53 Germline Mutations: A Li-Fraumeni Syndrome Consortium Effort. Breast Cancer Res Treat (2012) 1333:1125–30. doi: 10.1007/s10549-012-1993-9
151. Daly MB, Pilarski R, Yurgelun MB, Berry MP, Buys SS, Dickson P, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J Natl Compr Canc Netw (2020) 184:380–91. doi: 10.6004/jnccn.2020.0017
152. Brooks-Wilson AR, Kaurah P, Suriano G, Leach S, Senz J, Grehan N, et al. Germline E-Cadherin Mutations in Hereditary Diffuse Gastric Cancer: Assessment of 42 New Families and Review of Genetic Screening Criteria. J Med Genet (2004) 417:508–17. doi: 10.1136/jmg.2004.018275
153. Kaurah P, MacMillan A, Boyd N, Senz J, De Luca A, Chun N, et al. Founder and Recurrent CDH1 Mutations in Families With Hereditary Diffuse Gastric Cancer. Jama (2007) 29721:2360–72. doi: 10.1001/jama.297.21.2360
154. Khanna KK, Chenevix-Trench G. ATM and Genome Maintenance: Defining Its Role in Breast Cancer Susceptibility. J Mammary Gland Biol Neoplasia (2004) 93:247–62. doi: 10.1023/B:JOMG.0000048772.92326.a1
155. Li DM, Sun H. TEP1, Encoded by a Candidate Tumor Suppressor Locus, Is a Novel Protein Tyrosine Phosphatase Regulated by Transforming Growth Factor Beta. Cancer Res (1997) 5711:2124–9.
156. Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, et al. High Cumulative Risks of Cancer in Patients With PTEN Hamartoma Tumour Syndrome. J Med Genet (2013) 504:255–63. doi: 10.1136/jmedgenet-2012-101339
157. Nakanishi C, Yamaguchi T, Iijima T, Saji S, Toi M, Mori T, et al. Germline Mutation of the LKB1/STK11 Gene With Loss of the Normal Allele in an Aggressive Breast Cancer of Peutz-Jeghers Syndrome. Oncology (2004) 675-6:476–9. doi: 10.1159/000082933
158. Beggs AD, Latchford AR, Vasen HF, Moslein G, Alonso A, Aretz S, et al. Peutz-Jeghers Syndrome: A Systematic Review and Recommendations for Management. Gut (2010) 597:975–86. doi: 10.1136/gut.2009.198499
159. Gupta S, Provenzale D, Llor X, Halverson AL, Grady W, Chung DC, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 2.2019. J Natl Compr Canc Netw (2019) 179:1032–41. doi: 10.6004/jnccn.2019.0044
160. Moyer VA. Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer in Women: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med (2014) 1604:271–81. doi: 10.7326/m13-2747
161. Balmaña J, Sanz J, Bonfill X, Casado A, Rué M, Gich I, et al. Genetic Counseling Program in Familial Breast Cancer: Analysis of Its Effectiveness, Cost and Cost-Effectiveness Ratio. Int J Cancer (2004) 1124:647–52. doi: 10.1002/ijc.20458
162. Antoniou AC, Hardy R, Walker L, Evans DG, Shenton A, Eeles R, et al. Predicting the Likelihood of Carrying a BRCA1 or BRCA2 Mutation: Validation of BOADICEA, BRCAPRO, IBIS, Myriad and the Manchester Scoring System Using Data From UK Genetics Clinics. J Med Genet (2008) 457:425–31. doi: 10.1136/jmg.2007.056556
163. Hull LE, Haas JS, Simon SR. Provider Discussions of Genetic Tests With U.S. Women at Risk for a BRCA Mutation. Am J Prev Med (2018) 542:221–28. doi: 10.1016/j.amepre.2017.10.015
164. He Jie CW, Ni L, Hongbing S, Jiang L, Yong W, Jing L, et al. China Guideline for the Screening and Early Detection of Female Breast Cancer(2021, Beijing). Chin J Oncol (2021) 43(4):357–82. doi: 10.3760/cma.j.cn112152-20210119-00061
165. Weir HK, Thun MJ, Hankey BF, Ries LA, Howe HL, Wingo PA, et al. Annual Report to the Nation on the Status of Cancer, 1975-2000, Featuring the Uses of Surveillance Data for Cancer Prevention and Control. J Natl Cancer Inst (2003) 9517:1276–99. doi: 10.1093/jnci/djg040
166. Burke W, Daly M, Garber J, Botkin J, Kahn MJ, Lynch P, et al. Recommendations for Follow-Up Care of Individuals With an Inherited Predisposition to Cancer. II. BRCA1 and BRCA2. Cancer Genetics Studies Consortium. Jama (1997) 27712:997–1003. doi: 10.1001/jama.277.12.997
167. Davis JG. Predictive Genetic Tests: Problems and Pitfalls. Ann N Y Acad Sci (1997) 833:42–6. doi: 10.1111/j.1749-6632.1997.tb48591.x
168. Evans DG, Graham J, O’Connell S, Arnold S, Fitzsimmons D. Familial Breast Cancer: Summary of Updated NICE Guidance. BMJ Br Med J (2013) 346:f3829. doi: 10.1136/bmj.f3829
169. Hartge P, Struewing JP, Wacholder S, Brody LC, Tucker MA. The Prevalence of Common BRCA1 and BRCA2 Mutations Among Ashkenazi Jews. Am J Hum Genet (1999) 644:963–70. doi: 10.1086/302320
170. Vos JR, de Bock GH, Teixeira N, van der Kolk DM, Jansen L, Mourits MJ, et al. Proven non-Carriers in BRCA Families Have an Earlier Age of Onset of Breast Cancer. Eur J Cancer (2013) 499:2101–6. doi: 10.1016/j.ejca.2013.02.018
171. Zhao Q, Yang J, Li L, Cao D, Yu M, Shen K. Germline and Somatic Mutations in Homologous Recombination Genes Among Chinese Ovarian Cancer Patients Detected Using Next-Generation Sequencing. J Gynecol Oncol (2017) 284:e39. doi: 10.3802/jgo.2017.28.e39
172. Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, et al. A Study of Over 35,000 Women With Breast Cancer Tested With a 25-Gene Panel of Hereditary Cancer Genes. Cancer (2017) 12310:1721–30. doi: 10.1002/cncr.30498
173. Manchanda R, Patel S, Gordeev VS, Antoniou AC, Smith S, Lee A, et al. Cost-Effectiveness of Population-Based BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, PALB2 Mutation Testing in Unselected General Population Women. J Natl Cancer Inst (2018) 1107:714–25. doi: 10.1093/jnci/djx265
174. Thompson ER, Rowley SM, Li N, McInerny S, Devereux L, Wong-Brown MW, et al. Panel Testing for Familial Breast Cancer: Calibrating the Tension Between Research and Clinical Care. J Clin Oncol (2016) 3413:1455–9. doi: 10.1200/jco.2015.63.7454
175. Costantino JP, Gail MH, Pee D, Anderson S, Redmond CK, Benichou J, et al. Validation Studies for Models Projecting the Risk of Invasive and Total Breast Cancer Incidence. J Natl Cancer Inst (1999) 9118:1541–8. doi: 10.1093/jnci/91.18.1541
176. Gail MH, Brinton LA, Byar DP, Corle DK, Green SB, Schairer C, et al. Projecting Individualized Probabilities of Developing Breast Cancer for White Females Who Are Being Examined Annually. J Natl Cancer Inst (1989) 8124:1879–86. doi: 10.1093/jnci/81.24.1879
177. Amir E, Freedman OC, Seruga B, Evans DG. Assessing Women at High Risk of Breast Cancer: A Review of Risk Assessment Models. J Natl Cancer Inst (2010) 10210:680–91. doi: 10.1093/jnci/djq088
178. Claus EB, Risch N, Thompson WD. Autosomal Dominant Inheritance of Early-Onset Breast Cancer. Implications for Risk Prediction. Cancer (1994) 733:643–51. doi: 10.1002/1097-0142(19940201)73:3<643::aid-cncr2820730323>3.0.co;2-5
179. Lindor NM, Johnson KJ, Harvey H, Shane Pankratz V, Domchek SM, Hunt K, et al. Predicting BRCA1 and BRCA2 Gene Mutation Carriers: Comparison of PENN II Model to Previous Study. Fam Cancer (2010) 94:495–502. doi: 10.1007/s10689-010-9348-3
180. Berry DA, Parmigiani G, Sanchez J, Schildkraut J, Winer E. Probability of Carrying a Mutation of Breast-Ovarian Cancer Gene BRCA1 Based on Family History. J Natl Cancer Inst (1997) 893:227–38. doi: 10.1093/jnci/89.3.227
181. Parmigiani G, Berry D, Aguilar O. Determining Carrier Probabilities for Breast Cancer-Susceptibility Genes BRCA1 and BRCA2. Am J Hum Genet (1998) 621:145–58. doi: 10.1086/301670
182. Berry DA, Iversen ES Jr., Gudbjartsson DF, Hiller EH, Garber JE, Peshkin BN, et al. BRCAPRO Validation, Sensitivity of Genetic Testing of BRCA1/BRCA2, and Prevalence of Other Breast Cancer Susceptibility Genes. J Clin Oncol (2002) 2011:2701–12. doi: 10.1200/jco.2002.05.121
183. Frank TS, Deffenbaugh AM, Reid JE, Hulick M, Ward BE, Lingenfelter B, et al. Clinical Characteristics of Individuals With Germline Mutations in BRCA1 and BRCA2: Analysis of 10,000 Individuals. J Clin Oncol (2002) 206:1480–90. doi: 10.1200/jco.2002.20.6.1480
184. Antoniou AC, Pharoah PD, McMullan G, Day NE, Stratton MR, Peto J, et al. A Comprehensive Model for Familial Breast Cancer Incorporating BRCA1, BRCA2 and Other Genes. Br J Cancer (2002) 861:76–83. doi: 10.1038/sj.bjc.6600008
185. Antoniou AC, Pharoah PP, Smith P, Easton DF. The BOADICEA Model of Genetic Susceptibility to Breast and Ovarian Cancer. Br J Cancer (2004) 918:1580–90. doi: 10.1038/sj.bjc.6602175
186. Evans DGR, Eccles DM, Rahman N, Young K, Bulman M, Amir E, et al. A New Scoring System for the Chances of Identifying a BRCA1/2 Mutation Outperforms Existing Models Including BRCAPRO. J Med Genet (2004) 416:474–80. doi: 10.1136/jmg.2003.017996
187. Evans DG, Young K, Bulman M, Shenton A, Wallace A, Lalloo F. Probability of BRCA1/2 Mutation Varies With Ovarian Histology: Results From Screening 442 Ovarian Cancer Families. Clin Genet (2008) 734:338–45. doi: 10.1111/j.1399-0004.2008.00974.x
Keywords: family history, familial breast cancer, gene mutations, genetic syndromes, screening, genetic counseling
Citation: Shen L, Zhang S, Wang K and Wang X (2021) Familial Breast Cancer: Disease Related Gene Mutations and Screening Strategies for Chinese Population. Front. Oncol. 11:740227. doi: 10.3389/fonc.2021.740227
Received: 12 July 2021; Accepted: 12 November 2021;
Published: 01 December 2021.
Edited by:
Tianhui Chen, University of Chinese Academy of Sciences, ChinaReviewed by:
Marco Lucioni, University of Pavia, ItalyAkira Hirasawa, Okayama University, Japan
Zhi-Gang Zhuang, Shanghai First Maternity and Infant Hospital, China
Copyright © 2021 Shen, Zhang, Wang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaochen Wang, d2FuZ3hpYW9jaGVuQHpqdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship