 Mojdeh Soltani
Mojdeh Soltani Yue Zhao
Yue Zhao Zhijia Xia
Zhijia Xia Mazdak Ganjalikhani Hakemi
Mazdak Ganjalikhani Hakemi Alexandr V. Bazhin
Alexandr V. Bazhin- 1Department of Immunology, Faculty of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
- 2Department of General, Visceral, and Transplant Surgery, Ludwig-Maximilians-University Munich, Munich, Germany
- 3German Cancer Consortium (DKTK), Partner Site Munich, Munich, Germany
Despite recent advancements in the treatment of hematologic malignancies and the emergence of newer and more sophisticated therapeutic approaches such as immunotherapy, long-term overall survival remains unsatisfactory. Metabolic alteration, as an important hallmark of cancer cells, not only contributes to the malignant transformation of cells, but also promotes tumor progression and metastasis. As an immune-escape mechanism, the metabolic adaptation of the bone marrow microenvironment and leukemic cells is a major player in the suppression of anti-leukemia immune responses. Therefore, metabolic rewiring in leukemia would provide promising opportunities for newer therapeutic interventions. Several therapeutic agents which affect essential bioenergetic pathways in cancer cells including glycolysis, β-oxidation of fatty acids and Krebs cycle, or anabolic pathways such as lipid biosynthesis and pentose phosphate pathway, are being tested in various types of cancers. So far, numerous preclinical or clinical trial studies using such metabolic agents alone or in combination with other remedies such as immunotherapy are in progress and have demonstrated promising outcomes. In this review, we aim to argue the importance of metabolic alterations and bioenergetic pathways in different types of leukemia and their vital roles in disease development. Designing treatments based on targeting leukemic cells vulnerabilities, particularly in nonresponsive leukemia patients, should be warranted.
Introduction
Hematologic malignancies are heterogeneous disorders caused by the carcinogenic transformation of hematopoietic cells. Leukemias are the most common set of these malignancies characterized by rapid and aggressive production of abnormal white blood cells (WBCs) that are not functional and efficient immune cells. This state finally eventuates to impairment of the bone marrow ability to produce sufficient and beneficial blood cells like platelets, red blood cells (RBCs), and normal WBCs because of space occupation (1–3). According to the American Cancer Society, about 60,530 new cases and approximately 23,100 deaths of leukemia were estimated in the United States in 2020 (4). Despite significant improvements in its treatment and the advent of newer and more efficient therapies such as immunotherapeutic approaches, long-term survival remains unsatisfactory and more investigation in this field is still a requisite. There are several categories of leukemia based on various criteria, but according to the most well-known one, leukemia can be divided into lymphocytic and myelogenous (or myeloid) groups (1).
Metabolic alteration is a significant hallmark of cancer cells which enables them to fulfill their requirements. Actually, metabolic adaptations not only contribute to cancer cells transformation, but also promote tumor progression (5, 6). It seems that metabolic rewiring is also a critical implement for leukemic cells so that metabolic properties of these cells can remarkably influence the disease progress and effective response to treatments. Indeed, both cell-intrinsic and extrinsic factors including oncogenic mutations and some aspects of the bone marrow microenvironment (BMM) significantly affect leukemic cells’ metabolism. The bone marrow and thymus, where the leukemia and lymphoma cells expands, are hypoxic areas with specific metabolic condition (7–9). Metabolic characteristics of leukemia cells, particularly leukemic stem cells (LSCs) that are usually different from normal hematopoietic stem cells (HSCs), actively contribute to the initiation and maintenance of malignancy. Furthermore, metabolic adaptations in the BMM and leukemic cells have a critical role in the suppression and eradication of anti-leukemia immune responses (10, 11). Regarding these issues, metabolic vulnerabilities in leukemia provide promising opportunities for therapeutic intervention. In this review, we try to discuss the importance of metabolic regulations and bioenergetic pathways in leukemia pathogenesis and their vital roles in disease development. In addition, designing of treatment based on targeting leukemic cells vulnerabilities, particularly in nonresponsive leukemia patients, is considered.
Classification of Leukemia and Common Therapies
According to several references, leukemias can be classified as acute and chronic as well as myelocytic and lymphocytic based on the degree of cell differentiation and the predominant type of cells involved (12). Acute myeloid leukemia (AML) which accounts for approximately 33% of leukemia new cases in the US, is the most frequent and worst prognostic type. According to the French–American–British classification, AML is divided into eight subtypes (M0–M7) (13). It occurs because of some genetic abnormalities in myeloblasts, which leads to bone marrow function failure eventually. A combination of cytarabine plus anthracycline cytotoxic chemotherapy has been the standard therapy of AML since the past three decades (14, 15). Despite advances in AML therapy, its prognosis is still poor, especially for older leukemia patients. Therefore, along with chemotherapeutic strategies, some immunotherapeutic approaches such as immune checkpoint blockade and multi-specific T cell-engaging antibodies, are under investigation (16–18). Chronic myeloid leukemia (CML) is a clonal myeloproliferative disease that accounts for about 15% of adult leukemia cases (13). It is characterized by aggressive proliferation of dysfunctional myeloid-derived cells and is classified into three stages including the chronic phase, accelerated phase, and blastic crisis (19). The overexpression of BCR-ABL1 fusion oncogene, which is resulted from a reciprocal translocation between chromosome 9 and 22 (Philadelphia chromosome) in HSCs, eventually gives rise to a constitutively active non-receptor tyrosine kinase activity. This incident is considered as the main origin point of the disease, especially in the chronic phase (20, 21). Hence, tyrosine kinase inhibitors (TKIs) which are classified into three generations, are the main standard treatment and have remarkably improved the outcomes of CML patients in the chronic phase. Nevertheless, almost 50% of CML patients suffer a relapse of disease post-TKI therapy due to the presence and drug-resistance of TKI-resistant CML stem cells (22, 23).
Acute lymphoblastic leukemia (ALL) is known as the most common leukemia in pediatrics with an approximate prevalence of 80% childhood leukemias, however, its adult‐onset form mostly involves males age >70 years (24, 25). ALL arises from the transformation of hematopoietic B cell or T cell progenitors with chromosomal abnormalities or genetic alterations. Not only genetic but also environmental predisposing factors, like childhood infections, will contribute to the initiation and progression of ALL. Although the 5-year overall survival (OS) of pediatric ALL patients has increased from 31% in 1975 to nearly 70% in 2009 and even to 90% in recent years, only 25% of patients older than 50 years old live longer than 5 years after diagnosis. That shows urgent needs for further improvements for its treatment particularly the adult form (25). Accordingly, frontline treatment of ALL includes four phases during 2–3 years: induction, consolidation, intensification, and long-term maintenance. Asparaginase therapy, as a frontline approach to cytoreduction, is administered in all cases of pediatric and even most cases of adult ALL and this issue declare the importance of cellular metabolism in its pathogenesis and treatment. Asparaginase is considered a critical component used for the treatment of childhood ALL and can give rise to complete remission in 40%–60% of patients, while the 5-year OS in older patients (>60 years old) is still poor, even less than 20% (25–27). The last type of leukemia is chronic lymphocytic leukemia (CLL) that is considered as the most common adult leukemia accounting for approximately 40% of all cases in adults. It results from an overproduction of abnormal B cell lymphocytes in the bone marrow that accumulate in the bloodstream and homing tissues. Even though it is known as slow-progressing leukemia, CLL is somehow incurable, and drug resistance or relapse is often presented in CLL patients (28).
The General Importance of Metabolic Pathway in Cancer Therapy
To fulfill the bioenergetic and biosynthetic needs, some alterations occur in the energy metabolism of cancer cells. These metabolic shifts are mainly triggered by various mechanisms which instigate signaling pathways and eventually manipulate the expression of some metabolism-related genes (29). Cancer cells generally have three metabolic hallmarks including increased glycolysis, glutaminolysis, and lipogenic pathways (30). It has been reported that the phosphoinositol 3-kinase (PI3K)-AKT signaling pathway, which activates mammalian target of rapamycin (mTOR), hypoxia-inducible factor 1 (HIF1) and other transcription factors like c-Myc, p53, or Oct1, has a critical role in these metabolic adaptations (29, 30). Cellular metabolism is mainly controlled by mTORC master regulator that affects the translation and transcription of metabolic genes, such as peroxisome proliferator activated receptor γ coactivator-1 α (PGC-1α), sterol regulatory element-binding protein 1/2 (SREBP1/2), and hypoxia inducible factor-1 α (HIF-1α). This regulator complex not only promotes glycolytic activity that contributes to cell growth but also, interestingly, can regulate mitochondrial metabolism. Thus, representing an important regulator in mitochondrial function (31–33). Fortunately, these metabolic rewirings of cancer cells have provided promising drug targets during the past decades. Several therapeutic agents which affect either requisite bioenergetic pathways or anabolic pathways in leukemia cells are being applied in various types of cancers. Numerous preclinical studies or clinical trials using these metabolic agents alone or in combination with other remedies such as immunotherapy are in progress and have demonstrated promising outcomes (5).
Metabolic Differences of Normal Hematopoietic and Leukemic Cells
Metabolic differences between normal hematopoietic and leukemic cells can provide new therapeutic windows to treat leukemia patients. HSC self-renewal and lineage differentiation potency are not only influenced by cytokines and transcription factors but also regulated by cell metabolism (7, 10). In the dormant state, low levels of ROS and ATP production due to the presence of anaerobic glycolysis were observed in HSCs. However, when switching to an active state, HSCs usually utilize oxidative metabolism through elevated levels of oxidative phosphorylation (OXPHOS) and fatty acids oxidation (FAO) due to a higher energy demand (34, 35). Actually, because of the low oxygen microenvironment of bone marrow, HIF-1s are stabilized and promote a glycolytic phenotype in HSCs mostly through the stimulation of the pyruvate dehydrogenase kinase (PDK) 2 and 4 as an active preventer of the TCA cycle (8, 36). Additionally, asymmetric division of HSCs which is requisite for the production of eternal daughter cells and committed progenitors is considerably dependent on PML-peroxisome proliferator-activated receptor delta (PPARδ)-FAO (37). According to previous studies, it seems that although LSCs may have some similar metabolic properties to normal HSCs, there are also some differences that exist. Lagadinou et al. have demonstrated that LSCs have a higher expression of BCL-2 and thus, contain lower ROS levels than HSCs (38). Some other investigations have demonstrated metabolic differences between leukemic cells and their normal counterparts. Zhong et al. have reported that T-ALL leukemic cells show increased OXPHOS in comparison to normal HSCs (39). Increased free fatty acids utilization and energy production by OXPHOS has been detected in CLL cells unlike normal B-lymphocytes (40). Interestingly, AML cells have more mitochondrial mass but lower respiratory chain complex activities and spare respiration in comparison to normal CD34+ cells (41, 42). On the other hand, a high level of glucose consumption is considered a conserved feature of LSCs in most hematological malignancies, regardless of genetic defects or types of them. Upregulation of glycolysis has been indicated in several kinds of leukemia including AML, ALL, CLL, and multiple myeloma (43–46). Moreover, it has been indicated that increased glycolysis correlates with drug resistance in both leukemia patients and cell lines (47). Oncogenic drivers in human leukemia including MYC and RAS are major operative factors in the metabolic rewiring of leukemia cells. For instance, MYC overexpression is considered a significant hallmark of B-cell and T-cell ALL and also Burkitt’s lymphoma, which can actively promote some metabolic pathways such as glycolysis, mitochondrial biogenesis, glutaminolysis, lipid, and nucleotide biosynthesis (48–52).
Importance of Cellular Metabolic Pathways in Myeloid Disorders
Glucose Related Metabolic Pathways
Regarding the critical role of glycolysis pathway in the growth and proliferation of cancer cells, myeloid leukemia cells also upregulate this metabolic pathway. Numerous genetic studies in mouse models have implicated the importance of glycolysis and high expression of glycolysis-associated enzymes in the initiation and maintenance of myeloid malignancies. They have proved that deletion of glycolytic enzymes such as pyruvate kinase (PKM2) or lactate dehydrogenase A (LDHA) can considerably extenuate the initiation and maintenance of murine CML and AML (53). Raised glycolytic activity not only gives rise to a decreased level of autophagy leading to more aggressive leukemia, but also is associated with drug resistance (54, 55). In AML cells, higher mTORC1 activity and FLT3-ITD-mediated signaling contribute to the continuance of glycolysis and leukemic cells survival and therefore, render therapeutic sensitivity to glycolytic inhibition (Figure 1) (56, 57). 2-Deoxyglucose, as an inhibitor of the glycolysis pathway, has shown beneficial effects in AML treatment (47, 58). Moreover, leukemic cells are significantly dependent on the pentose phosphate pathway (PPP) which provides ribonucleotides for nucleic acid synthesis and NADPH for lipid synthesis. Redox homeostasis and glucose-6-phosphate dehydrogenase (G6PD) overexpression are the key regulators of this pathway and correlate with an adverse prognosis in AML (Figure 1). Therefore, inhibition of G6PD activity can be a promising therapeutic target in AML and its beneficial effects have been demonstrated in FLT3-ITD-mutated AML (56, 59, 60). Additionally, synergic effects of G6PD and 6-phosphogluconate dehydrogenase (6PGD) inhibition with cytarabine to produce anti-leukemic activities in AML have been proven (42). BCR‐ABL fusion oncogene in CML also increases glucose transport and renders cells more glycolytic compared with normal counterparts (61). It has been demonstrated that continuous exposure of K562, a CML cell line, with imatinib which decreases glucose uptake and inactivates glycolytic enzymes, can lead to induction of autophagy in CML cells by mechanisms involving activation of AMPK and suppression of S6K1 (62).
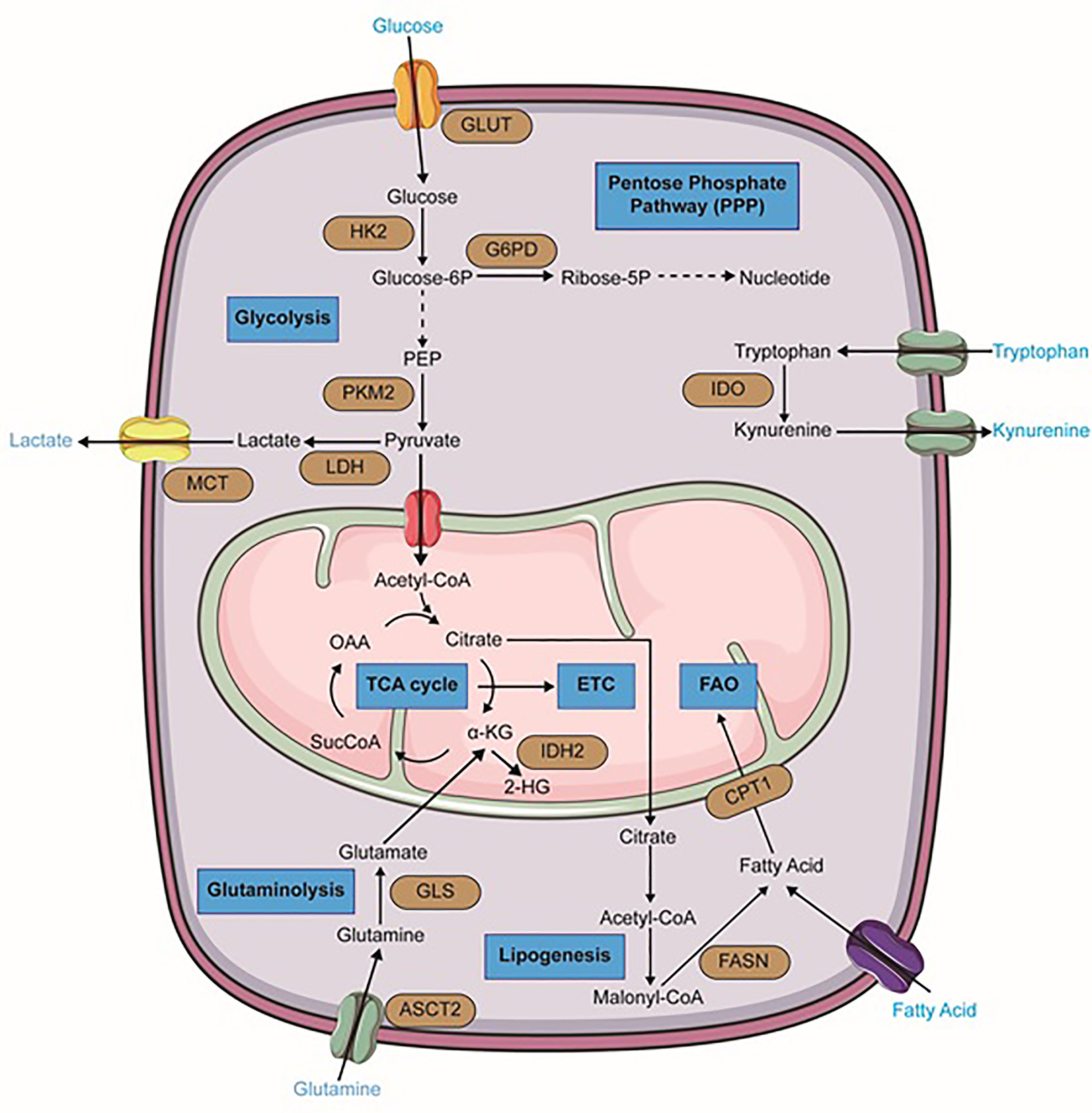
Figure 1 Metabolic alterations in leukemia cells. Important reactions of the metabolic pathway in leukemia cells are summarized in the schematic, including glycolysis, glutaminolysis, lipogenesis, TCA cycle, fatty acid oxidation (FAO), pentose phosphate pathway (PPP) (blue). Dominant nutrient transporters and metabolic enzymes are emphasized (brown). GLUT, glucose transporter; HK2, hexokinase-2; PKM2, pyruvate kinase isozymes M2; LDH, lactate dehydrogenase; MCT, monocarboxylate transporters; G6PD, glucose-6-phosphate dehydrogenase; ASCT2, glutamine transporter encoded by Solute Carrier Family 1 Member 5 gene; GLS, glutaminase; FASN, fatty acid synthase; CPT1, carnitine palmitoyltransferase I; IDH2, isocitrate dehydrogenase 2; IDO, Indoleamine-pyrrole 2,3-dioxygenase; α-KG, alpha-ketoglutarate; SucCoA, Succinyl-CoA; OAA, oxaloacetate. The figure was produced with the assistance of Servier Medical Art (https://smart.servier.com).
OXPHOS and mitochondrial metabolism also have a significant role in myeloid leukemia. According to Krtić et al, oxygen consumption rate and mitochondrial mass are increased in AML cells compared with normal hematopoietic progenitors (63). Furthermore, amplification of mitochondrial DNA (mtDNA) levels in AML has been indicated in various studies (64, 65). Noteworthily, different AML cell lines exhibit a different preference on glycolysis or mitochondrial OXPHOS for energy demand. For instance, NB4 was proved to be a “glycolytic” cell line while THP-1 was identified to be dependent on OXPHOS, even though they are all AML cell lines (66). Further mechanism investigation revealed that AMPK is responsible for the bioenergetic production difference, highlighting that the oncogene background of leukemia cells, at least partly, determines their metabolism preference. Furthermore, leukemia continuously reprograms to adapt to environmental pressures and alteration of growth conditions, resulting in the ratio between glycolysis and OXPHOS are continuously changing (67). Due to higher mitochondrial biogenesis and higher dependency on mitochondrial oxidation in leukemia myeloblasts, it has been indicated that metformin suppresses leukemia cell survival, proliferation, and clonogenic activity in both AML and CML (68, 69). The compound CPI-613, as an inhibitor of pyruvate dehydrogenase (PDH) and α-KG dehydrogenase (KDH), has clinical benefits through decreasing oxygen consumption rate in AML cells (70). An unexpected high remission rate has been observed in relapsed and refractory AML patients after co-administration of CPI-613 with high-dose Ara-C (71). Several OXPHOS or ETC complexes inhibitors including Tigecycline, Metformin, 2’3’-Dideoxycytidine, IACS-010759, and A2-32-01 have revealed clinical benefits in AML and CML (63, 65, 72, 73). Metformin, as a widely used anti-diabetic drug, has interestingly exhibited anti-leukemic activities in some leukemias such as AML. However, regarding its pharmacokinetics and maximum efficient dose, it is usually administered in association with other remedies but not alone in clinical settings. Regarding that AML cells are strongly dependent on mitochondrial metabolism, metformin is administered in doses which could inhibit ETC complex I (69, 74). It has been proven that in order to eliminate disease persistence in CML patients, a combination of tigecycline and imatinib can serve as a suitable clinical approach (75).
One of the most important and well-known metabolism-related hallmarks of AML is a mutation of isocitrate dehydrogenases. These are crucial enzymes of the TCA cycle. In their wild-type form, these isoforms oxidatively decarboxylate isocitrate to 2-KG and generate NADPH. However, their mutant forms oxidize NADPH and produce the oncometabolite 2-hydroxyglutarate (2-HG) from α-KG (Figure 1) (76–78). This alteration has been found approximately in 20% of AML patients and has been proven to be a promising therapeutic point. In 2018, ivosidenib (formerly known as AG120), an allosteric IDH1R132 inhibitor, was firstly approved by the FDA for patients with relapsed or refractory IDH1-mutated AML. Additionally, it is considered as front-line therapy for newly diagnosed patients who are 75 years or older and also some patients who are ineligible to receive intensive chemotherapy (79, 80). Enasidenib (formerly known as AG-221) that has gained FDA approval for IDH2 mutant AML patients, is a small molecule that selectively inhibits mIDH2 (IDH2R140Q and IDH2R172K) via binding to its allosteric site and prevents the conformation change requiring for the catalytic action and the R-2-hydroxyglutarate (R2HG) production. Its clinical benefits in AML treatment have been demonstrated in both in-vitro studies and clinical trials (80–82). Furthermore, Vorasidenib (AG-881) is under investigation in phase I for the treatment of AML patients with a mutation in IDH1 and/or IDH2 (83).
Amino Acids and Protein Related Metabolic Pathways
Amino acids not only serve as essential building blocks of protein biosynthesis but also are considered as a significant source of carbon and nitrogen on nucleotide synthesis and therefore they contribute to cancer cells’ growth and proliferation. According to numerous studies, it seems that three amino acids glutamine, arginine, and tryptophan have prominent roles in hematological malignancies particularly in myeloid leukemia. The importance of branched−chain amino acids (BCAAs) metabolism in myeloid leukemia has also been studied. Accordingly, in several leukemic cell lines, glutamine depletion can induce significant apoptosis (84–86). AML primary cells have indicated a higher level of glutamine dependence than normal CD34+ cells because this amino acid controls OXPHOS in these cells (87). Small molecule inhibitors such as BPTES and CB-839 that are glutaminase (GLS) inhibitors, can lead to cell proliferation arrest and apoptosis in AML and BCR–ABL-positive CML cells (88, 89). CB-839 can also inhibit glutathione production and cause accumulation of mitochondrial ROS and eventually apoptotic cell death in AML (90). Recent research showed that high-risk MDS patients can benefit from the combination of CB-839 and 5-azacitidine due to the rapid reduction of BM blasts (91). Moreover, GLS inhibition and BCL-2 inhibitor Venetoclax have displayed synergic effects in AML (Figure 1) (86).
Various studies also described a strong dependence of myeloid leukemia cells on arginine. Arginine is a semi-essential amino acid that is involved in many biochemical processes and can be considered as a potential therapeutic cancer target (92–94). Because of defects in the arginine-recycling pathway enzymes like argininosuccinate synthase and ornithine transcarbamylase, AML blast cells are mostly dependent on arginine uptake by the CAT-1 and CAT-2B arginine transporters. In addition, the plasma arginine levels are significantly higher in AML patients compared with healthy counterparts. Hence, BCT-100, a PEGylated human recombinant arginase that can deplete extracellular arginine, has resulted in proliferation arrest, apoptosis, and decreased AML engraftment in vivo. Generally, the cultivation of AML blasts or AML cell lines in an arginine-free media can lead to their apoptosis (94).
Tryptophan metabolism and its conversion to kynurenine, have an important role in a variety of malignant tissues, such as ovarian cancer, melanoma, and head and neck cancer (95). Increased kynurenine/tryptophan ratio in AML patient sera which negatively correlates with their OS, also indicates its importance in myeloid leukemia (96). In fact, in more than 50% of the cases tested at diagnosis, AML blasts constitutively express IDO (Indoleamine-2,3-dioxygenase) and so can promote the formation of regulatory T cells (Tregs) (97).
leucine, isoleucine, and valine are known as BCAAs synthesized by the BCAA transaminases 1 (BCAT1) and 2 (BCAT2) in a reversible transamination reaction. A quantitative expression proteomics analysis detected significant BCAT1 overexpressed in AML LSCs but not in the non-LSC bulk population. A high level of BCAT1 is involved in AML biology (98). Furthermore, Hattori et al. have indicated highly elevated BCAA levels in CML-initiating cells isolated from a BC-CML mouse model using amino acid-specific fluorescent markers. They also have shown that BCAT1 inhibition impairs CML blast crisis propagation in vitro and in vivo (99).
Lipid Related Metabolic Pathways
In a general perspective, lipids are considered as central players in cancer biology due to their critical roles in cell growth and proliferation. Therefore, extensive rewiring of lipid metabolism, which is driven by both oncogenic and tumor microenvironmental cues, is a significant hallmark of cancer cells. Indeed, these metabolic reprogramming and specific lipid profiles of cancer cells are so important that can be regarded as disease biomarkers, with diagnostic, prognostic, and predictive values. Lipids and their metabolism are emerging as promising tools and targets for anti-cancer therapies and numerous natural or synthetic compounds targeting lipid metabolism have been discovered with curative effects against cancer (100, 101). Interestingly, bone marrow-resident LSCs express higher fatty acid transporter CD36 than normal HSCs and hence, they can induce lipolysis in BM adipocytes to fuel FAO in leukemic cells. It seems to be that BM-resident adipocytes remarkably impair the anti-leukemia effect of various chemotherapeutic agents and obese mice compared to normal-weight counterparts show higher rates of relapse after chemotherapy (102). It has been proven that cell growth and survival of AML monocytes are strongly supported by BM adipocytes, which promote their fatty acid β-oxidation (103). Firstly, a study in 2010 showed that pharmacological inhibition of the carnitine palmitoyltransferase 1 (CPT1) which catalyzes one of the rate-limiting steps in FAO, enhanced AML sensitivity to apoptosis (Figure 1) (104). Moreover, other pharmacologic agents that inhibit fatty acid oxidation, such as Etomoxir or Ranolazine, restrain AML cell proliferation and sensitize human leukemia blasts to apoptosis (104). Even some more common lipid targeting drugs like statins are also clinically useful in AML treatment in combination with chemotherapy. Some studies have demonstrated that statin treatment can enhance complete remission rates in favorable-risk AML groups, but had a less clear role in high-risk AML (105, 106).
Lipids are not only a source of energy to produce ATP, NAD(P)H, and building blocks for cell membrane, but also are essential signaling molecules. This group of lipids including prostaglandins, leukotrienes, and eicosanoids has a critical role in several processes like inflammation, and carcinogenesis (107). Chen and colleagues have demonstrated that mRNAs for both Alox5 and Alox15 (arachidonic acid lipoxygenase enzymes) are highly expressed in CML stem cells and their overexpression is vital for their survival. Additionally, they showed that in vivo administration of PD146176 (an Alox15 inhibitor) and/or zileuton (an Alox5 inhibitor) can diminish the relapse of CML disease in WT CML model mice (108, 109). Another study has indicated that increased expression of the enzyme Gdpd3 (lysophospholipase D), which activates lysophospholipid metabolism, can eventually lead to stem cell quiescence and TKI resistance in CML. These observations suggest that targeting Gdpd3 or other enzymes that are involved in lysophospholipid metabolism can contribute in reducing CML disease relapse (110).
General Alterations of Metabolites in Lymphoid Leukemia
Glucose Related Metabolic Pathways
Although different hematological malignancies harbor individual genetical alterations and locate in a diverse microenvironment, ALL shows a conserved symptom of high consumption of glucose as myeloid leukemia does (111). B precursor ALL lymphoblasts exhibit a preferential upregulation of genes related to glycolysis compared to the TCA cycle, indicating the dominant position of aerobic glycolysis in glucose metabolism of ALL (111). However, in contrast to ALL, CLL shows a relatively low uptake of 2-Deoxy-2-[18F] fluoroglucose (FDG) and low sensitivity of positron emission tomography-FDG, demonstrating that the glycolytic pathway might not be a critical role in CLL cells’ metabolism (112, 113). As the glucose transporter, GLUT promotes the uptake of glucose extracellularly in ALL cells, and after deletion, a decrease in proliferation and a limited degree of apoptosis was observed in B cell ALL lymphoblasts harboring the BCR-ABL1 tyrosine kinase fusion oncogene (114). Moreover, inhibition of rate-limiting enzymes that catalyze the metabolic intermediates during glycolysis, such as HK2, PKM2, and LDHA, induces apoptosis, blocks proliferation, and even attenuates drug resistance in ALL cells. Therefore, opens a potential therapeutic window for ALL treatment by targeting crucial enzymes in the glycolysis pathway (114, 115). Numerous oncogenic pathways, such as MYC, PI3K/Akt, AMPK, and Notch, have been reported to participate in the glycolysis procedure in lymphoid leukemia, upregulating the expression of glucose transporters and glycolytic enzymes or balancing glycolysis and mitochondrial metabolism (116–118).
The PPP, which branches from glycolysis, produces NADPH and ribose-5-phosphate after an oxidative phase and plays an unreplaceable role in lipids generation and redox homeostasis in lymphoid leukemia (119). G6PD, the first enzyme in the PPP pathway, can serve as a prognostic biomarker of ALL (120). Furthermore, in B cell ALL, the serine/threonine-protein phosphatase 2A (PP2A) switches glucose carbon utilization from glycolysis to the PPP to cope with oxidative stress, implicating the possibility to remove the gatekeeper function of PP2A in the PPP by inhibiting this molecule in ALL (121).
Despite the roles of aerobic glycolysis as a predominant pathway for providing a source of energy and anabolic precursors for tumorigenesis in most hematopoietic malignancies, some subsets of leukemia, like CLL, satisfy from increasing energy demands during growth by upregulating mitochondrial OXPHOS activity. For instance, an increase in the number of mitochondria, the total mitochondrial mass, and mitochondrial biogenesis was evidenced in CLL cells, in comparison to normal B lymphocytes (122). Elevated mitochondrial oxidative activity generates a proper high level of ROS, contributing to tumor progression by mediating the tumor microenvironment and protecting cells from apoptosis. When compared with normal B cells, the level of mitochondria-derived ROS in CLL cells is higher due to a more active mitochondrial activity, thereby promoting the progression of leukemia (40, 122). Metformin, an inhibitor of mitochondrial complex I, has been proved to suppress cell cycle progression and cell proliferation via disruption of mitochondrial OXPHOS in CLL and ALL (123, 124). Furthermore, a synergetic antileukemia effect has been observed when combining metformin with daunorubicin or vincristine, which are extensively used to treat leukemia clinically. This has revealed that, revealing that targeting mitochondrial OXPHOS could be a potent adjuvant therapy strategy for lymphoid leukemia treatment (125, 126).
Amino Acids and Protein Related Metabolic Pathways
Glutamine provides a rich resource of carbon and nitrogen, which can be used to produce amino acids, nucleotides, and fuel mitochondrial activity via offering α-KG. Thus, many cancer cells become addicted to glutamine and rely heavily on glutaminolysis for rapid growth. Likewise, ALL cells show an active utilization of glutamine for leukemogenesis, while suppression of glutaminolysis enhances the antileukemic effects in mice harboring T cell ALL (127). Genetic or pharmacological targeting ASCT2 molecule, a transporter of neutral amino acids, especially glutamine, decreases leukemia initiation and maintenance driven by the oncogene MLL-AF9 or Pten deficiency (128). Furthermore, glutaminase (GLS), An enzyme that catalyzes glutamine into glutamate, has proved to be a potential therapeutic target in the treatment of ALL (129, 130).
Asparagine synthetase (ASNA) catalyzes the conversion from aspartate to asparagine in most tissues. However, due to the low expression of ASNA, ALL cells are auxotrophic for asparagine and highly sensitive to asparaginase treatment, which degrades extracellular asparagine systematically and blocks the cell growth of ALL cells (131, 132). Taking advantage of this unusual metabolic dependency on an exogenous supply of asparagine in ALL cells, asparaginase has become one of the standard therapeutic regimens in ALL treatment. Asparaginase already exhibited a synergetic and profound antileukemia effect when combining with chemotherapies for ALL treatment in clinical practice (133, 134). Intriguingly, asparaginase diminishes not only asparagine but also glutamine in the intracellular pool (130). Moreover, asparaginase reduces glucose uptake and glycolysis and hampers pyrimidine synthesis in ALL cells (135). Altogether, asparaginase can fulfill its antileukemia function in a comprehensive method.
Arginine is a semi-essential amino acid, which is critical for the synthesis of protein, nitric oxide, and other amino acids. Arginine can be synthesized from citrulline intracellularly while taking up exogenous arginine is also necessary to meet the cellular demands in leukemias. Increasing studies have shown the potential therapeutic benefit of pegylated-human-arginase I (peg-Arg I) in ALL treatment. By depleting the existence of arginine and starving ALL cells of it, peg-Arg I induces a decreased progression of the cell cycle via impairing the expression of cyclin D3 (136). Further study suggested the central role of phosphorylation of the eukaryotic-translation-initiation factor 2 alpha signaling in the antileukemia effects induced by peg-Arg I (137). Interestingly, although endogenous or genetic upregulation of argininosuccinate synthase or ornithine transcarbamylase rescues ALL survival from a moderately low arginine condition, these enzymes cannot prevent the cytotoxic effects of peg-Arg I towards ALL (138).
Lipid Related Metabolic Pathways
Lipids serve as fundamental building blocks for the construction of membranes. Cancer cells satisfy the high demands of lipids for the biosynthesis of membrane structure via synthesizing fatty acids themselves, rather than absorbing lipids from the blood. Fatty acid synthase (FASN), a large enzyme with multiple catalytic domains, carboxylates malonyl-CoA into fatty acid chains. An elevated level of FASN was evidenced in drug-resistant ALL and correlated with a poor prognosis, while inhibition of FASN increases apoptosis rates and overcomes dexamethasone resistance (139). Moreover, not only the suppression of FASN inhibits ALL cells growth but also produces a remarkable impairment of PD-L1 expression, indicating the potential role of FASN inhibitors in antileukemia immunotherapy (140).
FAO in the mitochondria degrades fatty acids and generates acetyl-CoA for OXPHOS and energy production in cancer cells. The transportation of fatty acids into mitochondria for FAO requires the assistance of CPT, an enzyme located in the outer membrane of mitochondria. CLL cells exhibit elevated levels of CPT1 and CPT2, while inhibition of CPT induces compromised FAO activity due to ineffective transportation of fatty acids into mitochondria, subsequently leading to massive CLL cell death (141).
Lipoprotein lipase (LPL), a water-soluble enzyme that hydrolyzes triglycerides in lipoproteins, has found to be abnormally expressed in CLL cells (142). Moreover, the elevated expression of LPL is associated with a poor prognosis and an aggressive disease stage of CLL (142, 143). LPL displays its oncogenic function via storing lipids in cytoplasmic vacuoles and fueling fatty acids for energy production, reprograming CLL cells to utilize fatty acids as the major energy resource (144). Furthermore, this unique metabolic feature of CLL cells is driven by STAT3, an oncogenic pathway that endows CLL cells’ survival advantages by binding and activating the promotors of LPL (145, 146). Additionally, further study has revealed that LPL in aggressive CLL cells generates activating ligands for the nuclear receptor PPARα and allows fatty acids to be employed as fuel (147). Thus, STAT3-mediated aberrant LPL in CLL can serve as an ideal targeted marker for blocking lipid metabolism.
Impact of Metabolic Regulation on Anti-Leukemia Immune Responses
Several research have revealed that altered cellular metabolism not only changes the phenotype of leukemia cells but also impacts the BMM greatly, resulting in the initiation, progression, and metastasis of leukemia (148). As a dynamic and complex biological tissue, the bone marrow niche influences the anti-leukemia immune responses significantly via cellular or noncellular components in the tumor microenvironment (TME) (Figure 2). For instance, by depriving essential nutrients from the bone marrow niche, leukemia cells fabricate a long-term nutrients deficiency microenvironment, resulting in the dysfunction of immunocytes, leading to a suppression of antileukemia immune responses. Although the current understanding of the relationship between metabolic regulation in the leukemia-resided BMM and antileukemia immunity is still incomplete, a deep investigation of this field will undoubtedly open a therapeutic avenue for leukemia immunotherapy.
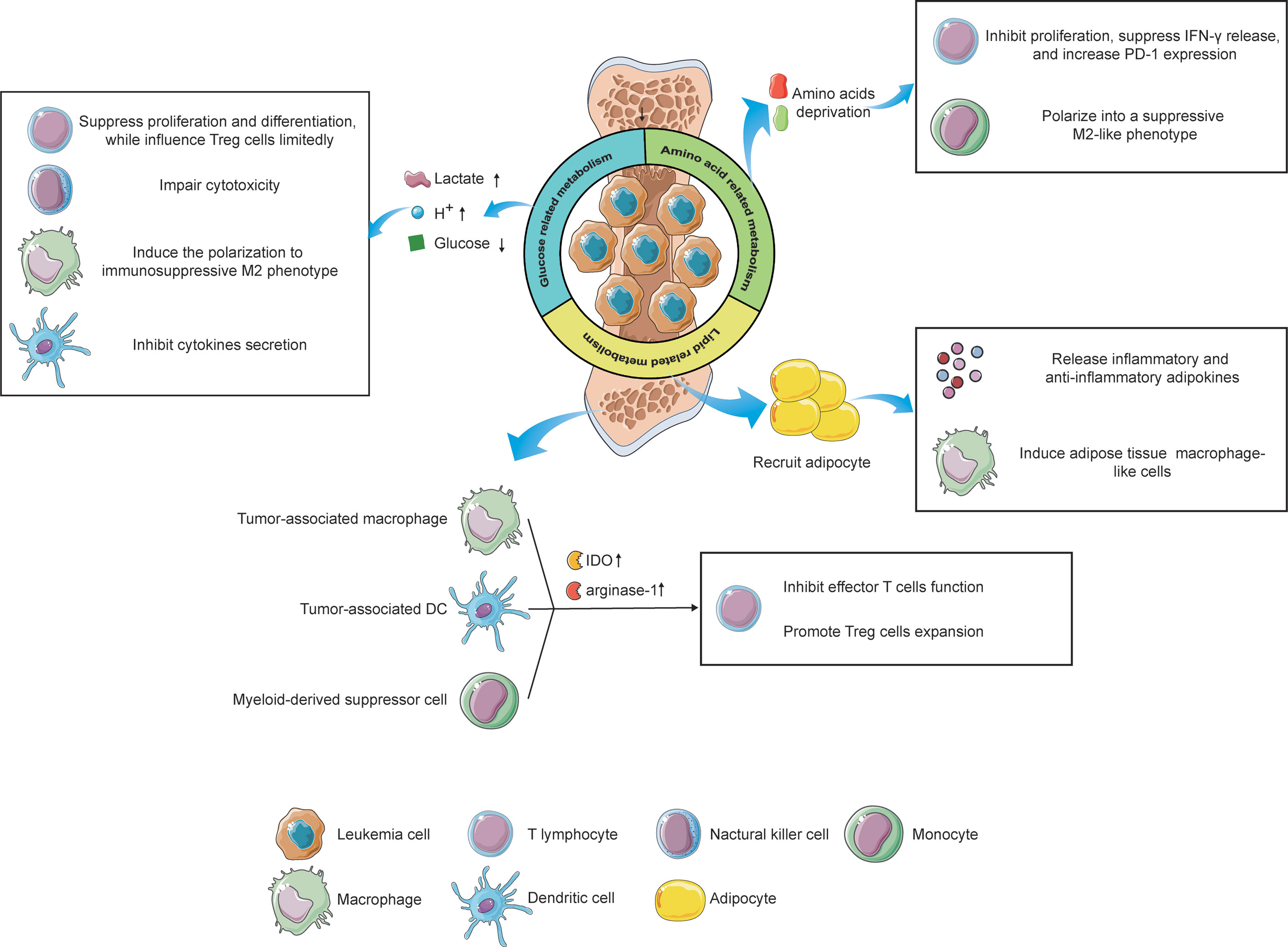
Figure 2 The influence of metabolic alterations of leukemia cells and immune cells on the immune responses under the microenvironment. Reprogramming glucose-related metabolism impacts immunity via releasing lactate and hydrogen ions, which create an acidic microenvironment, as well as depriving of extracellular glucose, resulting in an immunosuppressive status of filtrating immune cells. Furthermore, leukemia cells also destitute the amino acids in the microenvironment, leading to the inactivation of T lymphocytes and phenotype shift of monocytes. Lastly, by recruiting adipocytes in the BMM via the lipid-related pathway, leukemia cells reveal a potential effect on immunological regulation. Immune cells, like tumor-associated macrophages, tumor-associated DCs, and MDSCs can also impact the T cells’ function via upregulating the expression of IDO and arginine-1, contributing to an immune-tolerant microenvironment. The figure was produced with the assistance of Servier Medical Art (https://smart.servier.com).
As the preferential glucose metabolism of leukemia, aerobic glycolysis generates and secretes a high level of extracellular lactate and hydrogen ions, these products in turn, acidify the microenvironment. The acidic environment is beneficial to the invasion of leukemia cells, while detrimental to immune cells that target leukemia cells (149). Previous studies exhibited that a high level of extracellular lactate suppresses the differentiation from monocytes to dendritic cells (DC) and inhibits the cytokine secretion of DCs and effector T cells (150, 151). Furthermore, elevated lactate in the microenvironment attenuates the secretion of lactate from T cells because the transportation of lactate depends on the difference between intracellular and extracellular concentration in T cells, leading to the accumulation of intracellular lactate in T cells, which inhibits the cytotoxic T cell function (152). Additionally, an acidic microenvironment induces the polarization of macrophages from M1 to immunosuppressive M2 phenotype, which supports leukemia angiogenesis and metastasis (149). Meanwhile, a low-pH microenvironment also impairs the cytotoxicity of NK cells. Huge consumption of glucose by leukemia cells causes glucose deficiency in the microenvironment under limited blood supplement, inhibiting the proliferation and differentiation of tumor-infiltrating lymphocytes via glucose competence (153). However, glucose competition has limited effects on regulatory T (Treg) cells due to the predominant role of lipids metabolism in the energy production of Treg cells. Thus, under glucose-deprived conditions, Treg cells still exert consistent immunosuppressive functions while effector T cells show a relatively weakened antileukemia capability, which is favorable for the progression of leukemia (154). Taken together, neutralizing the acidic BMM and suppressing glucose deprivation of leukemia cells can serve as novel strategies against leukemias, prompting immune surveillance and clearance.
Amino acids provide building blocks for the synthesis of proteins and other compoundsnecessary for structure construction, signaling transduction, and etc. Deprivation of amino acids in the BMM by leukemia cells profoundly impacts the immune cells, leading to an immunosuppressive microenvironment eventually. For instance, IDO was found to be highly expressed and abnormally activated in leukemias, exhibiting its inhibitory effects on T cells via tryptophan deprivation (155). Moreover, leukemia cells can induce immune tolerance by increasing the expansion of Treg cells in an IDO-dependent manner (155, 156). In addition to leukemia cells, mesenchymal stem cells (MSCs), critical contributors to tissue inflammation homeostasis, were evidenced to utilize tryptophan-depleting IDO to suppress T cells via STAT1 glycosylation. Interfering with STAT1 activity in MSCs impacts IDO upregulation, further impeding T cell suppression (157). Arginine, a non-essential amino acid, also participates in the regulation of immune responses. Enhanced arginine metabolism in AML blasts contributes to the secretion of arginase II in the microenvironment and a high concentration of arginase II in the plasma has been evidenced in patients with AML (158). The low arginine microenvironment created by AML blasts acts as an immune brake resulting in T cells anergy, including impaired proliferation, reduced IFN-γ release, and up-regulation of PD-1 (159). Furthermore, AML blasts show an arginase-mediated ability to polarize monocytes into a suppressive M2-like phenotype and hamper the proliferation and differentiation of human CD34+ progenitors (158). Another key amino acid, glutamine, was also reported to be involved in immunological regulation. Depletion of glutamine impedes proliferation and cytokine production of T cells and this cannot be salvaged by offering biosynthetic precursors of glutamine (160). Paradoxically, a recent study revealed that tumor-infiltrating CD8+ T cells, under glutamine-restricted conditions, exhibit a boost in proliferation with an increased expression of Ki67, whereas T cells exhaustion was limited (161). Therefore, an integrated understanding of the impacts of glutamine metabolism on the BMM, even on immunological regulation towards leukemias, is urgently needed to clarify the underlying mechanism by which glutamine influences immune responses.
The BMM is an area filled with numerous adipocytes. When tumor cells migrating from other tissues are located in the BMM, adipocytes will be recruited and deliver lipids to tumor cells via the CD36 receptor (162). In addition, these adipocytes secrete a set of inflammatory adipokines, such as leptin, tumor necrosis factor-alpha (TNF-α), and IL-6, as well as an anti-inflammatory adipokine, called adiponectin, to modulate the immunological microenvironment (163). Furthermore, adipocytes have been found to induce the differentiation of bone marrow precursors into adipose tissue macrophage-like cells via releasing exosome-sized, lipid-filled vesicles (164). These results implicit lipids metabolism regulated by adipocytes might be involved in antileukemia immune responses. Besides, the existence of lipid intermediates in ascites of ovarian cancer patients induced a proliferation arrest of ascites-derived lymphocytes in the G0/G1 phase, implying that targeting lipid metabolism in the microenvironment can serve as a therapeutic option to struggle against tumors via regulation of immune responses (165). However, our current understanding concerning the relationship between lipid metabolism and antileukemia immunity remains elusive and in-depth investigations correlated with this field are encouraged to broaden the knowledge.
Nevertheless, not only does the reprogramming metabolic wiring of leukemia cells influences the leukemic niche but also the alteration of metabolism in some immune cells impacts the BMM, contributing to an immunosuppressive microenvironment that favors the progression of leukemia (Figure 2). As we mentioned above, acidification polarizes infiltrating macrophages into the tumor-supporting M2 phenotype, also known as “ tumor-associated macrophages” (TAMs). TAMs exhibit atendency of upregulation of transcription factor HIF1α, which in turn enhances the expression of vascular endothelial growth factor (VEGF) and arginase-1, underlining the tumor-favoring function of TAMs (166). Furthermore, an elevated IDO expression has been evidenced in TAMs (167). TAMs induce the proliferation of Treg cells via tryptophan catabolite, leading to the suppression of T cells function and immune-tolerant microenvironment (168). Another immunosuppressive cell phenotype, tumor-associated DCs (TADCs), has also been reported to participate in the modulation of the microenvironment via metabolic alterations. By enhancing the expression of arginase-1 and IDO, which delete the arginine and tryptophan in the TME, TADCs attenuate the tumor-fighting function of effector T lymphocytes, supporting the immune evasion of leukemia cells (169). The role of myeloid-derived suppressor cells (MDSCs) in the regulation of the TME via metabolic rewiring also could not be ignored. It has been reported that DMSCs overexpress IDO to catalyze tryptophan into kynurenine, suppressing T cells function and promoting Treg cells generation (170). These facts together imply that the metabolic reprogramming of immune cells also play a pivotal role in the modulation of the BMM, highlighting the promising possibility to target immune cells metabolism for leukemia treatment.
Exploiting of Metabolism Targeting Treatments in Leukemia
As discussed above, altered metabolism is essential in hematologic malignancies to overcome survival stress and adjust to the diverse microenvironment. Thus, therapy against leukemias by targeting reprogramming metabolic molecules seems to be an ideal option to struggle against hematological malignancies and the selection of specific biomarkers is critical in the development of targeted therapeutic agents (171). Recently, researchers are dedicated to identifying novel metabolism-targeting drugs. Metabolic enzymes and the components within the glucose, amino acid, and fatty acid metabolic pathways, are regarded as attractive targets (172), and some of them have shown inspiring results in clinical trials, as shown in Table 1.
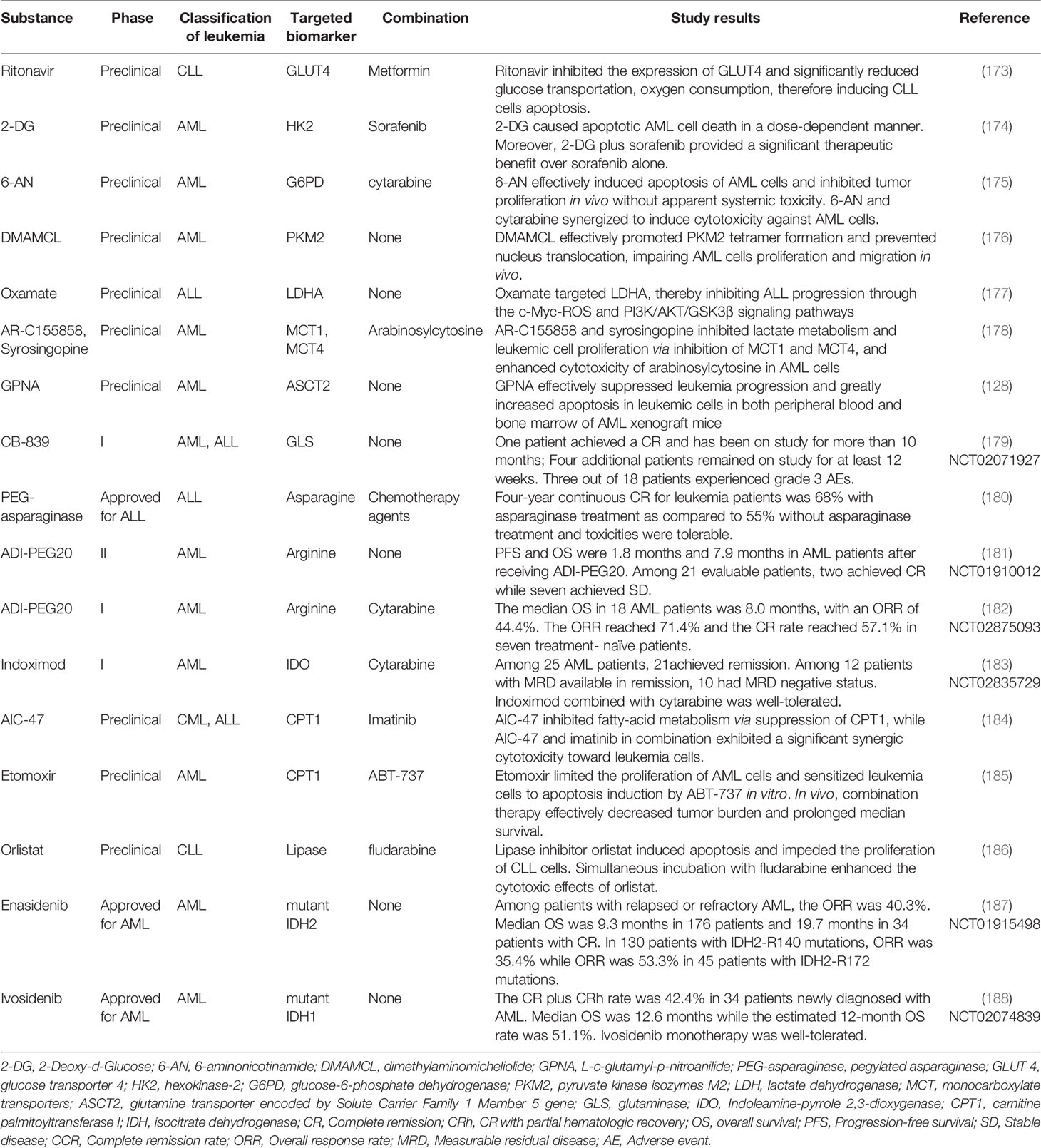
Table 1 Metabolism-targeted agents and their antileukemia effects in vitro and in vivo.
Targeting Glycolysis
Generally, most of the glucose consumed by leukemia cells is catabolized through glycolysis. Thus, preventing glucose uptake and glycolysis will be an effective strategy to struggle against leukemia. GLUT mediates the glucose uptake and transportation of leukemia cells. By inhibiting GLUT4, protease inhibitor ritonavir exhibits similar toxic effects on CLL cells as glucose deprivation does in vitro (173). Furthermore, when co-incubating with metformin, an inhibitor of mitochondrial complex I activity, ritonavir-resistant CLL cells are sensitized, resulting in the elimination of CLL cells via perturbation of glucose metabolism. Promising antileukemia results have also been evidenced by targeting rate-limiting enzymes related to glycolysis and the PPP, such as HK2 (174), G6PD (175), PKM2 (176). Leukemia cells prefer shunting pyruvate into lactate production to adjust to rapid energy consumption rather than into the TCA cycle for further OXPHOS, providing a probability to target LDH to inhibit the production of lactate in leukemia cells. The LDHA inhibitor oxamate fulfills its antileukemia effects by arresting ALL cells in G0/G1 phase and inducing apoptosis (177). Further mechanism exploration revealed that blocking LDHA also leads to the inactivation of the c-Myc and PI3K signaling pathway, suggesting that targeting LDH is an ideal selection to achieve antitumor benefits when applying in vitro. Last but not least, prohibiting monocarboxylate transporters (MCTs), the transporter of lactate, will results in the accumulation of glycolytic end-products including lactate and H+, which consequently, lead to intracellular acidification and apoptosis. A recent study demonstrated that AR-C155858 and syrosingopine, inhibitors of MCT1 and MCT4, exert an anti-proliferative effect and enhance AML cells’ sensitivity to chemotherapeutic agents (178). Altogether, the results highlight the important role of targeting the glycolysis pathway in the treatment of leukemia. Regretfully, clinical results concerning glycolysis-related inhibitors to cure leukemia are rare up to now so far. The bio-safe and efficacy of glycolysis-related inhibitors for leukemia treatment are eager to explore clinically.
Targeting Amino Acid Metabolism
The current development of targeted agents against glutamine metabolism of cancer cells focuses on membrane glutamine transporter inhibition, GLS inhibition, and glutamine depletion. ASCT2 mediates the entry of glutamine and other amino acids into cells. ASCT2 inhibitor GPNA showed potential in decreasing survival and promoting apoptosis of AML cells in vitro. In AML mice, GPNA treatment effectively suppressed leukemia progression. Attenuated splenomegaly and hepatomegaly, and reduced AML cells infiltration were observed. Notably, the impact of GPNA on normal blood cell formation was limited (128). As a selective and irreversible inhibitor of GLS, CB-839 has shown ideal antileukemia effects on AML and ALL cells in vivo (129). Furthermore, a completed phase I clinical trial also evidenced the potential role of CB-839 to combat leukemias (NCT02071927) (179). Eighteen patients, including AML (n=16), ALL (n=1) and mixed leukemia (n=1), were enrolled in this trial. Results showed that CB-839 was well tolerated in advanced leukemia patients. One AML patient achieved a complete remission (CR) and has been on study for more than 10 months, while four additional patients remained on study for at least 12 weeks. Another Phase Ib/II clinical trial has demonstrated that CB-839 is safe and effective in combination with azacitidine in patients with advanced myelodysplastic syndrome (MDS) (NCT03047993) (91). Among ten patients with MDS, seven patients achieved CR, two patients had stable disease (SD), while one patient had no response, suggesting that GLS inhibition may induce a robust effect against hematopoietic malignancies synergetically when combining with other conventional treatments.
Asparaginase has been approved by FDA as the frontline therapeutic agent used in ALL. A previous clinical trial revealed that 357 patients with ALL reached a 68% of four-year continuous CR rate after receiving asparaginase treatment, while the rate was only 55% in patients without asparaginase (180). The overall one-sided logrank test favors the asparaginase group over the control in ALL treatment. Toxicities related to asparaginase were manageable.
Many types of cancer cells, including AML disease, depend on the exogenous supply of arginine due to the defects in arginine synthetase (189, 190). Therefore, arginine depletion is a possible therapeutic option for leukemia cure. A phase II clinical trial reported that two patients reached CR while seven patients achieved SD among 21 argininosuccinate synthetase (ASS)-deficient AML patients after receiving arginine deiminase (ADI-PEG20) (NCT01910012) (181). The median progression-free survival (PFS) and overall survival (OS) were 1.8 months and 7.9 months, respectively. Clinical outcomes and further transcriptome sequencing implied that ADI-PEG20 monotherapy is required but not sufficient for ASS-deficient leukemia treatment. A rational combination with other targeted or conventional drugs may improve the efficacy of the antileukemia response as a whole. Recently, another phase I study combining ADI-PEG20 and low-dose cytarabine for the treatment of AML has completed and encouraging outcomes have been observed (NCT02875093) (182). Results revealed that the overall response rate (ORR) in 18 patients was 44.4%, with a median OS of 8.0 months. In seven treatment-naive patients, the ORR was 71.4% and the CR rate was 57.1%. Moreover, patients suffered limited fatal side effects after receiving the combination of ADI-PEG20 and cytarabine. Taken together, ADI-PEG20 shows the superior clinical performance when cooperating with conventional clinical agents for AML therapy, and the underlying mechanism of combination warrants further explorations.
Increased IDO activity results in T cells inactivation and immunosuppression (191). Even though IDO inhibitors are mainly being tested in solid tumors, clinical trials have also started to test this potential biomarker in AML (NCT02835729) (183). Among 25 AML patients who received more than one dose of indoximod, a small-molecule inhibitor of the IDO pathway, 21 AML patients attained disease remission. Furthermore, ten patients experience measurable residual disease (MRD) negative among twelve patients with MRD available in remission. When combining with standard AML induction therapy, indoximod is well tolerated. These promising results indicated that indoximod could serve as an ideal targeted agent to combat leukemia by inhibiting IDO activity.
Targeting Fatty Acid Metabolism
To meet the exquisite demands of cell growth and proliferation, leukemia cells exhibit high dependence on FA metabolism, making it possible to target this metabolic vulnerability to eliminate leukemia cells (192). Currently, FAO inhibitors are being tested in preclinical trials. Imatinib induces the inactivation of BCR-ABL, thereby suppressing glycolysis in CML cells. However, compensatory FAO activation rescued glucose-independent CML cells’ survival after imatinib treatment. AIC-47 suppressed the expression of CPT 1 and directly fatty acid metabolism, contributing to the complementary attack on CML energy metabolism when combining with imatinib (184). Moreover, another CPT 1 inhibitor, etomoxir, has been reported to exhibit significant antiproliferative effects on AML cells and sensitize AML cells to apoptosis induced by ABT-737 in vitro (185). Although FAO inhibitors have not been practiced clinically, preclinical studies already reveal the feasibility of appliance of FAO inhibitors in leukemia treatment.
In CLL, LPL has been identified as a specific biomarker, which indicates an unfavorable prognosis in patients (193, 194). Via targeting LPL molecule, orlistat induced apoptosis of CLL cells, while limited cytotoxicity was demonstrated in healthy B cells and peripheral blood mononuclear cells (186). Moreover, the antileukemia effects of orlistat on CLL cells strengthened when incubated with fludarabine, simultaneously.
Targeting Isocitrate Dehydrogenase Mutation
Retrospective studies claim that the IDH mutation was associated specifically with a poorer prognosis in AML (195–197). Currently, ivosidenib and enasidenib, inhibitors of IDH1 and IDH2, have become the first FDA-approved metabolism targeting drugs for AML. In a phase I/II trial, the efficacy of enasidenib was examined in patients with mutant-IDH2 advanced myeloid malignancies (187). The median overall survival among patients with relapsed/refractory AML was 9.3 months while patients who reached CR or partial remission had a median survival of 19.7 months or 14.4 months, respectively. The ORR for all relapsed/refractory AML patients was 40.3%, with a median response duration of 5.8 months. This study proved that daily enasidenib therapy induces antileukemia responses and is well-tolerated in patients who failed in conventional treatment previously. Ivosidenib, a mutant IDH1 specific inhibitor, has been evaluated in a phase I trial (NCT02074839) (188). Thirty four patients with AML were enrolled in this trial and their median overall survival was 12.6 months with a median follow-up period of 23.5 months. CR plus CR with partial hematologic recovery rate was 42.4%. The most commonly reported therapy-related serious adverse event was IDH differentiation syndrome. These results demonstrated that enasidenib and ivosidenib induce durable remissions and are safe in mutant IDH AML treatment.
Conclusion and Prospectives
In this review, we summarized the alteration of metabolic events supplementing building blocks and energy for rapid proliferation, drug resistance, self-renewal, etc. during leukemogenesis. Deep explorations into leukemia metabolomics have extended a novel therapeutic path through which targeting leukemia metabolic vulnerabilities exerts prominent antileukemia effects. Furthermore, the importance of cross-talk between leukemia cells and immunocytes has been highlighted in the text, especially under complicated BMM conditions. Since metabolism participates in immunological regulation and usually results in an immunosuppressive microenvironment favoring the occurrence and development of leukemia cells, metabolic interventions could serve as a potential option to improve immune response toward leukemia, particularly when combined with immune regulatory agents. Although clinical benefits were evidenced in patients with leukemia by targeting metabolic molecules, the effectiveness is still far from satisfying and cytotoxic side effects are still unignorable. Thus, exploring leukemia-specific metabolic dependencies to identify potential biomarkers with limited toxicities toward normal cells will be the future research focus. Moreover, different subsets of leukemia cells harbor individual genetic and biological features, therefore exhibiting different metabolism mechanisms, which means targeting metabolic vulnerabilities should be subgroup-specific, even individual-specific, to achieve the greatest effect. The application of accurate metabolic targeting requires sophisticated new techniques to elucidate the complexity and dynamicity of remodeling of metabolic wiring. Additionally, LSCs are hypothesized to be responsible for the conventional treatment resistance and leukemia relapse. Compared with normal leukemia cells, LSCs reveal a different metabolic preference, emphasizing that a comprehensive understanding of the metabolic status of LSCs will also provide us novel insights into therapy against drug-resistant or relapsed leukemia. Last but not least, a rational combination therapeutic strategy, for instance, integrating targeted agents related to metabolic pathways and conventional chemotherapies, is worth further exploration clinically and may improve the cure rate of leukemia comprehensively.
Author Contributions
MGH and AB contributed to conception and design of the study. MS, YZ, and ZX wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lyengar V, Shimanovsky A. Leukemia, in StatPearls. Treasure Island (FL: StatPearls Publishing Copyright © 2021, StatPearls Publishing LLC (2021).
3. Davis AS, Viera AJ, Mead MD. Leukemia: An Overview for Primary Care. Am Fam Phys (2014) 89(9):731–8.
4. (2020). Available at: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf.
5. Galluzzi L, Kepp O, Vander Heiden MG, Kroemer G. Metabolic Targets for Cancer Therapy. Nat Rev Drug Discov (2013) 12(11):829–46. doi: 10.1038/nrd4145
6. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
7. Ito K, Suda T. Metabolic Requirements for the Maintenance of Self-Renewing Stem Cells. Nat Rev Mol Cell Biol (2014) 15(4):243–56. doi: 10.1038/nrm3772
8. Simsek T, Kocabas F, Zheng J, De Berardinis RJ, Mahmoud AI, Olson EN, et al. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell (2010) 7(3):380–90. doi: 10.1016/j.stem.2010.07.011
9. Hale LP, Braun RD, Gwinn WM, Greer PK, Dewhirst MW. Hypoxia in the Thymus: Role of Oxygen Tension in Thymocyte Survival. Am J Physiol Heart Circulatory Physiol (2002) 282(4):H1467–77. doi: 10.1152/ajpheart.00682.2001
10. Karigane D, Takubo K. Metabolic Regulation of Hematopoietic and Leukemic Stem/Progenitor Cells Under Homeostatic and Stress Conditions. Int J Hematol (2017) 106(1):18–26. doi: 10.1007/s12185-017-2261-x
11. Mougiakakos D. The Induction of a Permissive Environment to Promote T Cell Immune Evasion in Acute Myeloid Leukemia: The Metabolic Perspective. Front Oncol (2019) 9:1166. doi: 10.3389/fonc.2019.01166
12. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood J Am Soc Hematol (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
13. (2020). Available at: https://www.msdmanuals.com/professional/hematology-and-oncology/leukemias/overview-of-leukemia.
14. Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med (2015) 373(12):1136–52. doi: 10.1056/NEJMra1406184
15. Dombret H, Gardin C. An Update of Current Treatments for Adult Acute Myeloid Leukemia. Blood J Am Soc Hematol (2016) 127(1):53–61. doi: 10.1182/blood-2015-08-604520
16. Hofmann S, Schubert M-L, Wang L, He B, Neuber B, Dreger P, et al. Chimeric Antigen Receptor (CAR) T Cell Therapy in Acute Myeloid Leukemia (AML). J Clin Med (2019) 8(2):200. doi: 10.3390/jcm8020200
17. Jitschin R, Saul D, Braun M, Tohumeken S, Völkl S, Kischel R, et al. CD33/CD3-Bispecific T-Cell Engaging (BiTE®) Antibody Construct Targets Monocytic AML Myeloid-Derived Suppressor Cells. J Immunother Cancer (2018) 6(1):1–6. doi: 10.1186/s40425-018-0432-9
18. Kansara R, Speziali C. Immunotherapy in Hematologic Malignancies. Curr Oncol (2020) 27(s2):124–31. doi: 10.3747/co.27.5117
19. Apperley JF. Chronic Myeloid Leukaemia. Lancet (2015) 385(9976):1447–59. doi: 10.1016/S0140-6736(13)62120-0
20. Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused Transcript of Abl and Bcr Genes in Chronic Myelogenous Leukaemia. Nature (1985) 315(6020):550–4. doi: 10.1038/315550a0
21. Cilloni D, Saglio G. Molecular Pathways: Bcr-Abl. Clin Cancer Res (2012) 18(4):930–7. doi: 10.1158/1078-0432.CCR-10-1613
22. Radich JP, Mauro MJ. Tyrosine Kinase Inhibitor Treatment for Newly Diagnosed Chronic Myeloid Leukemia. Hematol Oncol Clinics North Am (2017) 31(4):577–87. doi: 10.1016/j.hoc.2017.04.006
23. Braun TP, Eide CA, Druker BJ. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell (2020) 37(4):530–42. doi: 10.1016/j.ccell.2020.03.006
24. Pui C-H, Relling MV, Downing JR. Acute Lymphoblastic Leukemia. N Engl J Med (2004) 350(15):1535–48. doi: 10.1056/NEJMra023001
25. Malard F, Mohty M. Acute Lymphoblastic Leukaemia. Lancet (2020) 395(10230):1146–62. doi: 10.1016/S0140-6736(19)33018-1
26. Pession A, Valsecchi MG, Masera G, Kamps WA, Magyarosy E, Rizzari C, et al. Long-Term Results of a Randomized Trial on Extended Use of High Dose L-Asparaginase for Standard Risk Childhood Acute Lymphoblastic Leukemia. J Clin Oncol (2005) 23(28):7161–7. doi: 10.1200/JCO.2005.11.411
27. Dores GM, Devesa SS, Curtis RE, Linet MS, Morton LM. Acute Leukemia Incidence and Patient Survival Among Children and Adults in the United States, 2001-2007. Blood J Am Soc Hematol (2012) 119(1):34–43. doi: 10.1182/blood-2011-04-347872
28. Jacque N, Leblond V. Chronic Lymphocytic Leukemia. Presse Med (2019) 48(7-8 Pt 1):807–15. doi: 10.1016/j.lpm.2019.07.019
29. Cairns RA, Harris IS, Mak TW. Regulation of Cancer Cell Metabolism. Nat Rev Cancer (2011) 11(2):85–95. doi: 10.1038/nrc2981
30. Ward PS, Thompson CB. Metabolic Reprogramming: A Cancer Hallmark Even Warburg did Not Anticipate. Cancer Cell (2012) 21(3):297–308. doi: 10.1016/j.ccr.2012.02.014
31. Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol Cell (2010) 39(2):171–83. doi: 10.1016/j.molcel.2010.06.022
32. de la Cruz López KG, Toledo Guzmán ME, Sánchez EO, Carrancá AG. Mtorc1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front Oncol (2019) 9(1373). doi: 10.3389/fonc.2019.01373
33. Nepstad I, Hatfield KJ, Grønningsæter IS, Reikvam H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int J Mol Sci (2020) 21(8):2907. doi: 10.3390/ijms21082907
34. Folmes CD, Dzeja PP, Nelson TJ, Terzic A. Metabolic Plasticity in Stem Cell Homeostasis and Differentiation. Cell Stem Cell (2012) 11(5):596–606. doi: 10.1016/j.stem.2012.10.002
35. Maryanovich M, Zaltsman Y, Ruggiero A, Goldman A, Shachnai L, Zaidman SL, et al. An MTCH2 Pathway Repressing Mitochondria Metabolism Regulates Haematopoietic Stem Cell Fate. Nat Commun (2015) 6(1):1–9. doi: 10.1038/ncomms8901
36. Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, et al. Regulation of Glycolysis by Pdk Functions as a Metabolic Checkpoint for Cell Cycle Quiescence in Hematopoietic Stem Cells. Cell Stem Cell (2013) 12(1):49–61. doi: 10.1016/j.stem.2012.10.011
37. Ito K, Carracedo A, Weiss D, Arai F, Ala U, Avigan DE, et al. A PML–PPAR-δ Pathway for Fatty Acid Oxidation Regulates Hematopoietic Stem Cell Maintenance. Nat Med (2012) 18(9):1350–8. doi: 10.1038/nm.2882
38. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell (2013) 12(3):329–41. doi: 10.1016/j.stem.2012.12.013
39. Zhong W, Yi Q, Xu B, Li S, Wang T, Liu F, et al. ORP4L is Essential for T-Cell Acute Lymphoblastic Leukemia Cell Survival. Nat Commun (2016) 7(1):1–14. doi: 10.1038/ncomms12702
40. Rozovski U, Hazan-Halevy I, Barzilai M, Keating MJ, Estrov Z. Metabolism Pathways in Chronic Lymphocytic Leukemia. Leuk Lymphoma (2016) 57(4):758–65. doi: 10.3109/10428194.2015.1106533
41. Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, et al. AML Cells Have Low Spare Reserve Capacity in Their Respiratory Chain That Renders Them Susceptible to Oxidative Metabolic Stress. Blood J Am Soc Hematol (2015) 125(13):2120–30. doi: 10.1182/blood-2014-08-594408
42. Stuani L, Sabatier M, Sarry J-E. Exploiting Metabolic Vulnerabilities for Personalized Therapy in Acute Myeloid Leukemia. BMC Biol (2019) 17(1):1–17. doi: 10.1186/s12915-019-0670-4
43. Boag J, Beesley AH, Firth MJ, Freitas JR, Ford J, Hoffmann K, et al. Altered Glucose Metabolism in Childhood Pre-B Acute Lymphoblastic Leukaemia. Leukemia (2006) 20(10):1731–7. doi: 10.1038/sj.leu.2404365
44. Kominsky DJ, Klawitter J, Brown JL, Boros LG, Melo JV, Eckhardt SG, et al. Abnormalities in Glucose Uptake and Metabolism in Imatinib-Resistant Human BCR-ABL–positive Cells. Clin Cancer Res (2009) 15(10):3442–50. doi: 10.1158/1078-0432.CCR-08-3291
45. Herst PM, Howman RA, Neeson PJ, Berridge MV, Ritchie DS. The Level of Glycolytic Metabolism in Acute Myeloid Leukemia Blasts at Diagnosis is Prognostic for Clinical Outcome. J Leuk Biol (2011) 89(1):51–5. doi: 10.1189/jlb.0710417
46. Maiso P, Huynh D, Moschetta M, Sacco A, Aljawai Y, Mishima Y, et al. Metabolic Signature Identifies Novel Targets for Drug Resistance in Multiple Myeloma. Cancer Res (2015) 75(10):2071–82. doi: 10.1158/0008-5472.CAN-14-3400
47. Chen W-L, Wang J-H, Zhao A-H, Xu X, Wang Y-H, Chen T-L, et al. A Distinct Glucose Metabolism Signature of Acute Myeloid Leukemia With Prognostic Value. Blood J Am Soc Hematol (2014) 124(10):1645–54. doi: 10.1182/blood-2014-02-554204
48. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of Glucose Transporter 1 and Glycolytic Gene Expression by C-Myc. J Biol Chem (2000) 275(29):21797–800. doi: 10.1074/jbc.C000023200
49. Dalla F, Dalla Favera R, RC G. Translocation and Rearrangements of the C-Myc Oncogene Locus in Human Undifferentiated B-Cell Lymphomas. Science (1983) 219(4587):963–7. doi: 10.1126/science.6401867
50. Erikson J, Finger L, Sun L, ar-Rushdi A, Nishikura K, Minowada J, et al. Deregulation of C-Myc by Translocation of the Alpha-Locus of the T-Cell Receptor in T-Cell Leukemias. Science (1986) 232(4752):884–6. doi: 10.1126/science.3486470
51. Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, et al. Myc Stimulates Nuclearly Encoded Mitochondrial Genes and Mitochondrial Biogenesis. Mol Cell Biol (2005) 25(14):6225–34. doi: 10.1128/MCB.25.14.6225-6234.2005
52. Liu Y-C, Li F, Handler J, Ran C, Huang L, Xiang Y, et al. Global Regulation of Nucleotide Biosynthetic Genes by C-Myc. PloS One (2008) 3(7):e2722. doi: 10.1371/journal.pone.0002722
53. Wang Y-H, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, et al. Cell-State-Specific Metabolic Dependency in Hematopoiesis and Leukemogenesis. Cell (2014) 158(6):1309–23. doi: 10.1016/j.cell.2014.07.048
54. Watson AS, Riffelmacher T, Stranks A, Williams O, De Boer J, Cain K, et al. Autophagy Limits Proliferation and Glycolytic Metabolism in Acute Myeloid Leukemia. Cell Death Discov (2015) 1(1):1–10. doi: 10.1038/cddiscovery.2015.8
55. Song K, Li M, Xu X, Xuan L, Huang G, Liu Q. Resistance to Chemotherapy is Associated With Altered Glucose Metabolism in Acute Myeloid Leukemia. Oncol Lett (2016) 12(1):334–42. doi: 10.3892/ol.2016.4600
56. Poulain L, Sujobert P, Chapuis N. High Mtorc1 Activity Drives Glycolysis Addiction and Sensitivity to G6PD Inhibition in Acute Myeloid Leukemia Cells. Leukemia (2017) 31(11):2326–35. doi: 10.1038/leu.2017.81
57. Ju H, Zhan G, Huang A, Sun Y, Wen S, Yang J, et al. ITD Mutation in FLT3 Tyrosine Kinase Promotes Warburg Effect and Renders Therapeutic Sensitivity to Glycolytic Inhibition. Leukemia (2017) 31(10):2143–50. doi: 10.1038/leu.2017.45
58. Larrue C, Saland E, Vergez F, Serhan N, Delabesse E, Mansat-De Mas V, et al. Antileukemic Activity of 2-Deoxy-D-Glucose Through Inhibition of N-Linked Glycosylation in Acute Myeloid Leukemia With FLT3-ITD or C-KIT Mutations. Mol Cancer Ther (2015) 14(10):2364–73. doi: 10.1158/1535-7163.MCT-15-0163
59. Chen Y, Xu Q, Ji D, Wei Y, Chen H, Li T, et al. Inhibition of Pentose Phosphate Pathway Suppresses Acute Myelogenous Leukemia. Tumor Biol (2016) 37(5):6027–34. doi: 10.1007/s13277-015-4428-5
60. Gregory MA, D’Alessandro A, Alvarez-Calderon F, Kim J, Nemkov T, Adane B, et al. ATM/G6PD-Driven Redox Metabolism Promotes FLT3 Inhibitor Resistance in Acute Myeloid Leukemia. Proc Natl Acad Sci (2016) 113(43):E6669–78. doi: 10.1073/pnas.1603876113
61. Bentley J, Walker I, McIntosh E, Whetton AD, Owen-Lynch PJ, Baldwin SA. Glucose Transport Regulation by P210 Bcr–Abl in a Chronic Myeloid Leukaemia Model. Br J Haematol (2001) 112(1):212–5. doi: 10.1046/j.1365-2141.2001.02428.x
62. Hirao T, Yamaguchi M, Kikuya M, Chibana H, Ito K, Aoki S. Altered Intracellular Signaling by Imatinib Increases the Anti-Cancer Effects of Tyrosine Kinase Inhibitors in Chronic Myelogenous Leukemia Cells. Cancer Sci (2018) 109(1):121–31. doi: 10.1111/cas.13442
63. Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell (2011) 20(5):674–88. doi: 10.1016/j.ccr.2011.10.015
64. Boultwood J, Fidler C, Mills KI, Frodsham PM, Kusec R, Gaiger A, et al. Amplification of Mitochondrial DNA in Acute Myeloid Leukaemia. Br J Haematol (1996) 95(2):426–31. doi: 10.1046/j.1365-2141.1996.d01-1922.x
65. Liyanage SU, Hurren R, Voisin V, Bridon G, Wang X, Xu CJ, et al. Leveraging Increased Cytoplasmic Nucleoside Kinase Activity to Target mtDNA and Oxidative Phosphorylation in AML. Blood J Am Soc Hematol (2017) 129(19):2657–66. doi: 10.1182/blood-2016-10-741207
66. Suganuma K, Miwa H, Imai N, Shikami M, Gotou M, Goto M, et al. Energy Metabolism of Leukemia Cells: Glycolysis Versus Oxidative Phosphorylation. Leuk Lymphoma (2010) 51(11):2112–9. doi: 10.3109/10428194.2010.512966
67. Zheng J. Energy Metabolism of Cancer: Glycolysis Versus Oxidative Phosphorylation (Review). Oncol Lett (2012) 4(6):1151–7. doi: 10.3892/ol.2012.928
68. Vakana E, Altman JK, Glaser H, Donato NJ, Platanias LC. Antileukemic Effects of AMPK Activators on BCR-ABL–expressing Cells. Blood J Am Soc Hematol (2011) 118(24):6399–402. doi: 10.1182/blood-2011-01-332783
69. Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, et al. The LKB1/AMPK Signaling Pathway has Tumor Suppressor Activity in Acute Myeloid Leukemia Through the Repression of mTOR-Dependent Oncogenic mRNA Translation. Blood J Am Soc Hematol (2010) 116(20):4262–73. doi: 10.1182/blood-2010-02-269837
70. Pardee TS, Lee K, Luddy J, Maturo C, Rodriguez R, Isom S, et al. A Phase I Study of the First-in-Class Antimitochondrial Metabolism Agent, CPI-613, in Patients With Advanced Hematologic Malignancies. Clin Cancer Res (2014) 20(20):5255–64. doi: 10.1158/1078-0432.CCR-14-1019
71. Pardee TS, Anderson RG, Pladna KM, Isom S, Ghiraldeli LP, Miller LD, et al. A Phase I Study of CPI-613 in Combination With High-Dose Cytarabine and Mitoxantrone for Relapsed or Refractory Acute Myeloid Leukemia. Clin Cancer Res (2018) 24(9):2060–73. doi: 10.1158/1078-0432.CCR-17-2282
72. Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, et al. Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell (2015) 27(6):864–76. doi: 10.1016/j.ccell.2015.05.004
73. Molina JR, Sun Y, Marszalek JR. An Inhibitor of Oxidative Phosphorylation Exploits Cancer Vulnerability. Nat Med (2018) 24(7):1036–46. doi: 10.1038/s41591-018-0052-4
74. Scotland S, Saland E, Skuli N, de Toni F, Boutzen H, Micklow E, et al. Mitochondrial Energetic and AKT Status Mediate Metabolic Effects and Apoptosis of Metformin in Human Leukemic Cells. Leukemia (2013) 27(11):2129–38. doi: 10.1038/leu.2013.107
75. Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, et al. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Therapy-Resistant Chronic Myeloid Leukemia Stem Cells. Nat Med (2017) 23(10):1234–40. doi: 10.1038/nm.4399
76. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-Associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature (2009) 462(7274):739–44. doi: 10.1038/nature08617
77. Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, et al. Acquired Mutations in the Genes Encoding IDH1 and IDH2 Both are Recurrent Aberrations in Acute Myeloid Leukemia: Prevalence and Prognostic Value. Blood J Am Soc Hematol (2010) 116(12):2122–6. doi: 10.1182/blood-2009-11-250878
78. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations is a Neomorphic Enzyme Activity Converting α-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell (2010) 17(3):225–34. doi: 10.1016/j.ccr.2010.01.020
79. Norsworthy KJ, Luo L, Hsu V, Gudi R, Dorff SE, Przepiorka D, et al. FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia With an Isocitrate Dehydrogenase-1 Mutation. Clin Cancer Res (2019) 25(11):3205–9. doi: 10.1158/1078-0432.CCR-18-3749
80. Cerchione C, Romano A, Daver N, DiNardo C, Jabbour EJ, Konopleva M, et al. IDH1/IDH2 Inhibition in Acute Myeloid Leukemia. Front Oncol (2021) 11:345. doi: 10.3389/fonc.2021.639387
81. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood J Am Soc Hematol (2017) 130(6):722–31. doi: 10.1182/blood-2017-04-779405
82. Amatangelo MD, Quek L, Shih A, Stein EM, Roshal M, David MD, et al. Enasidenib Induces Acute Myeloid Leukemia Cell Differentiation to Promote Clinical Response. Blood J Am Soc Hematol (2017) 130(6):732–41. doi: 10.1182/blood-2017-04-779447
83. Yen K, Konteatis Z, Sui Z, Artin E, Dang L, Straley K, et al. Abstract B126: AG-881, a Brain Penetrant, Potent, Pan-Mutant IDH (mIDH) Inhibitor for Use in mIDH Solid and Hematologic Malignancies. J Mol Targets Cancer Ther (2018) 17(1 Supplement):B126. AACR. doi: 10.1158/1535-7163.TARG-17-B126
84. Goto M, Miwa H, Shikami M, Tsunekawa-Imai N, Suganuma K, Mizuno S, et al. Importance of Glutamine Metabolism in Leukemia Cells by Energy Production Through TCA Cycle and by Redox Homeostasis. Cancer Invest (2014) 32(6):241–7. doi: 10.3109/07357907.2014.907419
85. Polet F, Corbet C, Pinto A, Illan LR, Martherus R, Bol V, et al. Reducing the Serine Availability Complements the Inhibition of the Glutamine Metabolism to Block Leukemia Cell Growth. Oncotarget (2016) 7(2):1765. doi: 10.18632/oncotarget.6426
86. Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting Glutaminolysis has Antileukemic Activity in Acute Myeloid Leukemia and Synergizes With BCL-2 Inhibition. Blood J Am Soc Hematol (2015) 126(11):1346–56. doi: 10.1182/blood-2015-01-621870
87. Willems L, Jacque N, Jacquel A, Neveux N, Maciel TT, Lambert M, et al. Inhibiting Glutamine Uptake Represents an Attractive New Strategy for Treating Acute Myeloid Leukemia. Blood J Am Soc Hematol (2013) 122(20):3521–32. doi: 10.1182/blood-2013-03-493163
88. Sontakke P, Koczula KM, Jaques J, Wierenga ATJ, Brouwers-Vos AZ, Pruis M, et al. Hypoxia-Like Signatures Induced by BCR-ABL Potentially Alter the Glutamine Uptake for Maintaining Oxidative Phosphorylation. PLoS One (2016) 11(4):e0153226. doi: 10.1371/journal.pone.0153226
89. Parlati F, Bromley-Dulfano S, Demo S, Janes J, Gross M, Lewis E, et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Hematological Malignances. Am Soc Hematol Wash DC (2013) 122(21):4226. doi: 10.1182/blood.V122.21.4226.4226
90. Gregory MA, Nemkov T, Reisz JA, Zaberezhnyy V, Hansen KC, D'Alessandro A, et al. Glutaminase Inhibition Improves FLT3 Inhibitor Therapy for Acute Myeloid Leukemia. Exp Hematol (2018) 58:52–8. doi: 10.1016/j.exphem.2017.09.007
91. Guerra V, Dinardo CD, Konopleva M, Burger JA, Borthakur G, Jabbour E, et al. Interim Results From a Phase Ib/II Clinical Study of the Glutaminase Inhibitor Telaglenastat (CB-839) in Combination With Azacitidine in Patients With Advanced Myelodysplastic Syndrome (MDS). Am Soc Clin Oncol (2019) 37(15_suppl):7037–7. doi: 10.1200/JCO.2019.37.15_suppl.7037
92. Wheatley DN. Arginine Deprivation and Metabolomics: Important Aspects of Intermediary Metabolism in Relation to the Differential Sensitivity of Normal and Tumour Cells. Semin Cancer Biol (2005) 15:247–53. Elsevier. doi: 10.1016/j.semcancer.2005.04.002
93. Morris SM Jr. Arginine Metabolism: Boundaries of Our Knowledge. J Nutr (2007) 137(6):1602S–9S. doi: 10.1093/jn/137.6.1602S
94. Mussai F, Egan S, Higginbotham-Jones J, Perry T, Beggs A, Odintsova E, et al. Arginine Dependence of Acute Myeloid Leukemia Blast Proliferation: A Novel Therapeutic Target. Blood J Am Soc Hematol (2015) 125(15):2386–96. doi: 10.1182/blood-2014-09-600643
95. Munn DH, Mellor AL. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol (2016) 37(3):193–207. doi: 10.1016/j.it.2016.01.002
96. Mabuchi R, Hara T, Matsumoto T, Shibata Y, Nakamura N, Nakamura H, et al. High Serum Concentration of L-Kynurenine Predicts Unfavorable Outcomes in Patients With Acute Myeloid Leukemia. Leuk Lymphoma (2016) 57(1):92–8. doi: 10.3109/10428194.2015.1041388
97. Fukuno K, Hara T, Tsurumi H, Shibata Y, Mabuchi R, Nakamura N, et al. Expression of Indoleamine 2, 3-Dioxygenase in Leukemic Cells Indicates an Unfavorable Prognosis in Acute Myeloid Leukemia Patients With Intermediate-Risk Cytogenetics. Leuk Lymphoma (2015) 56(5):1398–405. doi: 10.3109/10428194.2014.953150
98. Raffel S, Falcone M, Kneisel N, Hansson J, Wang W, Lutz C, et al. BCAT1 Restricts αkg Levels in AML Stem Cells Leading to IDH Mut-Like DNA Hypermethylation. Nature (2017) 551(7680):384–8. doi: 10.1038/nature24294
99. Hattori A, Tsunoda M, Konuma T, Kobayashi M, Nagy T, Glushka J, et al. Cancer Progression by Reprogrammed BCAA Metabolism in Myeloid Leukaemia. Nature (2017) 545(7655):500–4. doi: 10.1038/nature22314
100. Butler L, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, et al. Lipids and Cancer: Emerging Roles in Pathogenesis, Diagnosis and Therapeutic Intervention. Adv Drug Deliv Rev (2020) 159:245–93. doi: 10.1016/j.addr.2020.07.013
101. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and Lipolysis: The Pathways Exploited by the Cancer Cells to Acquire Fatty Acids. Prog Lipid Res (2013) 52(4):585–9. doi: 10.1016/j.plipres.2013.08.005
102. Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell (2016) 19(1):23–37. doi: 10.1016/j.stem.2016.06.001
103. Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K, et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res (2017) 77(6):1453–64. doi: 10.1158/0008-5472.CAN-16-1645
104. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic Inhibition of Fatty Acid Oxidation Sensitizes Human Leukemia Cells to Apoptosis Induction. J Clin Invest (2010) 120(1):142–56. doi: 10.1172/JCI38942
105. Advani AS, Li H, Michaelis LC, Medeiros BC, Liedtke M, List AF, et al. Report of the Relapsed/Refractory Cohort of SWOG S0919: A Phase 2 Study of Idarubicin and Cytarabine in Combination With Pravastatin for Acute Myelogenous Leukemia (AML). Leuk Res (2018) 67:17–20. doi: 10.1016/j.leukres.2018.01.021
106. Kornblau SM, Banker DE, Stirewalt D, Shen D, Lemker E, Verstovsek S, et al. Blockade of Adaptive Defensive Changes in Cholesterol Uptake and Synthesis in AML by the Addition of Pravastatin to Idarubicin+ High-Dose Ara-C: A Phase 1 Study. Blood (2007) 109(7):2999–3006. doi: 10.1182/blood-2006-08-044446
107. Serhan CN. Pro-Resolving Lipid Mediators are Leads for Resolution Physiology. Nature (2014) 510(7503):92–101. doi: 10.1038/nature13479
108. Chen Y, Hu Y, Zhang H, Peng C, Li S. Loss of the Alox5 Gene Impairs Leukemia Stem Cells and Prevents Chronic Myeloid Leukemia. Nat Genet (2009) 41(7):783–92. doi: 10.1038/ng.389
109. Chen Y, Peng C, Abraham SA, Shan Y, Guo Z, Desouza N, et al. Arachidonate 15-Lipoxygenase is Required for Chronic Myeloid Leukemia Stem Cell Survival. J Clin Invest (2014) 124(9):3847–62. doi: 10.1172/JCI66129
110. Naka K, Ochiai R, Matsubara E, Kondo C, Yang K-M, Hoshii T, et al. The Lysophospholipase D Enzyme Gdpd3 is Required to Maintain Chronic Myelogenous Leukaemia Stem Cells. Nat Commun (2020) 11(1):1–15. doi: 10.1038/s41467-020-18491-9
111. Boag JM, Beesley AH, Firth MJ, Freitas JR, Ford J, Hoffmann K, et al. Altered Glucose Metabolism in Childhood Pre-B Acute Lymphoblastic Leukaemia. Leukemia (2006) 20(10):1731–7. doi: 10.1038/sj.leu.2404365
112. Papajík T, et al. 2-[18F]Fluoro-2-Deoxy-D-Glucose Positron Emission Tomography/Computed Tomography Examination in Patients With Chronic Lymphocytic Leukemia may Reveal Richter Transformation. Leuk Lymphoma (2014) 55(2):314–9. doi: 10.3109/10428194.2013.802313
113. Jerusalem G, Beguin Y, Najjar F, Hustinx R, Fassotte MF, Rigo P, et al. Positron Emission Tomography (PET) With 18F-Fluorodeoxyglucose (18F-FDG) for the Staging of Low-Grade non-Hodgkin's Lymphoma (NHL). Ann Oncol (2001) 12(6):825–30. doi: 10.1023/A:1011169332265
114. Liu T, Kishton RJ, Macintyre AN, Gerriets VA, Xiang H, Liu X, et al. Glucose Transporter 1-Mediated Glucose Uptake is Limiting for B-Cell Acute Lymphoblastic Leukemia Anabolic Metabolism and Resistance to Apoptosis. Cell Death Dis (2014) 5(10):e1470. doi: 10.1038/cddis.2014.431"10.1038/cddis.2014.431
115. Wang YH, Israelsen WJ, Lee D, Yu VWC, Jeanson NT, Clish CB, et al. Cell-State-Specific Metabolic Dependency in Hematopoiesis and Leukemogenesis. Cell (2014) 158(6):1309–23. doi: 10.1016/j.cell.2014.07.048
116. Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab (2016) 23(4):649–62. doi: 10.1016/j.cmet.2016.03.008
117. Moreno C. Chronic Lymphocytic Leukemia and the Warburg Effect. Blood (2015) 125(22):3368–9. doi: 10.1182/blood-2015-04-636332
118. Zhang B, Zhou P, Li X, Shi Q, Li D, Ju X. Bitterness in Sugar: O-GlcNAcylation Aggravates Pre-B Acute Lymphocytic Leukemia Through Glycolysis via the PI3K/Akt/c-Myc Pathway. Am J Cancer Res (2017) 7(6):1337–49.
119. Ferretti A, Lichen LI, Di Vito M. Pentose Phosphate Pathway Alterations in Multi-Drug Resistant Leukemic T-Cells: 31P NMR and Enzymatic Studies. Anticancer Res (1993) 13(4):867–72.
120. Devi GS, Prasad MH, Reddy PP, DN R. Leukocyte Glucose 6 Phosphate Dehydrogenase (G6PD) as Prognostic Indicator in Children With Acute Lymphocytic Leukemia. Indian J Exp Biol (1995) 33(11):829–32.
121. Xiao G, Chan LN, Klemm L, Braas D, Chen Z, Geng H, et al. B-Cell-Specific Diversion of Glucose Carbon Utilization Reveals a Unique Vulnerability in B Cell Malignancies. Cell (2018) 173(2):470–84.e18. doi: 10.1016/j.cell.2018.02.048
122. Jitschin R, Hofmann AD, Bruns H, Gießl A, Bricks J, Berger J, et al. Mitochondrial Metabolism Contributes to Oxidative Stress and Reveals Therapeutic Targets in Chronic Lymphocytic Leukemia. Blood (2014) 123(17):2663–72. doi: 10.1182/blood-2013-10-532200
123. Bruno S, Ledda B, Tenca C, Ravera S, Orengo AM, Mazzarello AN, et al. Metformin Inhibits Cell Cycle Progression of B-Cell Chronic Lymphocytic Leukemia Cells. Oncotarget (2015) 6(26):22624–40. doi: 10.18632/oncotarget.4168
124. Biondani G, Peyron JF. Metformin, an Anti-Diabetic Drug to Target Leukemia. Front Endocrinol (Lausanne) (2018) 9:446. doi: 10.3389/fendo.2018.00446
125. Pan J, Chen C, Jin Y, Fuentes-Mattei E, Velazquez-Tores G, Benito JM, et al. Differential Impact of Structurally Different Anti-Diabetic Drugs on Proliferation and Chemosensitivity of Acute Lymphoblastic Leukemia Cells. Cell Cycle (2012) 11(12):2314–26. doi: 10.4161/cc.20770
126. Yi Y, Gao L, Wu M, Ao J, Zhang C, Wang X, et al. Metformin Sensitizes Leukemia Cells to Vincristine via Activation of AMP-Activated Protein Kinase. J Cancer (2017) 8(13):2636–42. doi: 10.7150/jca.19873
127. Herranz D, Ambesi-Impiombato A, Sudderth J, Sánchez-Martín M, Belver L, Tosello V, et al. Metabolic Reprogramming Induces Resistance to Anti-NOTCH1 Therapies in T Cell Acute Lymphoblastic Leukemia. Nat Med (2015) 21(10):1182–9. doi: 10.1038/nm.3955
128. Ni F, Yu W-M, Li Z, Graham DK, Jin L, Kang S, et al. Critical Role of ASCT2-Mediated Amino Acid Metabolism in Promoting Leukaemia Development and Progression. Nat Metab (2019) 1(3):390–403. doi: 10.1038/s42255-019-0039-6
129. Gregory MA, Nemkov T, Park HJ, Zaberezhnyy V, Gehrke S, Adane B, et al. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin Cancer Res (2019) 25(13):4079–90. doi: 10.1158/1078-0432.CCR-18-3223
130. Chan WK, Horvath TD, Tan L, Link T, Harutyunyan KG, Pontikos MA, et al. Glutaminase Activity of L-Asparaginase Contributes to Durable Preclinical Activity Against Acute Lymphoblastic Leukemia. Mol Cancer Ther (2019) 18(9):1587–92. doi: 10.1158/1535-7163.MCT-18-1329
131. Akagi T, Yin D, Kawamata N, Bartram CR, Hofmann W-K, Wolf I, et al. Methylation Analysis of Asparagine Synthetase Gene in Acute Lymphoblastic Leukemia Cells. Leukemia (2006) 20(7):1303–6. doi: 10.1038/sj.leu.2404216
132. Broome JD. Evidence That the L-Asparaginase of Guinea Pig Serum is Responsible for its Antilymphoma Effects. I. Properties of the L-Asparaginase of Guinea Pig Serum in Relation to Those of the Antilymphoma Substance. J Exp Med (1963) 118(1):99–120. doi: 10.1084/jem.118.1.99”10.1084/jem.118.1.99
133. Clavell LA, Gelber RD, Cohen HJ, Hitchcock-Bryan S, Cassady JR, Tarbell NJ, et al. Four-Agent Induction and Intensive Asparaginase Therapy for Treatment of Childhood Acute Lymphoblastic Leukemia. N Engl J Med (1986) 315(11):657–63. doi: 10.1056/NEJM198609113151101
134. Sallan SE, Hitchcock-Bryan S, Gelber R, Cassady JR, Frei E III, Nathan DG. Influence of Intensive Asparaginase in the Treatment of Childhood non-T-Cell Acute Lymphoblastic Leukemia. Cancer Res (1983) 43(11):5601–7.
135. Hermanova I, Arruabarrena-Aristorena A, Valis K, Nuskova H, Alberich-Jorda M, Fiser K, et al. Pharmacological Inhibition of Fatty-Acid Oxidation Synergistically Enhances the Effect of L-Asparaginase in Childhood ALL Cells. Leukemia (2016) 30(1):209–18. doi: 10.1038/leu.2015.213
136. Hernandez CP, Morrow K, Lopez-Barcons LA, Zabaleta J, Sierra R, Velasco C, et al. Pegylated Arginase I: A Potential Therapeutic Approach in T-ALL. Blood (2010) 115(25):5214–21. doi: 10.1182/blood-2009-12-258822
137. Morrow K, Hernandez CP, Raber P, Del Valle L, Wilk AM, Majumdar S, et al. Anti-Leukemic Mechanisms of Pegylated Arginase I in Acute Lymphoblastic T-Cell Leukemia. Leukemia (2013) 27(3):569–77. doi: 10.1038/leu.2012.247
138. De Santo C, Booth S, Vardon A, Cousins A, Tubb V, Perry T, et al. The Arginine Metabolome in Acute Lymphoblastic Leukemia can be Targeted by the Pegylated-Recombinant Arginase I BCT-100. Int J Cancer (2018) 142(7):1490–502. doi: 10.1002/ijc.31170
139. Ghaeidamini Harouni M, Rahgozar S, Rahimi SB, Safavi A, Ghodousi ES. Fatty Acid Synthase, a Novel Poor Prognostic Factor for Acute Lymphoblastic Leukemia Which can be Targeted by Ginger Extract. Sci Rep (2020) 10(1):14072. doi: 10.1038/s41598-020-70839-9"10.1038/s41598-020-70839-9
140. Cioccoloni G, Aquino A, Notarnicola M, Caruso MG, Bonmassar E, Zonfrillo M, et al. Fatty Acid Synthase Inhibitor Orlistat Impairs Cell Growth and Down-Regulates PD-L1 Expression of a Human T-Cell Leukemia Line. J Chemother (2020) 32(1):30–40. doi: 10.1080/1120009X.2019.1694761
141. Liu PP, Liu J, Jiang W-Q, Carew JS, Ogasawara MA, Pelicano H, et al. Elimination of Chronic Lymphocytic Leukemia Cells in Stromal Microenvironment by Targeting CPT With an Antiangina Drug Perhexiline. Oncogene (2016) 35(43):5663–73. doi: 10.1038/onc.2016.103
142. Heintel D, Kienle D, Shehata M, Kröber A, Kroemer E, Schwarzinger I, et al. High Expression of Lipoprotein Lipase in Poor Risk B-Cell Chronic Lymphocytic Leukemia. Leukemia (2005) 19(7):1216–23. doi: 10.1038/sj.leu.2403748
143. Oppezzo P, Vasconcelos Y, Settegrana C, Jeannel D, Vuillier F, Legarff-Tavernier M, et al. The LPL/ADAM29 Expression Ratio is a Novel Prognosis Indicator in Chronic Lymphocytic Leukemia. Blood (2005) 106(2):650–7. doi: 10.1182/blood-2004-08-3344
144. Rozovski U, Grgurevic S, Bueso-Ramos C, Harris DM, Li P, Liu Z, et al. Aberrant LPL Expression, Driven by STAT3, Mediates Free Fatty Acid Metabolism in CLL Cells. Mol Cancer Res (2015) 13(5):944–53. doi: 10.1158/1541-7786.MCR-14-0412
145. Li P, Harris D, Liu Z, Rozovski U, Ferrajoli A, Wang Y, et al. STAT3-Activated GM-Csfrα Translocates to the Nucleus and Protects CLL Cells From Apoptosis. Mol Cancer Res (2014) 12(9):1267–82. doi: 10.1158/1541-7786.MCR-13-0652-T
146. Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, et al. STAT3 is Constitutively Phosphorylated on Serine 727 Residues, Binds DNA, and Activates Transcription in CLL Cells. Blood (2010) 115(14):2852–63. doi: 10.1182/blood-2009-10-230060
147. Spaner DE, Lee E, Shi Y, Wen F, Li Y, Tung S, et al. PPAR-Alpha is a Therapeutic Target for Chronic Lymphocytic Leukemia. Leukemia (2013) 27(5):1090–9. doi: 10.1038/leu.2012.329
148. Duarte D, Hawkins ED, Lo Celso C. The Interplay of Leukemia Cells and the Bone Marrow Microenvironment. Blood (2018) 131(14):1507–11. doi: 10.1182/blood-2017-12-784132
149. Xu B, Hu R, Liang Z, Chen T, Chen J, Hu Y, et al. Metabolic Regulation of the Bone Marrow Microenvironment in Leukemia. Blood Rev (2021) 48:100786. doi: 10.1016/j.blre.2020.100786
150. Gottfried E, Kunz-Schughart LA, Ebner S, Mueller-Klieser W, Hoves S, Andreesen R, et al. Tumor-Derived Lactic Acid Modulates Dendritic Cell Activation and Antigen Expression. Blood (2006) 107(5):2013–21. doi: 10.1182/blood-2005-05-1795
151. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory Effect of Tumor Cell-Derived Lactic Acid on Human T Cells. Blood (2007) 109(9):3812–9. doi: 10.1182/blood-2006-07-035972
152. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: A Metabolic Key Player in Cancer. Cancer Res (2011) 71(22):6921–5. doi: 10.1158/0008-5472.CAN-11-1457
153. Ecker C, Riley JL. Translating In Vitro T Cell Metabolic Findings to In Vivo Tumor Models of Nutrient Competition. Cell Metab (2018) 28(2):190–5. doi: 10.1016/j.cmet.2018.07.009
154. De Rosa V, Di Rella F, Di Giacomo A, Matarese G. Regulatory T Cells as Suppressors of Anti-Tumor Immunity: Role of Metabolism. Cytokine Growth Factor Rev (2017) 35:15–25. doi: 10.1016/j.cytogfr.2017.04.001
155. Curti A, Trabanelli S, Onofri C, Aluigi M, Salvestrini V, Ocadlikova D, et al. Indoleamine 2,3-Dioxygenase-Expressing Leukemic Dendritic Cells Impair a Leukemia-Specific Immune Response by Inducing Potent T Regulatory Cells. Haematologica (2010) 95(12):2022–30. doi: 10.3324/haematol.2010.025924
156. Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of Tryptophan Catabolism by Human Leukemic Cells Results in the Conversion of CD25- Into CD25+ T Regulatory Cells. Blood (2007) 109(7):2871–7. doi: 10.1182/blood-2006-07-036863
157. Jitschin R, Böttcher M, Saul D, Lukassen S, Bruns H, Loschinski R, et al. Inflammation-Induced Glycolytic Switch Controls Suppressivity of Mesenchymal Stem Cells via STAT1 Glycosylation. Leukemia (2019) 33(7):1783–96. doi: 10.1038/s41375-018-0376-6
158. Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, et al. Acute Myeloid Leukemia Creates an Arginase-Dependent Immunosuppressive Microenvironment. Blood (2013) 122(5):749–58. doi: 10.1182/blood-2013-01-480129
159. Mussai F, Wheat R, Sarrou E, Booth S, Stavrou V, Fultang L, et al. Targeting the Arginine Metabolic Brake Enhances Immunotherapy for Leukaemia. Int J Cancer (2019) 145(8):2201–8. doi: 10.1002/ijc.32028
160. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine Uptake and Metabolism are Coordinately Regulated by ERK/MAPK During T Lymphocyte Activation. J Immunol (2010) 185(2):1037–44. doi: 10.4049/jimmunol.0903586
161. Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the Antitumor Activity of CD8(+) T Cells via Glutamine Restriction. Cancer Sci (2018) 109(12):3737–50. doi: 10.1111/cas.13827
162. Mukherjee A, Chiang C-Y, Daifotis HA, Nieman KM, Fahrmann JF, Lastra RR, et al. Adipocyte-Induced FABP4 Expression in Ovarian Cancer Cells Promotes Metastasis and Mediates Carboplatin Resistance. Cancer Res (2020) 80(8):1748–61. doi: 10.1158/0008-5472.CAN-19-1999
163. Samimi A, Ghanavat M, Shahrabi S, Azizidoost S, Saki N. Role of Bone Marrow Adipocytes in Leukemia and Chemotherapy Challenges. Cell Mol Life Sci (2019) 76(13):2489–97. doi: 10.1007/s00018-019-03031-6
164. Flaherty SE 3rd, Grijalva A, Xu X, Ables E, Nomani A, Ferrante AW Jr. A Lipase-Independent Pathway of Lipid Release and Immune Modulation by Adipocytes. Science (2019) 363(6430):989–93. doi: 10.1126/science.aaw2586
165. Wefers C, Duiveman-de Boer T, Zusterzeel PLM, Massuger LFAG, Fuchs D, Torensma R, et al. Different Lipid Regulation in Ovarian Cancer: Inhibition of the Immune System. Int J Mol Sci (2018) 19(1):273. doi: 10.3390/ijms19010273
166. Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
167. Zhao Q, Kuang D-M, Wu Y, Xiao X, Li X-F, Li T-J, et al. Activated CD69+ T Cells Foster Immune Privilege by Regulating IDO Expression in Tumor-Associated Macrophages. J Immunol (2012) 188(3):1117–24. doi: 10.4049/jimmunol.1100164
168. Li Y, Wan YY, Zhu B. Immune Cell Metabolism in Tumor Microenvironment. Adv Exp Med Biol (2017) 1011:163–96. doi: 10.1007/978-94-024-1170-6_5
169. Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J Immunol (2015) 194(7):2985–91. doi: 10.4049/jimmunol.1403134
170. Zoso A, Mazza EMC, Bicciato S, Mandruzzato S, Bronte V, Serafini P, et al. Human Fibrocytic Myeloid-Derived Suppressor Cells Express IDO and Promote Tolerance via Treg-Cell Expansion. Eur J Immunol (2014) 44(11):3307–19. doi: 10.1002/eji.201444522
171. Zhao Y, Baldin AV, Isayev O, Werner J, Zamyatnin AA Jr., Bazhin AV. Cancer Vaccines: Antigen Selection Strategy. Vaccines (Basel) (2021) 9(2):85. doi: 10.3390/vaccines9020085
172. Rashkovan M, Ferrando A. Metabolic Dependencies and Vulnerabilities in Leukemia. Genes Dev (2019) 33(21-22):1460–74. doi: 10.1101/gad.326470.119
173. Adekola KU, Aydemir SD, Ma S, Zhou Z, Rosen ST, Shanmugam M. Investigating and Targeting Chronic Lymphocytic Leukemia Metabolism With the Human Immunodeficiency Virus Protease Inhibitor Ritonavir and Metformin. Leuk Lymphoma (2015) 56(2):450–9. doi: 10.3109/10428194.2014.922180
174. Ju HQ, Zhan G, Huang A, Sun Y, Wen S, Yang J, et al. ITD Mutation in FLT3 Tyrosine Kinase Promotes Warburg Effect and Renders Therapeutic Sensitivity to Glycolytic Inhibition. Leukemia (2017) 31(10):2143–50. doi: 10.1038/leu.2017.45
175. Poulain L, Sujobert P, Zylbersztejn F, Barreau S, Stuani L, Lambert M, et al. High Mtorc1 Activity Drives Glycolysis Addiction and Sensitivity to G6PD Inhibition in Acute Myeloid Leukemia Cells. Leukemia (2017) 31(11):2326–35. doi: 10.1038/leu.2017.81
176. Li J, Li S, Guo J, Li Q, Long J, Ma C, et al. Natural Product Micheliolide (MCL) Irreversibly Activates Pyruvate Kinase M2 and Suppresses Leukemia. J Med Chem (2018) 61(9):4155–64. doi: 10.1021/acs.jmedchem.8b00241
177. Yu H, Yin Y, Yi Y, Cheng Z, Kuang W, Li R, et al. Targeting Lactate Dehydrogenase A (LDHA) Exerts Antileukemic Effects on T-Cell Acute Lymphoblastic Leukemia. Cancer Commun (Lond) (2020) 40(10):501–17. doi: 10.1002/cac2.12080
178. Saulle E, Spinello I, Quaranta MT, Pasquini L, Pelosi E, Iorio E, et al. Targeting Lactate Metabolism by Inhibiting MCT1 or MCT4 Impairs Leukemic Cell Proliferation, Induces Two Different Related Death-Pathways and Increases Chemotherapeutic Sensitivity of Acute Myeloid Leukemia Cells. Front Oncol (2020) 10:621458. doi: 10.3389/fonc.2020.621458
179. Konopleva MY, Flinn IW, Wang ES, DiNardo CD. Phase 1 Study: Safety and Tolerability of Increasing Doses of Cb-839, an Orally-Administered Small Molecule Inhibitor of Glutaminase. Acute Leuk (2015) 100:378–9.
180. Amylon MD, Shuster J, Pullen J, Berard C, Link MP, Wharam M, et al. Intensive High-Dose Asparaginase Consolidation Improves Survival for Pediatric Patients With T Cell Acute Lymphoblastic Leukemia and Advanced Stage Lymphoblastic Lymphoma: A Pediatric Oncology Group Study. Leukemia (1999) 13(3):335–42. doi: 10.1038/sj.leu.2401310
181. Tsai HJ, Jiang SS, Hung W-C, Borthakur G, Lin S-F, Pemmaraju N, et al. A Phase II Study of Arginine Deiminase (ADI-PEG20) in Relapsed/Refractory or Poor-Risk Acute Myeloid Leukemia Patients. Sci Rep (2017) 7(1):11253. doi: 10.1038/s41598-017-10542-4
182. Tsai HJ, Hsiao H-H, Hsu Y-T, Liu Y-C, Kao H-W, Liu T-C, et al. Phase I Study of ADI-PEG20 Plus Low-Dose Cytarabine for the Treatment of Acute Myeloid Leukemia. Cancer Med (2021) 10(9):2946–55. doi: 10.1002/cam4.3871
183. Emadi A, Duong V, Pantin J, Imran M, Koka R, Singh Z, et al. Indoximod Combined With Standard Induction Chemotherapy Is Well Tolerated and Induces a High Rate of Complete Remission With MRD-Negativity in Patients With Newly Diagnosed AML: Results From a Phase 1 Trial. J Blood (2018) 132:332. doi: 10.1182/blood-2018-99-117433
184. Shinohara H, Kumazaki M, Minami Y, Ito Y, Sugito N, Kuranaga Y, et al. Perturbation of Energy Metabolism by Fatty-Acid Derivative AIC-47 and Imatinib in BCR-ABL-Harboring Leukemic Cells. Cancer Lett (2016) 371(1):1–11. doi: 10.1016/j.canlet.2015.11.020
185. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic Inhibition of Fatty Acid Oxidation Sensitizes Human Leukemia Cells to Apoptosis Induction. J Clin Invest (2010) 120(1):142–56. doi: 10.1172/JCI38942
186. Pallasch CP, Schwamb J, Königs S, Schulz A, Debey S, Kofler D, et al. Targeting Lipid Metabolism by the Lipoprotein Lipase Inhibitor Orlistat Results in Apoptosis of B-Cell Chronic Lymphocytic Leukemia Cells. Leukemia (2008) 22(3):585–92. doi: 10.1038/sj.leu.2405058
187. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood (2017) 130(6):722–31. doi: 10.1182/blood-2017-04-779405
188. Roboz GJ, DiNardo CD, Stein EM, de Botton S, Mims AS, Prince GT, et al. Ivosidenib Induces Deep Durable Remissions in Patients With Newly Diagnosed IDH1-Mutant Acute Myeloid Leukemia. Blood (2020) 135(7):463–71. doi: 10.1182/blood.2019002140
189. Qiu F, Huang J, Sui M. Targeting Arginine Metabolism Pathway to Treat Arginine-Dependent Cancers. Cancer Lett (2015) 364(1):1–7. doi: 10.1016/j.canlet.2015.04.020
190. Mussai F, Egan S, Higginbotham-Jones J, Perry T, Beggs A, Odintsova E, et al. Arginine Dependence of Acute Myeloid Leukemia Blast Proliferation: A Novel Therapeutic Target. Blood (2015) 125(15):2386–96. doi: 10.1182/blood-2014-09-600643
191. Jitschin R, Braun M, Büttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-Cells Induce IDOhi CD14+HLA-DRlo Myeloid-Derived Suppressor Cells That Inhibit T-Cell Responses and Promote TRegs. Blood (2014) 124(5):750–60. doi: 10.1182/blood-2013-12-546416
192. Shedding Light on Fat Dependence in AML. Cancer Discov (2016) 6(12):OF8. doi: 10.1158/2159-8290.CD-NB2016-128
193. Bilban M, Heintel D, Scharl T, Woelfel T, Auer MM, Porpaczy E, et al. Deregulated Expression of Fat and Muscle Genes in B-Cell Chronic Lymphocytic Leukemia With High Lipoprotein Lipase Expression. Leukemia (2006) 20(6):1080–8. doi: 10.1038/sj.leu.2404220
194. Oppezzo P, Vasconcelos Y, Settegrana C, Jeannel D, Vuillier F, Legarff-Tavernier M, et al. The LPL/ADAM29 Expression Ratio is a Novel Prognosis Indicator in Chronic Lymphocytic Leukemia. Blood (2005) 106(2):650–7. doi: 10.1182/blood-2004-08-3344
195. Marcucci G, Maharry K, Wu Y-Z, Radmacher MD, Mrózek K, Margeson D, et al. IDH1 and IDH2 Gene Mutations Identify Novel Molecular Subsets Within De Novo Cytogenetically Normal Acute Myeloid Leukemia: A Cancer and Leukemia Group B Study. J Clin Oncol (2010) 28(14):2348–55. doi: 10.1200/JCO.2009.27.3730
196. Boissel N, Nibourel O, Renneville A, Huchette P, Dombret H, Preudhomme C. Differential Prognosis Impact of IDH2 Mutations in Cytogenetically Normal Acute Myeloid Leukemia. Blood (2011) 117(13):3696–7. doi: 10.1182/blood-2010-11-320937
Keywords: acute lymphocytic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, chronic myeloid leukemia, cellular metabolism, immunometabolism
Citation: Soltani M, Zhao Y, Xia Z, Ganjalikhani Hakemi M and Bazhin AV (2021) The Importance of Cellular Metabolic Pathways in Pathogenesis and Selective Treatments of Hematological Malignancies. Front. Oncol. 11:767026. doi: 10.3389/fonc.2021.767026
Received: 30 August 2021; Accepted: 20 October 2021;
Published: 10 November 2021.
Edited by:
Franco Fais, University of Genoa, ItalyCopyright © 2021 Soltani, Zhao, Xia, Ganjalikhani Hakemi and Bazhin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandr V. Bazhin, YWxleGFuZHIuYmF6aGluQG1lZC51bmktbXVlbmNoZW4uZGU=; Mazdak Ganjalikhani Hakemi, bWdoYWtlbWlAbWVkLm11aS5hYy5pcg==
†These authors have contributed equally to this work