 Yilin Chen
Yilin Chen Jing Zou
Jing Zou Fanjun Cheng
Fanjun Cheng Weiming Li
Weiming Li- Department of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
The therapeutic landscape for chronic myeloid leukemia (CML) has improved significantly with the approval of tyrosine kinase inhibitors (TKIs) for therapeutic use. Most patients with optimal responses to TKIs can have a normal life expectancy. Treatment-free remission (TFR) after discontinuing TKI has increasingly become a new goal for CML treatment. However, TKI only “control“ CML, and relapse after discontinuation has become a key factor hindering patient access to attempt TFR. In this study, we reviewed studies on TKI discontinuation, including both first and second-generation TKI. We also reviewed predictors of relapse, new monitoring methods, and strategies targeting leukemic stem cells.
Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disease characterized by the BCR-ABL fusion gene as a result of t (9;22) (q34;q11) translocation (1). Tyrosine kinase inhibitors (TKIs) have revolutionized the treatment of CML and significantly improved the outcomes for CML patients. Currently, several TKIs are approved for the treatment of CML: imatinib, dasatinib, nilotinib, bosutinib, ponatinib (2–6). Most CML patients who benefit from TKIs have a life expectancy similar to that of the general population (7, 8). However, the adverse events and TKI costs associated with lifelong treatment considerably affect affected patients’ quality of life and dependence on TKI, ultimately affecting treatment response (9–11). An attempt at treatment discontinuation can be considered in some patients to mitigate the adverse events or reduce treatment costs. In addition, female patients of childbearing potential can benefit from stoppi ng TKI to lessen the risk of teratogenic effects (12). Results from multiple trials suggest that patients with a stable deep molecular response (DMR) can safely discontinue TKI without relapse, but close monitoring is recommended (13, 14). Treatment-free remission (TFR) is increasingly becoming a therapeutic goal for patients with DMR (15, 16). However, more studies analyzing predictive factors, new monitoring methods, new strategies targeting stem cells are required to improve current TFR rates. The purpose of this review is to provide updated data on TKI discontinuation studies and analyze predictive factors, monitoring methods, and novel strategies to improve TFR.
Outcomes With First- and Second-Generation TKI Discontinuation
The pioneering TFR trials with imatinib demonstrated the feasibility of TKI discontinuation. With the advent of second-generation TKI discontinuation trials, data regarding TFR are increasingly available. As clinical trials of TFR discontinuation show a favorable TFR rate, many patients in the real world can attempt to stop TKI to achieve TFR. Here, we mainly present the data on discontinuation imatinib, dasatinib, and nilotinib discontinuation in clinical trials and the real world (Tables 1 and 2). In addition, we discuss the feasibility of the second TKI discontinuation attempt after an unsuccessful attempt.
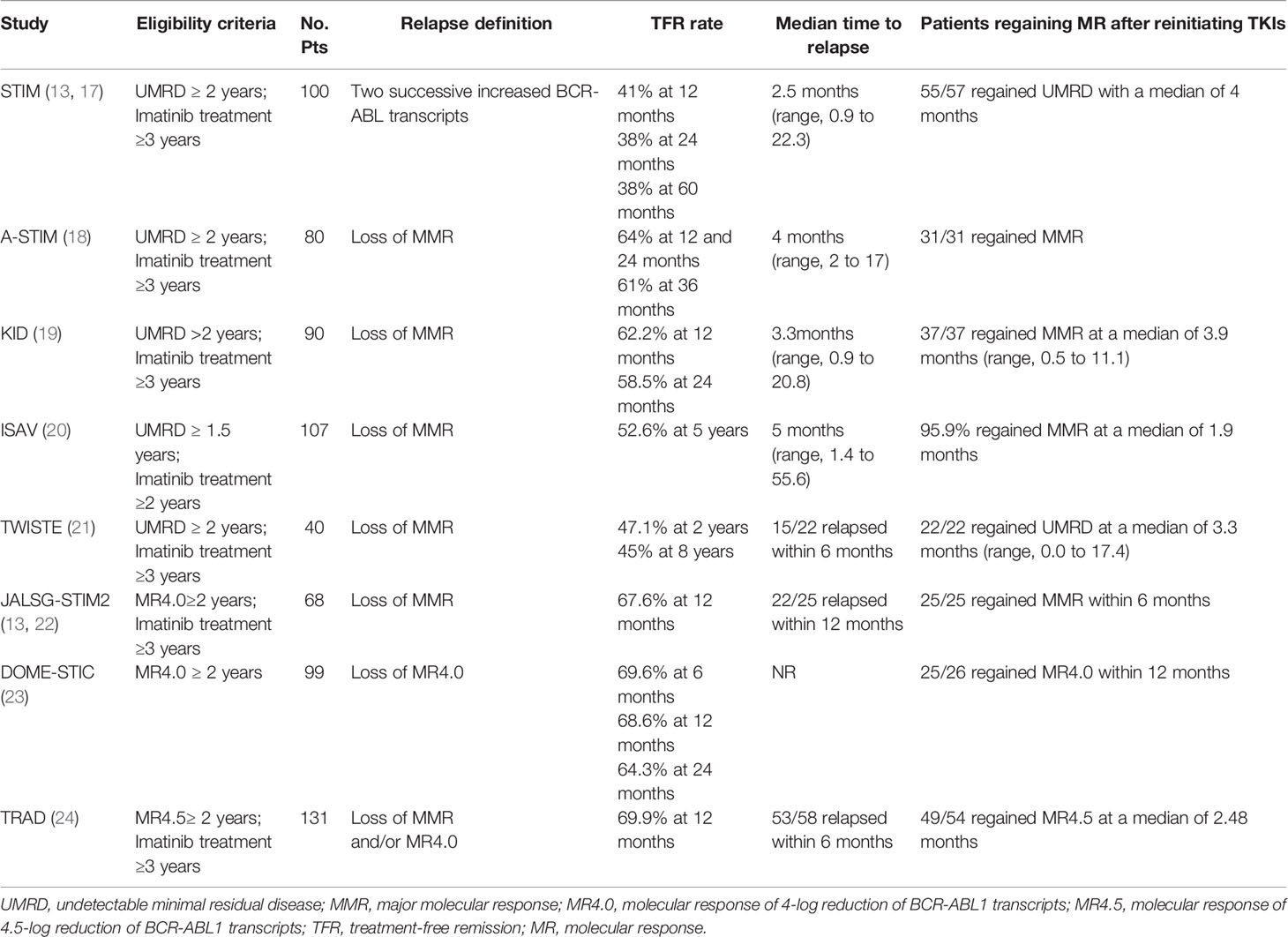
Table 1 Studies of Imatinib Discontinuation.
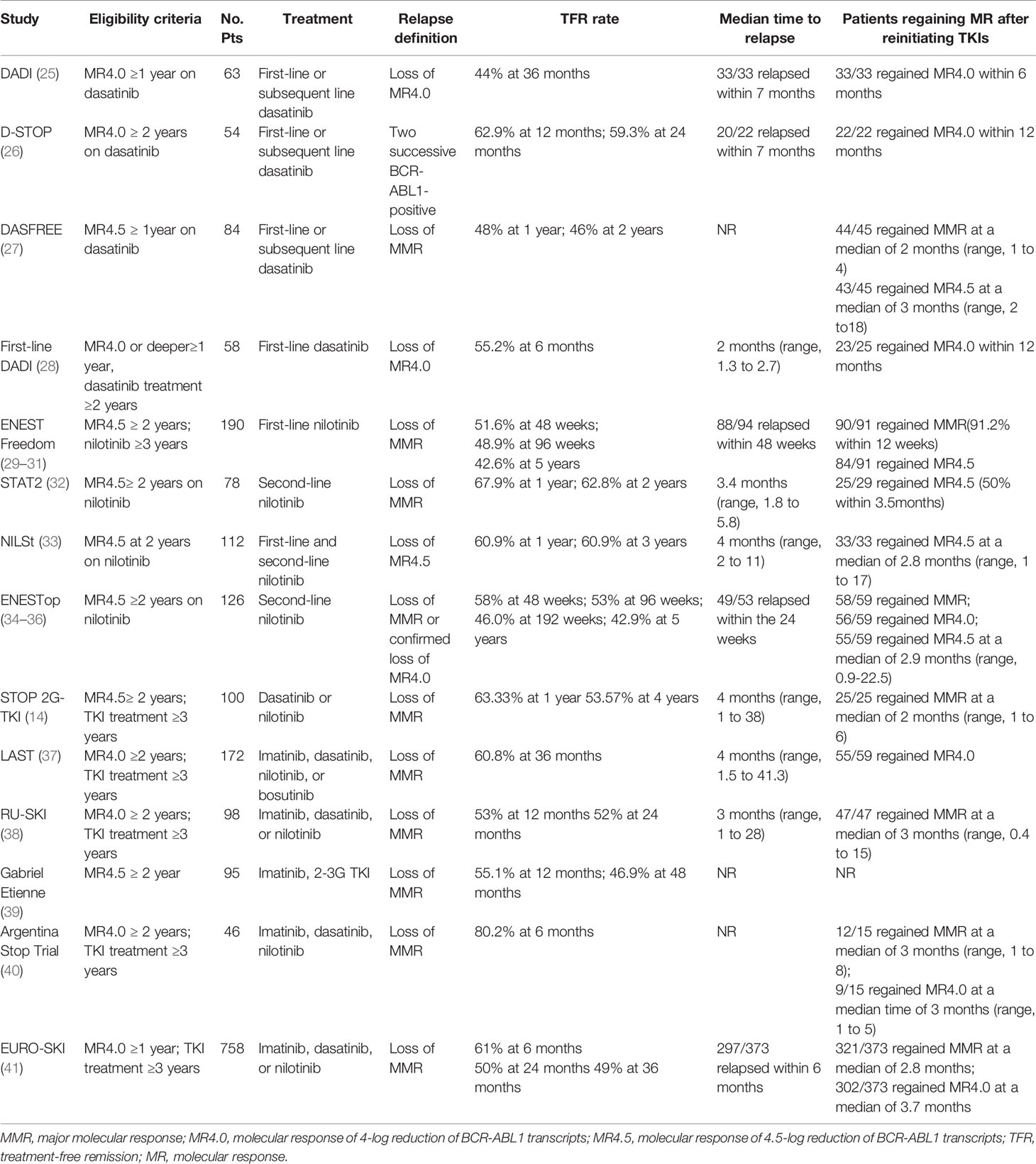
Table 2 Studies of dasatinib and nilotinib discontinuation.
Imatinib Discontinuation
In the prospective, multicenter Stop Imatinib (STIM) trial, 100 patients with undetectable minimal residual disease (UMRD) for more than two years with imatinib were enrolled. Molecular relapse was defined as a significant increase in BCR-ABL transcripts during two consecutive assessments. The estimated molecular relapse-free survival (MRFS) was 41% and 38% at 12 months and 24 months (17). In the Long-Term Follow-Up of the STIM1 study, the MRFS rate was 38% at 60 months. 80% of patients displayed relapsed in the first 6 months. 96.5% (55/57) patients achieved a second UMRD with a median of 4 months of retreatment (13). With longer follow‐up intervals, it became clear that low levels of transcripts were detectable in some patients and did not increase further. In some studies, molecular recurrence was defined as the loss of MMR. The A-STIM study estimated the TFR rates as 64% at 12 and 24 months and 61% at 36 months (18). The KID study reported similar TFR rates of 62.5% and 58.5%, at 12 and 24 months as in the A-STIM study (19). In studies with longer follow-ups, the 5-year MRFS rate in the ISAV study was 47.4%, and the 8-year MRFS rate in the TWISTER study was 45% (20, 21). These results suggest that some patients can achieve a long-term safe and stable response after discontinuation of imatinib. Due to difficulty achieving UMRD in many laboratories, DMR (MR4.0 or MR4.5) for two years has increasingly become an inclusion criterion in subsequent studies, JALSG-STIM213, DOMESTIC, and the TRAD study, which reported similar 12-month TFR rates of 67.6%, 68.6%, and 69.9%, respectively (22–24).
Molecular relapse commonly occurred in the first six months after imatinib withdrawal (13, 18, 24). In the TWISTER study with long-term follow-up, the latest relapse was detected 27 months after stopping imatinib (21). A recent study showed the residual rate of molecular recurrence after two years of discontinuing imatinib was estimated to be 18% (42). Even though some patients with a stable DMR or deeper molecular response can achieve TFR after imatinib discontinuation, long-term molecular follow-up remains mandatory for CML patients in TFR.
Dasatinib Discontinuation
The DADI trial was a prospective multicenter trial investigating the safety and efficacy of discontinuing first-line or subsequent dasatinib (25). 63 patients taking dasatinib and with confirmed stable DMR (MR4.0) for at least one year tried to stop dasatinib. The estimated overall TFR was 44% at 36 months. Most patients experienced molecular relapses within the first seven months after discontinuation. All relapsed patients achieved molecular response after retreatment with dasatinib. The presence of imatinib resistance was a significant risk factor for molecular relapse. In the D-STOP trial, 54 patients with steady DMR for two years had discontinued dasatinib. The estimated treatment-free survival at 12-month and 24 months were 62.9% and 59.3%, respectively (26). Moreover, in the DASFREE study, 84 patients discontinued first and second-line of dasatinib therapy; 48% and 46% of all patients were found with maintained TFR at one and two years. Patients with the first and second-line treatment showed comparable TFR rates at two years, 51%, and 42%, respectively. The TFR rate was the same for patients resistant to prior TKI and intolerant to prior TKI, both at 44% (27). The first-line DADI trial assessed the safety and efficacy of discontinuing first-line dasatinib; 58 patients with DMR for one year discontinued dasatinib after receiving first-line dasatinib for at least 24 months. The TFR rate at 6 months was 55.2% (28). These findings indicate that discontinuation of first-or subsequent-line dasatinib after a sustained DMR was feasible.
Nilotinib Discontinuation
The ENESTfreedom study was the first single-arm, phase 2 clinical trial to evaluate the possibility of stopping first-line nilotinib. 190 patients with at least two years of DMR (MR4.5) of frontline nilotinib therapy were enrolled. Patients restarted nilotinib following the loss of MMR. After stopping nilotinib, 98 (51.6%), and 93 patients (48.9%) remained in MMR at 48 and 96 weeks, respectively (29, 30). At the 5-year data cut-off, 81/190 patients (42.6%) were still in sustained TFR, while 90/91 patients (98.9%) regained MMR after restarting nilotinib treatment (31). Several clinical studies have evaluated the possibility of stopping second-line nilotinib. In the STAT2 trial, 78 patients with stable DMR (at least MR4.5) after 2-year consolidation therapy with nilotinib attempted to discontinue nilotinib. The TFR rates at 12 and 24 months were 67.9% and 62.8%, respectively (32). Similarly, patients who achieved MR4.5 after first-line imatinib or nilotinib therapy were given nilotinib consolidation for up to 24 months in the NILSt trial, and 87 of these patients proceeded to discontinuation of nilotinib. The TFR rate at 1 and 3 years both was 60.9% after nilotinib discontinuation (33). In the ENESTop study, 126 patients with sustained MR4.5 after second-line nilotinib treatment entered the TFR phase. The TFR rates at 48, 96, and 192 weeks were 58%, 53%, and 46.0%, respectively. Most patients requiring nilotinib retreatment rapidly regained MMR, MR4.0, or MR4.5 (34, 35). The long-term follow-up results showed a 5-year TFR rate of 42.9% (36), comparable to the results of the ENESTfreedom study (31). These results illustrate the long-term durability of TFR in patients with first or second-line nilotinib. In addition, the subgroup analysis of the ENESTop study showed that patients switching to nilotinib due to intolerance, resistance, and physician preference had a similar TFR rate at 48 weeks: 30 of 51 (58.8%), 16 of 30 (53.3%), and 27 of 44 (61.4%), respectively (43). Accordingly, first-line resistant or intolerant patients who discontinue second-line TKI still have the chance to achieve TFR.
In the ENSTnd study, with ≥10 years follow-up in newly diagnosed CML patients, the estimated cumulative rates of TFR eligibility [estimated using ENESTfreedom criteria (31)] with nilotinib 300-mg twice-daily, nilotinib 400-mg twice-daily, and imatinib, respectively, at 5 years were 20.9%, 20.6%, and 11.0% and 10 years were 48.6%, 47.3%, and 29.7% (44). The JALSG CML212 study compared the achievement of MR4.5 in newly diagnosed CML patients between nilotinib and dasatinib (45). The MR4.5 rates by 12, 24, and 36 months were 25.6%, 37.4%, and 40.5% in the nilotinib arm and 23.4%, 36.6%, and 44.5% in the dasatinib arm, respectively, with no significant difference, indicating nilotinib and dasatinib were equally effective for CML-CP patients in achieving MR4.5. Among patients reaching TFR eligibility, approximately 50% maintained their responses after TKI discontinuation, and dasatinib, nilotinib TKIs did not increase the overall TFR success rate (Tables 1, 2). Therefore, it can be speculated that only 5% and 10% of newly diagnosed CML patients achieved TFR at 5 years of treatment with imatinib, nilotinib (or dasatinib), compared with 15% and 24% at 10 years of treatment.
Multiple TKIs
The STOP 2G- TKI study estimated the safety of second-generation TKI discontinuation in CML patients receiving dasatinib or nilotinib (14). All patients included were treated for at least three years and had two years of stable MR4.5. TFR rates at 12 and 48 months were 63.33% and 53.57%, respectively. The LAST study presented a TFR rate of 60.8% at 36 months. The median time to molecular recurrence was the same as that in STOP -2G TKI, both 4 months (37). In the RU-SKI study, 98 patients treated with first or second-generation TKI were included. Survival without MMR loss at 12 and 24 months after TKI discontinuation was 53% and 52%, respectively (38). An observational study by Gabriel Etienne et al. exhibited MRFS rates of 51.8% and 43.8% at 12 and 60 months, respectively, similar to the RU-SKI study (39). Furthermore, the Argentina Stop Trial (AST) trial recently included 46 patients also showed a high MRFS at 6 months of 80.2% (40). Evidence from these trials demonstrates that patients with DMR can achieve favorable MRFS. The EURO-SKI study enrolled the largest number of patients (n=758) treated with imatinib, nilotinib, or dasatinib who achieved confirmed DMR for at least one year. The MRFS rate of these patients was 61%, 50%, 49% at 6, 24 months, and 49% at 36 months (41). With 72 months follow-up, 12 out of 111 patients (10.8%) who were in TFR at 36 months, subsequently lost MMR. Interestingly, 1% (1/98) of patients at MR4.0 at 36 months relapsed, yet the risk of relapse for those not at MR4.0 was 85% (11/13), indicating the molecular response at 36 months after TKI discontinuation was highly predictive of molecular relapse (46). The results suggested that the frequency of continued molecular monitoring after three years may depend on the molecular status. The ELN recommendations suggest continuous measurement of BCR-ABL1 levels every 3 months (16). BCR-ABL1 levels should be monitored every 3 months for patients not in MR4.0, while those in MR4.0 may only need an evaluation of BCR-ABL1 levels once or twice a year.
Second Attempt to TKI Discontinuation
An increasing body of evidence suggests that approximately one-half of CML patients with steady DMR can successfully discontinue TKI, and patients with molecular relapse can regain DMR rapidly after restarting TKI (13, 27, 31). Several studies explored the feasibility of the second discontinuation on patients with molecular relapse after the first attempt of TKI discontinuation. In the TRAD study, 32 patients with MR4.5 for 12 months with dasatinib treatment after failed imatinib discontinuation attempted to achieve a second TFR. After dasatinib discontinuation, the estimated TFR rate was 20.4% at 6 months (24). In the KID study, 15/23 patients (62.1%) who attempted second imatinib discontinuation experienced molecular relapse after a median of 2.9 months (47). In the RE-STIM study, 70 patients reattempted TKI discontinuation after a first unsuccessful attempt. The TFR rates at 12, 24, and 36 months were 48%, 42%, and 35%, respectively. Patients who lost MR4.5 later than the median time (>3 months) after the first TKI discontinuation experienced a remarkably lower rate of molecular relapses after the second attempt than others (48). In the enlarged RE-STIM study, 106 patients were enrolled with a median follow-up of 41 months after the second discontinuation. The TFR rates were 44.3%, 38.5%, and 33.2% at 24, 36, and 48 months after the second TKI discontinuation. The speed of molecular relapse after the first TKI discontinuation remained significantly associated with the second TFR (49). Those results showed that some patients who failed the first discontinuation could safely and successfully discontinue TKIs a second time, especially for patients who relapsed later than the median time during a first discontinuation attempt. Further investigations on the predictors that can identify patients for second TKI discontinuation and new strategies to improve the TFR rate are required.
Real‐World TKI Discontinuation
In recent years, several studies have reported the outcome of TKI withdrawal in patients with CML outside clinical trials. A study from a Spanish research group described the outcomes of 236 patients after TKI discontinuation in clinical practice. The TFR rate at 4 years was 64%. TKI treatment duration less than 5 years and MR4.5 duration shorter than 4 years were both associated with a higher incidence of molecular recurrence (50). Another observational study by an Italian research group reported an estimated TFR at 12 months of 69% for patients discontinuing first- and second-line TKI, including 68% for those with imatinib, and 73% for those with second-generation TKI (51). A Swedish group reported that 62.2% of patients discontinuing TKI in clinical practice remained in TFR at the last follow-up (median follow-up time 1.6 years), consistent with the Spanish and Italian studies (52). A single-institution retrospective study also showed that 65% (65/100) patients maintained MR4.5 after a median follow-up of 30 months after discontinuation of TKI. MR4.5 duration for at least six years before discontinuation has been associated with a considerably low risk of loss of MR4.5 (53). A retrospective study assessed first, second, and third attempts to stop TKIs in patients in the real world (54); 28 out of a total of 53 patients (53.4%) achieved sustained TFR after the first attempt; subsequently, 4 of the 10 patients (37.5%) who attempted the second discontinuation successfully achieved TFR. Patients with molecular relapse achieved MMR soon after restarting TKI. All six experienced a loss of MR4.5 after the third attempt to stop TKI. Consistent with the RE-STIM study, loss of MR4.5 at 3 months was an important predictive factor for achievement of second TFR. The above studies suggest that TKI discontinuation in CML is common and feasible outside of clinical trials. Importantly, second successful TKI discontinuation can still be achieved in appropriately selected patients in clinical practice.
De-Escalation TKI Dose – An Alternative Strategy
Several clinical studies have evaluated whether TKI dose reductions can maintain molecular responses. In a study assessing whether high-dose imatinib could be safely reduced to a standard dose without increasing the risk of losing DMR, MMR was maintained in 90% of 68 patients (61/68) with 400 mg imatinib (55). In the NILO-RED study, 67 patients with MMR or deeper molecular response switched from a standard to low dose Nilotinib. The 12-month probability of survival without MMR loss was 97% (56). The non-randomized DESTINY trial assessed the efficacy of a novel approach characterized by halving the dose of a TKI for 12 months and subsequently discontinuing the TKI completely for a further 24 months (57). 174 patients who received TKI for at least 3 years and achieved MMR at least for 12 months were included, of which 125 patients were in the MR4.0 cohort and 49 patients were in the MMR cohort. During the 12 months of half-dose therapy, 2% of patients with MR4.0 and 19% with MMR experienced molecular recurrence. All relapsing patients rapidly regained MMR or better within 4 months of resumption of full-dose TKI. These results indicated the feasibility of lower TKI doses in maintaining responses in patients with stable responses. Fassoni et al. applied a simple mathematical model that describes the time course of TKI response in CML as a dynamic process (58). Their findings showed that dose reduction retained the long-term efficacy in patients who achieved stable molecular remission.
Furthermore, long-term follow-up of the DESTINY trial presented 2-year MRFS rates of 72% and 36% for the MR4.0 and MMR groups after TKI discontinuation (59). Relapse-free survival in the DESTINY trial appeared to be better than in the EURO-SKI study, with 24-month relapse-free rates of 50% (41). Some studies had similar results as the DESTINY trial, with the reduced dose group having a higher TFR rate than the full-dose group. A retrospective study compared the efficacy of TKI discontinuation in patients with reduced and standard doses. Higher TFR rates were observed in the low-dose group at 12 (80% vs.56.8%, P=0.03) and 60 months (58.8% vs. 47.5%, P=0.14) (60). Another recent study evaluated the efficacy and safety of TKI dose reduction in 246 patients who reached MMR due to TKI intolerance in the real world. The 3-year MRFS in MMR and MR4.0 were 94.1% and 87.1%. Interestingly, of the 94 patients in MMR, 54.2% achieved a molecular response level of MR4.0 or higher with a lowered dose. 76 patients discontinued TKI after 1.76 years of low dose treatment, and the 2-year TFR rate in these patients was 74.1% (61). Consistent with the DESTINY trial, a TKI step-down strategy before treatment discontinuation may be a promising strategy to improve TFR.
A possible mechanism underlying the improved TFR and response in the reduced group may be altered immune response against leukemia induced by TKI de-escalation (62). In a recent study, an ordinary differential equation model including an antileukemic immunologic effect was used to assess the predictive impact of different immunological configurations on TKI discontinuation. Dose optimization could be considered for class C patients since they could achieve TFR only if an optimal balance between leukemia abundance and immunologic activation were achieved before treatment cessation (63). In the updated DESTINY study, BCR-ABL1IS values monitored during dose reduction were strongly associated with individual relapse risk after TKI discontinuation. It was recommended that after a 12-month dose reduction period, patients should only stop treatment if they are below MMR and have a negative/low BCR-ABL1 slope; patients with a high slope should return to full-dose TKI therapy because of the high likelihood of relapse (64). The changes in BCR-ABL1 kinetics changes during dose reduction may help clinicians decide whether to stop TKI treatment after the dose reduction period.
Predictive Factors of Molecular Recurrence
Identifying predictive factors of molecular recurrence contributes to the clinical prediction of a successful attempt and the ability to sustain TFR. Here, we discuss the clinical, immunological, and other factors that may help identify patients suitable for TKI discontinuation.
Clinical Indicators
During the Stop Imatinib (STIM1) study, patients with low or intermediate Sokal risk scores and IM duration longer than 54 months experienced a considerably lower rate of molecular recurrence (13). The TKI treatment duration was also associated with a favorable prognosis in other studies (23, 27, 43, 50). The EURO-SKI trial documented that longer DMR durations were associated with an increased probability of TFR (41). The recent TRAD study further reported that one additional year of MR4.0 duration decreased the risk of TFR failure by 14.0% and proposed six years as the shortest imatinib duration (65). Several studies reported the depth of molecular response at the study baseline could predict TFR (22, 25, 32). In this regard, the ENESTfreedom study described patients maintaining MR4.5 at 48 weeks experienced a better TFR rate at 5 years than patients lacking MR4.5 (86.0% for patients in MR4.5 vs. 33.3% for patients in MR4.0 and 20.0% for patients in MMR), suggesting a stable MR4.5 may be a potential predictor of successful TFR (31). In the DADI trial, imatinib resistance was a significant risk factor for molecular relapse (25). Nevertheless, patients switching to nilotinib due to imatinib intolerance and resistance in the ENESTop study interestingly experienced a similar TFR rate at 48 weeks (58.8%vs. 53.3%) (43). Furthermore, in the multiple, TKIs discontinuation study, first-line 2-3G TKIs compared to imatinib were significant predictors of MRFS (39). Imatinib withdrawal syndrome was associated with a higher probability of sustained MMR than patients without withdrawal syndrome (79.5% vs. 49.2%, P=0.003) in the KID study (19). However, no association between withdrawal syndrome development and the rate of molecular relapses was found in the RU-SKI study (38). These results substantiate patients with deeper molecular responses and a longer duration of molecular responses before stopping TKI had higher rates of TFR.
Several studies reported that patients with e14a2 transcripts had a higher probability of maintaining TFR than those with e13a2 transcripts (66, 67). In a recent study, BCR-ABL1 levels at the first month after TKI discontinuation were associated with successful TFR. Moreover, the slopes obtained with the values at baseline at 1 and 2 months were also significantly different between patients with and without sustained MMR (68). In another study, the time required to halve the initial BCR-ABL1 transcripts value was the strongest independent predictor of sustained TFR. Patients with halving times less than 9.35 days had a significantly higher TFR rate than those with halving times greater than 21.85 days (80% vs.4%, P<0.01) (69). Digital PCR (dPCR) is a technique that promises to achieve highly accurate absolute nucleic acid quantification with higher precision and improved daily reproducibility compared to real-time PCR (RT-PCR) (70). In the ISAV study, age and dPCR results were significant predictors of molecular recurrence (20). Those results corroborate the critical importance of the BCR-ABL1 transcript type and the kinetics of BCR-ABL1 decline on long-term outcomes. In addition, BCR-ABL1 levels monitored by dPCR, if available at TKI discontinuation, should be taken into account in selecting patients suitable for discontinuation.
Immunological Indicators
After discontinuation of TKI therapy, relapse depends significantly on an individual’s leukemia-specific immune response (63). Immunological indicators are also emerging as predictive biomarkers of molecular relapse. In the DADI trial, high NK-cell counts, including CD3– CD56+ and CD16+ CD56+ cells, and high counts of NK-cell large granular lymphocytes (CD57+ 56+) were significantly associated with TFR (25). During the three years follow-up of the D-STOP study, CD3- CD56+ NK, CD16+ CD56+ NK, and CD57+ CD56+ NK large granular lymphocyte (NK-LGL), CD8+ CD4- cytotoxic T cell, and CD57+ CD3+ T-LGL cell numbers in patients with TFR were transiently elevated after 12 months but returned to basal levels after 24-month dasatinib consolidation. Silent responses of the T/NK subsets to dasatinib throughout consolidation therapy were significant for maintaining TFR (26). In an immunological study within the EURO-SKI study, the MRFS rate was higher in patients with a higher relative proportion of NK cells than the median, compared with patients with lower NK-cell proportion (73% vs. 51% at 6 months, P=0.02). Moreover, patients with higher than median CD56bright NK cells exhibited a decreased MRFS at 6 months (52% vs 70%, P=0.14) (71). In the IMMUNOSTIM study, the CD56dim NK cell count was an independent prognostic factor of TFR (72). It is widely acknowledged that NK cell functions are under the control of surface inhibitory and activating receptors, such as Natural killer group 2(NKG2) and killer immunoglobulin-like receptors (KIR). Patients homozygous for KIR A haplotype experienced an increased cumulative TFR (73). In another study, the KIR2DL5B-positive genotype was independently related to a delayed second DMR after TKI restart (74). KIR2DS3 was also showed to be more frequent in patients who relapsed after TKI discontinuation (75). HLA polymorphisms have been observed to be associated with TFR (76). Natural killer group 2D receptor (NKG2D) is an activating receptor expressed on NK cells. NKG2D HNK1/HNK1 (high-cytotoxic activity-related allele on NKG2D hb-1) haplotype has been associated with the faster acquisition of MR4.5 (77). NKG2A downregulation by dasatinib enhanced NK cell cytotoxicity and accelerated molecular responses (78). These results demonstrated that NKG2D gene polymorphisms and NKG2A might serve as biomarkers for predicting TFR following dasatinib treatment.
Intriguingly, in the EURO-SKI trial, increased level of CD86 receptor, the ligand of CTLA-4 on plasmacytoid dendritic cells was associated with CD8+ CTLs exhaustion and higher risk of relapse after TKI cessation (79). Patients with CXorf48-specific CTL-negative displayed an increased relapse rate compared to patients with CXorf48-specific CTL-positive (63.6% vs. 0%), indicating CXorf48 could be a promising therapeutic target of Leukemic stem cells (LSCs) to achieve TFR (80). In a recent study, a notable increase in unconventional CD8± T cells expressing TCRγβ+ was observed in patients with TFR (75). Monocytic myeloid-derived suppressor cells (Mo-MDSCs) were concomitantly decreased in patients who achieved TFR (81). Such evidence showed that both immune suppressors and effectors in immunobiology contribute to underlying successful TFR.
Other Indicators
Several studies have explored the effect of genetic factors on the successful discontinuation of TKI. Downregulation of Plasma miR-215 and, microRNA-148b has been associated with successful discontinuation of imatinib (82, 83). The results from whole-exome sequencing revealed variants in genes CYP1B1, ALPK2, and IRF1 in patients with relapse and one variant in gene PARP9 in patients without relapse (84). In a EURO-SKI sub-trial, patients with high transcript levels of the ABCG2 efflux transporter underwent a higher risk of relapse (85).
The New Technology of Molecular Monitoring
New monitoring technology may contribute to the identification of patients who are eligible to discontinue TKI in the future. In recent years, digital PCR (dPCR) has emerged as one of the most promising tools. Some recent findings indicated that dPCR is more efficient than RQ-PCR for monitoring MRD in CML and contributes to selecting patients more compatible with TFR (37, 86–88). In the study by Nicolini et al., low levels of BCR-ABL1 were assessed by dPCR in 175 patients at the time of discontinuation of imatinib. The duration of TKI (≥74.8 months) and dPCR (≥ 0.0023%(IS)) were the two identified predictive factors of molecular recurrence (86). Colafigli, G et al. have reported that dPCR-positive patients presented significantly increased risks of molecular recurrence compared to dPCR-negative patients (50%vs.14%, P=0.026) (87). In another study, patients with a dPCR < 0.468 experienced a significantly higher TFR rate at 2 years than patients with dPCR ≥ 0.468 (83% vs. 52% P = 0.0017). Nevertheless, RT-qPCR was unable to identify patients with a higher risk of relapse after TKI discontinuation (88). In the LAST study, dPCR was administered in patients with undetectable BCR-ABL1 by RQ-PCR. The molecular recurrence rate for patients with detectable BCR-ABL1 by RQ-PCR, for undetectable BCR-ABL1 by RQ-PCR but detectable by dPCR, and for undetectable BCR-ABL1 by both dPCR and RQ-PCR were 50.0%, 64.3%, and 10.3%, respectively (P≤ 0.001). Accordingly, detection of BCR-ABL1 by RQ-PCR or dPCR at the time of TKI discontinuation predicted a higher risk of molecular recurrence (37).
Novel Strategies Targeting LSCs
Although most patients with CML-CP achieve a good response with TKI, approximately half of them can successfully discontinue TKI (13, 34, 89). Disease progression and relapse after TKI discontinuation is a conundrum that remains unresolved. Evidence from several studies demonstrated that TKIs act mainly on highly proliferating leukemic cells but have modest effects on CML stem cells (LSCs), which can survive using kinase-independent mechanisms (90, 91). Single-cell RNA sequencing analysis used to distinguish between Bcr-Abl-positive and negative stem cells showed that malignant stem cell populations with quiescence-related genetic features persisted after treatment (92) (Figure 1). More emphasis should be placed on kinase-independent mechanisms and targeting quiescent, insensitive LSCs to achieve long-term survival in patients. In recent years, many researchers sought to further study LSCs, and many promising novel strategies targeting LSCs have been developed, aiming to eliminate LSCs and improve outcomes of patients (Figure 2).
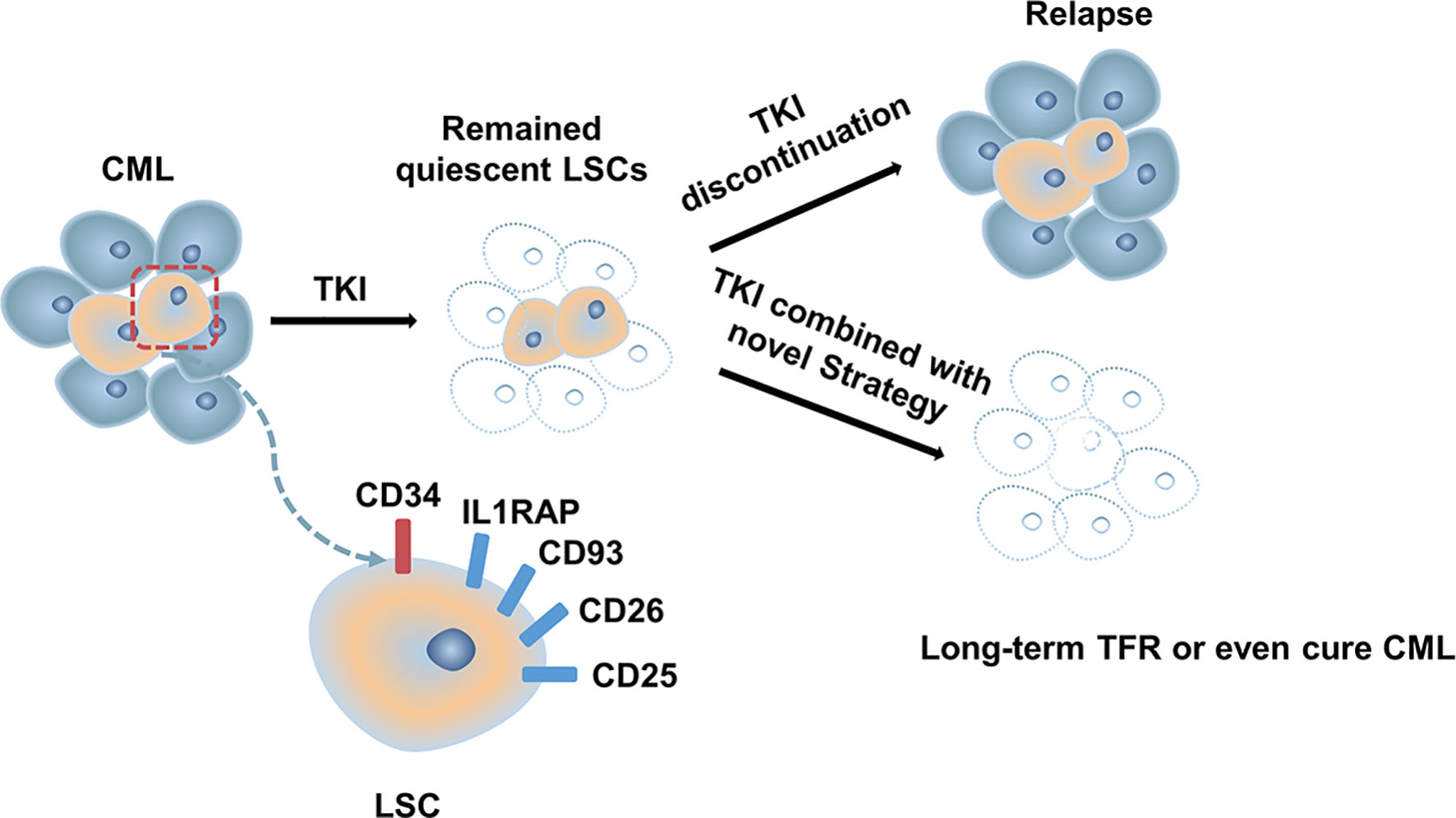
Figure 1 Model for the treatment of CML.
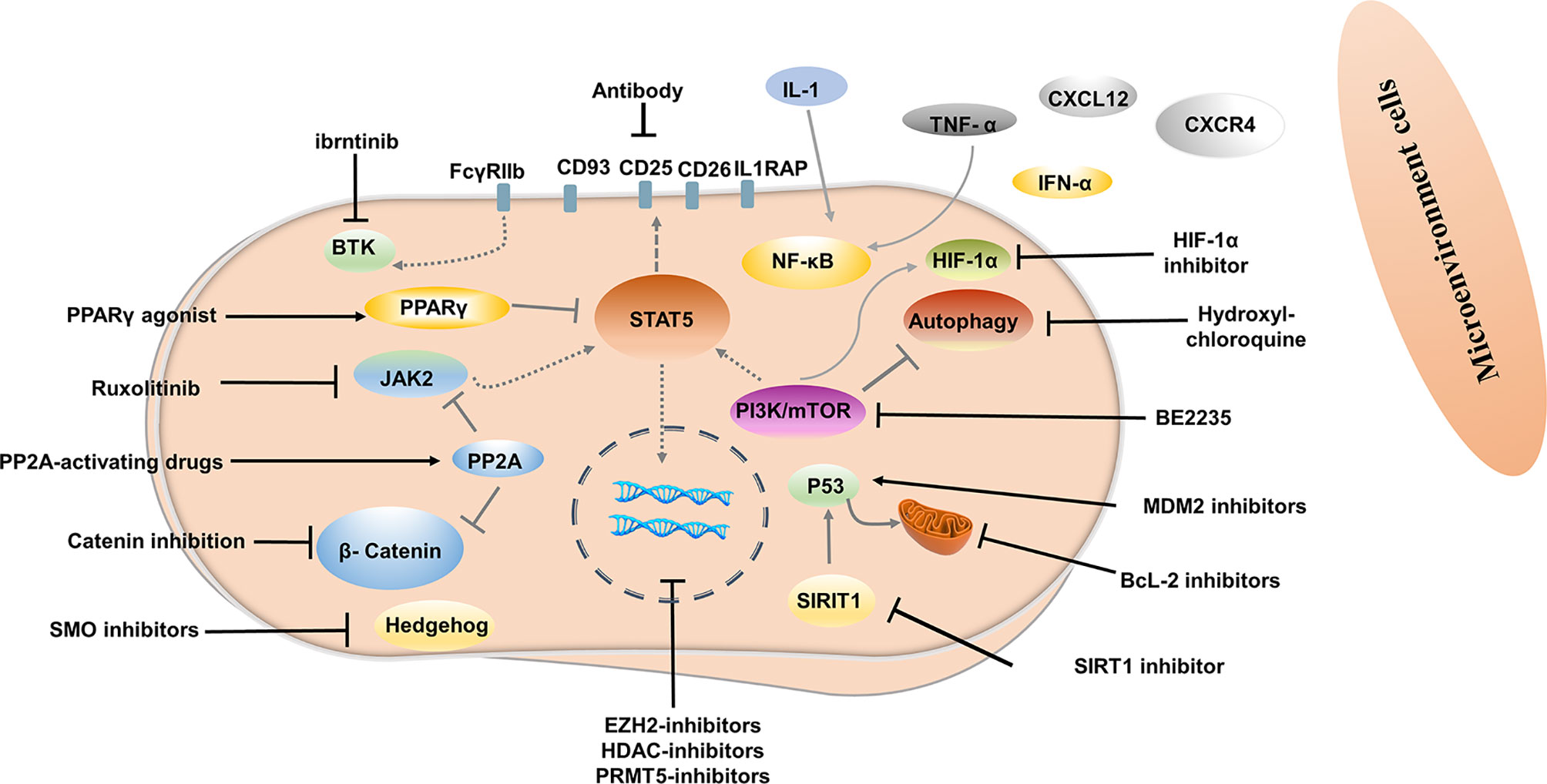
Figure 2 Novel strategies targeting CML leukemia stem cells.
Targeting Bone Marrow Microenvironment
Signals from the bone marrow microenvironment play a crucial role in quiescent LSCs escaping TKI therapy (93). The C-X-C motif chemokine ligand 12 (CXCL12) is a major chemoattractant for the homing process and plays a major role in their localization to regulatory niches of LSCs and normal HSCs (94). Disturbed expression of the C-X-C chemokine receptor type 4 (CXCR4) in CML LSCs or CXCL12 targeting in CML LSCs can affect the homing process. Moreover, up-regulation of CXCR4 by TKI contributed to the migration of LSCs to the bone marrow stroma and promoted the survival of quiescent LSCs (95). CXCR4 inhibitor, plerixafor, in conjunction with TKI has also been shown to override drug resistance (96). CXCR4 antagonist BKT140 combined with TKI was found to overcome the protection of bone marrow stroma and reduce the growth of CML cells both in vitro and in vivo (97). Deletion of CXCL12 from mesenchymal stem cells increased the elimination of LSC by TKI treatment (98). Novel therapy interrupting the CXCR4-CXCL12 axis may weaken the protective role of the bone marrow microenvironment, enhance the sensibility of CML cells to TKI, and even eliminate LSC.
Evidence has shown that autocrine tumor necrosis factor-α (TNF-α) production in LSCs promotes their survival by inducing NFκB/p65 activity, independent of BCR-ABL kinase. TNF-α inhibitor combined with TKI induced significantly higher levels of apoptosis of LSCs compared to either treatment alone, as TNF-α inhibitor showed no off-target inhibition of BCR-ABL kinase activity (99). In another study, TNFα antibody infliximab combined with TKI impaired LSCs growth (100). Moreover, TNF-α signaling was found to mediate expansion and increased expression of CXCL1 in 6C3+ stromal progenitors, and higher expression CXCL1 signaling through CXCR2 enhanced growth capacity and self-renewal of LSCs. Interestingly, CXCR2 inhibitor in combination with TKI remarkably impaired the long-term regenerative capacity of LSCs in vivo, with minimal impact on the total peripheral blood count of normal mice (101). IL-1 signaling has also been found to contribute to the overexpression of inflammatory mediators in CML LSCs, indicating that blocking IL-1 signaling can modulate the inflammatory environment. Combined treatment with IL-1RA for IL-1 blockade significantly enhanced LSC elimination compared with TKI alone, which may be associated with additional inhibition of NF-κB signaling (102). Indeed, before the advent of Imatinib, IFNα was used as first-line therapy and was more active against primitive CML progenitor cells than imatinib treatment, which preferentially targeted more mature, differentiated CML progenitor cells (103). In addition, results from an in vivo study showed that IFN-α activated dormant stem cells, sensitizing them to the killing effects of subsequent therapeutic agents (104), thus supporting IFN-α combined with a TKI may be a promising strategy to improve outcome. The interim analysis results in phase III clinical study of TIGER (CML V) showed that the upfront accession of Peg-IFN to Nilotinib further increased MR4.0 and MR4.5 rates, which may translate into higher TFR rates (105). In addition, the MRFS rate was significantly higher in patients treated with IFN for a longer period before initiating TKI and in patients using IFN after stopping TKI (106, 107).
Targeting LSCs via Molecular Pathways
Targeting key genes required to regulate LSCs survival but not normal hematopoietic stem cells (HSCs) is an important strategy to inhibit LSCs. Several signal pathways are involved in the regulation of survival and proliferation of LSCs, including Wnt/β-catenin, Hedgehog, MAPK/MNK1/2, mTOR, PTEN, PP2A, Alox5, JAK/STAT, SIRT1, and others (108–115). Inhibitors or agonists targeting some of these signal pathways have been developed and investigated. For example, inhibition of β-catenin by C82, deregulation of the hedgehog by smoothened antagonist LDE225, PP2A-activating drugs, JAK2/STAT5 inhibition ruxolitinib has been shown to reduced survival of LSCs (116–119). A Peroxisome Proliferator-Activated Receptor gamma (PPAR-g) agonist, Pioglitazone is currently used as an antidiabetic agent without hypoglycemic effects in healthy humans. Pioglitazone has been reported to alter the quiescent state of LSCs by reducing STAT5 transcription, thus sensitizing them to TKI (120). In another study, LSCs apoptosis triggered by PPARγ agonist rosiglitazone is related to increased expression of Scd1, Pten, and p53 (121). In the ACTIM phase 2 clinical trial, the cumulative incidence of MR4.5 was 56% in patients who yielded MMR but did not achieve MR4.5 after 12 months of pioglitazone combined with imatinib treatment (122). In a recent study, FcγRIIb was demonstrated to be upregulated in primary CML stem cells, with BTK as a major downstream mediator. Targeting the Bcr-Abl-FcγRIIb-BTK axis by ibrutinib combined with TKI remarkably enhanced apoptosis in quiescent LSCs and thus contributed to eradicating LSCs, suggesting that combining TKI therapy along with BTK inhibition could be a potential approach targeting LSCs (123). Hypoxia-inducible factor-1α (HIF-1α) is a key regulator of the cellular and systemic adaptation to low oxygen (124). In murine models of CML, HIF1α has been documented to have a critical role in the survival and proliferation of LSCs. Deletion of HIF-1α inhibited CML proliferation by impeding cell cycle procession and inducing apoptosis in LSCs (125). In vivo and in vitro studies have demonstrated that HIF-1 inhibitors reduce the survival and growth of CML cells and decrease the sustenance of LSCs, but without serious effects on non-CML hematopoietic cells (126).
Targeting LSCs via Bcl-2 and P53 Modulation
The splice variants encoded by BCL2 family genes have pro-and anti-apoptotic functions that contribute to leukemogenesis, CML progression, TKI resistance (127). BCL-2 is overwhelmingly expressed in LSCs and is further increased when patients advance to blast crisis (128). Emerging evidence has shown that combining targeting of BCL-2 by BCL-2/BCL-XL or pan-BCL-2 inhibitors and BCR-ABL tyrosine kinase can enhance the eradication of quiescent LSCs (128–130). Venetoclax, a BCL-2 -selective inhibitor, has shown potent activity in inhibiting the growth of BCL-2-dependent hematological cancers but spares platelets, thus avoiding pronounced thrombocytopenia caused by BCL-XL inhibition (131). Preclinical studies showed significant synergistic effects between venetoclax and TKI on eradicating CD34+CD38−, CD34+CD38+, and quiescent stem/progenitor CD34+ cells (132). In a recent retrospective study, nine CML-BP patients treated with venetoclax in combination with TKIs experienced an overall response rate of 75% and overall survival of 10.9 months (133).
P53 is crucial in tumor suppression, which can activate the pro-apoptotic BCL-2 family BAX, PUMA, NOXA, and BID and antagonize the anti-apoptotic proteins BCL-2 and BCL-XL triggering apoptosis (134, 135). Targeting p53 in combination with TKI is emerging as a potential strategy to eliminate CML LSCs. SIRT1 is, in fact, an important p53 regulator and is overexpressed in LSCs. As mitochondrial respiration is not affected by TKI treatment, in vitro and in vivo deletion of SIRT1 inhibited expression of mitochondrial genes and enhanced sensitivity to TKI (136). Inhibiting of SIRT1 or SIRT1 knockdown also increased apoptosis in LSCs and reduced their growth by activating p53 (137). In recent years, activation of p53 by inhibiting MDM2 (the E3 ligase of p53) in combination with TKI has been investigated. The results showed that TKI in combination with MDM2 inhibitor markedly induces apoptosis of LSCs and enhances the efficacy of TKI by inducing pro-apoptotic and suppressing anti-apoptotic Bcl-2 proteins (138–140).
Targeting Autophagy in LSCs
Autophagy is a lysosomal-mediated, self-degrading process involved in maintaining cellular homeostasis by recycling and decomposing impaired or senescent organelles through the formation of autophagosomes (141, 142). Autophagy may also promote tumor survival by assisting tumor cells to adapt to metabolic stress and evade apoptosis induced by anticancer drugs (143). In CML, autophagy has been documented to be induced in TKI-treated LSCs, which express higher levels than differentiated cells and act as a survival mechanism (144, 145). Several studies have shown that lysosomotropic agent hydroxychloroquine (HCQ) eliminated CML cells and enhanced LSCs to TKI-mediated apoptosis (146, 147). Intriguingly, Lys05, a highly potent lysosomotropic agent, reduced LSCs quiescence and targeted xenografted LSCs combined with TKI treatment (148). Autophagy inhibitors were also among the first compounds to be evaluated in the clinic. The CHOICES study, which explored the safety and efficacy of imatinib (IM) and hydroxychloroquine (HCQ) compared with IM alone, showed MMR rates were 92% and 80% (P=0.21) in patients with IM/HCQ and IM at 12 months, while DMR/MMR rates were75% and 66.7% in patients with IM/HCQ and IM at 24 months (149).
Epigenetic Targeting
Besides the acquisition of genetic lesions, LSCs also exhibit epigenetic dysregulation and reprogramming. Recently, epigenetic therapies have been demonstrated to effectively eliminate LSCs and provide a potential cure for CML in combination with TKI. EZH2, a histone methyltransferase, is a polycomb repressive complex 2 (PRC2) component and is overexpressed in CML LSCs (150). PRC2 is dysregulated in LSCs and coupled with extensive reprogramming of H3K27me3 targets, resulted in altered dependency of the survival in LSCs on EZH2 compared to normal cells (151). In another study, EZH2 inhibitors inhibited colony formation of both human LSC and LSCs with the T315I mutation but spared HSCs. EZH2 deletion also markedly reduced leukemic cells, delayed disease progression, and prolonged survival compared to the control in vivo (152). EZH2 inhibitors in combination with TKI led to significantly increased apoptosis and a significant reduction in colony formation in LSCs compared to TKI treatment alone, even in the undivided “TKI-persistent” cells, which may be associated with the reactivation of pro-apoptotic targets and/or promotion of apoptosis downstream of p53 by overcoming BCL6 and EZH2-mediated inhibition of p53 upstream (151).
Histone deacetylase inhibitors (HDACi), a group of promising anticancer agents, can induce apoptosis in nonproliferating cancer cell lines by modulating gene expression through increased histone lysine acetylation (153). HDACi combined with TKI resulted in significantly increased apoptosis of quiescent CML CD34+ cells highly resistant to TKI, which may be associated with downregulation of HOX-, MYC- and WNT-related genes and the reduced expression of E2F-regulated genes (154). In another study, HDACi JSL-1 combined with TKI enhanced the elimination of LSCs and sensitized LSC cells to TKI through γ-catenin-independent mechanisms (155). Chidamide, a novel selective HDACi, markedly reduced the transcript levels of Bcr-Abl and β-catenin and induced apoptosis in LSC when combined with TKI, but exhibited little toxicity towards normal CD34+ progenitor cells (156). Protein Arginine Methyltransferase (PRMT5) was overexpressed in human CML CD34+ cells. Targeting PRMT5 with the small-molecule inhibitor PJ-68 reduced the survival and renewal capacity of LSCs by suppressing the Wnt/β-catenin pathway and increased levels of negative regulators p15INK4B and p27KIP1 (157). These findings suggest that epigenetics-based therapies may have a potential role in eradicating LSCs.
Targeting LSCs via Surface Markers
Since LSCs and HSCs express similar cell surface markers, additional markers that can distinguish LSCs from HSCs have been investigated, providing an opportunity to prioritize the use of antibodies against LSCs. IL1RAP was identified as a unique cell surface biomarker distinguishing Ph (+) from Ph (-) LSCs by FISH (158). Targeting IL1RAP by antibodies and novel CAR T-cell therapy has been found to exert anti-leukemic effects in vivo and in vitro through specific killing, with no severe negative effects on normal HSC (159, 160). CD26 (DPPV) has also proved to be a novel, specific biomarker for CML LSCs, which is promising for the diagnosis and targeted treatment of CML (161–163). In a recent study, a venetoclax-loaded immunoliposome remarkably induced apoptosis in CD26+ cells in both stem cells and progenitor cells population (164). However, DPPIV blocker vildagliptin in combination with nilotinib did not exert a synergy effect, indicating insignificant effects of co-administration (165). CD25 is a novel STAT5-dependent marker of LSCs, whose expression is upregulated by the PI3K/mTOR blocker BEZ235. In addition, BEZ235 produced synergistic antitumor effects on CML cells when combined with nilotinib or ponatinib (166). Recently, CD93 has also been identified as a novel marker on LSC and persisted in patients with molecular recurrence after TKI discontinuation (167, 168). CD25 and, CD93 are potentially promising targets for LSCs-eradicating immunotherapies. These significant findings support the approach of targeting surface antigens of LSCs for LSCs elimination.
Targeted agents targeting the above molecules or pathways have shown promising efficacy in eliminating LSCs by enhancing the killing effect of TKI on LSCs in vitro and vivo studies. Several promising strategies have also entered clinical trials, and some preliminary results showed that TKIs in combination with IFN-α, JAK2 inhibitors, PPAR-γ agonists, BCL-2 inhibitors, and lysosomotropic agents have the potential to improve treatment response in CML (169, 170).
Conclusions
In clinical studies and the real world, some patients who achieve a stable DMR can successfully discontinue first- or second-generation TKI. However, caution should be taken in MMR patients attempting TFR outside of clinical trials. There remains a possibility for patients who fail a first TFR to discontinue TKI with close monitoring. Patients with a deeper molecular response and longer molecular response duration before stop TKI have decreased risk of molecular relapse. Some immunological indicators such as NK cell counts and NKG2D can also contribute to identifying patients suitable for TKI discontinuation. Developing models that incorporate relevant predictors to predict the likelihood of maintaining TFR is clinically important. Since TKI de-escalation may alter the immune response against leukemia and preserve the long-term efficacy of standard dose TKI therapy while reduce adverse events, the de-escalation approach to TFR may be a promising strategy aimed to improve TFR. New molecular monitoring techniques and novel strategies contributing to the eradication of LSCs are currently under evaluation and are expected to yield preferable outcomes for improving TFR. Importantly, there is hope to expand the TFR population and improve TFR in clinical practice shortly.
Author Contributions
YC, JZ, FC, and WL contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to express their gratitude to Home for Researchers (www.home-for-researchers.com) for the expert linguistic services provided.
References
1. Apperley JF. Chronic Myeloid Leukaemia. Lancet (2015) 385(9976):1447–59. doi: 10.1016/S0140-6736(13)62120-0
2. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N Engl J Med (2017) 376(10):917–27. doi: 10.1056/NEJMoa1609324
3. Cortes JE, Saglio G, Kantarjian HM, Baccarani M, Mayer J, Boque C, et al. Final 5-Year Study Results of DASISION: The Dasatinib Versus Imatinib Study in Treatment-Naive Chronic Myeloid Leukemia Patients Trial. J Clin Oncol (2016) 34(20):2333–40. doi: 10.1200/jco.2015.64.8899
4. Hughes TP, Saglio G, Larson RA, Kantarjian HM, Kim DW, Issaragrisil S, et al. Long-Term Outcomes in Patients With Chronic Myeloid Leukemia in Chronic Phase Receiving Frontline Nilotinib Versus Imatinib: Enestnd 10-Year Analysis. Blood (2019) 134(Supplement_1):2924–24. doi: 10.1182/blood-2019-128761
5. Chuah C, Koh LP, Numbenjapon T, Zang DY, Ong KH, Do YR, et al. Efficacy and Safety of Bosutinib Versus Imatinib for Newly Diagnosed Chronic Myeloid Leukemia in the Asian Subpopulation of the Phase 3 BFORE Trial. Int J Hematol (2021) 114(1):65–78. doi: 10.1007/s12185-021-03144-4
6. Andrews C, Lipton J. The Role of Ponatinib in Chronic Myeloid Leukemia in the Era of Treatment Free Remission. Leukemia Lymphoma (2019) 60(13):3099–101. doi: 10.1080/10428194.2019.1665667
7. Bower H, Bjorkholm M, Dickman PW, Hoglund M, Lambert PC, Andersson TML. Life Expectancy of Patients With Chronic Myeloid Leukemia Approaches the Life Expectancy of the General Population. J Clin Oncol (2016) 34(24):2851–7. doi: 10.1200/Jco.2015.66.2866
8. Hehlmann R, Lauseker M, Saussele S, Pfirrmann M, Krause S, Kolb HJ, et al. Assessment of Imatinib as First-Line Treatment of Chronic Myeloid Leukemia: 10-Year Survival Results of the Randomized CML Study IV and Impact of non-CML Determinants. Leukemia (2017) 31(11):2398–406. doi: 10.1038/leu.2017.253
9. Thanarajasingam G, Minasian LM, Baron F, Cavalli F, De Claro RA, Dueck AC, et al. Beyond Maximum Grade: Modernising the Assessment and Reporting of Adverse Events in Haematological Malignancies. Lancet Haematol (2018) 5(11):E563–98. doi: 10.1016/S2352-3026(18)30051-6
10. Efficace F, Cannella L. The Value of Quality of Life Assessment in Chronic Myeloid Leukemia Patients Receiving Tyrosine Kinase Inhibitors. Hematol-Am Soc Hemat (2016) 2016(1):170–9. doi: 10.1182/asheducation-2016.1.170
11. Dusetzina SB, Winn AN, Abel GA, Huskamp HA, Keating NL. Cost Sharing and Adherence to Tyrosine Kinase Inhibitors for Patients With Chronic Myeloid Leukemia. J Clin Oncol (2014) 32(4):306–11. doi: 10.1200/Jco.2013.52.9123
12. Abruzzese E, Turkina AG, Apperley JF, Bondanini F, de Fabritiis P, Kim DW, et al. Pregnancy Management in CML Patients: To Treat or Not to Treat? Report of 224 Outcomes of the European Leukemia Net (ELN) Database. Blood (2019) 134(Supplement_1):498. doi: 10.1182/blood-2019-124430
13. Etienne G, Guilhot J, Rea D, Rigal-Huguet F, Nicolini F, Charbonnier A, et al. Long-Term Follow-Up of the French Stop Imatinib (STIM1) Study in Patients With Chronic Myeloid Leukemia. J Clin Oncol (2017) 35(3):298–305. doi: 10.1200/Jco.2016.68.2914
14. Rea D, Nicolini FE, Tulliez M, Guilhot F, Guilhot J, Guerci-Bresler A, et al. Discontinuation of Dasatinib or Nilotinib in Chronic Myeloid Leukemia: Interim Analysis of the STOP 2g-TKI Study. Blood (2017) 129(7):846–54. doi: 10.1182/blood-2016-09-742205
15. Shah NP. NCCN Guidelines Updates: Discontinuing TKI Therapy in the Treatment of Chronic Myeloid Leukemia. J Natl Compr Canc Ne (2019) 17(5):611–3. doi: 10.6004/jnccn.2019.5013
16. Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 Recommendations for Treating Chronic Myeloid Leukemia. Leukemia (2020) 34(4):966–84. doi: 10.1038/s41375-020-0776-2
17. Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F, et al. Discontinuation of Imatinib in Patients With Chronic Myeloid Leukaemia Who Have Maintained Complete Molecular Remission for at Least 2 Years: The Prospective, Multicentre Stop Imatinib (STIM) Trial. Lancet Oncol (2010) 11(11):1029–35. doi: 10.1016/S1470-2045(10)70233-3
18. Rousselot P, Charbonnier A, Cony-Makhoul P, Agape P, Nicolini FE, Varet B, et al. Loss of Major Molecular Response As a Trigger for Restarting Tyrosine Kinase Inhibitor Therapy in Patients With Chronic-Phase Chronic Myelogenous Leukemia Who Have Stopped Imatinib After Durable Undetectable Disease. J Clin Oncol (2014) 32(5):424–30. doi: 10.1200/JCO.2012.48.5797
19. Lee SE, Choi SY, Song HY, Kim SH, Choi MY, Park JS, et al. Imatinib Withdrawal Syndrome and Longer Duration of Imatinib Have a Close Association With a Lower Molecular Relapse After Treatment Discontinuation: The KID Study. Haematologica (2016) 101(6):717–23. doi: 10.3324/haematol.2015.139899
20. Diral E, Mori S, Antolini L, Abruzzese E, Le Coutre P, Martino B, et al. Increased Tumor Burden in Patients With Chronic Myeloid Leukemia After 36 Months of Imatinib Discontinuation. Blood (2020) 136(19):2237–40. doi: 10.1182/blood.2019004371
21. Ross DM, Pagani IS, Shanmuganathan N, Kok CH, Seymour JF, Mills AK, et al. Long-Term Treatment-Free Remission of Chronic Myeloid Leukemia With Falling Levels of Residual Leukemic Cells. Leukemia (2018) 32(12):2572–9. doi: 10.1038/s41375-018-0264-0
22. Takahashi N, Tauchi T, Kitamura K, Miyamura K, Saburi Y, Hatta Y, et al. Deeper Molecular Response is a Predictive Factor for Treatment-Free Remission After Imatinib Discontinuation in Patients With Chronic Phase Chronic Myeloid Leukemia: The JALSG-STIM213 Study. Int J Hematol (2018) 107(2):185–93. doi: 10.1007/s12185-017-2334-x
23. Fujisawa S, Ueda Y, Usuki K, Kobayashi H, Kondo E, Doki N, et al. Feasibility of the Imatinib Stop Study in the Japanese Clinical Setting: Delightedly Overcome CML Expert Stop TKI Trial (DOMEST Trial). Int J Clin Oncol (2019) 24(4):445–53. doi: 10.1007/s10147-018-1368-2
24. Kim DDH, Bence-Bruckler I, Busque L, Forrest DL, Savoie L, Keating MM, et al. The Canadian Tyrosine Kinase Inhibitor Discontinuation Trial With Imatinib Discontinuation As a First Attempt and With Dasatinib Discontinuation As a Second Attempt of Treatment-Free Remission: Results of 4 Years of Follow-Up. Blood (2019) 134(Supplement_1):1644. doi: 10.1182/blood-2019-128283
25. Okada M, Imagawa J, Tanaka H, Nakamae H, Hino M, Murai K, et al. Final 3-Year Results of the Dasatinib Discontinuation Trial in Patients With Chronic Myeloid Leukemia Who Received Dasatinib as a Second-Line Treatment. Clin Lymphoma Myeloma Leukemia (2018) 18(5):353–60. doi: 10.1016/j.clml.2018.03.004
26. Kumagai T, Nakaseko C, Nishiwaki K, Yoshida C, Ohashi K, Takezako N, et al. Silent Nk/T Cell Reactions to Dasatinib During Sustained Deep Molecular Response Before Cessation are Associated With Longer Treatment-Free Remission. Cancer Sci (2020) 111(8):2923–34. doi: 10.1111/cas.14518
27. Shah NP, Garcia-Gutierrez V, Jimenez-Velasco A, Larson S, Saussele S, Rea D, et al. Dasatinib Discontinuation in Patients With Chronic-Phase Chronic Myeloid Leukemia and Stable Deep Molecular Response: The DASFREE Study. Leukemia Lymphoma (2020) 61(3):650–9. doi: 10.1080/10428194.2019.1675879
28. Kimura S, Imagawa J, Murai K, Hino M, Kitawaki T, Okada M, et al. Treatment-Free Remission After First-Line Dasatinib Discontinuation in Patients With Chronic Myeloid Leukaemia (First-Line DADI Trial): A Single-Arm, Multicentre, Phase 2 Trial. Lancet Haematol (2020) 7(3):E218–E25. doi: 10.1016/S2352-3026(19)30235-2
29. Hochhaus A, Masszi T, Giles FJ, Radich JP, Ross DM, Casares MTG, et al. Treatment-Free Remission Following Frontline Nilotinib in Patients With Chronic Myeloid Leukemia in Chronic Phase: Results From the ENESTfreedom Study. Leukemia (2017) 31(7):1525–31. doi: 10.1038/leu.2017.63
30. Ross DM, Masszi T, Casares MTG, Hellmann A, Stentoft J, Conneally E, et al. Durable Treatment-Free Remission in Patients With Chronic Myeloid Leukemia in Chronic Phase Following Frontline Nilotinib: 96-Week Update of the ENESTfreedom Study. J Cancer Res Clin (2018) 144(5):945–54. doi: 10.1007/s00432-018-2604-x
31. Radich JP, Hochhaus A, Masszi T, Hellmann A, Stentoft J, Casares MTG, et al. Treatment-Free Remission Following Frontline Nilotinib in Patients With Chronic Phase Chronic Myeloid Leukemia: 5-Year Update of the ENESTfreedom Trial. Leukemia (2021) 35(5):1344–55. doi: 10.1038/s41375-021-01205-5
32. Takahashi N, Nishiwaki K, Nakaseko C, Aotsuka N, Sano K, Ohwada C, et al. Treatment-Free Remission After Two-Year Consolidation Therapy With Nilotinib in Patients With Chronic Myeloid Leukemia: STAT2 Trial in Japan. Haematologica (2018) 103(11):1835–42. doi: 10.3324/haematol.2018.194894
33. Nagafuji K, Matsumura I, Shimose T, Kawaguchi T, Kuroda J, Nakamae H, et al. Cessation of Nilotinib in Patients With Chronic Myelogenous Leukemia Who Have Maintained Deep Molecular Responses for 2 Years: A Multicenter Phase 2 Trial, Stop Nilotinib (NILSt). Int J Hematol (2019) 110(6):675–82. doi: 10.1007/s12185-019-02736-5
34. Mahon FX, Boquimpani C, Kim DW, Benyamini N, Clementino NCD, Shuvaev V, et al. Treatment-Free Remission After Second-Line Nilotinib Treatment in Patients With Chronic Myeloid Leukemia in Chronic Phase. Ann Intern Med (2018) 168(7):461–70. doi: 10.7326/M17-1094
35. Hughes TP, Boquimpani C, Takahashi N, Benyamini N, Clementino NCD, Shuvaev V, et al. ENESTop 192-Week Results: Treatment-Free Remission (TFR) in Patients (Pts) With Chronic Myeloid Leukemia in Chronic Phase (CML-CP) After Stopping Second-Line (2L) Nilotinib (NIL). J Clin Oncol (2019) 37(15):7005. doi: 10.1200/JCO.2019.37.15_suppl.7005
36. Mahon FX, Clementino ND, Fominykh M, Lipton JH, Turkina AG, Moiraghi B, et al. ENESTop 5-Year Update: Durability of Treatment-Free Remission Following Second-Line Nilotinib and Exploratory Analysis of Molecular Response Regain After Nilotinib Re-Initiation in Patients With Chronic Myeloid Leukemia. Blood (2020) 136(Supplement 1):29–30. doi: 10.1182/blood-2020-136250
37. Atallah E, Schiffer CA, Radich JP, Weinfurt KP, Zhang MJ, Pinilla-Ibarz J, et al. Assessment of Outcomes After Stopping Tyrosine Kinase Inhibitors Among Patients With Chronic Myeloid Leukemia A Nonrandomized Clinical Trial. JAMA Oncol (2021) 7(1):42–50. doi: 10.1001/jamaoncol.2020.5774
38. Petrova A, Chelysheva E, Shukhov O, Bykova A, Nemchenko I, Gusarova G, et al. Withdrawal Syndrome After Tyrosine Kinase Inhibitor Discontinuation in Patients With Chronic Myeloid Leukemia in the Russian Prospective Study RU-SKI. Clin Lymphoma Myeloma Leukemia (2020) 20(5):E267–71. doi: 10.1016/j.clml.2019.12.019
39. Etienne G, Dulucq S, Bauduer F, Adiko D, Lifermann F, Dagada C, et al. Incidences of Deep Molecular Responses and Treatment-Free Remission in De Novo CP-CML Patients. Cancers (2020) 12(9):2521. doi: 10.3390/cancers12092521
40. Pavlovsky C, Varela AI, Fernandez II, Sanchez MB, Custidiano MD, Freitas J, et al. High Level of Successful TKI Discontinuation in Chronic Myeloid Leukemia (CML) Patients: Preliminary Results of AST-Argentina Stop Trial. Blood (2020) 136(Supplement 1):3–5. doi: 10.1182/blood-2020-142416
41. Saussele S, Richter J, Guilhot J, Gruber FX, Hjorth-Hansen H, Almeida A, et al. Discontinuation of Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukaemia (EURO-SKI): A Prespecified Interim Analysis of a Prospective, Multicentre, non-Randomised, Trial. Lancet Oncol (2018) 19(6):747–57. doi: 10.1016/S1470-2045(18)30192-X
42. Rousselot P, Loiseau C, Delord M, Cayuela JM, Spentchian M. Late Molecular Recurrences in Patients With Chronic Myeloid Leukemia Experiencing Treatment-Free Remission. Blood Adv (2020) 4(13):3034–40. doi: 10.1182/bloodadvances.2020001772
43. Hughes TP, Boquimpani CM, Takahashi N, Benyamini N, Clementino NCD, Shuvaev V, et al. Treatment-Free Remission in Patients With Chronic Myeloid Leukemia in Chronic Phase According to Reasons for Switching From Imatinib to Nilotinib: Subgroup Analysis From ENESTop. Blood (2016) 128(22):792. doi: 10.1182/blood.V128.22.792.792
44. Kantarjian HM, Hughes TP, Larson RA, Kim DW, Issaragrisil S, le Coutre P, et al. Long-Term Outcomes With Frontline Nilotinib Versus Imatinib in Newly Diagnosed Chronic Myeloid Leukemia in Chronic Phase: ENESTnd 10-Year Analysis. Leukemia (2021) 35(2):440–53. doi: 10.1038/s41375-020-01111-2
45. Matsumura I, Ohtake S, Atsuta Y, Kurata M, Minami Y, Takahashi N, et al. Nilotinib Vs. Dasatinib in Achieving MR4.5 for Newly Diagnosed Chronic Myeloid Leukemia: Results of the Prospective Randomized Phase 3 Study, JALSG Cml212. Blood (2020) 136(Supplement 1):40–1. doi: 10.1182/blood-2020-134168
46. Richter J, Lubking A, Soderlund S, Lotfi K, Markevarn B, Sjalander A, et al. Molecular Status 36 Months After TKI Discontinuation in CML is Highly Predictive for Subsequent Loss of MMR-Final Report From AFTER-SKI. Leukemia (2021) 35(8):2416–8. doi: 10.1038/s41375-021-01173-w
47. Lee SE, Park JS, Do YR, Kim SH, Zang DY, Oh S, et al. Second Imatinib Discontinuation Outcomes in Patients Regaining Durable Deep Molecular Response in the Korean Imatinib Discontinuation Study; KID Study. Blood (2020) 136(Supplement 1):51–2. doi: 10.1182/blood-2020-139253
48. Legros L, Nicolini FE, Etienne G, Rousselot P, Rea D, Giraudier S, et al. Second Tyrosine Kinase Inhibitor Discontinuation Attempt in Patients With Chronic Myeloid Leukemia. Cancer (2017) 123(22):4403–10. doi: 10.1002/cncr.30885
49. Legros L, Nicolini FE, Etienne G, Rousselot P, Rea D, Giraudier S, et al. The TKI-Free Duration After a First Discontinuation Attempt That Failed in CP CML Patients Is a Predictive Factor of TKI-Free Remission After a Second Attempt. Blood (2019) 134(Supplement_1):28. doi: 10.1182/blood-2019-123719
50. Hernandez-Boluda JC, Pereira A, Pastor-Galan I, Alvarez-Larran A, Savchuk A, Puerta JM, et al. Feasibility of Treatment Discontinuation in Chronic Myeloid Leukemia in Clinical Practice: Results From a Nationwide Series of 236 Patients. Blood Cancer J (2018) 8(10):91. doi: 10.1038/s41408-018-0125-0
51. Fava C, Rege-Cambrin G, Dogliotti I, Cerrano M, Berchialla P, Dragani M, et al. Observational Study of Chronic Myeloid Leukemia Italian Patients Who Discontinued Tyrosine Kinase Inhibitors in Clinical Practice. Haematologica (2019) 104(8):1589–96. doi: 10.3324/haematol.2018.205054
52. Flygt H, Sandin F, Dahlen T, Dremaine A, Lubking A, Markevarn B, et al. Successful Tyrosine Kinase Inhibitor Discontinuation Outside Clinical Trials - Data From the Population-Based Swedish Chronic Myeloid Leukaemia Registry. Br J Haematol (2021) 193(5):915–21. doi: 10.1111/bjh.17392
53. Chamoun K, Kantarjian H, Atallah R, Gonzalez GN, Issa GC, Rios MB, et al. Tyrosine Kinase Inhibitor Discontinuation in Patients With Chronic Myeloid Leukemia: A Single-Institution Experience. J Hematol Oncol (2019) 12(1):1. doi: 10.1186/s13045-018-0686-1
54. Ureshino H, Kamachi K, Nishioka A, Okamoto S, Katsuya H, Yoshimura M, et al. Subsequent Attempt Tyrosine Kinase Inhibitor Discontinuation in Patients With Chronic Myeloid Leukemia; a Single Institute Experience. Hematol Oncol (2021) 39(4):549–57. doi: 10.1002/hon.2896
55. Michel C, Burchert A, Hochhaus A, Saussele S, Neubauer A, Lauseker M, et al. Imatinib Dose Reduction in Major Molecular Response of Chronic Myeloid Leukemia: Results From the German Chronic Myeloid Leukemia-Study IV. Haematologica (2019) 104(5):955–62. doi: 10.3324/haematol.2018.206797
56. Rea D, Cayuela JM, Dulucq S, Etienne G. Molecular Responses After Switching From a Standard-Dose Twice-Daily Nilotinib Regimen to a Reduced-Dose Once-Daily Schedule in Patients With Chronic Myeloid Leukemia: A Real Life Observational Study (NILO-RED). Blood (2017) 130(Supplement 1):318. doi: 10.1182/blood.V130.Suppl_1.318.318
57. Clark RE, Polydoros F, Apperley JF, Milojkovic D, Pocock C, Smith G, et al. De-Escalation of Tyrosine Kinase Inhibitor Dose in Patients With Chronic Myeloid Leukaemia With Stable Major Molecular Response (DESTINY): An Interim Analysis of a non-Randomised, Phase 2 Trial. Lancet Haematol (2017) 4(7):E310–6. doi: 10.1016/S2352-3026(17)30066-2
58. Fassoni AC, Baldow C, Roeder I, Glauche I. Reduced Tyrosine Kinase Inhibitor Dose is Predicted to be as Effective as Standard Dose in Chronic Myeloid Leukemia: A Simulation Study Based on Phase III Trial Data. Haematologica (2018) 103(11):1825–34. doi: 10.3324/haematol.2018.194522
59. Clark RE, Polydoros F, Apperley JF, Milojkovic D, Rothwell K, Pocock C, et al. De-Escalation of Tyrosine Kinase Inhibitor Therapy Before Complete Treatment Discontinuation in Patients With Chronic Myeloid Leukaemia (DESTINY): A non-Randomised, Phase 2 Trial. Lancet Haematol (2019) 6(7):E375–83. doi: 10.1016/S2352-3026(19)30094-8
60. Cayssials E, Torregrosa-Diaz J, Gallego-Hernanz P, Tartarin F, Systchenko T, Maillard N, et al. Low-Dose Tyrosine Kinase Inhibitors Before Treatment Discontinuation do Not Impair Treatment-Free Remission in Chronic Myeloid Leukemia Patients: Results of a Retrospective Study. Cancer (2020) 126(15):3438–47. doi: 10.1002/cncr.32940
61. Claudiani S, Apperley JF, Szydlo R, Khan A, Nesr G, Hayden C, et al. TKI Dose Reduction can Effectively Maintain Major Molecular Remission in Patients With Chronic Myeloid Leukaemia. Br J Haematology (2021) 193(2):346–55. doi: 10.1111/bjh.17286
62. Austin GM, Knight K, Bell J, Carter A, Heartin E, Watson D, et al. The Effect on Lymphocyte Subsets of Decreasing/Stopping Tyrosine Kinase Inhibitor Therapy in Chronic Myeloid Leukaemia: Data From the DESTINY Trial. Br J Haematology (2019) 185(4):791–3. doi: 10.1111/bjh.15629
63. Hahnel T, Baldow C, Guilhot J, Guilhot F, Saussele S, Mustjoki S, et al. Model-Based Inference and Classification of Immunologic Control Mechanisms From TKI Cessation and Dose Reduction in Patients With CML. Cancer Res (2020) 80(11):2394–406. doi: 10.1158/0008-5472.Can-19-2175
64. Gottschalk A, Glauche I, Cicconi S, Clark RE, Roeder I. Molecular Monitoring During Dose Reduction Predicts Recurrence After TKI Cessation in CML. Blood (2020) 135(10):766–9. doi: 10.1182/blood.2019003395
65. Kim DDH, Novitzky-Basso I, Kim TS, Atenafu EG, Forrest D, Savoie L, et al. Optimal Duration of Imatinib Treatment/Deep Molecular Response for Treatment-Free Remission After Imatinib Discontinuation From a Canadian Tyrosine Kinase Inhibitor Discontinuation Trial. Br J Haematology (2021) 193(4):779–91. doi: 10.1111/bjh.17447
66. Claudiani S, Apperley JF, Gale RP, Clark R, Szydlo R, Deplano S, et al. E14a2 BCR-ABL1 Transcript is Associated With a Higher Rate of Treatment-Free Remission in Individuals With Chronic Myeloid Leukemia After Stopping Tyrosine Kinase Inhibitor Therapy. Haematologica (2017) 102(8):E297–E9. doi: 10.3324/haematol.2017.168740
67. D’Adda M, Farina M, Schieppati F, Borlenghi E, Bottelli C, Cerqui E, et al. The E13a2 BCR-ABL Transcript Negatively Affects Sustained Deep Molecular Response and the Achievement of Treatment-Free Remission in Patients With Chronic Myeloid Leukemia Who Receive Tyrosine Kinase Inhibitors. Cancer (2019) 125(10):1674–82. doi: 10.1002/cncr.31977
68. Di Giusto S, Toffoletti E, Bonifacio M, Binotto G, Miggiano MC, Calistri E, et al. BCR-ABL1 Levels at First Month After TKI Discontinuation Predict Subsequent Maintenance of Treatment-Free Remission: A Study From the "Gruppo Triveneto LMC". Blood (2020) 136(Supplement 1):9–10. doi: 10.1182/blood-2020-135968
69. Shanmuganathan N, Pagani IS, Ross DM, Park S, Yong ASM, Braley JA, et al. Early BCR-ABL1 Kinetics are Predictive of Subsequent Achievement of Treatment-Free Remission in Chronic Myeloid Leukemia. Blood (2021) 137(9):1196–207. doi: 10.1182/blood.2020005514
70. Hindson CM, Chevillet JR, Briggs HA, Gallichotte EN, Ruf IK, Hindson BJ, et al. Absolute Quantification by Droplet Digital PCR Versus Analog Real-Time PCR. Nat Methods (2013) 10(10):1003–5. doi: 10.1038/nmeth.2633
71. Ilander M, Olsson-Stromberg U, Schlums H, Guilhot J, Bruck O, Lahteenmaki H, et al. Increased Proportion of Mature NK Cells is Associated With Successful Imatinib Discontinuation in Chronic Myeloid Leukemia. Leukemia (2017) 31(5):1108–16. doi: 10.1038/leu.2016.360
72. Rea D, Henry G, Khaznadar Z, Etienne G, Guilhot F, Nicolini F, et al. Natural Killer-Cell Counts are Associated With Molecular Relapse-Free Survival After Imatinib Discontinuation in Chronic Myeloid Leukemia: The IMMUNOSTIM Study. Haematologica (2017) 102(8):1368–77. doi: 10.3324/haematol.2017.165001
73. Caocci G, Greco M, Littera R, Lai S, Galimberti S, Barate C, et al. Killer Immunoglobulin-Like Receptors Can Predict Tki Treatment-Free Remission in Chronic Myeloid Leukemia Patients. Haematologica (2015) 100:234–5. doi: 10.1016/j.exphem.2015.08.004
74. Dumas PY, Berard E, Breal C, Dulucq S, Rea D, Nicolini F, et al. Killer Immunoglobulin-Like Receptor Genotypes and Chronic Myeloid Leukemia Outcomes After Imatinib Cessation for Treatment-Free Remission. Cancer Med (2019) 8(11):4976–85. doi: 10.1002/cam4.2371
75. Vigon L, Luna A, Galan M, Rodriguez-Mora S, Fuertes D, Mateos E, et al. Identification of Immunological Parameters as Predictive Biomarkers of Relapse in Patients With Chronic Myeloid Leukemia on Treatment-Free Remission. J Clin Med (2021) 10(1):42. doi: 10.3390/jcm10010042
76. Ureshino H, Shindo T, Tanaka H, Saji H, Kimura S. HLA Polymorphisms Are Associated With Treatment-Free Remission Following Discontinuation of Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia. Mol Cancer Ther (2021) 20(1):142–9. doi: 10.1158/1535-7163.Mct-20-0336
77. Hara R, Onizuka M, Matsusita E, Kikkawa E, Nakamura Y, Matsushita H, et al. NKG2D Gene Polymorphisms are Associated With Disease Control of Chronic Myeloid Leukemia by Dasatinib. Int J Hematol (2017) 106(5):666–74. doi: 10.1007/s12185-017-2294-1
78. Chang MC, Cheng HI, Hsu K, Hsu YN, Kao CW, Chang YF, et al. NKG2A Down-Regulation by Dasatinib Enhances Natural Killer Cytotoxicity and Accelerates Effective Treatment Responses in Patients With Chronic Myeloid Leukemia. Front Immunol (2019) 9:3152. doi: 10.3389/fimmu.2018.03152
79. Schutz C, Inselmann S, Sausslele S, Dietz CT, Muller MC, Eigendorff E, et al. Expression of the CTLA-4 Ligand CD86 on Plasmacytoid Dendritic Cells (pDC) Predicts Risk of Disease Recurrence After Treatment Discontinuation in CML. Leukemia (2017) 31(4):829–36. doi: 10.1038/leu.2017.9
80. Matsushita M, Ozawa K, Suzuki T, Nakamura M, Nakano N, Kanchi S, et al. CXorf48 is a Potential Therapeutic Target for Achieving Treatment-Free Remission in CML Patients. Blood Cancer J (2017) 7(9):e601. doi: 10.1038/bcj.2017.84
81. Irani YD, Hughes A, Clarson J, Kok CH, Shanmuganathan N, White DL, et al. Successful Treatment-Free Remission in Chronic Myeloid Leukaemia and its Association With Reduced Immune Suppressors and Increased Natural Killer Cells. Br J Haematology (2020) 191(3):433–41. doi: 10.1111/bjh.16718
82. Ohyashiki JH, Ohtsuki K, Mizoguchi I, Yoshimoto T, Katagiri S, Umezu T, et al. Downregulated microRNA-148b in Circulating PBMCs in Chronic Myeloid Leukemia Patients With Undetectable Minimal Residual Disease: A Possible Biomarker to Discontinue Imatinib Safely. Drug Des Dev Ther (2014) 8:1151–9. doi: 10.2147/Dddt.S66812
83. Ohyashiki K, Umezu T, Katagiri S, Kobayashi C, Azuma K, Tauchi T, et al. Downregulation of Plasma miR-215 in Chronic Myeloid Leukemia Patients With Successful Discontinuation of Imatinib. Int J Mol Sci (2016) 17(4):570. doi: 10.3390/ijms17040570
84. Smirnikhina SA, Lavrov AV, Chelysheva EY, Adilgereeva EP, Shukhov OA, Turkina A, et al. Whole-Exome Sequencing Reveals Potential Molecular Predictors of Relapse After Discontinuation of the Targeted Therapy in Chronic Myeloid Leukemia Patients. Leukemia Lymphoma (2016) 57(7):1669–76. doi: 10.3109/10428194.2015.1132420
85. Rinaldetti S, Pfirrmann M, Manz K, Guilhot J, Dietz C, Panagiotidis P, et al. Effect of ABCG2, OCT1, and ABCB1 (MDR1) Gene Expression on Treatment-Free Remission in a EURO-SKI Subtrial. Clin Lymphoma Myeloma Leukemia (2018) 18(4):266–71. doi: 10.1016/j.clml.2018.02.004
86. Nicolini FE, Dulucq S, Boureau L, Cony-Makhoul P, Charbonnier A, Escoffre-Barbe M, et al. Evaluation of Residual Disease and TKI Duration Are Critical Predictive Factors for Molecular Recurrence After Stopping Imatinib First-Line in Chronic Phase CML Patients. Clin Cancer Res (2019) 25(22):6606–13. doi: 10.1158/1078-0432.Ccr-18-3373
87. Colafigli G, Scalzulli E, Porrazzo M, Diverio D, Loglisci MG, Latagliata R, et al. Digital Droplet PCR at the Time of TKI Discontinuation in Chronic-Phase Chronic Myeloid Leukemia Patients is Predictive of Treatment-Free Remission Outcome. Hematol Oncol (2019) 37(5):652–4. doi: 10.1002/hon.2650
88. Bernardi S, Malagola M, Zanaglio C, Polverelli N, Eke ED, DAdda M, et al. Digital PCR Improves the Quantitation of DMR and the Selection of CML Candidates to TKIs Discontinuation. Cancer Med (2019) 8(5):2041–55. doi: 10.1002/cam4.2087
89. Imagawa J, Tanaka H, Okada M, Nakamae H, Hino M, Murai K, et al. Discontinuation of Dasatinib in Patients With Chronic Myeloid Leukaemia Who Have Maintained Deep Molecular Response for Longer Than 1 Year (DADI Trial): A Multicentre Phase 2 Trial. Lancet Haematol (2015) 2(12):E528–35. doi: 10.1016/S2352-3026(15)00196-9
90. Hamilton A, Helgason GV, Schemionek M, Zhang B, Myssina S, Allan EK, et al. Chronic Myeloid Leukemia Stem Cells are Not Dependent on Bcr-Abl Kinase Activity for Their Survival. Blood (2012) 119(6):1501–10. doi: 10.1182/blood-2010-12-326843
91. Corbin AS, Agarwal A, Loriaux M, Cortes J, Deininger MW, Druker BJ. Human Chronic Myeloid Leukemia Stem Cells are Insensitive to Imatinib Despite Inhibition of BCR-ABL Activity. J Clin Invest (2011) 121(3):396–409. doi: 10.1172/Jci35721
92. Giustacchini A, Thongjuea S, Barkas N, Woll PS, Povinelli BJ, Booth CAG, et al. Single-Cell Transcriptomics Uncovers Distinct Molecular Signatures of Stem Cells in Chronic Myeloid Leukemia. Nat Med (2017) 23(6):692. doi: 10.1038/nm.4336
93. Shah M, Bhatia R. Preservation of Quiescent Chronic Myelogenous Leukemia Stem Cells by the Bone Marrow Microenvironment. Adv Exp Med Biol (2018) 1100:97–110. doi: 10.1007/978-3-319-97746-1_6
94. Kim JA, Shim JS, Lee GY, Yim HW, Kim TM, Kim M, et al. Microenvironmental Remodeling as a Parameter and Prognostic Factor of Heterogeneous Leukemogenesis in Acute Myelogenous Leukemia. Cancer Res (2015) 75(11):2222–31. doi: 10.1158/0008-5472.Can-14-3379
95. Jin LH, Tabe Y, Konoplev S, Xu YY, Leysath CE, Lu HB, et al. CXCR4 Up-Regulation by Imatinib Induces Chronic Myelogenous Leukemia (CML) Cell Migration to Bone Marrow Stroma and Promotes Survival of Quiescent CML Cells. Mol Cancer Ther (2008) 7(1):48–58. doi: 10.1158/1535-7163.Mct-07-0042
96. Weisberg E, Azab AK, Manley PW, Kung AL, Christie AL, Bronson R, et al. Inhibition of CXCR4 in CML Cells Disrupts Their Interaction With the Bone Marrow Microenvironment and Sensitizes Them to Nilotinib. Leukemia (2012) 26(5):985–90. doi: 10.1038/leu.2011.360
97. Beider K, Darash-Yahana M, Blaier O, Koren-Michowitz M, Abraham M, Wald H, et al. Combination of Imatinib With CXCR4 Antagonist BKT140 Overcomes the Protective Effect of Stroma and Targets CML In Vitro and In Vivo. Mol Cancer Ther (2014) 13(5):1155–69. doi: 10.1158/1535-7163.Mct-13-0410
98. Agarwal P, Isringhausen S, Li H, Paterson AJ, He JB, Gomariz A, et al. Mesenchymal Niche-Specific Expression of Cxcl12 Controls Quiescence of Treatment-Resistant Leukemia Stem Cells. Cell Stem Cell (2019) 24(5):769–84.e6. doi: 10.1016/j.stem.2019.11.013
99. Gallipoli P, Pellicano F, Morrison H, Laidlaw K, Allan EK, Bhatia R, et al. Autocrine TNF-Alpha Production Supports CML Stem and Progenitor Cell Survival and Enhances Their Proliferation. Blood (2013) 122(19):3335–9. doi: 10.1182/blood-2013-02-485607
100. Herrmann O, Kuepper MK, Butow M, Costa IG, Appelmann I, Beier F, et al. Infliximab Therapy Together With Tyrosine Kinase Inhibition Targets Leukemic Stem Cells in Chronic Myeloid Leukemia. BMC Cancer (2019) 19(1):658. doi: 10.1186/s12885-019-5871-2
101. Agarwal P, Li H, Choi K, Hueneman K, He JB, Welner RS, et al. TNF-Alpha-Induced Alterations in Stromal Progenitors Enhance Leukemic Stem Cell Growth via CXCR2 Signaling. Cell Rep (2021) 36(2):109386. doi: 10.1016/j.celrep.2021.109386
102. Zhang B, Chu S, Agarwal P, Campbell VL, Hopcroft L, Jorgensen HG, et al. Inhibition of Interleukin-1 Signaling Enhances Elimination of Tyrosine Kinase Inhibitor-Treated CML Stem Cells. Blood (2016) 128(23):2671–82. doi: 10.1182/blood-2015-11-679928
103. Angstreich GR, Matsui W, Huff CA, Vala MS, Barber J, Hawkins AL, et al. Effects of Imatinib and Interferon on Primitive Chronic Myeloid Leukaemia Progenitors. Br J Haematology (2005) 130(3):373–81. doi: 10.1111/j.1365-2141.2005.05606.x
104. Essers MAG, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFN Alpha Activates Dormant Haematopoietic Stem Cells. vivo Nat (2009) 458(7240):904–8. doi: 10.1038/nature07815
105. Hochhaus A, Saussele S, Baerlocher GM, Brummendorf TH, Burchert A, La Rosee P, et al. Nilotinib Vs Nilotinib Plus Pegylated Interferon-Alpha2b Induction and Nilotinib or Pegylated Interferon-Alpha2b Maintenance Therapy for Newly Diagnosed BCR-ABL Plus Chronic Myeloid Leukemia Patients in Chronic Phase: Interim Analysis of the Tiger (CML V)-Study. Blood (2018) 132(Supplement 1):460. doi: 10.1182/blood-2018-99-112119
106. Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Yeung DT, et al. Safety and Efficacy of Imatinib Cessation for CML Patients With Stable Undetectable Minimal Residual Disease: Results From the TWISTER Study. Blood (2013) 122(4):515–22. doi: 10.1182/blood-2013-02-483750
107. Jun K, Ya-zhen Q, Xiao-su Z, Hong-Xia S, Yue-Yun L, Kai-yan L, et al. Interferon-Alpha may Help Prevent Molecular Relapse of Chronic Myeloid Leukemia After the Discontinuation of Tyrosine Kinase Inhibitors. Ther Adv Hematol (2021) 12:2040620720986643. doi: 10.1177/2040620720986643
108. Hu Y, Chen Y, Douglas L, Li S. Beta-Catenin is Essential for Survival of Leukemic Stem Cells Insensitive to Kinase Inhibition in Mice With BCR-ABL-Induced Chronic Myeloid Leukemia. Leukemia (2009) 23(1):109–16. doi: 10.1038/leu.2008.262
109. Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al. Expansion of Bcr-Abl-Positive Leukemic Stem Cells is Dependent on Hedgehog Pathway Activation. Cancer Cell (2008) 14(3):238–49. doi: 10.1016/j.ccr.2008.08.003
110. Lim S, Saw TY, Zhang M, Janes MR, Nacro K, Hill J, et al. Targeting of the MNK-Eif4e Axis in Blast Crisis Chronic Myeloid Leukemia Inhibits Leukemia Stem Cell Function. P Natl Acad Sci USA (2013) 110(25):E2298–307. doi: 10.1073/pnas.1301838110
111. Vakana E, Platanias LC. AMPK in BCR-ABL Expressing Leukemias. Regulatory Effects and Therapeutic Implications. Oncotarget (2011) 2(12):1322–8. doi: 10.18632/oncotarget.413
112. Peng C, Chen YY, Yang ZF, Zhang HJ, Osterby L, Rosmarin AG, et al. PTEN is a Tumor Suppressor in CML Stem Cells and BCR-ABL-Induced Leukemias in Mice. Blood (2010) 115(3):626–35. doi: 10.1182/blood-2009-06-228130
113. Pippa R, Odero MD. The Role of MYC and PP2A in the Initiation and Progression of Myeloid Leukemias. Cells-Basel (2020) 9(3):544. doi: 10.3390/cells9030544
114. Chen YY, Li DG, Li SG. The Alox5 Gene is a Novel Therapeutic Target in Cancer Stem Cells of Chronic Myeloid Leukemia. Cell Cycle (2009) 8(21):3488–92. doi: 10.4161/cc.8.21.9852
115. Hantschel O, Warsch W, Eckelhart E, Kaupe I, Grebien F, Wagner KU, et al. BCR-ABL Uncouples Canonical JAK2-STAT5 Signaling in Chronic Myeloid Leukemia. Nat Chem Biol (2012) 8(3):285–93. doi: 10.1038/Nchembio.775
116. Zhou H, Mak PY, Mu H, Mak DH, Zeng Z, Cortes J, et al. Combined Inhibition of Beta-Catenin and Bcr-Abl Synergistically Targets Tyrosine Kinase Inhibitor-Resistant Blast Crisis Chronic Myeloid Leukemia Blasts and Progenitors. Vitro vivo Leukemia (2017) 31(10):2065–74. doi: 10.1038/leu.2017.87
117. Irvine DA, Zhang B, Kinstrie R, Tarafdar A, Morrison H, Campbell VL, et al. Deregulated Hedgehog Pathway Signaling is Inhibited by the Smoothened Antagonist LDE225 (Sonidegib) in Chronic Phase Chronic Myeloid Leukaemia. Sci Rep-Uk (2016) 6:25476. doi: 10.1038/srep25476
118. Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, et al. PP2A-Activating Drugs Selectively Eradicate TKI-Resistant Chronic Myeloid Leukemic Stem Cells. J Clin Invest (2013) 123(10):4144–57. doi: 10.1172/Jci68951
119. Gallipoli P, Cook A, Rhodes S, Hopcroft L, Wheadon H, Whetton AD, et al. JAK2/STAT5 Inhibition by Nilotinib With Ruxolitinib Contributes to the Elimination of CML CD34(+) Cells In Vitro and In Vivo. Blood (2014) 124(9):1492–501. doi: 10.1182/blood-2013-12-545640
120. Prost S, Relouzat F, Spentchian M, Ouzegdouh Y, Saliba J, Massonnet G, et al. Erosion of the Chronic Myeloid Leukaemia Stem Cell Pool by PPAR Gamma Agonists. Nature (2015) 525(7569):380–3. doi: 10.1038/nature15248
121. Zhang HJ, Li HW, Ho N, Li DG, Li SG. Scd1 Plays a Tumor-Suppressive Role in Survival of Leukemia Stem Cells and the Development of Chronic Myeloid Leukemia. Mol Cell Biol (2012) 32(10):1776–87. doi: 10.1128/Mcb.05672-11
122. Rousselot P, Prost S, Guilhot J, Roy L, Etienne G, Legros L, et al. Pioglitazone Together With Imatinib in Chronic Myeloid Leukemia: A Proof of Concept Study. Cancer (2017) 123(10):1791–9. doi: 10.1002/cncr.30490
123. Parting O, Langer S, Kuepper MK, Wessling C, SG Li, Braunschweig T, et al. Therapeutic Inhibition of Fc Gamma RIIb Signaling Targets Leukemic Stem Cells in Chronic Myeloid Leukemia. Leukemia (2020) 34(10):2635–47. doi: 10.1038/s41375-020-0977-8
124. Majmundar AJ, Wong WHJ, Simon MC. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol Cell (2010) 40(2):294–309. doi: 10.1016/j.molcel.2010.09.022
125. Zhang HJ, Li HW, Xi HLS, Li SG. HIF1 Alpha is Required for Survival Maintenance of Chronic Myeloid Leukemia Stem Cells. Blood (2012) 119(11):2595–607. doi: 10.1182/blood-2011-10-387381
126. Cheloni G, Tanturli M, Tusa I, DeSouza NH, Shan Y, Gozzini A, et al. Targeting Chronic Myeloid Leukemia Stem Cells With the Hypoxia-Inducible Factor Inhibitor Acriflavine. Blood (2017) 130(5):655–65. doi: 10.1182/blood-2016-10-745588
127. Jaiswal S, Traver D, Miyamoto T, Akashi K, Lagasse E, Weissman IL. Expression of BCR/ABL and BCL-2 in Myeloid Progenitors Leads to Myeloid Leukemias. P Natl Acad Sci USA (2003) 100(17):10002–7. doi: 10.1073/pnas.1633833100
128. Goff DJ, Recart AC, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, et al. A Pan-BCL2 Inhibitor Renders Bone-Marrow-Resident Human Leukemia Stem Cells Sensitive to Tyrosine Kinase Inhibition. Cell Stem Cell (2013) 12(3):316–28. doi: 10.1016/j.stem.2012.12.011
129. Kuroda J, Kimura S, Andreeff M, Ashihara E, Kamitsuji Y, Yokota A, et al. ABT-737 is a Useful Component of Combinatory Chemotherapies for Chronic Myeloid Leukaemias With Diverse Drug-Resistance Mechanisms. Br J Haematology (2008) 140(2):181–90. doi: 10.1111/j.1365-2141.2007.06899.x
130. Mak DH, Wang RY, Schober WD, Konopleva M, Cortes J, Kantarjian H, et al. Activation of Apoptosis Signaling Eliminates CD34(+) Progenitor Cells in Blast Crisis CML Independent of Response to Tyrosine Kinase Inhibitors. Leukemia (2012) 26(4):788–94. doi: 10.1038/leu.2011.285
131. Leverson JD, Phillips DC, Mitten MJ, Boghaert ER, Diaz D, Tahir SK, et al. Exploiting Selective BCL-2 Family Inhibitors to Dissect Cell Survival Dependencies and Define Improved Strategies for Cancer Therapy. Sci Transl Med (2015) 7(279):279ra40. doi: 10.1126/scitranslmed.aaa4642
132. Carter BZ, Mak PY, Mu H, Zhou HS, Mak DH, Schober W, et al. Combined Targeting of BCL-2 and BCR-ABL Tyrosine Kinase Eradicates Chronic Myeloid Leukemia Stem Cells. Sci Transl Med (2016) 8(355):355ra117. doi: 10.1126/scitranslmed.aag1180
133. Maiti A, Franquiz MJ, Ravandi F, Cortes JE, Jabbour EJ, Sasaki K, et al. Venetoclax and BCR-ABL Tyrosine Kinase Inhibitor Combinations: Outcome in Patients With Philadelphia Chromosome-Positive Advanced Myeloid Leukemias. Acta Haematologica (2020) 143(6):567–73. doi: 10.1159/000506346
134. Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct Activation of Bax by P53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science (2004) 303(5660):1010–4. doi: 10.1126/science.1092734
135. Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G. Apoptosis as Anticancer Mechanism: Function and Dysfunction of its Modulators and Targeted Therapeutic Strategies. Aging (Albany NY) (2016) 8(4):603–19. doi: 10.18632/aging.100934
136. Abraham A, Qiu SW, Chacko BK, Li H, Paterson A, He JB, et al. SIRT1 Regulates Metabolism and Leukemogenic Potential in CML Stem Cells. J Clin Invest (2019) 129(7):2685–701. doi: 10.1172/Jci127080
137. Li L, Wang LS, Li L, Wang ZQ, Ho YW, McDonald T, et al. Activation of P53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination With Imatinib. Cancer Cell (2012) 21(2):266–81. doi: 10.1016/j.ccr.2011.12.020
138. Peterson LF, Mitrikeska E, Giannola D, Lui Y, Sun H, Bixby D, et al. P53 Stabilization Induces Apoptosis in Chronic Myeloid Leukemia Blast Crisis Cells. Leukemia (2011) 25(5):761–9. doi: 10.1038/leu.2011.7
139. Carter BZ, Mak PY, Mak DH, Ruvolo VR, Schober W, McQueen T, et al. Synergistic Effects of P53 Activation via MDM2 Inhibition in Combination With Inhibition of Bcl-2 or Bcr-Abl in CD34(+) Proliferating and Quiescent Chronic Myeloid Leukemia Blast Crisis Cells. Oncotarget (2015) 6(31):30487–99. doi: 10.18632/oncotarget.5890
140. Carter BZ, Mak PY, Mu H, Wang XM, Tao WJ, Mak DH, et al. Combined Inhibition of MDM2 and BCR-ABL1 Tyrosine Kinase Targets Chronic Myeloid Leukemia Stem/Progenitor Cells in a Murine Model. Haematologica (2020) 105(5):1274–84. doi: 10.3324/haematol.2019.219261
141. Yun CW, Lee SH. The Roles of Autophagy in Cancer. Int J Mol Sci (2018) 19(11):3466. doi: 10.3390/ijms19113466
142. Rahman MA, Saha SK, Rahman MS, Uddin MJ, Uddin MS, Pang MG, et al. Molecular Insights Into Therapeutic Potential of Autophagy Modulation by Natural Products for Cancer Stem Cells. Front Cell Dev Biol (2020) 8:283. doi: 10.3389/fcell.2020.00283
143. Rubinsztein DC, Codogno P, Levine B. Autophagy Modulation as a Potential Therapeutic Target for Diverse Diseases. Nat Rev Drug Discov (2012) 11(9):709–30. doi: 10.1038/nrd3802
144. Rothe K, Lin HY, Lin KBL, Leung A, Wang HM, Malekesmaeili M, et al. The Core Autophagy Protein ATG4B is a Potential Biomarker and Therapeutic Target in CML Stem/Progenitor Cells. Blood (2014) 123(23):3622–34. doi: 10.1182/blood-2013-07-516807
145. Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting Autophagy Potentiates Tyrosine Kinase Inhibitor-Induced Cell Death in Philadelphia Chromosome-Positive Cells, Including Primary CML Stem Cells. J Clin Invest (2009) 119(5):1109–23. doi: 10.1172/JCI35660
146. Cea M, Cagnetta A, Nencioni A, Gobbi M, Patrone F. New Insights Into Biology of Chronic Myeloid Leukemia: Implications in Therapy. Curr Cancer Drug Targets (2013) 13(7):711–23. doi: 10.2174/15680096113139990085
147. Karvela M, Baquero P, Kuntz EM, Mukhopadhyay A, Mitchell R, Allan EK, et al. ATG7 Regulates Energy Metabolism, Differentiation and Survival of Philadelphia-Chromosome-Positive Cells. Autophagy (2016) 12(6):936–48. doi: 10.1080/15548627.2016.1162359
148. Baquero P, Dawson A, Mukhopadhyay A, Kuntz EM, Mitchell R, Olivares O, et al. Targeting Quiescent Leukemic Stem Cells Using Second Generation Autophagy Inhibitors. Leukemia (2019) 33(4):981–94. doi: 10.1038/s41375-018-0252-4
149. Horne GA, Stobo J, Kelly C, Mukhopadhyay A, Latif AL, Dixon-Hughes J, et al. A Randomised Phase II Trial of Hydroxychloroquine and Imatinib Versus Imatinib Alone for Patients With Chronic Myeloid Leukaemia in Major Cytogenetic Response With Residual Disease. Leukemia (2020) 34(7):1775–86. doi: 10.1038/s41375-019-0700-9
150. Rinke J, Chase A, Cross NCP, Hochhaus A, Ernst T. EZH2 in Myeloid Malignancies. Cells-Basel (2020) 9(7):1639. doi: 10.3390/cells9071639
151. Scott MT, Korfi K, Saffrey P, Hopcroft LEM, Kinstrie R, Pellicano F, et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov (2016) 6(11):1248–57. doi: 10.1158/2159-8290.Cd-16-0263
152. Xie HF, Peng C, Huang JL, Li BE, Kim W, Smith EC, et al. Chronic Myelogenous Leukemia-Initiating Cells Require Polycomb Group Protein Ezh2. Cancer Discov (2016) 6(11):1237–47. doi: 10.1158/2159-8290.Cd-15-1439
153. Falkenberg KJ, Johnstone RW. Histone Deacetylases and Their Inhibitors in Cancer, Neurological Diseases and Immune Disorders. Nat Rev Drug Discovery (2014) 13(9):673–91. doi: 10.1038/nrd4360
154. Zhang B, Strauss AC, Chu S, Li M, Ho YW, Shiang KD, et al. Effective Targeting of Quiescent Chronic Myelogenous Leukemia Stem Cells by Histone Deacetylase Inhibitors in Combination With Imatinib Mesylate. Cancer Cell (2010) 17(5):427–42. doi: 10.1016/j.ccr.2010.03.011
155. Jin YL, Yao YW, Chen L, Zhu XH, Jin B, Shen YY, et al. Depletion of Gamma-Catenin by Histone Deacetylase Inhibition Confers Elimination of CML Stem Cells in Combination With Imatinib. Theranostics (2016) 6(11):1947–62. doi: 10.7150/thno.16139
156. He BL, Wang Q, Liu XL, Lu ZY, Han JC, Pan CY, et al. A Novel HDAC Inhibitor Chidamide Combined With Imatinib Synergistically Targets Tyrosine Kinase Inhibitor Resistant Chronic Myeloid Leukemia Cells. BioMed Pharmacother (2020) 129:110390. doi: 10.1016/j.biopha.2020.110390
157. Jin YL, Zhou JF, Xu F, Jin B, Cui LJ, Wang Y, et al. Targeting Methyltransferase PRMT5 Eliminates Leukemia Stem Cells in Chronic Myelogenous Leukemia. J Clin Invest (2016) 126(10):3961–80. doi: 10.1172/Jci85239
158. Jaras M, Johnels P, Hansen N, Agerstam H, Tsapogas P, Rissler M, et al. Isolation and Killing of Candidate Chronic Myeloid Leukemia Stem Cells by Antibody Targeting of IL-1 Receptor Accessory Protein. P Natl Acad Sci USA (2010) 107(37):16280–5. doi: 10.1073/pnas.1004408107
159. Agerstam H, Hansen N, von Palffy S, Sanden C, Reckzeh K, Karlsson C, et al. IL1RAP Antibodies Block IL-1-Induced Expansion of Candidate CML Stem Cells and Mediate Cell Killing in Xenograft Models. Blood (2016) 128(23):2683–93. doi: 10.1182/blood-2015-11-679985
160. Warda W, Larosa F, Da Rocha MN, Trad R, Deconinck E, Fajloun Z, et al. CML Hematopoietic Stem Cells Expressing IL1RAP Can Be Targeted by Chimeric Antigen Receptor-Engineered T Cells. Cancer Res (2019) 79(3):663–75. doi: 10.1158/0008-5472.Can-18-1078
161. Herrmann H, Sadovnik I, Cerny-Reiterer S, Rulicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) Defines Leukemic Stem Cells (LSC) in Chronic Myeloid Leukemia. Blood (2014) 123(25):3951–62. doi: 10.1182/blood-2013-10-536078
162. Bocchia M, Sicuranza A, Abruzzese E, Iurlo A, Sirianni S, Gozzini A, et al. Residual Peripheral Blood CD26(+) Leukemic Stem Cells in Chronic Myeloid Leukemia Patients During TKI Therapy and During Treatment-Free Remission. Front Oncol (2018) 8:194. doi: 10.3389/fonc.2018.00194
163. Ruiz MS, Sanchez MB, Bonecker S, Furtado C, Koile D, Yankilevich P, et al. Mirnome Profiling of LSC-Enriched CD34(+)CD38(-)CD26(+) Fraction in Ph+ CML-CP Samples From Argentinean Patients: A Potential New Pharmacogenomic Tool. Front Pharmacol (2021) 11:612573. doi: 10.3389/fphar.2020.612573
164. Houshmand M, Garello F, Stefania R, Gaidano V, Cignetti A, Spinelli M, et al. Targeting Chronic Myeloid Leukemia Stem/Progenitor Cells Using Venetoclax-Loaded Immunoliposome. Cancers (2021) 13(6):1311. doi: 10.3390/cancers13061311
165. Willmann M, Sadovnik I, Eisenwort G, Entner M, Bernthaler T, Stefanzl G, et al. Evaluation of Cooperative Antileukemic Effects of Nilotinib and Vildagliptin in Ph+ Chronic Myeloid Leukemia. Exp Hematol (2018) 57:50–9. doi: 10.1016/j.exphem.2017.09.012
166. Sadovnik I, Hoelbl-Kovacic A, Herrmann H, Eisenwort G, Cerny-Reiterer S, Warsch W, et al. Identification of CD25 as STAT5-Dependent Growth Regulator of Leukemic Stem Cells in Ph+ CML. Clin Cancer Res (2016) 22(8):2051–61. doi: 10.1158/1078-0432.Ccr-15-0767
167. Kinstrie R, Horne GA, Morrison H, Irvine D, Munje C, Castaneda EG, et al. CD93 is Expressed on Chronic Myeloid Leukemia Stem Cells and Identifies a Quiescent Population Which Persists After Tyrosine Kinase Inhibitor Therapy (Vol 17, Pg 487, 2019). Leukemia (2020) 34(7):1975–. doi: 10.1038/s41375-020-0721-4
168. Herrmann H, Sadovnik I, Eisenwort G, Rulicke T, Blatt K, Herndlhofer S, et al. Delineation of Target Expression Profiles in CD34(+)/CD38(-) and CD34(+)/CD38(+) Stem and Progenitor Cells in AML and CML. Blood Adv (2020) 4(20):5118–32. doi: 10.1182/bloodadvances.2020001742
169. Soverini S, De Santis S, Monaldi C, Bruno S, Mancini M. Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort? Int J Mol Sci (2021) 22(13):7093. doi: 10.3390/ijms22137093
Keywords: chronic myeloid leukemia, tyrosine kinase inhibitors, leukemia stem cells, discontinuation, treatment-free remission
Citation: Chen Y, Zou J, Cheng F and Li W (2021) Treatment-Free Remission in Chronic Myeloid Leukemia and New Approaches by Targeting Leukemia Stem Cells. Front. Oncol. 11:769730. doi: 10.3389/fonc.2021.769730
Received: 02 September 2021; Accepted: 11 October 2021;
Published: 28 October 2021.
Edited by:
Haojian Zhang, Wuhan University, ChinaReviewed by:
Leland Metheny, University Hospitals of Cleveland, United StatesMonica Bocchia, University of Siena, Italy
Copyright © 2021 Chen, Zou, Cheng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiming Li, bGVlOTM3QDEyNi5jb20=