 Valentina Diehl1
Valentina Diehl1 Lisa Sophie Huber1
Lisa Sophie Huber1 Jonel Trebicka2
Jonel Trebicka2 Malgorzata Wygrecka3
Malgorzata Wygrecka3 Renato V. Iozzo4
Renato V. Iozzo4 Liliana Schaefer1*
Liliana Schaefer1*- 1Institute of Pharmacology and Toxicology, Goethe University, Frankfurt, Germany
- 2Department of Internal Medicine I, Goethe University, Frankfurt, Germany
- 3Center for Infection and Genomics of the Lung, Member of the German Center for Lung Research, University of Giessen and Marburg Lung Center, Giessen, Germany
- 4Department of Pathology, Anatomy and Cell Biology and the Translational Cellular Oncology Program, Sidney Kimmel Cancer Center, Sidney Kimmel Medical College at Thomas Jefferson University, Philadelphia, PA, United States
The complex and adaptive nature of malignant neoplasm constitute a major challenge for the development of effective anti-oncogenic therapies. Emerging evidence has uncovered the pivotal functions exerted by the small leucine-rich proteoglycans, decorin and biglycan, in affecting tumor growth and progression. In their soluble forms, decorin and biglycan act as powerful signaling molecules. By receptor-mediated signal transduction, both proteoglycans modulate key processes vital for tumor initiation and progression, such as autophagy, inflammation, cell-cycle, apoptosis, and angiogenesis. Despite of their structural homology, these two proteoglycans interact with distinct cell surface receptors and thus modulate distinct signaling pathways that ultimately affect cancer development. In this review, we summarize growing evidence for the complex roles of decorin and biglycan signaling in tumor biology and address potential novel therapeutic implications.
1 Introduction
1.1 Extracellular Matrix Guidance in Tumor Initiation and Progression
Over the past decades, it has become apparent that tumorigenesis is not merely the result of accumulated DNA mutations, but tumor development and progression also depend on the context in which malignant cells subsist (1–3). The tumor microenvironment (TME) is a permissive infrastructure primarily composed of non-malignant resident cells, cancer-associated fibroblasts, macrophages, and a variety of inflammatory and immune cells (4–7), pericytes (8, 9), and cells of the tumor-associated vascular system (10, 11). Moreover, the TME includes the extracellular matrix (ECM), an acellular structure with, as has become evident by now, unique tumor-specific characteristics (12, 13).
The ECM forms an intricate, highly ordered, 3D meshwork of large macromolecules, the components of which have been considered in the past solely as structural scaffolds that enable cellular adhesion and migration. However, a vast body of emerging evidence has led to a paradigmatic shift implicating ECM proteins in various signaling pathways and cellular processes including autophagy, inflammation, proliferation, survival, cell morphology, and motility (14–22). To date, several ECM components are increasingly appreciated for mediating and modulating cellular signaling (18, 23–26), and presumably even inter-organ cross-talk (27). Importantly, changes in the ECM are known to lead to, or to correlate with several diseases such as cardiovascular and skeletal disorders, fibrosis and importantly cancer (24, 28–30). The observations that link the ECM to cancer are as diverse as the different macromolecules that comprise the ECM. The impact of the biochemical composition of the ECM on tumor progression (12, 31–33), the ECM-dependent development of metastatic niches (34, 35), and the signaling between the tumor-adjacent ECM and the different players of the TME have been reviewed in detail (36, 37). In this review, we will focus on two proteoglycans of the ECM, biglycan and decorin. We will discuss their implications in selected pathways that are linked to the hallmarks of cancer, with special emphasis on autophagy and inflammation, whose contribution to tumorigenesis is increasingly recognized. Lastly, we will critically discuss recent advances and future perspectives in therapeutic targeting of these proteoglycans for cancer therapy.
1.2 Inflammation as an Emerging Hallmark of Cancer
Tumorigenesis is a multi-factorial process that is characterized by specific biological capabilities that are acquired during the development of tumors. These hallmarks of cancer include evading apoptosis, increased angiogenesis, insensitivity to anti-growth signals, and tissue invasion and metastasis (38). In addition to these established hallmarks of cancer, the vital roles of inflammation and the immune response have gained recognition as emerging hallmarks and enabling characteristics of cancer (31, 39, 40). Intricate interactions between cells of the tumor proper, surrounding stromal cells, and inflammatory cells result in an inflammatory TME. Indeed, chronic tissue inflammation has been linked to a heightened risk for malignant transformation, and additionally, sustained inflammation is assumed to cause pre-cancerous lesions (41–43). Moreover, it is well-established that the TME of most tumors is characterized by the presence of highly abundant inflammatory cells and inflammatory mediators, irrespective of the stages of tumor progression (44). Generally, there are two different types of cancer-associated inflammation. One can exert tissue-protective, anti-tumoral functions; the second on the other hand, is linked to pro-tumorigenic effects, presumably in a context and spatiotemporal-dependent manner (45).
Historically, cancer-associated inflammation was attributed to an attempt by the host immune system to eliminate the tumor and provide tissue-protection. Indeed, for some tumor types, there is increasing evidence for an acute immune response against the tumor, and the primary tumor development depends on the ability of tumor cells to escape the immune-mediated destruction (46, 47). This concept has been exploited for the development and application of immuno-therapies that reinstall or enhance the patient’s immune response towards recognizing and eradicating cancer cells (48, 49). Conversely, the tumor-associated inflammatory response can also facilitate tumorigenesis by providing an inflammatory milieu. This “cancer-promoting inflammation” relies on secretion of growth factors and enzymes to sustain proliferation, inhibit cell death, promote epithelial-mesenchymal transition (EMT), facilitate angiogenesis, invasion, and metastasis (39, 45, 50). In line with this, chronic inflammation has been linked to cancer-promoting effects in so-called ‘hot’ tumors, which are densely infiltrated by immune cells and are often unresponsive to immuno-therapy (44, 45, 51).
On a molecular level, constitutively active NF-κB (nuclear factor kappa-light-chain-enhancer of activated B-cells) signaling is considered to be one of many possible factors that promote chronic inflammation in the context of tumorigenesis (52). NF-κB downstream signaling results in expression of inflammatory cytokines, adhesion molecules, the inducible nitric oxide synthase and angiogenic factors. Inducers of NF-κB include cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin 1 beta (IL-1β). The activation of the NF-κB pathway is mediated through Toll-like receptors (TLRs) that act in concert with their adaptor molecules and co-receptors (53–55). Another signaling pathway that links inflammatory signaling to tumorigenesis includes the NOD-like receptor protein 3 (NLRP3) inflammasome, which activates NOD-like receptors or AIM2-like receptors and ultimately promotes the release of the inflammatory cytokines via activating Caspase 1, however, its precise function in tumor biology remains controversial (56–58).
In summary, inflammation and immune responses can critically contribute to the cancer onset and determine the outcome of the disease progression. Therefore, inflammation can be considered an enabling characteristic of cancer (39). Investigating the role of proteoglycans in mediating these cancer-associated inflammatory processes may provide novel insights in tumor biology.
1.3 Emerging Implications of Autophagy in Tumorigenesis
Autophagy has an ambivalent role in cancer development and progression. Depending on several factors, such as the tumor progression state, the TME and the genetic context, it can exert opposite outcomes and promote or decrease tumorigenesis (59–61). On one hand, before the onset of malignant transformation and tumorigenesis, the cytoprotective effect of autophagy can promote tumor suppression (62–65). On the other hand, evidence suggest autophagy to promote the survival of cancer cells under certain stress conditions, including therapy-induced stress. Moreover, various cancer types were shown to be linked to a high autophagy (66), such as cancers associated with RAS (Ras GTPase, Rat sarcoma) mutations (67, 68), or pancreatic cancer (66, 69), which have been reported to exhibit a high autophagic dependency. Additionally, the upregulation of autophagy is considered to be a frequent side effect of cancer therapies. Therefore, diverse autophagy inhibitors were suggested to increase the efficiency of therapeutics by reducing the tumor cells autophagy-dependent adaptive response mechanisms (70–72). However, increased therapy efficiencies have has also been reported upon activation of autophagy (73). Furthermore, unconventional secretion, endosomal and exosomal pathways have been shown to link autophagy to affecting cancer metastasis, immune responses and shaping the TME (74). Additionally, cross-talk between autophagy and the EMT was reported (75). In this context, autophagy induction was shown to impair EMT (76, 77), which is a critical contributor to metastasis. Other studies, however, showed that treatment-induced autophagy could exert cyto-protective functions and promote melanoma cell EMT (78). In line with this, knockdown of Beclin-1, which is required for autophagosome formation, reduced EMT of colon cancer cells (79). Notably, autophagy activation was also associated with increased anoikis resistance and enhanced metastasis in several tumor models (80, 81).
Several autophagy inhibitors and inducers are currently being evaluated as treatment options in cancer therapies (82, 83), however, the ambiguous roles of autophagy in tumor biology and the highly context-dependent outcomes of modulating autophagy in different cancer types and progression stages impede the translation to clinical applications. A deeper understanding of how proteoglycans influence autophagy in cancer might allow more precise indications for autophagy inhibitors and inducers, and enable the development of adjuvant therapies.
1.4 Structural Characteristics of Small Leucine-Rich Proteoglycans
Proteoglycans are complex molecules that are embedded in the ECM and comprise a protein core and one or more glycosaminoglycan (GAG) chains, which are covalently tethered to the protein core. The different types of GAG chains can be formed by heparan sulfate, chondroitin sulfate/dermatan sulfate (CS/DS), or keratan sulfate (84–86). In mammalian cells, more than 40 proteoglycans have been described, which are classified according to their localization into extra-, peri-, and intracellular, as well as plasma membrane-associated proteoglycans (85). The divers and manifold biological roles of proteoglycans can be attributed to their variability and modularity, as their protein cores and the heterogeneous GAG chains can be variously modified. Proteoglycans critically provide structure to the ECM, are key receptors to diverse signaling stimuli, such as growth factors, cytokines, chemokines, and important regulators at the intersection of the cell and matrix (87, 88).
The complex and multifaceted regulatory functions of proteoglycans are well exemplified by their largest protein family, the small leucine-rich proteoglycans (SLRPs), which are ubiquitously expressed and highly abundant in the ECM (89). After being secreted into the pericellular space SLRPs are incorporated into the ECM. They are composed of a relatively small protein core of about 40–60 kDa that contains 10–12 motifs of leucine-rich repeats (LRRs) and is flanked by characteristic cysteine-rich clusters with defined spacing. Additionally, the heterogeneous GAG chains are post-translationally attached to the protein core (85, 90–92). To date, there are 18 known SLRPs, which are classified into five distinct classes, based on their N-terminal cysteine-rich clusters, evolutionary conservation, sequence homology, and GAG chains (85). The LRRs of the protein core, which give rise to the characteristic solenoid structure of SLRPs, contain a conserved hydrophobic motif wherein distinct leucines can be substituted by other hydrophobic amino acids (90). Irrespective of their classification, all SLRPs share common biological functions, and most importantly, their capability to interact with various cell surface receptors such as receptor tyrosine kinases (RTKs) and TLRs, thus mediating divers downstream signaling events that regulate vital cellular pathways and processes that are also implicated in cancer, including inflammation and autophagy (17). SLRPs can be released upon tissue stress or injury and can act as damage-associated molecular patterns (DAMPs) (93, 94).
Importantly, an emerging body of evidence points to the vital implications of the SLRPs decorin and biglycan in several hallmarks of cancer and regulating tumor-associated pathways, such apoptosis, proliferation, angiogenesis, inflammation, and autophagy (17, 18, 87, 93–95). In the following sections, we will summarize and discuss the major signaling functions of biglycan and decorin in normal physiology and in the context of cancer.
2 Decorin Function In Tumorigenesis
Decorin is a well-characterized, prototypical member of the SLRP family and ubiquitously expressed in most tissues (96). It is mainly known for its vital functions in inflammation, innate immunity, wound healing, fibrotic diseases, angiogenesis, autophagy and cancer, where it is found in the stroma of several cancer types (97–100). Decorin, whose eponym derives from its high affinity for collagen fibrils and thus able to “decorate” collagen fibrils (92, 101, 102), was the first proteoglycan that was associated with regulating the cell cycle by inhibiting transforming growth factor β (TGF-β) signaling (103). This has led to a paradigmatic shift in matrix biology research, since proteoglycans have previously been described only as structural components. Indeed, decorin deficiency in mice leads to a permissive environment favoring tumorigenesis and EMT (104, 105). The diverse functions can be attributed to its ability to interact with numerous components of the ECM and cellular receptors, thereby inducing intracellular signaling cascades. Decorin has the ability to acts as a Pan-RTK inhibitor by engaging numerous RTKs, including the epidermal growth factor receptor (EGFR), the vascular endothelial growth factor receptor 2 (VEGFR2) and the mesenchymal-epithelial transition factor (Met) receptor (Figure 1), and can induce caveosomal internalization and degradation of the RTKs, thus restraining angiogenesis. Therefore, and because of its capability to inhibit cancer growth by sequestrating TGF-β, decorin has also been described as “the guardian from the matrix” (99).
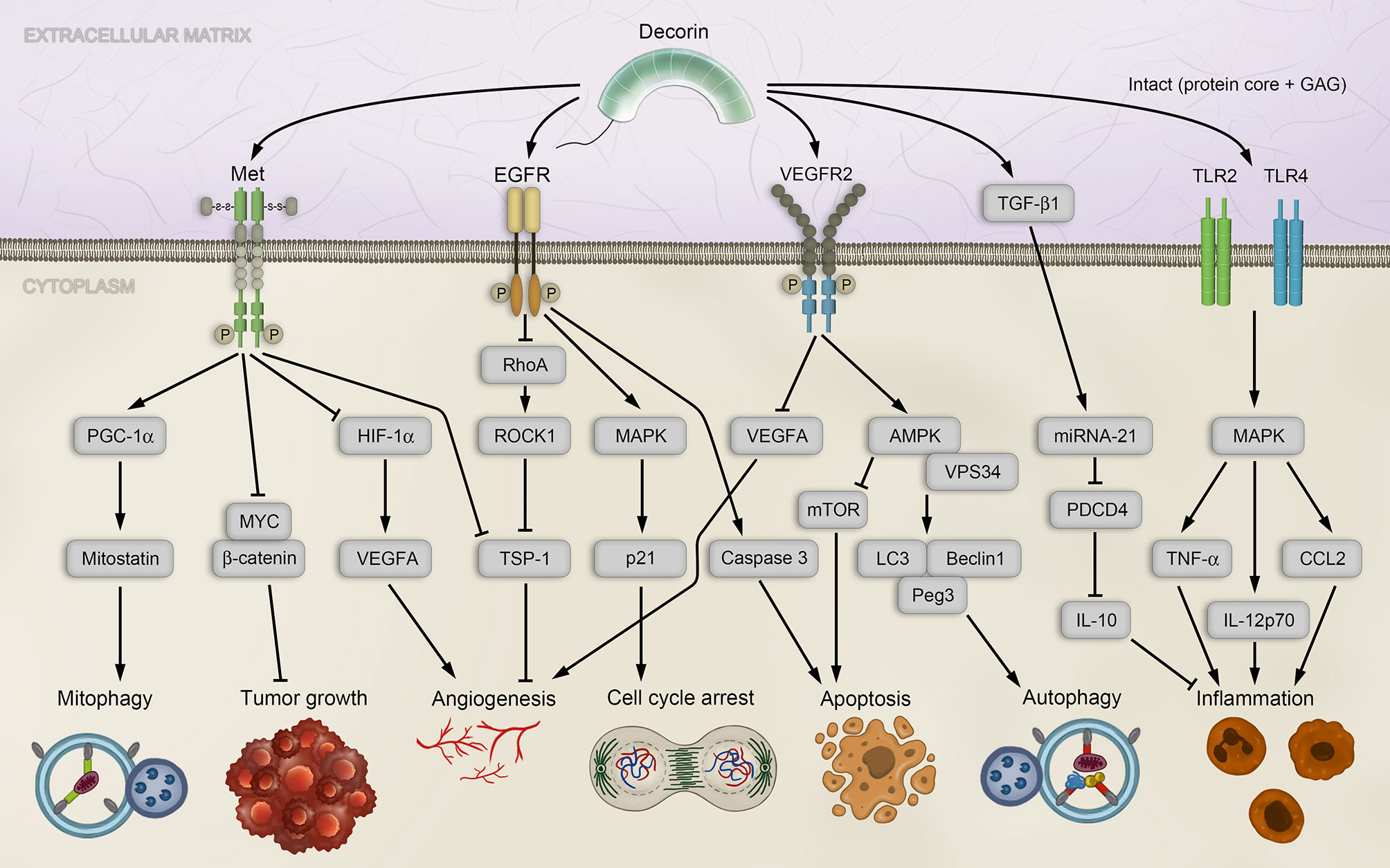
Figure 1 Decorin-mediated signaling affects diverse pathways associated with tumorigenesis. Decorin activates mitophagy in breast carcinoma cells by binding to the Met receptor and activating PGC-1α, leading to accumulation of Mitostatin. Further, via Met receptor signaling, decorin inhibits tumor growth by down-regulating β-catenin and MYC, and inhibiting angiogenesis by repressing HIF-1α and VEGFA. Angiogenesis is also modulated by decorin through EGFR in a signaling cascade that employs Rho and ROCK1, leading to upregulation of the anti-angiogenic effector TSP-1. EGFR phosphorylation by decorin induces intracellular MAPK, resulting in enhanced expression of p21 and cell cycle arrest, and in release of Caspase 3 and apoptosis. Decorin-mediated VEGFR2 signaling inhibits angiogenesis and induces autophagy in endothelial cells by inhibiting mTOR, activating VPS34, Beclin-1, LC3 and Peg3. Decorin regulates inflammation in a PDCD4-dependent manner through miRNA-21, or by TLR2/4 signaling. AMPK, AMP-activated protein kinase; CCL, chemokine C–C motif-ligand; EGFR, epidermal growth factor receptor; GAG, glycosaminoglycan; HIF-1α, hypoxia-inducible factor 1α; IL, interleukin; LC3, microtubule-associated protein 1A/1B-light chain 3; MAPK, mitogen-activated protein kinase; Met, mesenchymal-epithelial transition factor; miRNA, microRNA; mTOR, mammalian target of rapamycin; MYC, myelocytomatosis oncogene protein; PDCD4, programmed cell death protein 4; Peg3, paternally expressed 3; PGC-1α, peroxisome proliferator-activated receptor-γ co-activator 1-α; Rho, RAS homolog family member A; ROCK1, Rho-associated coiled-coil kinase 1; TGF-β1, transforming growth factor β isoform 1; TLR, Toll-like receptor; TNF-α, tumor necrosis factor alpha; TSP-1, thrombospondin-1; VEGFA, vascular endothelial growth factor A; VEGFR2, vascular endothelial growth factor receptor 2; VPS34, vacuolar protein sorting 34.
2.1 Decorin Acts as a Versatile Tumor Suppressor
One of the most significant RTKs, whose downstream signaling is affected by decorin, is the Met receptor pathway (106–108). Met receptor-mediated signaling is vital for embryonic development and upregulation of the receptor has been linked to tumorigenesis and metastasis (109–112). Direct binding of decorin to the Met receptor results in tumor suppression by phosphorylation of Met tyrosine sites (106), leading to recruitment of the proto-oncogene CBL (Casitas B-lineage Lymphoma) and subsequent proteosomal degradation of the Met receptor (113). This prevents binding of the natural ligand of the Met receptor, HGF (hepatocyte growth factor), which can contribute to tumor survival, growth, angiogenesis, and metastasis under pathologic conditions (112). The antagonistic effect of decorin on Met signaling results in down-regulation of the major oncogene β-catenin and its downstream effector MYC (myelocytomatosis oncogene protein), both involved in the pathologies of various cancers (107, 114), and thus promotes an anti-tumorigenic activity of decorin (Figure 1).
Apart from engaging the Met receptor, decorin can attenuate tumor growth as a monomeric proteoglycan (115). To date, the protein interacting network centered on decorin is vast and expanding (116), with numerous growth factors, receptors, and various matrix molecules. The interaction of decorin with TGF-β causes strong inhibition of proliferation in various cancer cell lines, presumably by decorin-binding activity on TGF-β isoform 1 (TGF-β1), thereby sequestrating it in the ECM and limiting its bioactivity (117). Mechanistically, EGFR phosphorylation by decorin induces intracellular MAPK (mitogen-activated protein kinase) signaling that results in enhanced expression of the cyclin-dependent kinase inhibitor p21, triggering cell cycle arrest, and in release of Caspase 3 to promote apoptosis (99, 108, 118, 119) (Figure 1). Moreover, decorin competes with the natural ligand of EGFR, the epidermal growth factor (EGF), and the decorin-EGFR interaction results in internalization and degradation of the receptor via caveolar endocytosis (120), thus controlling tumor growth (121–123).
2.2 Decorin Regulates Angiogenesis via Receptor Tyrosine Kinase Signaling
A central issue of tumorigenesis is angiogenesis, an intrinsic process required for supplying oxygen and nutrients to the growing tumor through vessels newly formed from preexisting large vessels. The role of decorin in regulating angiogenesis is ambivalent, as decorin promotes angiogenesis by facilitating endothelial cell adhesion and migration in a non-malignant state (108, 124, 125). Likewise, it was shown in healthy, non-tumorigenic models that decorin protects endothelial cells from hypoglycemia and promotes angiogenesis through IGF-1R (insulin-like growth factor type 1 receptor) signaling (126). Conversely, multiple independent studies have clearly demonstrated an angiostatic effect of decorin in the setting of cancer and its down-regulation correlates with the degree of tumor vascularization in various cancer types (99, 100, 108, 127).
The interaction of decorin with VEGFR2 constitutes the most significant contribution to impairing tumor angiogenesis (98, 99) (Figure 1). By competing with its canonical¨ ligand VEGFA (vascular endothelial growth factor A), decorin acts as a partial antagonist of VEGFR2 which is specifically located on the surface of endothelial cells to facilitate their angiogenic activity (128–130). Additionally, decorin decreases the proteolytic cleavage of matrix-bound VEGFA in the ECM by inhibiting matrix-metalloprotease activities and expression (99, 107, 131, 132).
Another contributing mechanism of decorin-mediated angiostasis is the recent observation that soluble decorin can reduce VGFA levels by evoking its catabolic degradation via autophagy (133) (see below). In parallel to suppressing these pro-angiogenic effectors via VEGFR2 signaling, angiogenesis can be modulated by decorin through EGFR. The decorin-EGFR interaction interferes with the signaling cascade that employs Rho (RAS homolog family member A) and the Rho-associated coiled-coil kinase 1 (ROCK1), leading to upregulation of anti-angiogenic effectors, such as thrombospondin-1 (TSP-1) and tissue inhibitor of metalloproteinases-3 (TIMP-3) (108, 131, 132) (Figure 1).
Additionally, decorin inhibits angiogenesis by impairing Met signaling, because attenuation of Met signaling causes an induction of TIMP-3. In parallel, hypoxia-inducible factor 1α (HIF-1α) is being repressed and degraded and Met-induced VEGFA expression is inhibited (107, 131). Regulated by a negative feedback loop, loss of HIF-1α in turn inhibits Met expression (131). Collectively, these observations posit the small leucine-rich proteoglycan decorin as an important component of the angiostatic network and the cross-talk between and endothelial and tumor cells.
2.3 Decorin Activates Endothelial Autophagy and Cancer Cell Mitophagy
The anti-tumor activities of decorin additionally include the regulation of the autophagy and mitophagy pathways. In vascular endothelial cells, a decorin-induced indirect induction of autophagy was demonstrated by promoting the formation of autophagic initiation complexes and by down-regulation of autophagy inhibitors (134–136). Decorin interaction with VEGFR2 induces the AMP-activated protein kinase (AMPK) and initiates a signaling cascade to activate VPS34 (vacuolar protein sorting 34) and to inhibit mTOR (mammalian target of rapamycin). Decorin evokes sustained autophagy by transcriptional activation of Peg3 (Paternally expressed gene 3), an imprinted gene expressed exclusively from the paternal allele (137), and that is often silenced in several tumor types by promoter hypomethylation (138, 139). This leads to accumulation of Beclin-1 and LC3 (microtubule-associated protein 1A/1Blight chain 3), which are required for autophagosome formation (130, 136, 140). As Peg3+ progenitor cells participate in vascular remodeling (141), and as Peg3 represents a marker for a subset of vessel-associated endothelial progenitors (142), it is possible that decorin may additionally affect angiogenesis via Peg3-endothelial stem cells interactions.
A recently uncovered implication of decorin in regulating mitophagy has been reported, which linked decorin directly to affecting the catabolic process of mitophagy within the tumor proper. In line with this, decorin directly activated mitophagy in breast carcinoma cells trough Met receptor activation and subsequent mobilization of PGC-1α (Peroxisome proliferator-activated receptor-γ co-activator 1-α) (143), accompanied by a decorin-mediated mRNA stabilization accumulation of Mitostatin (143) (Figure 1). Thus, decorin can affect the cellular energy production and indirectly cell death/apoptosis by evoking autophagic catabolism of mitochondria.
2.4 Decorin Regulates Inflammation and the Innate Immune Response
Extensive studies using models for tissue stress and injuries have demonstrated the well-established roles of decorin, and likewise biglycan, in regulating inflammatory and immune responses by interaction with TLR2 and TLR4 (144). There are two mechanisms for decorin-dependent control of inflammation and tumor growth that are based on either stimulation of PDCD4 (programmed cell death protein 4) expression, or on it translational repression (145). PDCD4 is known to promote the inflammatory response by activating NF-κB and suppressing the anti-inflammatory mediator interleukin 10 (IL-10) expression (146). PDCD4 expression is increased by decorin acting as an endogenous ligand of TLR2/4 and stimulating production of proinflammatory molecules, including PDCD4 (145). In a second mechanism, decorin down-regulates the pool of bioactive TGF-β1, which, as mentioned before, can be sequestered by decorin in the ECM. TGF-β1 in turn is an inducer of oncogenic microRNA (miRNA)-21 (147), which functions as a translational repressor of PDCD4 (148). Therefore, decorin indirectly regulates the levels of PDCD4 (145) (Figure 1).
3 Biglycan Is a Matrix-Derived Signaling Molecule in Cancer
Biglycan, another member of class I SLRPs and structurally homologous to decorin, is a ubiquitously expressed ECM protein (23, 85, 149). The 42 kDa protein core consists of 10 LRRs as well as two N-terminal, covalently bound, tissue-specific CS/DS type GAG chains (23, 150). Through its protein core and GAG chains, biglycan interacts with other ECM proteins like collagen types I-IV and elastin, thereby providing stability and organization in tissues, as well as bone composition (151–156). Although biglycan is tightly bound to the ECM under physiological conditions, the proteoglycan is released from its ECM-bound state during tissue stress and injury through cleavage by proteases (87, 157–160). Additionally, biglycan can be synthesized de novo in macrophages during inflammation and is released into circulation (161). In its soluble state, biglycan can be found in the bloodstream in many acute or chronic inflammatory disease acting as a DAMP (93, 159). Biglycan holds great potential as biomarker as deregulated levels of soluble biglycan are detected in a variety of inflammatory and chronic disease, such lupus nephritis (144), diabetes (162, 163), fibrosis (164), renal diseases (159), and cancer (165), biglycan holds great potential as biomarker.
Just as decorin, biglycan has proven to be a versatile matrix-derived signaling molecule that can trigger induction of several pathways involved in tumorigenesis via its complex and diverse interactions with both growth factors/cytokines and cell surface receptors (18, 23, 157).
3.1 Mechanisms of Biglycan-Induced Inflammation
Under physiological conditions, in its soluble and intact form, biglycan acts as a DAMP and engages in the innate immunity by binding TLR2 and/or TLR4, mimicking gram negative and positive microbial responses and triggering an inflammatory cell response (93, 157). This results in induction of NF-κB and Erk (extracellular signal-regulated kinase) signaling pathways leading to increased levels pro-inflammatory cytokines and chemokines. Most importantly, these include TNF-α and IL-1β, as well as C-C motif ligand (CCL) and C-X-C motif ligand (CXCL) proteins and ultimately result in recruitment of immune cells such as neutrophils, T-cells, B-cells and macrophages into the inflamed regions, infiltrating tissues and organs (23, 161).
The biglycan-induced downstream signaling and its outcome depend on the selective utilization of either TLR2 or TLR4, or both, as well as on the interactions with their distinct co-receptors and adaptor molecules (157, 161). For instance, while TLR4/2 interaction with MyD88 specifically induces TNF-α, IL-1β, CXCL1, CCL2, and CCL20, interaction of biglycan with TLR4 and TRIF (TIR-domain-containing adapter-inducing interferon-β) results in expression of CCL5 and CXCL10 (161) (Figure 2). Moreover, biglycan interaction with only TLR2 and MyD88 regulates the expression of HSP70 (heat shock protein 70), which binds to NADPH oxidase (NOX)2, and thereby impairs the inhibitory function of NOX2 on IL-1β expression (166) (Figure 2). The repertoire of co-receptors employed by TLR2/4 was extended by the discovery of the co-receptor CD14, a high-affinity ligand of biglycan, whose deficiency resulted in abrogated NF-κB, MAPK and ERK signaling and consequent absence of biglycan-induced increase of cytokine expression in macrophages (167).
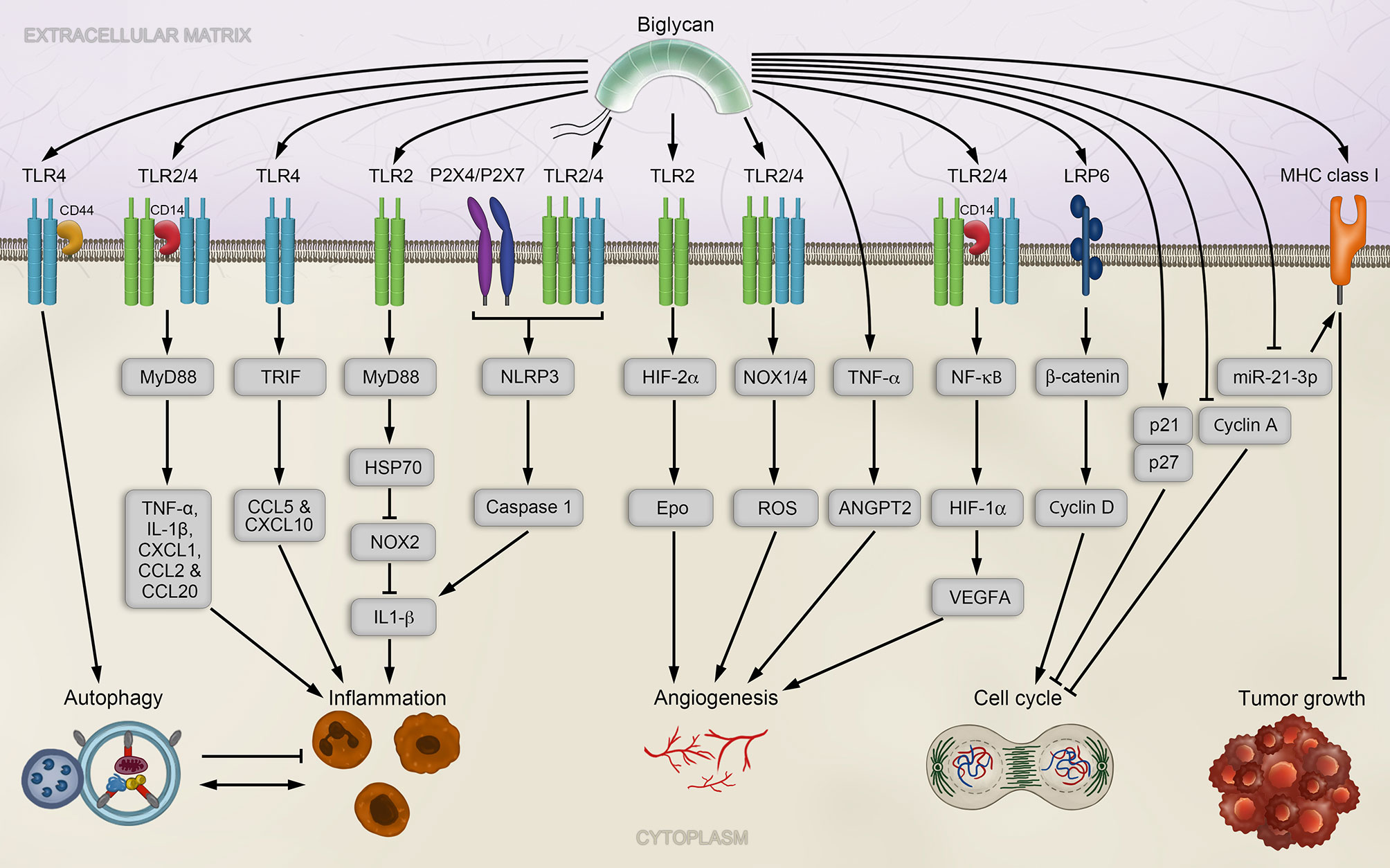
Figure 2 Biglycan signaling and implications in tumorigenesis. Soluble biglycan induces TLR4/CD44mediated autophagy in macrophages and TLR2/4/CD14-mediated inflammation via the adaptor molecule MyD88. Inflammation is also trigger by biglycan binding to TLR4 via TRIF, or TLR 2 via MyD88. Biglycan-mediated clustering of TLR2/4 with the P2X4/P2X7 receptors, which activates the NLRP3 inflammasome, consequently leading to the maturation of IL-1β. In endothelial cells, biglycan promotes angiogenesis in a TLR2/4-dependent manner by enhancing HIF-1α activity and VEGFA expression. Biglycan further affects ROS production via TLR2 and TLR4 in a NOX1- and NOX4-dependent manner. Biglycan stabilizes HIF-2α via TLR2 interaction and leads to of Epo synthesis. Biglycan knockout impaired tumor by repressing TNF-α and ANGPT2 signaling in a cancer mouse model. Additionally, biglycan can induces a cell cycle arrest by increasing the expression p27 and p21 and decreasing Cyclin A levels. By increasing MHC complex I expression via miR-21-3p, biglycan exerts anti-tumoral activities.ANGPT2, angiopoietin 2; CCL, chemokine C–C motif-ligand; CD, cluster of differentiation; CXCL, chemokine C-X-C motif-ligand; Epo, erythropoietin; HIF-1/2α, hypoxia-inducible factor 1/2α; HSP70, heat shock protein 70; IL-1β, interleukin 1 beta; LRP6, low-density lipoprotein receptor-related protein 6; MHC, major histocompatibility complex; miR, microRNA; MyD88, myeloid differentiation primary response 88; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B-cells; NLRP3, NOD-like receptor protein 3; NOX, NADPH oxidase; ROS, reactive oxygen species; TLR2/4, Toll-like receptor 2/4; TNF-α, tumor necrosis factor alpha; TRIF, TIR-domain-containing adapter-inducing interferon-β; VEGFA, vascular endothelial growth factor A.
Additionally, biglycan mediates clustering of TLR2/4 and the purigenic receptors P2X4/P2X7 (168) (Figure 2). This enables a cross-talk signaling between both receptor types and allows biglycan to autonomously activate the NLRP3 inflammasome assembly, which is followed by Caspase 1 activation and processing of pro-IL-1β and pro-IL-18 to their mature forms (168).
Biglycan signaling bridges innate and adaptive immune responses as it is involved not only in recruitment of innate immune cells, most importantly macrophages, but also induces synthesis of chemoattractants of B cells and T helper (Th) cells. Mechanistically, the biglycan/TLR4 interaction mediates signaling to sphingosine kinase-1 to regulate the synthesis of macrophage chemoattractants (169). In macrophages and dendritic cells, biglycan/TLR2/4 signaling evokes the expression of a key chemoattractant of B-cells, namely CXCL13 (144). Additionally, biglycan regulates the expression of CXCL9 and CXCL10 via TLR4 and TRIF to drive recruitment of Th1 and Th17 cells into the kidney (170). The signaling axis of biglycan and TLR2/4 also enhances antigen-specific T cell activation, potentially in a MyD88/TRIF-dependent manner, and induces autoimmune perimyocarditis (171).
The above-mentioned roles of biglycan in regulating inflammatory signaling and immune response, and in particular the recruitment of immune cells, posit several key connections between biglycan-mediated signaling and cancer-associated inflammation. For instance, the significant role of NLRP3 inflammasome activation in cancer is indisputable but ambivalent and complex (58). Thus, biglycan-mediated NLRP3 inflammasome activation provides an additional regulatory mechanism contributing to its complex regulation. Moreover, CCL2 and CCL5, whose expression is regulated in a biglycan-dependent manner, are known contributors that promote immune cells infiltration at the tumor site, metastasis, and angiogenesis (172–175). In addition, Th1 cells promote synthesis of pro-inflammatory factors, such as IL-12, IL-2, IFNγ and TNFα, thus driving inflammation. Tumor-infiltrating Th1 and Th17 cells have also been identified in various cancers inducing both, pro-tumoral and anti-tumoral immunity (176–178). In line with this, it is possible that biglycan-driven recruitment of immune cells could promote an inflammatory milieu that facilitates tumorigenesis, but also support immuno-surveillance by enhanced Th1, Th17 and macrophage recruitment in some cases with anti-tumorigenic effects of biglycan. These diverse implications of biglycan in triggering and modulating inflammation and the immune response suggest an important role for this proteoglycan in the development of tumor-associated inflammation.
3.2 Biglycan as a Regulator of Autophagy
In addition to the various biological roles of biglycan in inflammation, this proteoglycan can cause a switch between inflammation and autophagy in macrophages by selectively binding either CD14 or CD44 with equally high affinities causing an opposing biological outcome. While the interaction of biglycan and CD14, in an TLR2/4-dependent manner, results in an increase of pro-inflammatory signaling and the polarization of M1 macrophages (179), the interaction between biglycan and CD44 counteracts these events and results in the polarization of M2 macrophages as well as in an increase of autophagy-flux and anti-inflammatory effects (180) (Figure 2). The unexpected biglycan-induced opposing switch between inflammation and autophagy is solely caused by fluctuations in interaction partners. This process determines whether the downstream signaling will ultimately lead to chronic inflammation, fibrosis and possibly facilitate tumorigenesis, or promote regeneration. These findings underline how tightly those molecular processes, inflammation, and autophagy, are regulated in non-malignant tissues and presumably also in cancer. Accordingly, we presume that a subtle modulation of either one may have major impact on the other, a key factor affecting their therapeutic targeting. The described regulation of immunity responses by biglycan could, for example, affect the resolution of early pre-cancerous lesions, or promote their tumorigenic development. The cause of this switch is yet to be established and future studies will be needed to unravel the precise function of the biglycan-binding epitopes of CD14 and CD44. While these observations were made in non-malignant cells, the fact that biglycan mediates M2 macrophage polarization through autophagy induction suggests that biglycan could be involved in tumor-associated macrophage (TAM) activation during tumor progression at late stages. This is particularly important as sustained biglycan overexpression is linked to enhanced M2 macrophage numbers in ischemia reperfusion injury (180).
Emerging evidence over the past years has demonstrated the crucial role of autophagy in regulating cancer-associated inflammation, and vice versa. For example, autophagy critically affects inflammation by influencing the development, homeostasis, and survival of immune cells, including macrophages, neutrophils and lymphocytes (181, 182). In the future, it will be vital to investigate the mechanisms that regulate the intricate interplay of autophagy and inflammation. Therefore, this recent discovery of CD44 as an additional co-receptor for TLR4 and a high-affinity ligand for biglycan provide novel and valuable insights of possible cross-talk mechanisms between autophagy and inflammation (179, 180).
3.3 Mechanisms of Biglycan-Mediated Angiogenesis
Angiogenesis is an important hallmark of cancer and vital for tumor growth and metastasis, as it provides vessels for blood supply and allows transformed cells to enter the circulation (39). Despite their structural homology, the roles of decorin and biglycan in neovascularization are rather opposing. The main mechanism of biglycan signaling employs TLR2 and TLR4 receptors and results in pro-tumorigenic effects, including the production of cytokines and growth factors that ultimately promote tumor angiogenesis. For instance, biglycan-induced, upregulated VEGFA expression promotes neovascularization and cancer growth in colon cancer cells (183). Moreover, biglycan stimulation of human umbilical vein endothelial cells results in increased tubular formation capacity (184). On a molecular basis, biglycan stimulation of endothelial cells results in increased interaction between NF-κB and the HIF-1α promoter in a TLR2/4-dependent manner, enhanced HIF-1α levels and activity, and consequently increased VEGFA expression (184) (Figure 2). These findings support a promoting role of biglycan in cancer development and progression, likely by promoting and activating tumor angiogenesis (184).
An additional mechanism that links biglycan signaling to angiogenesis regulation is the enhanced production of reactive oxygen species (ROS). ROS can be produced by NOX enzymes and result in enhanced angiogenesis and tumor growth by increased NF-κB and VEGF expression (185). Due to the ability of biglycan to affect ROS production via TLR2 and TLR4 in a NOX1- and NOX4-dependent manner (166, 168) (Figure 2), it is very likely that biglycan potentially influences tumor angiogenesis via ROS level regulation.
Alternatively, angiogenesis in cancer could be affected by biglycan through the stabilization HIF-2α, which is induced by TLR2 interaction with biglycan and results in the induction of erythropoietin (Epo) synthesis and polycythemia (186) (Figure 2). Changes in HIF-2α expression levels have been described in a variety of solid tumors and are linked to TAMs and HIF-2α is, similar to biglycan, associated with late tumor progression stages (187).
In a mouse model of breast cancer with tumor-bearing mice, knockout of biglycan in the stroma inhibited metastasis to the lung, impaired tumor angiogenesis and normalized tumor vasculature by repressing TNF-α and angiopoietin 2 (ANGPT2) signaling (188) (Figure 2).
Several studies have focused on the role of biglycan in colon cancer (183, 189, 190). For instance, it was shown that, in colon cancer cells, inhibiting biglycan results in increased expression of pro-apoptotic effectors and is linked to suppressed NF-κB pathway activity. On the other hand, biglycan overexpression does the opposite effect (190). Moreover, biglycan-mediated chemotherapy resistance in colon cancer cells occurs by activating NF-κB signaling (190). Considering the well-established, TLR2/4-dependent function of biglycan in NF-κB signaling, the fact that TLRs have been linked to tumorigenesis of multiple tumor types (191, 192), and the known implications of the NF-κB pathway in the development of chemotherapy resistance in cancers (193, 194), it is convincing that biglycan may promote resistance mechanisms by regulating the NF-κB pathway.
3.4 The Role of Biglycan in Promoting Cancer Cell Proliferation, Invasion, and Metastasis
Already a decade ago, a study in osteoprogenitor cells and bone fracture healing models revealed the ability of biglycan to enhance canonical Wnt signaling and to interact with the Wnt co-receptor low-density lipoprotein receptor-related protein 6 (LRP6) (195). Wnt signaling is considered to be a major pathway regulating development and has also been closely linked to carcinogenesis, particularly in the context of colorectal cancer (196, 197). Therefore, these findings suggested a potential function of biglycan in tumorigenesis based on its ability to affect cancer cell proliferation via Wnt signaling modulation. More recently, a link between biglycan and Wnt/LRP6 signaling was additionally reported in the context of osteosarcoma. Specifically, biglycan favors MG63 osteosarcoma cell proliferation via a LPR6/β-catenin/IGF-1R signaling axis and functions in a positive feedback loop regulating osteosarcoma growth (198). By binding to LRP6, biglycan inhibits the destruction of β-catenin, an inducer of Cyclin D1 expression. Increased Cyclin D1 levels, in turn, activate IGF-1R and ultimately result in enhanced biglycan secretion (198).
Another observation that links biglycan signaling to the regulation of cancer cell proliferation was reported in a study conducted in HCT116 colon cancer cells. In this cellular context, suppression of biglycan expression via short hairpin RNA (shRNA) decreases cancer cell proliferation and causes cell cycle arrest at the G0/G1 phase, accompanied by decreased levels of cell cycle associated proteins, including Cyclin A and Cyclin D1 (189). In contrast, expression levels of p21 and p27 are markedly increased in the cancer cells of the control shRNA group (189). Furthermore, the decreased biglycan levels suppressed colon cancer cell migration and invasion, and induced apoptosis in a p38 and MAPK-dependent manner (189).
In the context of gastric cancer, significantly increased tissue biglycan levels have been linked to lymph node metastasis and depth of tumor invasion, both in vivo and in vitro (199). In vitro, investigation of endothelial cell migration, invasion and tube formation ability was positively linked to biglycan levels (199). In vivo, biglycan induced focal adhesion kinase (FAK) phosphorylation, suggesting an oncogenic function of biglycan in gastric cancer metastasis in a FAK signaling-dependent manner (199). In line with this, biglycan was recently shown to modulate gastric cancer aggressive features as cell survival, migration, and angiogenesis and biglycan knockout gastric cancer cells showed increased levels of PARP1 (Poly [ADP-ribose] polymerase 1) and Caspase 3 cleavage (200).
Additional roles of biglycan in cancer cell proliferation, migration and metastasis were reported in tumor endothelial cells, where biglycan mediated tumor cell migration via TLR2/TLR4/NF-κB/Erk1/2 (201), in melanoma cells, where biglycan increased invasiveness by enhancing integrin-β1 expression (202), and in endometrial cancer cells, where knockdown of biglycan reduced migration, tubular formation and metastasis (203). Collectively, these studies point to a pro-tumorigenic role of biglycan proteoglycan in both mesenchymal (osteosarcoma) and epithelial (colon and breast carcinomas) malignancies.
3.5 Anti-Tumorigenic Effects Mediated by Biglycan
The vast majority of research on biglycan in cancer indicate its pro-tumorigenic function. However, several studies have also linked anti-tumorigenic effects to higher biglycan expression levels. Tumor suppressive effects have, for instance, been reported in pancreatic cancer cell lines, where treatment with exogenous biglycan leads to an up-regulation of cyclin-dependent kinase inhibitor p27, down-regulation of Cyclin A and PCNA (proliferating cell nuclear antigen), and cell cycle arrest (204) (Figure 2).
Similarly, treatment with exogenous biglycan of bladder cancer cells inhibits cell proliferation and high biglycan levels correlate with prolonged survival of bladder cancer patients (205), and superior patient prognosis of patients with diffuse large B-cell lymphoma (206). In this context, CD40+ tumors exhibit increased levels of biglycan that additionally correlate with the number of infiltrating macrophages and CD4+ and CD8+ T-cells. In vitro experiments have demonstrated that CD40 signaling can enhance antigen presentation from malignant B-cells, thus inducing an autologous immune response (207, 208). In line with this, higher biglycan levels could be linked to a high intra-tumoral inflammatory reaction that may result in an increased tumor response, and thereby a better prognosis (206).
In an in vitro model of oncogenic transformation, the role of biglycan in the initiation and maintenance of neoplastic transformation was investigated (209). In this study, the HER-2-mediated oncogenic transformation caused silencing of biglycan gene expression, while reconstitution of biglycan expression led to an impaired proliferation and migration of oncogenic transformed cells (209). Therefore, HER2-mediated silencing of biglycan expression may promote tumor cell proliferation and migration and provides a putative therapeutic target for the treatment of HER2+ tumor cells (209). Moreover, restoration of biglycan in HER2 fibroblasts decreases their tumorigenic potential when compared to HER2 cells with low biglycan levels (210). Biglycan restoration is linked to an enhanced immune cell responses and increases numbers of immune effector cells in tumors and peripheral blood, possibly resulting from up-regulated major histocompatibility complex (MHC) class I surface antigens, and decreased expression levels of TGF-β and the TGF-β receptor 1 (210). The overexpression of biglycan in HER2 cells additionally mediates an upregulation of decorin, which also elevates MHC class I surface expression in biglycan negative HER2 cells, suggesting a putative synergistic action of both SLRPs (210). Most recently, miR-21-3p mediates down-regulation of MHC class I surface antigens, interferes with the expression of immune-modulating effectors and promotes immune suppression in HER-2/neu cells (211). Induced overexpression of biglycan in HER‐2/neu cells increases MHC class I expression and decreases miR‐21‐3p, highlighting the vital role of biglycan in the MHC class I‐driven immune escape in tumor cells (211) (Figure 2).
4 Therapeutic Implications and Future Perspectives
Cancer development and progression are multifaceted processes that involve an intricate interplay of genetic and environmental factors. Transformed cells continuously interact with their surroundings, mostly constituted by ECM components. Therefore, proteoglycans, key constituents of the tumor stroma, are increasingly recognized as crucial contributors in shaping the cancer microenvironment by regulating inflammatory and immune responses, affecting autophagy, mediating angiogenesis, and modulating cellular signaling pathways to control cell proliferation, migration and metastasis. All these processes can ultimately either promote resolution and regeneration, or lead to tumor development and disease progression. The diverse implications of proteoglycans in these processes highlight their broad potential as therapeutic targets in cancer therapy. Indeed, the characteristic expression patterns of proteoglycans and their interaction partners in diverse tumor types have been exploited as biomarkers for prognosis and indication for therapy efficiencies.
The well-known pan-tyrosine kinase inhibiting capabilities of decorin have made the SLRP an attractive target for cancer therapies. Although the underlying molecular functions of decorin are complex and not yet fully understood, the evidence favor an antitumorigenic role of decorin. This notion holds great potential of clinical relevance and has led to a variety of studies focused on expressing decorin to attenuate tumorigenic growth (17, 212). Delivery of decorin can be mediated via adenoviral vectors or by systemic administration of decorin proteoglycan, protein core or fragments (212). Numerous studies have showed that expression of exogenous decorin can suppress tumor growth, in particular for cancer types that depend on RTK signaling, such as EGFR, Met and IGF-1R (212). Delivery of decorin via adenoviral vectors into the tumor cells has been demonstrated to inhibit tumor growth of colon, lung, and squamous cell carcinoma (122, 213). Other decorin gene therapy approaches have been successfully tested in various types of cancer including models of breast cancer (214), glioblastoma (215, 216), prostate tumors (213). These studies have largely confirmed the therapeutic capability of decorin in suppressing tumor development and/or growth (212). Despite the promising evidence from in vivo and in vitro studies, and although decorin is unlikely to exhibit toxic side effects, the translation into a clinical drug has not been finalized. One major challenge that limits the development of decorin-based clinical drugs are technical limitations in proteoglycan mass production (217). Future improvements in production, delivery and efficiency will improve the rationale for applying decorin in the treatment of cancer.
Over the past few years, our understanding of how biglycan mediated signaling in the state of health and during disease progression has significantly improved. In particular, the recent evidence of the fine-tuned biglycan-mediated regulation of the inflammatory response, which is orchestrated via distinct receptor, co-receptor, and adaptor molecules, constitutes an important step towards understanding how biglycan signaling can induce different, and often conflicting, downstream signaling outcomes (218). The biglycan-mediated switch between inflammation and autophagy, depending on the respective co-receptor (167, 180), is of particular interest, but raises several questions that await further investigations. These questions include for example, the identification of the binding stoichiometry of biglycan and its co-receptors CD14 and CD44, of the selection mechanisms that govern the choice of the co-receptors and of the CD14 and CD44 binding motifs. The answers will ultimately enable the translation of the current knowledge of biglycan-mediated signaling into pharmacological interventions, as the ability to precisely modulate the biglycan-induced downstream signaling could be a promising therapeutic approach.
In contrast to decorin, which is associated with mainly anti-tumorigenic effects and where administration of the proteoglycan provides a potent strategy for cancer therapy, biglycan is rather linked to pro-tumorigenic effects. However, TLRs have crucial physiological functions, inhibitory targeting of biglycan-mediated signaling cascades requires further studies to gain precise knowledge on the co-receptors and downstream mechanisms of biglycan/TLR signaling to ensure selective targeting in the context of cancer. In cases where tumor growth is linked to decreased levels of biglycan, delivering biglycan could be beneficial to promote immunogenicity. For example, in the context of HER2 cells, where increased biglycan expression is linked to tumor suppression, the recent discovery of the biglycan/miR-21-3p/MHC class I axis provides an interesting and innovative therapeutic concept for HER2+ cancers by administration of biglycan (211). This could increase immunogenicity mediated by a strong infiltration with effector T cells, macrophages, and support inflammation by the production of pro-inflammatory factors, thus inhibit the escape from immune surveillance. Lastly, it is of note that the regulation of tumorigenesis likely depends on the synergistic and timely action of several components of the ECM and thus it is indispensable to broaden our knowledge on the intricate interplay of these proteoglycans in normal and malignant physiology.
In spite of the rapid progress in this field of research, there are still several outstanding questions. For example, given the cell-dependent context of decorin and biglycan in evoking autophagy in tumorigenesis, at what stage do these proteoglycans play an active role in either suppressing or promoting tumor development? Moreover, do the downstream signaling effects of these autophagic modulators occur independently or are they synergistically coordinated in the tumor stroma? Is decorin vs biglycan more critical in regulating disease-altering autophagy in certain types of malignancies over others? We feel that further investigations into these inquiries are required to better decipher the aberrant matrix remodeling and clarify the disease-driving impact of the ECM in carcinogenesis.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The authors’ laboratories were supported by the German Research Council (SFB 1039, project B02, SFB 1177, 259130777, project E02, all to LS; and the CardioPulmonary Institute (CPI), EXC 2026, Project ID: 390649896 (to LS and MW); WY119/1-3 (to MW), the Else Kroner-Fresenius-Foundation (to MW), and the German Center for Lung Research (to MW);¨ European Union’s Horizon 2020 research and innovation program’s MICROBPREDICT study (No 825694), European Union’s Horizon 2020 Research and Innovation Program GALAXY (No. 668031) and Societal Challenges LIVERHOPE (Health, demographic change, and well-being, No. 731875) the German Research Council (SFB TRR57, CRC1382), Cellex Foundation (PREDICT) all to JT. The original research in Dr. Iozzo laboratory was supported in part by National Institutes of Health Grants RO1 CA39481 and RO1 CA245311.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors apologize to those researchers whose work could not be cited due to space limitation.
Glossary

References
1. DeClerck YA, Pienta KJ, Woodhouse EC, Singer DS, Mohla S. The Tumor Microenvironment at a Turning Point—Knowledge Gained Over the Last Decade, and Challenges and Opportunities Ahead: A White Paper From the NCI TME Network. Cancer Res (2017) 77:1051–9. doi: 10.1158/0008-5472.CAN-16-1336
2. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79:4557–66. doi: 10.1158/0008-5472.CAN-18-3962
3. Werb Z, Lu P. The Role of Stroma in Tumor Development. Cancer J (2015) 21:250. doi: 10.1097/PPO.0000000000000127
4. Noy R, Pollard JW. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
5. Lewis CE, Harney AS, Pollard JW. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell (2016) 30:18–25. doi: 10.1016/j.ccell.2016.05.017
6. Santi A, Kugeratski FG, Zanivan S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics (2018) 18:1700167. doi: 10.1002/pmic.201700167
7. Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, et al. The Role of Tumor Stroma in Cancer Progression and Prognosis: Emphasis on Carcinoma-Associated Fibroblasts and Non-Small Cell Lung Cancer. J Thorac Oncol (2011) 6:209–17. doi: 10.1097/JTO.0b013e3181f8a1bd
8. Palucka AK, Coussens LM. The Basis of Oncoimmunology. Cell (2016) 164:1233–47. doi: 10.1016/j.cell.2016.01.049
9. Picoli CC, Gonçalves BÔ, Santos GS, Rocha BG, Costa AC, Resende RR, et al. Pericytes Cross-Talks Within the Tumor Microenvironment. Biochim Biophys Acta (BBA)-Reviews Cancer (2021) 41:188608. doi: 10.1016/j.bbcan.2021.188608
10. Fukumura D, Duda DG, Munn LL, Jain RK. Tumor Microvasculature and Microenvironment: Novel Insights Through Intravital Imaging in Pre-Clinical Models. Microcirculation (2010) 17:206–25. doi: 10.1111/j.1549-8719.2010.00029.x
11. Forster JC, Harriss-Phillips WM, Douglass MJ, Bezak E. A Review of the Development of Tumor Vasculature and Its Effects on the Tumor Microenvironment. Hypoxia (2017) 5:21–32. doi: 10.2147/HP.S133231
13. Henke E, Nandigama R, Ergün S. Extracellular Matrix in the Tumor Microenvironment and Its Impact on Cancer Therapy. Front Mol Biosci (2020) 6:160. doi: 10.3389/fmolb.2019.00160
14. Neill T, Schaefer L, Iozzo RV. Instructive Roles of Extracellular Matrix on Autophagy. Am J Pathol (2014) 184:2146–53. doi: 10.1016/j.ajpath.2014.05.010
15. Schaefer L, Dikic I. Autophagy: Instructions From the Extracellular Matrix. Matrix Biol (2021) 101:1–11. doi: 10.1016/j.matbio.2021.06.002
16. Hastings JF, Skhinas JN, Fey D, Croucher DR, Cox TR. The Extracellular Matrix as a Key Regulator of Intracellular Signalling Networks. Br J Pharmacol (2019) 176:82–92. doi: 10.1111/bph.14195
17. Schaefer L, Tredup C, Gubbiotti MA, Iozzo RV. Proteoglycan Neofunctions: Regulation of Inflammation and Autophagy in Cancer Biology. FEBS J (2017) 284:10–26. doi: 10.1111/febs.13963
18. Moreth K, Iozzo RV, Schaefer L. Small Leucine-Rich Proteoglycans Orchestrate Receptor Crosstalk During Inflammation. Cell Cycle (2012) 11:2084–91. doi: 10.4161/cc.20316
19. Miller AE, Hu P, Barker TH. Feeling Things Out: Bidirectional Signaling of the Cell–ECM Interface, Implications in the Mechanobiology of Cell Spreading, Migration, Proliferation, and Differentiation. Adv Healthcare Mater (2020) 9:1901445. doi: 10.1002/adhm.201901445
20. Raines EW. The Extracellular Matrix can Regulate Vascular Cell Migration, Proliferation, and Survival: Relationships to Vascular Disease. Int J Exp Pathol (2000) 81:173–82. doi: 10.1046/j.1365-2613.2000.00155.x
21. Sorokin L. The Impact of the Extracellular Matrix on Inflammation. Nat Rev Immunol (2010) 10:712–23. doi: 10.1038/nri2852
22. Rozario T, DeSimone DW. The Extracellular Matrix in Development and Morphogenesis: A Dynamic View. Dev Biol (2010) 341:126–40. doi: 10.1016/j.ydbio.2009.10.026
23. Nastase MV, Young MF, Schaefer L. Biglycan: A Multivalent Proteoglycan Providing Structure and Signals. J Histochem Cytochem (2012) 60:963–75. doi: 10.1369/0022155412456380
24. Gullberg D, Paraskevi H, Schaefer L, Tenni R, Theocharis A, Winberg JO. Extracellular Matrix: Pathobiology and Signaling (Walter De Gruyter). Boston/Berlin: Walter de Gruyter (2012).
25. Merline R, Schaefer RM, Schaefer L. The Matricellular Functions of Small Leucine-Rich Proteoglycans (Slrps). J Cell Commun Signaling (2009) 3:323–35. doi: 10.1007/s12079-009-0066-2
26. Zeng-Brouwers J, Pandey S, Trebicka J, Wygrecka M, Schaefer L. Communications via the Small Leucine-Rich Proteoglycans: Molecular Specificity in Inflammation and Autoimmune Diseases. J Histochem Cytochem (2020) 68:887–906. doi: 10.1369/0022155420930303
27. Schulz M, Diehl V, Trebicka J, Wygrecka M, Schaefer L. Biglycan in Hepatorenal Crosstalk Biglycan: A Regulator of Hepatorenal Inflammation and Autophagy. Matrix Biol (2021) 100–101:150–61. doi: 10.1016/j.matbio.2021.06.001
28. Ortiz C, Schierwagen R, Schaefer L, Klein S, Trepat X, Trebicka J. Extracellular Matrix Remodeling in Chronic Liver Disease. Curr Tissue Microenviron Rep (2021) 2:1–12. doi: 10.1007/s43152-021-00030-3
29. Bonnans C, Chou J, Werb Z. Remodelling the Extracellular Matrix in Development and Disease. Nat Rev Mol Cell Biol (2014) 15:786–801. doi: 10.1038/nrm3904
30. Iozzo RV, Gubbiotti MA. Extracellular Matrix: The Driving Force of Mammalian Diseases. Matrix Biol (2018) 71:1–9. doi: 10.1016/j.matbio.2018.03.023
31. Pickup MW, Mouw JK, Weaver VM. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep (2014) 15:1243–53. doi: 10.15252/embr.201439246
32. Paolillo M, Schinelli S. Extracellular Matrix Alterations in Metastatic Processes. Int J Mol Sci (2019) 20:4947. doi: 10.3390/ijms20194947
33. Brassart-Pasco S, Brézillon S, Brassart B, Ramont L, Oudart JB, Monboisse JC. Tumor Microenvironment: Extracellular Matrix Alterations Influence Tumor Progression. Front Oncol (2020) 10:397. doi: 10.3389/fonc.2020.00397
34. Oskarsson T, Massagué J. Extracellular Matrix Players in Metastatic Niches. EMBO J (2012) 31:254–6. doi: 10.1038/emboj.2011.469
35. Zhuyan J, Chen M, Zhu T, Bao X, Zhen T, Xing K, et al. Critical Steps to Tumor Metastasis: Alterations of Tumor Microenvironment and Extracellular Matrix in the Formation of Pre-Metastatic and Metastatic Niche. Cell Biosci (2020) 10:1–9. doi: 10.1186/s13578-020-00453-9
36. Yamauchi M, Gibbons DL, Zong C, Fradette JJ, Bota-Rabassedas N, Kurie JM. Fibroblast Heterogeneity and Its Impact on Extracellular Matrix and Immune Landscape Remodeling in Cancer. Matrix Biol (2020) 91:8–18. doi: 10.1016/j.matbio.2020.05.001
37. Nazemi M, Rainero E. Cross-Talk Between the Tumor Microenvironment, Extracellular Matrix, and Cell Metabolism in Cancer. Front Oncol (2020) 10:239. doi: 10.3389/fonc.2020.00239
38. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
39. Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
40. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-Related Inflammation, the Seventh Hallmark of Cancer: Links to Genetic Instability. Carcinogenesis (2009) 30:1073–81. doi: 10.1093/carcin/bgp127
41. Dunn GP, Old LJ, Schreiber RD. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
42. Hibino S, Kawazoe T, Kasahara H, Itoh S, Ishimoto T, Sakata-Yanagimoto M, et al. Inflammation-Induced Tumorigenesis and Metastasis. Int J Mol Sci (2021) 22:5421. doi: 10.3390/ijms22115421
43. Castle PE, Hillier SL, Rabe LK, Hildesheim A, Herrero R, Bratti MC, et al. An Association of Cervical Inflammation With High-Grade Cervical Neoplasia in Women Infected With Oncogenic Human Papillomavirus (HPV). Cancer Epidemiol Prev Biomarkers (2001) 10:1021–7.
44. Balkwill F, Charles KA, Mantovani A. Smoldering and Polarized Inflammation in the Initiation and Promotion of Malignant Disease. Cancer Cell (2005) 7:211–7. doi: 10.1016/j.ccr.2005.02.013
45. Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025
46. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat Rev Cancer (2002) 3:991–8. doi: 10.1038/ni1102-991
47. Dirnhofer S, Zippelius A. Cancer Immunology, Inflammation, and Tolerance: An Introduction. Eur J Pathol (2019) 474:405–6. doi: 10.1007/s00428-019-02547-3
48. Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat Med (2018) 24:541–50. doi: 10.1038/s41591-018-0014-x
49. Kumar A, Swain CA, Shevde LA. Informing the New Developments and Future of Cancer Immunotherapy. Cancer Metastasis Rev (2021) 40:549–62. doi: 10.1007/s10555-021-09967-1
50. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-Related Inflammation. Nature (2008) 454:436–44. doi: 10.1038/nature07205
51. Hou J, Karin M, Sun B. Targeting Cancer-Promoting Inflammation—Have Anti-Inflammatory Therapies Come of Age? Nat Rev Clin Oncol (2021) 18:261–79. doi: 10.1038/s41571-020-00459-9
52. Taniguchi K, Karin M. NF-κB, Inflammation, Immunity and Cancer: Coming of Age. Nat Rev Immunol (2018) 18:309–24. doi: 10.1038/nri.2017.142
53. Sabroe I, Parker L, Dower S, Whyte M. The Role of TLR Activation in Inflammation. J Pathol: A J Pathological Soc Great Britain Ireland (2008) 214:126–35. doi: 10.1002/path.2264
54. Sun SC, Ley SC. New Insights Into NF-κB Regulation and Function. Trends Immunol (2008) 29:469–78. doi: 10.1016/j.it.2008.07.003
55. Bryant CE, Gay NJ, Heymans S, Sacre S, Schaefer L, Midwood KS. Advances in Toll-Like Receptor Biology: Modes of Activation by Diverse Stimuli. Crit Rev Biochem Mol Biol (2015) 50:359–79. doi: 10.3109/10409238.2015.1033511
56. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 Inflammasome in Cancer. Mol Cancer (2018) 17:1–13. doi: 10.1186/s12943-018-0900-3
57. Sharma BR, Kanneganti TD. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat Immunol (2021) 22:550–9. doi: 10.1038/s41590-021-00886-5
58. Hamarsheh S, Zeiser R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front Immunol (2020) 11:1444. doi: 10.3389/fimmu.2020.01444
59. Li X, He S, Ma B. Autophagy and Autophagy-Related Proteins in Cancer. Mol Cancer (2020) 19:1–16. doi: 10.1186/s12943-019-1085-0
60. Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F. Autophagy and Cancer Stem Cells: Molecular Mechanisms and Therapeutic Applications. Cell Death Differ (2019) 26:690–702. doi: 10.1038/s41418-019-0292-y
61. Garg M. Epithelial Plasticity, Autophagy and Metastasis: Potential Modifiers of the Crosstalk to Overcome Therapeutic Resistance. Stem Cell Rev Rep (2020) 16:503–10. doi: 10.1007/s12015-019-09945-9
62. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy Suppresses Tumorigenesis Through Elimination of P62. Cell (2009) 137:1062–75. doi: 10.1016/j.cell.2009.03.048
63. Liu W, Meng Y, Zong C, Zhang S, Wei L. Autophagy and Tumorigenesis. In: Autophagy: Biology and Diseases. Singapore: Springer (2020). p. 12.
64. Mainz L, Rosenfeldt MT. Autophagy and Cancer–Insights From Mouse Models. FEBS J (2018) 285:792–808. doi: 10.1111/febs.14274
65. Cianfanelli V, Fuoco C, Lorente M, Salazar M, Quondamatteo F, Gherardini PF, et al. AMBRA1 Links Autophagy to Cell Proliferation and Tumorigenesis by Promoting C-Myc Dephosphorylation and Degradation. Nat Cell Biol (2015) 17:20–30. doi: 10.1038/ncb3072
66. Amaravadi RK, Kimmelman AC, Debnath J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov (2019) 9:1167–81. doi: 10.1158/2159-8290.CD-19-0292
67. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras Requires Autophagy to Maintain Oxidative Metabolism and Tumorigenesis. Genes Dev (2011) 25:460–70. doi: 10.1101/gad.2016311
68. Seton-Rogers S. Eliminating Protective Autophagy in KRAS-Mutant Cancers. Nat Rev Cancer (2019) 19:247–7. doi: 10.1038/s41568-019-0137-5
69. Bryant KL, Der CJ. Blocking Autophagy to Starve Pancreatic Cancer. Nat Rev Mol Cell Biol (2019) 20:265–5. doi: 10.1038/s41580-019-0120-8
70. Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A. Blocked Autophagy Sensitizes Resistant Carcinoma Cells to Radiation Therapy. Cancer Res (2008) 68:1485–94. doi: 10.1158/0008-5472.CAN-07-0562
71. Liu D, Yang Y, Liu Q, Wang J. Inhibition of Autophagy by 3-MA Potentiates Cisplatin-Induced Apoptosis in Esophageal Squamous Cell Carcinoma Cells. Med Oncol (2011) 28:105–11. doi: 10.1007/s12032-009-9397-3
72. Shingu T, Fujiwara K, Bögler O, Akiyama Y, Moritake K, Shinojima N, et al. Inhibition of Autophagy at a Late Stage Enhances Imatinib-Induced Cytotoxicity in Human Malignant Glioma Cells. Int J Cancer (2009) 124:1060–71. doi: 10.1002/ijc.24030
73. Galluzzi L, Bravo-San Pedro JM, Demaria S, Formenti SC, Kroemer G. Activating Autophagy to Potentiate Immunogenic Chemotherapy and Radiation Therapy. Nat Rev Clin Oncol (2017) 14:247–58. doi: 10.1038/nrclinonc.2016.183
74. Papandreou ME, Tavernarakis N. Crosstalk Between Endo/Exocytosis and Autophagy in Health and Disease. Biotechnol J (2020) 15:1900267. doi: 10.1002/biot.201900267
75. Chen HT, Liu H, Mao MJ, Tan Y, Mo XQ, Meng XJ, et al. Crosstalk Between Autophagy and Epithelial-Mesenchymal Transition and Its Application in Cancer Therapy. Mol Cancer (2019) 18:1–19. doi: 10.1186/s12943-018-0930-x
76. Catalano M, D’Alessandro G, Lepore F, Corazzari M, Caldarola S, Valacca C, et al. Autophagy Induction Impairs Migration and Invasion by Reversing EMT in Glioblastoma Cells. Mol Oncol (2015) 9:1612–25. doi: 10.1016/j.molonc.2015.04.016
77. Lv Q, Wang W, Xue J, Hua F, Mu R, Lin H, et al. DEDD Interacts With PI3KC3 to Activate Autophagy and Attenuate Epithelial–Mesenchymal Transition in Human Breast Cancer. Cancer Res (2012) 72:3238–50. doi: 10.1158/0008-5472.CAN-11-3832
78. Bao Y, Ding Z, Zhao P, Li J, Chen P, Zheng J, et al. Autophagy Inhibition Potentiates the Anti-EMT Effects of Alteronol Through TGF-β/Smad3 Signaling in Melanoma Cells. Cell Death Dis (2020) 11:1–10. doi: 10.1038/s41419-020-2419-y
79. Shen H, Yin L, Deng G, Guo C, Han Y, Li Y, et al. Knockdown of Beclin-1 Impairs Epithelial-Mesenchymal Transition of Colon Cancer Cells. J Cell Biochem (2018) 119:7022–31. doi: 10.1002/jcb.26912
80. Zhu HD, Liu L, Deng H, Li ZB, Sheng JQ, He XX, et al. Astrocyte Elevated Gene 1 (AEG-1) Promotes Anoikis Resistance and Metastasis by Inducing Autophagy in Hepatocellular Carcinoma. J Cell Physiol (2020) 235:5084–95. doi: 10.1002/jcp.29377
81. Peng YF, Shi YH, Ding ZB, Ke AW, Gu CY, Hui B, et al. Autophagy Inhibition Suppresses Pulmonary Metastasis of HCC in Mice via Impairing Anoikis Resistance and Colonization of HCC Cells. Autophagy (2013) 9:2056–68. doi: 10.4161/auto.26398
82. Liu T, Zhang J, Li K, Deng L, Wang H. Combination of an Autophagy Inducer and an Autophagy Inhibitor: A Smarter Strategy Emerging in Cancer Therapy. Front Pharmacol (2020) 11:408. doi: 10.3389/fphar.2020.00408
83. Cordani M, Somoza Á, Tafani M, Dando I, Kumar S. Novel Cancer Treatments Based on Autophagy Modulation. Front Pharmacol (2021) 12:650559. doi: 10.3389/fphar.2021.650559
84. Ruoslahti E. Structure and Biology of Proteoglycans. Annu Rev Cell Biol (1988) 4:229–55. doi: 10.1146/annurev.cb.04.110188.001305
85. Iozzo RV, Schaefer L. Proteoglycan Form and Function: A Comprehensive Nomenclature of Proteoglycans. Matrix Biol (2015) 42:11–55. doi: 10.1016/j.matbio.2015.02.003
86. Esko JD, Kimata K, Lindahl U. Proteoglycans and Sulfated Glycosaminoglycans. In: Essentials of Glycobiology, 2nd edition. La Jolla, California: Cold Spring Harbor Laboratory Press (2009) p. 1–27.
87. Schaefer L, Schaefer RM. Proteoglycans: From Structural Compounds to Signaling Molecules. Cell Tissue Res (2010) 339:237–46. doi: 10.1007/s00441-009-0821-y
88. Neill T, Schaefer L, Iozzo RV. Decoding the Matrix: Instructive Roles of Proteoglycan Receptors. Biochemistry (2015) 54:4583–98. doi: 10.1021/acs.biochem.5b00653
89. Iozzo RV. The Family of the Small Leucine-Rich Proteoglycans: Key Regulators of Matrix Assembly and Cellular Growth. Biochem Mol Biol (1997) 32:141–74. doi: 10.3109/10409239709108551
90. Matsushima N, Miyashita H, Kretsinger RH. Sequence Features, Structure, Ligand Interaction, and Diseases in Small Leucine Rich Repeat Proteoglycans. J Cell Commun Signaling (2021) 1:1–13. doi: 10.1007/s12079-021-00616-4
91. Huxley-Jones J, Robertson DL, Boot-Handford RP. On the Origins of the Extracellular Matrix in Vertebrates. Matrix Biol (2007) 26:2–11. doi: 10.1016/j.matbio.2006.09.008
92. Hocking AM, Shinomura T, McQuillan DJ. Leucine-Rich Repeat Glycoproteins of the Extracellular Matrix. Matrix Biol (1998) 17:1–19. doi: 10.1016/S0945-053X(98)90121-4
93. Schaefer L. Complexity of Danger: The Diverse Nature of Damage-Associated Molecular Patterns. J Biol Chem (2014) 289:35237–45. doi: 10.1074/jbc.R114.619304
94. Frey H, Schroeder N, Manon-Jensen T, Iozzo RV, Schaefer L. Biological Interplay Between Proteoglycans and Their Innate Immune Receptors in Inflammation. FEBS J (2013) 280:2165–79. doi: 10.1111/febs.12145
95. Iozzo RV, Sanderson RD. Proteoglycans in Cancer Biology, Tumour Microenvironment and Angiogenesis. J Cell Mol Med (2011) 15:1013–31. doi: 10.1111/j.1582-4934.2010.01236.x
96. Iozzo RV. The Biology of the Small Leucine-Rich Proteoglycans: Functional Network of Interactive Proteins. J Biol Chem (1999) 274:18843–6. doi: 10.1074/jbc.274.27.18843
97. Iozzo RV, Moscatello DK, McQuillan DJ, Eichstetter I. Decorin is a Biological Ligand for the Epidermal Growth Factor Receptor. J Biol Chem (1999) 274:4489–92. doi: 10.1074/jbc.274.8.4489
98. Järveläinen H, Sainio A, Wight TN. Pivotal Role for Decorin in Angiogenesis. Matrix Biol (2015) 43:15–26. doi: 10.1016/j.matbio.2015.01.023
99. Neill T, Schaefer L, Iozzo RV. Decorin: A Guardian From the Matrix. Am J Pathol (2012) 181:380–7. doi: 10.1016/j.ajpath.2012.04.029
100. Salomaki HH, Sainio AO, Söderström M, Pakkanen S, Laine J, Jarvelainen HT. Differential Expression of Decorin by Human Malignant and Benign Vascular Tumors. J Histochem Cytochem (2008) 56:639–46. doi: 10.1369/jhc.2008.950287
101. Vogel KG, Paulsson M, Heinegård D. Specific Inhibition of Type I and Type II Collagen Fibrillogenesis by the Small Proteoglycan of Tendon. Biochem J (1984) 223:587–97. doi: 10.1042/bj2230587
102. Scott JE. Proteoglycan-Fibrillar Collagen Interactions. Biochem J (1988) 252:313–23. doi: 10.1042/bj2520313
103. Yamaguchi Y, Mann DM, Ruoslahti E. Negative Regulation of Transforming Growth Factor-βby the Proteoglycan Decorin. Nature (1990) 346:281–4. doi: 10.1038/346281a0
104. Iozzo RV, Chakrani F, Perrotti D, McQuillan DJ, Skorski T, Calabretta B, et al. Cooperative Action of Germ-Line Mutations in Decorin and P53 Accelerates Lymphoma Tumorigenesis. Proc Natl Acad Sci (1999) 96:3092–7. doi: 10.1073/pnas.96.6.3092
105. Mao L, Yang J, Yue J, Chen Y, Zhou H, Fan D, et al. Decorin Deficiency Promotes Epithelial-Mesenchymal Transition and Colon Cancer Metastasis. Matrix Biol (2021) 95:1–14. doi: 10.1016/j.matbio.2020.10.001
106. Goldoni S, Humphries A, Nyström A, Sattar S, Owens RT, McQuillan DJ, et al. Decorin Is a Novel Antagonistic Ligand of the Met Receptor. J Cell Biol (2009) 185:743–54. doi: 10.1083/jcb.200901129
107. Buraschi S, Pal N, Tyler-Rubinstein N, Owens RT, Neill T, Iozzo RV. Decorin Antagonizes Met Receptor Activity and Down-Regulates β-Catenin and Myc Levels. J Biol Chem (2010) 285:42075–85. doi: 10.1074/jbc.M110.172841
108. Neill T, Schaefer L, Iozzo RV. Oncosuppressive Functions of Decorin. Mol Cell Oncol (2015) 2:e975645. doi: 10.4161/23723556.2014.975645
109. Boccaccio C, Comoglio PM. Invasive Growth: A MET-Driven Genetic Programme for Cancer and Stem Cells. Nat Rev Cancer (2006) 6:637–45. doi: 10.1038/nrc1912
110. Chen K, Ma X, Nehme A, Wei J, Cui Y, Cui Y, et al. Time-Delimited Signaling of MET Receptor Tyrosine Kinase Regulates Cortical Circuit Development and Critical Period Plasticity. Mol Psychiatry (2020) 26:1–14. doi: 10.1038/s41380-019-0635-6
111. Lai AZ, Abella JV, Park M. Crosstalk in Met Receptor Oncogenesis. Trends Cell Biol (2009) 19:542–51. doi: 10.1016/j.tcb.2009.07.002
112. Gao CF, Woude GFV. HGF/SF-Met Signaling in Tumor Progression. Cell Res (2005) 15:49–51. doi: 10.1038/sj.cr.7290264
113. Taher TE, Tjin EP, Beuling EA, Borst J, Spaargaren M, Pals ST. C-Cbl Is Involved in Met Signaling in B Cells and Mediates Hepatocyte Growth Factor-Induced Receptor Ubiquitination. J Immunol (2002) 169:3793–800. doi: 10.4049/jimmunol.169.7.3793
114. Yeh E, Cunningham M, Arnold H, Chasse D, Monteith T, Ivaldi G, et al. A Signalling Pathway Controlling C-Myc Degradation That Impacts Oncogenic Transformation of Human Cells. Nat Cell Biol (2004) 6:308–18. doi: 10.1038/ncb1110
115. Goldoni S, Owens RT, McQuillan DJ, Shriver Z, Sasisekharan R, Birk DE, et al. Biologically Active Decorin Is a Monomer in Solution. J Biol Chem (2004) 279:6606–12. doi: 10.1074/jbc.M310342200
116. Gubbiotti MA, Vallet SD, Ricard-Blum S, Iozzo RV. Decorin Interacting Network: A Comprehensive Analysis of Decorin-Binding Partners and Their Versatile Functions. Matrix Biol (2016) 55:7–21. doi: 10.1016/j.matbio.2016.09.009
117. Zhang W, Ge Y, Cheng Q, Zhang Q, Fang L, Zheng J. Decorin is a Pivotal Effector in the Extracellular Matrix and Tumour Microenvironment. Oncotarget (2018) 9:5480–91. doi: 10.18632/oncotarget.23869
118. Santra M, Reed CC, Iozzo RV. Decorin Binds to a Narrow Region of the Epidermal Growth Factor (EGF) Receptor, Partially Overlapping But Distinct From the EGF-Binding Epitope. J Biol Chem (2002) 277:35671–81. doi: 10.1074/jbc.M205317200
119. Seidler DG, Goldoni S, Agnew C, Cardi C, Thakur ML, Owens RT, et al. Decorin Protein Core Inhibits In Vivo Cancer Growth and Metabolism by Hindering Epidermal Growth Factor Receptor Function and Triggering Apoptosis via Caspase-3 Activation. J Biol Chem (2006) 281:26408–18. doi: 10.1074/jbc.M602853200
120. Zhu JX, Goldoni S, Bix G, Owens RT, McQuillan DJ, Reed CC, et al. Decorin Evokes Protracted Internalization and Degradation of the Epidermal Growth Factor Receptor via Caveolar Endocytosis. J Biol Chem (2005) 280:32468–79. doi: 10.1074/jbc.M503833200
121. Csordas G, Santra M, Reed CC, Eichstetter I, McQuillan DJ, Gross D, et al. Sustained Down-Regulation of the Epidermal Growth Factor Receptor by Decorin: A Mechanism for Controlling Tumor Growth In Vivo. J Biol Chem (2000) 275:32879–87. doi: 10.1074/jbc.M005609200
122. Reed CC, Gauldie J, Iozzo RV. Suppression of Tumorigenicity by Adenovirus-Mediated Gene Transfer of Decorin. Oncogene (2002) 21:3688–95. doi: 10.1038/sj.onc.1205470
123. Sainio AO, Järveläinen HT. Decorin-Mediated Oncosuppression–a Potential Future Adjuvant Therapy for Human Epithelial Cancers. Br J Pharmacol (2019) 176:5–15. doi: 10.1111/bph.14180
124. Schönherr E, Sunderkötter C, Schaefer L, Thanos S, Grässel S, Oldberg Å, et al. Decorin Deficiency Leads to Impaired Angiogenesis in Injured Mouse Cornea. J Vasc Res (2004) 41:499–508. doi: 10.1159/000081806
125. Mohan RR, Tovey JC, Sharma A, Schultz GS, Cowden JW, Tandon A. Targeted Decorin Gene Therapy Delivered With Adeno-Associated Virus Effectively Retards Corneal Neovascularization In Vivo. PloS One (2011) 6:e26432. doi: 10.1371/journal.pone.0026432
126. Lai J, Chen F, Chen J, Ruan G, He M, Chen C, et al. Overexpression of Decorin Promoted Angiogenesis in Diabetic Cardiomyopathy via Igf1r-Akt-Vegf Signaling. Sci Rep (2017) 7:1–11. doi: 10.1038/srep44473
127. Grant DS, Yenisey C, Rose RW, Tootell M, Santra M, Iozzo RV. Decorin Suppresses Tumor Cell-Mediated Angiogenesis. Oncogene (2002) 21:4765–77. doi: 10.1038/sj.onc.1205595
128. Mustonen T, Alitalo K. Endothelial Receptor Tyrosine Kinases Involved in Angiogenesis. J Cell Biol (1995) 129:895–8. doi: 10.1083/jcb.129.4.895
129. Khan GA, Girish GV, Lala N, Di Guglielmo GM, Lala PK. Decorin is a Novel Vegfr-2-Binding Antagonist for the Human Extravillous Trophoblast. Mol Endocrinol (2011) 25:1431–43. doi: 10.1210/me.2010-0426
130. Goyal A, Neill T, Owens RT, Schaefer L, Iozzo RV. Decorin Activates AMPK, an Energy Sensor Kinase, to Induce Autophagy in Endothelial Cells. Matrix Biol (2014) 34:46–54. doi: 10.1016/j.matbio.2013.12.011
131. Neill T, Painter H, Buraschi S, Owens RT, Lisanti MP, Schaefer L, et al. Decorin Antagonizes the Angiogenic Network: Concurrent Inhibition of Met, Hypoxia Inducible Factor 1αVascular Endothelial Growth Factor A, and Induction of Thrombospondin-1 and TIMP3. J Biol Chem (2012) 287:5492–506. doi: 10.1074/jbc.M111.283499
132. Neill T, Jones HR, Crane-Smith Z, Owens RT, Schaefer L, Iozzo RV. Decorin Induces Rapid Secretion of Thrombospondin-1 in Basal Breast Carcinoma Cells via Inhibition of Ras Homolog Gene Family, Member A/Rho-Associated Coiled-Coil Containing Protein Kinase 1. FEBS J (2013) 280:2353–68. doi: 10.1111/febs.12148
133. Neill T, Chen CG, Buraschi S, Iozzo RV. Catabolic Degradation of Endothelial VEGFA via Autophagy. J Biol Chem (2020) 295:6064–79. doi: 10.1074/jbc.RA120.012593
134. Buraschi S, Neill T, Goyal A, Poluzzi C, Smythies J, Owens RT, et al. Decorin Causes Autophagy in Endothelial Cells via Peg3. Proc Natl Acad Sci (2013) 110:E2582–91. doi: 10.1073/pnas.1305732110
135. Buraschi S, Neill T, Iozzo RV. Decorin Is a Devouring Proteoglycan: Remodeling of Intracellular Catabolism via Autophagy and Mitophagy. Matrix Biol (2019) 75:260–70. doi: 10.1016/j.matbio.2017.10.005
136. Torres A, Gubbiotti MA, Iozzo RV. Decorin-Inducible Peg3 Evokes Beclin 1-Mediated Autophagy and Thrombospondin 1-Mediated Angiostasis. J Biol Chem (2017) 292:5055–69. doi: 10.1074/jbc.M116.753632
137. Relaix F, Wei XJ, Wu X, Sassoon DA. Peg3/Pw1 Is an Imprinted Gene Involved in the TNF-NFκB Signal Transduction Pathway. Nat Genet (1998) 18:287–91. doi: 10.1038/ng0398-287
138. Kohda T, Asai A, Kuroiwa Y, Kobayashi S, Aisaka K, Nagashima G, et al. Tumour Suppressor Activity of Human Imprinted Gene PEG3 in a Glioma Cell Line. Genes to Cells (2001) 6:237–47. doi: 10.1046/j.1365-2443.2001.00412.x
139. Dowdy SC, Gostout BS, Shridhar V, Wu X, Smith DI, Podratz KC, et al. Biallelic Methylation and Silencing of Paternally Expressed Gene 3 (PEG3) in Gynecologic Cancer Cell Lines. Gynecologic Oncol (2005) 99:126–34. doi: 10.1016/j.ygyno.2005.05.036
140. Neill T, Sharpe C, Owens RT, Iozzo RV. Decorin-Evoked Paternally Expressed Gene 3 (PEG3) is an Upstream Regulator of the Transcription Factor EB (TFEB) in Endothelial Cell Autophagy. J Biol Chem (2017) 292:16211–20. doi: 10.1074/jbc.M116.769950
141. Dierick F, Héry T, Hoareau-Coudert B, Mougenot N, Monceau V, Claude C, et al. Resident PW1+ Progenitor Cells Participate in Vascular Remodeling During Pulmonary Arterial Hypertension. Circ Res (2016) 118:822–33. doi: 10.1161/CIRCRESAHA.115.307035
142. Malinverno M, Corada M, Ferrarini L, Formicola L, Marazzi G, Sassoon D, et al. Peg3/PW1 Is a Marker of a Subset of Vessel Associated Endothelial Progenitors. Stem Cells (2017) 35:1328–40. doi: 10.1002/stem.2566
143. Neill T, Torres A, Buraschi S, Owens RT, Hoek JB, Baffa R, et al. Decorin Induces Mitophagy in Breast Carcinoma Cells via Peroxisome Proliferator-Activated Receptor γ Coactivator-1α(PGC-1α) and Mitostatin. J Biol Chem (2014) 289:4952–68. doi: 10.1074/jbc.M113.512566
144. Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, et al. The Proteoglycan Biglycan Regulates Expression of the B Cell Chemoattractant CXCL13 and Aggravates Murine Lupus Nephritis. J Clin Invest (2010) 120:4251–72. doi: 10.1172/JCI42213
145. Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhão JG, et al. Signaling by the Matrix Proteoglycan Decorin Controls Inflammation and Cancer Through PDCD4 and MicroRNA-21. Sci Signaling (2011) 4:ra75–5. doi: 10.1126/scisignal.2001868
146. Yasuda M, Schmid T, Rübsamen D, Colburn NH, Irie K, Murakami A. Downregulation of Programmed Cell Death 4 by Inflammatory Conditions Contributes to the Generation of the Tumor Promoting Microenvironment. Mol Carcinog (2010) 49:837–48. doi: 10.1002/mc.20660
147. Davis BN, Hilyard AC, Lagna G, Hata A. SMAD Proteins Control DROSHA-Mediated microRNA Maturation. Nature (2008) 454:56–61. doi: 10.1038/nature07086
148. Asangani IA, Rasheed SA, Nikolova D, Leupold J, Colburn N, Post S, et al. MicroRNA-21 (miR-21) Post-Transcriptionally Downregulates Tumor Suppressor PDCD4 and Stimulates Invasion, Intravasation and Metastasis in Colorectal Cancer. Oncogene (2008) 27:2128–36. doi: 10.1038/sj.onc.1210856
149. Bianco P, Fisher LW, Young MF, Termine JD, Robey PG. Expression and Localization of the Two Small Proteoglycans Biglycan and Decorin in Developing Human Skeletal and non-Skeletal Tissues. J Histochem Cytochem (1990) 38:1549–63. doi: 10.1177/38.11.2212616
150. Choi H, Johnson T, Pal S, Tang L, Rosenberg L, Neame P. Characterization of the Dermatan Sulfate Proteoglycans, DS-PGI and DS-PGII, From Bovine Articular Cartilage and Skin Isolated by Octyl-Sepharose Chromatography. J Biol Chem (1989) 264:2876–84. doi: 10.1016/S0021-9258(19)81694-0
151. Schönherr E, Witsch-Prehm P, Harrach B, Robenek H, Rauterberg J, Kresse H. Interaction of Biglycan With Type I Collagen. J Biol Chem (1995) 270:2776–83. doi: 10.1074/jbc.270.6.2776
152. Wiberg C, Hedbom E, Khairullina A, Lamandé SR, Oldberg Å, Timpl R, et al. Biglycan and Decorin Bind Close to the N-Terminal Region of the Collagen VI Triple Helix. J Biol Chem (2001) 276:18947–52. doi: 10.1074/jbc.M100625200
153. Wiberg C, Heinegard D, Wenglen C, Timpl R, Moergelin M. Biglycan Organizes Collagen VI Into Hexagonal-Like Networks Resembling Tissue Structures. J Biol Chem (2002) 277:49120–6. doi: 10.1074/jbc.M206891200
154. Wiberg C, Klatt AR, Wagener R, Paulsson M, Bateman JF, Heinegard D, et al. Complexes of Matrilin-1 and Biglycan or Decorin Connect Collagen Vi Microfibrils to Both Collagen Ii and Aggrecan. J Biol Chem (2003) 278:37698–704. doi: 10.1074/jbc.M304638200
155. Reinboth B, Hanssen E, Cleary EG, Gibson MA. Molecular Interactions of Biglycan and Decorin With Elastic Fiber Components: Biglycan Forms a Ternary Complex With Tropoelastin and Microfibril-Associated Glycoprotein 1. J Biol Chem (2002) 277:3950–7. doi: 10.1074/jbc.M109540200
156. Douglas T, Heinemann S, Bierbaum S, Scharnweber D, Worch H. Fibrillogenesis of Collagen Types I, II, and III With Small Leucine-Rich Proteoglycans Decorin and Biglycan. Biomacromolecules (2006) 7:2388–93. doi: 10.1021/bm0603746
157. Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, et al. The Matrix Component Biglycan Is Proinflammatory and Signals Through Toll-Like Receptors 4 and 2 in Macrophages. J Clin Invest (2005) 115:2223–33. doi: 10.1172/JCI23755
158. Schaefer L, Iozzo RV. Biological Functions of the Small Leucine-Rich Proteoglycans: From Genetics to Signal Transduction. J Biol Chem (2008) 283:21305–9. doi: 10.1074/jbc.R800020200
159. Hsieh LTH, Nastase MV, Zeng-Brouwers J, Iozzo RV, Schaefer L. Soluble Biglycan as a Biomarker of Inflammatory Renal Diseases. Int J Biochem Cell Biol (2014) 54:223–35. doi: 10.1016/j.biocel.2014.07.020
160. Moreth K, Frey H, Hubo M, Zeng-Brouwers J, Nastase MV, Hsieh LTH, et al. Biglycan-Triggered TLR-2-and TLR-4-Signaling Exacerbates the Pathophysiology of Ischemic Acute Kidney Injury. Matrix Biol (2014) 35:143–51. doi: 10.1016/j.matbio.2014.01.010
161. Zeng-Brouwers J, Beckmann J, Nastase MV, Iozzo RV, Schaefer L. De Novo Expression of Circulating Biglycan Evokes an Innate Inflammatory Tissue Response via MyD88/TRIF Pathways. Matrix Biol (2014) 35:132–42. doi: 10.1016/j.matbio.2013.12.003
162. Schaefer L, Raslik I, Gröne HJ, Schönherr E, Macakova K, Ugorcakova J, et al. Small Proteoglycans in Human Diabetic Nephropathy: Discrepancy Between Glomerular Expression and Protein Accumulation of Decorin, Biglycan, Lumican, and Fibromodulin. FASEB J (2001) 15:559–61. doi: 10.1096/fj.00-0493fje
163. Bolton K, Segal D, Walder K. The Small Leucine-Rich Proteoglycan, Biglycan, Is Highly Expressed in Adipose Tissue of Psammomys Obesus and Is Associated With Obesity and Type 2 Diabetes. Biologics: Targets Ther (2012) 6:67–72. doi: 10.2147/BTT.S27925
164. Ciftciler R, Ozenirler S, Yucel AA, Cengiz M, Erkan G, Buyukdemirci E, et al. The Importance of Serum Biglycan Levels as a Fibrosis Marker in Patients With Chronic Hepatitis B. J Clin Lab Anal (2017) 31:e22109. doi: 10.1002/jcla.22109
165. Zhao SF, Yin XJ, Zhao WJ, Liu LC, Wang ZP. Biglycan as a Potential Diagnostic and Prognostic Biomarker in Multiple Human Cancers. Oncol Lett (2020) 19:1673–82. doi: 10.3892/ol.2020.11266
166. Hsieh LTH, Frey H, Nastase MV, Tredup C, Hoffmann A, Poluzzi C, et al. Bimodal Role of NADPH Oxidases in the Regulation of Biglycan-Triggered IL-1βSynthesis. Matrix Biol (2016) 49:61–81. doi: 10.1016/j.matbio.2015.12.005
167. Roedig H, Nastase MV, Frey H, Moreth K, Zeng-Brouwers J, Poluzzi C, et al. Biglycan is a New High-Affinity Ligand for CD14 in Macrophages. Matrix Biol (2019) 77:4–22. doi: 10.1016/j.matbio.2018.05.006
168. Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, et al. Biglycan, a Danger Signal That Activates the NLRP3 Inflammasome via Toll-Like and P2X Receptors. J Biol Chem (2009) 284:24035–48. doi: 10.1074/jbc.M109.014266
169. Hsieh LTH, Nastase MV, Roedig H, Zeng-Brouwers J, Poluzzi C, Schwalm S, et al. Biglycan-And Sphingosine Kinase-1 Signaling Crosstalk Regulates the Synthesis of Macrophage Chemoattractants. Int J Mol Sci (2017) 18:595. doi: 10.3390/ijms18030595
170. Nastase MV, Zeng-Brouwers J, Beckmann J, Tredup C, Christen U, Radeke HH, et al. Biglycan, a Novel Trigger of Th1 and Th17 Cell Recruitment Into the Kidney. Matrix Biol (2018) 68:293–317. doi: 10.1016/j.matbio.2017.12.002
171. Popovic ZV, Wang S, Papatriantafyllou M, Kaya Z, Porubsky S, Meisner M, et al. The Proteoglycan Biglycan Enhances Antigen-Specific T Cell Activation Potentially via MyD88 and TRIF Pathways and Triggers Autoimmune Perimyocarditis. J Immunol (2011) 187:6217–26. doi: 10.4049/jimmunol.1003478
172. Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 Inhibition Accelerates Breast Cancer Metastasis by Promoting Angiogenesis. Nature (2014) 515:130–3. doi: 10.1038/nature13862
173. Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-Induced Chemokine Cascade Promotes Breast Cancer Metastasis by Enhancing Retention of Metastasis-Associated Macrophages. J Exp Med (2015) 212:1043–59. doi: 10.1084/jem.20141836
174. Hao Q, Vadgama JV, Wang P. CCL2/CCR2 Signaling in Cancer Pathogenesis. Cell Commun Signaling (2020) 18:1–13. doi: 10.1186/s12964-020-00589-8
175. Aldinucci D, Borghese C, Casagrande N. The CCL5/CCR5 Axis in Cancer Progression. Cancers (2020) 12:1765. doi: 10.3390/cancers12071765
176. Gnerlich JL, Mitchem JB, Weir JS, Sankpal NV, Kashiwagi H, Belt BA, et al. Induction of Th17 Cells in the Tumor Microenvironment Improves Survival in a Murine Model of Pancreatic Cancer. J Immunol (2010) 185:4063–71. doi: 10.4049/jimmunol.0902609
177. Najafi S, Mirshafiey A. The Role of T Helper 17 and Regulatory T Cells in Tumor Microenvironment. Immunopharmacol Immunotoxicol (2019) 41:16–24. doi: 10.1080/08923973.2019.1566925
178. Renaude E, Kroemer M, Loyon R, Binda D, Borg C, Guittaut M, et al. The Fate of Th17 Cells Is Shaped by Epigenetic Modifications and Remodeled by the Tumor Microenvironment. Int J Mol Sci (2020) 21:1673. doi: 10.3390/ijms21051673
179. Roedig H, Nastase MV, Wygrecka M, Schaefer L. Breaking Down Chronic Inflammatory Diseases: The Role of Biglycan in Promoting a Switch Between Inflammation and Autophagy. FEBS J (2019) 286:2965–79. doi: 10.1111/febs.14791
180. Poluzzi C, Nastase MV, Zeng-Brouwers J, Roedig H, Hsieh LTH, Michaelis JB, et al. Biglycan Evokes Autophagy in Macrophages via a Novel CD44/Toll-Like Receptor 4 Signaling Axis in Ischemia/Reperfusion Injury. Kidney Int (2019) 95:540–62. doi: 10.1016/j.kint.2018.10.037
181. Deretic V, Levine B. Autophagy Balances Inflammation in Innate Immunity. Autophagy (2018) 14:243–51. doi: 10.1080/15548627.2017.1402992
182. Matsuzawa-Ishimoto Y, Hwang S, Cadwell K. Autophagy and Inflammation. Annu Rev Immunol (2018) 36:73–101. doi: 10.1146/annurev-immunol-042617-053253
183. Xing X, Gu X, Ma T, Ye H. Biglycan Up-Regulated Vascular Endothelial Growth Factor (VEGF) Expression and Promoted Angiogenesis in Colon Cancer. Tumor Biol (2015) 36:1773–80. doi: 10.1007/s13277-014-2779-y
184. Hu L, Hx W, Jf Li, Lp Su, Yan M, Li C, et al. Biglycan Stimulates VEGF Expression in Endothelial Cells by Activating the TLR Signaling Pathway. Mol Oncol (2016) 10:1473–84. doi: 10.1016/j.molonc.2016.08.002
185. Xia C, Meng Q, Liu LZ, Rojanasakul Y, Wang XR, Jiang BH. Reactive Oxygen Species Regulate Angiogenesis and Tumor Growth Through Vascular Endothelial Growth Factor. Cancer Res (2007) 67:10823–30. doi: 10.1158/0008-5472.CAN-07-0783
186. Frey H, Moreth K, Hsieh LTH, Zeng-Brouwers J, Rathkolb B, Fuchs H, et al. A Novel Biological Function of Soluble Biglycan: Induction of Erythropoietin Production and Polycythemia. Glycoconjugate J (2017) 34:393–404. doi: 10.1007/s10719-016-9722-y
187. Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, et al. The Expression and Distribution of the Hypoxia-Inducible Factors HIF-1αand HIF-2αin Normal Human Tissues, Cancers, and Tumor-Associated Macrophages. Am J Pathol (2000) 157:411–21. doi: 10.1016/S0002-9440(10)64554-3
188. Cong L, Maishi N, Annan DA, Young MF, Morimoto H, Morimoto M, et al. Inhibition of Stromal Biglycan Promotes Normalization of the Tumor Microenvironment and Enhances Chemotherapeutic Efficacy. Breast Cancer Res (2021) 23:1–17. doi: 10.1186/s13058-021-01423-w
189. Xing X, Gu X, Ma T. Knockdown of Biglycan Expression by RNA Interference Inhibits the Proliferation and Invasion of, and Induces Apoptosis in, the HCT116 Colon Cancer Cell Line. Mol Med Rep (2015) 12:7538–44. doi: 10.3892/mmr.2015.4383
190. Liu B, Xu T, Xu X, Cui Y, Xing X. Biglycan Promotes the Chemotherapy Resistance of Colon Cancer by Activating NF-κB Signal Transduction. Mol Cell Biochem (2018) 449:285–94. doi: 10.1007/s11010-018-3365-1
191. Shatz M, Menendez D, Resnick MA. The Human TLR Innate Immune Gene Family is Differentially Influenced by DNA Stress and P53 Status in Cancer Cells. Cancer Res (2012) 72:3948–57. doi: 10.1158/0008-5472.CAN-11-4134
192. Korneev KV, Atretkhany KSN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA. TLR-Signaling and Proinflammatory Cytokines as Drivers of Tumorigenesis. Cytokine (2017) 89:127–35. doi: 10.1016/j.cyto.2016.01.021
193. Li F, Sethi G. Targeting Transcription Factor Nf-κb to Overcome Chemoresistance and Radioresistance in Cancer Therapy. Biochim Biophys Acta (BBA)-Reviews Cancer (2010) 1805:167–80. doi: 10.1016/j.bbcan.2010.01.002
194. Sakamoto K, Maeda S. Targeting NF-κB for Colorectal Cancer. Expert Opin Ther Targets (2010) 14:593–601. doi: 10.1517/14728221003769903
195. Berendsen AD, Fisher LW, Kilts TM, Owens RT, Robey PG, Gutkind JS, et al. Modulation of Canonical Wnt Signaling by the Extracellular Matrix Component Biglycan. Proc Natl Acad Sci (2011) 108:17022–7. doi: 10.1073/pnas.1110629108
196. Clements WM, Wang J, Sarnaik A, Kim OJ, MacDonald J, Fenoglio-Preiser C, et al. β-Catenin Mutation Is a Frequent Cause of Wnt Pathway Activation in Gastric Cancer. Cancer Res (2002) 62:3503–6.
197. Zhan T, Rindtorff N, Boutros M. Wnt Signaling in Cancer. Oncogene (2017) 36:1461–73. doi: 10.1038/onc.2016.304
198. Aggelidakis J, Berdiaki A, Nikitovic D, Papoutsidakis A, Papachristou DJ, Tsatsakis AM, et al. Biglycan Regulates MG63 Osteosarcoma Cell Growth Through a LPR6/β-Catenin/IGFR-IR Signaling Axis. Front Oncol (2018) 8:470. doi: 10.3389/fonc.2018.00470
199. Hu L, Yt D, Jf Li, Lp Su, Yan M, Zg Z, et al. Biglycan Enhances Gastric Cancer Invasion by Activating FAK Signaling Pathway. Oncotarget (2014) 5:1885–96. doi: 10.18632/oncotarget.1871
200. Pinto F, Santos-Ferreira L, Pinto MT, Gomes C, Reis CA. The Extracellular Small Leucine-Rich Proteoglycan Biglycan Is a Key Player in Gastric Cancer Aggressiveness. Cancers (2021) 13:1330. doi: 10.3390/cancers13061330
201. Maishi N, Ohba Y, Akiyama K, Ohga N, Hamada Ji, Nagao-Kitamoto H, et al. Tumour Endothelial Cells in High Metastatic Tumours Promote Metastasis via Epigenetic Dysregulation of Biglycan. Sci Rep (2016) 6:1–13. doi: 10.1038/srep28039
202. Andrlová H, Mastroianni J, Madl J, Kern JS, Melchinger W, Dierbach H, et al. Biglycan Expression in the Melanoma Microenvironment Promotes Invasiveness via Increased Tissue Stiffness Inducing Integrin-β1 Expression. Oncotarget (2017) 8:42901–16. doi: 10.18632/oncotarget.17160
203. Sun H, Wang X, Zhang Y, Che X, Liu Z, Zhang L, et al. Biglycan Enhances the Ability of Migration and Invasion in Endometrial Cancer. Arch Gynecol Obstetrics (2016) 293:429–38. doi: 10.1007/s00404-015-3844-5
204. Weber CK, Sommer G, Michl P, Fensterer H, Weimer M, Gansauge F, et al. Biglycan is Overexpressed in Pancreatic Cancer and Induces G1-Arrest in Pancreatic Cancer Cell Lines. Gastroenterology (2001) 121:657–67. doi: 10.1053/gast.2001.27222
205. Niedworok C, Röck K, Kretschmer I, Freudenberger T, Nagy N, Szarvas T, et al. Inhibitory Role of the Small Leucine-Rich Proteoglycan Biglycan in Bladder Cancer. PloS One (2013) 8:e80084. doi: 10.1371/journal.pone.0080084
206. Rydström K, Joost P, Ehinger M, Edén P, Jerkeman M, Cavallin-Ståhl E, et al. Gene Expression Profiling Indicates That Immunohistochemical Expression of CD40 Is a Marker of an Inflammatory Reaction in the Tumor Stroma of Diffuse Large B-Cell Lymphoma. Leukemia Lymphoma (2012) 53:1764–8. doi: 10.3109/10428194.2012.666541
207. Schultze JL, Cardoso AA, Freeman GJ, Seamon MJ, Daley J, Pinkus GS, et al. Follicular Lymphomas can be Induced to Present Alloantigen Efficiently: A Conceptual Model to Improve Their Tumor Immunogenicity. Proc Natl Acad Sci (1995) 92:8200–4. doi: 10.1073/pnas.92.18.8200
208. French RR, Chan HC, Tutt AL, Glennie MJ. CD40 Antibody Evokes a Cytotoxic T-Cell Response That Eradicates Lymphoma and Bypasses T-Cell Help. Nat Med (1999) 5:548–53. doi: 10.1038/8426
209. Recktenwald CV, Leisz S, Steven A, Mimura K, Müller A, Wulfänger J, et al. HER-2/Neu-Mediated Down-Regulation of Biglycan Associated With Altered Growth Properties. J Biol Chem (2012) 287:24320–9. doi: 10.1074/jbc.M111.334425
210. Subbarayan K, Leisz S, Wickenhauser C, Bethmann D, Massa C, Steven A, et al. Biglycan-Mediated Upregulation of MHC Class I Expression in HER-2/Neu-Transformed Cells. Oncoimmunology (2018) 7:e1373233. doi: 10.1080/2162402X.2017.1373233
211. Subbarayan K, Massa C, Lazaridou MF, Ulagappan K, Seliger B. Identification of a Novel miR-21-3p/TGF-β Signaling-Driven Immune Escape via the MHC Class I/biglycan Axis in Tumor Cells. Clin Trans Med (2021) 11:e306. doi: 10.1002/ctm2.306
212. Neill T, Schaefer L, Iozzo RV. Decorin as a Multivalent Therapeutic Agent Against Cancer. Adv Drug Deliv Rev (2016) 97:174–85. doi: 10.1016/j.addr.2015.10.016
213. Xu W, Neill T, Yang Y, Hu Z, Cleveland E, Wu Y, et al. The Systemic Delivery of an Oncolytic Adenovirus Expressing Decorin Inhibits Bone Metastasis in a Mouse Model of Human Prostate Cancer. Gene Ther (2015) 22:247–56. doi: 10.1038/gt.2014.110
214. Yang Y, Xu W, Neill T, Hu Z, Wang CH, Xiao X, et al. Systemic Delivery of an Oncolytic Adenovirus Expressing Decorin for the Treatment of Breast Cancer Bone Metastases. Hum Gene Ther (2015) 26:813–25. doi: 10.1089/hum.2015.098
215. Ständer M, Naumann U, Dumitrescu L, Heneka M, Löschmann P, Gulbins E, et al. Decorin Gene Transfer-Mediated Suppression of TGF-βSynthesis Abrogates Experimental Malignant Glioma Growth In Vivo. Gene Ther (1998) 5:1187–94. doi: 10.1038/sj.gt.3300709
216. Ma HI, Hueng DY, Shui HA, Han JM, Wang CH, Lai YH, et al. Intratumoral Decorin Gene Delivery by AAV Vector Inhibits Brain Glioblastomas and Prolongs Survival of Animals by Inducing Cell Differentiation. Int J Mol Sci (2014) 15:4393–414. doi: 10.3390/ijms15034393
217. Järvinen TA, Prince S. Decorin: A Growth Factor Antagonist for Tumor Growth Inhibition. BioMed Res Int (2015) 2015:1–11. doi: 10.1155/2015/654765
Keywords: extracellular matrix, proteoglycan, autophagy, inflammation, angiogenesis, cancer, toll-like receptor
Citation: Diehl V, Huber LS, Trebicka J, Wygrecka M, Iozzo RV and Schaefer L (2021) The Role of Decorin and Biglycan Signaling in Tumorigenesis. Front. Oncol. 11:801801. doi: 10.3389/fonc.2021.801801
Received: 25 October 2021; Accepted: 09 November 2021;
Published: 30 November 2021.
Edited by:
Martin Götte, University of Münster, GermanyReviewed by:
Dragana Nikitovic, University of Crete, GreeceKornelia Baghy, Semmelweis University, Hungary
Copyright © 2021 Diehl, Huber, Trebicka, Wygrecka, Iozzo and Schaefer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liliana Schaefer, c2NoYWVmZXJAbWVkLnVuaS1mcmFua3VydC5kZQ==