 Jennifer A. Jiménez
Jennifer A. Jiménez Elizabeth R. Lawlor
Elizabeth R. Lawlor Costas A. Lyssiotis
Costas A. Lyssiotis- 1Department of Molecular & Integrative Physiology, University of Michigan Medical School, Ann Arbor, MI, United States
- 2Rogel Cancer Center, University of Michigan Medical School, Ann Arbor, MI, United States
- 3Department of Pediatrics, University of Washington, Seattle, WA, United States
- 4Seattle Children’s Research Institute, Seattle, WA, United States
- 5Department of Internal Medicine, Division of Gastroenterology and Hepatology, University of Michigan Medical School, Ann Arbor, MI, United States
Primary bone sarcomas, including osteosarcoma (OS) and Ewing sarcoma (ES), are aggressive tumors with peak incidence in childhood and adolescence. The intense standard treatment for these patients consists of combined surgery and/or radiation and maximal doses of chemotherapy; a regimen that has not seen improvement in decades. Like other tumor types, ES and OS are characterized by dysregulated cellular metabolism and a rewiring of metabolic pathways to support the biosynthetic demands of malignant growth. Not only are cancer cells characterized by Warburg metabolism, or aerobic glycolysis, but emerging work has revealed a dependence on amino acid metabolism. Aside from incorporation into proteins, amino acids serve critical functions in redox balance, energy homeostasis, and epigenetic maintenance. In this review, we summarize current studies describing the amino acid metabolic requirements of primary bone sarcomas, focusing on OS and ES, and compare these dependencies in the normal bone and malignant tumor contexts. We also examine insights that can be gleaned from other cancers to better understand differential metabolic susceptibilities between primary and metastatic tumor microenvironments. Lastly, we discuss potential metabolic vulnerabilities that may be exploited therapeutically and provide better-targeted treatments to improve the current standard of care.
1. Introduction: Bone sarcoma and metabolism
Primary bone cancers encompass approximately 0.2% of all malignant tumors, with osteosarcoma (OS), Ewing sarcoma (ES), and chondrosarcoma representing 90-95% of these diagnoses (1). ES and OS are predominately cancers of childhood and adolescence. While survival rates for other childhood malignancies have improved steadily in recent decades, the five-year survival rate for ES and OS patients presenting with metastatic disease at diagnosis has not improved and remains near 30% (2). Furthermore, for patients with localized disease, the standard of care for these highly aggressive bone tumors consists of high-dose chemotherapy, surgery, and radiation. This leads to significant long-term toxicities that contribute to both decreased life expectancy and diminished quality of life (2).
Throughout life, normal bone metabolism and growth are dependent on maintaining a balance between the constant and coincident processes of bone formation and bone resorption. Disruption to this balance in bone remodeling often occurs during normal aging when bone resorption can exceed bone formation, resulting in osteoporosis (3). In contrast, during adolescence, the time at which ES and OS most commonly arise, rapid growth requires that bone formation, or osteogenesis, exceed the level of bone resorption. Normal bone remodeling is a high-energy process that is dependent on cellular metabolic processes, and the central cellular mediators that carry out the processes of bone building and bone destruction are osteoblasts and osteoclasts, respectively. While it is appreciated that the skeleton is a metabolically active organ, and studies investigating the bioenergetic properties of bone can be traced back over 60 years (4–6), until recently, the metabolic dependencies of bone homeostasis had been relatively unexplored.
Dysregulated metabolism is recognized as a hallmark of cancer (7), where the rewiring of metabolic pathways is required for supporting the biosynthetic demands of highly proliferative malignant cells (8). Early interest in the field of cancer metabolism focused on glucose metabolism, with more recent work revealing the importance of amino acids in cancer progression (9). Amino acids play a central role, not only in protein synthesis, but also energy production, nucleotide synthesis, and maintenance of redox homeostasis (10, 11). More importantly, advancements have been made in the feasibility of targeting of amino acid metabolic pathways that are rewired in cancer, which may provide a therapeutic vulnerability (11). This has led to the preclinical and clinical development of therapeutic drugs, such as amino acid degrading enzymes, amino acid transporter inhibitors, and those that target de novo amino acid biosynthetic pathways. However, determination and therapeutic exploitation of metabolic vulnerabilities in primary bone tumors has lagged relative to advances for other tumor types.
In this review, we provide an overview of the amino acid metabolic pathways that are central in the “normal” bone microenvironment, primarily focused on the bioenergetics of the osteoblasts and osteoclasts that maintain bone homeostasis. We further contrast amino acid dependencies in the normal bone environment with the amino acid requirements of primary bone malignancies, focused mainly on OS and ES. Lastly, we provide a summary of therapeutically targetable pathways that are currently under study.
2. Metabolic rewiring in cancer
2.1. Amino acid metabolism
Cellular metabolism describes the series of biochemical reactions that generate energy and biomolecules needed to sustain homeostasis and function. The balance between catabolic and anabolic processes allows cells to both harness and utilize energy, as well as synthesize and break down macromolecules as appropriate to sustain growth. As an example, the catabolism of glucose provides a major energy source for the generation of adenosine triphosphate (ATP), a principal energy currency for the maintenance of cellular homeostasis, through glycolysis and mitochondrial oxidative phosphorylation (OXPHOS) (Figure 1).
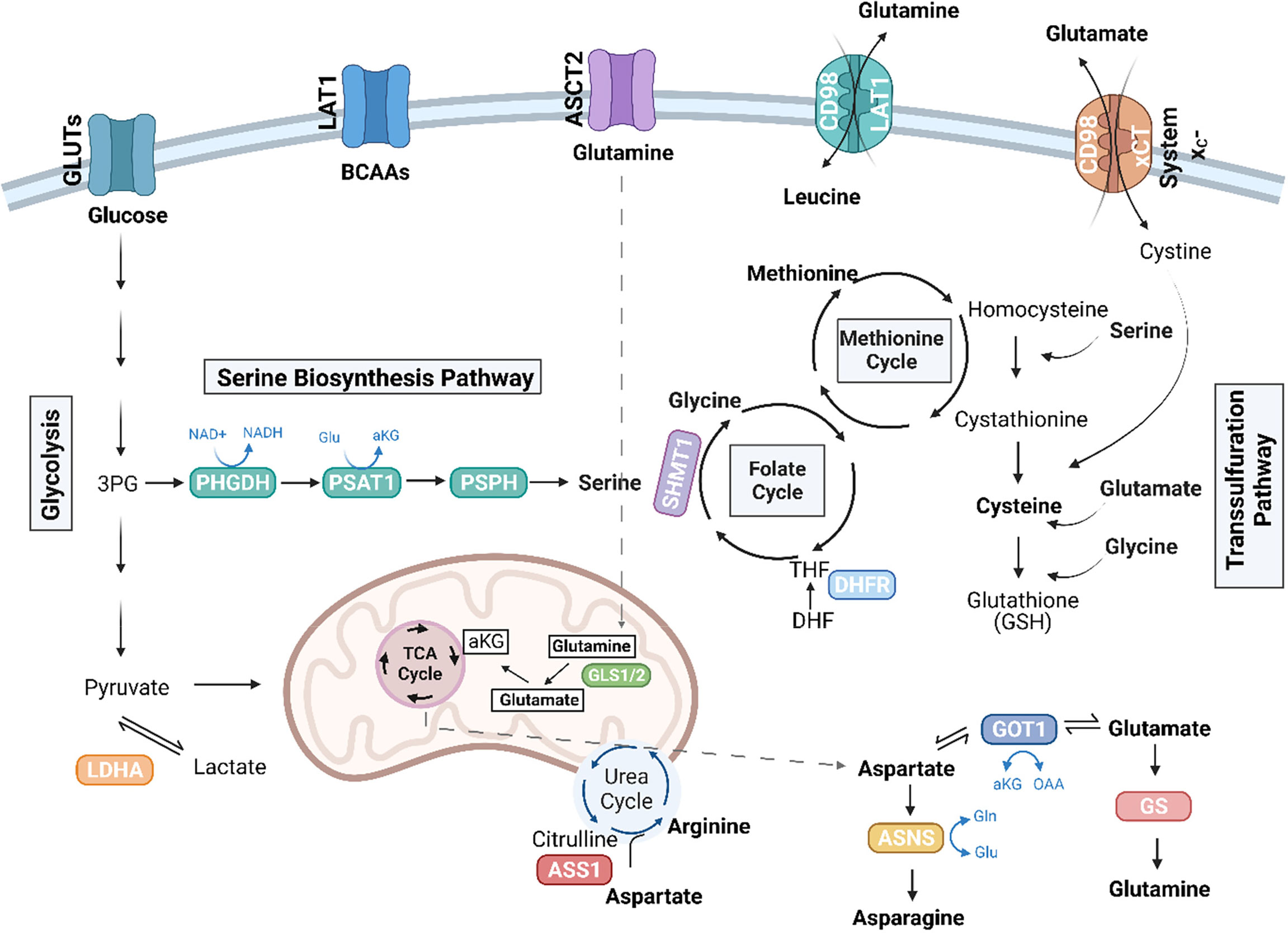
Figure 1 Amino acid pathways rewired in cancer. Illustration depicting amino acid pathways linked in an intricate metabolic network. Essential amino acids, including the branched chain amino acids (BCAAs), leucine, isoleucine, and valine, are imported from the extracellular environment, in part via the LAT1 (SLC7A5) transporter. Glutamine (Gln) can be conditionally essential and is imported through ASCT2 (SLC1A5). Glutaminase enzymes (GLS1/2) convert glutamine to glutamate (Glu), which can further be converted to alpha-ketoglutarate (αKG) to enter the TCA cycle. The TCA cycle also generates intermediates, which give rise to non-essential amino acids, including aspartate and arginine. The de novo synthesis of arginine involves the conversion of citrulline to aspartate by argininosuccinate synthetase 1 (ASS1). TCA-cycle derived aspartate is converted to asparagine via ASNS, and GOT1 can convert aspartate to glutamate, which is further converted to glutamine by glutamine synthetase (GS). Serine, another non-essential amino acid, can be imported or synthesized de novo in the serine biosynthesis pathway (SBP), which diverts the 3P-glycerate (3PG) intermediate of glycolysis to generate serine via the enzymes, phosphoglycerate dehydrogenase (PHGDH), phosphoserine aminotransferase (PSAT1), and phosphoserine phosphatase (PSPH). Serine can further be converted to glycine by serine hydroxymethyltransferase (SHMT) (cytosolic SHMT1 isozyme shown above) to generate one-carbon units to fuel the folate cycle and the methionine cycles. Dihydrofolate reductase (DHFR) in the folate cycle catalyzes the reduction of dihydrofolate to tetrahydrofolate (THF). Cysteine is another non-essential amino acid that can be synthesized in the transsulfuration pathway or imported as cystine via the cystine/glutamate antiporter, system xC-. The SBP, transsulfuration pathway, along with ASS1 and glutamine metabolism have been implicated in sarcoma biology. Figure was created with BioRender.com.
Apart from glucose, proliferating cells also exhibit a significant demand for amino acids, which, in part, are required to support protein synthesis. Essential amino acids, including the branched chain amino acids (BCAAs) leucine, isoleucine, and valine, cannot be synthesized within the cell and instead require import from the extracellular environment. Non-essential amino acids, including the amino acids glutamine, glutamate, alanine, proline, and aspartate, on the other hand, can by synthesized de novo providing cells with more flexibility when these amino acids become scarce (Figure 1). Aside from their proteinogenic function, amino acids have also been shown to play a role in energy production. Glutamine, the most abundant amino acid in circulation, can sustain the TCA cycle (Figure 1). Through glutaminolysis, the enzyme glutaminase (GLS) converts glutamine to glutamate, which can then be converted to alpha-ketoglutarate (αKG) to enter the TCA cycle. Glutamine also serves as a precursor for other non-essential amino acids, including aspartate, alanine, arginine, and proline. Additionally, non-essential amino acids participate in the regulation of signaling pathways and maintenance of cellular redox homeostasis; for example cysteine, glycine, and glutamate constitute the tripeptide and major antioxidant glutathione (GSH) (11).
2.2. Amino acid metabolism in cancer
The observation that cancer cells exhibit a rewiring of metabolic processes can be traced back to the work of Otto Warburg, with his finding that cancer cells preferentially use glycolysis, even in the presence of oxygen. This process is termed aerobic glycolysis and is known colloquially as the Warburg effect (12). In tumors and proliferative cells, glucose uptake is high. Aerobic glycolysis provides glycolytic intermediates necessary for anabolic reactions that generate building blocks to sustain biomass accumulation in cells (13). This metabolism is reenforced by glutamine, which is used to fuel the TCA cycle for energy production and also supplies carbon and nitrogen for the generation of biosynthetic precursors such as nucleotides, non-essential amino acids, and fatty acids (14, 15).
Beyond glucose and glutamine, cancers often become auxotrophic for various nutrients. This includes a dependence on an exogenous supply of non-essential amino acids, due to the loss of expression of key enzymes necessary for the synthesis of that amino acid, such as in the case of asparagine, arginine, and glutamine (11, 16, 17). In fact, one of the most successful amino acid-directed therapies targets the de novo synthesis of asparagine. It was found in the 1970s that the use of bacterial asparaginase (ASNase) could cure children with pediatric acute lymphoblastic leukemia (ALL) when used as a single agent or as a combination therapy, due to low expression of asparagine synthetase (ASNS) (Figure 1), the enzyme that converts aspartate to asparagine in leukemic blasts (18).
Cancers also depend on other de novo amino acid biosynthetic pathways and exhibit upregulated expression of enzymes in these pathways. For example, expression of phosphoglycerate dehydrogenase (PHGDH), the rate-limiting enzyme in the serine biosynthetic pathway (SBP), is upregulated in breast cancer and melanoma due to genomic amplification of the PHGDH locus. Further, this is essential for sustaining oncogenic growth (19, 20). Studies have demonstrated that the effect of inhibition of PHGDH in cancer cells cannot be fully rescued by extracellular serine alone, suggesting that these tumors depend on other intermediate products of the SBP (Figure 1) (20). A comprehensive review on amino acid metabolism in cancer, more broadly, is beyond the scope of the current review, and readers are directed to prior published reviews for recent summaries of this complex field of study (9, 10, 21, 22). While research is growing in this area, the investigation of amino acid requirements in primary bone sarcomas has just recently received due attention, and these new findings are detailed in the sections that follow.
3. Metabolism in normal bone homeostasis
3.1. Cycle of bone remodeling
The bone is a dynamic organ, with processes that maintain high turnover for constant bone remodeling. These cycles of building and destroying bone are commonly referred to as bone formation and bone resorption, respectively. The biological process of bone remodeling is required to maintain proper calcium homeostasis, respond to injury, and promote growth and development (23). In children, the rate of bone formation exceeds the level of bone resorption, supporting the growth of bone through adolescence. These two processes achieve a balance in adolescence, and eventually the scale tips toward a net loss of bone, which is associated with ageing (Figure 2). Defects in this cycle of bone turnover can contribute to diseases of bone such as osteoporosis, Paget’s disease of bone, osteogenesis imperfecta, and cancer, all of which interfere with the body’s normal bone-recycling processes (3, 24).
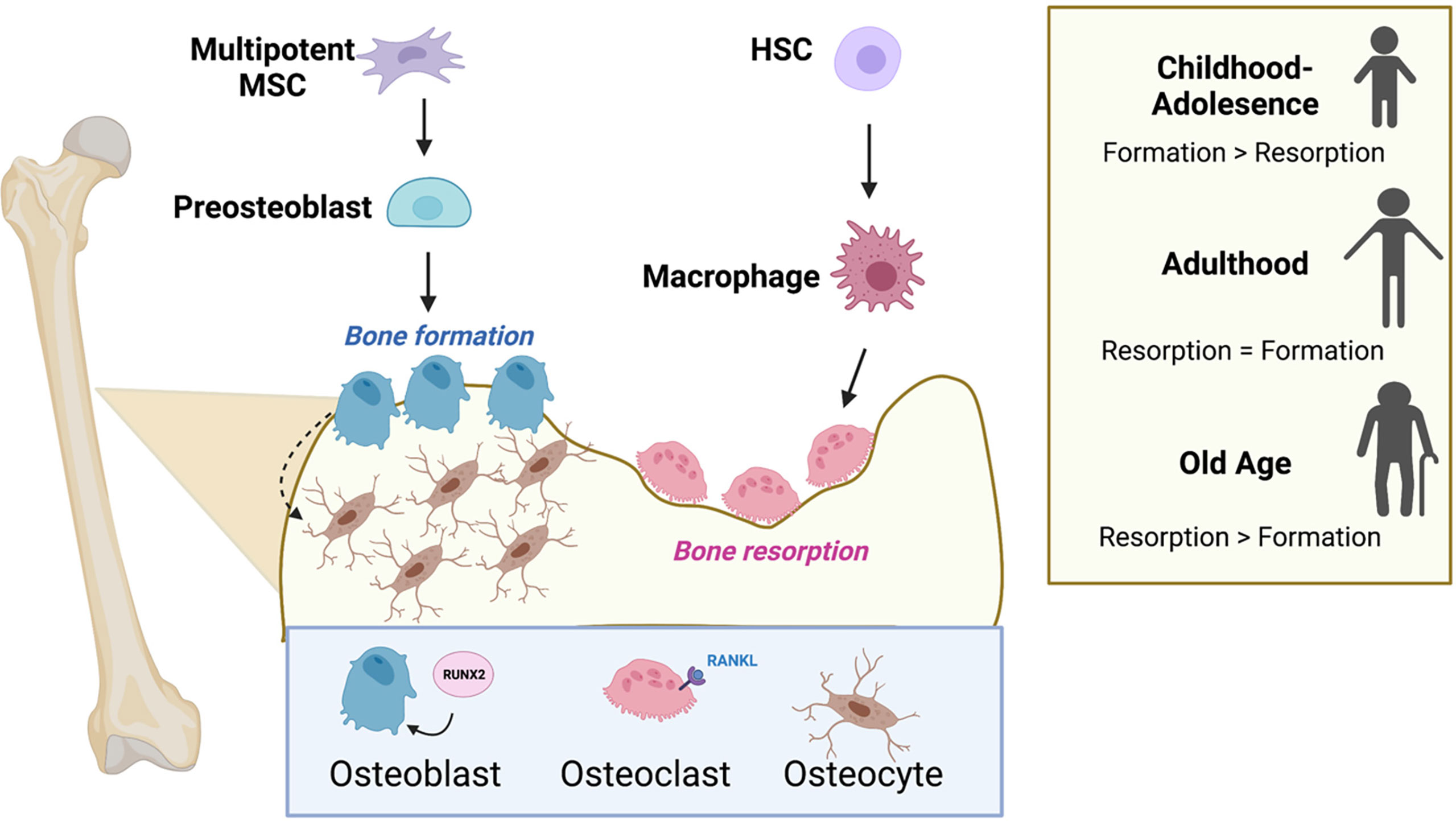
Figure 2 Osteoblasts, osteoclasts, and the cycle of bone remodeling. Bone remodeling balances the competing actions of the bone-building osteoblasts and the osteoclasts that promote bone resorption. Osteoblasts and osteocytes originate from common precursor bone marrow mesenchymal stem cells (MSCs), while osteoclasts are multinucleated and originate from hematopoietic stem cells (HSCs) in the bone marrow. Successful differentiation of progenitor cells into osteoblasts and osteoclasts requires the transcription factor, RUNX2 and RANK-ligand (RANKL), respectively. The level of bone formation versus bone resorption is dynamic and changes with age. Formation exceeds resorption during adolescence, and the scale tips the opposite direction favoring resorption in old age. Figure was created with BioRender.com.
The chief cell types regulating bone homeostasis are the bone-building osteoblasts and the bone-destroying osteoclasts, which together make up ~5% of cells in the bone. Osteoblasts and osteocytes originate from common precursor bone marrow mesenchymal stem cells (23). Osteoclasts are multinucleated cells that originate from hematopoietic progenitors in the bone marrow and result from the fusion of myeloid lineage monocytes (osteoclast precursor cells) (Figure 2). The cycle of bone remodeling consists of two main phases, the formation of bone, whereby osteoblasts function to produce and secrete alpha-1 type-1 collagen and mineralize bone; and bone resorption, where osteoclasts residing in resorption bays digest bone by secreting proteases and acids to promote bone degradation (3). Osteocytes are terminally differentiated osteoblasts consisting of 90-95% of bone and are the longest living bone cells. They are encased in the matrix of mature bone and function by secreting matrix proteins and help to regulate osteoblast and osteoclast activity.
The complex signaling and molecular mechanisms that govern bone remodeling have been extensively studied. In brief, successful differentiation of progenitor cells into osteoblasts and osteoclasts is controlled, respectively, by the transcription factor RUNX2 (25) and the cytokine RANK-ligand (RANKL), a member of the tumor necrosis factor (TNF) superfamily (26) (Figure 2). Osteoblast function is regulated by numerous autocrine and paracrine signals. In particular, osteoblasts respond to a range of stimuli, including insulin-growth factors (IGF) (27), platelet-derived growth factor (PDGF) (28), TGF-β (29), and bone morphogenetic proteins (BMP) (30), as well as numerous classical hormones such as parathyroid and thyroid hormones (31). Similarly, osteoclast function is regulated by hormones including calcitonin (32), thyroid and parathyroid hormones (33, 34), IGF-1 (35), and PDGF (36). Osteoclasts and osteoblasts are the most metabolically active cells in the normal bone environment, but little is known about the bioenergetic properties of osteocytes. Emerging work suggests that osteoblasts and osteoclasts must acquire the proper nutrients and amino acids to sustain their differentiation and execute their proper functions in orchestration of bone remodeling. In the sections that follow, we describe the cell-type specific metabolic programs operative for osteoblast and osteoclast function.
3.2. Energy metabolism of the osteoblast
Bone formation is an energy-intensive process, and the importance of glucose metabolism in bone and osteoblast lineage cells has long been appreciated from studies of bone explants and isolated bone osteoblastic cells (4–6). Specialized osteoblast cells needed to build bone require the generation of high levels of ATP (23). Osteoblasts express 3 glucose transporters (GLUT1, GLUT3, and GLUT4) to facilitate glucose uptake (37–39). In vitro studies, including metabolic tracing with murine calvarial cells, showed that osteoblast differentiation under aerobic conditions increases glucose consumption and lactate accumulation, and that aerobic glycolysis accounts for 80% of ATP production in mature osteoblasts (Figure 3) (40, 41). The majority of the remaining 20% of glucose-derived carbon is metabolized in the mitochondria through respiration.
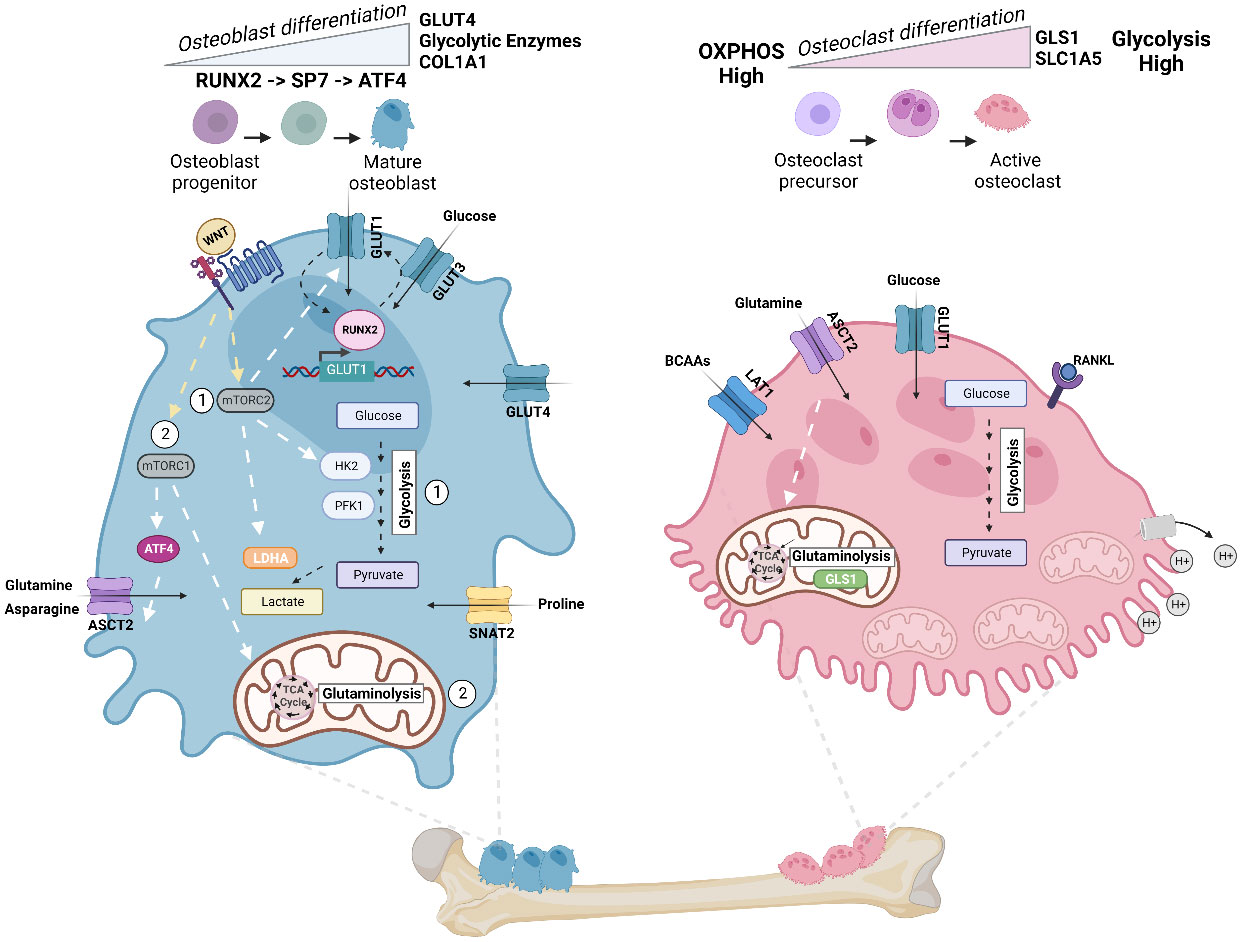
Figure 3 Energy metabolism in osteoblast and osteoclast. Metabolic pathways rewired in osteoblast (Left) and osteoclast (Right) during cellular differentiation. Left: In mature osteoblasts, glycolysis accounts for 80% of ATP production. Osteoblast precursors sequentially express the transcription factor RUNX2, followed by Sp7 (osterix (Osx)), and collagen-promoting ATF4 at late stages of differentiation. Illustration depicts the feed-forward mechanism where high intercellular glucose inhibits the proteasomal degradation of RUNX2, and RUNX2 increases the levels of GLUT1 transcription. WNT signaling regulates osteoblast activity and has been shown to (1) activate glycolytic enzymes (HK2 and PFK1), GLUT1, and LDHA via mTORC2 and (2) promote glutaminolysis and TCA cycle activation via mTORC1 and downstream regulation of ATF4 and glutamine transporter ASCT2 (SLC1A5). Import of asparagine via ASCT2 and proline via SNAT2 (SLC38A2) also facilitates osteoblast differentiation. Right: To dissolve bone and degrade collagen, osteoclasts require the generation of protons (H+) by hydrolyzing ATP. Thus, osteoclasts exhibit a significant demand for ATP and contain a high number of mitochondria. ATP demand is met by upregulating both glycolysis and glutaminolysis, and differentiation increases glycolysis and expression of glumaminase 1 (GLS1) and ASCT2. Mid-late stage osteoclast differentiation is also dependent on increased branched chain amino acid (BCAA) pools. Figure was created with BioRender.com.
The dependence of osteoblasts on aerobic glycolysis is intimately tied to normal differentiation of these specialized cells from mesenchymal precursors. Osteoblasts differentiate through sequential stages, which are associated with regulated expression of distinct transcription factors and markers. Early osteoblast precursors express the transcription factor RUNX2, while later stages of differentiation are activated by the expression of Sp7, also called osterix (Osx). At late stages of differentiation, mature osteoblasts activate expression of activating transcription factor 4 (ATF4), a master transcriptional regulator of amino acid metabolism and stress-response pathways. ATF4 activation promotes an increase in collagen type I (COL1A1) synthesis, as well as other matrix proteins such as integrin-binding sialoprotein (IBSP) and osteocalcin (OCN) (42, 43) (Figure 3). Studies by Wei and colleagues illustrated that a feed forward mechanism exists between the glucose transporter GLUT1 and the osteoblast transcription factor RUNX2 (44). High levels of intracellular glucose inhibit the proteasomal degradation of RUNX2, and RUNX2 increases the levels of GLUT1 by binding to and activating the Glut1 promoter (Figure 3). This promotes the uptake of glucose, which in turn enhances osteoblast differentiation and bone formation. The researchers show that GLUT1 is responsible for the majority of glucose uptake in osteoblasts, and that this enhanced uptake is necessary for osteoblast differentiation (44). During differentiation in vitro, GLUT4 expression, but not that of GLUT1 or GLUT3, is upregulated. GLUT4 is further induced by insulin-stimulated glucose uptake, revealing that other glucose transporters can contribute to cellular metabolism that supports osteoblast differentiation (38). Moreover, mechanistic data by Esen and colleagues define an axis where Wnt3a contributes to aerobic glycolysis in osteoblasts via an mTORC2-AKT axis downstream of Rac Family Small GTPase 1 (RAC1). In this pathway, WNT-LRP signaling promotes bone formation through the induction of key glycolytic enzymes, including GLUT1, Hexokinase 2 (HK2), Phosphofructokinase-1 (PFK1), and Lactate Dehydrogenase A (LDHA) (Figure 3) (45). More recent work has also found that, in the presence of glucose, NR2F1, a nuclear hormone receptor and transcription factor, promotes osteoblast survival and osteogenic differentiation via AKT/ERK signaling; while in the absence of glucose, expression of NR2F1 is reduced, and activation of this pathway is inhibited (46)
Glutamine serves as an additional fuel source for osteoblasts during osteogenesis (47). There, glutamine is oxidized in the TCA cycle, which contributes to energy production in the mitochondria in osteoblast precursors (48). Indeed, Karner et al. demonstrated that glutamine is required for osteoblast function in response to WNT signaling (48). They show using cultures of osteoblast precursors and in vivo mouse models that WNT, via mTORC1, stimulates catabolism of glutamine in the TCA cycle, which lowers intracellular glutamine and activates the general control nonderepressible 2 (GCN2)-mediated integrated stress response (ISR) (Figure 3). This WNT-mTORC1-GCN2 axis was found to be important for maintaining protein translation during the process of bone anabolism by activating ATF4, which promotes the transcription of genes important for amino acid uptake and biosynthesis (48). More recently, Shen and colleagues found that ASCT2 (SLC1A5) is a target of ATF4, downstream of mTORC1, and is the primary transporter of glutamine in response to WNT in osteoblasts (49). Building on this knowledge, Sharma et al. further found, using genetic and metabolic techniques, that SLC1A5 is required for osteoblast proliferation, differentiation, and extracellular matrix synthesis by facilitating the import of glutamine and, to a lesser extent, asparagine for de novo non-essential amino acid synthesis and protein production (50). Aside from glutamine and asparagine, osteoblast differentiation increases import of the amino acid proline via the neutral amino acid transporter, SNAT2 (SLC38A2), thereby facilitating the incorporation of proline into proline-rich osteoblast proteins, such as RUNX2, OSX, OCN, and COL1A1 (51).
3.3. Energy metabolism of the osteoclast
Osteoclasts are highly motile cells. Their differentiation, as well as their function in bone resorption, are ATP-demanding processes. As such, osteoclasts have more mitochondria per surface area than virtually any other cell type to support the need for high levels of ATP (52). The function of osteoclasts in dissolving bone and degrading collagen requires the generation of protons (H+) by hydrolyzing ATP in order to support acidification and bone resorption (Figure 3) (23). Like osteoblasts, osteoclasts require an upregulation of glycolysis and increase in glucose uptake via GLUT1 for their differentiation and function. Depriving osteoclasts of glucose inhibits their bone-resorbing activity (53).
Furthermore, osteoclast differentiation was also found to be dependent on glutamine, as expression of the glutamine transporter SLC1A5 and the first enzyme in the breakdown of glutamine, glutaminase 1 (GLS1), increased during differentiation (Figure 3). Depletion of glutamine or inhibition of ASCT2 repressed osteoclast differentiation and function (53). The regulation of glycolysis in osteoclasts is under the control of HIF1a, while c-MYC regulates expression of SLC1A5 and GLS1 (53, 54). More recently, Ozaki et al. demonstrated that, aside from glutamine, osteoclast differentiation is negatively regulated by the neutral amino acid transporter, LAT1 (SLC7a5) in mice (55). Further, all three BCAAs, particularly valine, as well as the enzymatic activity of branched-chain aminotransferase 1 (BCAT1) are increased during RANKL-induced mid-late stage osteoclast differentiation (56). Demonstrating a complex relationship between the maintenance of amino acid pools, osteoclastogenesis, and bone homeostasis.
Consistent with prior work, Li et al. demonstrate that osteoclasts require both glycolysis and oxidative phosphorylation for proper function (57). However, metabolic dependencies differ between osteoclast differentiation and active bone resorption. As a notable example, it has been shown that the formation of osteoclasts requires OXPHOS, while bone resorption exhibits a greater dependence on glycolysis (Figure 3) (52).
3.4. ATF4 plays a central role in physiologic bone remodeling
Although studies surrounding the bioenergetics of normal bone homeostasis have been relatively limited, the importance of glucose and glutamine utilization by osteoblasts and osteoclasts has been established. As detailed above, osteoblasts demonstrate a dependence on aerobic glycolysis that is also observed in cancer cells, and both osteoblasts and osteoclasts have a preferential need for upregulating glycolysis for cell differentiation and function. While there is logic to the role of aerobic glycolysis in cellular function, the reasons for this phenomenon have not been fully elucidated in the context of differentiation and require further study. One can speculate that this may be a result of a dependency on glycolytic intermediates to fuel biosynthetic reactions, such as de novo amino acid synthesis. Furthermore, the role of other metabolic pathways in osteoclast and osteoblast differentiation and function also remains unanswered. One notable example is the role of ATF4. Osteoblasts upregulate expression of ATF4 in the terminal differentiation process (42). ATF4 has been shown to regulate osteogenic genes and its phosphorylation by RSK2 is needed for differentiation (58). ATF4 enhances bone formation by activating amino acid import and collagen synthesis, and studies have shown that implementing a high-protein diet in mouse models can overcome the effect of ATF4 loss on skeletal formation (58, 59). ATF4 expression in osteoblasts has been demonstrated to be regulated, at least in part, via an mTORC1-GCN2-ATF4 axis (48, 60). Interestingly, mTORC1 has been shown to play a role in the ATF4-dependent regulation of serine and glycine biosynthesis and downstream collagen production (61), suggesting another downstream pathway that would explain osteoblast dependence on ATF4 signaling.
4. Malignant bone metabolism
4.1. Primary bone malignancies
4.1.1. Osteosarcoma
Osteosarcomas (OS)s are the most common primary malignant bone sarcomas, representing 40-50% of bone tumors (62). OS is characterized by a bimodal age distribution with peak incidence in children and adolescents, and a median age of 18, as well as a smaller second peak incidence in individuals over 60 years of age. The worldwide incidence of OS is 1-3 cases annually per million individuals (63). Over 75% of OS cases occur in the metaphysis of the long bones, most commonly in the distal femur, proximal tibia, and humerus (64). The current standard of care for OS includes a combination of chemotherapy, which may include methotrexate, cisplatin, doxorubicin, ifosfamide, and etoposide, as well as surgery. OS cells are not readily responsive to radiation, so it does not play a major role in treatment (65). Due to limited success of new biologically targeted therapies, chemotherapeutic agents have been part of OS treatment for decades, and the standard of care has largely been unchanged. Commonly occurring genetic lesions or pathway alterations have not been identified in OS, though mutations in p53 and Rb have been observed along with significant cases of aneuploidy (66). However, to this date, no clear molecular signatures have been identified that have led to successful targeted therapies (64).
4.1.2. Ewing sarcoma
Ewing sarcoma (ES) is the second most common pediatric bone tumor, with an incidence of 1 case per 1.5 million in the population and presents most commonly during adolescence. ES can arise both in the bone and in soft tissue sites, most commonly in the pelvis and proximal long bones of the body (67). Current treatment regimens include chemotherapy consisting of doxorubicin, etoposide, cyclophosphamide, vincristine, and ifosfamide, as well as local control with surgery and/or radiation (67). ES is characterized by the presence of EWS::ETS fusion proteins, most commonly EWS::FLI1, which results from a chromosomal t(11;22) translocation (67). These fusion oncogenes act as the tumor-initiating event, leading to malignant transformation. EWS::FLI1 acts as an aberrant transcription factor that promotes widespread epigenetic and transcriptional reprogramming through its function at enhancers and gene promoters (68–70). Aside from the presence of the EWS::FLI1 fusion protein, ES tumors are otherwise genomically quiet, with few other recurrent mutations (71–73). Thus far, efforts to target EWS::FLI1 itself have been largely unsuccessful due to its lack of enzymatic activity and disordered structure (67). Thus far, efforts to target EWS : FLI1 itself have been largely unsuccessful due to its lack of enzymatic activity and disordered structure.
Given the lack of therapeutically targetable mutations in primary bone sarcomas, characterization of metabolic dependencies may identify opportunities for novel therapeutic strategies. To date, investigation of the metabolic properties of sarcomas has been limited, but early studies have begun to provide insights. Studies of sarcoma patients identified that their tumors displayed high rates of glycolytic activity and lactate production (74). As a result, fluorodeoxyglucose positron emission tomography (FDG–PET) imaging is used clinically as a staging tool for several diverse types of cancers, including OS and ES, where it can be used to identify tumors at the primary site and skeletal and lymph node metastases. This radiographic tool can in some cases also be used to follow disease progression and to predict patient outcome (75, 76). More recently, data have emerged around the rewiring of other metabolic pathways in sarcomas. In the following sections we will focus on and summarize the current understanding of amino acid pathways dependencies in OS and ES.
4.2. Amino acid dependencies in osteosarcoma and Ewing sarcoma
4.2.1 The serine biosynthetic pathway as a key dependency
The most extensively studied amino acid biosynthetic pathway in sarcomas is the SBP. As a non-essential amino acid, serine can be taken up extracellularly or synthesized de novo. The SBP utilizes the 3-phosphoglycerate (3-PG) intermediate of glycolysis as the carbon backbone to generate serine de novo via three enzymatic steps carried out by the enzymes PHGDH, phosphoserine aminotransferase (PSAT1), and phosphoserine phosphatase (PSPH) (Figure 1). PHGDH expression is high in both OS and ES (77–81), and high PHGDH correlates with poor survival in both of these cancers, suggesting a clear dependency on the SBP. The SBP contributes to a broad metabolic hub. In addition to its proteinogenic role, serine has numerous additional biological and metabolic functions. For example, the conversion of serine to glycine by serine hydroxymethyltransferase (SHMT) (isozymes SHMT1, cytosol; and SHMT2, mitochondria) generates one-carbon units that enter two main pathways: the folate cycle and the methionine cycle. These pathways are important for nucleotide synthesis and the generation of the methyl donor S-Adenosyl Methionine (SAM), respectively (Figure 1). Glycine and cysteine, derived from the transsulfuration pathway, are also utilized to synthesize GSH. Thus, the sum of metabolites produced by these interconnected networks contribute to de novo purine and pyrimidine synthesis, maintenance of GSH and NADPH pools for redox homeosis, as well as the production of methyl groups to fuel DNA and histone methylation reactions (82). Preferential use of the SBP allows cancer cells to take advantage of the pathway for the generation of biomass and to promote cell growth and division.
Methotrexate is a dihydrofolate reductase (DHFR) inhibitor (Figure 1) and has been part of the standard of care for OS for over 40 years. DHFR is a critical enzyme in the folate cycle, downstream of the SBP, which catalyzes the reduction of dihydrofolate to tetrahydrofolate (THF), important for DNA synthesis and methylation. These recent mechanistic insights on the role of the SBP and interrelated pathways provide clarity on longstanding empirically determined chemotherapy regimens utilizing methotrexate.
In primary bone sarcomas, the SBP has received extensive attention, given the high levels of PHGDH that are observed in both OS and ES (77–81, 83). Rathore et al. recently explored the effect of PHGDH inhibition in OS cell lines and xenograft models and provided preclinical evidence for the combination of PHGDH inhibition with inhibition of mTORC1. Using metabolomic and lipidomic profiling, the authors identified the accumulation of fatty acids, BCAAs, SAM, and methionine in OS following inhibition of PHGDH using the inhibitor NCT-503 (81). They were able to show that PHGDH inhibition led to increased expression of SLC7A5 (LAT1) and SLC3A2 (CD98) transporters that drive transport of leucine into the lysosome, leading to mTORC1 activation (Figure 4). Given this finding, the researchers then demonstrated that combining NCT-503 with non-rapalog mTORC1 inhibitor perhexiline led to tumor reduction in xenograft models (81). These studies are consistent with prior work, which also demonstrated that mTORC1 regulates OS cell proliferation partly via the regulation of serine/glycine metabolism (83).
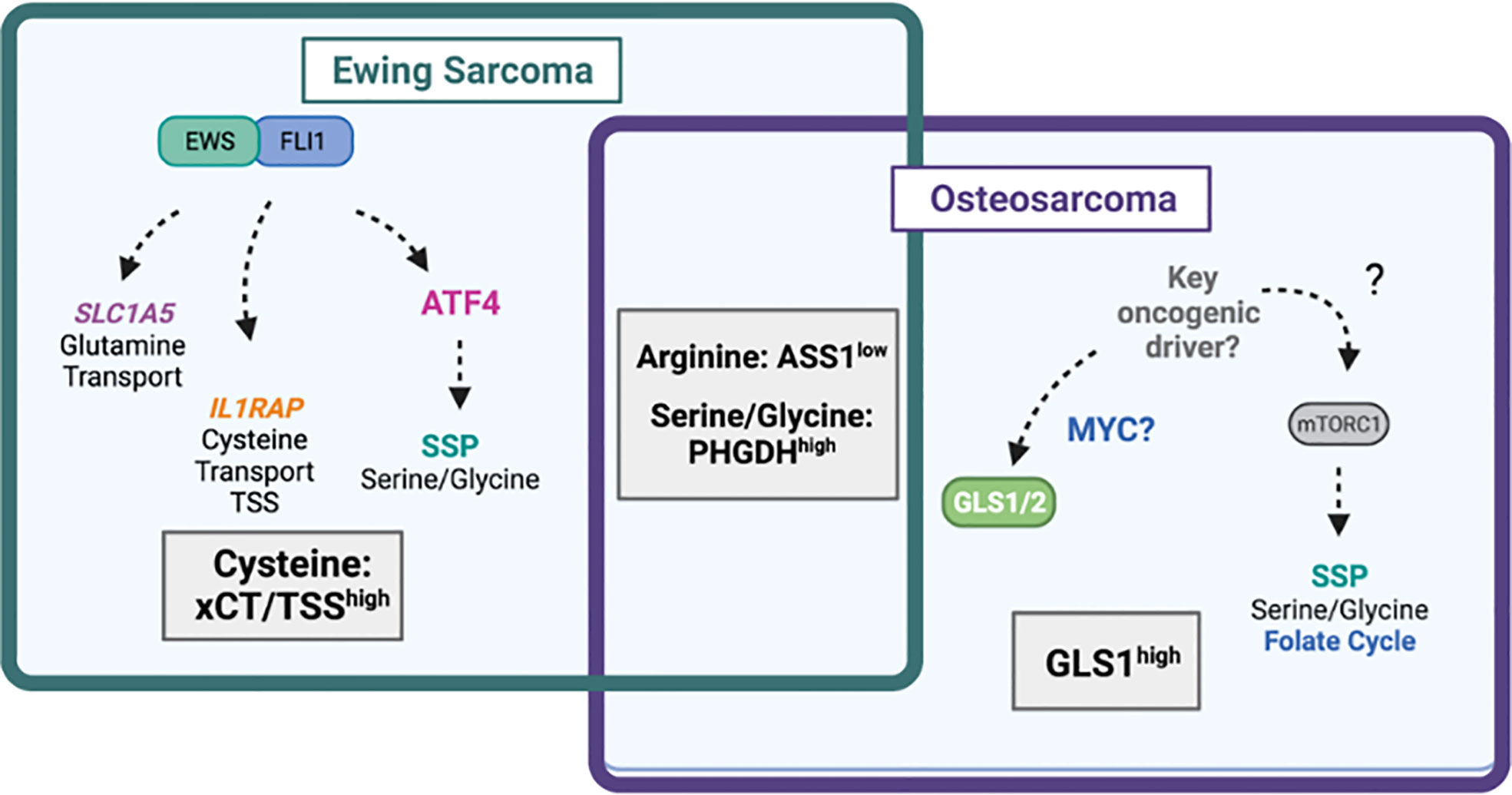
Figure 4 Amino acid dependencies in Ewing sarcoma and osteosarcoma. Venn-diagram depicting shared and unique amino acid vulnerabilities in Ewing sarcoma and osteosarcoma. The EWS::FLI1 fusion protein has been shown to directly regulate expression of glutamine transporter SLC1A5 (ASCT2) leading to high glutamine import. EWS::FLI1 also regulates expression of IL1RAP and downstream cysteine availability via the transsulfuration pathway (TSS) and xCT (SLC7A11). EWS::FLI1 indirectly regulates the serine biosynthesis pathway (SSP) via ATF4. In osteosarcoma, the SSP also drives tumorgenicity through undefined mechanisms likely involving mTORC1. The mitochondrial enzyme GLS1, which may be under the control of MYC is highly expressed in osteosarcoma and predicts worse survival. Diagram was created with BioRender.com.
Numerous studies have focused on defining the role of the SBP in ES (77–80). This work has shown that PHGDH is overexpressed in ES cell lines and primary patient tumors, and that ES cells are highly dependent on the SBP. Furthermore, several groups, including our own, have demonstrated that the EWS : FLI1 fusion oncogene regulates expression of key enzymes in the SBP, PHGDH, PSAT1, and PSPH, as well as their metabolic products (77, 78, 80, 84).
In cancers without amplification of the PHGDH locus, such as lung cancer, activation of the SBP can be mediated through upregulation of ATF4 (85–88). Studies from our lab showed that EWS : FLI1 is able to transcriptionally regulate expression of ATF4 through direct promoter binding, and that ATF4 expression can also be regulated by the scaffolding protein menin, which also has an oncogenic role in ES (Figure 4) (84, 89). However, studies have been inconsistent on the predominate function of the SBP is in supporting ES tumorgenicity. Some evidence suggests that inhibition of the SBP in ES affects redox homeostasis and leads to the accumulation of ROS and DNA damage (78). Other work has implicated PHGDH loss in decreasing 2-hydroxyglutarate levels, increasing total histone-3 levels, and enhancing methylation of histone H3 at lysine (K) 9 and K27. It should be noted that responses varied among the cell lines evaluated (77). These studies suggest that the SBP may serve an oncogenic role in ES by influencing multiple downstream metabolic programs. In either case, recent work by Issaq and colleagues showed that PHGDH inhibition is a targetable vulnerability, where combination with inhibition of nicotinamide phosphoribosyltransferase (NAMPT), which blocks synthesis of the PHGDH substrate NAD+, had a synergistic effect in vitro and in in vivo xenograft models (80).
While the mechanism of SBP activity in ES can be attributed to the direct and indirect effects EWS : FLI1 fusion-mediated metabolic reprogramming, the mechanism by which the SBP is activated in OS is much less clear, likely due to the lack of known oncogenic drivers. Wildtype p53 has been shown to reduce PHGDH in melanoma (90). The majority of OSs exhibit mutations in p53, suggesting a potential mechanism of SBP regulation (91). Importantly, PHGDH inhibition has been reported to induce cell death in ES (77), but not in OS where PHGDH inhibition resulted in cytostasis (81). Whether this represents unique difference between the tumor types or technical experimental variables requires further study. Further, ES cell lines exhibit varying sensitivities to serine/glycine withdrawal (79), while uniformly showing reduced growth with genetic PHGDH loss of function. Additionally, the effectiveness of PHGDH inhibition in OS cells does not increase their sensitivity to serine/glycine depletion (81). This suggests that ES and OS cells may be more dependent on the de novo synthesis of serine than the extracellular import of the amino acid.
4.2.2 Glutamine—a conditionally essential amino acid
Although glutamine is a non-essential amino acid, many tumors rely on extracellular glutamine for survival, therefore classifying it as a conditionally essential amino acid (14). As the most abundant amino acid in the plasma, glutamine is found at levels between 500 and 750µM. In addition to its proteinogenic functions, glutamine also participates in the synthesis of other non-essential amino acids, as well as acting as an essential carbon and nitrogen donor for the synthesis of nucleotides, GSH, and glycosylation precursors.
Numerous studies have identified a dependence of OS and ES cells on glutamine and glutaminolysis. In cancer cells, the rate-limiting enzyme for glutaminolysis is GLS, the mitochondrial enzyme responsible for the conversion of glutamine to glutamate, which can exist as one of two isoforms, GLS1 and GLS2. Molecularly, research has shown that MYC-induced glutaminolysis occurs specifically in MYC-dependent OS cells, but not in osteocytes (Figure 4) (92). Work in ES has demonstrated that EWS::FLI1 positively regulates expression of the glutamine transporter ASCT2 (SLC1A5), potentially via the direct binding of the fusion to a regulatory sequence in the SLC1A5 gene (Figure 4) (78). Clinical studies with OS patients have shown that GLS1 is highly expressed in OS tumor tissue, which predicts poor survival (Figure 4). The effect of GLS1 inhibition was recently evaluated in OS cell lines using metabolic flux analysis and tracing studies. These studies demonstrated that pharmacologic inhibition of GLS1 with CB-839, combined with electron transport chain (ETC) inhibitor metformin, reduced primary tumor growth and metastasis by inhibiting glycolytic and TCA cycle activity (93). Furthermore, GLS1 expression decreases with neoadjuvant chemotherapy, suggesting that GLS1 may be predictive of OS treatment response (94). Recent studies have also demonstrated success in therapeutically targeting GLS in cell line and murine models of soft tissue sarcomas (95).
4.2.3 Arginine—a case of auxotrophy in sarcomas
Arginine can be synthesized de novo by the action of the argininosuccinate synthetase 1 (ASS1) enzyme, which converts citrulline and aspartate to arginine (Figure 1). Arginine auxotrophy can result from the silencing of ASS1 in several tumor types, including sarcomas (96). In cancers such as lymphomas, bladder cancer, and prostate cancer, diminished ASS1 expression can be due to methylation of the ASS1 promoter (97). In the case of sarcomas, a 2016 study found that almost 90% of sarcoma tumors have low ASS1 expression; however, ASS1 promoter methylation is not observed in sarcomas. Specifically, Bean et al. evaluated 701 bone or soft tissue sarcomas by immunohistochemistry analysis for ASS1 expression and found that most primary bone tumors (87.2%, 34/39) and soft tissue tumors (86.4%, 572/662) had low ASS1 expression. 3 of 10 OS tumors evaluated were determined to be ASS1 positive, while 1 of 7 ES tumors evaluated were ASS1 positive (98). By characterizing select ASS1Low cell lines, including OS and ES cell lines, the authors showed that arginine deprivation with pegylated arginine deaminase (ADI-PEG20) led to cytostasis and a dependence on autophagy. Combining ADI-PEG20 with chloroquine, an autophagy inhibitor, led to necroptotic and apoptotic cell death. The authors tested this synthetic lethal targeting strategy in vivo using subcutaneous xenografts with MNNG/HOS OS cells and showed that combining ADI-PEG20 with chloroquine led to the greatest reduction in tumor growth, relative to single arm treatments (98).
4.2.4 Transsulfuration pathway and cysteine metabolism
Cysteine is another non-essential amino acid, which can be acquired from the extracellular space through several transport mechanisms or synthesized de novo in the transsulfuration pathway from serine and methionine (Figure 1). Cysteine is the rate-limiting substrate for the synthesis of GSH, which is critical for the maintenance of cellular redox homeostasis. A drop in GSH can lead to the accumulation of reactive oxygen species (ROS) and/or oxidized membrane phospholipids (lipid ROS). An inability to manage ROS leads to the oxidation of various biomolecules and can culminate in cell death. Thus, cancer cells employ a variety of mechanisms to mitigate ROS burden. As it relates to cysteine, many types of cancers will upregulate SLC7A11, which encodes for xCT, the rate-limiting subunit of the cystine-glutamate antiporter system xC-. In a recent study, application of 3D cultures with ES cells in proteomic and translatomic screens found that the EWS : FLI1 fusion oncoprotein directly induces expression of system xC- and the transsulfuration pathway, via induction of IL1RAP expression, a cell surface IL-1 receptor co-receptor. The researchers found that IL1RAP directly binds system xC- to increase its cystine importing activity, and that EWS::FLI1 regulates IL1RAP expression through enhancer activation (Figure 4). IL1RAP was found to be highly expressed in ES and important for promoting resistance to anoikis, detachment-induced cell death, and metastasis (99). Although little is known about the role of cysteine and GSH metabolism in OS, it has been demonstrated that OS cell are dependent on methionine, suggesting that this may be in part to maintain cysteine levels (100).
5. Bone microenvironment and metastasis
5.1. Metabolism in the bone tumor microenvironment
The function of non-malignant cells in mesenchymal tumors has been less studied when compared to epithelial cancers of adulthood (101). Additionally, a distinguishing feature of sarcomas is that the distinction between malignant and stromal cells can be unclear, due to the mesenchymal origin of the tumor cells themselves (101). Primary bone sarcomas grow in specialized and complex bone microenvironment that are highly vascularized and contain osteoclasts, osteoblasts, osteocytes, stromal cells (MSCs, fibroblasts), vascular cells (endothelial cells and pericytes), immune cells (macrophages, lymphocytes) (102, 103), and a mineralized extracellular matrix (ECM) (104). This is fertile soil for tumor cells to hijack growth promoting pathways such as cytokines, chemokines, and growth factors in the bone microenvironment (65, 105). Remodeling and resorption of bone is a key feature of primary bone malignancies like OS and ES, leading to an osteolytic bone environment that provides space for growth of the tumor (101, 106, 107).The metabolic crosstalk in and among these cell types, and how it influences and is influenced by tumor cells, are important outstanding questions that are beginning to be addressed.
MSCs have been shown to favorably impact OS progression and contribute to osteolysis (108, 109). Several studies have centered around the role of lactate in promoting an acidic microenvironment. Lactate released by both cancer cells and cells in the TME plays a prominent role in tumor progression, by increasing angiogenesis, motility, and the migration of cancer cells (110). Studies in OS models have demonstrated that lactate derived from MSCs feeds OS (111). MSCs are induced by adjacent OS cells to undergo Warburg metabolism and increase lactate secretion and expression of the lactate exporter monocarboxylate transporter 4 (MCT4). Lactate from MSCs feeds OS, and OS cells import lactate via monocarboxylate transporter 1 (MCT1). This indirectly increases mitochondrial biogenesis and promotes the migration of OS cells (111). The mechanisms linking lactate metabolism to mitochondrial biogenesis and cell migration remain exciting avenues for further exploration.
Insights into the metabolic functions of the bone TME can also be gleaned from cancers that metastasize to the bone, such as breast cancer, prostate cancer, and multiple myeloma (112). For example, Pollari et al. show that serine from breast cancer cells in the metastatic bone microenvironment contributes to the formation of bone resorbing human osteoclasts and may contribute to osteolysis (113). Additionally, similar to studies in OS, bone metastatic cancer cells have also been shown to release lactate via MCT4, which is taken up by MCT1 in osteoclasts to fuel OXPHOS and collagen resorption (114). In both of these cases, the activity of osteoclasts leads to bone resorption, creating a more fertile metastatic niche. Interestingly, studies in multiple myeloma have shown that tumor cells prevent the differentiation of bone marrow stromal cells into osteoblasts in part by depleting glutamine. Finally, osteoblast differentiation increased expression of ASNS and asparagine could rescue osteoblast differentiation after glutamine depletion (115). This examination of amino acid cross talk in the primary tumor of OS and ES is clearly incomplete. Further exploration of this complex interaction with tumor-supporting cells in the bone microenvironment will be central to determining the effectiveness of metabolic therapies and predicting potential resistance mechanisms in sarcomas.
5.2. Primary versus metastatic metabolism
The ability to starve cancer cells through dietary restriction and/or the use of targeted therapies has garnered extensive attention recently (22). However, the cancer context in which these therapies are evaluated is key to determining their therapeutic potential. Further, it is evident that the metabolic dependencies of the primary tumor differ from those of metastatic lesions. For instance, in breast cancer models, researchers have demonstrated that lung and brain metastases are more sensitive to targeting of the SBP than the primary tumor site, in part due to SBP-dependent activation of mTORC1 (116–119). In the brain, the low availability of serine and glycine makes brain metastases more sensitive to inhibition of PHGDH (118). More recently, work also utilizing breast cancer models has suggested that heterogeneous or low PHGDH protein expression at the primary tumor site is associated with increased metastatic potential. This study shed light on distinct regulation of PHGDH transcript and protein and demonstrated a non-catalytic role for PHGDH in directly interacting with the glycolytic enzyme phosphofructokinase resulting in aberrant protein glycosylation (120). Though PHGDH expression is high in both OS and ES and drives tumorgenicity, the impact of potential heterogeneity in transcript and protein expression on primary growth and metastatic dissemination has not been explored. A deep understanding of metabolic dependencies in the tumor niche in bone for OS and ES compared to common sites of metastasis, such as the lung, is lacking and warrants further study.
Studies have begun to look at metabolic differences associated with metastasis in bone sarcomas. Using metabolic profiling and metabolic flux analysis in high and low metastatic cell lines, recent work has suggested that metastatic OS cells activate pathways such as arginine, GSH, and fatty acid metabolism (121–123). Additionally, GLS1 has recently been shown to play a role in promoting metastasis in OS (93). Parallel studies in ES have been more limited, though as discussed above, recent work has implicated cysteine metabolism downstream of IL1RAP in promoting metastasis (99). This work would benefit greatly from studies in additional models of spontaneous metastasis, as the current models do not readily metastasize before mice succumb to primary tumor burden (124).
6. Therapeutic applications targeting amino acid metabolism in sarcomas
The current standard of care for sarcoma, consisting of surgery, chemotherapy and radiation has improved the life expectancy for patients, but survival has since plateaued without meaningful improvements in 30 years. Therefore, new therapies as well as diagnostic and prognostic tools are needed. As it relates to metabolism, there are new therapies under clinical evaluation for sarcomas. As previously discussed, preclinical studies showed that ~90% of sarcomas have low expression of ASS1, making these cells more dependent on extracellular sources of arginine (98). Treatment of ASS1Low sarcoma cell lines with PEGylated arginine deiminase, ADI-PEG20, sensitized cells to cell death induced by the chemotherapeutic agents, gemcitabine and docetaxel. This prompted a Phase II clinical trial that started in May 2018 in which PEGylated arginine deiminase, ADI-PEG 20, was used in combination with gemcitabine and docetaxel for the treatment of bone and soft tissue sarcoma, including OS, ES, and small cell lung cancer (NCT03449901) (98, 125). However, challenges in the clinical use of ADI-PEG20 are likely based on preclinical observations. These may include acquired resistance to ADI-PEG20 by upregulation of ASS1 (98) or rewiring of other metabolic pathways, such an increased dependence on glutamine anaplerosis and the SBP upon arginine starvation (126). Targeting these compensatory pathways with addition of a GLS inhibitor or PHGDH inhibitor has been found to result in synthetic lethality in several models, including leiomyosarcoma (126).
Telaglenastat (CB-839) is a potent orally bioavailable GLS inhibitor that has exhibited anti-tumor activity in breast cancer and lymphoma (127, 128). Current clinical trials are evaluating its efficacy in hematologic and solid cancers (129). A recent metabolomics study tested the effect of GLS inhibition with CB-839 in undifferentiated pleomorphic sarcoma (UPS) and soft tissue sarcomas and found that GLS inhibition causes tumor cell death (95). These findings are promising for the translation of CB-839 to the clinic, especially for OS patients who exhibit high GLS1 activity (93, 94). However, the use of GLS inhibitors needs to be carefully evaluated. In some contexts, GLS dependency may be a phenomenon of cancer cells in vitro, where tumors are able to acquire compensatory mechanisms when GLS is inhibited in vivo (130, 131).
Given the dependency of numerous cancers on the SBP and PHGDH expression, PHGDH inhibition is also a promising therapeutic vulnerability. In fact, several small molecule enzymatic inhibitors against PHGDH are under preclinical development (116, 132, 133); however, none are yet ready for clinical examination (134).
Metabolomics technology has allowed for the profiling of cancer-associated metabolites, which could provide insight into tumor specific biomarkers. While this has led to progress in identifying cancer-specific prognostic and diagnostic biomarkers in other cancer types (135), there are only a few studies that have focused on sarcoma metabolomics (136). Early work in 2011 used liquid chromatography-tandem mass spectrometry (LC-MS/MS) in formalin-fixed and paraffin-embedded (FFPE) patient tissue from soft tissue sarcomas, including leiomyosarcoma, liposarcoma, and synovial sarcoma, and showed that 8 metabolites exhibited differential abundances between soft tissue sarcoma samples and control tissue (137). Researchers have also investigated liposarcoma cell lines in vitro, revealing high consumption of nucleosides and amino acids such as arginine, glutamine, and serine. Through in vitro and in vivo models, their studies further implicate nucleoside salvage pathway activity as a potential metabolic biomarker predicting response to gemcitabine (138).
More recent work has shifted to metabolomics analysis of patient serum in search of prognostic biomarkers. This work has identified depletion of citrulline, a precursor for arginine synthesis, in high risk metastatic soft tissue sarcoma patients (139). Very few studies have focused on OS and ES patients, specifically. Work by Jia et al. used LC/MS to evaluate changes in plasma amino acid levels between 23 sarcoma patients, including OS and ES, compared to 30 healthy control subjects and identified 4 amino acids or amino acid derivatives (glutamine, sarcosine, homoproline and citrulline) that decreased and 3 (carnosine, lysine, and glutamic acid) that increased in sarcoma patients (140). One of the first metabolic profiling studies in OS utilized LC/MS to investigate the serum and urine of OS patients compared to benign tumor patients and healthy controls and identified down-regulated lipid metabolism and upregulated amino acid metabolism among the pathways impacted (141). Though these patient-centered studies are preliminary, this and future work in this area may provide leads or insights that will subsequentially require further evaluation in larger cohorts of patients to determine the utility of metabolic profiling. The hope is that the identification of bone sarcoma-specific metabolic biomarkers will allow this technology to be used in the future for the diagnosis and monitoring of sarcoma patients (142).
7. Conclusions and future directions
In recent years, the exploration of dysregulated amino acid metabolism pathways has garnered increased attention in sarcomas, with exciting new findings on pathways that may serve as tumor-specific vulnerabilities. As discussed above, the potential for basic discoveries in sarcoma metabolism to lead to clinical translation is exemplified by the identification of dysregulated arginine metabolism in bone sarcoma cells and subsequent inclusion of sarcoma patients in a Phase II clinical trial (NCT03449901) that is testing ADI-PEG20 in combination with chemotherapy (98). There is also clear precedent for success including amino acid targeting agents as adjuvant therapies in other cancers. ASNase, which depletes circulating asparagine, has been used in pediatric patients with acute lymphoblastic leukemia since the 1970s. Other amino acid pathways are being evaluated in clinical trials, such as indoleamine-2,3-dioxygenase-1 (IDO1) inhibitors, epacadostat and indoximod, for tryptophan catabolism in cervical cancer and glioblastoma (143–145). Additionally, activation of the SBP has been demonstrated to predict poor prognosis in both ES and OS and to be regulated by an EWS : FLI1-ATF4 axis in ES (77–81, 83). Preclinically, inhibitors of biosynthetic enzymes, such as the SBP enzyme PHGDH are under development for a wide array of cancers (116, 132, 133), and drugs to target amino acid transporters, such as LAT1 (SLC7A5), are also being explored (146, 147).
Primary pediatric bone sarcomas arise during a time in development when normal physiologic processes direct bone remodeling that is required for bone growth. This remodeling relies on the activation of transcriptional and metabolic pathways that support high-energy demands, including the increased import and synthesis of amino acids (42, 48). Transcription factors such as ATF4 are activated during osteoblast differentiation in order to maintain amino acid pools and downstream amino acid-dependent products, like collagen (42, 58, 59). Moreover, distinct metabolic programs and dependencies are evident across heterogeneous cell types in the sarcoma tumor niche, including osteoblasts, osteoclasts, and tumor cells, adding to the complexity of dissecting metabolic dependencies in the bone tumor ecosystem. Further, emerging work in sarcoma and other cancer types, has shown that amino acid metabolism differs among distinct tumor cell subpopulations, and that individual metabolic pathways may serve as biomarkers of metastatic potential (93, 99, 119, 120). Intratumor metabolic heterogeneity and differential amino acid requirements among tumor cells, microenvironment components, and between between primary and metastatic sites are poorly understood in the context of primary bone tumors and filling these gaps in knowledge should be a priority for investigation in sarcomas.
Given the dynamic nature of metabolic requirements during bone differentiation, the effect of amino acid targeting therapies on normal developmental processes will require thoughtful consideration, especially in the context of pediatric cancer patients who are still growing. Many other questions will also need to be addressed when investigating metabolic dependencies in the bone tumor niche. For example, the precise cell of origin for both OS and ES is still unclear and highly debated (62, 148), and the timing of genetic and signaling events during development which govern malignant transformation are not well understood. As such, it is not possible to directly compare metabolism in tumor cells to the normal precursor cell from which they arose. Furthermore, how dysregulated metabolism contributes to oncogenesis within cells undergoing transformation in the context of a bone microenvironment is not known. Nevertheless, given recent exciting advances in our understanding of bone and cancer metabolic programs in general, and of distinct amino acid dependencies in OS and ES, the promise of targeting metabolic processes in these tumors is high. Based on precedent with metabolic therapies and dynamic compensatory mechanisms, it is likely that combination approaches and targeting co-vulnerabilities will serve as the most viable option for patients.
Author contributions
JAJ, EL and CL contributed to the conception and design of the review. JAJ wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Support for this work was provided by the following sources: National Institute of Health: F31 CA254079 (JJ), R01 CA218116 (EL), R37CA237421, R01CA248160, R01CA244931 (CL).
Conflict of interest
CL has received consulting fees from Astellas Pharmaceuticals, Odyssey Therapeutics, and T-Knife Therapeutics, and is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, and targeting the GOT1-pathway as a therapeutic approach (US Patent No: 2015126580-A1, 05/07/2015; US Patent No: 20190136238, 05/09/2019; International Patent No: WO2013177426-A2, 04/23/2015).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Palmerini E, Righi A, Staals EL. Rare primary malignant bone sarcomas. Cancers (Basel) (2020) 12(11):3092. doi: 10.3390/cancers12113092
2. Molina ER, Chim LK, Barrios S, Ludwig JA, Mikos AG. Modeling the tumor microenvironment and pathogenic signaling in bone sarcoma. Tissue Eng Part B Rev (2020) 26(3):249–71. doi: 10.1089/ten.teb.2019.0302
3. Hadjidakis DJ, Androulakis II. Bone remodeling. Ann N Y Acad Sci (2006) 1092:385–96. doi: 10.1196/annals.1365.035
4. Borle AB, Nichols N, Nichols G Jr. Metabolic studies of bone in vitro. i. normal bone. J Biol Chem (1960) 235:1206–10. doi: 10.1016/S0021-9258(18)69506-7
5. Cohn DV, Forscher BK. Aerobic metabolism of glucose by bone. J Biol Chem (1962) 237:615–8. doi: 10.1016/S0021-9258(18)60342-4
6. Peck WA, Birge SJ Jr., Fedak SA. Bone cells: Biochemical and biological studies after enzymatic isolation. Science (1964) 146(3650):1476–7. doi: 10.1126/science.146.3650.1476
7. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
8. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science (2020) 368(6487):eaaw5473. doi: 10.1126/science.aaw5473
9. Lieu EL, Nguyen T, Rhyne S, Kim J. Amino acids in cancer. Exp Mol Med (2020) 52(1):15–30. doi: 10.1038/s12276-020-0375-3
10. Choi BH, Coloff JL. The diverse functions of non-essential amino acids in cancer. Cancers (Basel) (2019) 11(5):675. doi: 10.3390/cancers11050675
11. Vettore L, Westbrook RL, Tennant DA. New aspects of amino acid metabolism in cancer. Br J Cancer (2020) 122(2):150–6. doi: 10.1038/s41416-019-0620-5
12. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11(5):325–37. doi: 10.1038/nrc3038
13. Whitburn J, Edwards CM. Metabolism in the tumour-bone microenvironment. Curr Osteoporos Rep (2021) 19(5):494–499. doi: 10.1007/s11914-021-00695-7
14. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci (2010) 35(8):427–33. doi: 10.1016/j.tibs.2010.05.003
15. Liberti MV, Locasale JW. The warburg effect: How does it benefit cancer cells? Trends Biochem Sci (2016) 41(3):211–8. doi: 10.1016/j.tibs.2015.12.001
16. Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des (2008) 14(11):1049–57. doi: 10.2174/138161208784246199
17. Lomelino CL, Andring JT, McKenna R, Kilberg MS. Asparagine synthetase: Function, structure, and role in disease. J Biol Chem (2017) 292(49):19952–8. doi: 10.1074/jbc.R117.819060
18. Clavell LA, Gelber RD, Cohen HJ, Hitchcock-Bryan S, Cassady JR, Tarbell NJ, et al. Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. N Engl J Med (1986) 315(11):657–63. doi: 10.1056/NEJM198609113151101
19. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet (2011) 43(9):869–74. doi: 10.1038/ng.890
20. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011) 476(7360):346–50. doi: 10.1038/nature10350
21. Wei Z, Liu X, Cheng C, Yu W, Yi P. Metabolism of amino acids in cancer. Front Cell Dev Biol (2020) 8:603837. doi: 10.3389/fcell.2020.603837
22. Butler M, van der Meer LT, van Leeuwen FN. Amino acid depletion therapies: Starving cancer cells to death. Trends Endocrinol Metab (2021) 32(6):367–81. doi: 10.1016/j.tem.2021.03.003
23. Motyl KJ, Guntur AR, Carvalho AL, Rosen CJ. Energy metabolism of bone. Toxicol Pathol (2017) 45(7):887–93. doi: 10.1177/0192623317737065
24. Datta HK, Ng WF, Walker JA, Tuck SP, Varanasi SS. The cell biology of bone metabolism. J Clin Pathol (2008) 61(5):577–87. doi: 10.1136/jcp.2007.048868
25. Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell (1997) 89(5):747–54. doi: 10.1016/S0092-8674(00)80257-3
26. Hsu H, Lacey DL, Dunstan CR, Solovyev I, Colombero A, Timms E, et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc Natl Acad Sci U.S.A. (1999) 96(7):3540–5. doi: 10.1073/pnas.96.7.3540
27. Canalis E, Pash J, Varghese S. Skeletal growth factors. Crit Rev Eukaryot Gene Expr (1993) 3(3):155–66.
28. Rydziel S, Shaikh S, Canalis E. Platelet-derived growth factor-AA and -BB (PDGF-AA and -BB) enhance the synthesis of PDGF-AA in bone cell cultures. Endocrinology (1994) 134(6):2541–6. doi: 10.1210/endo.134.6.8194480
29. Canalis E, Pash J, Gabbitas B, Rydziel S, Varghese S. Growth factors regulate the synthesis of insulin-like growth factor-I in bone cell cultures. Endocrinology (1993) 133(1):33–8. doi: 10.1210/endo.133.1.8319580
30. Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors (2004) 22(4):233–41. doi: 10.1080/08977190412331279890
31. Rizzoli R, Poser J, Burgi U. Nuclear thyroid hormone receptors in cultured bone cells. Metabolism (1986) 35(1):71–4. doi: 10.1016/0026-0495(86)90098-3
32. Warshawsky H, Goltzman D, Rouleau MF, Bergeron JJ. Direct in vivo demonstration by radioautography of specific binding sites for calcitonin in skeletal and renal tissues of the rat. J Cell Biol (1980) 85(3):682–94. doi: 10.1083/jcb.85.3.682
33. Teti A, Rizzoli R, Zambonin Zallone A. Parathyroid hormone binding to cultured avian osteoclasts. Biochem Biophys Res Commun (1991) 174(3):1217–22. doi: 10.1016/0006-291X(91)91551-M
34. Abu EO, Bord S, Horner A, Chatterjee VK, Compston JE. The expression of thyroid hormone receptors in human bone. Bone (1997) 21(2):137–42. doi: 10.1016/S8756-3282(97)00097-5
35. Hou P, Sato T, Hofstetter W, Foged NT. Identification and characterization of the insulin-like growth factor I receptor in mature rabbit osteoclasts. J Bone Miner Res (1997) 12(4):534–40. doi: 10.1359/jbmr.1997.12.4.534
36. Zhang Z, Chen J, Jin D. Platelet-derived growth factor (PDGF)-BB stimulates osteoclastic bone resorption directly: The role of receptor beta. Biochem Biophys Res Commun (1998) 251(1):190–4. doi: 10.1006/bbrc.1998.9412
37. Zoidis E, Ghirlanda-Keller C, Schmid C. Stimulation of glucose transport in osteoblastic cells by parathyroid hormone and insulin-like growth factor I. Mol Cell Biochem (2011) 348(1-2):33–42. doi: 10.1007/s11010-010-0634-z
38. Li Z, Frey JL, Wong GW, Faugere MC, Wolfgang MJ, Kim JK, et al. Glucose transporter-4 facilitates insulin-stimulated glucose uptake in osteoblasts. Endocrinology (2016) 157(11):4094–103. doi: 10.1210/en.2016-1583
39. Donat A, Knapstein PR, Jiang S, Baranowsky A, Ballhause TM, Frosch KH, et al. Glucose metabolism in osteoblasts in healthy and pathophysiological conditions. Int J Mol Sci (2021) 22(8):4120. doi: 10.3390/ijms22084120
40. Guntur AR, Gerencser AA, Le PT, DeMambro VE, Bornstein SA, Mookerjee SA, et al. Osteoblast-like MC3T3-E1 cells prefer glycolysis for ATP production but adipocyte-like 3T3-L1 cells prefer oxidative phosphorylation. J Bone Miner Res (2018) 33(6):1052–65. doi: 10.1002/jbmr.3390
41. Lee WC, Ji X, Nissim I, Long F. Malic enzyme couples mitochondria with aerobic glycolysis in osteoblasts. Cell Rep (2020) 32(10):108108. doi: 10.1016/j.celrep.2020.108108
42. Karner CM, Long F. Glucose metabolism in bone. Bone (2018) 115:2–7. doi: 10.1016/j.bone.2017.08.008
43. Shen L, Hu G, Karner CM. Bioenergetic metabolism in osteoblast differentiation. Curr Osteoporos Rep (2022) 20(1):53–64. doi: 10.1007/s11914-022-00721-2
44. Wei J, Shimazu J, Makinistoglu MP, Maurizi A, Kajimura D, Zong H, et al. Glucose uptake and Runx2 synergize to orchestrate osteoblast differentiation and bone formation. Cell (2015) 161(7):1576–91. doi: 10.1016/j.cell.2015.05.029
45. Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab (2013) 17(5):745–55. doi: 10.1016/j.cmet.2013.03.017
46. Lee E, Park SY, Moon JY, Ko JY, Kim TK, Im GI. Metabolic switch under glucose deprivation leading to discovery of NR2F1 as a stimulus of osteoblast differentiation. J Bone Miner Res (2022) 37(7):1382–99. doi: 10.1002/jbmr.4565
47. Lee WC, Guntur AR, Long F, Rosen CJ. Energy metabolism of the osteoblast: Implications for osteoporosis. Endocr Rev (2017) 38(3):255–66. doi: 10.1210/er.2017-00064
48. Karner CM, Esen E, Okunade AL, Patterson BW, Long F. Increased glutamine catabolism mediates bone anabolism in response to WNT signaling. J Clin Invest (2015) 125(2):551–62. doi: 10.1172/JCI78470
49. Shen L, Sharma D, Yu Y, Long F, Karner CM. Biphasic regulation of glutamine consumption by WNT during osteoblast differentiation. J Cell Sci (2021) 134(1):jcs251645. doi: 10.1242/jcs.251645
50. Sharma D, Yu Y, Shen L, Zhang GF, Karner CM. SLC1A5 provides glutamine and asparagine necessary for bone development in mice. Elife (2021) 10:e71595. doi: 10.7554/eLife.71595
51. Shen L, Yu Y, Zhou Y, Pruett-Miller SM, Zhang GF, Karner CM. SLC38A2 provides proline to fulfill unique synthetic demands arising during osteoblast differentiation and bone formation. Elife (2022) 11:e76963. doi: 10.7554/eLife.76963.sa2
52. Lemma S, Sboarina M, Porporato PE, Zini N, Sonveaux P, Di Pompo G, et al. Energy metabolism in osteoclast formation and activity. Int J Biochem Cell Biol (2016) 79:168–80. doi: 10.1016/j.biocel.2016.08.034
53. Indo Y, Takeshita S, Ishii KA, Hoshii T, Aburatani H, Hirao A, et al. Metabolic regulation of osteoclast differentiation and function. J Bone Miner Res (2013) 28(11):2392–9. doi: 10.1002/jbmr.1976
54. Sugatani T, Hruska KA. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas akt is dispensable for cell survival in isolated osteoclast precursors. J Biol Chem (2005) 280(5):3583–9. doi: 10.1074/jbc.M410480200
55. Ozaki K, Yamada T, Horie T, Ishizaki A, Hiraiwa M, Iezaki T, et al. The l-type amino acid transporter LAT1 inhibits osteoclastogenesis and maintains bone homeostasis through the mTORC1 pathway. Sci Signal (2019) 12(589):eaaw3921. doi: 10.1126/scisignal.aaw3921
56. Go M, Shin E, Jang SY, Nam M, Hwang GS, Lee SY. BCAT1 promotes osteoclast maturation by regulating branched-chain amino acid metabolism. Exp Mol Med (2022) 54(6):825–33. doi: 10.1038/s12276-022-00775-3
57. Li B, Lee WC, Song C, Ye L, Abel ED, Long F. Both aerobic glycolysis and mitochondrial respiration are required for osteoclast differentiation. FASEB J (2020) 34(8):11058–67. doi: 10.1096/fj.202000771R
58. Chan WCW, Tan Z, To MKT, Chan D. Regulation and role of transcription factors in osteogenesis. Int J Mol Sci (2021) 22(11):5445. doi: 10.3390/ijms22115445
59. Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, et al. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metab (2006) 4(6):441–51. doi: 10.1016/j.cmet.2006.10.010
60. Hu G, Yu Y, Tang YJ, Wu C, Long F, Karner CM. The amino acid sensor Eif2ak4/GCN2 is required for proliferation of osteoblast progenitors in mice. J Bone Miner Res (2020) 35(10):2004–14. doi: 10.1002/jbmr.4091
61. Selvarajah B, Azuelos I, Plate M, Guillotin D, Forty EJ, Contento G, et al. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-beta1-induced collagen biosynthesis. Sci Signal (2019) 12(582):eaav3048. doi: 10.1126/scisignal.aav3048
62. Rathore R, Van Tine BA. Pathogenesis and current treatment of osteosarcoma: Perspectives for future therapies. J Clin Med (2021) 10(6):1182. doi: 10.3390/jcm10061182
63. Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational biology of osteosarcoma. Nat Rev Cancer (2014) 14(11):722–35. doi: 10.1038/nrc3838
64. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: Current treatment and a collaborative pathway to success. J Clin Oncol (2015) 33(27):3029–35. doi: 10.1200/JCO.2014.59.4895
65. Corre I, Verrecchia F, Crenn V, Redini F, Trichet V. The osteosarcoma microenvironment: A complex but targetable ecosystem. Cells (2020) 9(4):976. doi: 10.3390/cells9040976
66. Martin JW, Squire JA, Zielenska M. The genetics of osteosarcoma. Sarcoma (2012) 2012:627254. doi: 10.1155/2012/627254
67. Riggi N, Suva ML, Stamenkovic I. Ewing's sarcoma. N Engl J Med (2021) 384(2):154–64. doi: 10.1056/NEJMra2028910
68. Riggi N, knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suva ML, et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell (2014) 26(5):668–681.
69. Tomazou EM, Sheffield NC, Schimdl C, Schuster M, Schonegger A, Datlinger P, et al. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep (2015) 10(7):1082–1095.
70. Boulay G, Sandoval GJ, Schimdl C, Riggi N, Iyer S, Buisson R, Naigles B, et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell (2017) 171(1):163–178 e119.
71. Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet (2014) 10(7): e1004475.
72. Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, kurek KC, Calicchio ML, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov (2014) 4(11):1326–1341.
73. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, St. Jude Children's Research Hospital-Washington University Pediatric Cancer Genome and C. the International Cancer Genome, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov (2014) 4(11):1342–1353.
74. Shaw JH, Humberstone DM, Wolfe RR. Energy and protein metabolism in sarcoma patients. Ann Surg (1988) 207(3):283–9. doi: 10.1097/00000658-198803000-00010
75. Hawkins DS, Rajendran JG, Conrad EU 3rd, Bruckner JD, Eary JF. Evaluation of chemotherapy response in pediatric bone sarcomas by [F-18]-fluorodeoxy-D-glucose positron emission tomography. Cancer (2002) 94(12):3277–84. doi: 10.1002/cncr.10599
76. Harrison DJ, Parisi MT, Shulkin BL. The role of (18)F-FDG-PET/CT in pediatric sarcoma. Semin Nucl Med (2017) 47(3):229–41. doi: 10.1053/j.semnuclmed.2016.12.004
77. Tanner JM, Bensard C, Wei P, Krah NM, Schell JC, Gardiner J, et al. EWS/FLI is a master regulator of metabolic reprogramming in Ewing sarcoma. Mol Cancer Res (2017) 15(11):1517–30. doi: 10.1158/1541-7786.MCR-17-0182
78. Sen N, Cross AM, Lorenzi PL, Khan J, Gryder BE, Kim S, et al. EWS-FLI1 reprograms the metabolism of Ewing sarcoma cells via positive regulation of glutamine import and serine-glycine biosynthesis. Mol Carcinog (2018) 57(10):1342–57. doi: 10.1002/mc.22849
79. Svoboda LK, Teh SSK, Sud S, Kerk S, Zebolsky A, Treichel S, et al. Menin regulates the serine biosynthetic pathway in Ewing sarcoma. J Pathol (2018) 245(3):324–36. doi: 10.1002/path.5085
80. Issaq SH, Mendoza A, Kidner R, Rosales TI, Duveau DY, Heske CM, et al. EWS-FLI1-regulated serine synthesis and exogenous serine are necessary for Ewing sarcoma cellular proliferation and tumor growth. Mol Cancer Ther (2020) 19(7):1520–29. doi: 10.1158/1535-7163.MCT-19-0748
81. Rathore R, Caldwell KE, Schutt C, Brashears CB, Prudner BC, Ehrhardt WR, et al. Metabolic compensation activates pro-survival mTORC1 signaling upon 3-phosphoglycerate dehydrogenase inhibition in osteosarcoma. Cell Rep (2021) 34(4):108678. doi: 10.1016/j.celrep.2020.108678
82. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci (2014) 39(4):191–8. doi: 10.1016/j.tibs.2014.02.004
83. Wang DW, Wu L, Cao Y, Yang L, Liu W, E XQ, et al. A novel mechanism of mTORC1-mediated serine/glycine metabolism in osteosarcoma development. Cell Signal (2017) 29:107–14. doi: 10.1016/j.cellsig.2016.06.008
84. Jimenez JA, Apfelbaum AA, Hawkins AG, Svoboda LK, Kumar A, Ruiz RO, et al. EWS-FLI1 and menin converge to regulate ATF4 activity in Ewing sarcoma. Mol Cancer Res (2021) 19(7):1182–95. doi: 10.1158/1541-7786.MCR-20-0679
85. Ding J, Li T, Wang X, Zhao E, Choi JH, Yang L, et al. The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab (2013) 18(6):896–907. doi: 10.1016/j.cmet.2013.11.004
86. DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet (2015) 47(12):1475–81. doi: 10.1016/j.ccell.2019.01.018
87. Reina-Campos M, Linares JF, Duran A, Cordes T, L'Hermitte A, Badur MG, et al. Increased serine and one-carbon pathway metabolism by PKClambda/iota deficiency promotes neuroendocrine prostate cancer. Cancer Cell (2019) 35(3):385–400 e389. doi: 10.1016/j.ccell.2019.01.018
88. Xia Y, Ye B, Ding J, Yu Y, Alptekin A, Thangaraju M, et al. Metabolic reprogramming by MYCN confers dependence on the serine-Glycine-One-Carbon biosynthetic pathway. Cancer Res (2019) 79(15):3837–50. doi: 10.1158/0008-5472.CAN-18-3541
89. Svoboda LK, Bailey N, Van Noord RA, Krook MA, Harris A, Cramer C, et al. Tumorigenicity of Ewing sarcoma is critically dependent on the trithorax proteins MLL1 and menin. Oncotarget (2017) 8(1):458–71. doi: 10.18632/oncotarget.13444
90. Ou Y, Wang SJ, Jiang L, Zheng B, Gu W. p53 protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J Biol Chem (2015) 290(1):457–66. doi: 10.1074/jbc.M114.616359
91. Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep (2014) 7(1):104–12. doi: 10.1016/j.celrep.2014.03.003
92. Anso E, Mullen AR, Felsher DW, Mates JM, Deberardinis RJ, Chandel NS. Metabolic changes in cancer cells upon suppression of MYC. Cancer Metab (2013) 1(1):7. doi: 10.1186/2049-3002-1-7
93. Ren L, Ruiz-Rodado V, Dowdy T, Huang S, Issaq SH, Beck J, et al. Glutaminase-1 (GLS1) inhibition limits metastatic progression in osteosarcoma. Cancer Metab (2020) 8:4. doi: 10.1186/s40170-020-0209-8
94. Zhang SP, Li X, Li H, Sun XH, Yan XF. Significance of neoadjuvant chemotherapy (NACT) in limb salvage treatment of osteosarcoma and its effect on GLS1 expression. Eur Rev Med Pharmacol Sci (2018) 22(19):6538–44. doi: 10.26355/eurrev_201810_16068
95. Lee P, Malik D, Perkons N, Huangyang P, Khare S, Rhoades S, et al. Targeting glutamine metabolism slows soft tissue sarcoma growth. Nat Commun (2020) 11(1):498. doi: 10.1038/s41467-020-14374-1
96. Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, et al. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: a method for identifying cancers sensitive to arginine deprivation. Cancer (2004) 100(4):826–33. doi: 10.1002/cncr.20057
97. Delage B, Luong P, Maharaj L, O'Riain C, Syed N, Crook T, et al. Promoter methylation of argininosuccinate synthetase-1 sensitises lymphomas to arginine deiminase treatment, autophagy and caspase-dependent apoptosis. Cell Death Dis (2012) 3:e342. doi: 10.1038/cddis.2012.83
98. Bean GR, Kremer JC, Prudner BC, Schenone AD, Yao JC, Schultze MB, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death Dis (2016) 7(10):e2406. doi: 10.1038/cddis.2016.232
99. Zhang HF, Hughes CS, Li W, He JZ, Surdez D, El-Naggar AM, et al. Proteomic screens for suppressors of anoikis identify IL1RAP as a promising surface target in Ewing sarcoma. Cancer Discovery (2021) 11(11):2884–903. doi: 10.1158/2159-8290.CD-20-1690
100. Mecham JO, Rowitch D, Wallace CD, Stern PH, Hoffman RM. The metabolic defect of methionine dependence occurs frequently in human tumor cell lines. Biochem Biophys Res Commun (1983) 117(2):429–34. doi: 10.1016/0006-291X(83)91218-4
101. Ehnman M, Chaabane W, Haglund F, Tsagkozis P. The tumor microenvironment of pediatric sarcoma: Mesenchymal mechanisms regulating cell migration and metastasis. Curr Oncol Rep (2019) 21(10):90. doi: 10.1007/s11912-019-0839-6
102. Berghuis D, Santos SJ, Baelde HJ, Taminiau AH, Egeler RM, Schilham MW, et al. Pro-inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8(+) T-lymphocyte infiltration and affect tumour progression. J Pathol (2011) 223(3):347–57. doi: 10.1002/path.2819
103. Inagaki Y, Hookway E, Williams KA, Hassan AB, Oppermann U, Tanaka Y, et al. Dendritic and mast cell involvement in the inflammatory response to primary malignant bone tumours. Clin Sarcoma Res (2016) 6:13. doi: 10.1186/s13569-016-0053-3
104. Sivan U, De Angelis J, Kusumbe AP. Role of angiocrine signals in bone development, homeostasis and disease. Open Biol (2019) 9(10):190144. doi: 10.1098/rsob.190144
105. Tsagozis P, Gonzalez-Molina J, Georgoudaki AM, Lehti K, Carlson J, Lundqvist A, et al. Sarcoma tumor microenvironment. Adv Exp Med Biol (2020) 1296:319–48. doi: 10.1007/978-3-030-59038-3_20
106. Redini F, Heymann D. Bone tumor environment as a potential therapeutic target in Ewing sarcoma. Front Oncol (2015) 5:279. doi: 10.3389/fonc.2015.00279
107. Norregaard KS, Jurgensen HJ, Gardsvoll H, Engelholm LH, Behrendt N, Soe K. Osteosarcoma and metastasis associated bone degradation-a tale of osteoclast and malignant cell cooperativity. Int J Mol Sci (2021) 22(13):6865. doi: 10.3390/ijms22136865
108. Cortini M, Massa A, Avnet S, Bonuccelli G, Baldini N. Tumor-activated mesenchymal stromal cells promote osteosarcoma stemness and migratory potential via IL-6 secretion. PloS One (2016) 11(11):e0166500. doi: 10.1371/journal.pone.0166500
109. Avnet S, Di Pompo G, Chano T, Errani C, Ibrahim-Hashim A, Gillies RJ, et al. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-kappaB activation. Int J Cancer (2017) 140(6):1331–45. doi: 10.1002/ijc.30540
110. Taddei ML, Pietrovito L, Leo A, Chiarugi P. Lactate in sarcoma microenvironment: Much more than just a waste product. Cells (2020) 9(2):510. doi: 10.3390/cells9020510
111. Bonuccelli G, Avnet S, Grisendi G, Salerno M, Granchi D, Dominici M, et al. Role of mesenchymal stem cells in osteosarcoma and metabolic reprogramming of tumor cells. Oncotarget (2014) 5(17):7575–88. doi: 10.18632/oncotarget.2243
112. Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer (2011) 11(6):411–25. doi: 10.1038/nrc3055
113. Pollari S, Kakonen SM, Edgren H, Wolf M, Kohonen P, Sara H, et al. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res Treat (2011) 125(2):421–30. doi: 10.1007/s10549-010-0848-5
114. Lemma S, Di Pompo G, Porporato PE, Sboarina M, Russell S, Gillies RJ, et al. MDA-MB-231 breast cancer cells fuel osteoclast metabolism and activity: A new rationale for the pathogenesis of osteolytic bone metastases. Biochim Biophys Acta Mol Basis Dis (2017) 1863(12):3254–64. doi: 10.1016/j.bbadis.2017.08.030
115. Chiu M, Toscani D, Marchica V, Taurino G, Costa F, Bianchi MG, et al. Myeloma cells deplete bone marrow glutamine and inhibit osteoblast differentiation limiting asparagine availability. Cancers (Basel) (2020) 12(11):3267. doi: 10.3390/cancers12113267
116. Pacold ME, Brimacombe KR, Chan SH, Rohde JM, Lewis CA, Swier LJ, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol (2016) 12(6):452–8. doi: 10.1038/nchembio.2070
117. Sullivan MR, Mattaini KR, Dennstedt EA, Nguyen AA, Sivanand S, Reilly MF, et al. Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab (2019) 29(6):1410–1421 e1414. doi: 10.1016/j.cmet.2019.02.015
118. Ngo B, Kim E, Osorio-Vasquez V, Doll S, Bustraan S, Liang RJ, et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discovery (2020) 10(9):1352–73. doi: 10.1158/2159-8290.CD-19-1228
119. Rinaldi G, Pranzini E, Van Elsen J, Broekaert D, Funk CM, Planque M, et al. In vivo evidence for serine biosynthesis-defined sensitivity of lung metastasis, but not of primary breast tumors, to mTORC1 inhibition. Mol Cell (2021) 81(2):386–397 e387. doi: 10.1016/j.molcel.2020.11.027
120. Rossi M, Altea-Manzano P, Demicco M, Doglioni G, Bornes L, Fukano M, et al. PHGDH heterogeneity potentiates cancer cell dissemination and metastasis. Nature (2022) 605(7911):747–53. doi: 10.1038/s41586-022-04758-2
121. Ren L, Hong ES, Mendoza A, Issaq S, Tran Hoang C, Lizardo M, et al. Metabolomics uncovers a link between inositol metabolism and osteosarcoma metastasis. Oncotarget (2017) 8(24):38541–53. doi: 10.18632/oncotarget.15872
122. Dean DC, Shen S, Hornicek FJ, Duan Z. From genomics to metabolomics: emerging metastatic biomarkers in osteosarcoma. Cancer Metastasis Rev (2018) 37(4):719–31. doi: 10.1007/s10555-018-9763-8
123. Fritsche-Guenther R, Gloaguen Y, Kirchner M, Mertins P, Tunn PU, Kirwan JA. Progression-dependent altered metabolism in osteosarcoma resulting in different nutrient source dependencies. Cancers (Basel) (2020) 12(6):1371. doi: 10.3390/cancers12061371
124. Imle R, Kommoss FKF, Banito A. Preclinical In vivo modeling of pediatric sarcoma-promises and limitations. J Clin Med (2021) 10(8):1578. doi: 10.3390/jcm10081578
125. Prudner BC, Rathore R, Robinson AM, Godec A, Chang SF, Hawkins WG, et al. Arginine starvation and docetaxel induce c-Myc-Driven hENT1 surface expression to overcome gemcitabine resistance in ASS1-negative tumors. Clin Cancer Res (2019) 25(16):5122–34. doi: 10.1158/1078-0432.CCR-19-0206
126. Kremer JC, Prudner BC, Lange SES, Bean GR, Schultze MB, Brashears CB, et al. Arginine deprivation inhibits the warburg effect and upregulates glutamine anaplerosis and serine biosynthesis in ASS1-deficient cancers. Cell Rep (2017) 18(4):991–1004. doi: 10.1016/j.celrep.2016.12.077
127. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther (2014) 13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870
128. Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood (2015) 126(11):1346–56. doi: 10.1182/blood-2015-01-621870
129. Song M, Kim SH, Im CY, Hwang HJ. Recent development of small molecule glutaminase inhibitors. Curr Top Med Chem (2018) 18(6):432–43. doi: 10.2174/1568026618666180525100830
130. Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, et al. Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab (2016) 23(3):517–28. doi: 10.1016/j.cmet.2016.01.007
131. Biancur DE, Paulo JA, Malachowska B, Quiles Del Rey M, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun (2017) 8:15965. doi: 10.1038/ncomms15965
132. Mullarky E, Lucki NC, Beheshti Zavareh R, Anglin JL, Gomes AP, Nicolay BN, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U.S.A. (2016) 113(7):1778–83. doi: 10.1073/pnas.1521548113
133. Wang Q, Liberti MV, Liu P, Deng X, Liu Y, Locasale JW, et al. Rational design of selective allosteric inhibitors of PHGDH and serine synthesis with anti-tumor activity. Cell Chem Biol (2017) 24(1):55–65. doi: 10.1016/j.chembiol.2016.11.013
134. McNamee MJ, Michod D, Niklison-Chirou MV. Can small molecular inhibitors that stop de novo serine synthesis be used in cancer treatment? Cell Death Discovery (2021) 7(1):87. doi: 10.1038/s41420-021-00474-4
135. Schmidt DR, Patel R, Kirsch DG, Lewis CA, Vander Heiden MG, Locasale JW. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J Clin (2021) 71(4):333–58. doi: 10.3322/caac.21670
136. Pillozzi S, Bernini A, Palchetti I, Crociani O, Antonuzzo L, Campanacci D, et al. Soft tissue sarcoma: An insight on biomarkers at molecular, metabolic and cellular level. Cancers (Basel) (2021) 13(12):3044. doi: 10.3390/cancers13123044
137. Kelly AD, Breitkopf SB, Yuan M, Goldsmith J, Spentzos D, Asara JM. Metabolomic profiling from formalin-fixed, paraffin-embedded tumor tissue using targeted LC/MS/MS: application in sarcoma. PloS One (2011) 6(10):e25357. doi: 10.1371/journal.pone.0025357
138. Braas D, Ahler E, Tam B, Nathanson D, Riedinger M, Benz MR, et al. Metabolomics strategy reveals subpopulation of liposarcomas sensitive to gemcitabine treatment. Cancer Discovery (2012) 2(12):1109–17. doi: 10.1158/2159-8290.CD-12-0197
139. Miolo G, Di Gregorio E, Saorin A, Lombardi D, Scalone S, Buonadonna A, et al. Integration of serum metabolomics into clinical assessment to improve outcome prediction of metastatic soft tissue sarcoma patients treated with trabectedin. Cancers (Basel) (2020) 12(7):1983. doi: 10.3390/cancers12071983
140. Jia B, Wang W, Lin S, Shi L, Li Y, Gu Y, et al. The free amino acid profiles and metabolic biomarkers of predicting the chemotherapeutic response in advanced sarcoma patients. Clin Transl Oncol (2020) 22(12):2213–21. doi: 10.1007/s12094-020-02494-5
141. Zhang Z, Qiu Y, Hua Y, Wang Y, Chen T, Zhao A, et al. Serum and urinary metabonomic study of human osteosarcoma. J Proteome Res (2010) 9(9):4861–8. doi: 10.1021/pr100480r
142. Van Tine BA, Lyssiotis CA. In an era of ctDNA, is metabolomics the new kid on the block? Clin Cancer Res (2022) 28(8):1477–8. doi: 10.1158/1078-0432.CCR-21-4180
143. Hascitha J, Priya R, Jayavelu S, Dhandapani H, Selvaluxmy G, Sunder Singh S, et al. Analysis of Kynurenine/Tryptophan ratio and expression of IDO1 and 2 mRNA in tumour tissue of cervical cancer patients. Clin Biochem (2016) 49(12):919–24. doi: 10.1016/j.clinbiochem.2016.04.008
144. Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: From bench to bedside. Cancer Res (2017) 77(24):6795–811. doi: 10.1158/0008-5472.CAN-17-2285
145. Zhai L, Ladomersky E, Lauing KL, Wu M, Genet M, Gritsina G, et al. Infiltrating T cells increase IDO1 expression in glioblastoma and contribute to decreased patient survival. Clin Cancer Res (2017) 23(21):6650–60. doi: 10.1158/1078-0432.CCR-17-0120
146. Oda K, Hosoda N, Endo H, Saito K, Tsujihara K, Yamamura M, et al. L-type amino acid transporter 1 inhibitors inhibit tumor cell growth. Cancer Sci (2010) 101(1):173–9. doi: 10.1111/j.1349-7006.2009.01386.x
147. Enomoto K, Sato F, Tamagawa S, Gunduz M, Onoda N, Uchino S, et al. A novel therapeutic approach for anaplastic thyroid cancer through inhibition of LAT1. Sci Rep (2019) 9(1):14616. doi: 10.1038/s41598-019-51144-6
Keywords: tumor metabolism, sarcoma, Ewing sarcoma, amino acid metabolism, osteoclast, osteoblast
Citation: Jiménez JA, Lawlor ER and Lyssiotis CA (2022) Amino acid metabolism in primary bone sarcomas. Front. Oncol. 12:1001318. doi: 10.3389/fonc.2022.1001318
Received: 23 July 2022; Accepted: 19 August 2022;
Published: 05 October 2022.
Edited by:
Jiyeon Kim, University of Illinois at Chicago, United StatesReviewed by:
Dominique Heymann, Institut de Cancérologie de l’Ouest, FranceJonathan Coloff, University of Illinois at Chicago, United States
Copyright © 2022 Jiménez, Lawlor and Lyssiotis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth R. Lawlor, QmV0aC5MYXdsb3JAc2VhdHRsZWNoaWxkcmVucy5vcmc=; Costas A. Lyssiotis, Y2x5c3Npb3RAbWVkLnVtaWNoLmVkdQ==