 Meng Gao1,2†
Meng Gao1,2† Chengjie Mei1,2†
Chengjie Mei1,2† Yonghua Guo1,2Peng Xia1,2Hao Zhang1,2
Yonghua Guo1,2Peng Xia1,2Hao Zhang1,2 Yinyi Liu1,2Ye Yao1,2Xiang Jiang1,2
Yinyi Liu1,2Ye Yao1,2Xiang Jiang1,2 Yufeng Yuan1,2*
Yufeng Yuan1,2* Yawei Qian3*
Yawei Qian3*- 1Department of Hepatobiliary and Pancreatic Surgery, Zhongnan Hospital of Wuhan University, Wuhan, China
- 2Clinical Medicine Research Center for Minimally Invasive Procedure of Hepatobiliary & Pancreatic Diseases of Hubei Province, Wuhan, China
- 3Department of General Surgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, China
Intrahepatic cholangiocarcinoma (ICC) is one of the most commonly diagnosed malignancies worldwide, and the second most common primary liver tumor. The lack of effective diagnostic and treatment methods results in poor patient prognosis and high mortality rate. Atypical protein kinase C-ι (aPKC-ι) is highly expressed in primary and metastatic ICC tissues, and regulates epithelial mesenchymal transition (EMT) through the aPKC-ι/P-Sp1/Snail signaling pathway. Recent studies have correlated aberrant glucose metabolism with EMT. Given the vital role of FBP1 in regulating glucose metabolism in cancer cells, we hypothesized that aPKC-ι downregulates FBP1 in ICC cells through the Snai1 pathway, and enhances glycolysis and metastasis. We confirmed the ability of aPKC-ι promotes glycolysis, invasion and metastasis of cancer cells, and further demonstrated that FBP1 inhibits the malignant properties of ICC cells by antagonizing aPKC-ι. Our findings provide novel insights into the molecular mechanisms of ICC progression and metastasis, as well as a theoretical basis for exploring new treatment strategies.
Introduction
Intrahepatic cholangiocarcinoma (ICC) is a highly malignant tumor that originates from the epithelial cells of the intrahepatic secondary bile duct and its branches. It is the second most common primary liver tumor, and has poor prognosis (1) due to frequent metastasis and recalcitrance to radiotherapy and chemotherapy (2, 3). Although notable improvements have been made with different preclinical available cholangiocarcinoma models (4–6), such as the fibroblast growth factor receptor 2 inhibitor pemigatinib, the first approval of a molecularly targeted treatment in patients with advanced cholangiocarcinoma by US Food and Drug Administration, the prognosis of this disease remains unsatisfactory (7). Therefore, further studies on the molecular mechanism of invasion and metastasis and exploring a novel and effective therapeutic target of ICC are urgently needed (8).
Atypical protein kinase C-ι (aPKC-ι) is a promising target for multiple tumor types, such as non-small cell lung cancer, pancreatic ductal adenocarcinoma and hepatocellular carcinoma (9–11). A recent study showed that targeted silencing of aPKC-ι reversed TGF-β-induced epithelial mesenchymal transition (EMT) of non-small cell lung cancer cells, implying that aPKC-ι may function to promote EMT (12). In addition, it is also involved in tumor cell transformation, adhesion, movement, invasion and metastasis (13).
Fructose-1,6-bisphosphatase (FBP1) is one of the rate-limiting enzymes of gluconeogenesis (14), and its epigenetic regulation is critical to the glucose metabolism in cancer cells (15, 16). Loss of FBP1 in basal-like breast cancer cells significantly increased glucose uptake, lactate secretion and glycolysis rate, which was conducive to EMT and the maintenance of the basal-like phenotype (17, 18). It is also reported that decreased FBP1 expression regulating by miR-18a-5p promotes liver cancer cells migration and invasion (19). Glucose metabolism is a determinant of cancer cell invasion and metastasis. Cancer cells preferentially use glycolysis to produce energy in the hypoxic tumor mass, a phenomenon known as the “Warburg effect” (20–23). Glucose uptake is significantly enhanced in the cancer cells during EMT, which coincides with increased glycolysis (24, 25) and lactate secretion that promote tumor invasion and distant metastasis (26, 27). It is also reported that the reprogramming of tumor cell metabolism is a macroscopic change, and the change of ICC from oxidative phosphorylation to glycolysis provides favorable conditions for the proliferation of tumor cells (5–7).
The Snail family of transcriptional repressors, including Snai1, Snai2 (Slug) and Snai3 (Smuc), have been implicated in EMT during embryonic development as well as carcinogenesis. Snai1-mediated inhibition of the tumor suppressor microRNA let-7 is associated with poor prognosis in several cancers (28). In our previous studies, we found that high levels aPKC-ι in ICC cells correlated to increased glycolysis and lactate production (29). Given that FBP1 deletion promotes tumorigenesis in some cancers (30), our aim of this study was to elucidate the mechanistic relationship between PKC-ι, FBP1 and Snai1 in the regulation of glycolysis and metastasis of ICC cells.
Materials and Methods
Tissue Samples and Cell Culture
40 paired of ICC and para-cancerous tissue specimens were collected at the Zhongnan Hospital of Wuhan University (Hubei Province, China) after obtaining written consent from the patients. HCCC-9810 and RBE cell lines were obtained from the cell bank of the Chinese Academy of Sciences (Shanghai, China), and were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (GIBCO, US) at 37°C under 5% CO2. Cells in the logarithmic growth phase were selected for the experiments. The study was conducted according to the guidelines of the “Declaration of Helsinki” and approved by the Hospital Committee for the Protection of Human Subjects.
RNA Isolation and qRT-PCR
Total RNA was isolated form the cells and tissues using TRIzol reagent (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. A reverse transcription kit Vazyme, Hubei, China) was used to synthesize cDNA, and RT-PCR was performed using the SYBR Green PCR Kit (Vazyme, Hubei, China).
Histology and Immunohistochemistry (IHC)
The tumor tissue samples were fixed in 10% formalin, embedded in paraffin, and cut into 4μm-thick sections. After clearing with xylene and rehydrating with ethanol, the sections were incubated with 0.3% hydrogen peroxide to inactivate the endogenous peroxidases. The tissue sections were then incubated with the primary antibody, washed with PBS, and probed with the HRP-conjugated secondary antibody and streptavidin (Santa Cruz). Diaminobenzidine substrate was used for color development, followed by counterstaining with hematoxylin. The tissue microarray chips were probed with specific antibodies and digitally scanned.
Western Blotting
Western blotting was performed as previously described (31). The proteins extracted from the cells were quantified using the BCA protein assay method (Biyuntian, Wuhan, China), and equal amounts of protein per sample were separated via 6–15% SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, USA). After blocking with 5% skimmed milk, the membranes were incubated overnight with the primary antibodies at 4°C, washed thrice with TBST (10 minutes each), and probed with the secondary antibody at room temperature. The membranes were washed thrice with TBST and developed with ECL.
Glucose Uptake and Lactate Secretion Assay
The cells were seeded in a 6-well plate at the density of 5 × 105 cells per well, and cultured in the presence of EGF or 2-DG 48h. The glucose uptake was measured using a glucose uptake cell-based assay kit (Cayman Chemical) according to the manufacturer’s instructions. The cells were analyzed by flow cytometry. The conditioned media were centrifuged at 13000 g for 10 minutes at 4°C, and the lactate concentration in the clarified supernatants was measured using the L-lactate detection kit (Eton Biosciences) according to the manufacturer’s instructions. The absolute lactate level was calculated from the corresponding standard curve and normalized to the number of cells.
Seahorse XFp Metabolic Flux Analysis
The day before the experiment, ICC cells were planted on the Seahorse XF cell culture plate at 1×106 cells/well. Add the test solution prepared by Seahorse XF Base Medium to the plate, then use NaOH to adjust the pH to 7.4. 2 hours later, change the medium and place it in a carbon dioxide-free incubator for one hour. After adding mitochondrial inhibitors oligomycin, FCCP, and antimycin (AA) plus rotenone (AR), the baseline was measured four times in sequence. Then use the XFp extracellular analyzer (Agilent Technologies, USA) to analyze the oxygen consumption rate (OCR). Similarly, after adding Gluoose, Oligomycin, 2-DG, we can calculate the extracellular acidification rate (ECAR).
Chromatin Immunoprecipitation (ChIP)
EZ ChIP™ Chromatin Immunoprecipitation Kit (Millipore, Billerica, MA) was used for ChIP assay. The treated cells were cross-linked in 1% formaldehyde at 37°C for 10 minutes, and sonicated to obtain chromatin fragments of 200-1000 bp. The lysates were incubated overnight with 3μg anti-Snai1 or anti-IgG antibody (Millipore) at 4°C, followed by magnetic beads for 2h. The protein/DNA complex was eluted and cross-linked, and the immunoprecipitated DNA was analyzed by qRT-PCR.
Dual Luciferase Assay
Cells were co-transfected with FBP1-3’UTR-WT or -MUT reporter plasmid and Renilla luciferase vector using Lipofectamine 3000 reagent (Invitrogen). Luciferase activity was measured 48h later using the dual luciferase reporter gene detection system (Promega), and the data was normalized to Renilla luciferase activity.
Establishment of Subcutaneous Tumor and Lung Metastasis Model
The animal experiments were approved by the Institutional Animal Ethics Committee of Wuhan University. 10 of four-week-old female BALB/c nude mice were injected subcutaneously with 1×106 HCCC-9810 cells to induce subcutaneous tumors. The mice were sacrificed 4 weeks later for further analysis. The lung metastasis model was established in four-week-old female BALB/c nude mice by intravenous injection of 3×106 HCCC-9810 cells (n=5 for each group), and the lungs were harvested 4 weeks later for IHC (32).
Statistical Analysis
All data are expressed as the mean ± SD of at least three independent experiments. Two groups were compared by Student’s t test (SPSS statistical software package, version 12; SPSS Inc.). Overall survival rates were analyzed by the Kaplan-Meier method and Cox proportional hazards regression model. P <0.05 was considered statistically significant.
Results
FBP1 Expression Is Inversely Correlated With aPKC-ι in Intrahepatic Cholangiocarcinoma
Quantitative real-time PCR was employed to determine the expression levels of aPKC-ι and FBP1 in 40 paired ICC and adjacent nontumor tissues (Figure 1A). The results were confirmed by WB and IHC (Figures 1C, D). We found that aPKC-ι and FBP1 were both localized to the cytoplasm of cancer cells in IHC experiments. In addition, the expression level of aPKC-ι was significantly higher in tumor tissues compared to that in paired para-matched non-tumor specimens, while FBP1 expression was markedly lower. We next investigated whether the expression of aPKC-ι and FBP1 was associated with clinicopathological characteristics and prognosis. The clinical data of all ICC patients, including age, gender, tumor/non-tumor tissues, nodal invasion, tumor staging and tumor differentiation degree, are summarized in Table 1.
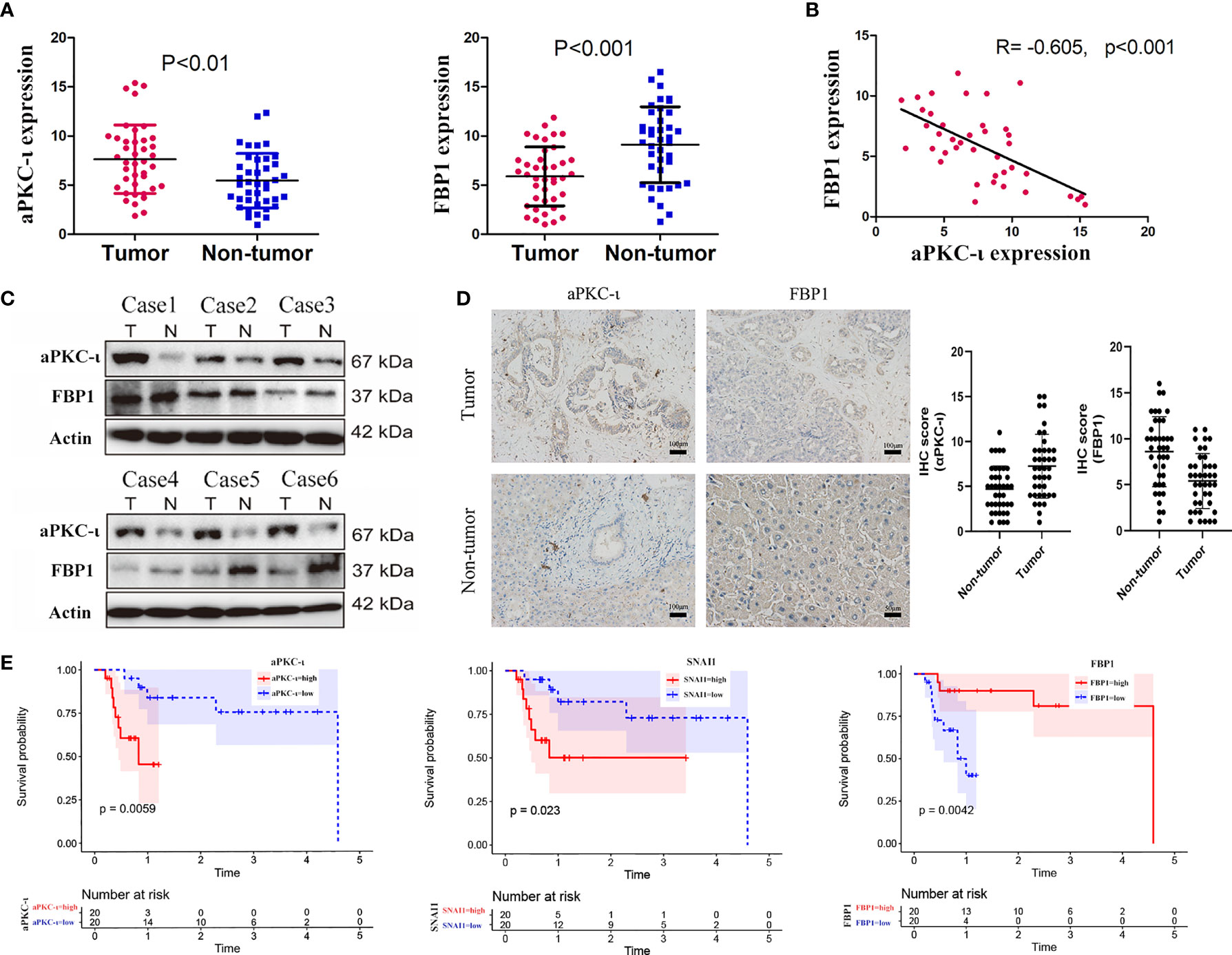
Figure 1 FBP1 expression is inversely correlated with aPKC-ι in ICC. (A) mRNA expression levels of aPKC-ι and FBP1 in 40 paired ICC and adjacent nontumor tissues (B). Pearson correlation analysis of aPKC-ι and FBP1 expression (R=-0.605,p<0.01). (C) Expression of aPKC-ι and FBP1 in 6 matched ICC samples. (D) IHC score and representative images showing in situ expression of aPKC-ι and FBP1 in ICC and normal bile duct tissues. (E) Overall survival rate in patient with aPKC-ι, FBP1 and SNAI1 expression.

Table 1 Clinicopathological parameters of ICC patients.
Overexpression of aPKC-ι was related to tumor–node–metastasis (TNM) stage III-IV (χ2 = 10.417, P=0.004), and medium/poor differentiation (χ2 = 10.157, P=0.005) in ICC patients. And, as expected, low expression of FBP1 was related to tumor–node–metastasis (TNM) stage III-IV (χ2 = 10.417, P=0.004), and medium/poor differentiation (χ2 = 6.144, P=0.034). We further investigated whether there was an correlation between aPKC-ι and FBP1 expression in ICC samples. As shown in Figure 1B, aPKC-ι level is significantly negatively associated with FBP1 (R=-0.605, p<0.01). Meanwhile, a Kaplan-Meier analysis indicated that patients with high expression of aPKC-ι displayed a shorter OS compared to those with low expression, while those with low FBP1 expression exhibited a shorter OS (Figure 1E). Multivariate Cox regression analyses also showed that aPKC-ι and FBP1 were independent prognostic factors for OS in ICC (Table 2). These above results suggested that aPKC-ι may interact with FBP1 and promote invasion and metastasis of ICC.
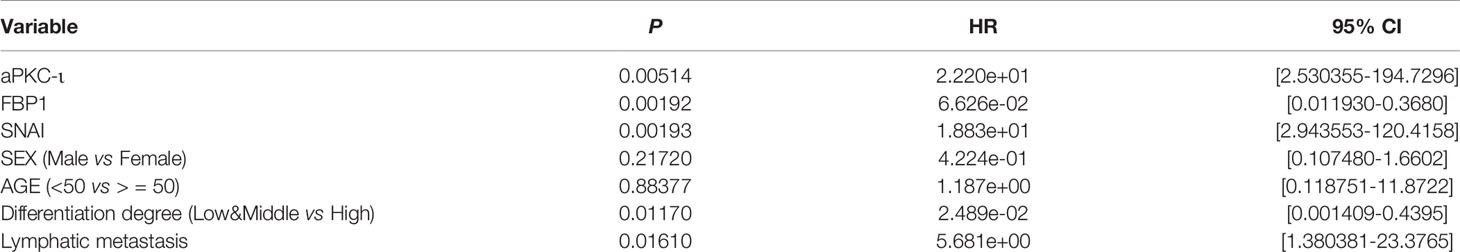
Table 2 Multivariate cox regression analyses.
Silencing aPKC-ι Attenuates Aerobic Glycolysis and Retards EMT-Like Changes in ICC Cells
Previous studies reported that aPKC-ι may induce EMT-like changes and promote metastasis in cancer cells (33). In this study, we first established two stable human ICC cell lines, HCCC-9810 and RBE, with down-regulated aPKC-ι expression levels in vitro by transfection with human aPKC-ι siRNA (Figure 2A). Along with aPKC-ι down-regulation, the EMT-like protein expression profiles in both HCCC-9810 and RBE cell lines were reversed compared with negative controls (Figure 2B), including up-regulation of the epithelial markers E-cadherin and down-regulation of mesenchymal marker Vimentin. These indicated that aPKC-ι promoted early invasion and metastasis of ICC cells by accelerating EMT. Consistent with above findings, ICC cells with down-regulated aPKC-ι also reversed EMT-like cellular characteristics, including decreased cell proliferation, migration and invasion as compared to negative controls (Figures 2C–E). To further assess the effects of aPKC-ι in ICC, metabolism-related indices were determined by glucose uptake and lactate secretion assay. We found that knocking down aPKC-ι deceased the rate of glucose uptake and lactate production in the ICC cells (Figures 2F, G), which was according with its role in the aberrant glucose metabolism of cancer cells. Thus, we speculated that aPKC-ι promoted EMT and aerobic glycolysis of ICC cells, which could provide a competitive environment for ICC cells invasion and migration.
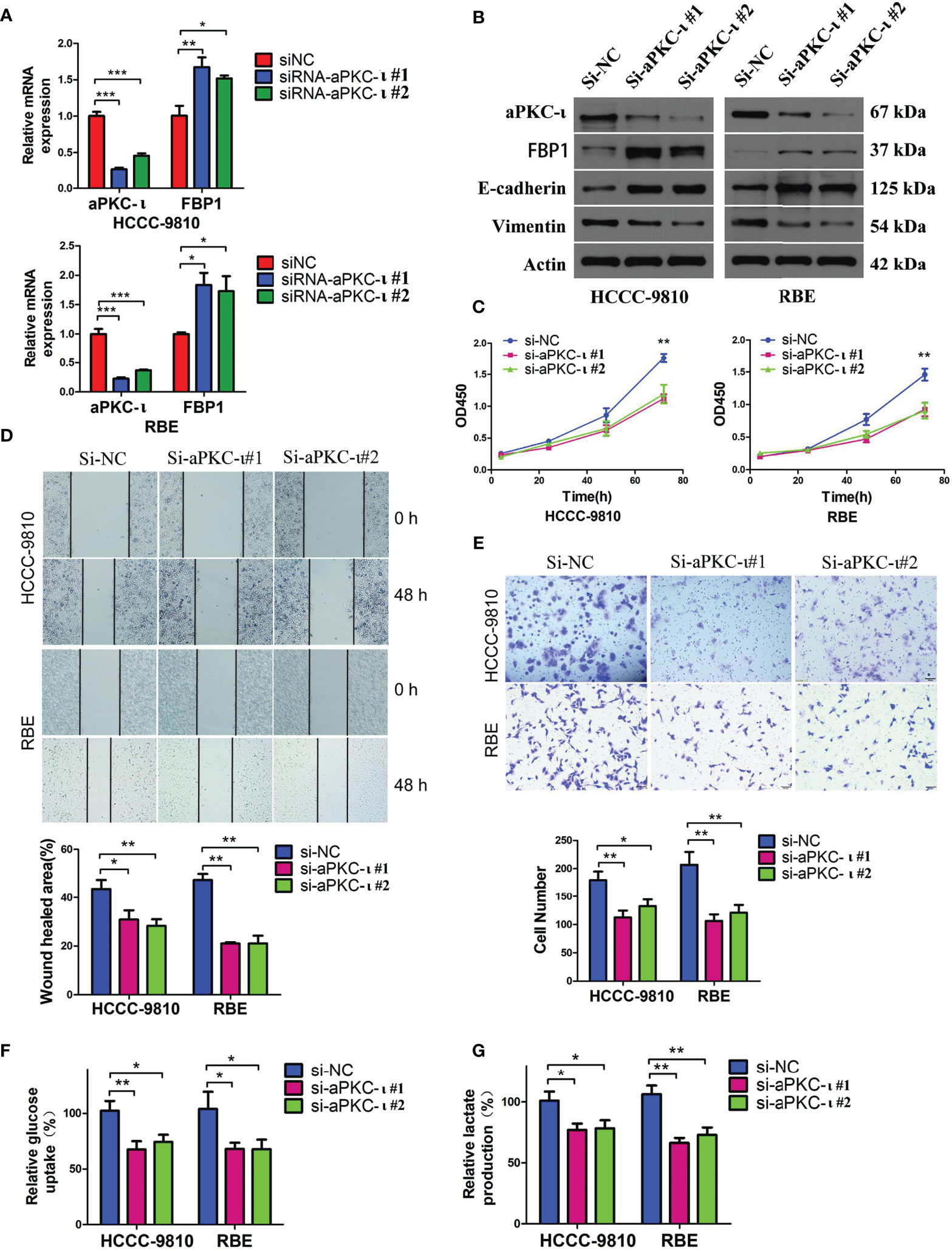
Figure 2 Down-regulated aPKC-ι reversed EMT-like changes and metabolic advantages in cancer cells. (A) FBP1 mRNA level in aPKC-ι knockdown HCCC-9810 and RBE cell lines. (B–E) Expression levels of EMT-related proteins including the epithelial markers E-cadherin and mesenchymal marker Vimentin, viability, and in vitro migration and invasion of control and aPKC-ι knockdown ICC cells. (F, G) Glucose uptake rate and lactate production after knocking down aPKC-ι in HCCC-9810 and RBE cell lines. * means P < 0.05, ** means P < 0.01, *** means P < 0.001.
FBP1 Is Crucial for aPKC-ι Induced EMT-Like Changes and Glycolysis in Human ICC Cells
To investigate the causal relationship between aPKC-ι and FBP1, we first suppressed aPKC-ι expression in two ICC cell lines. FBP1 mRNA and protein levels were up-regulated in aPKC-ι-deficient HCCC-9810 and RBE cells (Figures 2A, B), which was suggestive of an inhibitory effect of aPKC-ι on FBP1 mRNA and protein. We further determined whether FBP1 was regulated by aPKC-ι. The mRNA and protein levels of FBP1 were down-regulated in aPKC-ι-overexpressing ICC cells by transfecting with aPKC-ι-cDNA (Figures 3A, B). Therefore, FBP1 expressions were markedly increased both in mRNA and protein levels by transfecting aPKC-ι-cDNA ICC cells with FBP1-cDNA, while aPKC-ι levels were no significant changes as compared with negative control.
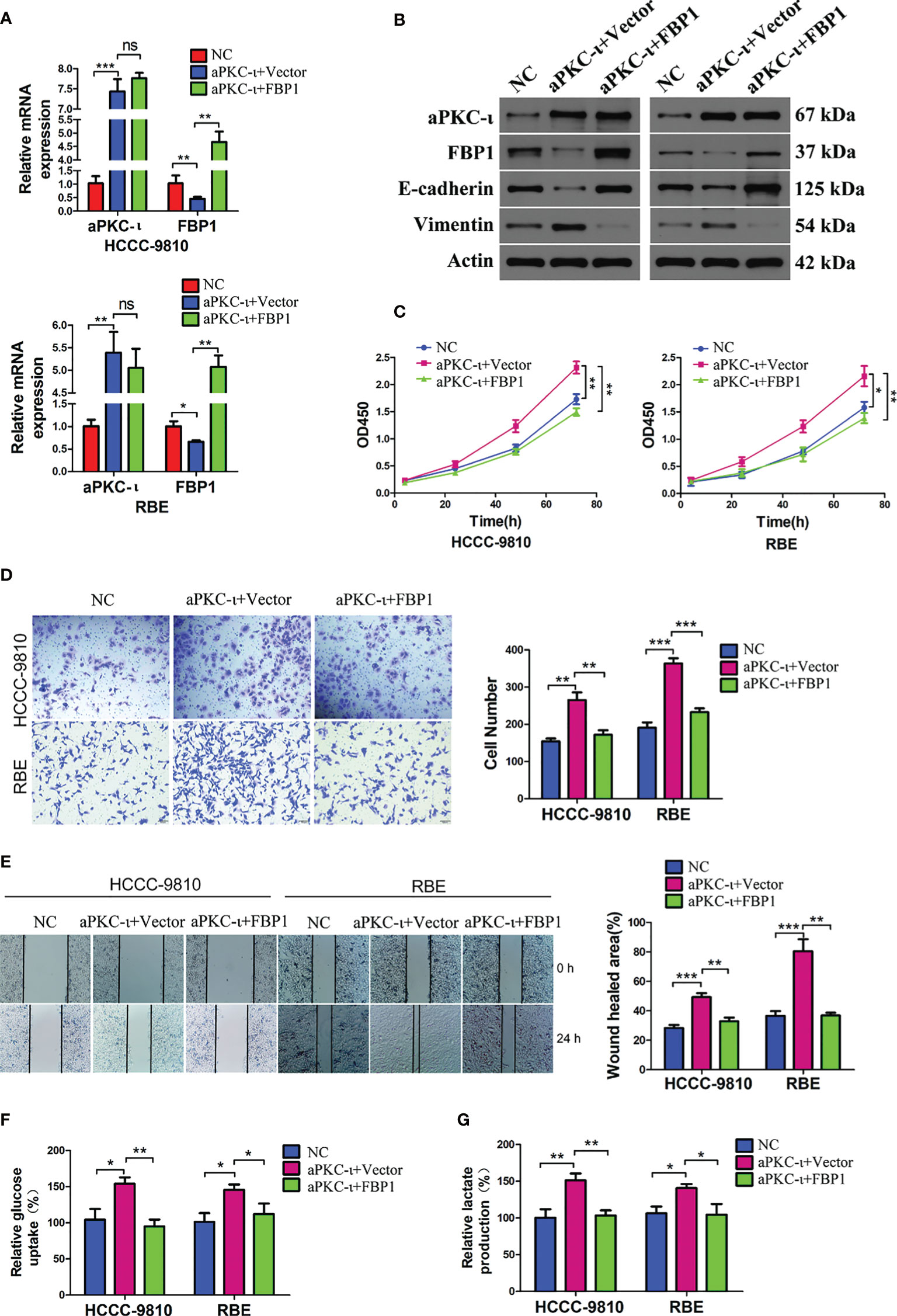
Figure 3 FBP1 antagonizes aPKC-ι and inhibits tumor progression. (A, B) The mRNA and protein levels of FBP1 were down-regulated in aPKC-ι-overexpressing ICC cells by transfecting with aPKC-ι-cDNA. (C–E) EMT-like changes were reversed compared with negative control groups, including cell proliferation, invasion, and migration. (F, G) Glucose uptake rate and lactate production in aPKC-ι/aPKC-ι + FBP1 groups. * means P < 0.05, ** means P < 0.01, *** means P < 0.001. ns, no statistical difference.
Following the up-regulation of FBP1 expression in aPKC-ι-overexpressing ICC cells, EMT-like changes were almost reversed compared with negative control groups, including the expression levels of EMT markers (Figure 3B) and cell proliferation, invasion, and migration (Figures 3C–E). In addition, along with the up-regulation of FBP1 expression, FBP1-cDNA transfection significantly decreased the relative glucose uptake rate and lactate production (Figures 3F, G). Those suggested that FBP1 could reverse the aPKC-ι induced EMT-like changes and normalize glucose metabolism in ICC cells.
aPKC-ι and FBP1 Play the Roles of Accelerator and Speed Limiter in the Process of Glycolysis
Along with koncking down aPKC-i, four key glycolytic enzymes (HK2, ENO1, PKM, LDHA) were decreased at the protein level (Figure 4A), which additionally supported that aPKC-i could enhance the glycolysis level of ICC. The cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were further measured in ICC cells (Figure 4B). According to ECAR and OCR curve chart (Figures 4C, D), the glycolysis level, glycolysis capacity and glycolysis capacity reserve were significantly increased in aPKC-i-overexpressing ICC cells compared to negative control, while OCR was decreased as compared with negative control. Following increased FBP1 expression in aPKC-i-overexpressing ICC cells, those glycolysis markers were markedly down-regulated, whereas the OCR was obviously up-regulated. The results reinforced that aPKC-i/FBP1 pathway palys a important role in glucose metabolism of ICC, which was consistent with our previous conclusions.
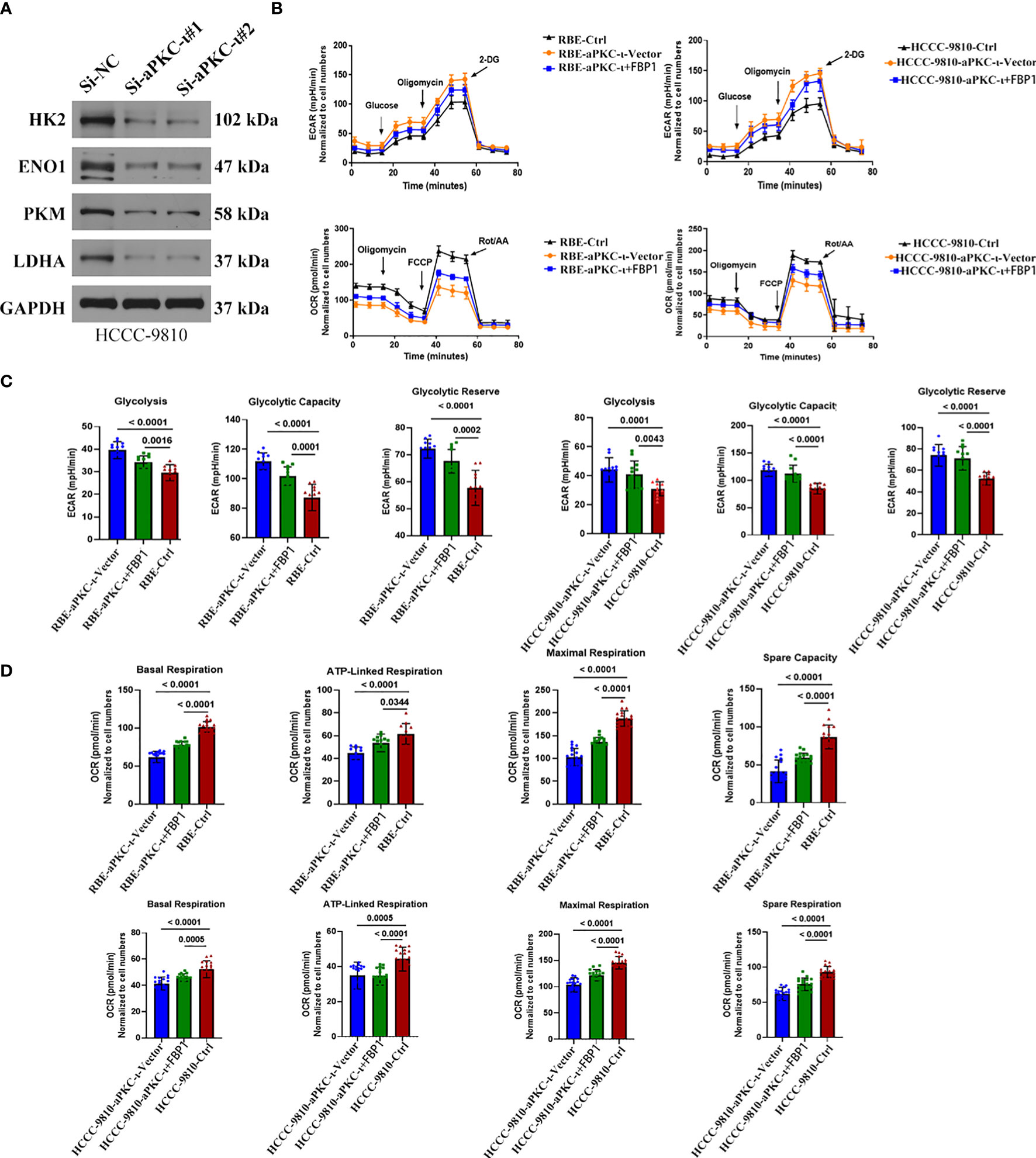
Figure 4 aPKC-ι and FBP1 play the roles of accelerator and speed limiter in the process of glycolysis. (A) The protein levels of HK2,ENO1,PKM,LDHA were down-regulated in aPKC-ι knockdown ICC cells. (B) Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of ICC cells which were transfected with aPKC-ι/aPKC-ι+FBP1 cDNA. (C, D) Glycolysis level, glycolysis capacity,glycolysis capacity,reserve basal respiration, ATP-linked respiration, maximum respiration, and spare respiration of ICC cells transfected with aPKC-ι/aPKC-ι+FBP1 cDNA.
FBP1 Antagonizes aPKC-ι and Inhibits Tumor Growth and Metastasis In Vivo
To further evaluate the role of FBP1 in ICC, a xenograft tumor model and a pulmonary metastasis tumor model were established. We subcutaneously injected aPKC-ι-overexpressing HCCC-9810 cells transfected with FBP1-cDNA or vector into nude mice. The volumes of tumors from aPKC-ι-overexpressing cells transfected with FBP1-cDNA were dramatically smaller compared with negative control groups (Figure 5A). Furthermore, the in situ expression of FBP1 and E-cadherin detected by IHC were significantly higher in FBP1-cDNA treatment groups, while Vimentin expression was lower (Figure 5B). The in situ expression of HK2,ENO1,PKM,LDHA detected by IHC was higher in FBP1-cDNA treatment groups (Figure 5C). Moreover, fewer metastatic nodules were determined in the treatment groups than in the untreated groups, suggesting that FBP1 could significantly inhibit lung metastases of ICC cells induced by aPKC-ι (Figure 5D). In conclusion, the assay confirmed that FBP1 played an important role in aPKC-ι-mediated ICC progression and metastasis in vivo.
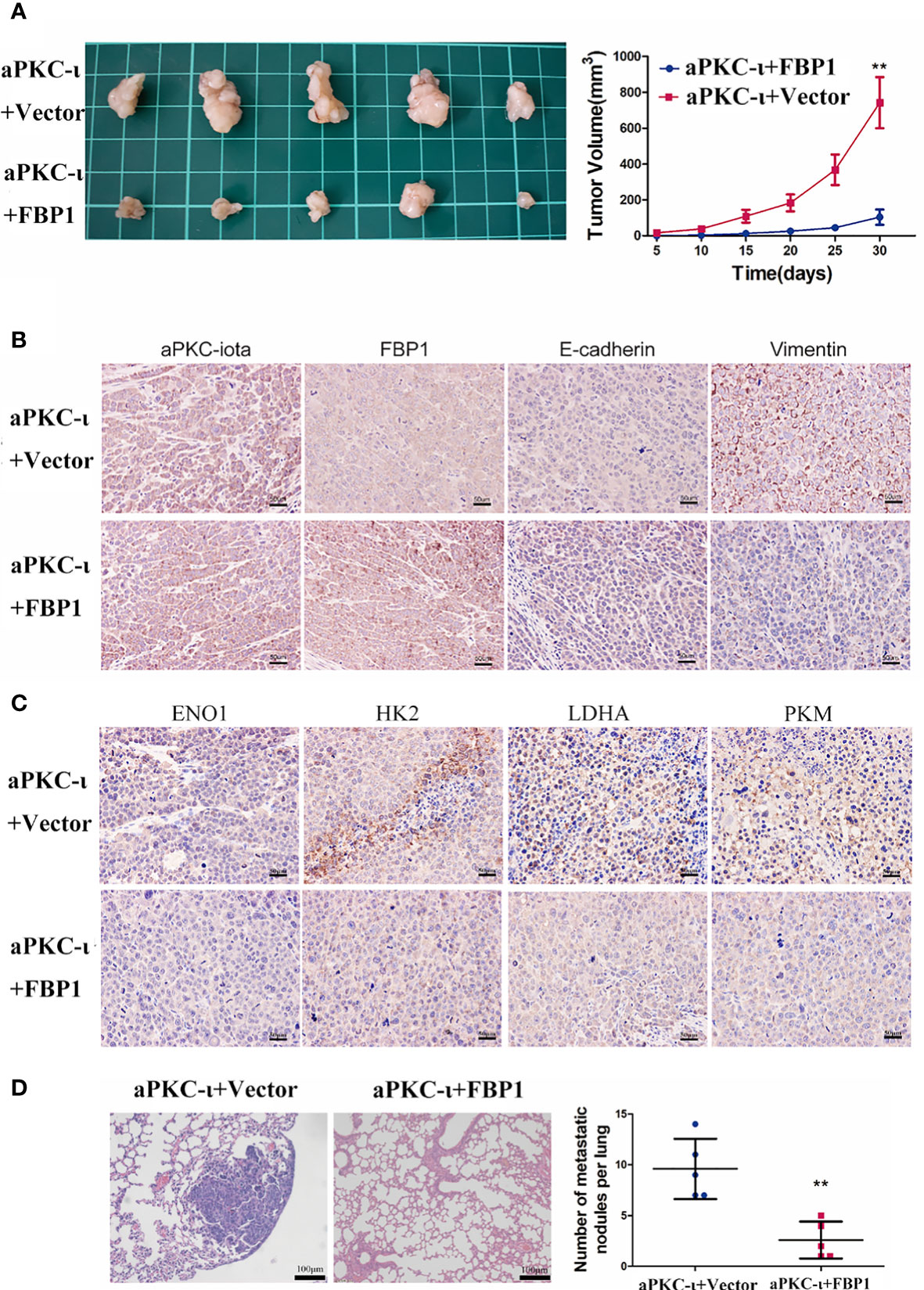
Figure 5 FBP1 antagonizes aPKC-ι and inhibits tumor growth and metastasis in vivo. (A) Tumor volume changes in nude mice over a 30-day period after injection of HCCC-9810 cells transfected with aPKC-ι/aPKC-ι + FBP1. (B) In situ expression of aPKC-ι, FBP1, E-cadherin and vimentin in the tumor tissues of the indicated groups. (C) In situ expression of HK2,ENO1,PKM,LDHA in the tumor tissues of the indicated groups (D) Representative images of HE-stained lung metastatic nodules from the indicated groups. ** means P < 0.01.
FBP1 Is a Direct Target of Snail in aPKC-ι Mediated EMT and “Warburg Effect”
To explore the underlying molecular mechanisms of decreased FBP1 by aPKC-ι, we first performed coimmunoprecipitation (CO-IP) experiments to assess whether aPKC-ι directly interacts with FBP1. Expression of aPKC-ι and FBP1 was not detected in the precipitated protein complex, suggesting that aPKC-ι indirectly regulated Snail through other ways. Snail, a very well-known transcription factor, was reported to be critical for epidermal growth factor–induced EMT of cancer cells (34). We noticed that nine consensus Snail-binding E-boxes domains (CAGGTG) were identified in the FBP1 promoter (17). Moreover, our previous studies had demonstrated that snail was also crucial for aPKC-ι-induced EMT-like changes in cholangiocarcinoma (29). Hence, we hypothesized that Snail may bind to the FBP1 promoter and is responsible for FBP1 repression in aPKC-ι-overexpressing ICC cells.
The expression of Snail was first examined by qRT-PCR in 40 pair-matched tumor specimens and non-tumor tissues. In agreement with existing results, higher Snail expression was in tumor samples (Figure 6A). Pearson correlation analysis showed a significant positive correlation between aPKC-ι and Snail expression (R=-0.643, p<0.001), whereas high Snail was related to low FBP1 (R=-0.484, p<0.01) (Figure 6B). In addition, ectopic expression of Snail in the HCCC-9810 and RBE cells markedly decreased FBP1 protein levels, suggesting that Snail is required for loss of FBP1 by aPKC-ι-mediated inhibition (Figure 6E).
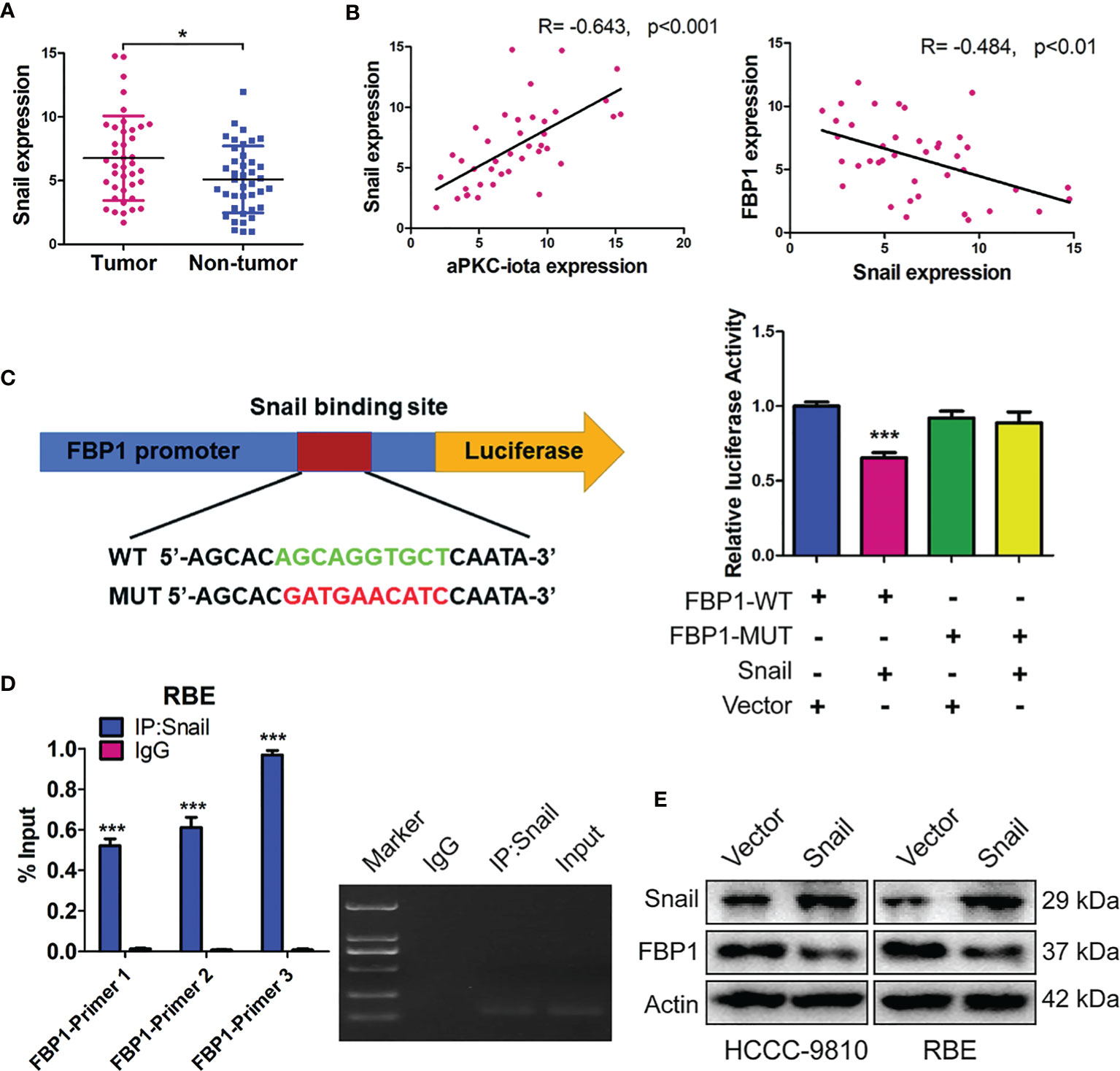
Figure 6 FBP1 is a direct target of Snail in aPKC-ι mediated EMT and “Warburg effect”. (A) Snai1 expression levels in 40 paired ICC and adjacent nontumor tissues. (B) Pearson correlation analysis showed a significant positive correlation between aPKC-ι and FBP1 expression (R=-0.643, p<0.001), whereas high Snail was related to low FBP1 (R=-0.484, p<0.01). (C, D) Snail significantly repressed the FBP1 promoter activity in dual luciferase assay and ChIP was performed in the RBE cells by using three sets of FBP1 primers and confirmed a direct physical interaction between FBP1 promoter and Snail. (E) Snai1 and FBP1 protein expression in HCCC-9810 and RBE cells transfected with Snai1.
To investigate whether Snail binds the promoter regions of FBP1, we constructed wild-type FBP1 (FBP1-WT) and mutant FBP1 (FBP1-MUT) eukaryotic expression vectors and generated ICC cells that stably expressed these two constructs. In dual luciferase assay, Snail significantly repressed the FBP1 promoter activity, indicated that Snail inhibited FBP1 transcription (Figure 6C). Therefore, chromatin immunoprecipitation (ChIP) was performed in the RBE cells by using three sets of FBP1 primers and confirmed a direct physical interaction between FBP1 promoter and Snail (Figure 6D). Taken together, these results implied that FBP1 is a direct target of Snail in ICC cells.
Discussion
As aPKC-ι, a polarization regulatory protein, regarded as human oncogene and potential therapeutic target in various epithelial cancers (35), we then found that aPKC-ι knockdown in ICC cells led to increase of E-cadherin, decreases of Vimentin expression and the abilities of migratory and invasive. These were in line with our previous studies indicating that aPKC-ι/Snail signaling pathway has a critical role in the regulation of EMT in cholangiocarcinoma in vitro and in vivo (29). The EMT constitutes a pivotal step in variety epithelial cancer cells invasion and metastasis, defined by loss of epithelial cell polarity and reorganization of the cytoskeleton (34). Therefore, a better understanding of EMT process regulating by aPKC-ι may hold a great promise for the development of novel therapeutic approaches to eradicate ICC that are currently resistant to conventional therapies.
Despite a high genetic diversity, cancer cells still exhibit a common set of characteristics. Aberrant metabolic reprogramming constitutes one of its important features during the tumor progression, which could enhance glucose uptake and lactate production of cancer cells through aerobic glycolysis to meet the rapid energy requirements for catabolism and anabolism (36). Recent studies indicated that extracellular matrix (ECM) of cancer cells is remodeled by aerobic glycolysis, which could also increase the expression of EMT-inducing transcription factor and decrease the pH value of tumor microenvironment (37). It had shown that an increased glycolytic metabolism can facilitate cancer cells EMT and promote tumor progression. In our current study, the content of secreted lactate, the end product of glycolysis, were significantly increased in the ICC cells and correlated positively with aPKC-ι expression. These results first demonstrated that aPKC-ι could facilitate aerobic glycolysis and contributed to EMT process in ICC. However, the underlying mechanisms that aPKC-ι affect the biological behavior of ICC cells, are still unclear.
Intracellular glucose homeostasis is regulated by catabolic glycolysis, aerobic oxidation of sugars, and anabolic gluconeogenesis. Until now, metabolites that accumulated via aerobic glycolysis were considered as a building blocks or fuel source for cancer cells proliferation (38). Several studies reported that metabolic intermediates of gluconeogenesis regulate aerobic breakdown of glucose in cancer cells (22, 39, 40). FBP1 catalyzes the decomposition of fructose-1,6-bisphosphate into 6-phosphate fructose and inorganic phosphate, and its epigenetic regulation in cancer cells is an underlying factor of aberrant glucose metabolism (14). Interestingly, the loss of FBP1 was recently determined to be a critical oncogenic event in breast cancer and renal cell carcinoma progression (17). Consistent with previous findings, we found that FBP1 expression is negatively correlated with aPKC-ι and the malignant progression in ICC. Moreover, upregulating FBP1 expression in aPKC-ι-overexpressing ICC cells reversed EMT-like changes and aberrant metabolic reprogramming. These results further support the critical role of FBP1 for aPKC-ι induced EMT-like changes and glycolysis in ICC.
To elucidate the specific mechanism by which aPKC-ι regulates FBP1 in ICC, co-IP experiment was performed and showed that aPKC-ι indirectly regulated FBP1 through other molecules. Snail, a transcriptional repressor, could induce EMT, allowing cancer cells with invasive properties (41). It also could regulate many genes involved in glucose metabolism, such as glucose phosphate isomerase and aldolase, suggesting that Snail participates in complex metabolic reprogramming in cancer (42). Furthermore, some studies have confirmed that the Snail transcriptional factor Snai1 represses FBP1 to accelerate EMT of various tumor cells (43). Since we had previously observed that Snail is a major downstream target of aPKC-ι in triggering EMT (29), we hypothesized that aPKC-ι promotes EMT and provides metabolic advantages of ICC cells by repressing FBP1 via Snail. As expected, we identified that Snail inhibited the transcription of FBP1 by directly binding to its promoter by dual luciferase assay and ChIP experiment. In addition, the expression level of Snail correlated positively with that of aPKC-ι, whereas correlated negatively with FBP1.
Oxidative metabolism is impaired in cancer cells due to the increase in glycolysis and oxygen consumption. The Warburg effect, wherein the tumor cells preferentially use glycolysis to produce energy under hypoxic conditions (44), protects the cells against metabolic stress, and promotes their invasion, migration and EMT (45). In the glycolytic pathway, lactate dehydrogenase A (LDHA) irreversibly catalyzes the conversion of pyruvate to lactate via the oxidative dehydrogenation of nicotinamide adenine dinucleotide (NADH) to NAD+ (46). As an important carcinogen, lactate is closely related to tumor growth, immune escape, angiogenesis and EMT (47). Existed data had shown that LDHA is highly expressed in various cancers and its expression is positively associated with tumor progression (48). Based on our findings therefore, we speculated that aPKC-ι/Snail-mediated repression of FBP1 increases glycolysis and lactate production in the ICC cells, which in turn upregulates aPKC-ι to form a positive feedback loop, eventually enhancing EMT, invasion and migration. Further mechanism studies are needed to investigate.
In conclusion, we found that aPKC-ι, Snail and loss of FBP1 are significantly correlated with poor prognosis in ICC patients. FBP1 played a crucial role in aPKC-ι induced EMT and glycolysis in ICC cells. We illustrated aPKC-ι was confirmed to inhibit FBP1 expression by increasing Snail binding to the FBP1 promoter, which is conducive to EMT and metastasis. Our results strongly suggest that aPKC-ι induces EMT and promotes glycolysis in human ICC cells through an aPKC-ι/Snail/FBP1 pathway. It is known that cells always take the most economical way to carry out physiological activities, which is well verified in our study. The polarization regulatory protein, aPKC-ι, simultaneously regulated EMT and metabolic reprogramming via the same signal pathway, which eventually led to more effective metastasis of ICC cells. The above results provide a new perspective to illustrate the mechanism of tumor progression.
Our and recent observations implied that glucose metabolism is not simply a consequence but rather affects polarized states of cancer cells. Nevertheless, the specific interaction of enzymes associated with glucose metabolism regulating EMT-like characteristics is still unknown. One emerging idea on how changes in glucose metabolism induce EMT-like phenotype is that mitochondrial derived reactive oxygen species could serve as signaling molecules (49). It will be of interest how interaction between EMT process and glucose metabolism. Targeting this pathway may abolish cancer metastasis and metabolic advantages. This strategy will generate an entirely effective approach for treating ICC.
Radical surgical resection remains to be the only potential curative treatment option for ICC in the near future. Limitations of current clinical trials include small sample size, combined analysis of cholangiocarcinoma and gallbladder cancer, and lack of randomization (50). Precision medicine will be advocated to improve outcomes for patients with ICC, which is highly genetically heterogeneous tumor. More recently, a variety of genetic mutations implicated in causing ICC have been identified, and future studies will continue to focus on targeting genetic aberrations. In addition, the efficacy and safety of immunotherapies have also been widely reported in ICC. Although immunotherapy has been disappointing now, it could become an important part of the treatment landscape in the future. The road is rough, but the beauty lies ahead.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by the Committee for the Protection of Human Subjects, Zhongnan Hospital of Wuhan University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Institutional Animal Ethics Committee of Wuhan University.
Author Contributions
MG and CM designed the learning concept and design. MG drafted the manuscript. CM is in charge of statistics. XJ, PX, HZ, YL, YG, and YY made significant contributions to the research program. YQ and YFY reviewed and approved the final version of this article. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Science Fund for Young Scholars (No. 81802442) and Scientific Research Project of Hubei Province Health commission (WJ2017Z007).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Razumilava N, Gores GJ. Cholangiocarcinoma. Lancet (London England) (2014) 383(9935):2168–79. doi: 10.1016/s0140-6736(13)61903-0
2. Zhang H, Yang T, Wu M, Shen F. Intrahepatic Cholangiocarcinoma: Epidemiology, Risk Factors, Diagnosis and Surgical Management. Cancer Lett (2016) 379(2):198–205. doi: 10.1016/j.canlet.2015.09.008
3. Gupta A, Dixon E. Epidemiology and Risk Factors: Intrahepatic Cholangiocarcinoma. Hepatobiliary Surg Nutr (2017) 6(2):101–4. doi: 10.21037/hbsn.2017.01.02
4. Rizzo A, Ricci AD, Brandi G. Pemigatinib: Hot Topics Behind the First Approval of a Targeted Therapy in Cholangiocarcinoma. Cancer Treat Res Commun (2021) 27:100337. doi: 10.1016/j.ctarc.2021.100337
5. Pant K, Richard S, Peixoto E, Gradilone SA. Role of Glucose Metabolism Reprogramming in the Pathogenesis of Cholangiocarcinoma. Front Med (2020) 7:113. doi: 10.3389/fmed.2020.00113
6. Fujiwara H, Tateishi K, Misumi K, Hayashi A, Igarashi K, Kato H, et al. Mutant IDH1 Confers Resistance to Energy Stress in Normal Biliary Cells Through PFKP-Induced Aerobic Glycolysis and AMPK Activation. Sci Rep (2019) 9(1):18859. doi: 10.1038/s41598-019-55211-w
7. Tang D, Xu L, Zhang M, Dorfman RG, Pan Y, Zhou Q, et al. Metformin Facilitates BG45−Induced Apoptosis via an Anti−Warburg Effect in Cholangiocarcinoma Cells. Oncol Rep (2018) 39(4):1957–65. doi: 10.3892/or.2018.6275
8. Florio AA, Ferlay J, Znaor A, Ruggieri D, Alvarez CS, Laversanne M, et al. Global Trends in Intrahepatic and Extrahepatic Cholangiocarcinoma Incidence From 1993 to 2012. Cancer (2020) 126(11):2666–78. doi: 10.1002/cncr.32803
9. Ma CQ, Yang Y, Wang JM, Du GS, Shen Q, Liu Y, et al. The Apkcι Blocking Agent ATM Negatively Regulates EMT and Invasion of Hepatocellular Carcinoma. Cell Death Dis (2014) 5(3):e1129. doi: 10.1038/cddis.2014.91
10. Justilien V, Fields AP. Ect2 Links the PKCiota-Par6alpha Complex to Rac1 Activation and Cellular Transformation. Oncogene (2009) 28(41):3597–607. doi: 10.1038/onc.2009.217
11. Garg R, Benedetti LG, Abera MB, Wang H, Abba M, Kazanietz MG. Protein Kinase C and Cancer: What We Know and What We do Not. Oncogene (2014) 33(45):5225–37. doi: 10.1038/onc.2013.524
12. Gunaratne A, Thai BL, Di Guglielmo GM. Atypical Protein Kinase C Phosphorylates Par6 and Facilitates Transforming Growth Factor β-Induced Epithelial-to-Mesenchymal Transition. Mol Cell Biol (2013) 33(5):874–86. doi: 10.1128/mcb.00837-12
13. Du GS, Qiu Y, Wang WS, Peng K, Zhang ZC, Li XS, et al. Knockdown on aPKC-ι Inhibits Epithelial-Mesenchymal Transition, Migration and Invasion of Colorectal Cancer Cells Through Rac1-JNK Pathway. Exp Mol Pathol (2019) 107:57–67. doi: 10.1016/j.yexmp.2018.11.007
14. Grasmann G, Smolle E, Olschewski H, Leithner K. Gluconeogenesis in Cancer Cells - Repurposing of a Starvation-Induced Metabolic Pathway? Biochim Biophys Acta Rev Cancer (2019) 1872(1):24–36. doi: 10.1016/j.bbcan.2019.05.006
15. Liu X, Wang X, Zhang J, Lam EK, Shin VY, Cheng AS, et al. Warburg Effect Revisited: An Epigenetic Link Between Glycolysis and Gastric Carcinogenesis. Oncogene (2010) 29(3):442–50. doi: 10.1038/onc.2009.332
16. Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, et al. Fructose-1,6-Bisphosphatase Opposes Renal Carcinoma Progression. Nature (2014) 513(7517):251–5. doi: 10.1038/nature13557
17. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by Snail-Mediated Repression Provides Metabolic Advantages in Basal-Like Breast Cancer. Cancer Cell (2013) 23(3):316–31. doi: 10.1016/j.ccr.2013.01.022
18. Cong J, Wang X, Zheng X, Wang D, Fu B, Sun R, et al. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis During Lung Cancer Progression. Cell Metab (2018) 28(2):243–55.e5. doi: 10.1016/j.cmet.2018.06.021
19. Gao S, Zhu D, Zhu J, Shen L, Zhu M, Ren X. miR-18a-5p Targets FBP1 to Promote Proliferation, Migration, and Invasion of Liver Cancer Cells and Inhibit Cell Apoptosis. Comput Math Methods Med (2021) 2021:3334065. doi: 10.1155/2021/3334065
20. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
21. Dang CV. Links Between Metabolism and Cancer. Genes Dev (2012) 26(9):877–90. doi: 10.1101/gad.189365.112
22. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science (New York NY) (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
23. Lu J, Tan M, Cai Q. The Warburg Effect in Tumor Progression: Mitochondrial Oxidative Metabolism as an Anti-Metastasis Mechanism. Cancer Lett (2015) 356(2 Pt A):156–64. doi: 10.1016/j.canlet.2014.04.001
24. Liu M, Quek LE, Sultani G, Turner N. Epithelial-Mesenchymal Transition Induction is Associated With Augmented Glucose Uptake and Lactate Production in Pancreatic Ductal Adenocarcinoma. Cancer Metab (2016) 4:19. doi: 10.1186/s40170-016-0160-x
25. Yu J, Li J, Chen Y, Cao W, Lu Y, Yang J, et al. Snail Enhances Glycolysis in the Epithelial-Mesenchymal Transition Process by Targeting FBP1 in Gastric Cancer. Cell Physiol Biochem (2017) 43(1):31–8. doi: 10.1159/000480314
26. Zhao J, Huang X, Xu Z, Dai J, He H, Zhu Y, et al. LDHA Promotes Tumor Metastasis by Facilitating Epithelial−Mesenchymal Transition in Renal Cell Carcinoma. Mol Med Rep (2017) 16(6):8335–44. doi: 10.3892/mmr.2017.7637
27. Hua Q, Mi B, Huang G. The Emerging Co-Regulatory Role of Long Noncoding RNAs in Epithelial-Mesenchymal Transition and the Warburg Effect in Aggressive Tumors. Crit Rev Oncol Hematol (2018) 126:112–20. doi: 10.1016/j.critrevonc.2018.03.028
28. Wang H, Chirshev E, Hojo N, Suzuki T, Bertucci A, Pierce M, et al. The Epithelial-Mesenchymal Transcription Factor SNAI1 Represses Transcription of the Tumor Suppressor miRNA Let-7 in Cancer. Cancers (2021) 13(6):1–18. doi: 10.3390/cancers13061469
29. Qian Y, Yao W, Yang T, Yang Y, Liu Y, Shen Q, et al. aPKC-ι/P-Sp1/Snail Signaling Induces Epithelial-Mesenchymal Transition and Immunosuppression in Cholangiocarcinoma. Hepatology (Baltimore Md) (2017) 66(4):1165–82. doi: 10.1002/hep.29296
30. Li F, Huangyang P, Burrows M, Guo K, Riscal R, Godfrey J, et al. FBP1 Loss Disrupts Liver Metabolism and Promotes Tumorigenesis Through a Hepatic Stellate Cell Senescence Secretome. Nat Cell Biol (2020) 22(6):728–39. doi: 10.1038/s41556-020-0511-2
31. Jiang X, Liu Y, Wang G, Yao Y, Mei C, Wu X, et al. Up-Regulation of CLIC1 Activates MYC Signaling and Forms a Positive Feedback Regulatory Loop With MYC in Hepatocellular Carcinoma. Am J Cancer Res (2020) 10(8):2355–70.
32. Massa A, Varamo C, Vita F, Tavolari S, Peraldo-Neia C, Brandi G, et al. Evolution of the Experimental Models of Cholangiocarcinoma. Cancers (2020) 12(8):1–31. doi: 10.3390/cancers12082308
33. Tian L, Deng Z, Xu L, Yang T, Yao W, Ji L, et al. Downregulation of ASPP2 Promotes Gallbladder Cancer Metastasis and Macrophage Recruitment via aPKC-ι/GLI1 Pathway. Cell Death Dis (2018) 9(11):1115. doi: 10.1038/s41419-018-1145-1
34. Goossens S, Vandamme N, Van Vlierberghe P, Berx G. EMT Transcription Factors in Cancer Development Re-Evaluated: Beyond EMT and MET. Biochim Biophys Acta Rev Cancer (2017) 1868(2):584–91. doi: 10.1016/j.bbcan.2017.06.006
35. Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI Activation by Atypical Protein Kinase C ι/λ Regulates the Growth of Basal Cell Carcinomas. Nature (2013) 494(7438):484–8. doi: 10.1038/nature11889
36. Ganapathy-Kanniappan S, Geschwind JF. Tumor Glycolysis as a Target for Cancer Therapy: Progress and Prospects. Mol Cancer (2013) 12:152. doi: 10.1186/1476-4598-12-152
37. Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, Autophagy, Necroptosis, and Cancer Metastasis. Mol Cancer (2015) 14:48. doi: 10.1186/s12943-015-0321-5
38. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: A Metabolic Key Player in Cancer. Cancer Res (2011) 71(22):6921–5. doi: 10.1158/0008-5472.Can-11-1457
39. Lunt SY, Vander Heiden MG. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
40. Vaupel P, Multhoff G. Revisiting the Warburg Effect: Historical Dogma Versus Current Understanding. J Physiol (2021) 599(6):1745–57. doi: 10.1113/jp278810
41. Kim NH, Cha YH, Lee J, Lee SH, Yang JH, Yun JS, et al. Snail Reprograms Glucose Metabolism by Repressing Phosphofructokinase PFKP Allowing Cancer Cell Survival Under Metabolic Stress. Nat Commun (2017) 8:14374. doi: 10.1038/ncomms14374
42. Wang Y, Shi J, Chai K, Ying X, Zhou BP. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets (2013) 13(9):963–72. doi: 10.2174/15680096113136660102
43. Liu GM, Li Q, Zhang PF, Shen SL, Xie WX, Chen B, et al. Restoration of FBP1 Suppressed Snail-Induced Epithelial to Mesenchymal Transition in Hepatocellular Carcinoma. Cell Death Dis (2018) 9(11):1132. doi: 10.1038/s41419-018-1165-x
44. Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci (2016) 41(3):211–8. doi: 10.1016/j.tibs.2015.12.001
45. Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P. The Warburg Effect: Insights From the Past Decade. Pharmacol Ther (2013) 137(3):318–30. doi: 10.1016/j.pharmthera.2012.11.003
46. Pathria G, Scott DA, Feng Y, Sang Lee J, Fujita Y, Zhang G, et al. Targeting the Warburg Effect via LDHA Inhibition Engages ATF4 Signaling for Cancer Cell Survival. EMBO J (2018) 37(20):1–17. doi: 10.15252/embj.201899735
47. Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, et al. Inhibition of Lactate Dehydrogenase A Induces Oxidative Stress and Inhibits Tumor Progression. Proc Natl Acad Sci U S A (2010) 107(5):2037–42. doi: 10.1073/pnas.0914433107
48. Urbańska K, Orzechowski A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int J Mol Sci (2019) 20(9):1–15. doi: 10.3390/ijms20092085
49. Laura AS, Navdeep SC. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol Cell (2012) 48(2):158–67. doi: 10.1016/j.molcel.2012.09.025
Keywords: aPKC-ι, intrahepatic cholangiocarcinoma, FBP1, Snai1, EMT, invasion, metastasis
Citation: Gao M, Mei C, Guo Y, Xia P, Zhang H, Liu Y, Yao Y, Jiang X, Yuan Y and Qian Y (2021) Loss of FBP1 by aPKC-ι/Snail Pathway-Mediated Repression Promotes Invasion and Aerobic Glycolysis of Intrahepatic Cholangiocarcinoma. Front. Oncol. 11:756419. doi: 10.3389/fonc.2021.756419
Received: 10 August 2021; Accepted: 15 November 2021;
Published: 02 December 2021.
Edited by:
Yunfei Xu, Shandong University, ChinaReviewed by:
Alessandro Rizzo, Sant’Orsola-Malpighi Polyclinic, ItalyKishor Pant, University of Minnesota Twin Cities, United States
Copyright © 2021 Gao, Mei, Guo, Xia, Zhang, Liu, Yao, Jiang, Yuan and Qian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yawei Qian, eXdlc3MxOTkwQDE2My5jb20=; Yufeng Yuan, eXVhbnlmMTk3MUB3aHUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship