 Yu-Ling Bin2
Yu-Ling Bin2 Hong-Sai Hu3
Hong-Sai Hu3 Feng Tian2
Feng Tian2 Zhen-Hua Wen2Mei-Feng Yang4
Zhen-Hua Wen2Mei-Feng Yang4 Ben-Hua Wu1Li-Sheng Wang1Jun Yao1
Ben-Hua Wu1Li-Sheng Wang1Jun Yao1 De-Feng Li1*
De-Feng Li1*- 1Department of Gastroenterology, Shenzhen People’s Hospital (The Second Clinical Medical College, Jinan University, The First Affiliated Hospital, Southern University of Science and Technology), Shenzhen, China
- 2Department of Rheumatology and Immunology, ZhuZhou Central Hospital, Zhuzhou, China
- 3Department of Gastroenterology, ZhuZhou Central Hospital, Zhuzhou, China
- 4Department of Hematology, Yantian District People’s Hospital, Shenzhen, China
Worldwide, gastric cancer (GC) represents the fifth most common cancer for incidence and the third leading cause of death in developed countries. Despite the development of combination chemotherapies, the survival rates of GC patients remain unsatisfactory. The reprogramming of energy metabolism is a hallmark of cancer, especially increased dependence on aerobic glycolysis. In the present review, we summarized current evidence on how metabolic reprogramming in GC targets the tumor microenvironment, modulates metabolic networks and overcomes drug resistance. Preclinical and clinical studies on the combination of metabolic reprogramming targeted agents and conventional chemotherapeutics or molecularly targeted treatments [including vascular endothelial growth factor receptor (VEGFR) and HER2] and the value of biomarkers are examined. This deeper understanding of the molecular mechanisms underlying successful pharmacological combinations is crucial in finding the best-personalized treatment regimens for cancer patients.
Introduction
Gastric cancer (GC) is currently the third leading cause of cancer-related death globally and varies significantly among different geographical areas, despite the overall morbidity and mortality are declining (1). Surgery is an effective option for the treatment of GC, while patients with advanced GC lose the best opportunity of surgery due to multiple metastasis (2). Compared with other primary tumors, GC with multiple metastases has higher tissue heterogeneity, which is caused by multiple specific gene clusters or gene mutations (3). Therefore, GC displays aggressive behavior and treatment resistance, bringing great difficulties for the development of molecular targeted drugs and individualized precise treatment. Moreover, based on the molecular classification of The Cancer Genome Atlas (TCGA), GC encompasses different molecular subtypes, such as Epstein–Barr virus (EBV 9%), microsatellite instability (MSI 22%), genomic stable (20%), and chromosomal instability (50%), and often exhibits a poor and unfavorable prognosis (4).
It has become clear enough that a single cancer hallmark (e.g., self-sufficiency in growth signals, insensitivity to antigrowth signals, evading apoptosis, limitless replicative potential, sustained angiogenesis, and tissue invasion and metastasis) cannot be used to globally define tumor alteration (5). As early as last century, Warburg found that owing to uninterrupted growth, tumor cells would reprogram their metabolism production network by circumventing mitochondrial oxidative phosphorylation and facilitating aerobic glycolysis to maintain the normal levels of ATP and NADH (6). Metabolic reprogramming, including the remodeling of glucose, lipid, glutamine, oxidative phosphorylation, and mitochondrial respiration (7), plays a pivotal role in the regulation of gene transcription, DNA damage repair, and metabolic enzymes, to transmit or release cytokines through signaling pathways in the tumor microenvironment (TME). Accumulating evidence indicates that cancer cells may transfer biologically functional molecules to their surrounding stromal cells by reprogramming metabolism, which facilitates cancer metastasis, drug resistance, and immunosuppression (8–10). If this series of cancer cells disorders are regarded as energy metabolism alteration, limiting energy currency ATP and redox currency NADH can be achieved by using small molecule drugs targeting energy metabolism or cutting off the metabolic pathway of energy supply. Similar to the Trojan horse effect, by targeting metabolic changes, we can identify potential new targets for accurate cancer treatment and design antitumor strategies to improve the concentration of drugs into cells. Therefore, metabolic reprogramming has become a promising target in cancer therapy, including refractory cancers such as GC.
Alterations in amino acid synthesis and catabolism, lipid biogenesis, and other pathways such as polyamine processing, are commonly seen in GC (11, 12). However, the development of GC and TME forms a complex loop, and the specific mechanism underlying its metabolic reprogramming remains largely unexplored. The present review outlines recent updates, addressing how bioenergetic metabolism reprogramming is involved in GC, aiming to better understand their role in the GC progression, which might help develop new therapeutic approaches by targeting GC metabolism.
Characteristics of Metabolic Reprogramming in GC
Malignant tumors have the common characteristics of high metabolism. However, epigenetic changes, tissue origin, differentiation status, and other internal and external factors such as oxygen and nutrients in tumor microcirculation result in a unique metabolic profile that distinguishes cancer cells from normal cells (Table 1). Reprogramming of the tumor metabolism includes upregulation of aerobic glycolysis, a strongly enhanced glutaminyl, and lipid accumulation in tumor cells, potentially providing energy and structural requirements for the development of cancer cells (Figures 1A, B) (23). However, effective stratification strategies and selection of predictive biomarkers for personalized medicine are currently limited. GC, as a heterogeneous disease, lacks specific symptoms in its early stages, leading to a delayed diagnosis with three-quarters of patients presenting with non-curable advanced disease (24). Moreover, the energy metabolism reprogramming of GC has its own characteristics due to the heterogeneity. For instance, six metabolites (alanine, α-ketoisocaproic acid, proline, glycerin acid, pantothenic acid, and adenosine) show varying expression levels between GC cell lines and a normal gastric epithelial cell line (25). In particular, genome-wide expression profiles have found that an intestinal subtype of gastric tumors is involved in glucose metabolism and glutamine metabolism-related gene, and glucose transport and glucan related to metabolic genes are enriched in the diffuse subtype of GC (26). Therefore, it is urgently necessary to integrate clinical, morphological, and molecular data by identifying key metabolic processes of GC for the patient stratification for personalized therapy.
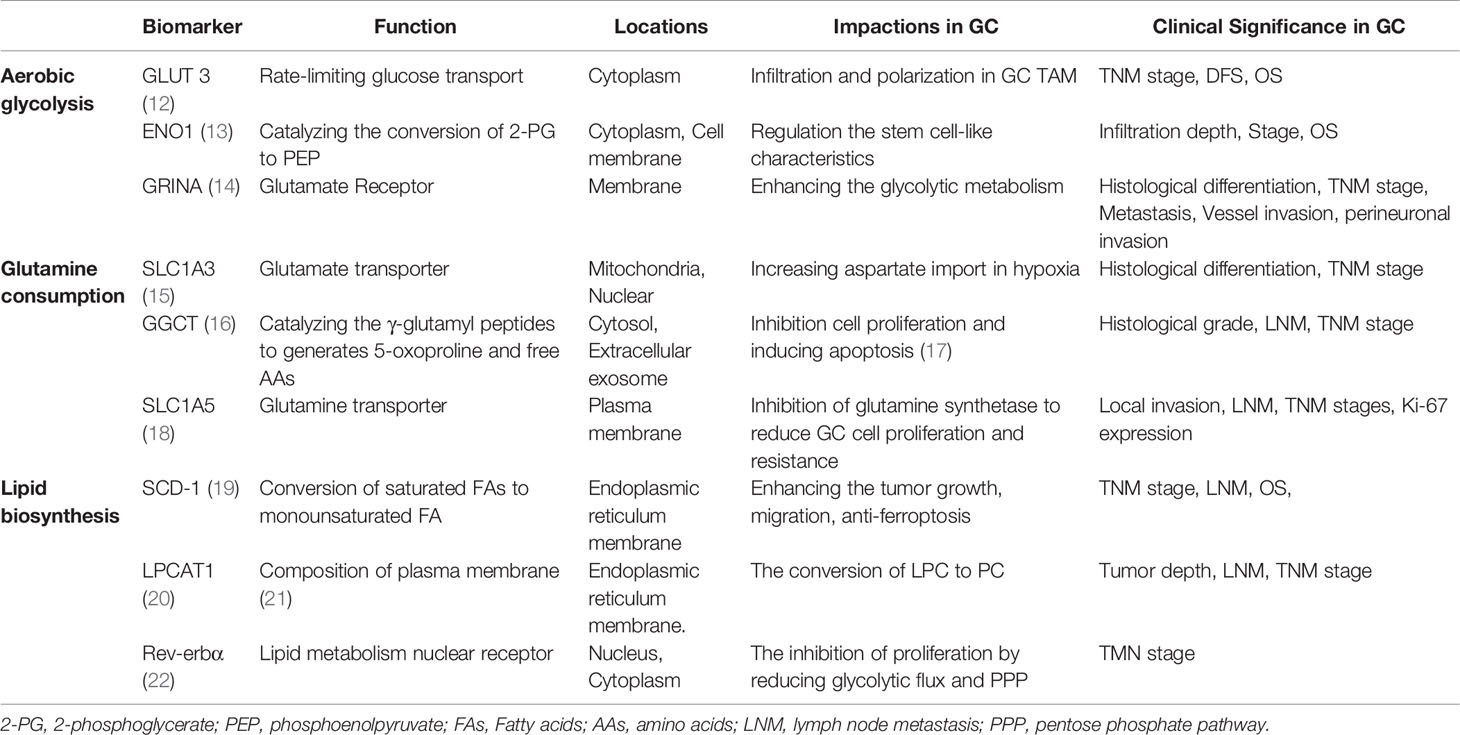
Table 1 Biomarker of metabolic reprogramming in GC.
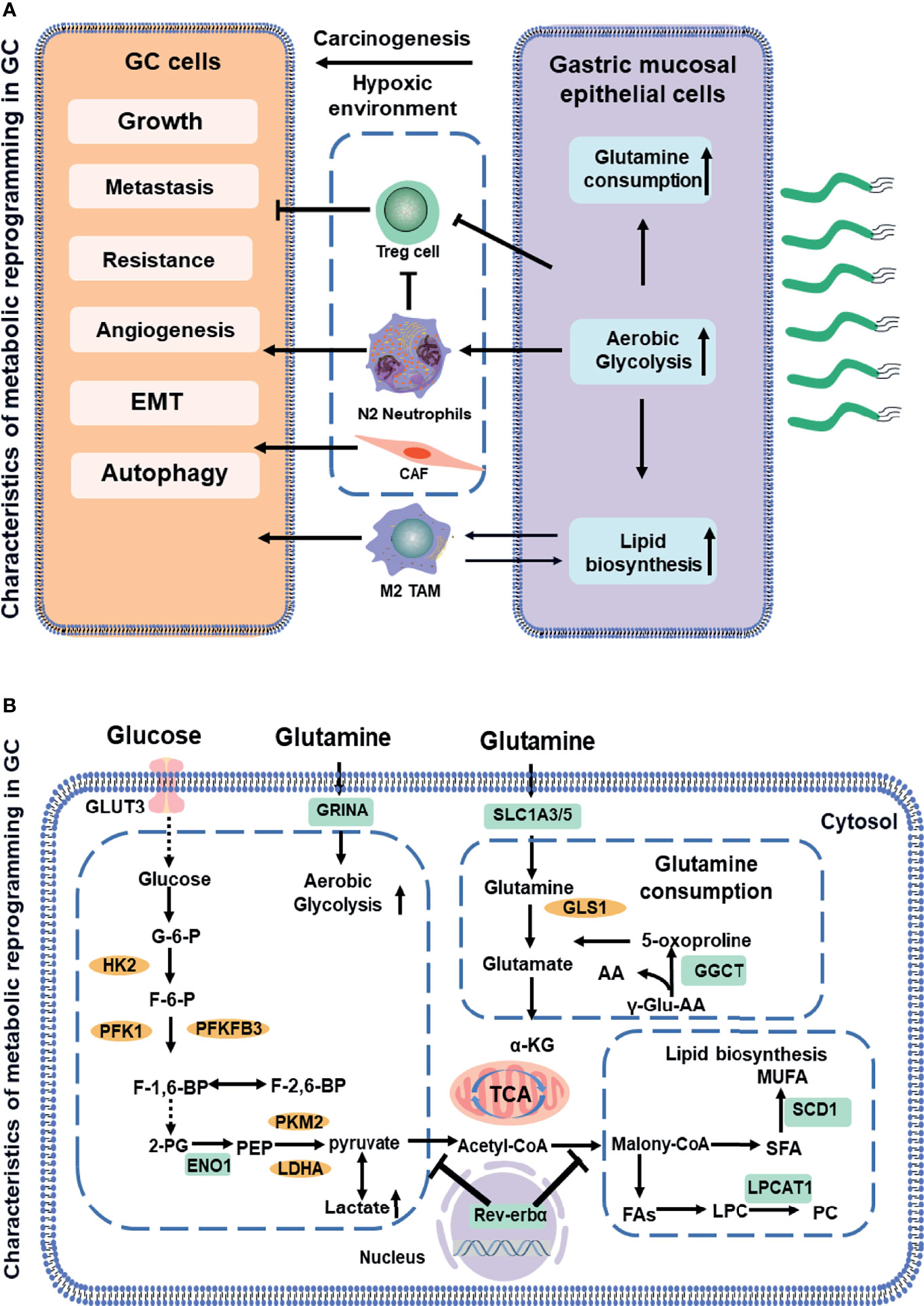
Figure 1 Schematic showing a comparative account of normal vs. cancer cell metabolic reprogramming (A). The association between aerobic glycolysis (Warburg effect) and the glutamine metabolism and fatty acids metabolism. Biomarkers in GC (indicated in green boxes) along with signaling molecules (orange circles). Next, the mitochondrial dysfunction or phenotypic alteration (B). AA, amino acid; CoA, coenzyme A; ENO1, enolase 1; F-6-P, fructose 6-phosphate; FA, fatty acids; G-6-P, glucose-6-phosphate; GGCT, glutamylcyclo transferase; GLUT3, glucose transporter3; GRINA, glutamate receptor; GLS, glutaminase1; HK2, hexokinase2; LDHA, lactate dehydrogenase; LPC, lysophosphatidylcholine; LPCAT1, lysophosphatidylcholine acyltransferase; MUFA, multiunsaturated fatty acid; PEP, phosphoenolpyruvate; PFK1, phosphofructokinase1; PC, phosphatidylcholine; PFKFB3, phosphofructokinase-2/fructose-2,6 bisphosphatase 3; PKM2, pyruvate kinase2; SFA, saturated fatty acids; SCD-1, stearoyl-CoA desaturase 1; TCA, tricarboxylic acid cycle. Dotted lines indicate the feed-back inhibition/regulation of some of the glycolytic enzymes by corresponding metabolites.
Aerobic Glycolysis
Aerobic glycolysis is the process of oxidation of glucose into pyruvate, followed by lactate production under normoxic conditions, which promotes glutaminolysis to satisfy the precursor requirements of nucleic acids (27). The upregulation of glycolysis is mostly due to the increased expressions of enzymes and transporters involved in glucose uptake, lactate production, and lactate secretion (28). Figure 1 outlines the stepwise process of glycolysis, including the substrates and enzymes of the pathway. The glucose uptake of cells largely depends on the concentration of membrane transport proteins collectively known as the glucose transporter (GLUT) family. Significantly, GLUT 3, acting as a biomarker to determine prognosis and immune infiltration in GC, not only potentially contributes to M2 subtype transition of macrophages in the TME by mediating glucose influx (12) but also is correlated with higher tumor–node–metastasis (TNM) stage and negative survival (29). Moreover, glycolytic enzyme Enolase 1 (ENO1), as a poor prognosis biomarker in GC (13), which is involved in hypoxia, increases glucose uptake and metabolism via upregulating GLUT3 and promoting the lactate production (30). The molecular mechanisms of metabolic reprogramming in GC have been applied in clinical practice. For example, a study consisting of 279 patients routinely staged in the absence of metastases on CT has identified previously unsuspected metastases in 7% of patients using F-18 fluorodeoxyglucose, which would likely not have been identified by conventional staging without PET-CT in 5% (31).
Glutamine
Glutamine, a new energy source for tumor cells, provides nitrogen and carbon sources that replenish tricarboxylic acid (TCA) cycle intermediates for the sake of nucleic acids. Glutamine is first converted to glutamate and ammonium by glutaminase (GLS). Subsequently, it is catalyzed by glutamate dehydrogenase (GDH) and converted to α- ketoglutarate (32). Then, α-ketoglutarate enters the TCA cycle, which provides energy and macromolecular intermediates, as seen in Figure 2. The combination of GLS1 and glutamyl cyclotransferase (GGCT) is highly sensitive and specific for detecting GC, which is strongly associated with histological grade, lymph node metastasis, and TNM stage (16). The SLC1 family (glutamate transporters) plays important roles in providing cells throughout the body with glutamate for metabolic purposes (33). For example, the loss of function of SLC1A3 (GLAST) and SLC1A5 (also known as ASCT2 or Na-dependent transmembrane transporter) has been implicated in the pathogenesis of GC. SLC1A3 is positively associated with the poor prognosis, and it provides a competitive advantage to GC, increasing aspartate import under the hypoxic condition (15). SLC1A5 is correlated with malignant features, such as deeper local invasion, higher lymph node metastasis, advanced TNM stages, and higher Ki-67 expression (18). However, the inhibition of glutamine synthetase remarkably reduces the proliferation and resistance of GC cells, suggesting that glutamine mediates GC growth and the therapeutic efficacy of targeted treatment (34). Interestingly, as a glutamate receptor, the N-methyl D-aspartate-associated protein 1 (GRINA) is involved in lipid and sterol synthesis (35), and it also modulates aerobic glycolysis and promotes tumor progression in GC (14).
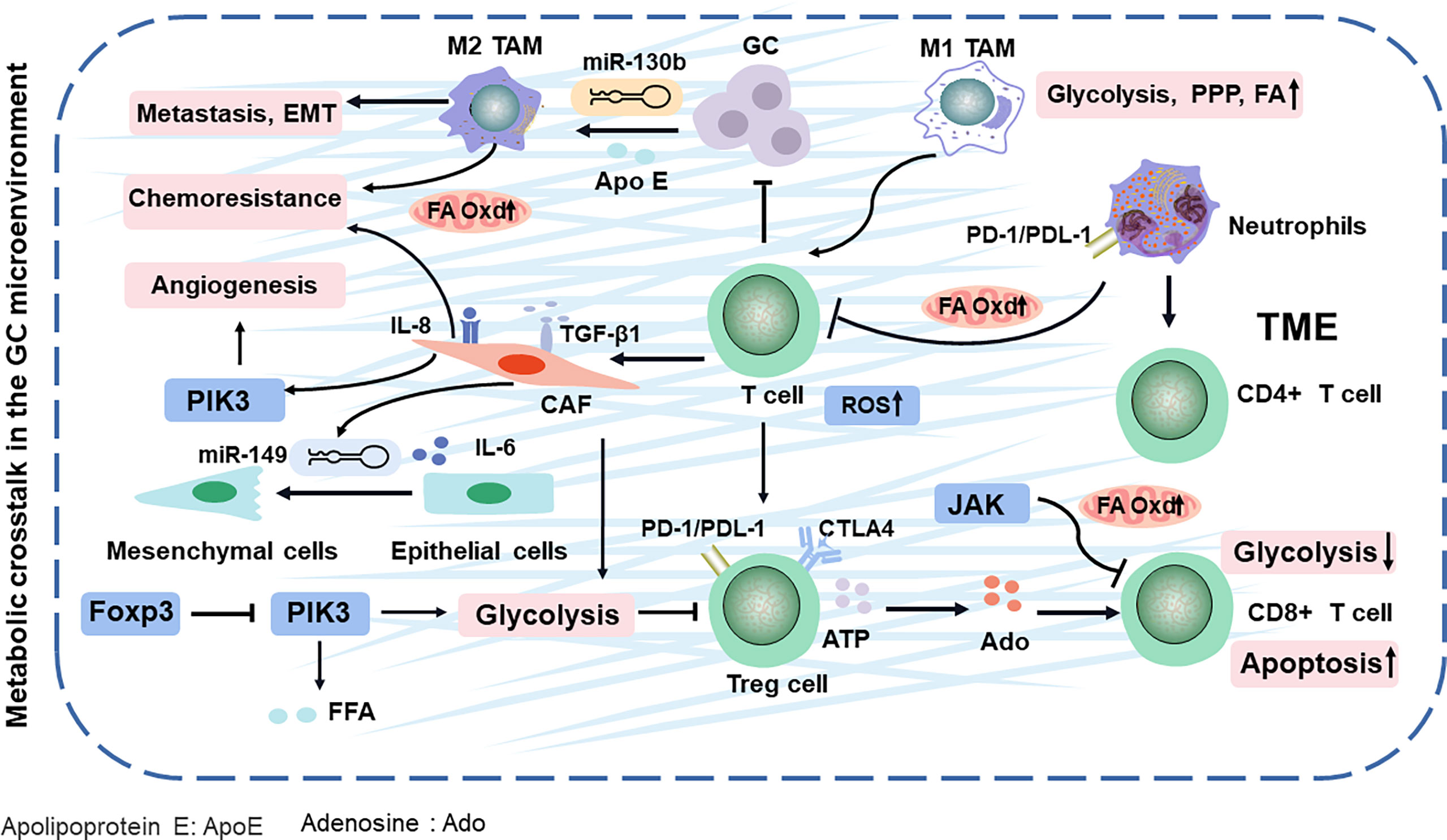
Figure 2 TME comprising the tumor cells and various stromal cells in GC. They evade immune surveillance during GC progression by balancing energy requirements and in TME. Finally, the metabolites of TME impacts cancer-specific or related phenotypes. Apo E, apolipoprotein E; Ado, adenosine; Oxd, oxidation; PPP, pentose phosphate pathway; ROS, reactive oxygen species; TAM, tumor-associated macrophages.
Fatty Acids
Fatty acids (FAs, as molecule signals and energy sources, are important as the basic backbone of many lipids and generally recognized as part of the metabolic landscape of cancer (36). The de novo FA synthesis pathway is enhanced to glucose and glutamine metabolism in tumor cells (Figure 3) (11). Strikingly, FA metabolisms, FA transport, and fat differentiation-related signatures are also highly activated in GC (26). Stearoyl-CoA desaturase 1 (SCD-1), which converts saturated FAs into monounsaturated FAs, is overexpressed and exhibits the ability to promote tumor growth, migration, and anti-ferroptosis in GC (19). Lysophosphatidylcholine acyltransferase 1 (LPCAT1) is involved in the metastasis and recurrence of GC (20), especially in converting lysophosphatidylcholine (LPC) to phosphatidylcholine (PC), which is positively correlated with tumor differentiation but negatively correlated with tumor depth, lymph node metastasis, and tumor stage in GC (37). interestingly, Rev-erbα (nuclear receptor subfamily 1 group D member 1) regulates lipid metabolism nuclear receptor, and it is not only associated with TMN stages but also its reduction causes GC progression by augmenting the glycolysis (22).
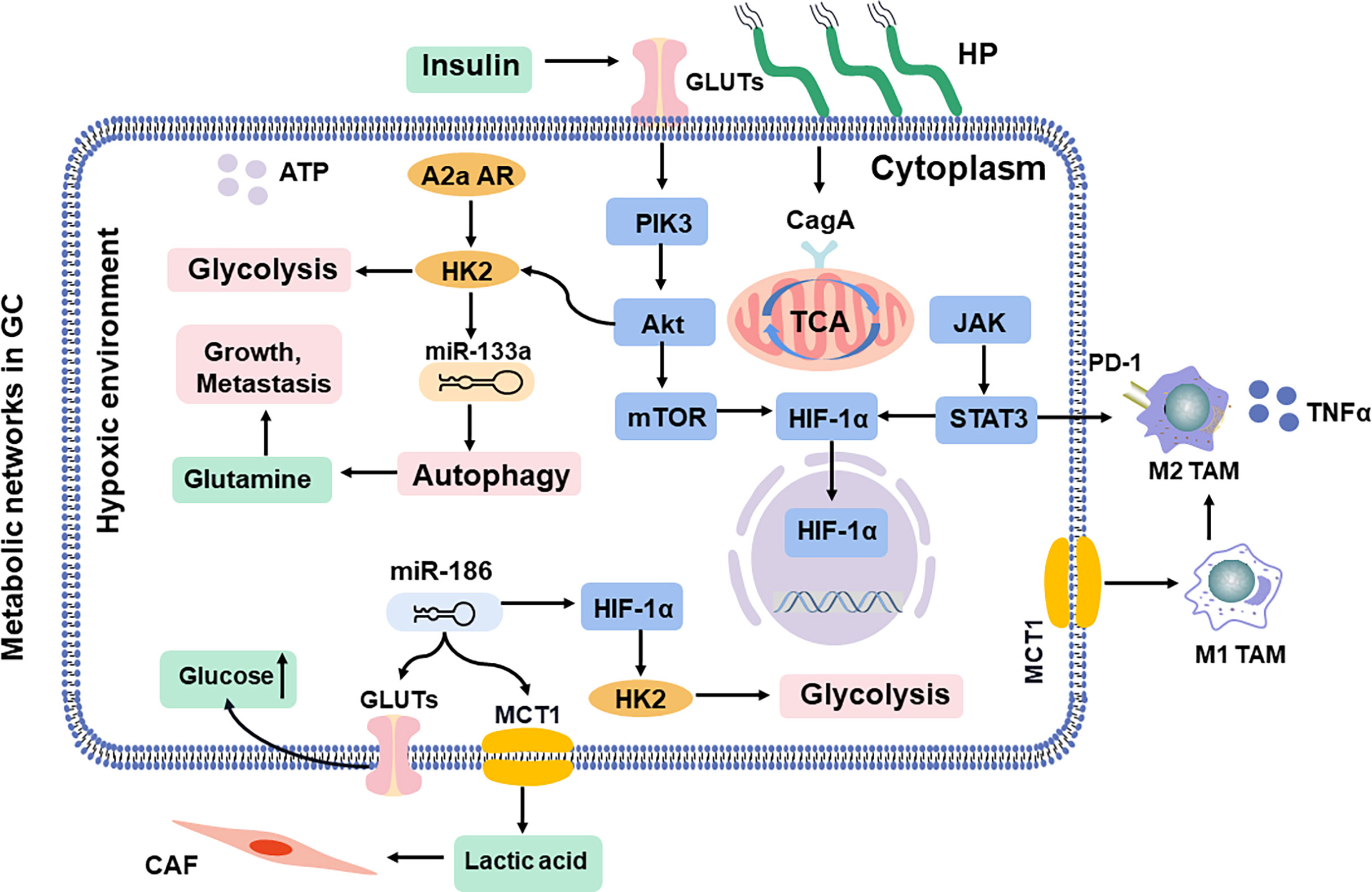
Figure 3 Shift in metabolic networks in GC. The metabolic intermediates of metabolic reprogramming are associated with diverse pathways in the cells inside and outside. HP, H. pylori; MCT, monocarboxylate channel transporter.
Based on the TCGA dataset, a signature consisting of seven glycolysis genes (STC1, CLDN9, EFNA3, ZBTB7A, NT5E, NUP50, and CXCR4) is established, demonstrating that an immunosuppressive TME can lead to poor prognosis in GC (38). All the above evidence displays different metabolic traits compared with the tumors from which they originate, enabling survival and growth in the new TME, and it selectively and dynamically adapts their metabolism at every step during the metastatic cascade, which creates a nutrient-rich microenvironment. These alterations are pivotal to the development and maintenance of the malignant phenotype of cancer cells in unfavorable TME or metastatic sites.
Metabolic Alteration in the GC Immune Microenvironment
TME (composed of the tumor cells, immune cells, and fibroblasts) releases various molecules or activates the metabolic reprogramming signaling in cancer cells to remodel surrounding areas (39), contributing to immune escape mechanisms and drug resistance with GC development (40). However, altered metabolism is not limited to cellular energetic pathways. For example, the metabolic programming of immune cells can affect antigen presentation, ultimately leading to the alteration of tumor immunity (Figure 2) (41). Especially, immune-infiltrating cells in the TME can play dual roles, either promoting or inhibiting tumor growth, in response to metabolic stresses and external signals.
T Cells
T cells have a natural ability to fight cancer cells in the TME. Yet, these cancer-fighting T cells are gradually exhausted and lose immunological memory potential (42). CD4+ T cells (helper T cells) and CD8+T cells (cytotoxic T cells) are the two broad functional groups of mature T cells (43). First, regulatory T (Treg) cells, the subsets of CD4+ T cells, are rapidly expanded upon encountering self-antigens expressed by cancer cells, and its accumulation in GC can decompose ATP to adenosine, then induce apoptosis, and inhibit the proliferation of CD8+ T cells, leading to immune inactivation and evasion (44). In addition, Treg cells can regulate transcription factor Foxp3 to restrain PIK3/Akt/mTOR signaling, which diminished glycolysis metabolism (45). Further research has demonstrated that Treg cells activate their lipid metabolism to support the survival (46). In addition, the accumulation of Treg cells in GC also activates the PI3K/Akt/mTOR pathway, which increases free fatty acids (FFAs) and generates an immunosuppressive TME, resulting in resistance to immunotherapy (47). The glycolysis and antitumor functions of CD8+ T cells can be inhibited by activating STAT3 to drive the FA oxidation (FAO) (48). These findings explain that the ratio of CD8+ T cells to Treg cells in the GC TME is an important factor for prognosis and clinical efficacies (49).
Neutrophils
Neutrophils, as an important component of the tumor-infiltrating immune cells, can release several cytokines [such as interleukin-1β (IL-1β), tumor necrosis factor alpha (TNF-α), and interferon gamma (IFN-γ)], which is mediated by multiple mediators, including cytokines, chemokines, lipids, and growth factors in TME (50). In GC, high-infiltration neutrophils have been associated with poor prognosis (51). Especially, neutrophils in GC inhibit the proliferation of CD4+ T cells and form a local immunosuppressive environment through the programmed cell death 1 (PD-1)/programmed cell death protein-L1 (PDL−1) pathway (52). They secrete a wide spectrum of factors, including matrix metalloproteinases and proinflammatory cytokines, to initiate carcinogenesis (53) (Figure 2). Neutrophils effectively suppress normal T-cell immunity and prolong their lifespan, contributing to the migration of GC (54). In GC, neutrophils are polarized to an N2 phenotype to promote tumor migration (53). Neutrophil is often discounted as purely glycolytic (55), while oxidative neutrophils use mitochondrial FAO to produce and suppress T cells in glucose-restricted TME (56). Evidently, these results show that targeting the lipid metabolic mechanism of neutrophils and T cells can synergize with antitumor immunity.
Tumor-Associated Macrophages
Tumor-associated macrophages (TAMs) include antitumor M1-like (M1-TAMs) or protumor M2-like (M2-TAMs) TAMs (57). Upon stimulation by IFN-γ or lipopolysaccharide (LPS), macrophages are polarized in the M1 phenotype, whereas M2 polarization can be achieved via incubation with IL-4 and IL-13 (58–60). The metabolic alterations of macrophage polarization can determine the phenotype and function of TAMs in promoting the cancer progression. Conversely, cancer cells can also utilize metabolic byproducts to manipulate TAMs to their benefits (61). For example, M2 macrophages are triggered by GC-derived mesenchymal stromal cells, promoting metastasis and EMT (62). Further research has found that M2 macrophage polarization from GC, involving the JAK2/STAT3 signaling pathway, is attenuated by blockading the secretion of IL-6/IL-8 (63). Most likely, M2 macrophages modulate lipid metabolism by deriving apolipoprotein E and then remodel the cytoskeleton to support migration in GC (64, 65). Especially, M2 macrophage can exacerbate the FA β-oxidation and promote the 5-fluorouracil (5-FU) chemoresistance in GC (66). The lipid restores the activity and substantially enhances the phagocytosis of TAMs, leading to promoted cytotoxic T-cell-mediated tumor regression in GC (67). In addition, miR-130b, the correspondent of the M2-TAMs in GC (68), is associated with lipid metabolism and 5-FU resistance and even can activate PI3K (69–71), which is potentially a new chemotherapeutic target by interfering immune cell metabolism in TAMs. Since TAMs have a high degree of plasticity, M2 macrophages can be repolarized to M1-TAMs. Therefore, reprogramming TAMs into antitumor activity is a new cancer treatment strategy.
Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs), a protective barrier of the tumor, activate metabolically reprogrammed TAMs (72, 73) and block T-cell penetration into tumor nests by secreting transforming growth factor beta 1 (TGF-β1) (74). It is nourished by TGF-β1, which then strongly promotes the metabolic switch from oxidative phosphorylation to aerobic glycolysis in highly metastatic GC (75, 76). Further studies show that the CAFs facilitate vasculogenic mimicry formation via metabolic pathways PI3K (77), which exacerbates the chemotherapeutical efficacy and prognosis of GC (78). MiR-149 links IL-6 to mediate the crosstalk between tumor cells and CAFs, leading to the enhanced epithelial-to-mesenchymal transition and stem-like properties, which alters the metabolism and allows GC cells to spread throughout the body (79, 80).
Helicobacter pylori Infection
Persistent Helicobacter pylori infection is well-known to affect the inflammatory TME and promote GC carcinogenesis (81). In addition to involving inflammatory activation, H. pylori participates in various cell types, including immune cells, gastric epithelium, glands, and stem cells (82). H. pylori activates, polarizes, and recruits macrophages to sustain a continuous supply of proinflammatory and protumorigenic cytokines [such as IL-1, IL-6, IL-1β, TNF-α, macrophage inflammatory protein-2 (MIP-2), and inducible nitric oxide synthase (iNOS)] (83), and inevitably, they alter the metabolism as key contributors to immune evasion. The above-mentioned studies involved harnessing metabolic byproducts and hijacking the functions of tumor-infiltrating immune cells, favoring an immunosuppressive phenotype (84), which impacts many malignancy features, including the expansion and survival of tumor cells, metastasis, and angiogenesis (85). These findings provide a rationale for metabolically targeting the TME, which may assist in improving tumor responsiveness to immune checkpoint blockade (ICB) therapies. Therefore, whether the dysregulated metabolism of TME is a cell-intrinsic program or competition with GC cells for limited nutrients needs to be further discussed.
Metabolic Networks in GC
The progression of GC involves a shared set of metabolic reprogramming pathways, which produce excess lactic acid to reduce the pH value in TME and acquire metabolic adaptations (Figure 3) (86, 87). This metabolic alteration in GC switches from oxidative phosphorylation to glycolysis concerned promoting EMT, tumor angiogenesis, and the metastatic colonization of distant organs, resulting in regulation of the invasion-metastasis cascade (80). In addition, some pathogens, such as H. pylori, further mediate an inflammatory environment and trigger the oncogenic pathway, leading to DNA damage in gastric mucosal epithelial cells, continuous accumulation of intracellular abnormal metabolites, and eventually malignant transformation (88, 89).
HIF-1α/ROS
The physiological gastrointestinal luminal epithelium is hypoxic (90), and tissue hypoxia induces metabolic reprogramming and may result in malignant transformation of gastric mucosal epithelial cells (91). Moreover, it even induces resistance to chemoradiotherapy, leading to therapeutic failure (92). Hypoxia-inducible factor-1 alpha (HIF-1α) controls the production of reactive oxygen species (ROS) in oxygen concentration, which supports the adaptation of tumor cells and mediates lactic acid efflux by the monocarboxylate channel transporter (MCT) to promote macrophage polarization in a hypoxic TME (93). In addition, insulin treatment induces glucose uptake and enhances the expression of GLUT1, which is accompanied by the apoptotic effect due to HIF-1α inhibition (94). MiR-186 is involved in the CAF formation (95), which regulates glucose uptake and lactate production via HIF-1α (96, 97). Approximately 70% of cases of H. pylori infection are involved in GC progression, which is responsible for persistent oxidative stress and DNA damage. Ultimately, HIF-1α promotes metabolic adaptation in a hypoxic environment (98). The cytotoxin-associated protein A (CagA) protein, one of the most important virulence factors of H. pylori, is localized in the mitochondria, where it subsequently results in a hypoxic condition in gastric epithelial cells and increases the HIF-1α activity (99). Then, the crosstalk between ROS and HIF-1α induces macrophage polarization via the Akt/mTOR pathway, which affects the progression of gastric lesions and state of infection (100).
PI3K/Akt/mTOR
The PI3K/Akt/mTOR pathway is frequently activated in promoting GC aggressiveness (101). It involves enhanced aerobic glycolysis (102) and then reshapes the immunosuppressive TAMs (103). Akt, as downstream of PI3K, is an important driver of the tumor glycolytic phenotype, which stimulates ATP production to increase GLUT expression and membrane translocation, phosphorylates key glycolytic enzymes, and thereby stimulates the signal transduction of the mTOR pathway (104). Especially, the PI3K/Akt pathway is significantly activated after H. pylori infection in tumor cells (105). Further studies indicate that CagA protein reduces cellular amino acids, and bolstering amino acid pools prevents mTOR inhibition (106). Moreover, CagA protein activates the PI3K/Akt pathway, induces glucose metabolism, and promotes GC cell proliferation (107). It has been reported that miR-133a blocks the autophagy to ruin the abnormal glutaminolysis via the Akt/mTOR pathway, further inhibiting the growth and metastasis of GC (80, 108). Moreover, the A2a adenosine receptor promotes the GC Warburg effect by enhancing PI3K/Akt/mTOR pathway in hypoxic TAMs (109, 110).
JAK/STAT
Janus kinase-signal transducer and activator of transcription (JAK/STAT) signaling, as the upstream of HIF-1α (111, 112), regulates survival and immunosuppression of GC cells and sustains inflammation in TAMs, including tumor cell recognition and tumor-driven immune escape (113–115), and it is essential in the activation of macrophages, natural killer (NK) cells, and T cells (116, 117). However, efforts to develop therapeutic STAT3 inhibitors have thus far been unsuccessful (118). Activated STAT3 upregulates energy metabolism by translocating mitochondria, which is critical for glutamate-induced cell proliferation (119). Under hypoxic conditions, STAT3 physically interacts with programmed cell death protein-L1 (PD-L1) and facilitates its nuclear translocation, enhancing the macrophage-derived TNFα-induced tumor necrosis in vivo, and correlates with chemotherapeutic drugs (120). Especially, H. pylori disrupts lipid rafts via JAK/STAT and thereby reduces cholesterol levels in infected gastric epithelial cells, allowing the bacteria to escape from the host inflammatory response (121). Infiltrated macrophages can release STAT3 to induce PD-L1 expression in GC, which helps tumor cells escape from cytotoxic T-cell killing and promotes the proliferation of tumor cells (122). Given that interference with STAT3 activity is an amplified signaling cascade by targeting these cytokines; it curbs the growth of GC and augments antitumor immunity (123).
Although these studies have proven many substantial crosstalks and numerous links in metabolic activities, how to allow cells to maximize growth and proliferation and activate chronically in cancer remains unknown. Beyond doubt, the precancerous lesions of gastric epithelial cells have abnormal metabolic energy, and there is a cross-relationship with the pathways mentioned above. Therefore, it seems to be more valuable to trace the heterogeneity of primary lesions and the changes in metabolic enzymes in the tumor progression. In addition, drugging a specific metabolic circuitry associated with malignancy may ultimately be efficient only on a fraction of GC cells, operating as selective pressure and favoring the rapid emergence of resistant cells.
The Strategies of Metabolic Reprogramming in GC
Nowadays, systemic chemotherapy is still the mainstay of treatment for advanced GC. A majority of patients do not benefit from monotherapy, such as 5-FU, due to frequent relapses caused by chemotherapy-resistant cancer clones. Therefore, the 5-year overall survival rate is only 20%–35% (124–126). Accumulating evidence showed that tumor cells, in order to adapt various toxic stimuli in the TME, are involved in the mechanism of self-defense or drug resistance, including enhancing DNA damage repair capacity, increasing efflux of drugs via upregulated resistance-associated proteins, and upregulating antiapoptotic proteins. However, this series of activities require a large amount of ATP supply (127). Therefore, metabolic reprogramming contributes to chemoresistance. The proposed metabolic mechanisms of drug resistance involve mainly in the increase in glucose and glutamine demand, glutaminolysis and glycolysis pathways activity, promotion of reduced nicotinamide adenine dinucleotide phosphate (NADPH) from the pentose phosphate pathway, activation of FAO, and upregulation of ornithine decarboxylase for polyamine production (128). Moreover, several genes are associated with metabolic reprogramming and drug resistance, such as GLUT1, LDHA, GAPDH, MCAM, and FAO (129–132).
Currently, recurrent therapeutic resistance presents revolutionary claims, and targeting the metabolic reprogramming, such as glycolytic inhibitor, could be a strategy of Trojan Horse, which highlights the novel combinational trials and their preclinical rationale. A combination of glycolysis inhibitor and 5-FU can synergistically enhance the cytotoxicity of resistant GC cells (133). Glycolysis negatively affects survival outcomes of metastatic GC patients treated with paclitaxel-ramucirumab therapy (134).
Molecularly Targeted Drugs
Human epidermal growth factor receptor 2 (HER2), an oncogenic tyrosine kinase, is overexpressed or amplified in 12%–20% of GC (135). Several strategies have been developed directly against HER2. However, drug resistance remains a major unresolved clinical problem (136). KU004, a HER2 inhibitor, inhibits the Warburg effect by the PI3K/Akt signaling pathway and suppresses hexokinase II (HK2), which mediates antitumor effect (137). Especially, the PI3K/Akt pathway induces targeted HER2 drug resistance in GC (138, 139). A glycolysis inhibitor MK2206 diminishes the trastuzumab resistance in HER2(+) GCs by attenuating the Warburg effect (139). Moreover, GATA6, the downstream of STAT3 (140), is involved in GC metabolic reprogramming, which may contribute to trastuzumab resistance (141). Further results indicate that Rhodium (III) complex 6, an effective STAT3 inhibitor (142), may be beneficial for targeting HER2 treatment of GC.
Aerobic glycolysis leads to the accumulation of lactate, which induces angiogenesis, an important process underlying tumor growth and metastasis (143). Ramucirumab, a vascular endothelial growth factor receptor (VEGFR) inhibitor, has shown limited benefits to GC due to metabolism activity (144). A further study suggested that glycolysis can negatively affect survival outcomes of metastatic GC patients treated with ramucirumab systemic therapy (134). Apatinib, another competitive inhibitor of VEGFR2, effectively suppresses glycolysis (145) and even induces the lipid metabolism in GC (146). The 2-deoxy-D-glucose, an inhibitor of glycolysis, can significantly reduce its angiogenic sprouting in tumor (147). PFKFB3 (glycolytic enzyme) not only regulates abnormal glycolytic metabolism in GC (148), and its inhibitors, PA-1 and PA-2, are potential antiangiogenic properties (149). Therefore, VEGFR inhibitor can be one of the cornerstones against angiogenesis therapies in GC subtypes, which represents an attractive therapeutic strategy to improve the efficacy of anti-GC treatments.
Immunotherapy
The cancer-immunity cycle (CIC) comprises a series of events that are required for immune-mediated control of tumor growth. Interruption of one or more steps of the CIC enables tumors to evade immunosurveillance. However, attempts to restore antitumor immunity by reactivating the CIC have had limited success thus far. The suppressive activity of Treg cells is mediated by several proteins present on the cell surface, such as the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and PD-1 (150), which induces cellular senescence and suppresses responder T cells through mediating accelerated glucose consumption (43). Immunotherapy, targeting the PD-1/PD-L1 and anticytotoxic lymphocyte antigen 4 (CTLA4) pathway, collectively named immune checkpoint inhibitor (ICI), by blocking Treg-mediated immunosuppression, derives durable remission and survival benefits for GC (151, 152). However, 50% of MSI-high GC are intrinsically resistant to PD-1 therapies (153). It is likely that continuous exposure to PD-1 antigen, which induces metabolic reprogramming of the T cell, induces T-cell exhaustion (154, 155). Diclofenac, a non-steroidal drug, turns out to inhibit the lactate transporters MCTs and improve T-cell killing, which improves the efficacy of anti-PD1 therapy (156). 6-Diazo-5-oxo-l-norleucine, a small molecule glutamine analog, increases infiltration of CD8+ T cells and sensitizes tumors to anti-PD1 therapy (157). Moreover, EBV-associated GC cells are treated with JAK2 inhibitor, PI3K inhibitor, and mTOR inhibitor, which arrests G0/G1, promotes the proliferation of T cells, and reduces the PD-L1 expression (158).
CTLA-4 represents a crucial immune checkpoint, the blockade of which can potentiate antitumor immunity. Limiting Treg cell metabolic competition in the TME may increase the effectiveness of immunotherapy (159). Especially, the effect of CTLA-4 blockade on the destabilization of T cells is dependent on T-cell glycolysis. Metformin is associated with decreased expression CTLA-4 of Treg cells, which induces glycolysis (160). Telaglenastat (CB-839), a potent GLS inhibitor, comminates with anti-PD1 or anti-CTLA4 antibodies, then increases tumor infiltration by effector T cells and improves the antitumor activity of these ICIs (161). Therefore, the combinational use of ICIs together with metabolic treatments to alleviate metabolic stress may improve the efficacy of immunotherapy.
Natural Compounds
Natural compounds, targeting the components of mitochondria, modulate metabolic abnormalities that are a consequence of immune cell dysfunction (162, 163). For example, salazosulfapyridine blocks cystine/glutamate exchange activity and mitigates the supply of cysteine to increase intracellular ROS production, thereby increasing the effect of anticancer drugs, such as cisplatin. Especially, its combination with 2-deoxyglucose significantly inhibits cell proliferation (164). Crocin, one of the main bioactive compounds of saffron, not only inhibits the EMT, migration, and invasion of GC cells through HIF-1α signaling (165) but also protects against malignant transformation by altering mitochondrial function (166, 167). The above-mentioned results show that natural compounds have great potential in regulating metabolic reprogramming. However, there are many kinds of natural compounds and different molecular pathways, and it is still necessary to establish a huge database and screen GC cell lines with metabolic phenotype for further studies.
To sum up, several metabolic inhibitors designed to target these pathways have been advanced into preclinical trials (Table 2). Anticancer effect or resistance can be revered by innovative anticancer treatments targeting metabolism. Depending on tumor type, not all patients benefit from metabolic reprogramming treatment and clinical responses, and the outcome on GC progression can be either positive or negative. Therefore, understanding the mechanisms of metabolic reprogramming can be a necessary tool to identify combinations of drugs that elude resistance and allow a better response for the patients.
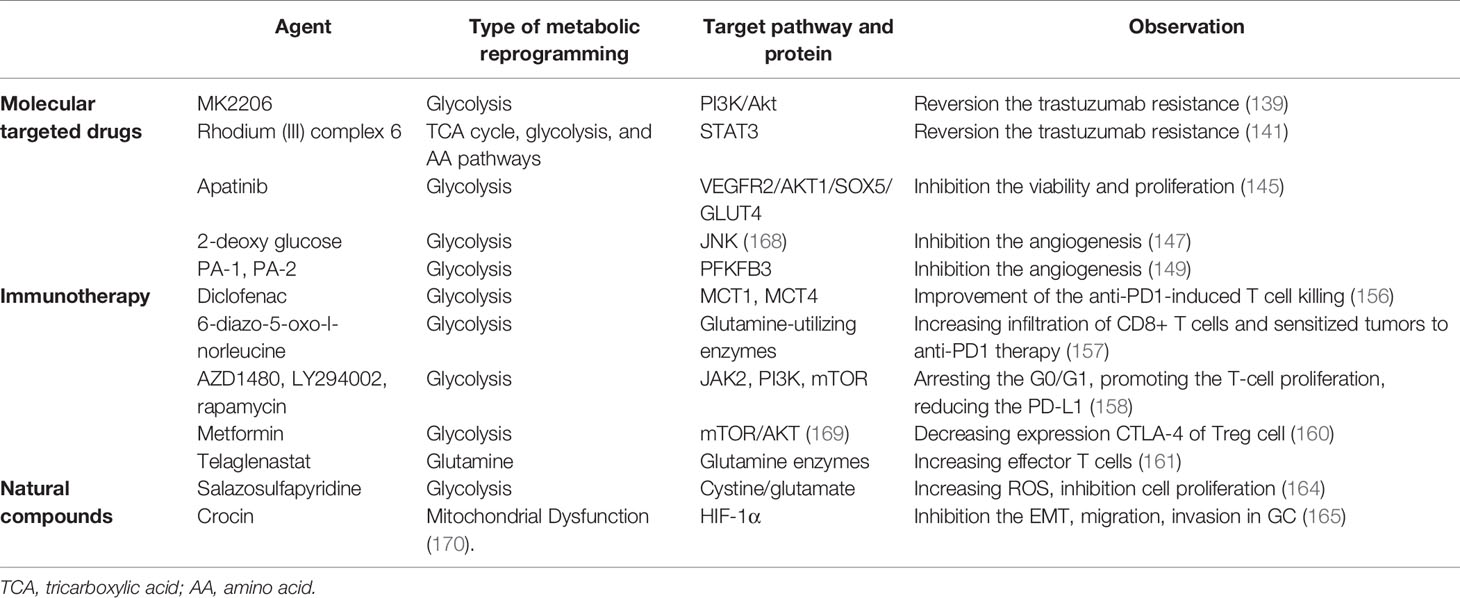
Table 2 Metabolic reprogramming drugs in GC.
Conclusion
Historically, the numerous metabolic reprogramming advances in distinguishing tumors from adjacent, non-malignant tissues and targeting these phenotypes indicate potential clinical applications. However, most cancer metabolism research has focused on phenotypes of clinically detectable tumors or experimental models derived from them, and the metabolic reprogramming of cancer cells is much more complex than first observed. Moreover, most metabolic changes are neutral or only slightly modify cancer cell fitness under stress (171). Certain pathways are essential for the progression of selected cancers and can be exploited therapeutically, and understanding GC metabolism and identifying liabilities require a sophisticated view of how metabolic phenotypes evolve.
The development of anticancer drugs in GC presents some challenges. First is the identification of accurate biomarkers that can predict the response to anticancer therapy. The second challenge is that metabolic reprogramming has emerged as a druggable target across GC, and the clinical development of combinatorial approaches should focus on how to maximize the efficacy. Third, most of the previous metabolic reprogramming studies to this point have been focused on alterations in the metabolism of glucose, glutamine, and lipid, while metabolic reprogramming also utilizes a great variety of other microelements (126). Taken together, understanding gene alterations in metabolic reprogramming is extremely important not only for GC diagnosis and prognosis but also for the development of potential targeted therapy. We should expand the research direction from the perspective of energy metabolism reprogramming.
Author Contributions
D-FL and Y-LB drafted the work or revised it critically for important intellectual content. H-SH, FT, ZHW, M-FY, B-HW, L-SW and JY contributed significantly to analysis and manuscript preparation. D-FL approved the final version to be published. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Guangdong Province (No. 2018A0303100024), Three Engineering Training Funds in Shenzhen (No. SYLY201718, No. SYJY201714, and No. SYLY201801), Technical Research and Development Project of Shenzhen (No. JCYJ20150403101028164, No. JCYC20170307100911479, and No. JCYJ20190807145617113), National Natural Science Foundation of China (No. 81800489), and the Natural Science Foundation of Hunan Province (No. 2021JJ70076), Technical Research and Development Project of Shenzhen (JCYJ20210324113802006).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Yuan L, Xu ZY, Ruan SM, Mo S, Qin JJ, Cheng XD, et al. Long Non-Coding RNAs Towards Precision Medicine in Gastric Cancer: Early Diagnosis, Treatment, and Drug Resistance. Mol Cancer (2020) 19:96. doi: 10.1186/s12943-020-01219-0
3. Li W, Ng JM, Wong CC, Ng EKW, Yu J. Molecular Alterations of Cancer Cell and Tumour Microenvironment in Metastatic Gastric Cancer. Oncogene (2018) 37:4903–20. doi: 10.1038/s41388-018-0341-x
4. Yan HHN, Siu HC, Law S, Ho SL, Yue SSK, Tsui WY, et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell (2018) 23:882–97.e11. doi: 10.1016/j.stem.2018.09.016
5. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
6. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic Reprogramming and Cancer Progression. Science (2020) 368(6487). doi: 10.1126/science.aaw5473
7. Ilkhani K, Bastami M, Delgir S, Safi A, Talebian S, Alivand MR. The Engaged Role of Tumor Microenvironment in Cancer Metabolism: Focusing on Cancer-Associated Fibroblast and Exosome Mediators. Anticancer Agents Med Chem (2020) 21(2):254–66. doi: 10.2174/1871520620666200910123428
8. Huang HC, Lin WR, Lim SN, Yeh CT, Yen TH, Alison MR, et al. Aldolase Triggers Metabolic Reprogramming in Colorectal Cancer in Hypoxia and Stiff Desmoplastic Microenvironments. Colloids Surf B Biointerfaces (2020) 190:110969. doi: 10.1016/j.colsurfb.2020.110969
9. Hu Y, Xu W, Zeng H, He Z, Lu X, Zuo D, et al. OXPHOS-Dependent Metabolic Reprogramming Prompts Metastatic Potential of Breast Cancer Cells Under Osteogenic Differentiation. Br J Cancer (2020) 123:1644–55. doi: 10.1038/s41416-020-01040-y
10. Sun L, Yang X, Huang X, Yao Y, Wei X, Yang S, et al. 2-Hydroxylation of Fatty Acids Represses Colorectal Tumorigenesis and Metastasis via the YAP Transcriptional Axis. Cancer Res (2020) 81(2):289–302. doi: 10.1158/0008-5472.CAN-20-1517
11. Lee JY, Nam M, Son HY, Hyun K, Jang SY, Kim JW, et al. Polyunsaturated Fatty Acid Biosynthesis Pathway Determines Ferroptosis Sensitivity in Gastric Cancer. Proc Natl Acad Sci USA (2020) 117:32433–42. doi: 10.1073/pnas.2006828117
12. Yao X, He Z, Qin C, Deng X, Bai L, Li G, et al. SLC2A3 Promotes Macrophage Infiltration by Glycolysis Reprogramming in Gastric Cancer. Cancer Cell Int (2020) 20:503. doi: 10.1186/s12935-020-01599-9
13. Yu M, Ozaki T, Sun D, Xing H, Wei B, An J, et al. HIF-1α-Dependent miR-424 Induction Confers Cisplatin Resistance on Bladder Cancer Cells Through Down-Regulation of Pro-Apoptotic UNC5B and SIRT4. J Exp Clin Cancer Res (2020) 39:108. doi: 10.1186/s13046-020-01613-y
14. Xu DH, Li Q, Hu H, Ni B, Liu X, Huang C, et al. Transmembrane Protein GRINA Modulates Aerobic Glycolysis and Promotes Tumor Progression in Gastric Cancer. J Exp Clin Cancer Res (2018) 37:308. doi: 10.1186/s13046-018-0974-1
15. Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, et al. Aspartate Is a Limiting Metabolite for Cancer Cell Proliferation Under Hypoxia and in Tumours. Nat Cell Biol (2018) 20:775–81. doi: 10.1038/s41556-018-0118-z
16. Jiang Z, Zhang C, Gan L, Jia Y, Xiong Y, Chen Y, et al. iTRAQ-Based Quantitative Proteomics Approach Identifies Novel Diagnostic Biomarkers That Were Essential for Glutamine Metabolism and Redox Homeostasis for Gastric Cancer. Proteomics Clin Appl (2019) 13:e1800038. doi: 10.1002/prca.201800038
17. Zhang W, Chen L, Xiang H, Hu C, Shi W, Dong P, et al. Knockdown of GGCT Inhibits Cell Proliferation and Induces Late Apoptosis in Human Gastric Cancer. BMC Biochem (2016) 17:19. doi: 10.1186/s12858-016-0075-8
18. Lu J, Chen M, Tao Z, Gao S, Li Y, Cao Y, et al. Effects of Targeting SLC1A5 on Inhibiting Gastric Cancer Growth and Tumor Development In Vitro and In Vivo. Oncotarget (2017) 8:76458–67. doi: 10.18632/oncotarget.19479
19. Wang C, Shi M, Ji J, Cai Q, Zhao Q, Jiang J, et al. Stearoyl-CoA Desaturase 1 (SCD1) Facilitates the Growth and Anti-Ferroptosis of Gastric Cancer Cells and Predicts Poor Prognosis of Gastric Cancer. Aging (Albany NY) (2020) 12:15374–91. doi: 10.18632/aging.103598
20. Wei C, Dong X, Lu H, Tong F, Chen L, Zhang R, et al. LPCAT1 Promotes Brain Metastasis of Lung Adenocarcinoma by Up-Regulating PI3K/AKT/MYC Pathway. J Exp Clin Cancer Res (2019) 38:95. doi: 10.1186/s13046-019-1092-4
21. Bi J, Ichu TA, Zanca C, Yang H, Zhang W, Gu Y, et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers Dependent on Membrane Lipid Remodeling. Cell Metab (2019) 30:525–38.e8. doi: 10.1016/j.cmet.2019.06.014
22. Tao L, Yu H, Liang R, Jia R, Wang J, Jiang K, et al. Rev-Erbα Inhibits Proliferation by Reducing Glycolytic Flux and Pentose Phosphate Pathway in Human Gastric Cancer Cells. Oncogenesis (2019) 8:57. doi: 10.1038/s41389-019-0168-5
23. Liu H, Luo J, Luan S, He C, Li Z. Long Non-Coding RNAs Involved in Cancer Metabolic Reprogramming. Cell Mol Life Sci (2019) 76:495–504. doi: 10.1007/s00018-018-2946-1
24. Tong H, Wang Y, Li Y, Liu S, Chi C, et al. Volatile Organic Metabolites Identify Patients With Gastric Carcinoma, Gastric Ulcer, or Gastritis and Control Patients. Cancer Cell Int (2017) 17:108. doi: 10.1186/s12935-017-0475-x
25. Jiang W, Zhou L, Lin S, Li Y, Xiao S, Liu J, et al. Metabolic Profiles of Gastric Cancer Cell Lines With Different Degrees of Differentiation. Int J Clin Exp Pathol (2018) 11:869–75.
26. Balakrishnan K, Ganesan K. Occurrence of Differing Metabolic Dysregulations, a Glucose Driven and Another Fatty Acid Centric in Gastric Cancer Subtypes. Funct Integr Genomics (2020) 20:813–24. doi: 10.1007/s10142-020-00753-w
27. Ganapathy-Kanniappan S. Molecular Intricacies of Aerobic Glycolysis in Cancer: Current Insights Into the Classic Metabolic Phenotype. Crit Rev Biochem Mol Biol (2018) 53:667–82. doi: 10.1080/10409238.2018.1556578
28. Orang AV, Petersen J, McKinnon RA, Michael MZ. Micromanaging Aerobic Respiration and Glycolysis in Cancer Cells. Mol Metab (2019) 23:98–126. doi: 10.1016/j.molmet.2019.01.014
29. Schlößer HA, Drebber U, Urbanski A, Haase S, Baltin C, Berlth F, et al. Glucose Transporters 1, 3, 6, and 10 Are Expressed in Gastric Cancer and Glucose Transporter 3 Is Associated With UICC Stage and Survival. Gastric Cancer (2017) 20:83–91. doi: 10.1007/s10120-015-0577-x
30. Park HS, Kim JH, Sun BK, Song SU, Suh W, Sung JH. Hypoxia Induces Glucose Uptake and Metabolism of Adipose−Derived Stem Cells. Mol Med Rep (2016) 14:4706–14. doi: 10.3892/mmr.2016.5796
31. Findlay JM, Antonowicz S, Segaran A, El Kafsi J, Zhang A, Bradley KM, et al. Routinely Staging Gastric Cancer With (18)F-FDG PET-CT Detects Additional Metastases and Predicts Early Recurrence and Death After Surgery. Eur Radiol (2019) 29:2490–8. doi: 10.1007/s00330-018-5904-2
32. Shen YA, Chen CL, Huang YH, Evans EE, Cheng CC, Chuang YJ, et al. Inhibition of Glutaminolysis in Combination With Other Therapies to Improve Cancer Treatment. Curr Opin Chem Biol (2021) 62:64–81. doi: 10.1016/j.cbpa.2021.01.006
33. Kanai Y, Clémençon B, Simonin A, Leuenberger M, Lochner M, Weisstanner M, et al. The SLC1 High-Affinity Glutamate and Neutral Amino Acid Transporter Family. Mol Aspects Med (2013) 34:108–20. doi: 10.1016/j.mam.2013.01.001
34. Ye J, Huang Q, Xu J, Huang J, Wang J, Zhong W, et al. Targeting of Glutamine Transporter ASCT2 and Glutamine Synthetase Suppresses Gastric Cancer Cell Growth. J Cancer Res Clin Oncol (2018) 144:821–33. doi: 10.1007/s00432-018-2605-9
35. Jiménez-González V, Ogalla-García E, García-Quintanilla M, García-Quintanilla A. Deciphering GRINA/Lifeguard1: Nuclear Location, Ca(2+) Homeostasis and Vesicle Transport. Int J Mol Sci (2019) 20:16. doi: 10.3390/ijms20164005
36. Koundouros N, Poulogiannis G. Reprogramming of Fatty Acid Metabolism in Cancer. Br J Cancer (2020) 122:4–22. doi: 10.1038/s41416-019-0650-z
37. Uehara T, Kikuchi H, Miyazaki S, Iino I, Setoguchi T, Hiramatsu Y, et al. Overexpression of Lysophosphatidylcholine Acyltransferase 1 and Concomitant Lipid Alterations in Gastric Cancer. Ann Surg Oncol (2016) 23(Suppl 2):S206–13. doi: 10.1245/s10434-015-4459-6
38. Yu S, Hu C, Cai L, Du X, Lin F, Yu Q, et al. Seven-Gene Signature Based on Glycolysis Is Closely Related to the Prognosis and Tumor Immune Infiltration of Patients With Gastric Cancer. Front Oncol (2020) 10:1778. doi: 10.3389/fonc.2020.01778
39. Oya Y, Hayakawa Y, Koike K. Tumor Microenvironment in Gastric Cancers. Cancer Sci (2020) 111:2696–707. doi: 10.1111/cas.14521
40. Brown TP, Ganapathy V. Lactate/GPR81 Signaling and Proton Motive Force in Cancer: Role in Angiogenesis, Immune Escape, Nutrition, and Warburg Phenomenon. Pharmacol Ther (2020) 206:107451. doi: 10.1016/j.pharmthera.2019.107451
41. Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer Metabolism: A Therapeutic Perspective. Nat Rev Clin Oncol (2017) 14:11–31. doi: 10.1038/nrclinonc.2016.60
42. Crowther MD, Svane IM, Met Ö. T-Cell Gene Therapy in Cancer Immunotherapy: Why It Is No Longer Just CARs on The Road. Cells (2020) 9(7):1588. doi: 10.3390/cells9071588
43. Wei J, Zheng W, Chapman NM, Geiger TL, Chi H. T Cell Metabolism in Homeostasis and Cancer Immunity. Curr Opin Biotechnol (2021) 68:240–50. doi: 10.1016/j.copbio.2021.02.003
44. Shi L, Feng M, Du S, Wei X, Song H, Yixin X, et al. Adenosine Generated by Regulatory T Cells Induces CD8(+) T Cell Exhaustion in Gastric Cancer Through A2aR Pathway. BioMed Res Int (2019) 2019:4093214. doi: 10.1155/2019/4093214
45. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-Like Receptor Signaling Balance T(reg) Cell Anabolic Metabolism for Suppression. Nat Immunol (2016) 17:1459–66. doi: 10.1038/ni.3577
46. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial Integrity Regulated by Lipid Metabolism Is a Cell-Intrinsic Checkpoint for Treg Suppressive Function. Cell Metab (2020) 31:422–37.e5. doi: 10.1016/j.cmet.2019.11.021
47. Kumagai S, Togashi Y, Sakai C, Kawazoe A, Kawazu M, Ueno T, et al. An Oncogenic Alteration Creates a Microenvironment That Promotes Tumor Progression by Conferring a Metabolic Advantage to Regulatory T Cells. Immunity (2020) 53:187–203.e8. doi: 10.1016/j.immuni.2020.06.016
48. Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8(+) T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab (2020) 31:148–61.e5. doi: 10.1016/j.cmet.2019.10.013
49. Sathe A, Grimes SM, Lau BT, Chen J, Suarez C, Huang RJ, et al. Single-Cell Genomic Characterization Reveals the Cellular Reprogramming of the Gastric Tumor Microenvironment. Clin Cancer Res (2020) 26:2640–53. doi: 10.1158/1078-0432.CCR-19-3231
50. Zhang Y, Guoqiang L, Sun M, Lu X. Targeting and Exploitation of Tumor-Associated Neutrophils to Enhance Immunotherapy and Drug Delivery for Cancer Treatment. Cancer Biol Med (2020) 17:32–43. doi: 10.20892/j.issn.2095-3941.2019.0372
51. Wang Y, Zhai J, Zhang T, Han S, Zhang Y, Yao X, et al. Tumor-Associated Neutrophils Can Predict Lymph Node Metastasis in Early Gastric Cancer. Front Oncol (2020) 10:570113. doi: 10.3389/fonc.2020.570113
52. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature (2012) 487:330–7. doi: 10.1038/nature11252
53. Zhang X, Shi H, Yuan X, Jiang P, Qian H, Xu W. Tumor-Derived Exosomes Induce N2 Polarization of Neutrophils to Promote Gastric Cancer Cell Migration. Mol Cancer (2018) 17:146. doi: 10.1186/s12943-018-0898-6
54. Wang TT, Zhao YL, Peng LS, Chen N, Chen W, Lv YP, et al. Tumour-Activated Neutrophils in Gastric Cancer Foster Immune Suppression and Disease Progression Through GM-CSF-PD-L1 Pathway. Gut (2017) 66:1900–11. doi: 10.1136/gutjnl-2016-313075
55. Injarabian L, Devin A, Ransac S, Marteyn BS. Neutrophil Metabolic Shift During Their Lifecycle: Impact on Their Survival and Activation. Int J Mol Sci (2019) 21(1):287. doi: 10.3390/ijms21010287
56. Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-Elicited Neutrophils Engage Mitochondrial Metabolism to Circumvent Nutrient Limitations and Maintain Immune Suppression. Nat Commun (2018) 9:5099. doi: 10.1038/s41467-018-07505-2
57. Chen J, Cao X, Li B, Zhao Z, Chen S, Lai SWT, et al. Warburg Effect Is a Cancer Immune Evasion Mechanism Against Macrophage Immunosurveillance. Front Immunol (2020) 11:621757. doi: 10.3389/fimmu.2020.621757
58. Genin M, Clement F, Fattaccioli A, Raes M, Michiels C. M1 and M2 Macrophages Derived From THP-1 Cells Differentially Modulate the Response of Cancer Cells to Etoposide. BMC Cancer (2015) 15:577. doi: 10.1186/s12885-015-1546-9
59. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
60. Zhou J, Bai W, Liu Q, Cui J, Zhang W. Intermittent Hypoxia Enhances THP-1 Monocyte Adhesion and Chemotaxis and Promotes M1 Macrophage Polarization via RAGE. BioMed Res Int (2018) 2018:1650456. doi: 10.1155/2018/1650456
61. Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
62. Li W, Zhang X, Wu F, Zhou Y, Bao Z, Li H, et al. Gastric Cancer-Derived Mesenchymal Stromal Cells Trigger M2 Macrophage Polarization That Promotes Metastasis and EMT in Gastric Cancer. Cell Death Dis (2019) 10:918. doi: 10.1038/s41419-019-2131-y
63. Odegaard JI, Chawla A. Alternative Macrophage Activation and Metabolism. Annu Rev Pathol (2011) 6:275–97. doi: 10.1146/annurev-pathol-011110-130138
64. Zheng P, Luo Q, Wang W, Li J, Wang T, Wang P, et al. Tumor-Associated Macrophages-Derived Exosomes Promote the Migration of Gastric Cancer Cells by Transfer of Functional Apolipoprotein E. Cell Death Dis (2018) 9:434. doi: 10.1038/s41419-018-0465-5
65. Ben Hassen C, Gutierrez-Pajares JL, Guimaraes C, Guibon R, Pinault M, Fromont G, et al. Apolipoprotein-Mediated Regulation of Lipid Metabolism Induces Distinctive Effects in Different Types of Breast Cancer Cells. Breast Cancer Res (2020) 22:38. doi: 10.1186/s13058-020-01276-9
66. Yu S, Li Q, Yu Y, Cui Y, Li W, Liu T, et al. Activated Hif1α of Tumor Cells Promotes Chemoresistance Development via Recruiting GDF15-Producing Tumor-Associated Macrophages in Gastric Cancer. Cancer Immunol Immunother (2020) 69:1973–87. doi: 10.1007/s00262-020-02598-5
67. Luo Q, Zheng N, Jiang L, Wang T, Zhang P, Liu Y, et al. Lipid Accumulation in Macrophages Confers Protumorigenic Polarization and Immunity in Gastric Cancer. Cancer Sci (2020) 111:4000–11. doi: 10.1111/cas.14616
68. Zhang Y, Meng W, Yue P, Li X. M2 Macrophage-Derived Extracellular Vesicles Promote Gastric Cancer Progression via a microRNA-130b-3p/MLL3/GRHL2 Signaling Cascade. J Exp Clin Cancer Res (2020) 39:134. doi: 10.1186/s13046-020-01626-7
69. Lan X, Wu L, Wu N, Chen Q, Li Y, Du X, et al. Long Noncoding RNA lnc-HC Regulates Pparγ-Mediated Hepatic Lipid Metabolism Through miR-130b-3p. Mol Ther Nucleic Acids (2019) 18:954–65. doi: 10.1016/j.omtn.2019.10.018
70. Chu H, Han N, Xu J. CMPK1 Regulated by miR-130b Attenuates Response to 5-FU Treatment in Gastric Cancer. Front Oncol (2021) 11:637470. doi: 10.3389/fonc.2021.637470
71. Miao Y, Zheng W, Li N, Su Z, Zhao L, Zhou H, et al. MicroRNA-130b Targets PTEN to Mediate Drug Resistance and Proliferation of Breast Cancer Cells via the PI3K/Akt Signaling Pathway. Sci Rep (2017) 7:41942. doi: 10.1038/srep41942
72. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
73. Melissari MT, Chalkidi N, Sarris ME, Koliaraki V, et al. Fibroblast Reprogramming in Gastrointestinal Cancer. Front Cell Dev Biol (2020) 8:630. doi: 10.3389/fcell.2020.00630
74. Li L, Wei JR, Dong J, Lin QG, Tang H, Jia YX, et al. Laminin γ2-Mediating T Cell Exclusion Attenuates Response to Anti-PD-1 Therapy. Sci Adv (2021) 7(6). doi: 10.1126/sciadv.abc8346
75. Kogure A, Naito Y, Yamamoto Y, Yashiro M, Kiyono T, Yanagihara K, et al. Cancer Cells With High-Metastatic Potential Promote a Glycolytic Shift in Activated Fibroblasts. PloS One (2020) 15:e0234613. doi: 10.1371/journal.pone.0234613
76. Li Q, Zhu CC, Ni B, Zhang ZZ, Jiang SH, Hu LP, et al. Lysyl Oxidase Promotes Liver Metastasis of Gastric Cancer via Facilitating the Reciprocal Interactions Between Tumor Cells and Cancer Associated Fibroblasts. EBioMedicine (2019) 49:157–71. doi: 10.1016/j.ebiom.2019.10.037
77. Kim HS, Won YJ, Shim JH, Kim HJ, Kim BS, Hong HN. Role of EphA2-PI3K Signaling in Vasculogenic Mimicry Induced by Cancer-Associated Fibroblasts in Gastric Cancer Cells. Oncol Lett (2019) 18:3031–8. doi: 10.3892/ol.2019.10677
78. Zhai J, Shen J, Xie G, Wu J, He M, Gao L, et al. Cancer-Associated Fibroblasts-Derived IL-8 Mediates Resistance to Cisplatin in Human Gastric Cancer. Cancer Lett (2019) 454:37–43. doi: 10.1016/j.canlet.2019.04.002
79. Li P, Shan JX, Chen XH, Zhang D, Su LP, Huang XY, et al. Epigenetic Silencing of microRNA-149 in Cancer-Associated Fibroblasts Mediates Prostaglandin E2/interleukin-6 Signaling in the Tumor Microenvironment. Cell Res (2015) 25:588–603. doi: 10.1038/cr.2015.51
80. Zhang X, Wang S, Wang H, Cao J, Huang X, Chen Z, et al. Circular RNA Circnrip1 Acts as a microRNA-149-5p Sponge to Promote Gastric Cancer Progression via the AKT1/mTOR Pathway. Mol Cancer (2019) 18:20. doi: 10.1186/s12943-018-0935-5
81. Kashyap D, Baral B, Jakhmola S, Singh AK, Jha HC. Helicobacter Pylori and Epstein-Barr Virus Coinfection Stimulates Aggressiveness in Gastric Cancer Through the Regulation of Gankyrin. mSphere (2021) e0075121. doi: 10.1128/mSphere.00751-21
82. Khosravi Y, Loke MF, Goh KL, Vadivelu J. Proteomics Analysis Revealed That Crosstalk Between Helicobacter Pylori and Streptococcus Mitis May Enhance Bacterial Survival and Reduces Carcinogenesis. Front Microbiol (2016) 7:1462. doi: 10.3389/fmicb.2016.01462
83. Navashenaq JG, Shabgah AG, Banach M, Jamialahmadi T, Penson PE, Johnston TP, et al. The Interaction of Helicobacter Pylori With Cancer Immunomodulatory Stromal Cells: New Insight Into Gastric Cancer Pathogenesis. Semin Cancer Biol (2021) S1044-579X(21):00248-0. doi: 10.1016/j.semcancer.2021.09.014
84. Gao S, Chen M, Wei W, Zhang X, Zhang M, Yao Y, et al. Crosstalk of mTOR/PKM2 and STAT3/c-Myc Signaling Pathways Regulate the Energy Metabolism and Acidic Microenvironment of Gastric Cancer. J Cell Biochem (2018). doi: 10.1002/jcb.26915
85. Ebron JS, Shankar E, Singh J, Sikand K, Weyman CM, Gupta S, et al. MiR-644a Disrupts Oncogenic Transformation and Warburg Effect by Direct Modulation of Multiple Genes of Tumor-Promoting Pathways. Cancer Res (2019) 79:1844–56. doi: 10.1158/0008-5472.CAN-18-2993
86. Ramapriyan R, Caetano MS, Barsoumian HB, Mafra ACP, Zambalde EP, Menon H, et al. Altered Cancer Metabolism in Mechanisms of Immunotherapy Resistance. Pharmacol Ther (2019) 195:162–71. doi: 10.1016/j.pharmthera.2018.11.004
87. Wang X, Che X, Yu Y, Cheng Y, Bai M, Yang Z, et al. Hypoxia-Autophagy Axis Induces VEGFA by Peritoneal Mesothelial Cells to Promote Gastric Cancer Peritoneal Metastasis Through an Integrin α5-Fibronectin Pathway. J Exp Clin Cancer Res (2020) 39:221. doi: 10.1186/s13046-020-01703-x
88. Yousefi B, Mohammadlou M, Abdollahi M, Salek Farrokhi A, Karbalaei M, Keikha M, et al. Epigenetic Changes in Gastric Cancer Induction by Helicobacter Pylori. J Cell Physiol (2019) 234:21770–84. doi: 10.1002/jcp.28925
89. Kim IJ, Lee J, Oh SJ, Yoon MS, Jang SS, Holland RL, et al. Helicobacter Pylori Infection Modulates Host Cell Metabolism Through VacA-Dependent Inhibition of Mtorc1. Cell Host Microbe (2018) 23:583–93.e8. doi: 10.1016/j.chom.2018.04.006
90. Taylor CT, Colgan SP. Hypoxia and Gastrointestinal Disease. J Mol Med (Berl) (2007) 85:1295–300. doi: 10.1007/s00109-007-0277-z
91. Wang J, Ni Z, Duan Z, Wang G, Li F. Altered Expression of Hypoxia-Inducible Factor-1α (HIF-1α) and Its Regulatory Genes in Gastric Cancer Tissues. PloS One (2014) 9:e99835. doi: 10.1371/journal.pone.0099835
92. Masoud GN, Li W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm Sin B (2015) 5:378–89. doi: 10.1016/j.apsb.2015.05.007
93. Zhang L, Li S. Lactic Acid Promotes Macrophage Polarization Through MCT-Hif1α Signaling in Gastric Cancer. Exp Cell Res (2020) 388:111846. doi: 10.1016/j.yexcr.2020.111846
94. Tanaka T, Kitajima Y, Miyake S, Yanagihara K, Hara H, Nishijima-Matsunobu A, et al. The Apoptotic Effect of HIF-1α Inhibition Combined With Glucose Plus Insulin Treatment on Gastric Cancer Under Hypoxic Conditions. PloS One (2015) 10:e0137257. doi: 10.1371/journal.pone.0137257
95. Vaupel P, Multhoff G. Hypoxia-/HIF-1α-Driven Factors of the Tumor Microenvironment Impeding Antitumor Immune Responses and Promoting Malignant Progression. Adv Exp Med Biol (2018) 1072:171–5. doi: 10.1007/978-3-319-91287-5_27
96. Sun P, Hu JW, Xiong WJ, Mi J. miR-186 Regulates Glycolysis Through Glut1 During the Formation of Cancer-Associated Fibroblasts. Asian Pac J Cancer Prev (2014) 15:4245–50. doi: 10.7314/APJCP.2014.15.10.4245
97. Liu L, Wang Y, Bai R, Yang K, Tian Z. MiR-186 Inhibited Aerobic Glycolysis in Gastric Cancer via HIF-1α Regulation. Oncogenesis (2016) 5:e224. doi: 10.1038/oncsis.2016.35
98. Canales J, Valenzuela M, Bravo J, Cerda-Opazo P, Jorquera C, Toledo H, et al. Helicobacter Pylori Induced Phosphatidylinositol-3-OH Kinase/mTOR Activation Increases Hypoxia Inducible Factor-1α to Promote Loss of Cyclin D1 and G0/G1 Cell Cycle Arrest in Human Gastric Cells. Front Cell Infect Microbiol (2017) 7:92. doi: 10.3389/fcimb.2017.00092
99. Lee DY, Jung DE, Yu SS, Lee YS, Choi BK, Lee YC. Regulation of SIRT3 Signal Related Metabolic Reprogramming in Gastric Cancer by Helicobacter Pylori Oncoprotein CagA. Oncotarget (2017) 8:78365–78. doi: 10.18632/oncotarget.18695
100. Lu Y, Rong J, Lai Y, Tao L, Yuan X, Shu X. The Degree of Helicobacter Pylori Infection Affects the State of Macrophage Polarization Through Crosstalk Between ROS and HIF-1α. Oxid Med Cell Longev (2020) 2020:5281795. doi: 10.1155/2020/5281795
101. Kou Y, Tong B, Wu W, Liao X, Zhao M. Berberine Improves Chemo-Sensitivity to Cisplatin by Enhancing Cell Apoptosis and Repressing PI3K/AKT/mTOR Signaling Pathway in Gastric Cancer. Front Pharmacol (2020) 11:616251. doi: 10.3389/fphar.2020.616251
102. Peng W, Huang W, Ge X, Xue L, Zhao W, Xue J. Type Iγ Phosphatidylinositol Phosphate Kinase Promotes Tumor Growth by Facilitating Warburg Effect in Colorectal Cancer. EBioMedicine (2019) 44:375–86. doi: 10.1016/j.ebiom.2019.05.015
103. Sun P, Meng LH. Emerging Roles of Class I PI3K Inhibitors in Modulating Tumor Microenvironment and Immunity. Acta Pharmacol Sin (2020) 41:1395–402. doi: 10.1038/s41401-020-00500-8
104. Ma L, Zong X. Metabolic Symbiosis in Chemoresistance: Refocusing the Role of Aerobic Glycolysis. Front Oncol (2020) 10:5. doi: 10.3389/fonc.2020.00005
105. Xie Y, Liu L. Analysis of Correlation Between HP Infection and Activation of PI3K/Akt Pathway in Mucosal Tissues of Gastric Cancer and Precancerous Lesions. Oncol Lett (2018) 16:5615–20. doi: 10.3892/ol.2018.9329
106. Li N, Tang B, Jia YP, Zhu P, Zhuang Y, Fang Y, et al. Helicobacter Pylori CagA Protein Negatively Regulates Autophagy and Promotes Inflammatory Response via C-Met-PI3K/Akt-mTOR Signaling Pathway. Front Cell Infect Microbiol (2017) 7:417. doi: 10.3389/fcimb.2017.00417
107. Xu L, Chen J, Jia L, Chen X, Awaleh Moumin F, Cai J. SLC1A3 Promotes Gastric Cancer Progression via the PI3K/AKT Signalling Pathway. J Cell Mol Med (2020) 24(24):14392–404. doi: 10.1111/jcmm.16060
108. Zhang X, Li Z, Xuan Z, Xu P, Wang W, Chen Z, et al. Novel Role of miR-133a-3p in Repressing Gastric Cancer Growth and Metastasis via Blocking Autophagy-Mediated Glutaminolysis. J Exp Clin Cancer Res (2018) 37:320. doi: 10.1186/s13046-018-0993-y
109. Shi L, Wu Z, Miao J, Du S, Ai S, Xu E, et al. Adenosine Interaction With Adenosine Receptor A2a Promotes Gastric Cancer Metastasis by Enhancing PI3K-AKT-mTOR Signaling. Mol Biol Cell (2019) 30:2527–34. doi: 10.1091/mbc.E19-03-0136
110. Yang L, Zhang W, Wang Y, Zou T, Zhang B, Xu Y, et al. Hypoxia-Induced miR-214 Expression Promotes Tumour Cell Proliferation and Migration by Enhancing the Warburg Effect in Gastric Carcinoma Cells. Cancer Lett (2018) 414:44–56. doi: 10.1016/j.canlet.2017.11.007
111. Li J, Shen J, Wang Z, Xu H, Wang Q, Chai S, et al. ELTD1 Facilitates Glioma Proliferation, Migration and Invasion by Activating JAK/STAT3/HIF-1α Signaling Axis. Sci Rep (2019) 9:13904. doi: 10.1038/s41598-019-50375-x
112. Zhao FL, Qin CF. EGF Promotes HIF-1α Expression in Colorectal Cancer Cells and Tumor Metastasis by Regulating Phosphorylation of STAT3. Eur Rev Med Pharmacol Sci (2019) 23:1055–62. doi: 10.26355/eurrev_201902_16993
113. Miao L, Qi J, Zhao Q, Wu QN, Wei DL, Wei XL, et al. Targeting the STING Pathway in Tumor-Associated Macrophages Regulates Innate Immune Sensing of Gastric Cancer Cells. Theranostics (2020) 10:498–515. doi: 10.7150/thno.37745
114. Shen J, Zhai J, You Q, Zhang G, He M, Yao X, et al. Cancer-Associated Fibroblasts-Derived VCAM1 Induced by H. Pylori Infection Facilitates Tumor Invasion in Gastric Cancer. Oncogene (2020) 39:2961–74. doi: 10.1038/s41388-020-1197-4
115. Wang X, Che X, Liu C, Fan Y, Bai M, Hou K, et al. Cancer-Associated Fibroblasts-Stimulated Interleukin-11 Promotes Metastasis of Gastric Cancer Cells Mediated by Upregulation of MUC1. Exp Cell Res (2018) 368:184–93. doi: 10.1016/j.yexcr.2018.04.028
116. Dodington DW, Desai HR, Woo M. JAK/STAT - Emerging Players in Metabolism. Trends Endocrinol Metab (2018) 29:55–65. doi: 10.1016/j.tem.2017.11.001
117. Owen KL, Brockwell NK, Parker BS. JAK-STAT Signaling: A Double-Edged Sword of Immune Regulation and Cancer Progression. Cancers (Basel) (2019) 11(12). doi: 10.3390/cancers11122002
118. Heppler LN, Frank DA. Inhibit Versus Destroy: Are PROTAC Degraders the Solution to Targeting Stat3? Cancer Cell (2019) 36:459–61. doi: 10.1016/j.ccell.2019.10.010
119. Li XR, Cheng X, Sun J, Xu YS, Chen N, Hao Y, et al. Acetylation-Dependent Glutamate Receptor GluR Signalosome Formation for STAT3 Activation in Both Transcriptional and Metabolism Regulation. Cell Death Discov (2021) 7:11. doi: 10.1038/s41420-020-00389-6
120. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-Mediated Gasdermin C Expression Switches Apoptosis to Pyroptosis in Cancer Cells and Facilitates Tumour Necrosis. Nat Cell Biol (2020) 22:1264–75. doi: 10.1038/s41556-020-0575-z
121. Morey P, Pfannkuch L, Pang E, Boccellato F, Sigal M, Imai-Matsushima A, et al. Helicobacter Pylori Depletes Cholesterol in Gastric Glands to Prevent Interferon Gamma Signaling and Escape the Inflammatory Response. Gastroenterology (2018) 154:1391–404.e9. doi: 10.1053/j.gastro.2017.12.008
122. Ju X, Zhang H, Zhou Z, et al. Tumor-Associated Macrophages Induce PD-L1 Expression in Gastric Cancer Cells Through IL-6 and TNF-α Signaling. Exp Cell Res (2020) 396:112315. doi: 10.1016/j.yexcr.2020.112315
123. Huynh J, Chand A, Gough D, Ernst M. Therapeutically Exploiting STAT3 Activity in Cancer - Using Tissue Repair as a Road Map. Nat Rev Cancer (2019) 19:82–96. doi: 10.1038/s41568-018-0090-8
124. Chon SH, Berlth F, Plum PS, Herbold T, Alakus H, Kleinert R, et al. Gastric Cancer Treatment in the World: Germany. Transl Gastroenterol Hepatol (2017) 2:53. doi: 10.21037/tgh.2017.05.07
125. Salati M, Di Emidio K, Tarantino V, Cascinu S. Second-Line Treatments: Moving Towards an Opportunity to Improve Survival in Advanced Gastric Cancer? ESMO Open (2017) 2:e000206. doi: 10.1136/esmoopen-2017-000206
126. Di Bartolomeo M, Morano F, Raimondi A, Miceli R, Corallo S, Tamborini E, et al. Prognostic and Predictive Value of Microsatellite Instability, Inflammatory Reaction and PD-L1 in Gastric Cancer Patients Treated With Either Adjuvant 5-FU/LV or Sequential FOLFIRI Followed by Cisplatin and Docetaxel: A Translational Analysis From the ITACA-S Trial. Oncologist (2020) 25:e460–8. doi: 10.1634/theoncologist.2019-0471
127. Russi S, Verma HK, Laurino S, Mazzone P, Storto G, Nardelli A, et al. Adapting and Surviving: Intra and Extra-Cellular Remodeling in Drug-Resistant Gastric Cancer Cells. Int J Mol Sci (2019) 20(15). doi: 10.3390/ijms20153736
128. Chen X, Chen S, Yu D. Metabolic Reprogramming of Chemoresistant Cancer Cells and the Potential Significance of Metabolic Regulation in the Reversal of Cancer Chemoresistance. Metabolites (2020) 10(7). doi: 10.3390/metabo10070289
129. Shimanishi M, Ogi K, Sogabe Y, Kaneko T, Dehari H, Miyazaki A, et al. Silencing of GLUT-1 Inhibits Sensitization of Oral Cancer Cells to Cisplatin During Hypoxia. J Oral Pathol Med (2013) 42:382–8. doi: 10.1111/jop.12028
130. Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, et al. Warburg Effect in Chemosensitivity: Targeting Lactate Dehydrogenase-A Re-Sensitizes Taxol-Resistant Cancer Cells to Taxol. Mol Cancer (2010) 9:33. doi: 10.1186/1476-4598-9-33
131. Cruz IN, Coley HM, Kramer HB, Madhuri TK, Safuwan NA, Angelino AR, et al. Proteomics Analysis of Ovarian Cancer Cell Lines and Tissues Reveals Drug Resistance-Associated Proteins. Cancer Genomics Proteomics (2017) 14:35–51. doi: 10.21873/cgp.20017
132. Tripathi SC, Fahrmann JF, Celiktas M, Aguilar M, Marini KD, Jolly MK, et al. MCAM Mediates Chemoresistance in Small-Cell Lung Cancer via the PI3K/AKT/SOX2 Signaling Pathway. Cancer Res (2017) 77:4414–25. doi: 10.1158/0008-5472.CAN-16-2874
133. Gao S, Song D, Liu Y, Yan H, Chen X. Helicobacter Pylori CagA Protein Attenuates 5-Fu Sensitivity of Gastric Cancer Cells Through Upregulating Cellular Glucose Metabolism. Onco Targets Ther (2020) 13:6339–49. doi: 10.2147/OTT.S230875
134. Ruzzo A, Graziano F, Bagaloni I, Di Bartolomeo M, Prisciandaro M, Aprile G, et al. Glycolytic Competence in Gastric Adenocarcinomas Negatively Impacts Survival Outcomes of Patients Treated With Salvage Paclitaxel-Ramucirumab. Gastric Cancer (2020) 23:1064–74. doi: 10.1007/s10120-020-01078-0
135. Wu S, Zhang Q, Zhang F, Meng F, Liu S, Zhou R, et al. HER2 Recruits AKT1 to Disrupt STING Signalling and Suppress Antiviral Defence and Antitumour Immunity. Nat Cell Biol (2019) 21:1027–40. doi: 10.1038/s41556-019-0352-z
136. Sung M, Tan X, Lu B, Golas J, Hosselet C, Wang F, et al. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-Dm1). Mol Cancer Ther (2018) 17:243–53. doi: 10.1158/1535-7163.MCT-17-0403
137. Tian C, Yuan Z, Xu D, Ding P, Wang T, Zhang L, et al. Inhibition of Glycolysis by a Novel EGFR/HER2 Inhibitor KU004 Suppresses the Growth of HER2+ Cancer. Exp Cell Res (2017) 357:211–21. doi: 10.1016/j.yexcr.2017.05.019
138. Su B, Huang T, Jin Y, Yin H, Qiu H, Yuan X. Apatinib Exhibits Synergistic Effect With Pyrotinib and Reverses Acquired Pyrotinib Resistance in HER2-Positive Gastric Cancer via Stem Cell Factor/C-Kit Signaling and Its Downstream Pathways. Gastric Cancer (2021) 24:352–67. doi: 10.1007/s10120-020-01126-9
139. Liu J, Pan C, Guo L, Wu M, Guo J, Peng S, et al. A New Mechanism of Trastuzumab Resistance in Gastric Cancer: MACC1 Promotes the Warburg Effect via Activation of the PI3K/AKT Signaling Pathway. J Hematol Oncol (2016) 9:76. doi: 10.1186/s13045-016-0302-1
140. Wu CS, Wei KL, Chou JL, Lu CK, Hsieh CC, Lin JM, et al. Aberrant JAK/STAT Signaling Suppresses TFF1 and TFF2 Through Epigenetic Silencing of GATA6 in Gastric Cancer. Int J Mol Sci (2016) 17(9). doi: 10.3390/ijms17091467
141. Chang J, Wang Q, Bhetuwal A, Liu W. Metabolic Pathways Underlying GATA6 Regulating Trastuzumab Resistance in Gastric Cancer Cells Based on Untargeted Metabolomics. Int J Med Sci (2020) 17:3146–64. doi: 10.7150/ijms.50563
142. Ma DL, Liu LJ, Leung KH, Chen YT, Zhong HJ, Chan DS, et al. Antagonizing STAT3 Dimerization With a Rhodium(III) Complex. Angew Chem Int Ed Engl (2014) 53:9178–82. doi: 10.1002/anie.201404686
143. Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, Mourón S, et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Rep (2016) 15:2705–18. doi: 10.1016/j.celrep.2016.05.052
144. Kim ST, Sa JK, Oh SY, Kim K, Hong JY, Kang WK, et al. Comprehensive Molecular Characterization of Gastric Cancer Patients From Phase II Second-Line Ramucirumab Plus Paclitaxel Therapy Trial. Genome Med (2021) 13:11. doi: 10.1186/s13073-021-00826-w
145. Chen L, Cheng X, Tu W, Qi Z, Li H, Liu F, et al. Apatinib Inhibits Glycolysis by Suppressing the VEGFR2/AKT1/SOX5/GLUT4 Signaling Pathway in Ovarian Cancer Cells. Cell Oncol (Dordr) (2019) 42:679–90. doi: 10.1007/s13402-019-00455-x
146. Zhao L, Peng Y, He S, Li R, Wang Z, Huang J, et al. Apatinib Induced Ferroptosis by Lipid Peroxidation in Gastric Cancer. Gastric Cancer (2021) 24:642–54. doi: 10.1007/s10120-021-01159-8
147. Kunhiraman H, Edatt L, Thekkeveedu S, Poyyakkara A, Raveendran V, Kiran MS, et al. 2-Deoxy Glucose Modulates Expression and Biological Activity of VEGF in a SIRT-1 Dependent Mechanism. J Cell Biochem (2017) 118:252–62. doi: 10.1002/jcb.25629
148. Jin X, Qiao L, Fan H, Liao C, Zheng J, Wang W, et al. Long Non-Coding RNA MSC-AS1 Facilitates the Proliferation and Glycolysis of Gastric Cancer Cells by Regulating PFKFB3 Expression. Int J Med Sci (2021) 18:546–54. doi: 10.7150/ijms.51947
149. Abdali A, Baci D, Damiani I, Belloni F, De Dominicis C, Gelmi ML, et al. In Vitro Angiogenesis Inhibition With Selective Compounds Targeting the Key Glycolytic Enzyme PFKFB3. Pharmacol Res (2021) 168:105592. doi: 10.1016/j.phrs.2021.105592
150. Ohue Y, Nishikawa H. Regulatory T (Treg) Cells in Cancer: Can Treg Cells be a New Therapeutic Target? Cancer Sci (2019) 110:2080–9. doi: 10.1111/cas.14069
151. Kumar P, Saini S, Prabhakar BS. Cancer Immunotherapy With Check Point Inhibitor can Cause Autoimmune Adverse Events Due to Loss of Treg Homeostasis. Semin Cancer Biol (2020) 64:29–35. doi: 10.1016/j.semcancer.2019.01.006
152. Catanese S, Lordick F. Targeted and Immunotherapy in the Era of Personalised Gastric Cancer Treatment. Best Pract Res Clin Gastroenterol (2021) 50–51:101738. doi: 10.1016/j.bpg.2021.101738
153. Panda A, Mehnert JM, Hirshfield KM, Riedlinger G, Damare S, Saunders T, et al. Immune Activation and Benefit From Avelumab in EBV-Positive Gastric Cancer. J Natl Cancer Inst (2018) 110:316–20. doi: 10.1093/jnci/djx213
154. Daneshmandi S, Cassel T, Lin P, Higashi RM, Wulf GM, Boussiotis VA, et al. Blockade of 6-Phosphogluconate Dehydrogenase Generates CD8(+) Effector T Cells With Enhanced Anti-Tumor Function. Cell Rep (2021) 34:108831. doi: 10.1016/j.celrep.2021.108831
155. Yu X, Gao R, Li Y, Zeng C. Regulation of PD-1 in T Cells for Cancer Immunotherapy. Eur J Pharmacol (2020) 881:173240. doi: 10.1016/j.ejphar.2020.173240
156. Renner K, Bruss C, Schnell A, Koehl G, Becker HM, Fante M, et al. Restricting Glycolysis Preserves T Cell Effector Functions and Augments Checkpoint Therapy. Cell Rep (2019) 29:135–50.e9. doi: 10.1016/j.celrep.2019.08.068
157. Sharma NS, Gupta VK, Garrido VT, Hadad R, Durden BC, Kesh K, et al. Targeting Tumor-Intrinsic Hexosamine Biosynthesis Sensitizes Pancreatic Cancer to Anti-PD1 Therapy. J Clin Invest (2020) 130:451–65. doi: 10.1172/JCI127515
158. Sasaki S, Nishikawa J, Sakai K, Iizasa H, Yoshiyama H, Yanagihara M, et al. EBV-Associated Gastric Cancer Evades T-Cell Immunity by PD-1/PD-L1 Interactions. Gastric Cancer (2019) 22:486–96. doi: 10.1007/s10120-018-0880-4
159. Anti-CTLA4 Perturbs T(reg)-Based Immunosuppression in Glycolysis-Low Tumors. Cancer Discovery (2021). doi: 10.1158/2159-8290.Cd-rw2021-028
160. Kunisada Y, Eikawa S, Tomonobu N, Domae S, Uehara T, Hori S, et al. Attenuation of CD4(+)CD25(+) Regulatory T Cells in the Tumor Microenvironment by Metformin, a Type 2 Diabetes Drug. EBioMedicine (2017) 25:154–64. doi: 10.1016/j.ebiom.2017.10.009
161. Varghese S, Pramanik S, Williams LJ, Hodges HR, Hudgens CW, Fischer GM, et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol Cancer Ther (2021) 20:500–11. doi: 10.1158/1535-7163.MCT-20-0430
162. Chen H, Jiang T, Chen H, Su J, Wang X, Cao Y, et al. Brusatol Reverses Lipopolysaccharide-Induced Epithelial-Mesenchymal Transformation and Induces Apoptosis Through PI3K/Akt/NF-Кb Pathway in Human Gastric Cancer SGC-7901 Cells. Anticancer Drugs (2020) 32(4):394–404. doi: 10.1097/CAD.0000000000001022
163. Xiang S, Zhao Z, Zhang T, Zhang B, Meng M, Cao Z, et al. Triptonide Effectively Suppresses Gastric Tumor Growth and Metastasis Through Inhibition of the Oncogenic Notch1 and NF-κb Signaling Pathways. Toxicol Appl Pharmacol (2020) 388:114870. doi: 10.1016/j.taap.2019.114870
164. Takizawa K, Muramatsu K, Maruyama K, Urakami K, Sugino T, Kusuhara M, et al. Metabolic Profiling of Human Gastric Cancer Cells Treated With Salazosulfapyridine. Technol Cancer Res Treat (2020) 19:1533033820928621. doi: 10.1177/1533033820928621
165. Zhou Y, Xu Q, Shang J, Lu L, Chen G. Crocin Inhibits the Migration, Invasion, and Epithelial-Mesenchymal Transition of Gastric Cancer Cells via miR-320/KLF5/HIF-1α Signaling. J Cell Physiol (2019) 234:17876–85. doi: 10.1002/jcp.28418
166. Wu Z, Hui J. Crocin Reverses 1-Methyl-3-Nitroso-1-Nitroguanidine (MNNG)-Induced Malignant Transformation in GES-1 Cells Through the Nrf2/Hippo Signaling Pathway. J Gastrointest Oncol (2020) 11:1242–52. doi: 10.21037/jgo-20-406
167. Zhang Y, Zhu M, Krishna Mohan S, Hao Z. Crocin Treatment Promotes the Oxidative Stress and Apoptosis in Human Thyroid Cancer Cells FTC-133 Through the Inhibition of STAT/JAK Signaling Pathway. J Biochem Mol Toxicol (2021) 35:e22608. doi: 10.1002/jbt.22608
168. Xu Y, Wang Q, Zhang L, Zheng M. 2-Deoxy-D-Glucose Enhances TRAIL-Induced Apoptosis in Human Gastric Cancer Cells Through Downregulating JNK-Mediated Cytoprotective Autophagy. Cancer Chemother Pharmacol (2018) 81:555–64. doi: 10.1007/s00280-018-3526-7
169. Lu CC, Chiang JH, Tsai FJ, Hsu YM, Juan YN, Yang JS, et al. Metformin Triggers the Intrinsic Apoptotic Response in Human AGS Gastric Adenocarcinoma Cells by Activating AMPK and Suppressing mTOR/AKT Signaling. Int J Oncol (2019) 54:1271–81. doi: 10.3892/ijo.2019.4704
170. Li X, Liu Y, Cao A, Li C, Wang L, Wu Q, et al. Crocin Improves Endothelial Mitochondrial Dysfunction via GPx1/ROS/KCa3.1 Signal Axis in Diabetes. Front Cell Dev Biol (2021) 9:651434. doi: 10.3389/fcell.2021.651434
Keywords: gastric cancer, glycolysis, metabolic reprogramming, tumor microenvironment, drug resistance
Citation: Bin Y-L, Hu H-S, Tian F, Wen Z-H, Yang M-F, Wu B-H, Wang L-S, Yao J and Li D-F (2022) Metabolic Reprogramming in Gastric Cancer: Trojan Horse Effect. Front. Oncol. 11:745209. doi: 10.3389/fonc.2021.745209
Received: 21 July 2021; Accepted: 12 November 2021;
Published: 12 January 2022.
Edited by:
Elisa Giommoni, Careggi University Hospital, ItalyReviewed by:
Tania Fiaschi, Università degli Studi di Firenze, ItalyArun Upadhyay, Northwestern University, United States
Copyright © 2022 Bin, Hu, Tian, Wen, Yang, Wu, Wang, Yao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: De-Feng Li, bGRmODMwNzEyQDE2My5jb20=