 Christopher J. Fay
Christopher J. Fay Katherine C. Awh
Katherine C. Awh Cecilia A. Larocca
Cecilia A. Larocca- Department of Dermatology, Center for Cutaneous Oncology, Brigham and Women's Hospital, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA, United States
Cutaneous T cell lymphomas are a rare subset of non-Hodgkin’s lymphomas with predilection for the skin with immunosuppressive effects that drive morbidity and mortality. We are now appreciating that suppression of the immune system is an important step in the progression of disease. It should come as no surprise that therapies historically and currently being used to treat these cancers have immune modulating functions that impact disease outcomes. By understanding the immune effects of our therapies, we may better develop new agents that target the immune system and improve combinatorial treatment strategies to limit morbidity and mortality of these cancers. The immune modulating effect of therapeutic drugs in use and under development for cutaneous T cell lymphomas will be reviewed.
Capsule summary:
● Several therapeutic drugs in use and under development for cutaneous T cell lymphomas have immune modulatory effects
● Novel combinatorial treatment strategies have been developed based on the latest understanding of the favorable immune effects of individual therapies
1. Introduction
Cutaneous T cell lymphomas (CTCL) are a heterogeneous class of non-Hodgkin’s lymphomas that arise from skin tropic T cells, characterized by inflamed skin lesions with variable involvement of blood and lymph nodes. Mycosis fungoides (MF) and Sézary syndrome (SS) are the two most common clinical variants of CTCL with an incidence of approximately 10.2 per million persons in the United States (1). Patients with early stage MF (stage I-IIA) typically have an indolent disease course, but those with SS (Stage IV) have a 41% survival rate at five years (2).
The pathogenesis of CTCL remains unclear. Although environmental exposures and associations with infectious agents have been hypothesized, none have been definitively identified thus far (3). Furthermore, the oncogenic drivers of cutaneous T cell malignant transformation remain incompletely characterized given that genomic studies are largely derived from SS cohorts and the mutational landscape shows heterogeneous mutations and wide variation in chromosomal abnormalities (4). However, what is coming into focus is that the malignant T cell is a key driver of immunosuppression, reshaping the cutaneous microenvironment and broadly impairing cellular immunity, which is needed for an effective anti-tumor response (5). Please see Krejsgaard et al. for a comprehensive review of “malignant inflammation”—describing the mechanism by which malignant T cells remodel the immune environment from early to advanced disease via release of immunosuppressive cytokines, modulation of chemokines, and engagement with stromal and immune cells (6). In addition to directly affecting immune cell function, the malignant T cells can also impair the epidermal skin barrier (7). This allows bacteria such as Staphylococcus aureus to further influence the tumor microenvironment (TME). S. aureus, via release of enterotoxins, pore-forming α-toxins, and lipoproteins, suppresses anti-tumor immunity. Staphylococcal enterotoxins function as superantigens activating malignant T cells and inducing an immunosuppressive phenotype, characterized by upregulation of CD25, FOXP3, IL-17, and miR-155 expression (8). Staphylococcal α-toxin more easily causes apoptosis in benign CD4+ and CD8+ T cells and can interfere with the cytotoxic killing capacity of anti-tumor CD8+ T cells (8, 9). And lastly, bacterial lipoproteins promote a type 2 helper T cell (Th2)dominant milieu by activating toll-like receptors on keratinocytes to release pro-tumorigenic cytokines (8). In fact, use of antibiotics in patients with CTCL has led to remarkable clinical responses (9), a unique example of how targeting drivers of pro-tumorigenic inflammation can improve disease outcomes.
The immunosuppressive effects of malignant T cells can be reversed with skin-directed and systemic therapies (10–12). However, it is unclear whether this is mediated solely by depletion of malignant T cells or if therapies that independently augment the immune system toward a type 1 helper T cell (Th1) bias have greater efficacy. By understanding how past and emerging therapies can modulate the immune system we hope more effective multimodal and combination therapies will be developed.
We aim to provide a comprehensive and updated review of the immune modulating effects of all classes of therapeutics used and under development for CTCL (13). We caution that many drugs are incompletely understood in terms of their mechanism of cell death, direct impact on tumor cell function, and effect on different aspects of the immune system. Here, we report on the broadly accepted mechanism of action for each drug and primarily focus on the effect of treatment on the immune environment. In addition, the mechanistic rational for several combination therapies under investigation will be summarized. Favorable (Figure 1) and unfavorable immune modulatory effects are summarized in Table 1. Please see Garcia-Diaz et al. for a comprehensive review integrating how different signaling pathways targeted in CTCL interact, which is beyond the scope of the review (14).
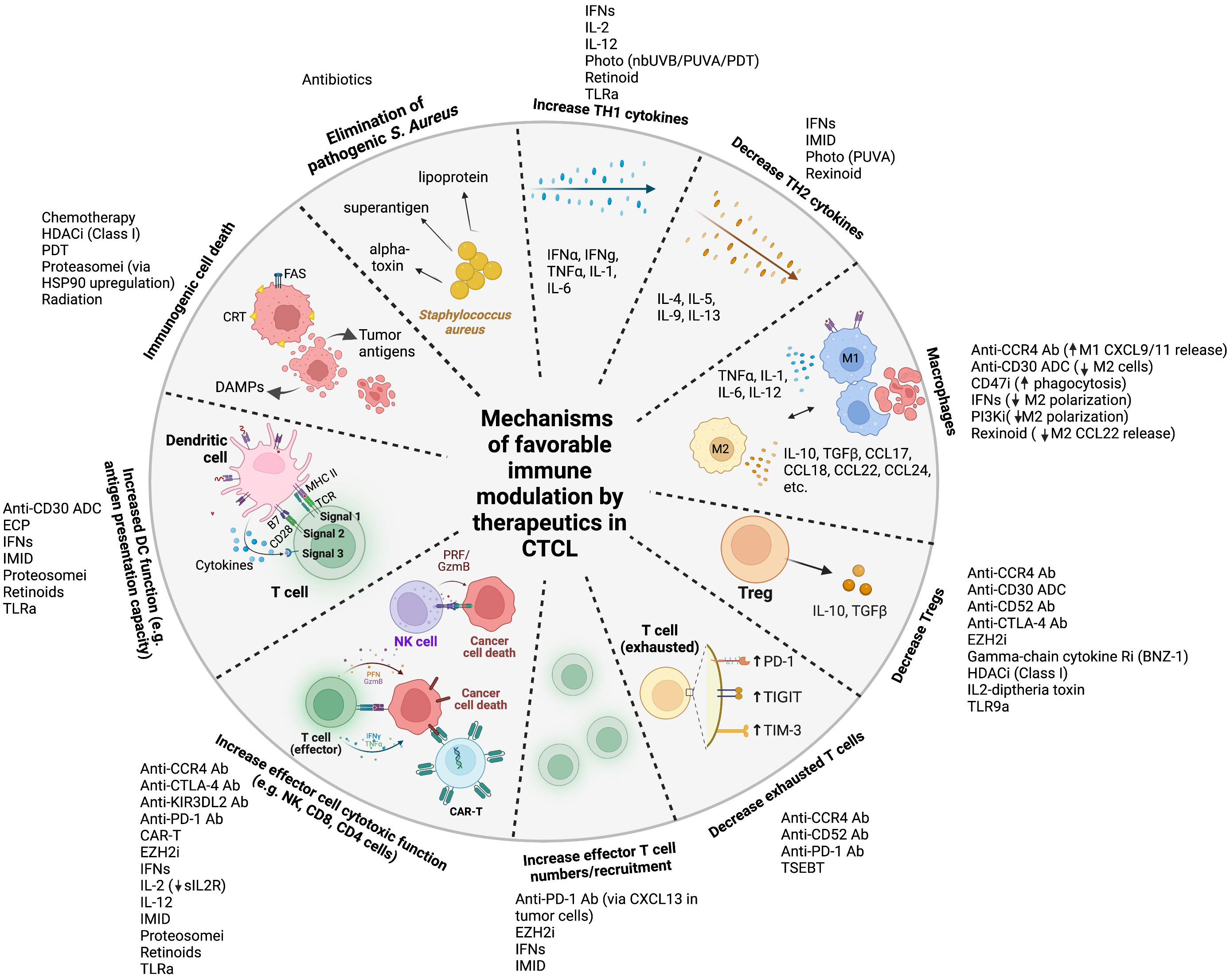
Figure 1 Mechanisms of favorable immune modulation by therapeutics in cutaneous T cell lymphoma Legend: ab, antibody; ADC, antibody-drug conjugate; anti-CTLA-4, anti-cytotoxic T-lymphocyte-associated protein 4; anti-PD-1, anti-programmed cell death protein 1; CAR, chimeric antigen receptor; CCR4, anti-C-C chemokine receptor 4; CD47i, cluster of differentiation 47 inhibitor; CRT, calreticulin; DAMPs, damage associated molecular patterns DC, dendritic cell; ECP, extracorporeal photopheresis; EZH2i, enhancer of zeste homolog 2 inhibitor; GzmB, granzyme B; HDACi, histone deacetylase inhibitor; IFNs, interferons; IL, interleukin; IMID, immunomodulator (lenalidomide); MHC, major histocompatibility complex; nbUVB, narrowband ultraviolet B; NK, natural killer; PI3Ki, phosphoinositide 3-kinase inhibitor; PRF, perforin; proteosomei, proteosome inhibitor; PUVA, psoralen plus ultraviolet A; sIL2R, soluble interleukin-2 receptor; TCR, T cell receptor; TGF, transforming growth factor; Th1, Type 1 T helper; Th2, Type 2 T helper; TIGIT, T cell immunoreceptor with Ig and ITIM domains; TIM-3, T cell immunoglobulin and mucin-domain containing-3; TLRa, toll like receptor agonist; TNF, tumor necrosis factor; TSEBT, total skin electron beam therapy.
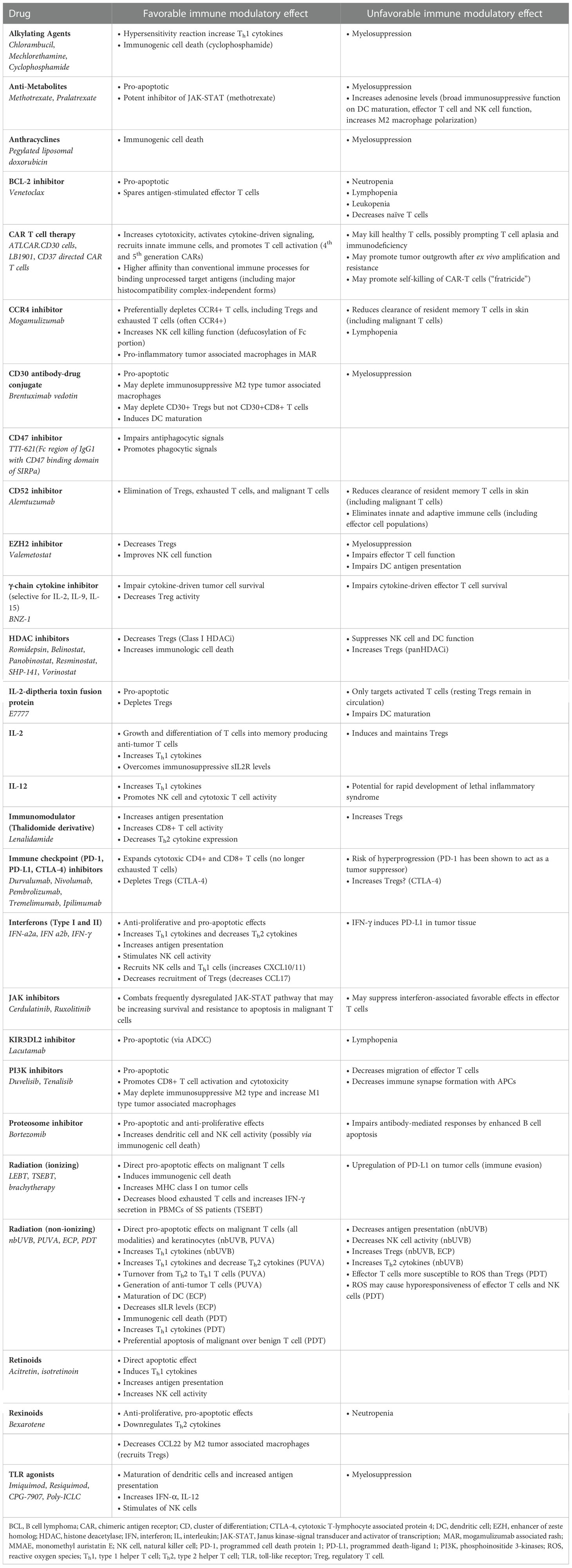
Table 1 Favorable and unfavorable immune modulatory effects of CTCL therapies.
A summary of pivotal clinical trials along with their response rates for all reviewed therapies are included in Tables 2–4 to help put into context the potential importance of observed immune modulatory effects. Notable responses are listed in the text. Unless otherwise stated the response rate (RR) is determined by the sum of the rates of complete and partial responders. Of note, definitions of complete and partial response vary within each study until the establishment of response criteria for MF/SS by Olsen et al. published in 2011 (56). In general, for assessment of skin response, a complete response (CR) requires 100% clearing of skin disease and a partial response (PR) requires ≥ 50% reduction in skin tumor burden as determined by the severity weight assessment tool (SWAT) (57) or its modification (mSWAT) (44) score compared with baseline, sustained for 4 weeks. Prior studies were based on change in body surface area affected. Later studies required global responses in the cutaneous and extracutaneous compartments, including blood, lymph node and viscera, as evaluated by CT scans and peripheral lymphocyte counts (56).
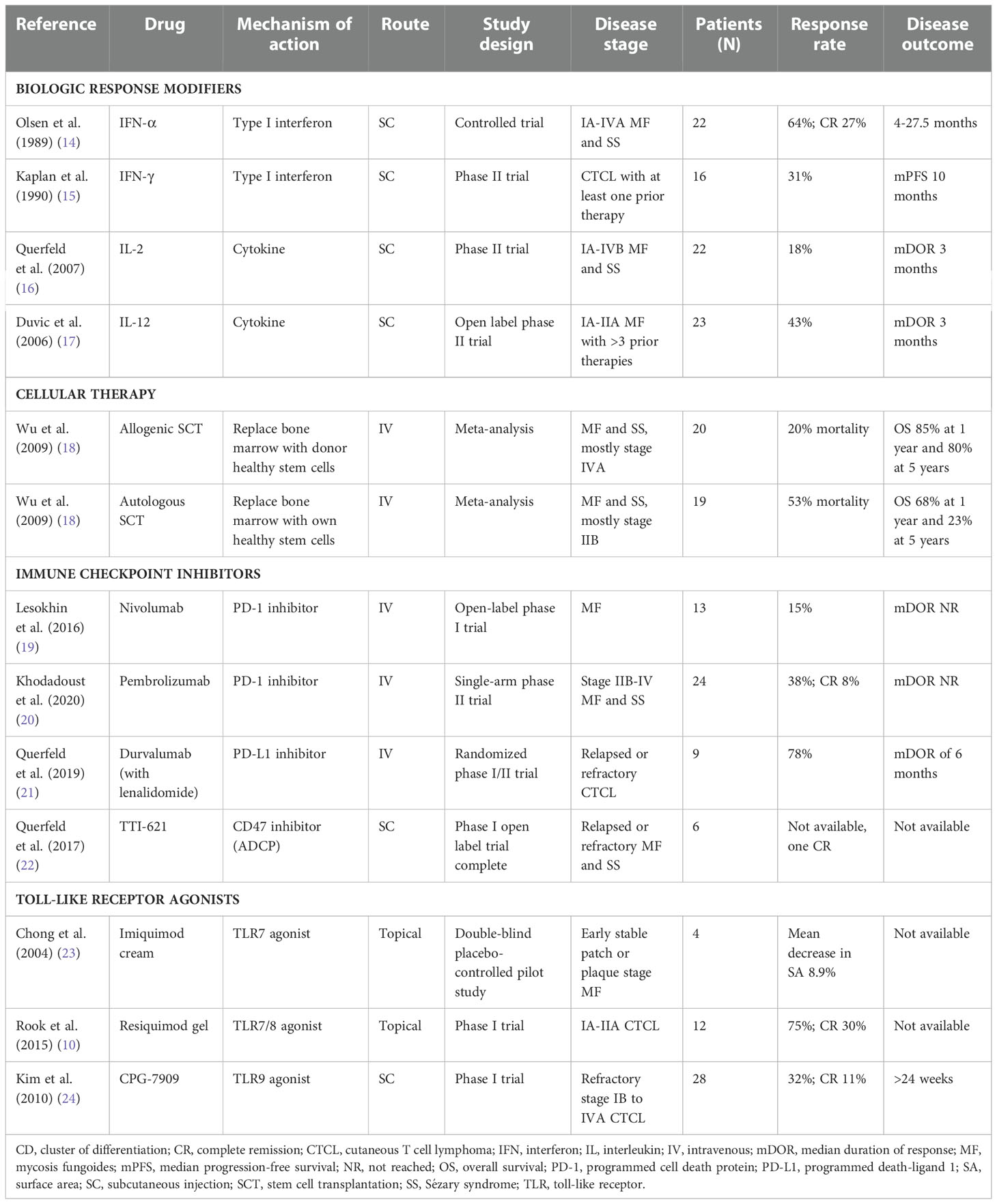
Table 2 Biologic response modifiers, cellular immunotherapy, immune checkpoint inhibitors, and toll-like receptor agonists for the treatment of CTCL.
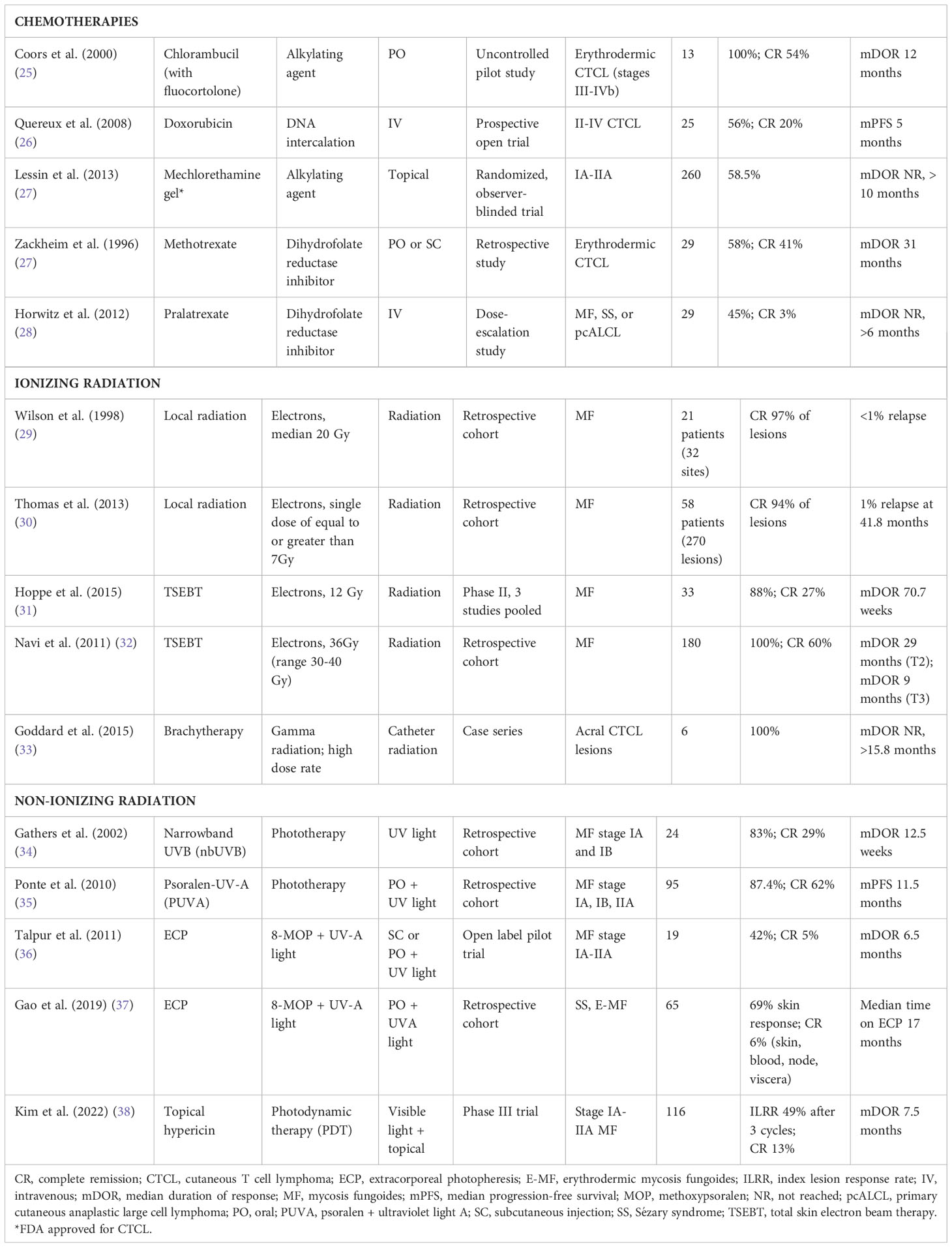
Table 3 Chemotherapies and radiotherapies for the treatment of CTCL.
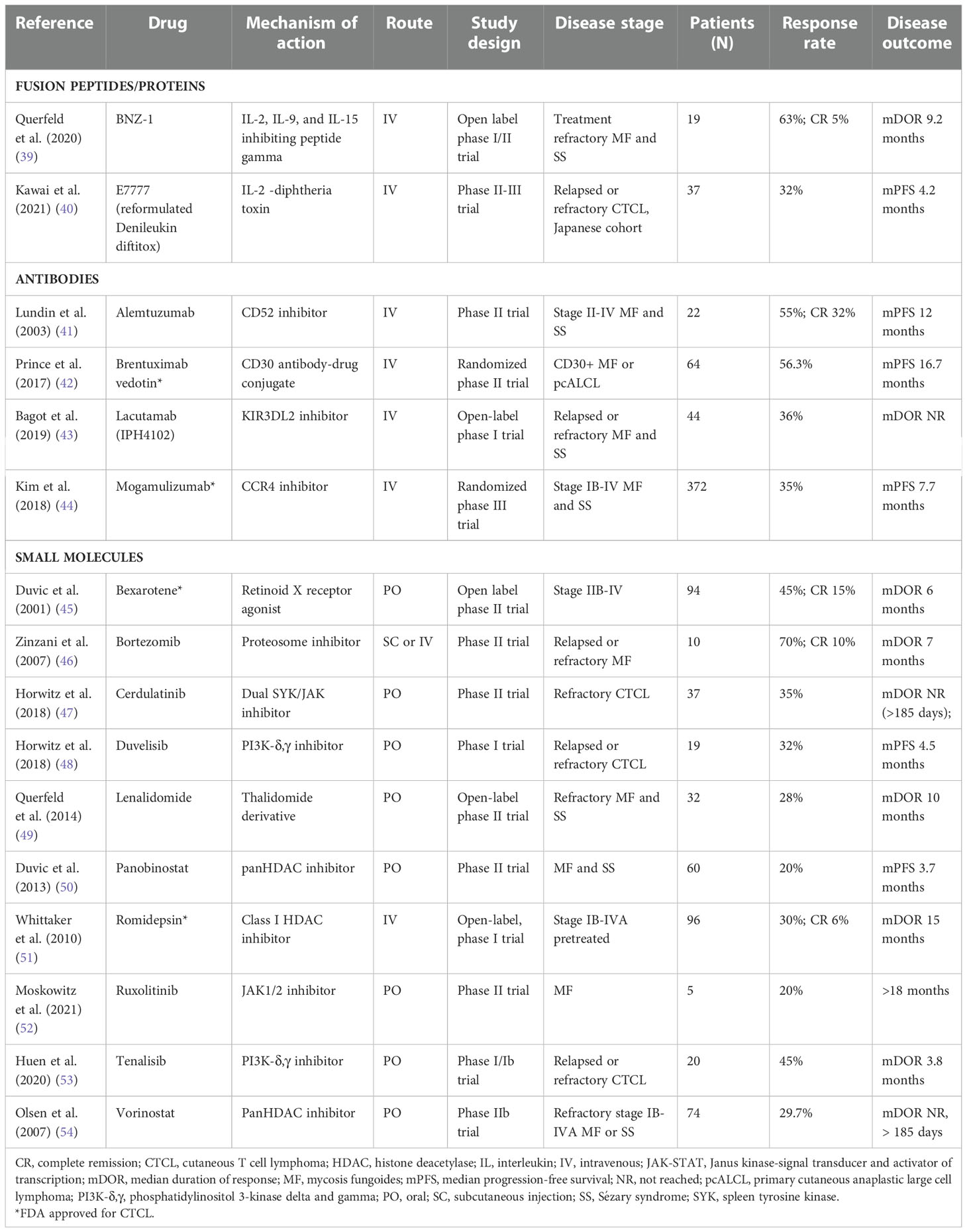
Table 4 Targeted therapies for the treatment of CTCL.
2. Biologic response modifiers
The first reported use of immune stimulating agents for the treatment of cancer was in 1891 by Coley, who used bacterial cell extracts to treat osteosarcoma (58). Since the discovery of the first interferon in 1957 and its later isolation in sufficient quantities from donated blood for treatment in the 1970s, interferon alpha (IFN-α) and its other subtypes (β and γ) have been tried in numerous cancers (58). It was quickly noted that lymphomas, including MF, were among the most responsive to this treatment modality (58).
2.1. Interferons
Interferons are pleiotropic cytokines with diverse effects on cellular function and immune response. There are three types of interferons, I (IFN-α and IFN−β), type II (IFN-γ), and type III interferons (IFN-λ). Type I and II interferons primarily activate the Janus kinase/signal transducer and activator of transcription (JAK-STAT) pathway to induce expression of interferon responsive genes by binding to IFN-response elements (59, 60). A variety of STAT dimers may be activated by these interferons, however the mechanisms driving particular STAT usage and specificity are not understood (61). These interferons have also been shown to activate the mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) signaling pathways, independent of STAT signaling. Surprisingly, activating mutations and signaling in these aforementioned pathways have all been implicated in the pathogenesis of MF/SS (62). Therefore, the cellular context and concurrent activation or absence of activation of other IFN-dependent signaling pathways may drive divergent biologic responses (e.g. mitogenic stimuli versus induction of apoptosis) as interferons have not been seen in practice to promote disease progression.
Only type I and II interferons have been used as cancer therapies. They exhibit direct anti-proliferative and pro-apoptotic effects on malignant cells (59, 60). Immunomodulating effects of interferons include an increase in Th1 cytokines and a decrease in Th2 cytokines (59). They have been shown to boost the anti-tumor immune response by activating dendritic cells to enhance tumor antigen presentation to CD4+ helper and CD8+ cytotoxic T cells and stimulate NK cell activity in vivo (59, 63). After IFN-α2a and IFN-γ treatment, there is a decrease in chemokine receptor ligands (CCLs) CCL17 and CCL18 and an increase in C-X-C motif chemokine ligands (CXCLs) CXCL10 and CXCL11 by M2 macrophages (64). This leads to a favorable TME as CCL17 is important for recruitment of T regulatory (Treg) cells and CXCL10/CXCL11 recruits NK cells and activated Th1 cells (65–67). In CTCL, interferon α2a, α2b, and γ are used as a monotherapy or in combination with numerous other therapies (Table 2) (15, 60, 68). SS patients treated with IFN-α have decreased IL-4 and IL-5 expression, cytokines that promote Th2 polarization (69). Interestingly, compared to chemotherapy-based regimens, IFN-α can induces longer-term remissions (70).
Some patients have an initial or acquired resistance to interferon therapy (60). The development of neutralizing antibodies, mutations in the JAK-STAT pathway, down-regulation of IFN receptors, deregulation of cell cycle control, apoptosis and signal transduction may explain IFN-α resistance (60, 71–74). Therefore, the malignant T cells largely develop intrinsic mechanisms of resistance to biologic response modifiers that are not overcome by favorable effects on the immune microenvironment. However, patients resistant to IFN-α may respond to interferon-γ (16).
2.2. Interleukin-2
IL-2 was first used in cancer immunotherapy in 1984 for the treatment of melanoma (75). IL-2 is secreted primarily by antigen stimulated CD4+ T cells, as well as by CD8+ T cells, NK cells, and activated dendritic cells (75). The desirable effects of IL-2 are the stimulation of growth and differentiation of T cells into memory producing anti-tumor T cells (75). It has been shown to increase release of IFN-α, TNF-α, IL-1, IL-5, and IL-6 (60). Furthermore, recombinant IL-2 can be used to overcome elevated levels of soluble IL-2 receptor (IL-2R) (76), which acts as an inhibitor of IL-2 and inhibits NK cell activity in vitro (77). However, IL-2, via a JAK-STAT3 pathway, can also induce and maintain Tregs which are characterized by their expression of the master transcription factor FOXP3 and their ability to suppress other T cells via both cell-contact dependent and independent mechanisms (5, 75). A study of IL-2 in MF/SS patients showed only an 18% partial response rate and mixed immune responses (17). A subset of patients treated with IL-2 had an expansion of CD25+ cells in CD3+CD4+ populations, but it was unclear whether this was an expansion of Tregs, malignant T cells or activated anti-tumor T cells.
2.3. Interleukin-12
IL-12 has also been investigated in MF in an open-label trial (Table 2) (23). IL-12 has been shown to increase the Th1 cytokine IFN-γ and promote NK cell and cytotoxic T cell activity (23, 78, 79). However, this recombinant protein is not readily available for clinical practice (68) and earlier studies showed high rates of toxicity (80). An intratumorally delivered DNA plasmid expressing IL-12 was also being evaluated in MF, but the trial was terminated due to company resource constraints (clinicaltrials.gov identifier NCT01579318).
3. Cellular immunotherapy
3.1. Chimeric antigen receptor T cell therapy
CARs are recombinant receptor proteins engineered to redirect the function of T lymphocytes (81). They are composed of an extracellular antigen binding moiety, extracellular hinge region, transmembrane domain, and intracellular T cell activation component (82). The CAR is anchored to the plasma membrane by the transmembrane domain, which fosters stability of antigen binding. The extracellular antigen binding moiety is linked to the intracellular signaling domain: upon CAR-antigen binding, the cell is activated and triggers cytotoxicity towards the targeted cancer cells. Tumor cells attempt to evade the immune system through a variety of mechanisms, such as decreased expression of major histocompatibility complex (MHC) class I molecules, which reduces the antigen presentation capacity (83). CAR T cells have many advantages in comparison to conventional immune approaches, including higher antigen affinity for binding unprocessed target antigens independent of MHC (82). Additionally, with their own costimulatory domains, CAR T cells can increase immune responses to apoptosis and lyse target cells (82). Recent advances have led to fourth and fifth generation CARs, which have a transgenic cytokine or intracellular domain of a cytokine receptor to increase cytotoxicity, activate cytokine-driven signaling, recruit innate immune cells for antigen-negative tumor cells, and promote T cell activation (84, 85). This therapy has been approved for and is successful in B cell hematopoietic tumors, among others.
Use of CAR T therapy for CTCL, however, has specific challenges. First, as healthy and malignant T cells share commonalities, this could inadvertently lead to CAR T cells killing healthy T cells, prompting T cell aplasia and immunodeficiency (82). There is no treatment for quickly remedying T cell depletion, unlike intravenous immunoglobulin for B cell aplasia, which makes this a more concerning adverse event in CTCL patients. Additionally, both healthy T cells and circulating tumor T cells may be simultaneously collected when the CAR product is being created. This contamination may promote both tumor outgrowth after ex vivo amplification and resistance (82, 86). In a patient with B-ALL, for instance, the CAR gene was accidentally introduced into a leukemic B cell during the T cell manufacturing which can induce resistance (87). With this kind of contamination occurring in CTCL, there can be an interaction between the “CAR tumor T cells” and the tumor antigen on their own surfaces. This creates a “loop situation” that hides the surface antigen, and the activity of the non-contaminated CAR T cells is hindered. Another possible complication of CAR T cell therapy in CTCL is fratricide, which is self-killing of CAR T cells when the target antigen is common between malignant T cells and T effector cells (82). To et al. has proposed ways to minimize these risks of CAR T cell therapy in CTCL, although it is clear that CTCL presents more obstacles than B cell tumors for application of this therapy (82). CAR T cell therapy in preclinical in vivo and in vitro studies has been promising, demonstrating anti-tumor activity, without apparent off-target effects (88). To date there are three ongoing phase I trials investigating the safety and efficacy of CAR T cell therapy in CTCL. One clinical trial targets CD30 (clinicaltrials.gov identifiers: NCT03602157). In this same trial, some participants will receive CAR T therapy that targets CD30 but also has been genetically modified to express CCR4, which is important in trafficking cells to the skin. Another clinical trial involves a CAR T named LB1901 that targets CD4 (clinicaltrials.gov NCT04712864). Lastly, there is a clinical trial for a CAR T that targets CD37 (clinicaltrials.gov NCT04136275).
3.2. Hematopoietic stem cell transplantation
Hematopoietic stem-cell transplantations may be autologous, which involves the patient’s own stem cells, or allogenic, which involves donor stem cells. Allogenic stem cell transplantation (alloSCT) was first performed in CTCL patients in the 1990s with hope of a potential cure (89). While no randomized controlled trials comparing the efficacy of alloSCT with conventional therapy in advanced CTCL have been performed (90), retrospective analyses have demonstrated long-term remission in advanced stage CTCL after alloSCT (91–94). It is worth mentioning that compared to alloSCT, autologous stem cell transplant (autoSCT) has significantly lower overall survival and shorter event-free survival, failing to show durable responses with progression noted as early as 100 days (18, 95). Interestingly, patients with alloSCT have shown multiple pieces of evidence supporting a “graft-versus-lymphoma/graft-versus-tumor” (GvL/GvT) effect thought to be mediated by donor T cells and NK cells: there is durable remission associated with chronic graft-versus-host disease, relapse or progression may be triggered with initiation of immunosuppressive treatment, and this relapse or progression upon initiation of immunosuppressive treatment responds to discontinuation of the drugs and to infusion of donor lymphocytes (95, 96). The success of alloSCT and not autoSCT to induce long-term remissions is the most striking evidence yet that by harnessing the immune system we may achieve our greatest success in the treatment of CTCL.
4. Chemotherapy
Several chemotherapeutic agents are recognized by the National Comprehensive Cancer Network for the treatment of MF/SS. These drugs include dihydrofolate reductase inhibitors (methotrexate and pralatrexate), anthracycline topoisomerase inhibitors (doxorubicin), nucleosides (gemcitabine), purine nucleoside phosphorylase inhibitors (forodesine), adenosine deaminase inhibitors (pentostatin), and alkylating agents (mechlorethamine and chlorambucil), among others (Table 3) (60). However, given the limited duration of response and side effect profile associated with chemotherapy, single agent doxorubicin and gemcitabine are typically employed in relapsed or refractory cases of MF/SS. Patients with visceral disease, aggressive non-MF/SS, or those who have failed multiple lines of therapy often receive multi-agent chemotherapy regimens employed for peripheral T cell lymphoma. We will review the most commonly used chemotherapeutic agents.
4.1. Alkylating agents
Alkylating agents, such as topical mechlorethamine (nitrogen mustard) and oral chlorambucil and cyclophosphamide, have been used in the treatment of MF for more than 50 years (60, 97, 98). These agents binding to DNA, namely at guanine residues, lead to strand breaks and inter-strand cross-linking, impairing DNA replication (60). Cyclophosphamide, in particular, has been able to induce immunogenic cell death (ICD) (see below). Nitrogen mustards can also generate reactive oxygen species (ROS) (99). Mechlorethamine has been shown to have direct apoptotic effects in malignant skin T cells, but it is not clear if it can cause ICD (100). Historically, mechlorethamine was formulated into aqueous solutions and used as a total body application, which was associated with high rates of delayed-type cutaneous hypersensitivity reactions (≤67%) (101, 102). Ensuing formulations used a petrolatum-based ointment for compounding, which was associated with fewer hypersensitivity reactions (103). While the development of a delayed hypersensitivity reaction limits its prolonged use, it is associated with a better prognosis (101). It is unclear whether the hypersensitivity reaction is itself therapeutic or instead a measure of an intact cell-mediated immunity, as reactivity to the contact allergen 2,4-dinitrochlorobenzene (DNCB) is also associated with better overall prognosis and diminishes with advancing stage of disease (104). It is possible that by inducing a hypersensitivity reaction there is a local increase in Th1 cytokines by tumor infiltrating lymphocytes, which could contribute to an anti-tumor response (103). In 2013, the U.S. Food and Drug Administration (FDA) approved a readily available mechlorethamine, 0.02%, gel (Valchor) for the treatment of MF-CTCL. The gel achieved a RR of 58.5% while the ointment achieved a RR of 47.7% in a non-inferiority study compared with mechlorethamine 0.02% compounded in ointment (53). Chlorambucil (2-12 mg daily) has been used in combination with systemic steroids, or in conjunction with ECP, biologic response modifiers, and/or phototherapy in several retrospective cohort studies and case series (51, 60). The main toxicity of chlorambucil, like cyclophosphamide, is myelosuppression (60).
4.2. Anthracycline topoisomerase inhibitor
Pegylated liposomal doxorubicin (Doxil) is an anthracycline topoisomerase inhibitor, with direct cytotoxic effects on tumor cells. Interestingly, in vivo and in vitro studies have found that anthracyclines may also have favorable immune modulatory effects, namely through inducing an immunogenic cell death (105, 106). Physiologic cell death is normally a non-immunogenic event, thereby preventing the development of autoimmunity (107). However, cancer cell death, as induced by radiotherapy or certain chemotherapies, namely anthracyclines, has been shown to promote an anti-tumor response (107). Doxil treated tumor cells, in in vitro and in vivo studies, are preferentially processed by antigen presenting cells, leading to dendritic cell maturation, and are capable of eliciting a cytotoxic CD8+ T cell response (106). This effect is partly due to release of immune activating molecules called damage associated molecular patterns (DAMPs). This includes translocation of calreticulin (CRT) to the cell surface and release of high mobility group box 1 protein (HMGB1), which both facilitate the recognition and engulfment of dying tumor cells by dendritic cells, via binding of TLR4 in the case of HMGB1 (106, 107). In MF/SS, various dosing regimens of Doxil have been evaluated with a RR ranging from 30% to 80% with CR rates of 20% to 60% (52, 108).
4.3. Anti-metabolites
Antimetabolites interfere with the cell cycle due to similarities with key cellular substrates or their cognate enzymes. Antimetabolites used for CTCL include gemcitabine, a pyrimidine antagonist, and folic acid antagonists methotrexate and pralatrexate. Gemcitabine is capable of inducing immunologic cell death (109). Methotrexate is a dihydrofolate reductase inhibitor, preventing the conversion of dihydrofolate to tetrahydrofolate, which is required for synthesis of thymidylate and purine nucleotides involved in DNA and RNA synthesis (110). Methotrexate also has immune modulatory functions, which are thought to be a result of inhibition of methionine synthetase and aminoimidazole-carboxamide ribonucleotide transformylase, which ultimately leads to an increase in adenosine (110). Adenosine in turn broadly suppresses effector NK and T cell function, impairs dendritic cell maturation and antigen presentation capacity, and enforces M2 polarization (111). In addition, methotrexate, through an unknown mechanism, is a potent inhibitor of the JAK-STAT pathway, as described above (112, 113). In a monotherapy study of low dose methotrexate (defined as doses <100 mg/week), there was an observed RR of 58% (17 of 29) with a 41% CR in patients with erythrodermic MF and a RR of 33% (20 of 60) with 12% CR in patients with plaque-stage MF (54). Pralatrexate, a novel anti-folate agent, selectively binds and is internalized into cells that express reduced folate carrier type 1 (RFC-1), for which it has a high affinity and is selectively expressed in tumor cells (114). In a dose finding study of pralatrexate in CTCL an overall RR of 45% was observed, with only one CR (55).
5. Immune checkpoint inhibitors
Immune checkpoint inhibitors (ICIs) have led to dramatic clinical responses in melanoma, which has attracted the exploration of their use in numerous other cancers, including Hodgkin’s and non-Hodgkin’s lymphomas (115).
5.1. Programmed death/programmed death ligand 1 targeting
Programmed death-ligand 1 (PD-L1) can be expressed by hematologic and non-hematologic cells; whereas programmed cell death protein 1 (PD-1), an inhibitory receptor, is expressed following antigen recognition on activated T cells, as well as Tregs, B cells, NK cells and other myeloid populations (116). Canonically, engagement of PD-1 on T cells by its ligand promotes phosphorylation of PD-1 tyrosine residues in the cytoplasmic region, which leads to binding of protein tyrosine phosphatases. This inhibits kinases downstream of T cell receptor (TCR)-CD28 signaling, including PI3K, Ras, extracellular signal-regulated kinase (ERK), and phospholipase C-γ (PLCγ) (116, 117). This results in decreased T cell activation, cytotoxic function, proliferation, survival, and cytokine secretion—enabling immune evasion by tumor cells. In cancer, tumor infiltrating lymphocytes often express high levels of PD-1, characteristic of an exhausted T cell phenotype that is unable to mount an effective anti-tumor response (118). PD-1 inhibition thereby unleashes T cells to affect tumor cell killing. However, the downstream effects of PD-1 inhibition on malignant T cells expressing PD-1 with or without PD-L1 is less clear.
Initial observations in MF/SS found elevated PD-L1 and decreased PD-1 expression on malignant T cells with advancing stages, leading to investigation of ICIs in MF/SS (119). In the phase II study of MF/SS (n=24), the RR was 38% (120). Of the nine responding patients, six had 90% or more improvement by mSWAT. There were no differences between responders and non-responders with respect to pre-treatment PD-1/PD-L1 expression on malignant T cells, IFN-γ/TGF-β gene signatures, or absolute numbers of CD4+ T cells, CD8+ T cells, Tregs, dendritic cells or tumor cells in the TME. However, single-cell spatial phenotyping of the TME from tissue microarrays obtained from skin biopsies of trial patients pre- and on-treatment revealed that responders compared to non-responders had cellular neighborhoods enriched for tumor with dendritic cells, tumor with CD4+ T cells, and decreased zones with Tregs (121). Among responders following PD-1 blockade, increased numbers of activated (ICOS+), proliferating (Ki-67+) CD4+ T cells with cytotoxic function (GZMB+), and CXCL13 expressing tumor cells were noted. Non-responders had greater Treg enriched neighborhoods, including greater numbers of activated (ICOS+) Tregs and a suppressive subset (IDO-1+) of CD8+ T cells. Importantly, they found that a simple metric, termed the SpatialScore, which calculates the physical distance ratio of each CD4+ T cell and its nearest tumor cell (“right” distance) relative to its nearest Treg (“left” distance), calculated on a per-cell basis, was predictive of responders versus non-responders pre-treatment. Among SS patients, responders were found to have lower levels of KIR3DL2 expression in malignant cells, which inhibits activation-induced cell death, pre-treatment. And following PD-1 blockade, responders had expanded peripheral blood CD8+ terminal effector and CD8+ effector memory T cell populations (122). Favorable responses to PD-1 inhibition have also been observed in MF cases with PD-L1 structural variants (123). Rare PD-L1 structural variants have been identified in some MF patients (20, 124), similar to PD-L1 3’ untranslated region disruptions reported in extranodal natural killer/T cell lymphomas and adult T cell leukemia lymphoma (125, 126). Disruption of PD-L1 3’ untranslated region in mice demonstrated elevated PD-L1 expression with immune evasion of E.G7-OVA T lymphoblast tumor cells (126). Use of anti-PD-L1 antibody in these mice promoted tumor regression and restored CD8+ cytotoxic T lymphocyte function.
Enthusiasm for ICIs in T cell lymphoma, however, is attenuated by the risk for disease hyperprogression through disinhibition of PD-1 expressing malignant T cells. In T cell lymphomas, PD-1 has been shown to act as a tumor suppressor via enhanced PTEN expression and decreased PI3K-Akt signaling (127). Risk of hyperprogression is likely diminished in MF/SS with mono- or bi-allelic loss of PDCD1, encoding PD-1 (120). Hyperprogression has been reported in peripheral T cell lymphoma (PTCL) and adult T cell leukemia/lymphoma (128, 129). In a phase II study of pembrolizumab in MF/SS a flare of erythroderma was noted exclusively among SS patients, which correlated with PD-1 tumor expression but did not prohibit continuation of ICI and did not correlate with disease responses or progression (120). While increased proliferation of Sézary cells has been reported following exposure to nivolumab in vitro (130), there was no increased percentages of proliferating Sézary cells among non-responders in the phase II trial of pembrolizumab (120).
ICIs are being investigated in combination with targeted therapies. A phase I/II study of nine patients with refractory and advanced CTCL using the PD-L1 inhibitor durvalumab with lenalidomide demonstrated clinical activity (21). Seven (78%) achieved a response with two having 90% or more improvement in mSWAT. The phase II portion is ongoing (NCT03011814). Given the importance of oncogenic PI3K signaling in PDCD1 deficient malignant T cells, the addition of PI3K inhibitors may mitigate the risk of hyperprogression in T cell lymphomas with PD-1 expression following PD-1 inhibition (NCT04652960). Furthermore, inhibition of PI3K signaling in tumor-associate myeloid cells (see PI3K inhibitor section) can improve responses to ICI in murine models (131). Lastly, nivolumab is being combined with brentuximab vedotin (discussed below), which has been shown to decrease highly activated/suppressive IRF4+ effector Tregs and tumor associated macrophages (132).
5.2. Cytotoxic T-lymphocyte-associated protein 4 targeting
Another common checkpoint target, CTLA-4, has had more limited exploration in CTCL. CTLA-4 is expressed soon after T cell activation, primarily in lymph nodes (priming phase), to regulate proliferation and can induce T cell anergy (133). This contrasts with PD-1, which inhibits T cells later in the immune response and primarily expressed in T cells in the peripheral tissue (peripheral tolerance) (133). CTLA-4 is also highly expressed on Tregs, where it is believed to increase Treg immunosuppressive function (134, 135). Therefore, CTLA-4 blockade is thought to increase the activity of anti-tumor effector T cells and inhibit Tregs. However, in clinical trials the impact of ipilimumab on Tregs is less clear, with some reports of increased levels of Tregs following treatment, and other studies showing decreased intra-tumoral Tregs (136, 137). In pre-clinical studies, ipilimumab was able to deplete Tregs via antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis (138). Two patients with MF/SS treated with ipilimumab have been reported. A patient receiving the CTLA-4 inhibitor ipilimumab for his advanced melanoma achieved complete MF regression, and another patient with SS treated with ipilimumab experienced rapid clinical response (139, 140). The patient with SS had a highly expressed CTLA-4 and CD28 gene fusion, a putative oncogenic driver. Jurkat cells engineered with this fusion construct proliferated faster than both cells with empty vector and cells with CTLA-4 overexpression (124). It is not clear if ipilimumab efficacy is limited to patients with this specific CTLA-4 fusion protein.
5.3. Cluster of differentiation 47 inhibitor
Another potential checkpoint target involves cluster of differentiation 47 (CD47). This transmembrane protein binds to signal regulatory protein alpha (SIRPα), leading to inhibition of macrophage phagocytosis (141). Tumor cells commonly overexpress CD47 to exploit this innate macrophage checkpoint. Therapeutic TTI-621 has a CD47 binding domain of SIRPα that is also associated with the Fc region of human IgG1 (22, 142). Therefore, this immune checkpoint inhibitor not only impairs anti-phagocytic signals but also promotes phagocytic signals through immunoglobulin G1 (IgG1) Fc linking with macrophage Fcγ receptors. A phase I study with TTI-621 intratumoral injections had encouraging results (143). Notably, out of the ten patients with paired assessments, eight (80%) achieved reduction in Composite Assessment of Index Lesion Severity (CAILS) scores in non-injected adjacent lesions. One additional patient had a CAILS score reduction in distal non-injected lesions.
6. Radiation
6.1. Ionizing radiation
Radiation is one of the oldest modalities of cancer therapy for CTCL (144). This skin-directed therapy includes local or total skin electron beam therapy (TSEBT) and brachytherapy. Brachytherapy is the administration of radiation through flexible catheters that can be secured within fully customized surface molds used for complex curved surfaces, such as the face (145). Ionizing radiation is the most effective single therapy for the treatment of MF, partly due to the unusually high radiosensitivity of malignant T cells (Table 2) (29–33, 144). Most often, ionizing radiation induces double-stranded DNA breaks leading to programmed cell death (146). Importantly, radiation can induce an immunogenic cell death. In addition, there is upregulation of MHC class I with tumor antigen peptides on tumor cells, leading to enhanced recognition of tumor cells by cytotoxic T lymphocytes (147, 148). Interestingly, TSEBT has also been used in patients with SS with improvement in leukemic disease in addition to the skin. A case series of three SS patients receiving low-dose (8-12 Gray) TSEBT while on stable combination therapy (interferon, bexarotene) saw a decrease in the percentage of Sézary cells in the blood along with decreased numbers of exhausted T cells and increased IFN- γ secretion by peripheral blood monocytes (PBMCs) (149). The ability of a skin-directed therapy to influence immune parameters in the blood, as well as decrease leukemic cells, may in part reflect generation of a systemic anti-tumor immune response.
Due to the immunomodulatory effects of radiation, “combination therapy” consisting of radiotherapy and immune checkpoint inhibitors may therefore induce long-term immune-mediated antitumor activity (148, 150). Several trials are currently underway to evaluate the safety and efficacy of combining TSEBT with ICI for the treatment of MF/SS (NCT03385226). Additionally, there is a trial combining TSEBT with mogamulizumab (see below), which requires NK cell mediated cellular toxicity for effective tumor cell killing (NCT04128072).
6.2. Non-ionizing radiation
Non-ionizing radiation, namely phototherapy, can have anti-proliferative and pro-apoptotic effects on T cells but can also drive favorable and unfavorable inflammatory response through several mechanisms. Ultraviolet radiation (UVR) is absorbed by chromophores in the skin (151). This includes trans-urocanic acid in the stratum corneum, cell membrane lipids, intracytoplasmic molecules such as melanin or amino acids, or most importantly by DNA nucleotides. UVR can cause apoptosis by direct chemical modification of DNA nucleotides or via phototoxic products that generate free radicals, reactive oxygen and nitrogen species leading to oxidative damage of DNA (151). It is unclear how the immune effects of ultraviolet A (UVA) and ultraviolet B (UVB) differ. However, a notable difference is that the wavelength of UVR determines the depth of radiation. The UVA spectrum penetrates the deepest and is between 320 and 400nm, whereas the UVB spectrum is between 280-320nm and penetrates only to the superficial dermis (152). UVB may be delivered as full-spectrum (broad-band UVB lamps 270-350nm, along with the shorter wavelengths from UVA spectrum) or delivered as small-spectrum (narrow-band UVB lamps 311-313nm). Narrow-band UVB (nbUVB) therapy was developed as a less harmful alternative to broad-band UVB and to photochemotherapy, both of which have significant side effects and carry a risk of carcinogenesis (153).
6.2.1. Narrowband UVB
nbUVB radiation works by creation of DNA photoproducts, namely pyrimidine dimers, which leads to replication arrest or apoptosis. Its efficacy in MF is likely due to its ability to induce apoptosis in T cells (154). Many studies evaluating the effects of UVB photobiology have been carried out following irradiation of immune cells or keratinocytes in vitro or following UVR of murine models (155). In these settings, both release of pro-inflammatory and immunosuppressive cytokines have been observed following irradiation. However, the majority of studies have observed immunosuppressive effects of nbUVB, including altered antigen presentation, Langerhans cell depletion, decreased activity of NK cells (156, 157), and generation of Tregs (147, 158–161). Despite these negative immune effects, nbUVB is one of the most commonly used and effective skin-directed therapies for MF (Table 3) (34).
6.2.2. Psoralen and ultraviolet A
PUVA is photochemotherapy consisting of taking the oral drug psoralen and then exposing the skin to UVA light. Psoralen forms DNA crosslinks following UVA exposure leading to DNA damage and apoptosis (162). Unlike nbUVB, the effect of PUVA has been studied in detail in an MF cohort leading to several major observations (163). The first is that PUVA can indeed reduce or eliminate malignant T cell as well as decrease benign T cells from the skin. However, this largely occurred in those with a “low” pre-treatment tumor burden (<10% tumor clone frequency, TCF, the percent of T cells that are malignant). In those with a “high” initial tumor burden (>20% TCF), clinical resolution of inflammation was independent of changes in total malignant or benign T cell numbers but due to a change in benign T cell populations based on characterization of the T cell repertoire by high throughout sequencing (HTS) of the T cell receptor (TCR). Bulk RNA sequencing coupled with this HTS TCR data suggested that PUVA may induce a turnover of benign T cell from Th2 to Th1 populations. Interestingly, a subset of benign T cells recruited post-PUVA treatment showed markers of TCR-dependent, antigen-specific activation-markers of anti-tumor T cells. Therefore, PUVA may lead to clinical responses by direct clearance of malignant T cells or indirectly via generation of an anti-tumor immune response. And, surprisingly, PUVA can lead to clinical responses through clearance of inflammation-producing benign T cells without reducing malignant T cells. Despite the later observation, PUVA has been successfully used in MF with long-term remissions possible (Table 3) (35, 164, 165). A number of retrospective cohort studies have evaluated PUVA in combination with IFN-α, but given the lack of a comparator arm it is difficult to assess the full benefit of the addition (166–171). In one randomized clinical trial comparing IFN-α and PUVA versus PUVA monotherapy in CTCL stages I and II, there was not a statistically significant difference in complete remission, but the combination therapy had increased progression-free time (172).
6.2.3. Extracorporeal photopheresis
ECP involves ex vivo treatment of a patient’s blood followed by retransfusion into the patient (173). ECP, typically used for MF/SS patients with a low blood burden of disease (Table 3) (25, 36), exposes peripheral blood (buffy coat) to UVA radiation following photosensitization with 8-methoxypsoralen (8-MOP) (173). It has direct pro-apoptotic as well as immune modulating effects (174). During the ECP process, platelets adherent to the ECP plate, inadvertently coated with fibrin, engage monocytes and induce them to mature into dendritic cells capable of presenting tumor antigens to generate an anti-tumor immune response (175). While the type of apoptosis induced in leukemic malignant T cells has not been further evaluated, in 8-MOP treated melanoma cell lines, photoactivation induced an immunogenic cell death (176). While ECP has been shown to eliminate alloreactive T cells in GVHD, it has also been reported to induce the generation of Tregs, a potentially unfavorable immune response in CTCL (177). Understanding the effect of ECP on Treg populations in CTCL in the past has been challenging given the difficulty of distinguishing malignant T cells expressing Treg markers from benign Tregs (178). Additionally, in murine models, ECP impairs effector T cell functions (179). Several retrospective and some prospective cohort studies have suggested that combination of interferon or bexarotene with ECP may lead to improved responses, although further studies are needed (60).
6.2.4. Photodynamic therapy
PDT involves the combination of a photosensitizing compound, such as porphyrins (e.g. 5-aminolevulinic acid, ALA) or non-porphyrins (e.g. hypercin), with administration of light of a certain wavelength (180, 181). These photosensitizers may preferentially accumulate in the mitochondria, lysosomes, endoplasmic reticulum (ER), Golgi apparatus, and/or plasma membrane. The light then activates the sensitizer, generating reactive oxygen species (ROS) (182). ROS can be generated by physiologic signaling pathways in T cells leading to activation, differentiation, proliferation, or apoptosis (183). However, high levels of ROS generated by photosensitizers, especially those leading to ER stress, potently induce immunogenic cell death (181). Following PDT, pro-inflammatory cytokines IL-1 and IL-6 are also increased (184). While inflammatory apoptosis of malignant T cells is favorable, Tregs are not as suspectable to ROS (185). In addition, ROS can cause hyporesponsiveness of effector T cells and NK cells (185). Therefore, generation of ROS can be a double-edged sword.
PDT has shown promise for the treatment of MF. Case series evaluating the use of PDT date to the 1990’s (186). Initial studies used ALA, but this was later replaced by methyl-aminolevulinate (MAL), which is thought to have better specificity given its lipophilic properties (186). Despite encouraging responses (Table 3) (26), the use of in office PDT for MF is impractical given the size of lamp and need for frequent treatments. Not surprisingly PDT in the past was mainly investigated in unilesional MF (186). Recently, a non-porphyrin photosensitizer, synthetic hypercin (SGX301), which is activated by visible fluorescent light in the 500-650mn range (yellow-red spectrum) has completed its pivotal phase 3 clinical study. In in vitro studies activated hypercidin caused greater apoptosis of malignant T cells over benign T cells (187). Notably, hypercidin is a potent inducer of immunogenic cell death (181).
7. Targeted therapies
7.1. Fusion peptides and proteins
7.1.1. E7777
The high affinity IL-2R (CD25) is overexpressed in the malignant T cells in CTCL (188). E7777 (formerly known as denileukin diftitox) is an engineered fusion protein combining IL-2 and the diphtheria toxin and binds to cells expressing the high affinity IL-2R, including Tregs and malignant T cells, causing endocytosis of the fusion protein (189). After endocytosis, ADP-ribosyltransferase activity of diphtheria toxin inhibits protein synthesis and causes cell death. Therefore, this therapy simultaneously decreases tumor burden and theoretically boosts the anti-tumor immune response by depleting immunosuppressive Tregs (190, 191). However, treatment has been shown to only target activated T cells, and thus resting Tregs remain in circulation (192). Furthermore, this treatment impairs dendritic cell maturation in vitro and in vivo, creating a tolerogenic dendritic cell phenotype, which induces T cell anergy instead of activated anti-tumor T cells (192). After being approved by the FDA in 1999, denileukin diftitox was taken off the market voluntarily in 2014 in order to create a more purified form with improved bioactivity to reduce the risk for systemic capillary leak syndrome (193, 194). E7777, a purified and reformulated denileukin diftitox, achieved a RR of 32% (six of 19 with PR and zero with CR) in patients with relapsed or refractory CTCL in a phase II trial (46). E7777 has also been investigated in a recently completed Phase III trial (clinicaltrials.gov identifier: NCT01871727), and application for FDA approval within the next year is anticipated after results are published. Resimmune, a similar fusion protein carrying cytotoxic diphtheria toxin, targets cells expressing the CD3ε surface molecule, which is found predominantly in T cells (195). In an inter-patient dose escalation trial in CTCL there was a 36% RR (nine out of 25) with four CRs (196). The modest response rates may be explained by its complex immune modulating function.
7.1.2. Selective γ-chain cytokine inhibitor
BNZ-1 an antagonist pegylated peptide that binds to the common gamma chain signaling receptor for certain cytokines, namely IL-2, IL-9, and IL-15, was investigated in MF/SS (45). It was hypothesized that inhibition of IL-2 and IL-15 would impair cytokine-driven tumor cell survival, inhibition of IL-2 and IL-9 would increase the anti-tumor immune response by decreasing Treg activity, and inhibition of IL-15 would create a beneficial anti-inflammatory effect (45). IL-15 is a putative autocrine survival factor found in elevated levels in MF skin samples and expressed by MF/SS cell lines (197). A phase I/II study of 19 patients with refractory MF/SS demonstrated positive results with 11 participants achieving PR and one CR; a phase 3 trial is being planned (45).
7.2. Monoclonal antibodies
7.2.1. Anti-C-C chemokine receptor 4 antibody
Mogamulizumab is a defucosylated, humanized, anti-CCR4 monoclonal antibody. CCR4 is the receptor for thymus- and activation-regulated chemokine (TARC/CCL17) and MDC/CCL22, a predominantly macrophage-derived chemokine (198). As mentioned, CCR4 is mainly expressed on CD4+ T cells (particularly memory T cells), CD4+CD25+FOXP3+ Treg cells, and only on a minority of CD8+ T cells (66). It is a putative marker for T cells with skin homing abilities, as it is thought to play a role in transendothelial diapedesis and epidermotropism (66). Not surprisingly, CCR4 is overexpressed in malignancies of skin homing T cells including MF, SS, and some peripheral T cell lymphomas (199). Mogamulizumab binds to the N terminal domain of CCR4, eliciting tumor cell killing via antibody-dependent cellular cytotoxicity (200). Defucosylation of the antibody’s Fc portion enhances binding-affinity of the Fc receptor on effector cells leading to more potent antibody dependent cellular cytotoxicity (ADCC).
In 2018, mogamulizumab was approved for relapsed or refractory MF/SS following a phase III clinical trial of 370 MF/SS patients, comparing mogamulizumab to vorinostat (50). Mogamulizumab achieved a global response of 35% compared to 6% in patients receiving vorinostat. The mogamulizumab group also had longer progression-free survival (median 7.7 months) compared to the vorinostat group (median 3.1 months). Infusion-related reactions, lymphopenia, drug rash [now termed mogamulizumab-associated rash (MAR)], diarrhea, and fatigue were the most common adverse events of any grade in patients receiving mogamulizumab (201). Immune-related adverse events, such as polymyositis, myocarditis, and hepatitis have also been observed, thought to be driven by Treg depletion (201). Recent retrospective cohorts have affirmed a high incidence of MAR in patients with CTCL (17 of 24, 68%) and found that it is associated with clinical responses, leading to the hypothesis that those with effective decreases in Tregs are more likely to experience MAR and achieve a clinical response (202). Patients who experience MAR and/or other immune related adverse events reportedly have higher rates of CR and PR as well as prolonged remissions of disease, in some cases over two years, giving credence to the theory that remodeling of the immune microenvironment contributes to short and long-term therapeutic efficacy (202, 203).
From the phase I/II study, mogamulizumab was observed to have a favorable effect on patients’ immune profile by reducing Treg cells and increasing NK cell killing function but no effect on the absolute number of NK and CD8+ T cell populations were noted at one month (204). However, a real world SS cohort found that mogamulizumab, in addition to depleting benign CD4+ T cells, Tregs, and malignant T cells, also decreased CD8+ T cells and NK cells at one month of treatment. Longer-term follow-up in this cohort revealed the emergence of “immune restoration” involving expansion of naïve and stem memory CD4+ subsets with almost complete disappearance of pre-treatment exhausted lymphocytes (PD-1+ or TIGIT+ CD4+ T cells) and activated Tregs (205). In skin biopsies taken from patients with MAR, an increase in interferon-stimulated gene IFI44L and macrophages expressing CXCL11 and CXCL9 coupled with expansion of new T cell clones expressing CXCR3 (the cognate receptor for CXCL9/11) and genes important for cytotoxic effector function was observed, shedding new insight on the potential role tumor associated macrophages may play in generating prolonged remissions from mogamulizumab (206). Despite favorable changes in the immune environment, disease relapse in the SS cohort appeared to be tied to the emergence of CCR4- malignant T cells (205).
7.2.2. Anti-CD52 antibody
Alemtuzumab, a monoclonal antibody, targets cells expressing CD52, which is expressed on T cells, B cells, monocytes, eosinophils, and a subset of dendritic cells (60). Alemtuzumab is primarily utilized for leukemic CTCL given its limited efficacy in clearing cutaneous disease. This is likely due to its primary mechanism of action, ADCC, which requires NK cells and neutrophils; these are present predominantly in the blood but not in the skin (207). Administration of alemtuzumab causes a depletion of lymphocytes, with depletion of Tregs and exhausted T cells being a favorable immune modulatory effect in CTCL (208). However, CD52 is broadly expressed in effector cells of the innate and adaptive immune response, important for anti-tumor immune responses and protection against infections (60). The overall RR in alemtuzumab is 55% with 32% of patients achieving a CR (47), though RR may be higher in select populations such as SS. Lower doses of alemtuzumab and anti-microbial prophylaxis have prevented many of the infectious complications seen in the earlier studies (47, 207, 209).
7.2.3. Anti-killer cell immunoglobulin-like receptor 3DL2 antibody
Lacutamab is a first-in-class humanized monoclonal antibody targeting inhibitory killer cell immunoglobulin-like receptor (KIR) KIR3DL2, which is aberrantly expressed in MF/SS and minor subpopulations of NK cells, CD8+ T Cells, and CD4+ T Cells (49, 210). In NK cells, KIR3DL2 is one of several inhibitory KIR receptors that recognizes MHC class I molecules (211, 212). Loss of MHC-I in tumor cells or decreased expression of inhibitory KIRs on NK cells leads to activation of NK cell-mediated killing. In MF/SS its function is less clear, but it appears to function as an inhibitory coreceptor—downmodulating CD3-dependent early signaling events following antigen recognition (213). Therefore, it is suspected that KIR3DL2 may protect malignant T cells from activation-induced cell death (AICD) (213). In pre-clinical studies of SS, lacutamab exerted an anti-tumor effect via ADCC, rather than through modulation of KIR signaling in malignant cells. A phase I clinical trial of 44 patients with relapsed or refractory cutaneous T cell lymphoma demonstrated 36% RR, predominantly in SS patients (49). Three patients experienced lymphopenia, which was the most common grade 3 or worse adverse event. The effect of lacutamab on NK cells in CTCL cohorts has not been evaluated. However, use of a related antibody directed against KIR2D inhibitory receptors (IPH2101) in multiple myeloma patients found unexpectedly that IPH2101 decreased the responsiveness of the NK cell. A phase II trial investigating lacutamab alone or in combination with chemotherapy in patients with advanced T cell lymphoma is underway (clinicaltrials.gov identifier: NCT03902184). Combination of lacutamab with PD-1 inhibition has been proposed, as stated above, given that SS responders to PD-1 inhibition had lower levels of KIR3DL2 (122).
7.2.4. CD30 antibody-drug conjugate
Brentuximab vedotin (BV) is an antibody-drug conjugate, consisting of the chimeric monoclonal antibody to CD30 linked to monomethyl auristatin E (MMAE), a potent anti-tubulin agent. It was approved by the FDA for CD30+ cutaneous lymphomas in 2017 (214, 215). Upon binding to CD30, the antibody-drug conjugate is internalized, and MMAE is released causing G2/M arrest and apoptosis (216). For this reason, it is used to treat CD30+ lymphoproliferative diseases, including patients with large cell transformation of MF, which often express CD30 on tumor cells (217). Early clinical trials with 28 MF patients showed a RR of 54% with 7% achieving a CR (218). A median duration of response was 32 weeks. This response was not correlated with CD30 expression. Kim et al. evaluated the use of BV in MF with variable CD30 expression and found an overall response rate of 70% with 20 PR and one CR; they found that median CD30max was higher in responders (13%) versus non-responders (3%) and least effective in cases with a CD30max of <5% (219). This study suggested that BV can be effective in MF even with relatively low to intermediate levels of CD30 (CD30max >5%). This may be attributable to the additional favorable immune modulatory effects, including depleting immunosuppressive M2 type tumor associated macrophages, inducing dendritic cell maturation, and promoting bystander cell death upon releasing MMAE (even among CD30- tumor cells) (214, 219). Further, BV was shown to deplete immunosuppressive inducible and primary CD30+ Tregs in a dose dependent manner in vitro and in vivo (132, 220). CD30+ CD8+ T cells, which play a role in anti-tumor immunity, however, were not affected by BV (132, 220). In a phase III clinical trial of 131 patients with CD30+ MF or primary cutaneous anaplastic large-cell lymphoma, BV was compared to physician’s choice of methotrexate or bexarotene (48). Among 48 MF patients, 50% achieved an objective global response lasting at least four months, outperforming the physician’s choice group.
7.3. Small molecules
7.3.1. B cell lymphoma 2 inhibitor
The BCL-2 family consists of pro-apoptotic and pro-survival proteins. Shifts in balance between these two protein groups lead to cell death or survival (221). Overexpression of pro-survival BCL-2 proteins allows for various cellular stresses to occur that would otherwise promote apoptosis in non-cancerous cells, and overexpression of BCL-2 in cancers is associated with resistance to therapy (222–224). Likewise, BCL-2 expression is observed in CTCL lines that are sensitive to venetoclax, a BCL-2 inhibitor already approved by the FDA to treat chronic lymphocytic leukemia, small lymphocytic lymphoma, and acute myeloid lymphoma (225–227). An investigator-initiated, single-arm, open label study of venetoclax closed after enrollment of four patients (clinicaltrials.gov identifier: NCT04171791). A single case was published of a SS participant with skin and blood involvement treated with venetoclax who achieved a near CR (228). Despite no immediate plans for additional trials of venetoclax in CTCL, this agent may be best considered as part of a combination therapy to sensitize cells to apoptosis without impeding immune cell function (227). Control of apoptosis is an important mechanism regulating immune cell development and response (229). Despite decreases in T cell counts with venetoclax, largely due to reductions in naïve T cells, antigen-stimulated effector T cells can survive and proliferate via upregulation of other pro-survival proteins, such as BCL-xL (229, 230).
7.3.2. Enhancer of zeste homolog 2 inhibitors
EZH2 is the enzymatic component of the polycomb repressive complex 2 (PRC2). PRC2 catalyzes trimethylation of lysine 27 of histone H3 (H3K27me3), which is deposited at CpG-dense promoters and is involved in gene transcriptional repression (231–233). EZH2 represses expression of tumor suppressor genes causing cell cycle progression and cell proliferation. Gain-of-function mutations in EZH2 have been associated with a variety of cancers, including T cell lymphoproliferative disorders (234–237). Additionally, there is a growing body of literature investigating the function EZH2 in the immune system, with many immune cells expressing EZH2 (238). Tumor infiltrating Tregs, for instance, appear to have EZH2 activity. Disruption of EZH2 activity in Tregs promotes pro-inflammatory functions and increases recruitment and function of CD8+ and CD4+ effector T cells (239). Inhibition of EZH2 in NK cells improves NK cell function (240). However, inhibition of EZH2 can impair effector T cell function and dendritic cell antigen presentation (240, 241). Currently, valemetostat (DS-3201b) a highly specific inhibition of EZH2 and EZH1 is being evaluated in a phase II trial for patients with relapsed or refractory peripheral T cell lymphoma, including non-MF/SS CTCL (NCT04703192). A phase I study of 15 patients with relapsed or refractory non-Hodgkin lymphomas, including PTCL, treated with valemetostat had a 53% RR (seven PR, one CR) (242, 243). Among the five patients with T cell lymphoma the RR was 80% (three PR, one CR).
7.3.3 Histone deacetylase inhibitors
There are 18 histone deacetylases (HDACs), which are categorized into four major classes: class I (HDAC1, 2, 3 and 8), class II (HDAC4, 5, 6, 7, 9, and 10), class III (aka sirtuins; SIRT1, 2, 3, 4, 5, 6, and 7), and class IV (HDAC11) (244). In general, histone deacetylation induces gene silencing. The clinical utility of HDAC inhibitors (HDACi) may include disruption of the epigenetic balance in malignant cells or activation of tumor suppressors, but other “off-target” effects include acetylation of transcription factors modulating their function. The current HDACi that are approved or under development are “pan-HDAC” inhibitors (inhibit HDACs in Class I, II, or IV), class specific inhibitors for either class I or class II, or inhibitors of a single HDAC protein. For example, vorinostat is a pan-HDACi and romidepsin inhibits class I HDACs. In multiple cancer models HDACi have been shown to affect several pathways, which favorably lead to cell cycle arrest, induction of apoptosis, and inhibition of angiogenesis (245). However, HDACi have pleiotropic immune modulating functions, which may have opposing effects on the anti-tumor response.
The “off-target” effects of HDACs on the immune system are diverse with both desirable and undesirable results. HDACs can have immunosuppressive effects. Several HDACs in class I, II, and III have been shown to inhibit TLR mediated expression of pro-inflammatory genes such as the p40 subunit of IL-12, IFN-β, and NF-κB activation (244). Class I HDACs have also been shown to inhibit MHC II expression, thereby decreasing antigen presentation. HDACs also increase PD-1L expression (246).
HDACs also have a multitude of immune activating functions. Class I HDACs have been shown to be important for activation of STAT1 and STAT2 dependent interferon signaling (247). Class IIa HDACs, notably HDAC7, have been shown to positively regulate TLR mediated expression of pro-inflammatory cytokines in macrophages, cytokines, and associated receptors for cytotoxic T lymphocyte function (248, 249). Class IV HDACs are thought to play a role in promoting antigen presentation and antigen specific T cell responses (250). Therefore, inhibition of these pathways could impair the interferon-mediated immune response to viral, bacterial, and fungal pathogens and have a negative effect on the adaptive immune response. In fact, mice treated with HDACi are more susceptible to bacterial and fungal pathogens (251).
Given the overlapping and opposing actions of HDACs on the immune system it is unclear if selective inhibition of a particular class of HDACs or an individual HDAC protein would be more beneficial in CTCL. HDACi have been shown to increase tumor immunogenicity, have both a positive and negative regulation of the innate and adaptive immune system via modulation of TLR and interferon signaling pathways, and divergent effects on Treg function (244). Inhibition of class I HDACs has been shown to decrease Treg populations and increase immunologic cell death via upregulation of FAS, FASL and TRAIL. However, pan-HDACi, likely due to inhibition of class II HDACs, have been shown to increase Tregs and their function (252). Resistance may be related to an inability to orchestrate a favorable TME or may be related to properties intrinsic to the tumor cell. At the single cell level, for instance, SS has been shown to have a high degree of heterogeneity with differing sensitivity and resistance to HDACi (253). Clinical trials of romidepsin have shown an overall RR of approximately 30% with 6% of patients achieving CRs (41). As suggested above, infections (58% of patients for romidepsin) were common adverse events (254). Vorinostat and romidepsin have been shown to suppress NK and dendritic cell function in patients with CTCL (255). In a patient with SS that effect could be overcome with addition of an immune activating agent, such as TLR agonists or IFN-α when evaluated in vitro (255, 256). Therefore, future rational combination therapies with an immune activating agent and HDACi may synergistically enhance the anti-tumor response and decrease the risk of infections. Vorinostat and romidepsin are both approved for use in CTCL. Additional HDACi have been investigated in CTCL including panobinostat with 20% RR (40), belinostat with 14% RR (257), topical SHAPE gel also known as SHP-141 with interim results of 32% showing mSWAT response at 6 months (258), and resminostat with no published results (259).
7.3.4. Immunomodulator
Lenalidomide is a derivative of thalidomide (260). It binds E3 ligase protein cereblon (CRBN) directing protein ubiquitination and degradation of the IL-2 transcriptional repressors Ikaros (IKZF1) and Aiolos (IKZF3) (260, 261). Through modulation of these transcription factors, and likely others, it is able to have diverse effects on the innate and adaptive immune system. These effects include an increase in antigen presentation capacity, expansion and increased activity of cytotoxic CD8+ T cells, and a decrease in Th2 cytokine expression (261). In a phase II study of lenalidomide in refractory MF/SS, there was an overall RR of 28%; all were PR. The median duration of response was 10 months (39). In these patients, while there was an overall decrease in malignant CD4+ T cells and an increase in CD8+ T cells in MF skin lesions, an increase in CD25+ and FOXP3+ staining was also observed (39). This increased T regulatory phenotype may be secondary to the increase in IL-2 induced by lenalidomide.
7.3.5. Janus kinase/signal transducer and activator of transcription inhibitors
The JAK-STAT pathway, which transduces signals from cytokines, interleukins and growth factors, has been shown to be dysregulated in both MF and SS (4, 262). The signaling begins with binding to the cell-surface receptor which results in dimerization and causes activation of JAK tyrosine kinases. Subsequently, activated JAKs phosphorylate specific tyrosine residues on the receptor and act as docking sites for STATs, which are cytoplasmic transcription factors (61). Once STAT proteins bind to the receptor, the JAKs phosphorylate the STAT proteins, which dimerize and translocate to the nucleus to induce expression of genes involved in inflammation. STAT4, which is required for Th1 differentiation and is activated downstream of IL-12 signaling, has decreased expression in the skin and blood of patients with advancing CTCL stage (262). STAT5, activated downstream by IL-2, IL-7 and IL-15 signaling, and STAT3, activated downstream by IL-2, IL-6, IL-7, IL-9, IL-10, IL-15 and IL-21, may be constitutively activated independent of cytokines in MF/SS (262–264), although there is conflicting data (265). Constitutively activated STAT3 can increase survival and resistance to apoptosis in malignant T cells by increasing the anti-apoptotic protein BCL-2 (266). Furthermore, activation of STAT3 has been shown to induce expression of immunosuppressive ligand PD-L1 (77). STAT5 induces anti-apoptotic proteins, cell cycle genes (cyclin D and c-myc) and IL-4, which promotes carcinogenesis (61). In addition, its activation induces microRNA-155 that degrades STAT4 further solidifying the Th2 bias in the microenvironment (262, 267, 268). Furthermore, STAT3 and 5 are also important for the function of Tregs (269). Interestingly, methotrexate has been found, using a luciferase-based transcriptional assay in a Drosophila model system where a library of 2000 small molecules were screened, to be a potent inhibitor of the JAK-STAT pathway at doses that are equivalent to that seen in patients taking low-dose oral methotrexate (113).
While inhibition of JAK appears to be an attractive therapeutic candidate, malignant T cells do not exist in an isolated environment. As mentioned in the biologic response modifiers section, type I and II interferons activate the JAK-STAT pathway, leading to expression of interferon response genes with anti-proliferative and pro-apoptotic effects on malignant T cells. It is possible that the favorable effects of interferon-associated JAK-STAT signaling are blunted by JAK inhibitors. TLR-stimulated plasmacytoid dendritic cells, known for their ability to produce high levels of IFN-α for instance, had robust inhibition of IFN-α production in response to JAK inhibitor tofacitinib in vitro (270). Furthermore, with constitutive activation of STATs downstream from JAK in some patients, there is controversy over the ability of JAK inhibition to overcome downstream activation.
The FDA approved JAK inhibitors, including tofacitinib and ruxolitinib, demonstrated anti-tumor properties in CTCL cell lines (271–273), which has subsequently led to recent clinical trials. In a phase II trial of patients with relapsed or refractory PTCL or CTCL receiving ruxolitinib, a 20% RR was achieved among the five evaluable patients with CTCL (42). A phase IIa study of 37 patients with CTCL and 61 patients with PTCL investigated a small molecule spleen tyrosine kinase (SYK)/JAK kinase inhibitor called cerdulatinib (274). SYK is a tyrosine kinase, with its signaling pathway thought to be an oncogenic driver in multiple cancers, including immature hematopoietic neoplasms with T cell differentiation (275). The RR was 35% in CTCL patients overall. In MF the RR was 45%, with 9% of these participants achieving CR (37). Patients with Sézary syndrome had a 17% RR with no CRs.
7.3.6. Phosphoinositide 3-kinase inhibitors
The PI3K-Akt pathway has been implicated in several cancers and is an important signal transduction pathway for immune cells (276, 277). There are three classes (I-III) of PI3Ks (278). Class I, the most relevant to oncology, consists of four isoforms: α, β, δ, and γ. (271) PI3K γ and PI3Kδ are expressed in hematopoietic cells, but PI3Kδ is the dominant isoform in T cells, mediating downstream signaling of the TCR, costimulatory and cytokine receptors (279). PI3Ks phosphorylate the 3-hydroxyl group of the inositol ring found in phosphatidylinositol lipid substrates (280). The 3-phosphoinositides (PIP3) mediate the function of multiple effector proteins that bind these lipids. In turn, PI3Ks play a role in intracellular vesicular transport, cell cycle progression, and cell growth, survival, and migration.
Activating mutations in the PI3K-Akt pathway and constitutive Akt activity have been observed in CTCL, supporting the importance of PI3K signaling for malignant T cell survival and proliferation (38). Duvelisib, a PI3Kγ and PI3Kδ inhibitor, was approved by the FDA in 2018 for refractory chronic lymphocytic leukemia and small lymphocytic leukemia and has demonstrated activity in T cell lymphoma cell lines (281, 282). In a phase 1 study of duvelisib for patients with relapsed or refractory PTCL or CTCL, a RR of 31.6% (six of 19) was observed in the CTCL cohort (38). Of note, PI3K inhibition may also affect the immune microenvironment. In PTCL patient-derived xenografts treated with duvelisib a shift in polarization from immunosuppressive M2 macrophages to an inflammatory M1 phenotype, which promotes anti-tumor responses, was observed (38). In other murine models, P13K inhibition of tumor associated myeloid cells was able to sensitize tumors to ICI (131). However, these murine models lack an intact immune system to understand the full effects of P13K inhibition on the microenvironment. For example, PI3K signaling is important for antigen- and chemokine-dependent effector cell trafficking to peripheral sites of inflammation through regulation of leukocyte function-associated antigen-1 (LFA-1) (279). LFA-1 is needed for transendothelial egress and establishment of immunological synapse with antigen presenting cells (279). Therefore, PI3K inhibition may limit migration of anti-tumor effector T cells to the skin and effective engagement with antigen presenting cells thereby hindering the anti-tumor response. However, treatment of CD8+ T cells ex vivo with duvelisib, leads to enhanced T cell activation and cytotoxicity (283). Therefore, the net result of PI3K inhibition is unclear. Tenalisib (RP6530), another dual PI3Kδ and PI3Kγ inhibitor, is also under investigation, with a recent phase I/Ib study of relapsed or refractory PTCL and CTCL and RR of 45% was observed (43). A phase I/II study evaluating tenalisib in combination with romidepsin was recently completed (NCT03770000). Duvelisib was also investigated in combination with romidepsin or bortezomib in a phase I/II trial of PTCL/CTCL, based on in vitro drug screens predicting synergy. The RR in CTCL was 46% with romidepsin and 28% with bortezomib; no CRs were observed in either group (284).
7.3.7. Proteosome inhibitor
Proteasomes are fundamental to the ATP-dependent proteolytic pathway in eukaryotic cells (285). In fact, most cellular protein degradation occurs through proteasome catalyzed degradation (286). The FDA approved the first proteasome inhibitor, bortezomib (PS-341), for multiple myeloma in 2003 (287, 288). Bortezomib is a modified dipeptidyl boronic acid that is a reversible inhibitor of the 26S proteasome specifically (287). Bortezomib has pro-apoptotic and anti-proliferative effects via inhibition of NF-kB and induction of the unfolded protein response (289). It has also been shown to increase tumor cell killing by dendritic cells, likely due to induction of immunogenic cell death (290), and by NK cells (291, 292). A phase II trial of 15 patients with MF and unspecified peripheral T cell lymphoma explored the anti-tumor properties of bortezomib (28). Among the 10 patients with MF the RR was 70%, with one CR and six PRs. Responses were remarkably durable, with the CR in remission at the latest follow up at 12 months and with the PRs lasting at least seven months.
7.3.8. Retinoids
Several retinoids have been used in the treatment of CTCL, including etretinate, acitretin, all trans retinoic acid (ATRA), alitretinoin and isotretinoin (293). Retinoids exert their biologic effect via binding and activation of the retinoic acid receptor (RAR) isoforms RAR α, β, and γ, which form a heterodimer with each other or with retinoid X receptors (RXRs), or, in the case of isotretinoin, through indirect effects on the RAR signaling pathway (293). They have anti-proliferative, pro-apoptotic effects in CTCL, as well as immune modulating effects (294). Retinoids can induce IFN-γ partly via IL-12 (295), upregulate Langerhans cell antigen presentation capacity and coreceptors required for T cell activation (296), and increase NK cell activity (60).
7.3.9. Rexinoid
Bexarotene, a novel rexinoid, is a ligand for the RXR isoforms: RXR α, β, and γ. RXR can form heterodimers with the RAR, vitamin D receptor, thyroid hormone receptor, peroxisome proliferator activator receptor, liver X receptor, and the farnesoid X receptor to induce gene transcription. Bexarotene has anti-proliferative and pro-apoptotic effects on malignant cells (60). It has been shown to downregulate Th2 cytokines (297) and limit migration of malignant T cells via decreased expression of CCR4 on T cells and decrease of E-selectin on endothelial cells (298). Additionally, bexarotene in MF patients reduces CCL22 production by M2-polarized tumor associated macrophages (299). CCL22, similar to CCL17, can recruit Tregs (65). Bexarotene was also shown in leukemic cell lines to increase expression of IL-2R, which may be beneficial if combined with E7777, which targets IL-2R expressing cells (discussed above) (300). Bexarotene induced an objective RR of 45% with 13% of patients achieving a CR (27). Bexarotene has been used in combination with many other treatments, including PUVA, interferons, ECP, and others, and while this combination therapy appears safe it is unclear if there is appreciable benefit (60, 301).
8. Toll-like receptor agonists
The use of toll-like receptor (TLR) agonists for the treatment of MF is a promising new area of investigation. Toll-like receptors are expressed on various immune cells and sense a variety of foreign molecules, sequences, and peptides to trigger innate immune response by increasing the function of antigen presenting cells (APCs) as well as the expression of type I interferons, which promote a Th1 immune response (302, 303). Upon stimulation of dendritic cells, as well as NK cells, by agonists of TLR7, TLR8 or TLR9, there is an increase in IFN-α, IL-12 and IL-15 expression and upregulation of costimulatory markers for improved antigen presentation (302–305).
Several TLR agonists have been evaluated over the years, however only imiquimod, a TLR7 agonist, is available clinically. While several case reports have supported the use of imiquimod to treat MF, a randomized placebo-controlled trial of imiquimod failed to show that it was better than a placebo cream (24, 306–311). It is unclear if inherent differences in the host immune system or of the malignant T cells drives this variable clinical response. Most recently, resiquimod gel, a TLR7 and 8 agonist completed a randomized placebo controlled phase II trial, but the results are not yet published (302). In the phase I trial, a notable 92% of patients had a 50% or greater improvement in mSWAT (11). In addition, resiquimod induced regression of untreated lesions in select cases, suggesting an abscopal effect. Responders were found to have increased numbers of benign T cells with increased effector function and increased activation of circulating dendritic cells (11).
In the past, a phase I study of intratumoral injection of a TLR9-activating class B CPG motif oligodeoxynucleotide (ODN) (CPG-7907) in lesions of MF was investigated and a decrease in CD25+FOXP3+ T cells and an increase of plasmacytoid dendritic cells was observed (312), but further clinical development is not planned (19, 313). Interestingly, when CPG-A and CPG-B ODNs were evaluated ex vivo on PBMCs of patients with SS in comparison to healthy volunteers, SS PBMCs had dampened pro-inflammatory responses to ODNs, and CPG-A, but not CPG-B, was capable of stimulating NK cell function (313). This may in part explain the poor performance of CPG-7907, but also highlight the importance of combining TLR agonists with other immune activating agents or one with direct cytotoxic ability to overcome the immunosuppressive effects of malignant T cells. Recently, a phase I/II trial investigating the anti-tumor response of poly-ICLC, a TLR3 agonist, in combination with tremelimumab and durvalumab in patients with relapsed, advanced cancers, including cutaneous T cell lymphoma (NCT02643303) has been completed.
9. Discussion
Cancer therapies used for the treatment of CTCL, old and new, may affect the immune compartment, whether intended or not. Despite not being the main target, the therapeutic efficacy of certain agents may be aided by favorably remodeling the microenvironment, including boosting anti-tumor effector cells. Advantageous immune effects of therapies used for CTCL include inducing immunogenic cell death (PDT, chemotherapy, radiation), maturation of dendritic cells/antigen presenting cells (lenalidomide, ECP, interferons, and TLR agonists), reduction of immunosuppressive M2 macrophages (BV, duvelisib) or Tregs (mogamulizumab, alemtuzumab, BV), elimination of exhausted T cells (mogamulizumab, pembrolizumab, alemtuzumab), increasing levels of Th1 cytokines (interferon, TLR agonists), and recruitment of cytotoxic CD4+ or CD8+ T cells (pembrolizumab), among others. While several agents may cause myelosuppression or lymphopenia, not all T cell populations are affected equally. Such is the case for venetoclax, which decreases naïve T cells but spares effector T cells making it an attractive candidate to combine with therapies that depend on T- or NK- cell cytotoxicity, and mogamulizumab which preferentially depletes CCR4+ T cells, including Treg cells. We also eagerly await the results of several novel drugs under investigation, summarized in Table 5. While we are not certain what will be the most effective therapeutic regimen, the current trend appears to be combination therapy based on the effects of tumor cell and microenvironment. An ideal combination therapy would debulk disease via effective and immunogenic killing of highly immunosuppressive tumor cells without causing broad immunosuppression, coupled with activation and education of the immune system to generate a potent anti-tumor immune response for long-term disease remission.
Table 5 Therapies under investigation for the treatment of CTCL.
There is a growing body of literature on the immune effects of drugs, but much remains unknown. An inherent challenge in understanding the immune effects of therapies is that they may be solely driven by the selective killing of malignant T cells, which are highly immunosuppressive in CTCL. Regardless, with successful eradication of suppressive malignant T cells, immune restoration is observed across several therapies. And through better understanding of the immune modulating effects of our therapies, superior combination therapies may be developed, and prolonged remissions realized, such as that achieved by allogenic stem cell transplantation.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributions
CF prepared the initial draft of the manuscript and participated in revisions. KA created the tables and participated in revisions. NL participated in revision. CL participated in conception of the review, prepared the initial draft, created the figure, and participated in revisions. All authors contributed to the article and approved the submitted version.
Funding
Department of Dermatology at Brigham and Women’s Hospital funded the journal submission fee.
Acknowledgments
We thank Brigham and Women’s Hospital and Dana-Farber Cancer Institute for valuing and supporting cutaneous T cell lymphoma research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Korgavkar K, Xiong M, Weinstock M. Changing incidence trends of cutaneous T cell lymphoma. JAMA Dermatol (2013) 149(11):1295–9. doi: 10.1001/jamadermatol.2013.5526
2. Kim YH, Liu HL, Mraz-Gernhard S, Varghese A, Hoppe RT. Long-term outcome of 525 patients with mycosis fungoides and sezary syndrome: Clinical prognostic factors and risk for disease progression. Arch Dermatol (2003) 139(7):857–66. doi: 10.1001/archderm.139.7.857
3. Mirvish ED, Pomerantz RG, Geskin LJ. Infectious agents in cutaneous T cell lymphoma. J Am Acad Dermatol (2011) 64(2):423–31. doi: 10.1016/j.jaad.2009.11.692
4. Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet (2015) 47(9):1011–9. doi: 10.1038/ng.3356
5. Abraham RM, Zhang Q, Odum N, Wasik MA. The role of cytokine signaling in the pathogenesis of cutaneous T cell lymphoma. Cancer Biol Ther (2011) 12(12):1019–22. doi: 10.4161/cbt.12.12.18144
6. Krejsgaard T, Lindahl LM, Mongan NP, Wasik MA, Litvinov IV, Iversen L, et al. Malignant inflammation in cutaneous T cell lymphoma-a hostile takeover. Semin Immunopathol (2017) 39(3):269–82. doi: 10.1007/s00281-016-0594-9
7. Gluud M, Pallesen EMH, Buus TB, Gjerdrum LMR, Lindahl LM, Kamstrup MR, et al. Malignant T cells induce skin barrier defects through cytokine-mediated Jak/Stat signalling in cutaneous T cell lymphoma. Blood (2022). doi: 10.1182/blood.2022016690
8. Fujii K. Pathogenesis of cutaneous T cell lymphoma: Involvement of staphylococcus aureus. J Dermatol (2022) 49(2):202–9. doi: 10.1111/1346-8138.16288
9. Lindahl LM, Willerslev-Olsen A, Gjerdrum LMR, Nielsen PR, Blumel E, Rittig AH, et al. Antibiotics inhibit tumor and disease activity in cutaneous T cell lymphoma. Blood (2019) 134(13):1072–83. doi: 10.1182/blood.2018888107
10. Guenova E, Watanabe R, Teague JE, Desimone JA, Jiang Y, Dowlatshahi M, et al. Th2 cytokines from malignant cells suppress Th1 responses and enforce a global Th2 bias in leukemic cutaneous T cell lymphoma. Clin Cancer Res (2013) 19(14):3755–63. doi: 10.1158/1078-0432.CCR-12-3488
11. Rook AH, Gelfand JM, Wysocka M, Troxel AB, Benoit B, Surber C, et al. Topical resiquimod can induce disease regression and enhance T cell effector functions in cutaneous T cell lymphoma. Blood (2015) 126(12):1452–61. doi: 10.1182/blood-2015-02-630335
12. Rook AH, Kubin M, Cassin M, Vonderheid EC, Vowels BR, Wolfe JT, et al. Il-12 reverses cytokine and immune abnormalities in sezary syndrome. J Immunol (1995) 154(3):1491–8.
13. Shalabi D, Bistline A, Alpdogan O, Kartan S, Mishra A, Porcu P, et al. Immune evasion and current immunotherapy strategies in mycosis fungoides (Mf) and sezary syndrome (Ss). Chin Clin Oncol (2019) 8(1):11. doi: 10.21037/cco.2019.01.01
14. Garcia-Diaz N, Piris MA, Ortiz-Romero PL, Vaque JP. Mycosis fungoides and sezary syndrome: An integrative review of the pathophysiology, molecular drivers, and targeted therapy. Cancers (Basel) (2021) 13(8):1931. doi: 10.3390/cancers13081931
15. Olsen EA, Rosen ST, Vollmer RT, Variakojis D, Roenigk HH Jr., Diab N, et al. Interferon Alfa-2a in the treatment of cutaneous T cell lymphoma. J Am Acad Dermatol (1989) 20(3):395–407. doi: 10.1016/s0190-9622(89)70049-9
16. Kaplan EH, Rosen ST, Norris DB, Roenigk HH Jr., Saks SR, Bunn PA Jr. Phase ii study of recombinant human interferon gamma for treatment of cutaneous T cell lymphoma. J Natl Cancer Inst (1990) 82(3):208–12. doi: 10.1093/jnci/82.3.208
17. Querfeld C, Rosen ST, Guitart J, Rademaker A, Foss F, Gupta R, et al. Phase ii trial of subcutaneous injections of human recombinant interleukin-2 for the treatment of mycosis fungoides and sezary syndrome. J Am Acad Dermatol (2007) 56(4):580–3. doi: 10.1016/j.jaad.2006.08.067
18. Wu PA, Kim YH, Lavori PW, Hoppe RT, Stockerl-Goldstein KE. A meta-analysis of patients receiving allogeneic or autologous hematopoietic stem cell transplant in mycosis fungoides and sezary syndrome. Biol Blood Marrow Transplant (2009) 15(8):982–90. doi: 10.1016/j.bbmt.2009.04.017
19. Kim YH, Girardi M, Duvic M, Kuzel T, Link BK, Pinter-Brown L, et al. Phase I trial of a toll-like receptor 9 agonist, pf-3512676 (Cpg 7909), in patients with treatment-refractory, cutaneous T cell lymphoma. J Am Acad Dermatol (2010) 63(6):975–83. doi: 10.1016/j.jaad.2009.12.052
20. Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase ib study. J Clin Oncol (2016) 34(23):2698–704. doi: 10.1200/JCO.2015.65.9789
21. Querfeld C, Zain J, Jovanovic-Talisman T, Wakefield DL, Kil S, Estephan R, et al. Phase 1/2 trial of anti-Pd-Ligand 1 (Durvalumab) +/- lenalidomide in patients with cutaneous T cell lymphoma: Preliminary results of phase 1 and correlative studies. Hematological Oncol (2019) 37(S2):519–20. doi: 10.1002/hon.205_2631
22. Querfeld C, Thompson J, Taylor M, Pillai R, Johnson LDS, Catalano T, et al. A single direct intratumoral injection of tti-621 (Sirpαfc) induces antitumor activity in patients with Relapsed/Refractory mycosis fungoides and sézary syndrome: Preliminary findings employing an immune checkpoint inhibitor blocking the Cd47 “Do not eat” signal. Blood (2017) 130:4076–. doi: 10.1182/blood.V130.Suppl_1.4076.4076
23. Duvic M, Sherman ML, Wood GS, Kuzel TM, Olsen E, Foss F, et al. A phase ii open-label study of recombinant human interleukin-12 in patients with stage ia, ib, or iia mycosis fungoides. J Am Acad Dermatol (2006) 55(5):807–13. doi: 10.1016/j.jaad.2006.06.038
24. Chong A, Loo WJ, Banney L, Grant JW, Norris PG. Imiquimod 5% cream in the treatment of mycosis fungoides–a pilot study. J Dermatolog Treat (2004) 15(2):118–9. doi: 10.1080/09546630310019373
25. Gao C, McCormack C, van der Weyden C, Goh MS, Campbell BA, Twigger R, et al. Prolonged survival with the early use of a novel extracorporeal photopheresis regimen in patients with sezary syndrome. Blood (2019) 134(16):1346–50. doi: 10.1182/blood.2019000765
26. Kim EJ, Mangold AR, DeSimone JA, Wong HK, Seminario-Vidal L, Guitart J, et al. Efficacy and safety of topical hypericin photodynamic therapy for early-stage cutaneous T cell lymphoma (Mycosis fungoides): The flash phase 3 randomized clinical trial. JAMA Dermatol (2022) 158(9):1031–9. doi: 10.1001/jamadermatol.2022.2749
27. Duvic M, Hymes K, Heald P, Breneman D, Martin AG, Myskowski P, et al. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T cell lymphoma: Multinational phase ii-iii trial results. J Clin Oncol (2001) 19(9):2456–71. doi: 10.1200/JCO.2001.19.9.2456
28. Zinzani PL, Musuraca G, Tani M, Stefoni V, Marchi E, Fina M, et al. Phase ii trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T cell lymphoma. J Clin Oncol (2007) 25(27):4293–7. doi: 10.1200/JCO.2007.11.4207
29. Wilson LD, Kacinski BM, Jones GW. Local superficial radiotherapy in the management of minimal stage ia cutaneous T cell lymphoma (Mycosis fungoides). Int J Radiat Oncol Biol Phys (1998) 40(1):109–15. doi: 10.1016/s0360-3016(97)00553-1
30. Thomas TO, Agrawal P, Guitart J, Rosen ST, Rademaker AW, Querfeld C, et al. Outcome of patients treated with a single-fraction dose of palliative radiation for cutaneous T cell lymphoma. Int J Radiat Oncol Biol Phys (2013) 85(3):747–53. doi: 10.1016/j.ijrobp.2012.05.034
31. Hoppe RT, Harrison C, Tavallaee M, Bashey S, Sundram U, Li S, et al. Low-dose total skin electron beam therapy as an effective modality to reduce disease burden in patients with mycosis fungoides: Results of a pooled analysis from 3 phase-ii clinical trials. J Am Acad Dermatol (2015) 72(2):286–92. doi: 10.1016/j.jaad.2014.10.014
32. Navi D, Riaz N, Levin YS, Sullivan NC, Kim YH, Hoppe RT. The Stanford university experience with conventional-dose, total skin electron-beam therapy in the treatment of generalized patch or plaque (T2) and tumor (T3) mycosis fungoides. Arch Dermatol (2011) 147(5):561–7. doi: 10.1001/archdermatol.2011.98
33. Goddard AL, Vleugels RA, LeBoeuf NR, O'Farrell DA, Cormack RA, Hansen JL, et al. Palliative therapy for recalcitrant cutaneous T cell lymphoma of the hands and feet with low-dose, high dose-rate brachytherapy. JAMA Dermatol (2015) 151(12):1354–7. doi: 10.1001/jamadermatol.2015.3028
34. Gathers RC, Scherschun L, Malick F, Fivenson DP, Lim HW. Narrowband uvb phototherapy for early-stage mycosis fungoides. J Am Acad Dermatol (2002) 47(2):191–7. doi: 10.1067/mjd.2002.120911
35. Ponte P, Serrao V, Apetato M. Efficacy of narrowband uvb vs. puva in patients with early-stage mycosis fungoides. J Eur Acad Dermatol Venereol (2010) 24(6):716–21. doi: 10.1111/j.1468-3083.2009.03500.x
36. Talpur R, Demierre MF, Geskin L, Baron E, Pugliese S, Eubank K, et al. Multicenter photopheresis intervention trial in early-stage mycosis fungoides. Clin Lymphoma Myeloma Leuk (2011) 11(2):219–27. doi: 10.1016/j.clml.2011.03.003
37. Horwitz SM, Feldman TA, Hess BT, Khodadoust MS, Kim YH, Munoz J, et al. The novel Syk/Jak inhibitor cerdulatinib demonstrates good tolerability and clinical response in a phase 2a study in Relapsed/Refractory peripheral T cell lymphoma and cutaneous T cell lymphoma. Blood (2018) 132(Supplement 1):1001–. doi: 10.1182/blood-2018-99-119944
38. Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, et al. Activity of the Pi3k-Delta,Gamma inhibitor duvelisib in a phase 1 trial and preclinical models of T cell lymphoma. Blood (2018) 131(8):888–98. doi: 10.1182/blood-2017-08-802470
39. Querfeld C, Rosen ST, Guitart J, Duvic M, Kim YH, Dusza SW, et al. Results of an open-label multicenter phase 2 trial of lenalidomide monotherapy in refractory mycosis fungoides and sezary syndrome. Blood (2014) 123(8):1159–66. doi: 10.1182/blood-2013-09-525915
40. Duvic M, Dummer R, Becker JC, Poulalhon N, Ortiz Romero P, Grazia Bernengo M, et al. Panobinostat activity in both bexarotene-exposed and -naive patients with refractory cutaneous T cell lymphoma: Results of a phase ii trial. Eur J Cancer (2013) 49(2):386–94. doi: 10.1016/j.ejca.2012.08.017
41. Whittaker SJ, Demierre MF, Kim EJ, Rook AH, Lerner A, Duvic M, et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T cell lymphoma. J Clin Oncol (2010) 28(29):4485–91. doi: 10.1200/JCO.2010.28.9066
42. Moskowitz AJ, Ghione P, Jacobsen E, Ruan J, Schatz JH, Noor S, et al. A phase 2 biomarker-driven study of ruxolitinib demonstrates effectiveness of Jak/Stat targeting in T cell lymphomas. Blood (2021) 138(26):2828–37. doi: 10.1182/blood.2021013379
43. Huen A, Haverkos BM, Zain J, Radhakrishnan R, Lechowicz MJ, Devata S, et al. Phase I/Ib study of tenalisib (Rp6530), a dual Pi3k Delta/Gamma inhibitor in patients with Relapsed/Refractory T cell lymphoma. Cancers (Basel) (2020) 12(8):2293. doi: 10.3390/cancers12082293
44. Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, et al. Phase iib multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T cell lymphoma. J Clin Oncol (2007) 25(21):3109–15. doi: 10.1200/JCO.2006.10.2434
45. Querfeld C, William BM, Sokol L, Akilov O, Poligone B, Zain J, et al. Co-Inhibition of il-2, il-9 and il-15 by the novel immunomodulator, bnz-1, provides clinical efficacy in patients with refractory cutaneous T cell lymphoma in a phase 1/2 clinical trial. Blood (2020) 136(Supplement 1):37. doi: 10.1182/blood-2020-143135
46. Kawai H, Ando K, Maruyama D, Yamamoto K, Kiyohara E, Terui Y, et al. Phase ii study of E7777 in Japanese patients with Relapsed/Refractory peripheral and cutaneous T cell lymphoma. Cancer Sci (2021) 112(6):2426–35. doi: 10.1111/cas.14906
47. Lundin J, Hagberg H, Repp R, Cavallin-Stahl E, Freden S, Juliusson G, et al. Phase 2 study of alemtuzumab (Anti-Cd52 monoclonal antibody) in patients with advanced mycosis Fungoides/Sezary syndrome. Blood (2003) 101(11):4267–72. doi: 10.1182/blood-2002-09-2802
48. Prince HM, Kim YH, Horwitz SM, Dummer R, Scarisbrick J, Quaglino P, et al. Brentuximab vedotin or physician's choice in Cd30-positive cutaneous T cell lymphoma (Alcanza): An international, open-label, randomised, phase 3, multicentre trial. Lancet (2017) 390(10094):555–66. doi: 10.1016/S0140-6736(17)31266-7
49. Bagot M, Porcu P, Marie-Cardine A, Battistella M, William BM, Vermeer M, et al. Iph4102, a first-in-Class anti-Kir3dl2 monoclonal antibody, in patients with relapsed or refractory cutaneous T cell lymphoma: An international, first-in-Human, open-label, phase 1 trial. Lancet Oncol (2019) 20(8):1160–70. doi: 10.1016/S1470-2045(19)30320-1
50. Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T cell lymphoma (Mavoric): An international, open-label, randomised, controlled phase 3 trial. Lancet Oncol (2018) 19(9):1192–204. doi: 10.1016/s1470-2045(18)30379-6
51. Coors EA, von den Driesch P. Treatment of erythrodermic cutaneous T cell lymphoma with intermittent chlorambucil and fluocortolone therapy. Br J Dermatol (2000) 143(1):127–31. doi: 10.1046/j.1365-2133.2000.03601.x
52. Quereux G, Marques S, Nguyen JM, Bedane C, D'Incan M, Dereure O, et al. Prospective multicenter study of pegylated liposomal doxorubicin treatment in patients with advanced or refractory mycosis fungoides or sezary syndrome. Arch Dermatol (2008) 144(6):727–33. doi: 10.1001/archderm.144.6.727
53. Lessin SR, Duvic M, Guitart J, Pandya AG, Strober BE, Olsen EA, et al. Topical chemotherapy in cutaneous T cell lymphoma: Positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0. 02% Gel Mycosis Fungoides. JAMA Dermatol (2013) 149(1):25–32. doi: 10.1001/2013.jamadermatol.541
54. Zackheim HS, Kashani-Sabet M, Hwang ST. Low-dose methotrexate to treat erythrodermic cutaneous T cell lymphoma: Results in twenty-nine patients. J Am Acad Dermatol (1996) 34(4):626–31. doi: 10.1016/s0190-9622(96)80062-4
55. Horwitz SM, Kim YH, Foss F, Zain JM, Myskowski PL, Lechowicz MJ, et al. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T cell lymphoma. Blood (2012) 119(18):4115–22. doi: 10.1182/blood-2011-11-390211
56. Olsen EA, Whittaker S, Kim YH, Duvic M, Prince HM, Lessin SR, et al. Clinical end points and response criteria in mycosis fungoides and sezary syndrome: A consensus statement of the international society for cutaneous lymphomas, the united states cutaneous lymphoma consortium, and the cutaneous lymphoma task force of the European organisation for research and treatment of cancer. J Clin Oncol (2011) 29(18):2598–607. doi: 10.1200/JCO.2010.32.0630
57. Stevens SR, Ke MS, Parry EJ, Mark J, Cooper KD. Quantifying skin disease burden in mycosis fungoides-type cutaneous T cell lymphomas: The severity-weighted assessment tool (Swat). Arch Dermatol (2002) 138(1):42–8. doi: 10.1001/archderm.138.1.42
58. Goldstein D, Laszlo J. The role of interferon in cancer therapy: A current perspective. CA Cancer J Clin (1988) 38(5):258–77. doi: 10.3322/canjclin.38.5.258
59. Brassard DL, Grace MJ, Bordens RW. Interferon-alpha as an immunotherapeutic protein. J leukocyte Biol (2002) 71:565–81.
60. Olsen EA, Rook AH, Zic J, Kim Y, Porcu P, Querfeld C, et al. Sezary syndrome: Immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the united states cutaneous lymphoma consortium (Usclc). J Am Acad Dermatol (2011) 64(2):352–404. doi: 10.1016/j.jaad.2010.08.037
61. Shuai K, Liu B. Regulation of jak-stat signalling in the immune system. Nat Rev Immunol (2003) 3(11):900–11. doi: 10.1038/nri1226
62. Park J, Yang J, Wenzel AT, Ramachandran A, Lee WJ, Daniels JC, et al. Genomic analysis of 220 ctcls identifies a novel recurrent gain-of-Function alteration in rltpr (P. Q575e). Blood (2017) 130(12):1430–40. doi: 10.1182/blood-2017-02-768234
63. Yoo EK, Cassin M, Lessin SR, Rook AH. Complete molecular remission during biologic response modifier therapy for sezary syndrome is associated with enhanced helper T type 1 cytokine production and natural killer cell activity. J Am Acad Dermatol (2001) 45(2):208–16. doi: 10.1067/mjd.2001.116345
64. Furudate S, Fujimura T, Kakizaki A, Hidaka T, Asano M, Aiba S. Tumor-associated M2 macrophages in mycosis fungoides acquire immunomodulatory function by interferon alpha and interferon gamma. J Dermatol Sci (2016) 83(3):182–9. doi: 10.1016/j.jdermsci.2016.05.004
65. Yoshie O, Matsushima K. Ccr4 and its ligands: From bench to bedside. Int Immunol (2015) 27(1):11–20. doi: 10.1093/intimm/dxu079
66. Wilcox RA. Mogamulizumab: 2 birds, 1 stone. Blood (2015) 125(12):1847–8. doi: 10.1182/blood-2015-02-625251
67. Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, et al. Cxcl9, Cxcl10, Cxcl11/Cxcr3 axis for immune activation - a target for novel cancer therapy. Cancer Treat Rev (2018) 63:40–7. doi: 10.1016/j.ctrv.2017.11.007
68. Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and sezary syndrome. Blood (2009) 114:4337–53. doi: 10.1182/blood-2009-07-202895
69. Suchin KR, Cassin M, Gottleib SL, Sood S, Cucchiara AJ, Vonderheid EC, et al. Increased interleukin 5 production in eosinophilic sezary syndrome: Regulation by interferon Alfa and interleukin 12. J Am Acad Dermatol (2001) 44(1):28–32. doi: 10.1067/mjd.2001.109853
70. Hughes CF, Khot A, McCormack C, Lade S, Westerman DA, Twigger R, et al. Lack of durable disease control with chemotherapy for mycosis fungoides and sezary syndrome: A comparative study of systemic therapy. Blood (2015) 125(1):71–81. doi: 10.1182/blood-2014-07-588236
71. Sun WH, Pabon C, Alsayed Y, Huang PP, Jandeska S, Uddin S, et al. Interferon-alpha resistance in a cutaneous T cell lymphoma cell line is associated with lack of Stat1 expression. Blood (1998) 91:570–6.
72. Tracey L, Spiteri I, Ortiz P, Lawler M, Piris MA, Villuendas R. Transcriptional response of T cells to ifn-alpha: Changes induced in ifn-Alpha-Sensitive and resistant cutaneous T cell lymphoma. J Interferon Cytokine Res (2004) 24(3):185–95. doi: 10.1089/107999004322917034
73. Tracey L, Villuendas R, Ortiz P, Dopazo A, Spiteri I, Lombardia L, et al. Identification of genes involved in resistance to interferon-alpha in cutaneous T cell lymphoma. Am J Pathol (2002) 161(5):1825–37. doi: 10.1016/s0002-9440(10)64459-8
74. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of ifn-gamma in tumor progression and regression: A review. biomark Res (2020) 8(1):49. doi: 10.1186/s40364-020-00228-x
75. Rosenberg SA. Il-2: The first effective immunotherapy for human cancer. J Immunol (2014) 192(12):5451–8. doi: 10.4049/jimmunol.1490019
76. Wasik MA, Vonderheid EC, Bigler RD, Marti R, Lessin SR, Polansky M, et al. Increased serum concentration of the soluble interleukin-2 receptor in cutaneous T cell lymphoma. Clin Prognostic Implications. Arch Dermatol (1996) 132(1):42–7. doi: 10.1001/archderm.1996.03890250052009
77. Dummer R, Posseckert G, Nestle F, Witzgall R, Burger M, Becker JC, et al. Soluble interleukin-2 receptors inhibit interleukin 2-dependent proliferation and cytotoxicity: Explanation for diminished natural killer cell activity in cutaneous T cell lymphomas in vivo? J Invest Dermatol (1992) 98(1):50–4. doi: 10.1111/1523-1747.ep12494223
78. Rook AH, Zaki MH, Wysocka M, Wood GS, Duvic M, Showe LC, et al. The role for interleukin-12 therapy of cutaneous T cell lymphoma. Ann N Y Acad Sci (2001) 941:177–84. doi: 10.1111/j.1749-6632.2001.tb03721.x
79. Zaki MH, Wysocka M, Everetts SE, Wang KS, French LE, Ritz J, et al. Synergistic enhancement of cell-mediated immunity by interleukin-12 plus interleukin-2: Basis for therapy of cutaneous T cell lymphoma. J Invest Dermatol (2002) 118(2):366–71. doi: 10.1046/j.1523-1747.2002.01646.x
80. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-Associated toxicity and interferon-gamma production. Blood (1997) 90(7):2541–8.
81. Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discovery (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548
82. To V, Evtimov VJ, Jenkin G, Pupovac A, Trounson AO, Boyd RL. Car-T cell development for cutaneous T cell lymphoma: Current limitations and potential treatment strategies. Front Immunol (2022) 13:968395. doi: 10.3389/fimmu.2022.968395
83. Cornel AM, Mimpen IL, Nierkens S. Mhc class I downregulation in cancer: Underlying mechanisms and potential targets for cancer immunotherapy. Cancers (Basel) (2020) 12(7):1760. doi: 10.3390/cancers12071760
84. Chmielewski M, Abken H. Trucks: The fourth generation of cars. Expert Opin Biol Ther (2015) 15(8):1145–54. doi: 10.1517/14712598.2015.1046430
85. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a jak-stat signaling domain mediates superior antitumor effects. Nat Med (2018) 24(3):352–9. doi: 10.1038/nm.4478
86. Alcantara M, Tesio M, June CH, Houot R. Car T cells for T cell malignancies: Challenges in distinguishing between therapeutic, normal, and neoplastic T cells. Leukemia (2018) 32(11):2307–15. doi: 10.1038/s41375-018-0285-8
87. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic b cell. Nat Med (2018) 24(10):1499–503. doi: 10.1038/s41591-018-0201-9
88. Zeng E, Zhan T, Wu Y, Chen M, Wang J, Zhang Y, et al. Preclinical analysis of an autologous Cd4-targeted chimeric antigen receptor T cell (Car-T) immunotherapy for relapsed or refractory peripheral T cell lymphoma (Ptcl) or cutaneous T cell lymphoma (Ctcl). J Clin Oncol (2021) 39(15_suppl):e14510–e. doi: 10.1200/JCO.2021.39.15_suppl.e14510
89. Molina A, Nademanee A, Arber DA, Forman SJ. Remission of refractory sezary syndrome after bone marrow transplantation from a matched unrelated donor. Biol Blood Marrow Transplant (1999) 5(6):400–4. doi: 10.1016/s1083-8791(99)70017-0
90. Schlaak M, Pickenhain J, Theurich S, Skoetz N, von Bergwelt-Baildon M, Kurschat P. Allogeneic stem cell transplantation versus conventional therapy for advanced primary cutaneous T cell lymphoma. Cochrane Database Syst Rev (2013). doi: 10.1002/14651858.CD008908.pub3
91. Duarte RF, Canals C, Onida F, Gabriel IH, Arranz R, Arcese W, et al. Allogeneic hematopoietic cell transplantation for patients with mycosis fungoides and sezary syndrome: A retrospective analysis of the lymphoma working party of the European group for blood and marrow transplantation. J Clin Oncol (2010) 28(29):4492–9. doi: 10.1200/JCO.2010.29.3241
92. Duvic M, Donato M, Dabaja B, Richmond H, Singh L, Wei W, et al. Total skin electron beam and non-myeloablative allogeneic hematopoietic stem-cell transplantation in advanced mycosis fungoides and sezary syndrome. J Clin Oncol (2010) 28(14):2365–72. doi: 10.1200/JCO.2009.25.8301
93. Introcaso CE, Leber B, Greene K, Ubriani R, Rook AH, Kim EJ. Stem cell transplantation in advanced cutaneous T cell lymphoma. J Am Acad Dermatol (2008) 58(4):645–9. doi: 10.1016/j.jaad.2007.12.021
94. Molina A, Zain J, Arber DA, Angelopolou M, O'Donnell M, Murata-Collins J, et al. Durable clinical, cytogenetic, and molecular remissions after allogeneic hematopoietic cell transplantation for refractory sezary syndrome and mycosis fungoides. J Clin Oncol (2005) 23(25):6163–71. doi: 10.1200/JCO.2005.02.774
95. Duarte RF, Schmitz N, Servitje O, Sureda A. Haematopoietic stem cell transplantation for patients with primary cutaneous T cell lymphoma. Bone Marrow Transplant (2008) 41(7):597–604. doi: 10.1038/sj.bmt.1705968
96. Schlaak M, Theurich S, Pickenhain J, Skoetz N, Kurschat P, von Bergwelt-Baildon M. Allogeneic stem cell transplantation for advanced primary cutaneous T cell lymphoma: A systematic review. Crit Rev Oncol Hematol (2013) 85(1):21–31. doi: 10.1016/j.critrevonc.2012.06.002
97. Haserick JR, Richardson JH, Grant DJ. Remission of lesions in mycosis fungoides following topical application of nitrogen mustard. Cleve Clin Q (1983) 50(2):91–5.
98. Highley MS, Landuyt B, Prenen H, Harper PG, De Bruijn EA. The nitrogen mustards. Pharmacol Rev (2022) 74(3):552–99. doi: 10.1124/pharmrev.120.000121
99. Jeelani R, Khan SN, Shaeib F, Kohan-Ghadr HR, Aldhaheri SR, Najafi T, et al. Cyclophosphamide and acrolein induced oxidative stress leading to deterioration of metaphase ii mouse oocyte quality. Free Radic Biol Med (2017) 110:11–8. doi: 10.1016/j.freeradbiomed.2017.05.006
100. Chang YT, Ignatova D, Hoetzenecker W, Pascolo S, Fassnacht C, Guenova E. Increased chlormethine-induced DNA double-stranded breaks in malignant T cells from mycosis fungoides skin lesions. JID Innov (2022) 2(1):100069. doi: 10.1016/j.xjidi.2021.100069
101. Vonderheid EC, Tan ET, Kantor AF, Shrager L, Micaily B, Van Scott EJ. Long-term efficacy, curative potential, and carcinogenicity of topical mechlorethamine chemotherapy in cutaneous T cell lymphoma. J Am Acad Dermatol (1989) 20(3):416–28. doi: 10.1016/s0190-9622(89)70051-7
102. Liner K, Brown C, McGirt LY. Clinical potential of mechlorethamine gel for the topical treatment of mycosis fungoides-type cutaneous T cell lymphoma: A review on current efficacy and safety data. Drug Des Devel Ther (2018) 12:241–54. doi: 10.2147/DDDT.S137106
103. Kim YH. Management with topical nitrogen mustard in mycosis fungoides. Dermatol Ther (2003) 16(4):288–98. doi: 10.1111/j.1396-0296.2003.01640.x
104. Vonderheid EC, Ekbote SK, Kerrigan K, Kalmanson JD, Van Scott EJ, Rook AH, et al. The prognostic significance of delayed hypersensitivity to dinitrochlorobenzene and mechlorethamine hydrochloride in cutaneous T cell lymphoma. J Invest Dermatol (1998) 110(6):946–50. doi: 10.1046/j.1523-1747.1998.00206.x
105. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med (2005) 202(12):1691–701. doi: 10.1084/jem.20050915
106. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi: 10.1038/nm1523
107. Tesniere A, Apetoh L, Ghiringhelli F, Joza N, Panaretakis T, Kepp O, et al. Immunogenic cancer cell death: A key-lock paradigm. Curr Opin Immunol (2008) 20(5):504–11. doi: 10.1016/j.coi.2008.05.007
108. Wollina U, Dummer R, Brockmeyer NH, Konrad H, Busch JO, Kaatz M, et al. Multicenter study of pegylated liposomal doxorubicin in patients with cutaneous T cell lymphoma. Cancer (2003) 98(5):993–1001. doi: 10.1002/cncr.11593
109. Hayashi K, Nikolos F, Lee YC, Jain A, Tsouko E, Gao H, et al. Tipping the immunostimulatory and inhibitory damp balance to harness immunogenic cell death. Nat Commun (2020) 11(1):6299. doi: 10.1038/s41467-020-19970-9
110. Chan ES, Cronstein BN. Methotrexate–how does it really work? Nat Rev Rheumatol (2010) 6(3):175–8. doi: 10.1038/nrrheum.2010.5
111. Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P, et al. Targeting adenosine in cancer immunotherapy to enhance T cell function. Front Immunol (2019) 10:925. doi: 10.3389/fimmu.2019.00925
112. Thomas S, Fisher KH, Snowden JA, Danson SJ, Brown S, Zeidler MP. Methotrexate is a Jak/Stat pathway inhibitor. PloS One (2015) 10(7):e0130078. doi: 10.1371/journal.pone.0130078
113. Thomas S, Fisher K, Snowden J, Danson S, Brown S, Zeidler M. Effect of methotrexate on Jak/Stat pathway activation in myeloproliferative neoplasms. Lancet (2015) 385 Suppl 1(Suppl 1):S98. doi: 10.1016/S0140-6736(15)60413-5
114. Visentin M, Unal ES, Zhao R, Goldman ID. The membrane transport and polyglutamation of pralatrexate: A new-generation dihydrofolate reductase inhibitor. Cancer Chemother Pharmacol (2013) 72(3):597–606. doi: 10.1007/s00280-013-2231-9
115. Wang Y, Wu L, Tian C, Zhang Y. Pd-1-Pd-L1 immune-checkpoint blockade in malignant lymphomas. Ann Hematol (2018) 97(2):229–37. doi: 10.1007/s00277-017-3176-6
116. Jiang Y, Chen M, Nie H, Yuan Y. Pd-1 and pd-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum Vaccin Immunother (2019) 15(5):1111–22. doi: 10.1080/21645515.2019.1571892
117. Ward SG. Cd28: A signalling perspective. Biochem J (1996) 318(Pt 2):361–77. doi: 10.1042/bj3180361
118. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific Cd8 T cells infiltrating the tumor express high levels of pd-1 and are functionally impaired. Blood (2009) 114(8):1537–44. doi: 10.1182/blood-2008-12-195792
119. Kantekure K, Yang Y, Raghunath P, Schaffer A, Woetmann A, Zhang Q, et al. Expression patterns of the immunosuppressive proteins pd-1/Cd279 and pd-L1/Cd274 at different stages of cutaneous T cell Lymphoma/Mycosis fungoides. Am J Dermatopathol (2012) 34(1):126–8. doi: 10.1097/DAD.0b013e31821c35cb
120. Khodadoust MS, Rook AH, Porcu P, Foss F, Moskowitz AJ, Shustov A, et al. Pembrolizumab in relapsed and refractory mycosis fungoides and sezary syndrome: A multicenter phase ii study. J Clin Oncol (2020) 38(1):20–8. doi: 10.1200/JCO.19.01056
121. Phillips D, Matusiak M, Gutierrez BR, Bhate SS, Barlow GL, Jiang S, et al. Immune cell topography predicts response to pd-1 blockade in cutaneous T cell lymphoma. Nat Commun (2021) 12(1):6726. doi: 10.1038/s41467-021-26974-6
122. Su T, Duran GE, Kwang AC, Ramchurren N, Fling SP, Kim YH, et al. Single-cell rna-sequencing reveals predictive features of response to pembrolizumab in sezary syndrome. Oncoimmunology (2022) 11(1):2115197. doi: 10.1080/2162402X.2022.2115197
123. Beygi S, Fernandez-Pol S, Duran G, Wang EB, Stehr H, Zehnder JL, et al. Pembrolizumab in mycosis fungoides with pd-L1 structural variants. Blood Adv (2021) 5(3):771–4. doi: 10.1182/bloodadvances.2020002371
124. Ungewickell A, Bhaduri A, Rios E, Reuter J, Lee CS, Mah A, et al. Genomic analysis of mycosis fungoides and sezary syndrome identifies recurrent alterations in Tnfr2. Nat Genet (2015) 47(9):1056–60. doi: 10.1038/ng.3370
125. Kataoka K, Miyoshi H, Sakata S, Dobashi A, Couronne L, Kogure Y, et al. Frequent structural variations involving programmed death ligands in Epstein-Barr virus-associated lymphomas. Leukemia (2019) 33(7):1687–99. doi: 10.1038/s41375-019-0380-5
126. Kataoka K, Shiraishi Y, Takeda Y, Sakata S, Matsumoto M, Nagano S, et al. Aberrant pd-L1 expression through 3'-utr disruption in multiple cancers. Nature (2016) 534(7607):402–6. doi: 10.1038/nature18294
127. Wartewig T, Kurgyis Z, Keppler S, Pechloff K, Hameister E, Ollinger R, et al. Pd-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature (2017) 552(7683):121–5. doi: 10.1038/nature24649
128. Bennani NN, Kim HJ, Pederson LD, Atherton PJ, Micallef IN, Thanarajasingam G, et al. Nivolumab in patients with relapsed or refractory peripheral T cell lymphoma: Modest activity and cases of hyperprogression. J Immunother Cancer (2022) 10(6):e004984. doi: 10.1136/jitc-2022-004984
129. Ratner L, Waldmann TA, Janakiram M, Brammer JE. Rapid progression of adult T cell leukemia-lymphoma after pd-1 inhibitor therapy. N Engl J Med (2018) 378(20):1947–8. doi: 10.1056/NEJMc1803181
130. Saulite I, Ignatova D, Chang YT, Fassnacht C, Dimitriou F, Varypataki E, et al. Blockade of programmed cell death protein 1 (Pd-1) in sezary syndrome reduces Th2 phenotype of non-tumoral T lymphocytes but may enhance tumor proliferation. Oncoimmunology (2020) 9(1):1738797. doi: 10.1080/2162402X.2020.1738797
131. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting Pi3kgamma in myeloid cells. Nature (2016) 539(7629):443–7. doi: 10.1038/nature20554
132. Grogan B, James R, Ulrich M, Gardai S, Heiser R. 696 brentuximab vedotin, a Cd30-directed antibody-drug conjugate, selectively depletes activated tregs in vitro and in vivo. J Immunother Cancer (2020) 8(Suppl 3):A418–A9. doi: 10.1136/jitc-2020-SITC2020.0696
133. Buchbinder EI, Desai A. Ctla-4 and pd-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
134. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
135. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. Ctla-4 control over Foxp3+ regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
136. Rowshanravan B, Halliday N, Sansom DM. Ctla-4: A moving target in immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
137. Hong MMY, Maleki Vareki S. Addressing the elephant in the immunotherapy room: Effector T cell priming versus depletion of regulatory T cells by anti-Ctla-4 therapy. Cancers (Basel) (2022) 14(6):1580. doi: 10.3390/cancers14061580
138. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-Ctla-4 antibodies of Igg2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res (2013) 1(1):32–42. doi: 10.1158/2326-6066.CIR-13-0013
139. Bar-Sela G, Bergman R. Complete regression of mycosis fungoides after ipilimumab therapy for advanced melanoma. JAAD Case Rep (2015) 1(2):99–100. doi: 10.1016/j.jdcr.2015.02.009
140. Sekulic A, Liang WS, Tembe W, Izatt T, Kruglyak S, Kiefer JA, et al. Personalized treatment of sezary syndrome by targeting a novel Ctla4:Cd28 fusion. Mol Genet Genomic Med (2015) 3(2):130–6. doi: 10.1002/mgg3.121
141. Oronsky B, Carter C, Reid T, Brinkhaus F, Knox SJ. Just eat it: A review of Cd47 and sirp-alpha antagonism. Semin Oncol (2020) 47(2-3):117–24. doi: 10.1053/j.seminoncol.2020.05.009
142. Huang Y, Ma Y, Gao P, Yao Z. Targeting Cd47: The achievements and concerns of current studies on cancer immunotherapy. J Thorac Dis (2017) 9(2):E168–E74. doi: 10.21037/jtd.2017.02.30
143. Querfeld C, Thompson JA, Taylor MH, DeSimone JA, Zain JM, Shustov AR, et al. Intralesional tti-621, a novel biologic targeting the innate immune checkpoint Cd47, in patients with relapsed or refractory mycosis fungoides or sezary syndrome: A multicentre, phase 1 study. Lancet Haematol (2021) 8(11):e808–e17. doi: 10.1016/S2352-3026(21)00271-4
144. Hoppe RT. Mycosis fungoides: Radiation therapy. Dermatol Ther (2003) 16(4):347–54. doi: 10.1111/j.1396-0296.2003.01647.x
145. Devlin P. Brachytherapy: Applications and techniques. 1st ed. Wilkins LW&, editor. Lippincott Williams: Wilkins in Philadelphia, PA. (2006), p. 420
146. Bartek J, Lukas J. Cell cycle. Order Destruction. Sci (2001) 294(5540):66–7. doi: 10.1126/science.1066237
147. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances mhc class I expression, and induces successful antitumor immunotherapy. J Exp Med (2006) 203(5):1259–71. doi: 10.1084/jem.20052494
148. Ruckert M, Deloch L, Fietkau R, Frey B, Hecht M, Gaipl US. Immune modulatory effects of radiotherapy as basis for well-reasoned radioimmunotherapies. Strahlenther Onkol (2018) 194(6):509–19. doi: 10.1007/s00066-018-1287-1
149. Durgin JS, Jariwala NN, Wysocka M, Zhang KK, Maity A, Benoit B, et al. Low-dose total skin electron beam therapy as part of a multimodality regimen for treatment of sezary syndrome: Clinical, immunologic, and molecular analysis. JAMA Dermatol (2021) 157(1):90–5. doi: 10.1001/jamadermatol.2020.3958
150. Elsayad K, Stadler R, Steinbrink K, Eich HT. Combined total skin radiotherapy and immune checkpoint inhibitors: A promising potential treatment for mycosis fungoides and sezary syndrome. J Dtsch Dermatol Ges (2020) 18(3):193–7. doi: 10.1111/ddg.14044
151. Bernard JJ, Gallo RL, Krutmann J. Photoimmunology: How ultraviolet radiation affects the immune system. Nat Rev Immunol (2019) 19(11):688–701. doi: 10.1038/s41577-019-0185-9
152. D'Orazio J, Jarrett S, Amaro-Ortiz A, Scott T. Uv radiation and the skin. Int J Mol Sci (2013) 14(6):12222–48. doi: 10.3390/ijms140612222
153. el-Ghorr AA, Norval M. Biological effects of narrow-band (311 nm Tl01) uvb irradiation: A review. J Photochem Photobiol B (1997) 38(2-3):99–106. doi: 10.1016/s1011-1344(96)07454-4
154. Ozawa M, Ferenczi K, Kikuchi T, Cardinale I, Austin LM, Coven TR, et al. 312-nanometer ultraviolet b light (Narrow-band uvb) induces apoptosis of T cells within psoriatic lesions. J Exp Med (1999) 189(4):711–8. doi: 10.1084/jem.189.4.711
155. Vieyra-Garcia PA, Wolf P. A deep dive into uv-based phototherapy: Mechanisms of action and emerging molecular targets in inflammation and cancer. Pharmacol Ther (2021) 222:107784. doi: 10.1016/j.pharmthera.2020.107784
156. Weitzen ML, Bonavida B. Mechanism of inhibition of human natural killer activity by ultraviolet radiation. J Immunol (1984) 133(6):3128–32.
157. Hersey P, MacDonald M, Henderson C, Schibeci S, D'Alessandro G, Pryor M, et al. Suppression of natural killer cell activity in humans by radiation from solarium lamps depleted of uvb. J Invest Dermatol (1988) 90(3):305–10. doi: 10.1111/1523-1747.ep12456090
158. Schwarz T. 25 years of uv-induced immunosuppression mediated by T cells-from disregarded T suppressor cells to highly respected regulatory T cells. Photochem Photobiol (2008) 84(1):10–8. doi: 10.1111/j.1751-1097.2007.00223.x
159. Kurimoto I, Streilein JW. Tumor necrosis factor-alpha impairs contact hypersensitivity induction after ultraviolet b radiation Via tnf-receptor 2 (P75). Exp Dermatol (1999) 8(6):495–500. doi: 10.1111/j.1600-0625.1999.tb00308.x
160. Shreedhar V, Giese T, Sung VW, Ullrich SE. A cytokine cascade including prostaglandin E2, il-4, and il-10 is responsible for uv-induced systemic immune suppression. J Immunol (1998) 160(8):3783–9.
161. Araneo BA, Dowell T, Moon HB, Daynes RA. Regulation of murine lymphokine production in vivo. ultraviolet radiation exposure depresses il-2 and enhances il-4 production by T cells through an il-1-Dependent mechanism. J Immunol (1989) 143(6):1737–44.
162. Rathod D, Muneer H, Masood S. Phototherapy. Treasure Island (FL): StatPearls Publishing (2022).
163. Vieyra-Garcia P, Crouch JD, O'Malley JT, Seger EW, Yang CH, Teague JE, et al. Benign T cells drive clinical skin inflammation in cutaneous T cell lymphoma. JCI Insight (2019) 4(1). doi: 10.1172/jci.insight.124233
164. Vieyra-Garcia P, Fink-Puches R, Porkert S, Lang R, Pochlauer S, Ratzinger G, et al. Evaluation of low-dose, low-frequency oral psoralen-uv-a treatment with or without maintenance on early-stage mycosis fungoides: A randomized clinical trial. JAMA Dermatol (2019) 155(5):538–47. doi: 10.1001/jamadermatol.2018.5905
165. Roupe G, Sandstrom MH, Kjellstrom C. Puva in early mycosis fungoides may give long-term remission and delay extracutaneous spread. Acta Derm Venereol (1996) 76(6):475–8. doi: 10.2340/0001555576475478
166. Lovgren ML, Scarisbrick JJ. Update on skin directed therapies in mycosis fungoides. Chin Clin Oncol (2019) 8(1):7. doi: 10.21037/cco.2018.11.03
167. Kuzel TM, Roenigk HH Jr., Samuelson E, Herrmann JJ, Hurria A, Rademaker AW, et al. Effectiveness of interferon Alfa-2a combined with phototherapy for mycosis fungoides and the sezary syndrome. J Clin Oncol (1995) 13(1):257–63. doi: 10.1200/JCO.1995.13.1.257
168. Chiarion-Sileni V, Bononi A, Fornasa CV, Soraru M, Alaibac M, Ferrazzi E, et al. Phase ii trial of interferon-Alpha-2a plus psolaren with ultraviolet light a in patients with cutaneous T cell lymphoma. Cancer (2002) 95(3):569–75. doi: 10.1002/cncr.10706
169. Rupoli S, Barulli S, Guiducci B, Offidani M, Mozzicafreddo G, Simonacci M, et al. Low dose interferon-alpha 2b combined with puva is an effective treatment of early stage mycosis fungoides: Results of a multicenter study. Haematologica (Roma) (1999) 84(9):809–13.
170. Nikolaou V, Siakantaris MP, Vassilakopoulos TP, Papadavid E, Stratigos A, Economidi A, et al. Puva plus interferon Alpha2b in the treatment of advanced or refractory to puva early stage mycosis fungoides: A case series. J Eur Acad Dermatol Venereol (2011) 25(3):354–7. doi: 10.1111/j.1468-3083.2010.03732.x
171. Wozniak MB, Tracey L, Ortiz-Romero PL, Montes S, Alvarez M, Fraga J, et al. Psoralen plus ultraviolet a +/- interferon-alpha treatment resistance in mycosis fungoides: The role of tumour microenvironment, nuclear transcription factor-kappab and T cell receptor pathways. Br J Dermatol (2009) 160(1):92–102. doi: 10.1111/j.1365-2133.2008.08886.x
172. Stadler R, Kremer A, Luger T, Sterry W. Prospective, randomized, multicentre clinical trial on the use of interferon a 2a plus puva versus puva monotherapy in patients with cutaneous T cell lymphoma, stages I and ii. J Clin Oncol (2006) 24(18_suppl):7541. doi: 10.1200/jco.2006.24.18_suppl.7541
173. Zic JA. Extracorporeal photopheresis in the treatment of mycosis fungoides and sezary syndrome. Dermatol Clin (2015) 33(4):765–76. doi: 10.1016/j.det.2015.05.011
174. Edelson RL. Mechanistic insights into extracorporeal photochemotherapy: Efficient induction of monocyte-to-Dendritic cell maturation. Transfus Apher Sci (2014) 50(3):322–9. doi: 10.1016/j.transci.2013.07.031
175. Durazzo TS, Tigelaar RE, Filler R, Hayday A, Girardi M, Edelson RL. Induction of monocyte-to-Dendritic cell maturation by extracorporeal photochemotherapy: Initiation Via direct platelet signaling. Transfus Apher Sci (2014) 50(3):370–8. doi: 10.1016/j.transci.2013.11.008
176. Tatsuno K, Yamazaki T, Hanlon D, Han P, Robinson E, Sobolev O, et al. Extracorporeal photochemotherapy induces bona fide immunogenic cell death. Cell Death Dis (2019) 10(8):578. doi: 10.1038/s41419-019-1819-3
177. Crocchiolo R, Cesana C, Girgenti D, Bertani G, Barba C, Liga G, et al. Tregs and gvhd prevention by extracorporeal photopheresis: Observations from a clinical trial. Exp Hematol Oncol (2021) 10(1):14. doi: 10.1186/s40164-021-00210-9
178. Rao V, Saunes M, Jorstad S, Moen T. Cutaneous T cell lymphoma and graft-Versus-Host disease: A comparison of in vivo effects of extracorporeal photochemotherapy on Foxp3+ regulatory T cells. Clin Immunol (2009) 133(3):303–13. doi: 10.1016/j.clim.2009.08.016
179. Perez MI, Edelson RL, John L, Laroche L, Berger CL. Inhibition of antiskin allograft immunity induced by infusions with photoinactivated effector T lymphocytes (Pet cells). Yale J Biol Med (1989) 62(6):595–609.
180. Dolmans DE, Fukumura D, Jain RK. Photodynamic therapy for cancer. Nat Rev Cancer (2003) 3(5):380–7. doi: 10.1038/nrc1071
181. Alzeibak R, Mishchenko TA, Shilyagina NY, Balalaeva IV, Vedunova MV, Krysko DV. Targeting immunogenic cancer cell death by photodynamic therapy: Past, present and future. J Immunother Cancer (2021) 9(1):e001926. doi: 10.1136/jitc-2020-001926
182. Adkins I, Fucikova J, Garg AD, Agostinis P, Spisek R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology (2014) 3(12):e968434. doi: 10.4161/21624011.2014.968434
183. Chen X, Song M, Zhang B, Zhang Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid Med Cell Longev (2016) 2016:1580967. doi: 10.1155/2016/1580967
184. Gollnick SO, Liu XN, Owczarczak B, Musser DA, Henderson BW. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer Res (1997) 57(18):3904–9.
185. Mougiakakos D, Johansson CC, Kiessling R. Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood (2009) 113(15):3542–5. doi: 10.1182/blood-2008-09-181040
186. Quereux G, Brocard A, Saint-Jean M, Peuvrel L, Knol AC, Allix R, et al. Photodynamic therapy with methyl-aminolevulinic acid for paucilesional mycosis fungoides: A prospective open study and review of the literature. J Am Acad Dermatol (2013) 69(6):890–7. doi: 10.1016/j.jaad.2013.07.047
187. Fox FE, Niu Z, Tobia A, Rook AH. Photoactivated hypericin is an anti-proliferative agent that induces a high rate of apoptotic death of normal, transformed, and malignant T lymphocytes: Implications for the treatment of cutaneous lymphoproliferative and inflammatory disorders. J Invest Dermatol (1998) 111(2):327–32. doi: 10.1046/j.1523-1747.1998.00278.x
188. Bagherani N, Smoller BR. An overview of cutaneous T cell lymphomas. F1000Res (2016) 5:1882. doi: 10.12688/f1000research.8829.1
189. Foss FM. Dab(389)Il-2 (Ontak): A novel fusion toxin therapy for lymphoma. Clin Lymphoma (2000) 1(2):110–6. doi: 10.3816/clm.2000.n.009
190. Litzinger MT, Fernando R, Curiel TJ, Grosenbach DW, Schlom J, Palena C. Il-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T cell immunity. Blood (2007) 110(9):3192–201. doi: 10.1182/blood-2007-06-094615
191. Gritzapis AD, Voutsas IF, Baxevanis CN. Ontak reduces the immunosuppressive tumor environment and enhances successful therapeutic vaccination in her-2/Neu-Tolerant mice. Cancer Immunol Immunother (2012) 61(3):397–407. doi: 10.1007/s00262-011-1113-4
192. Baur AS, Lutz MB, Schierer S, Beltrame L, Theiner G, Zinser E, et al. Denileukin diftitox (Ontak) induces a tolerogenic phenotype in dendritic cells and stimulates survival of resting treg. Blood (2013) 122(13):2185–94. doi: 10.1182/blood-2012-09-456988
193. Manoukian G, Hagemeister F. Denileukin diftitox: A novel immunotoxin. Expert Opin Biol Ther (2009) 9(11):1445–51. doi: 10.1517/14712590903348135
194. Avarbock AB, Loren AW, Park JY, Junkins-Hopkins JM, Choi J, Litzky LA, et al. Lethal vascular leak syndrome after denileukin diftitox administration to a patient with cutaneous Gamma/Delta T cell lymphoma and occult cirrhosis. Am J Hematol (2008) 83(7):593–5. doi: 10.1002/ajh.21180
195. Thompson J, Stavrou S, Weetall M, Hexham JM, Digan ME, Wang Z, et al. Improved binding of a bivalent single-chain immunotoxin results in increased efficacy for in vivo T cell depletion. Protein Eng (2001) 14(12):1035–41. doi: 10.1093/protein/14.12.1035
196. Frankel AE, Woo JH, Ahn C, Foss FM, Duvic M, Neville PH, et al. Resimmune, an anti-Cd3epsilon recombinant immunotoxin, induces durable remissions in patients with cutaneous T cell lymphoma. Haematologica (2015) 100(6):794–800. doi: 10.3324/haematol.2015.123711
197. Willerslev-Olsen A, Litvinov IV, Fredholm SM, Petersen DL, Sibbesen NA, Gniadecki R, et al. Il-15 and il-17f are differentially regulated and expressed in mycosis fungoides (Mf). Cell Cycle (2014) 13(8):1306–12. doi: 10.4161/cc.28256
198. Imai T, Nagira M, Takagi S, Kakizaki M, Nishimura M, Wang J, et al. Selective recruitment of Ccr4-bearing Th2 cells toward antigen-presenting cells by the cc chemokines thymus and activation-regulated chemokine and macrophage-derived chemokine. Int Immunol (1999) 11(1):81–8. doi: 10.1093/intimm/11.1.81
199. Ferenczi K, Fuhlbrigge RC, Pinkus J, Pinkus GS, Kupper TS. Increased Ccr4 expression in cutaneous T cell lymphoma. J Invest Dermatol (2002) 119(6):1405–10. doi: 10.1046/j.1523-1747.2002.19610.x
200. Niwa R, Shoji-Hosaka E, Sakurada M, Shinkawa T, Uchida K, Nakamura K, et al. Defucosylated chimeric anti-cc chemokine receptor 4 Igg1 with enhanced antibody-dependent cellular cytotoxicity shows potent therapeutic activity to T cell leukemia and lymphoma. Cancer Res (2004) 64(6):2127–33. doi: 10.1158/0008-5472.can-03-2068
201. Ogura M, Ishida T, Hatake K, Taniwaki M, Ando K, Tobinai K, et al. Multicenter phase ii study of mogamulizumab (Kw-0761), a defucosylated anti-cc chemokine receptor 4 antibody, in patients with relapsed peripheral T cell lymphoma and cutaneous T cell lymphoma. J Clin Oncol (2014) 32(11):1157–63. doi: 10.1200/JCO.2013.52.0924
202. Trum NA, Zain J, Martinez XU, Parekh V, Afkhami M, Abdulla F, et al. Mogamulizumab efficacy is underscored by its associated rash that mimics cutaneous T cell lymphoma: A retrospective single-centre case series. Br J Dermatol (2022) 186(1):153–66. doi: 10.1111/bjd.20708
203. Bonnet P, Battistella M, Roelens M, Ram-Wolff C, Herms F, Frumholtz L, et al. Association of autoimmunity and long-term complete remission in patients with sezary syndrome treated with mogamulizumab. Br J Dermatol (2019) 180(2):419–20. doi: 10.1111/bjd.17320
204. Ni X, Langridge T, Duvic M. Depletion of regulatory T cells by targeting cc chemokine receptor type 4 with mogamulizumab. Oncoimmunology (2015) 4(7):e1011524. doi: 10.1080/2162402X.2015.1011524
205. Roelens M, de Masson A, Andrillon A, Ram-Wolff C, Biard L, Boisson M, et al. Mogamulizumab induces long-term immune restoration and reshapes tumour heterogeneity in sezary syndrome. Br J Dermatol (2022) 186(6):1010–25. doi: 10.1111/bjd.21018
206. de Masson A, Darbord D, Dobos G, Boisson M, Roelens M, Ram-Wolff C, et al. Macrophage-derived Cxcl9 and Cxcl11, T cell skin homing, and disease control in mogamulizumab-treated ctcl patients. Blood (2022) 139(12):1820–32. doi: 10.1182/blood.2021013341
207. Clark RA, Watanabe R, Teague JE, Schlapbach C, Tawa MC, Adams N, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated ctcl patients. Sci Transl Med (2012) 4(117):117ra7. doi: 10.1126/scitranslmed.3003008
208. Havari E, Turner MJ, Campos-Rivera J, Shankara S, Nguyen TH, Roberts B, et al. Impact of alemtuzumab treatment on the survival and function of human regulatory T cells in vitro. Immunology (2014) 141(1):123–31. doi: 10.1111/imm.12178
209. Querfeld C, Mehta N, Rosen ST, Guitart J, Rademaker A, Gerami P, et al. Alemtuzumab for relapsed and refractory erythrodermic cutaneous T cell lymphoma: A single institution experience from the Robert h. Lurie Compr Cancer Center. Leuk Lymphoma (2009) 50(12):1969–76. doi: 10.3109/10428190903216770
210. Poszepczynska-Guigne E, Schiavon V, D'Incan M, Echchakir H, Musette P, Ortonne N, et al. Cd158k/Kir3dl2 is a new phenotypic marker of sezary cells: Relevance for the diagnosis and follow-up of sezary syndrome. J Invest Dermatol (2004) 122(3):820–3. doi: 10.1111/j.0022-202X.2004.22326.x
211. Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, et al. Recognition of hla-A3 and hla-A11 by Kir3dl2 is peptide-specific. Eur J Immunol (2004) 34(6):1673–9. doi: 10.1002/eji.200425089
212. Schmitt C, Marie-Cardine A, Bensussan A. Therapeutic antibodies to Kir3dl2 and other target antigens on cutaneous T cell lymphomas. Front Immunol (2017) 8:1010. doi: 10.3389/fimmu.2017.01010
213. Thonnart N, Caudron A, Legaz I, Bagot M, Bensussan A, Marie-Cardine A. Kir3dl2 is a coinhibitory receptor on sezary syndrome malignant T cells that promotes resistance to activation-induced cell death. Blood (2014) 124(22):3330–2. doi: 10.1182/blood-2014-09-598995
214. Muller P, Martin K, Theurich S, Schreiner J, Savic S, Terszowski G, et al. Microtubule-depolymerizing agents used in antibody-drug conjugates induce antitumor immunity by stimulation of dendritic cells. Cancer Immunol Res (2014) 2(8):741–55. doi: 10.1158/2326-6066.CIR-13-0198
215. Gopal AK, Ramchandren R, O'Connor OA, Berryman RB, Advani RH, Chen R, et al. Safety and efficacy of brentuximab vedotin for Hodgkin lymphoma recurring after allogeneic stem cell transplantation. Blood (2012) 120(3):560–8. doi: 10.1182/blood-2011-12-397893
216. Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, et al. Cac10-vcmmae, an anti-Cd30-Monomethyl auristatin e conjugate with potent and selective antitumor activity. Blood (2003) 102(4):1458–65. doi: 10.1182/blood-2003-01-0039
217. Saintes C, Saint-Jean M, Renaut JJ, Dreno B, Quereux G. Dramatic efficacy of brentuximab vedotin in two patients with epidermotropic cutaneous T cell lymphomas after treatment failure despite variable Cd30 expression. Br J Dermatol (2015) 172(3):819–21. doi: 10.1111/bjd.13337
218. Duvic M, Tetzlaff M, Clos AL, Gangar P, Talpur R. Phase ii trial of brentuximab vedotin for Cd30+ cutaneous T cell lymphomas and lymphoproliferative disorders. Blood (2013) 122(21):367–. doi: 10.1182/blood.V122.21.367.367
219. Kim YH, Tavallaee M, Sundram U, Salva KA, Wood GS, Li S, et al. Phase ii investigator-initiated study of brentuximab vedotin in mycosis fungoides and sezary syndrome with variable Cd30 expression level: A multi-institution collaborative project. J Clin Oncol (2015) 33(32):3750–8. doi: 10.1200/JCO.2014.60.3969
220. Heiser RA, Grogan BM, Manlove LS, Gardai SJ. Abstract 1789: Cd30+T regulatory cells, but not Cd30+Cd8 T cells, are impaired following brentuximab vedotin treatment in vitro and in vivo. Cancer Res (2018) 78:1789. doi: 10.1158/1538-7445.Am2018-1789
221. Cory S, Adams JM. The Bcl2 family: Regulators of the cellular life-or-Death switch. Nat Rev Cancer (2002) 2(9):647–56. doi: 10.1038/nrc883
222. Tan T-T, White E. Therapeutic targeting of death pathways in cancer: Mechanisms for activating cell death in cancer cells. Adv Exp Med Biol (2008) 615:81–104. doi: 10.1007/978-1-4020-6554-5_5
223. Izutsu K, Yamamoto K, Kato K, Ishikawa T, Fukuhara N, Terui Y, et al. Phase 1/2 study of venetoclax, a bcl-2 inhibitor, in Japanese patients with relapsed or refractory chronic lymphocytic leukemia and small lymphocytic lymphoma. Int J Hematol (2021) 113(3):370–80. doi: 10.1007/s12185-020-03024-3
224. Mathew R, Haribhai D, Kohlhapp F, Duggan R, Ellis P, Riehm JJ, et al. The bcl-2-Selective inhibitor venetoclax spares activated T cells during anti-tumor immunity. Blood (2018) 132(Supplement 1):3704. doi: 10.1182/blood-2018-99-113134
225. Dummer R, Michie SA, Kell D, Gould JW, Haeffner AC, Smoller BR, et al. Expression of bcl-2 protein and ki-67 nuclear proliferation antigen in benign and malignant cutaneous T cell infiltrates. J Cutan Pathol (1995) 22(1):11–7. doi: 10.1111/j.1600-0560.1995.tb00733.x
226. D'Aguanno S, Del Bufalo D. Inhibition of anti-apoptotic bcl-2 proteins in preclinical and clinical studies: Current overview in cancer. Cells (2020) 9(5):1287. doi: 10.3390/cells9051287
227. Cyrenne BM, Lewis JM, Weed JG, Carlson KR, Mirza FN, Foss FM, et al. Synergy of Bcl2 and histone deacetylase inhibition against leukemic cells from cutaneous T cell lymphoma patients. Blood (2017) 130(19):2073–83. doi: 10.1182/blood-2017-06-792150
228. King ALO, Mirza FN, Lewis JM, Carlson KR, Huntington S, Foss FM, et al. B-cell lymphoma 2 inhibitor venetoclax treatment of a patient with cutaneous T cell lymphoma. JAAD Case Rep (2021) 8:89–92. doi: 10.1016/j.jdcr.2020.12.025
229. Kohlhapp FJ, Haribhai D, Mathew R, Duggan R, Ellis PA, Wang R, et al. Venetoclax increases intratumoral effector T cells and antitumor efficacy in combination with immune checkpoint blockade. Cancer Discovery (2021) 11(1):68–79. doi: 10.1158/2159-8290.CD-19-0759
230. Murakami S, Suzuki S, Hanamura I, Yoshikawa K, Ueda R, Seto M, et al. Combining T cell-Based immunotherapy with venetoclax elicits synergistic cytotoxicity to b-cell lines in vitro. Hematol Oncol (2020) 38(5):705–14. doi: 10.1002/hon.2794
231. Ribeiro ML, Reyes-Garau D, Armengol M, Fernandez-Serrano M, Roue G. Recent advances in the targeting of epigenetic regulators in b-cell non-Hodgkin lymphoma. Front Genet (2019) 10:986. doi: 10.3389/fgene.2019.00986
232. Bracken AP, Helin K. Polycomb group proteins: Navigators of lineage pathways led astray in cancer. Nat Rev Cancer (2009) 9(11):773–84. doi: 10.1038/nrc2736
233. Morey L, Helin K. Polycomb group protein-mediated repression of transcription. Trends Biochem Sci (2010) 35(6):323–32. doi: 10.1016/j.tibs.2010.02.009
234. Sasaki D, Imaizumi Y, Hasegawa H, Osaka A, Tsukasaki K, Choi YL, et al. Overexpression of enhancer of zeste homolog 2 with trimethylation of lysine 27 on histone H3 in adult T cell Leukemia/Lymphoma as a target for epigenetic therapy. Haematologica (2011) 96(5):712–9. doi: 10.3324/haematol.2010.028605
235. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering Ezh2 (Tyr641) in follicular and diffuse Large b-cell lymphomas of germinal-center origin. Nat Genet (2010) 42(2):181–5. doi: 10.1038/ng.518
236. Visser HP, Gunster MJ, Kluin-Nelemans HC, Manders EM, Raaphorst FM, Meijer CJ, et al. The polycomb group protein Ezh2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol (2001) 112(4):950–8. doi: 10.1046/j.1365-2141.2001.02641.x
237. Raaphorst FM, van Kemenade FJ, Blokzijl T, Fieret E, Hamer KM, Satijn DP, et al. Coexpression of bmi-1 and Ezh2 polycomb group genes in reed-sternberg cells of hodgkin's disease. Am J Pathol (2000) 157(3):709–15. doi: 10.1016/S0002-9440(10)64583-X
238. Arenas-Ramirez N, Sahin D, Boyman O. Epigenetic mechanisms of tumor resistance to immunotherapy. Cell Mol Life Sci (2018) 75(22):4163–76. doi: 10.1007/s00018-018-2908-7
239. Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting Ezh2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep (2018) 23(11):3262–74. doi: 10.1016/j.celrep.2018.05.050
240. Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc Natl Acad Sci U.S.A. (2015) 112(52):15988–93. doi: 10.1073/pnas.1521740112
241. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting micrornas and Ezh2 Via glycolysis restriction. Nat Immunol (2016) 17(1):95–103. doi: 10.1038/ni.3313
242. Maruyama D, Tobinai K, Makita S, Ishida T, Kusumoto S, Ishitsuka K, et al. First-in-Human study of the Ezh1/2 dual inhibitor ds-3201b in patients with relapsed or refractory non-Hodgkin lymphomas — preliminary results. Blood (2017) 130:4070–. doi: 10.1182/blood.V130.Suppl_1.4070.4070
243. Izutsu K, Makita S, Nosaka K, Yoshimitsu M, Utsunomiya A, Kusumoto S, et al. An open-label, single-arm, phase 2 trial of valemetostat in relapsed or refractory adult T cell Leukemia/Lymphoma. Blood (2022). doi: 10.1182/blood.2022016862
244. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discovery (2014) 13(9):673–91. doi: 10.1038/nrd4360
245. Wozniak MB, Villuendas R, Bischoff JR, Aparicio CB, Martinez Leal JF, de la Cueva P, et al. Vorinostat interferes with the signaling transduction pathway of T cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T cell lymphoma. Haematologica (2010) 95(4):613–21. doi: 10.3324/haematol.2009.013870
246. Woods DM, Sodre AL, Villagra A, Sarnaik A, Sotomayor EM, Weber J. Hdac inhibition upregulates pd-1 ligands in melanoma and augments immunotherapy with pd-1 blockade. Cancer Immunol Res (2015) 3(12):1375–85. doi: 10.1158/2326-6066.CIR-15-0077-T
247. Nusinzon I, Horvath CM, Proc Natl Acad Sci USA. (2003) 100(25):14742–7. doi: 10.1073/pnas.2433987100
248. Kasler HG, Young BD, Mottet D, Lim HW, Collins AM, Olson EN, et al. Histone deacetylase 7 regulates cell survival and tcr signaling in Cd4/Cd8 double-positive thymocytes. J Immunol (2011) 186(8):4782–93. doi: 10.4049/jimmunol.1001179
249. Shakespear MR, Hohenhaus DM, Kelly GM, Kamal NA, Gupta P, Labzin LI, et al. Histone deacetylase 7 promotes toll-like receptor 4-dependent proinflammatory gene expression in macrophages. J Biol Chem (2013) 288(35):25362–74. doi: 10.1074/jbc.M113.496281
250. Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, et al. The histone deacetylase Hdac11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol (2009) 10(1):92–100. doi: 10.1038/ni.1673
251. Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, et al. Histone deacetylase inhibitors impair innate immune responses to toll-like receptor agonists and to infection. Blood (2011) 117(4):1205–17. doi: 10.1182/blood-2010-05-284711
252. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med (2007) 13(11):1299–307. doi: 10.1038/nm1652
253. Buus TB, Willerslev-Olsen A, Fredholm S, Blumel E, Nastasi C, Gluud M, et al. Single-cell heterogeneity in sezary syndrome. Blood Adv (2018) 2(16):2115–26. doi: 10.1182/bloodadvances.2018022608
254. Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase ii multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T cell lymphoma. J Clin Oncol (2009) 27(32):5410–7. doi: 10.1200/JCO.2008.21.6150
255. Kelly-Sell MJ, Kim YH, Straus S, Benoit B, Harrison C, Sutherland K, et al. The histone deacetylase inhibitor, romidepsin, suppresses cellular immune functions of cutaneous T cell lymphoma patients. Am J Hematol (2012) 87(4):354–60. doi: 10.1002/ajh.23112
256. Stephen S, Morrissey KA, Benoit BM, Kim EJ, Vittorio CC, Nasta SD, et al. Inhibition of cell-mediated immunity by the histone deacetylase inhibitor vorinostat: Implications for therapy of cutaneous T cell lymphoma. Am J Hematol (2012) 87(2):226–8. doi: 10.1002/ajh.22231
257. Sawas A, Radeski D, O'Connor OA. Belinostat in patients with refractory or relapsed peripheral T cell lymphoma: A perspective review. Ther Adv Hematol (2015) 6(4):202–8. doi: 10.1177/2040620715592567
258. Duvic M, Kim YH, LeBoeuf NR, Porcu P, Hastings J, Bassuner J, et al. A phase 2 randomized study of shape gel (Shp-141) in patients with early-stage cutaneous T cell lymphoma: Interim results. J Clin Oncol (2016) 34(15_suppl):7562–. doi: 10.1200/JCO.2016.34.15_suppl.7562
259. Stadler R, Scarisbrick JJ, Knobler R, Quaglino P, Borgmann M, Orlovius M, et al. Phase ii trial evaluating resminostat for maintenance treatment of patients with advanced stage (Stage iib–ivb) mycosis fungoides (Mf) or sézary syndrome (Ss): Resmain study. Eur J Cancer (2021) 156:S53–S4. doi: 10.1016/s0959-8049(21)00724-3
260. Davies F, Baz R. Lenalidomide mode of action: Linking bench and clinical findings. Blood Rev (2010) 24 Suppl 1:S13–9. doi: 10.1016/S0268-960X(10)70004-7
261. Kater AP, Tonino SH, Egle A, Ramsay AG. How does lenalidomide target the chronic lymphocytic leukemia microenvironment? Blood (2014) 124(14):2184–9. doi: 10.1182/blood-2014-05-578286
262. Netchiporouk E, Litvinov IV, Moreau L, Gilbert M, Sasseville D, Duvic M. Deregulation in stat signaling is important for cutaneous T cell lymphoma (Ctcl) pathogenesis and cancer progression. Cell Cycle (2014) 13(21):3331–5. doi: 10.4161/15384101.2014.965061
263. Nielsen M, Kaltoft K, Nordahl M, Ropke C, Geisler C, Mustelin T, et al. Constitutive activation of a slowly migrating isoform of Stat3 in mycosis fungoides: Tyrphostin Ag490 inhibits Stat3 activation and growth of mycosis fungoides tumor cell lines. Proc Natl Acad Sci U.S.A. (1997) 94(13):6764–9. doi: 10.1073/pnas.94.13.6764
264. Eriksen KW, Kaltoft K, Mikkelsen G, Nielsen M, Zhang Q, Geisler C, et al. Constitutive Stat3-activation in sezary syndrome: Tyrphostin Ag490 inhibits Stat3-activation, interleukin-2 receptor expression and growth of leukemic sezary cells. Leukemia (2001) 15(5):787–93. doi: 10.1038/sj.leu.2402093
265. Zhang Q, Nowak I, Vonderheid EC, Rook AH, Kadin ME, Nowell PC, et al. Activation of Jak/Stat proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic Large T cell lymphoma and sezary syndrome. Proc Natl Acad Sci U.S.A. (1996) 93(17):9148–53. doi: 10.1073/pnas.93.17.9148
266. Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. The role of Jak/Stat signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer (2015) 113(3):365–71. doi: 10.1038/bjc.2015.233
267. Kopp KL, Ralfkiaer U, Gjerdrum LM, Helvad R, Pedersen IH, Litman T, et al. Stat5-mediated expression of oncogenic mir-155 in cutaneous T cell lymphoma. Cell Cycle (2013) 12(12):1939–47. doi: 10.4161/cc.24987
268. Litvinov IV, Cordeiro B, Fredholm S, Odum N, Zargham H, Huang Y, et al. Analysis of Stat4 expression in cutaneous T cell lymphoma (Ctcl) patients and patient-derived cell lines. Cell Cycle (2014) 13(18):2975–82. doi: 10.4161/15384101.2014.947759
269. Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. Il-2 regulates Foxp3 expression in human Cd4+Cd25+ regulatory T cells through a stat-dependent mechanism and induces the expansion of these cells in vivo. Blood (2006) 108(5):1571–9. doi: 10.1182/blood-2006-02-004747
270. Boor PPC, de Ruiter PE, Asmawidjaja PS, Lubberts E, van der Laan LJW, Kwekkeboom J. Jak-inhibitor tofacitinib suppresses interferon Alfa production by plasmacytoid dendritic cells and inhibits arthrogenic and antiviral effects of interferon Alfa. Transl Res (2017) 188:67–79. doi: 10.1016/j.trsl.2016.11.006
271. McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood (2015) 126(4):508–19. doi: 10.1182/blood-2014-11-611194
272. da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and sezary syndrome. Nat Genet (2015) 47(12):1465–70. doi: 10.1038/ng.3442
273. Perez C, Gonzalez-Rincon J, Onaindia A, Almaraz C, Garcia-Diaz N, Pisonero H, et al. Mutated jak kinases and deregulated stat activity are potential therapeutic targets in cutaneous T cell lymphoma. Haematologica (2015) 100(11):e450–3. doi: 10.3324/haematol.2015.132837
274. Hamlin PA, Curnutte JT, Coffey GP, Michelson GC, Conley PB, Leeds JM, et al. A phase 2 study of the dual Syk/Jak inhibitor cerdulatinib demonstrates good tolerability and clinical response in Relapsed/Refractory peripheral T cell lymphoma and cutaneous T cell lymphoma. Blood (2019) 134(Supplement_1):466–. doi: 10.1182/blood-2019-123986
275. Zhang J-P, Gao Q, Chan A, Xiao W, El Daker S, Roshal M, et al. Aberrant syk expression as a potential therapeutic target in T/Nk cell neoplasms. Blood (2019) 134(Supplement_1):3978–. doi: 10.1182/blood-2019-130406
276. Hoxhaj G, Manning BD. The Pi3k-akt network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer (2020) 20(2):74–88. doi: 10.1038/s41568-019-0216-7
277. von Keudell G, Moskowitz AJ. The role of Pi3k inhibition in lymphoid malignancies. Curr Hematol Malig Rep (2019) 14(5):405–13. doi: 10.1007/s11899-019-00540-w
278. Jean S, Kiger AA. Classes of phosphoinositide 3-kinases at a glance. J Cell Sci (2014) 127:923–8. doi: 10.1242/jcs.093773
279. Johansen KH, Golec DP, Thomsen JH, Schwartzberg PL, Okkenhaug K. Pi3k in T cell adhesion and trafficking. Front Immunol (2021) 12:708908. doi: 10.3389/fimmu.2021.708908
280. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific Pi3k signalling. Nat Rev Mol Cell Biol (2010) 11(5):329–41. doi: 10.1038/nrm2882
281. Faia K, White K, Proctor J, Andrade P, Pink M, Rickles R, et al. High throughput in vitro combination sensitivity screen in hematologic malignancies with the phosphoinositide-3 kinase (Pi3k)-Δ,Γ inhibitor, duvelisib. J Clin Oncol8559- (2015) 33(15_suppl). doi: 10.1200/jco.2015.33.15_suppl.8559
282. Duvelisib (Copiktra, verastem, Inc.) for adult patients with relapsed or refractory chronic lymphocytic leukemia (Cll) or small lymphocytic lymphoma (Sll). Case Med Res (2018). doi: 10.31525/fda1-ucm621503.htm
283. Abu Eid R, Ahmad S, Lin Y, Webb M, Berrong Z, Shrimali R, et al. Enhanced therapeutic efficacy and memory of tumor-specific Cd8 T cells by ex vivo Pi3k-delta inhibition. Cancer Res (2017) 77(15):4135–45. doi: 10.1158/0008-5472.CAN-16-1925
284. Horwitz SM, Moskowitz AJ, Jacobsen ED, Mehta-Shah N, Khodadoust MS, Fisher DC, et al. The combination of duvelisib, a Pi3k-Δ,Γ inhibitor, and romidepsin is highly active in Relapsed/Refractory peripheral T cell lymphoma with low rates of transaminitis: Results of parallel multicenter, phase 1 combination studies with expansion cohorts. Blood (2018) 132(Supplement 1):683–. doi: 10.1182/blood-2018-99-115241
285. Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20s and 26s proteasomes. Annu Rev Biochem (1996) 65(1):801–47. doi: 10.1146/annurev.bi.65.070196.004101
286. Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, et al. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on mhc class I molecules. Cell (1994) 78(5):761–71. doi: 10.1016/s0092-8674(94)90462-6
287. Paramore A, Frantz S. Bortezomib. Nat Rev Drug Discovery (2003) 2(8):611–2. doi: 10.1038/nrd1159
288. Heidt S, Roelen DL, Vergunst M, Doxiadis II, FH C, Mulder A. Bortezomib affects the function of human b cells: Possible implications for desensitization protocols. Clin Transpl (2009), 70:387–92.
289. Verfaillie T, Salazar M, Velasco G, Agostinis P. Linking er stress to autophagy: Potential implications for cancer therapy. Int J Cell Biol (2010) 2010:930509. doi: 10.1155/2010/930509
290. Chang CL, Hsu YT, Wu CC, Yang YC, Wang C, Wu TC, et al. Immune mechanism of the antitumor effects generated by bortezomib. J Immunol (2012) 189(6):3209–20. doi: 10.4049/jimmunol.1103826
291. Lundqvist A, Yokoyama H, Smith A, Berg M, Childs R. Bortezomib treatment and regulatory T cell depletion enhance the antitumor effects of adoptively infused nk cells. Blood (2009) 113(24):6120–7. doi: 10.1182/blood-2008-11-190421
292. Armeanu S, Krusch M, Baltz KM, Weiss TS, Smirnow I, Steinle A, et al. Direct and natural killer cell-mediated antitumor effects of low-dose bortezomib in hepatocellular carcinoma. Clin Cancer Res (2008) 14(11):3520–8. doi: 10.1158/1078-0432.CCR-07-4744
293. Sokolowska-Wojdylo M, Lugowska-Umer H, Maciejewska-Radomska A. Oral retinoids and rexinoids in cutaneous T cell lymphomas. Postepy Dermatol Alergol (2013) 30(1):19–29. doi: 10.5114/pdia.2013.33375
294. Zhang C, Duvic M. Retinoids: Therapeutic applications and mechanisms of action in cutaneous T cell lymphoma. Dermatol Ther (2003) 16(4):322–30. doi: 10.1111/j.1396-0296.2003.01644.x
295. Fox FE, Kubin M, Cassin M, Niu Z, Trinchieri G, Cooper KD, et al. Retinoids synergize with interleukin-2 to augment ifn-gamma and interleukin-12 production by human peripheral blood mononuclear cells. J Interferon Cytokine Res (1999) 19(4):407–15. doi: 10.1089/107999099314117
296. Meunier L, Bohjanen K, Voorhees JJ, Cooper KD. Retinoic acid upregulates human langerhans cell antigen presentation and surface expression of hla-Dr and Cd11c, a beta 2 integrin critically involved in T cell activation. J Invest Dermatol (1994) 103(6):775–9. doi: 10.1111/1523-1747.ep12413014
297. Budgin JB, Richardson SK, Newton SB, Wysocka M, Zaki MH, Benoit B, et al. Biological effects of bexarotene in cutaneous T cell lymphoma. Arch Dermatol (2005) 141(3):315–21. doi: 10.1001/archderm.141.3.315
298. Richardson SK, Newton SB, Bach TL, Budgin JB, Benoit BM, Lin JH, et al. Bexarotene blunts malignant T cell chemotaxis in sezary syndrome: Reduction of chemokine receptor 4-positive lymphocytes and decreased chemotaxis to thymus and activation-regulated chemokine. Am J Hematol (2007) 82(9):792–7. doi: 10.1002/ajh.20952
299. Tanita K, Fujimura T, Sato Y, Lyu C, Kambayashi Y, Ogata D, et al. Bexarotene reduces production of Ccl22 from tumor-associated macrophages in cutaneous T cell lymphoma. Front Oncol (2019) 9:907. doi: 10.3389/fonc.2019.00907
300. Gorgun G, Foss F. Immunomodulatory effects of rxr rexinoids: Modulation of high-affinity il-2r expression enhances susceptibility to denileukin diftitox. Blood (2002) 100(4):1399–403. doi: 10.1182/blood-2002-01-0300
301. Panchal MR, Scarisbrick JJ. The utility of bexarotene in mycosis fungoides and sezary syndrome. Onco Targets Ther (2015) 8:367–73. doi: 10.2147/OTT.S61308
302. Huen AO, Rook AH. Toll receptor agonist therapy of skin cancer and cutaneous T cell lymphoma. Curr Opin Oncol (2014) 26(2):237–44. doi: 10.1097/CCO.0000000000000048
303. Morrison C, Baer MR, Zandberg DP, Kimball A, Davila E. Effects of toll-like receptor signals in T cell neoplasms. Future Oncol (2011) 7(2):309–20. doi: 10.2217/fon.10.185
304. Rook AH. The beauty of tlr agonists for ctcl. Blood (2012) 119(2):321–2. doi: 10.1182/blood-2011-11-391243
305. Manfrere CK, Torrealba MP, Miyashiro DR, Pereira NZ, Yoshikawa FSY, de MOL, et al. Profile of differentially expressed toll-like receptor signaling genes in the natural killer cells of patients with sezary syndrome. Oncotarget (2017) 8(54):92183–94. doi: 10.18632/oncotarget.21006
306. Soler-Machin J, Gilaberte-Calzada Y, Vera-Alvarez J, Coscojuela-Santaliestra C, Martinez-Morales J, Osan-Tello M. [Imiquimod in treatment of palpebral mycosis fungoides]. Arch Soc Esp Oftalmol (2006) 81(4):221–3. doi: 10.4321/s0365-66912006000400009
307. Dummer R, Urosevic M, Kempf W, Kazakov D, Burg G. Imiquimod induces complete clearance of a puva-resistant plaque in mycosis fungoides. Dermatology (2003) 207(1):116–8. doi: 10.1159/000070962
308. Onsun N, Ufacik H, Kural Y, Topcu E, Somay A. Efficacy of imiquimod in solitary plaques of mycosis fungoides. Int J Tissue React (2005) 27(4):167–72.
309. Martinez-Gonzalez MC, Verea-Hernando MM, Yebra-Pimentel MT, Del Pozo J, Mazaira M, Fonseca E. Imiquimod in mycosis fungoides. Eur J Dermatol (2008) 18(2):148–52. doi: 10.1684/ejd.2008.0352
310. Deeths MJ, Chapman JT, Dellavalle RP, Zeng C, Aeling JL. Treatment of patch and plaque stage mycosis fungoides with imiquimod 5% cream. J Am Acad Dermatol (2005) 52(2):275–80. doi: 10.1016/j.jaad.2004.04.049
311. Chiam LY, Chan YC. Solitary plaque mycosis fungoides on the penis responding to topical imiquimod therapy. Br J Dermatol (2007) 156(3):560–2. doi: 10.1111/j.1365-2133.2006.07599.x
312. Kim YH, Gratzinger D, Harrison C, Brody JD, Czerwinski DK, Ai WZ, et al. In situ vaccination against mycosis fungoides by intratumoral injection of a Tlr9 agonist combined with radiation: A phase 1/2 study. Blood (2012) 119(2):355–63. doi: 10.1182/blood-2011-05-355222
Keywords: cutaneous T cell lymphoma, mycosis fungoides, immune system, skin neoplasm, oncology
Citation: Fay CJ, Awh KC, LeBoeuf NR and Larocca CA (2023) Harnessing the immune system in the treatment of cutaneous T cell lymphomas. Front. Oncol. 12:1071171. doi: 10.3389/fonc.2022.1071171
Received: 15 October 2022; Accepted: 01 December 2022;
Published: 12 January 2023.
Edited by:
Jianjun Qiao, Zhejiang University, ChinaReviewed by:
Robert Gniadecki, University of Alberta, CanadaChalid Assaf, Helios Hospital Krefeld, Germany
Niels Odum, University of Copenhagen, Denmark
Brian Poligone, Rochester Skin Lymphoma Medical Group, PLLC, United States
Pietro Quaglino, University of Turin, Italy
Copyright © 2023 Fay, Awh, LeBoeuf and Larocca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia A. Larocca, Y2xhcm9jY2FAYndoLmhhcnZhcmQuZWR1