 Ayenachew Bezawork-Geleta
Ayenachew Bezawork-Geleta James Dimou2,3
James Dimou2,3 Matthew J. Watt
Matthew J. Watt- 1Department of Anatomy and Physiology, School of Biomedical Sciences, The University of Melbourne, Melbourne, VIC, Australia
- 2Department of Surgery, The University of Melbourne, Parkville, VIC, Australia
- 3Department of Neurosurgery, The Royal Melbourne Hospital, Parkville, VIC, Australia
A primary brain tumor glioblastoma is the most lethal of all cancers and remains an extremely challenging disease. Apparent oncogenic signaling in glioblastoma is genetically complex and raised at any stage of the disease’s progression. Many clinical trials have shown that anticancer drugs for any specific oncogene aberrantly expressed in glioblastoma show very limited activity. Recent discoveries have highlighted that alterations in tumor metabolism also contribute to disease progression and resistance to current therapeutics for glioblastoma, implicating an alternative avenue to improve outcomes in glioblastoma patients. The roles of glucose, glutamine and tryptophan metabolism in glioblastoma pathogenesis have previously been described. This article provides an overview of the metabolic network and regulatory changes associated with lipid droplets that suppress ferroptosis. Ferroptosis is a newly discovered type of nonapoptotic programmed cell death induced by excessive lipid peroxidation. Although few studies have focused on potential correlations between tumor progression and lipid droplet abundance, there has recently been increasing interest in identifying key players in lipid droplet biology that suppress ferroptosis and whether these dependencies can be effectively exploited in cancer treatment. This article discusses how lipid droplet metabolism, including lipid synthesis, storage, and use modulates ferroptosis sensitivity or tolerance in different cancer models, focusing on glioblastoma.
Introduction
Glioblastoma, designated as WHO grade 4, is the most common and most aggressive intra-axial brain tumor and has limited treatment options (1–3). The development of cancer therapies has improved outcomes for most malignancies, with the five-year survival of patients ranging from 25–95% (4–6). In contrast, glioblastoma remains overwhelmingly lethal, with only ~5% of patients alive five years after diagnosis (7).
The gold standard treatment of glioblastoma includes maximal safe surgical resection (8) combined with radiotherapy and temozolomide chemotherapy, which is referred to in neuro-oncology circles as the Stupp protocol (1, 2); however, relapse remains inevitable. Temozolomide (chemical name 3-methyl-4-oxoimidazo[5,1-d](1–3, 5)tetrazine-8-carboxamide (9)) has several advantages, such as oral administration, a favorable side-effect profile, evidence of blood–brain barrier penetration, acidic environment stability, and limited drug interaction-related toxicity. The original randomized controlled trial published by Stupp et al. (1, 2) showed that adding postoperative adjuvant temozolomide chemotherapy to surgery and radiotherapy increased the median survival of glioblastoma patients from 12.1 months to 14.6 months. Bevacizumab, a monoclonal antibody targeted to vascular endothelial growth factor A (VEGF-A), has been shown to offer some progression-free survival benefits in patients who develop recurrent glioblastoma (10). Another study has shown that epigenetic silencing of the DNA repair gene O-6-methylguanine-DNA methyltransferase (MGMT) offers a superior response to chemoresistance tumors to temozolomide (11). However, it has limited therapeutic efficacy due to recurrence.
For three decades, other strategies have been pursued to improve the survival outcomes of glioblastoma patients (12, 13). Considerable efforts have been made to catalog genetic aberrations and associated disrupted signaling pathways in glioblastoma for the purpose of developing novel targeted therapies. The first genome atlas study by The Cancer Genome Atlas (TCGA) Research Network uncovered 453 validated missense somatic mutations in glioblastoma (14). Subsequent systematic analysis at the genomic and transcriptomic levels showed that 71 mutated genes as significant pathogenic factors (14, 15). Histological and molecular analysis further revealed a landscape of tumor heterogeneity, which led to efforts to differentiate glioblastoma into distinct molecular subtypes (16, 17). An early attempt at such molecular classification by Verhaak et al. distinguished glioblastoma into four molecular subtypes: proneural, mesenchymal, classical, and neural (18). The classical subtype was characterized by amplification of epidermal growth factor receptor (EGFR) and associated with the upregulation of retinoblastoma (RB), sonic hedgehog (SHH), and notch signaling-related pathway genes. The classical type typically lacks of TP53 mutation. Conversely, mesenchymal subtype glioblastomas were characterized by high expression of chitinase 3 like 1 (CHI3L1) and tyrosine-protein kinase Met (MET), a high frequency of neurofibromin 1 (NF1) mutation/deletion, and low NF1 gene expression. Key proneural subtype markers are TP53 aberrations and metabolic enzyme isocitrate dehydrogenase 1 (IDH1) mutation and platelet-derived growth factor receptor alpha (PDGFRA). The neural subtype expressed neurofilament light chain (NEFL), gamma-aminobutyric acid type A receptor subunit alpha1 (GABRA1), synaptotagmin 1 (SYT1), and solute carrier family 12 member 5 (SLC12A5). It has been proposed that these specific subtypes of glioblastoma develop due to promutagenic aberrations in distinct cells of origin (18). However, due to the inclusion of mRNA from glioblastoma-associated stroma (nonmalignant cells) along with tumor cells in the Verhaak et al. transcriptome study, these four pathological subtypes subjected to further interrogation. Accordingly, Wang et al. (19) used stringent criteria to distinguish specific mRNA from peripheral nonmalignant cells by comparing the transcriptome of core versus peripheral glioblastoma surgery specimens and mRNA profile from glioblastoma cell culture and revealed the presence of only three pathological subtypes (i.e., classical, proneural, and mesenschymal subtypes). Subsequent studies also classify glioblastoma into different prognostic subtypes (20–22) using different multi-omics signatures.
Recently, the integration of cross-platform analyses coupling metabolomic profiling with genomics and proteomics has provided an in-depth understanding of the metabolic programming that occurs during tumor growth. It has identified key metabolic nodes specific to glioblastoma and their molecular context. Notable findings of mutations in the metabolic enzyme IDH1, representing an early event in gliomagenesis (22, 23), have led to an in-depth study of metabolic status across all grades and subtypes of glioma. However, it must be stressed that the presence of IDH1 mutation now precludes the formal pathological diagnosis of glioblastoma, as it is now genetically defined as “IDH-wildtype” in the most recent edition of the WHO Classification of CNS Tumors (24).
Further, it has been found that glucose, glutamine, and tryptophan metabolism play roles in glioblastoma progression and recurrence following surgery and chemotherapy (25–28). Glutamine metabolism changes in glioblastoma have been incorporated into the clinical practice of noninvasive metabolic imaging strategies for stratifying patients, monitoring treatment response, and prognostication (29–32). Advances in metabolic imaging modalities, including magnetic resonance spectroscopy (MRS), positron emission tomography (PET), single-photon emission computerized tomography (SPECT), mass spectrometry imaging (MSI), and fluorescence imaging have given researchers and clinicians unprecedented opportunities for in vivo measurements of glutamine metabolism and clinical management of gliomas (29–32). In particular, PET using 11C-glutamine allows noninvasive visualization of glioma and various malignant tumors in multiple organs and for subsequent monitoring of responses to clinical treatments (29).
Unfortunately, our ever-improving understanding of the molecular basis of glioblastoma initiation and progression has not yet translated to therapeutic success (17). This requires the interrogation of novel molecular signaling mechanisms and metabolic processes in glioblastoma and their exploitation for therapeutic progress. To this end, lipid metabolism and ferroptosis have only recently been explored and identified as key regulators in the initiation and maintenance of glioblastoma (33–35) and other cancers (36–38). Notable related discoveries and translations include the recently developed and characterized selective, irreversible, and potent fatty acid synthase (FASN) inhibitor IPI-9119 (39), as well as the identification of druggable targets, including ATP-citrate lyase (ACLY) (40) and the plasma membrane lipid importer CD36 (41). This review examines recent studies showing lipid droplets’ critical role in suppressing ferroptosis to promote tumorigenesis.
Features of ferroptosis
Ferroptosis is a recently discovered type of programmed cell death that exhibits unique morphological, biochemical and genetic features compared to previously characterized processes of cell death, including apoptosis, necrosis, and autophagy (42–44). Accordingly, ferroptosis does not depend on the mechanisms by which cancer cells frequently evade apoptosis, such as the activation of caspases. Apoptosis inhibitors (e.g., Z-VAD-FMK, BOC-D-FMK, wortmannin, and necrostatin-1) failed to protect cells from ferroptosis mediated cell death (42–44). Instead, ferroptosis is mainly embodied by accumulation iron, leading to lethal levels of lipid peroxidation. The typical cells in ferroptosis show extensive lipid peroxidative damage to biological membranes, shrunken mitochondria, and an increase in mitochondrial membrane density (45), indicating that execution of ferroptosis requires active mitochondrial function. Interestingly, the nucleus remains intact during ferroptosis, in contrast to other cell death mechanisms that lead to fragmented nuclei (43, 46, 47). Ferroptosis is also caused by a redox imbalance of cellular homeostasis, leading to the formation of a myriad of secondary byproducts, including breakdown products of lipid peroxides (e.g., malondialdehyde (MDA), 4-hydroxynonenal (4HNE), 4-hydroxyhexenal (4HHE), and 4-oxo-nonenal (4ONE)) and oxidized and modified proteins). This chain reaction may eventually lead to the breakdown of membrane integrity and the rupture of organelles and/or cell membranes and, ultimately, result in cell death (43–45, 48). In contrast, ferroptosis-mediated cell death can be modulated by the expression and activity of proteins that regulate the levels, transport, storage, and metabolism of iron, cystine, cysteine, GSH, glutamine, and selenium (43–45, 48). Antioxidant defense enzymes that repair oxidative damage to lipids, such as Glutathione peroxidase 4 (GPX4), are important inhibitors of ferroptosis (43–45, 48). Recently, it has been reported that Anti-TfR1 3B8 2A1 and anti-MDA 1F83 can be used to detect ferroptosis in both cell culture and xenograft tumor sections (49).
Although it was initially proposed to be the mechanism of neuronal death in many neurodegenerative diseases (43, 50, 51), emerging data indicate that ferroptosis plays an important role in cancer development and drug resistance (45). Therefore, inducing ferroptosis represents a potentially orthogonal approach to drug discovery to kill cancer cells that have developed resistance to apoptosis. Thus, a clearer understanding of ferroptosis in glioblastoma and its role in drug resistance may positively impact clinical practice (52, 53).
In and out of lipids: Lipid droplets as intracellular sources of fatty acids
Lipid droplets accumulate in cancers
Lipid droplets are fundamental in the storage and release of fatty acids. Lipid droplets are ubiquitously present organelles in many cells with a hydrophobic core surrounded by a single layer of phospholipids decorated with various sets of proteins. The hydrophobic core contains neutral lipids, including triacylglycerols, sterol esters, retinyl esters and 1-O-acylceramides (54–57). Emerging evidence indicated lipid droplet accumulation in several cancers (58, 59), especially in hypoxic cancer cells, and several studies have linked lipid droplet abundance with more aggressive tumor phenotype (34, 36, 58, 60). It has also shown lipid metabolism-dependent proliferation and survival of glioblastoma following radiation (61, 62), antiangiogenic (63) and ketone diet therapy (64). As such, they have recently been proposed to play direct roles in many cancers and considered a hallmark of fundamental tumor processes (58, 60, 65). The following sections discuss the possible roles of lipid droplet metabolism, including (1) fatty acid storage as neutral lipids, (2) lipolysis of neutral lipids, and (3) lipophagy in limiting or triggering ferroptosis.
De novo lipogenesis and ferroptosis
Lipogenesis (lipid acquisition) is an energy-expensive process of fatty acid synthesis and subsequent esterification to neutral lipids into lipid droplets. The primary process of synthesizing fatty acids from acetyl‐CoA subunits that are produced most commonly from carbohydrate catabolism is called de novo lipogenesis (DNL) (66). Acetyl-coenzyme A (acetyl-CoA) is a key metabolite precursor in DNL, and its abundance is closely monitored by cellular adaptive mechanisms. While glucose is the most common supplier of carbon units for DNL (Figure 1A), fructose is metabolized to glycerol and subsequently converted to a lipogenic substrate glycerol-3-phosphate (G3P) that can drive DNL.
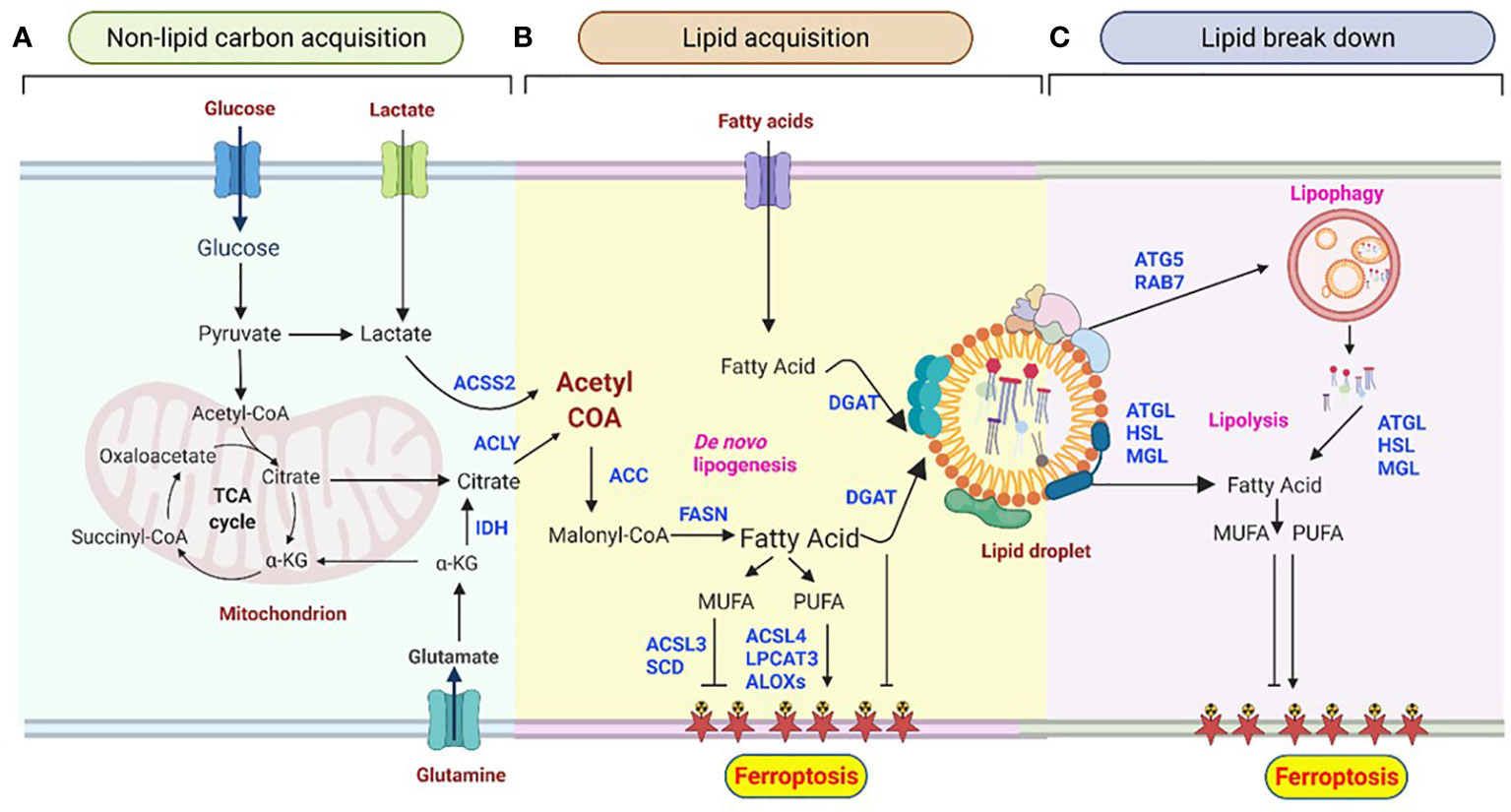
Figure 1 Lipid droplet metabolism is central to shaping the ferroptotic response. (A) Non-lipid carbon acquisition of carbon in cancer cells. Glucose- and glutamine-derived citrate, which results from increased glycolysis and glutaminolysis, is first converted to acetyl-coenzyme A (acetyl-CoA) by ATP-citrate lyase (ACLY). Acetyl-CoA can also be derived from acetate. (B) De novo lipogenesis (DNL) and esterification in ferroptosis. Acetyl-CoA is converted to malonyl-CoA by acetyl-CoA carboxylase (ACC) and condensed by fatty acid synthase (FAS). Acyl-CoA synthetase long-chain 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) mediate the production of polyunsaturated fatty acids (PUFAs), which are essential for the induction of ferroptosis. In contrast, acyl-CoA synthetase long-chain 3 (ACSL3) and stearoyl CoA desaturase (SCD) contribute to the synthesis of monounsaturated fatty acids (MUFAs), leading to ferroptosis resistance. Arachidonate lipoxygenases (ALOXs) catalyze the stereospecific insertion of oxygen into PUFAs, thereby promoting ferroptosis. Diacylglycerol acyltransferase (DGAT1/2)-mediated triglyceride synthesis and lipid droplet formation act as a sink for free fatty acids, thus preventing their peroxidation.(C) Lipid degradation in ferroptosis. The selective degradation of lipid droplets by member RAS oncogene family 7 (RAB7A)- and adipocyte triglyceride lipase (ATGL)-related lipophagy increases the production of free fatty acids for subsequent ferroptosis. Lipolysis may provide PUFAs, thus stimulating lipid peroxidation and sensitizing cells to ferroptosis. Lipases may also provide MUFAs that reduce the abundance of oxidizable PUFAs in membranes, thereby restricting lipid peroxidation and ferroptosis. Lipid droplets act as buffers of lipid flux and release, thereby emerging as master regulators of ferroptotic sensitivity.
Glutamine is another non-lipid metabolite that contributes carbon to lipogenic acetyl-CoA (Figure 1A) through two distinct pathways: Glutamine metabolism can produce citrate either through the tricarboxylic acid (TCA) cycle in mitochondria or by the reductive carboxylation of cytoplasmic α-ketoglutarate (αKG) to citrate by IDH1 (Figure 2) (67, 68). Interestingly, many cancer cells, including glioblastoma, generate 10–25% of their lipogenic acetyl-CoA from glutamine through reductively carboxylation (69), highlighting the general use of reductive carboxylation as the primary route to convert non-lipid carbon to lipids in cancer cells. In agreement with this, blood-borne glucose contribute minimal fraction to the acetyl-CoA pool of glioblastoma and brain metastases that originate from other tissues (70). In addition to a direct contribution of supplying a carbon unit, glucose and glutamine uptake promotes lipogenesis through transcriptional regulation in a sterol regulatory element-binding protein (SREBP) dependent manner in glioblastoma (71, 72).
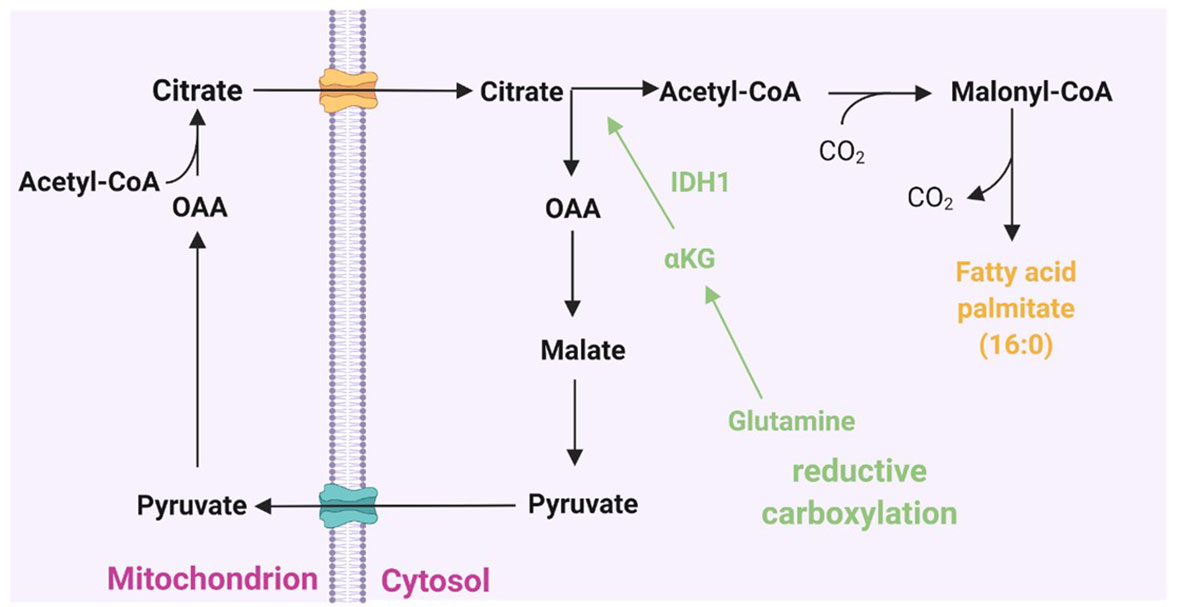
Figure 2 Cytoplasmic citrate pool and fatty acid synthesis. Acetyl-CoA couples with oxaloacetate (OAA) to form citrate at the beginning of the citric acid cycle. Citrate can then shuttle across the mitochondrial membrane to the cytoplasmic citrate pool. In the cytosol, citrate lyase splits citrate back into acetyl-CoA and OAA. The latter can then return to the mitochondrion. Acetyl-CoA is activated in the cytoplasm for incorporation into fatty acids by acetyl-CoA carboxylase to form malonyl-CoA and then to de novo fatty acid synthesis. Glutamine-derived α-ketoglutarate (α-KG) can add to the cytoplasmic citrate pool via reductive carboxylation to provide an alternative source of lipid synthesis.
Acetate, often dispensable for cells, has also been shown as a source of acetyl-CoA for DNL in cancer cells, especially primary glioblastomas and other cancer metastasis to brain (73, 74). In this case, three important processes need to be considered: acetate production, its intercellular import across the cell membrane, and the carbon fluxes from acetate to lipids. Imported acetate is converted to acetyl-CoA in a reaction catalyzed by acetyl-CoA synthetase 2 (ACSS2) (Figure 1A) (75). Thus, hypoxia-associated deficits in acetyl-CoA can be supplemented by increasing the use of free extracellular acetate (including plasma, interstitial fluid, and neighboring cells) and the intercellular pool of mainly acetate released from the deacetylation of histones and other cellular proteins (76). In line with acetate metabolism, apparent ACSS2 expression is associated with poor progenesis in glioblastoma patients (73, 74). Despite the fundamental importance of acetate and its vital roles to several intracellular and extracellular metabolism, little is known about the mechanisms and regulatory processes in acetate metabolism.
Cytoplasmic acetyl-CoA is then carboxylated by acetyl-CoA carboxylase (ACC) to produce malonyl-CoA (Figure 1B), which is further used to synthesizes medium-and long-chain fatty acids. The key three proteins involved in the initial fatty acyl chain biosynthesis, ACLY, ACC, and FASN, are commonly upregulated and activated in many cancers (36, 60, 65), and they are in the frontline of pre-clinical target evaluations, drug discovery pipelines, and clinical trials. For instance, the therapeutic targeting of FASN is currently being explored in a phase 2 clinical trial in patients with glioblastoma using the FASN inhibitor TVB-2640 in combination with bevacizumab (NCT03032484) (28).
De novo synthesized fatty acids are then further esterified to neutral lipids and stored in their nontoxic (inert, neutral) form in lipid droplets (Figure 1B). Esterification starts with the acylation of glycerol-3-phosphate with two fatty acids to produce phosphatidic acid (PA), followed by dephosphorylation to yields diacylglycerol (DAG). DAG then serves as a precursor for the synthesis of both phospholipids and acylation to yield triacylglycerols (TAGs), which is catalyzed by diacylglycerol acyltransferase 1 and 2 (DGAT1 and DGAT2) (77, 78). TAG is stored in lipid droplets as energy reservoir, supplying the cell with energy when required.
Lipid droplets also store 1-O-acylceramides and cholesteryl ester. Although some studies indicated a minimal abundance of1-O-acylceramides in tissue (0.25-1.3 per mg of mice tissue), the indication of its level during Western diet consumption (79) warrants special attention, mainly its correlation with cancer progression in obese glioblastoma patients. Cholesteryl ester is another neutral lipid stored in lipid droplets. Acyl-CoA acyltransferase, also known as acyl-Coenzyme A or cholesterol acyltransferase (SOAT1 and SOAT2), catalyzes the formation of cholesteryl esters, using cholesterol and long-chain fatty acyl-CoA as substrates. Both isoforms of acyl-CoA acyltransferase (ACAT1 and ACAT2) are highly expressed and post-translationally regulated in glioblastomas and other cancers, and their expression and activation levels correlate with patient survival (79–81). In vivo studies showed that both genetic silencing of ACAT1 or blocking its activity using inhibitors suppresses tumor growth (80, 82, 83). These suggest that targeting ACAT1 and cholesteryl ester synthesis may be a promising anticancer strategy.
The de novo synthesis of fatty acids can be extended to the formation of either saturated or unsaturated fatty acids. Unsaturated fatty acids can be further classified into monounsaturated fatty acids [MUFAs, with only one double bond, e.g., oleic acid (18:1)] and polyunsaturated fatty acids [PUFAs, with at least two double bonds, e.g., linoleic acid (18:2 n-6) and α-linolenic (18:3 n-3)]. Unsaturated fatty acids can bind membrane phospholipids in differential degrees that subject tumor cells to oxidative stress. Studies have revealed that an increase in the MUFA/PUFA ratio results in fewer peroxidation-susceptible targets, ultimately reducing the susceptibility of cancer cells to ferroptosis (84). On the other hand, an increase in the synthesis of PUFAs can promote subsequent lipid peroxidation in cancer cells under oxidative stress conditions, suggesting that the formation of acetyl-CoA derivatives (de novo synthesis) of these PUFA is necessary for producing ferroptosis death signals (43, 45). Similarly, the esterification of fatty acids into neutral lipids has been shown to deplete the substrates for lipid peroxidation and increase the resistance of cancer cells to ferroptosis-mediated cell death (34, 36, 60, 85).
Lipid peroxidation normally occurs at a slow rate because PUFAs are required to react with the free radicals generated by iron acyl-CoA synthase long-chain family member 4 (ACSL4). Using genetic ablation and pharmaceutical inhibition, it has been shown that ACSL4 is a rate-limiting enzyme to catalyze the conversion of long-chain PUFAs, such as arachidonic acid and eicosapentaenoic acid to PUFA-CoA, and ultimately this increases lipid peroxidation and decreases resistance to ferroptosis (86–88). Under oxidative or energetic stress, PUFAs, particularly arachidonoyl (AA, 20:4 n-6) and adrenic acid (AdA), are oxygenated by different classes of enzymes, including ACSL4, lysophosphatidylcholine acyltransferase (LPCAT), and 15-lipoxygenase (15-LOX/ALOX15), to generate PUFA-containing phospholipids and various bioactive lipids that modulate sensitivity to ferroptosis. In this regard, genetic or pharmacological inhibition of ACSL4 suppresses ferroptosis in glioblastoma (86). On the other hand, overexpression of ACSL4 was shown to increase the levels of ferroptosis markers, including 5-hydroxyeicosatetraenoic acid (HETE), 12-HETE, and 15-HETE (86), indicating a key role of ACSL4 in regulating ferroptosis and proliferation of glioma cells (89). Independent experimental evidence showed that suppressing miR-670-3p, which targets ACSL4, also modulates the sensitivity of glioblastoma cells to ferroptosis-mediated cell death (87, 90).
Esterification and ferroptosis
Blocking the esterification of fatty acyl-CoA prevents not only the formation of triglycerides but also modifies cell resistance to ferroptosis. It has been shown that in 3D tumor spheroids and in vivo, DGAT inhibition induces significant cytotoxic effects, especially when combined with dietary long-chain PUFAs (LC-PUFAs), implicating the combination of diet (LC-PUFAs) and DGAT inhibitor (DFATi) administration as a highly relevant therapeutic combination to induce ferroptosis (85). Mechanistically, DGATi administration inhibits lipid droplet formation resulting in the availability of more LC-PUFAs for peroxidation and ferroptotic cell death (85). In contrast, DGAT1 inhibition was recently reported to drive fatty acid-dependent oxidation in mitochondria generating high levels of reactive oxygen species, leading to apoptosis and significant growth inhibitory effects in glioblastoma (91). These contradictory reports are likely due to the difference in cell lineage, or a potential secondary effect associated with DGAT1 inhibition. Therefore, it will be relevant in the future to dissect the relative contributions of DGAT in different cancers, including glioblastoma, and the potential overlapping effects of DGAT inhibition on ferroptosis and apoptosis.
Lipolysis and lipophagy and their role in cell sensitivity to ferroptosis
Similar to lipid fluxes into lipid droplets that act as buffers, the release of fatty acids from storage controls the cancer cells’ fate, including ferroptosis. Lipid droplet breakdown occurs via two major mechanisms-lipolysis, and lipophagy (Figure 1C), which provide a substrate for lipid peroxidation during ferroptosis. Unlike lipogenesis, the lipolysis process enables a highly regulated release of fatty acids from TAGs and is mediated by three essential lipases: adipocyte triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL) (92–94). These three enzymes work together to promote the lipolysis of triglycerides and produce fatty acids and glycerol. ATGL catalyzes the first rate-limiting reaction whereby triglycerides are hydrolyzed to DAG. HSL then transforms DAG to monoacylglycerol (MAG), and MGL finally converts MAG to fatty acids and glycerol (93–96). However, the molecular regulation of the lipolysis of lipid droplet-containing triglycerides is complex. It involves a combination of subcellular localization, posttranslational modification (particularly phosphorylation), and protein-protein interactions (92, 97). For instance, hypoxia-induced lipid droplet-associated protein (HILPDA), also called hypoxia-induced gene-2 (HIG2), inhibits ATGL activity (98). Experimental evidence showed the correlation of the HIG2 expression with the glioma tumor grade and glioblastoma patient prognosis (34, 99).
Catabolism of lipid droplets also occur via lipophagy, a form of selective (macro)autophagy. Lipophagy is a process that tags specific lipid droplets and traffic whole lipid droplets to lysosomes for bulk degradation (Figure 1C). Lysosomal localized hydrolytic enzymes, such as triglycerides and cholesteryl ester hydrolase lysosomal acid lipase (LAL) (100), liberate neutral lipids stored in lipid droplet and generate free fatty acids and cholesterol in the cell. Although mechanistically lipolysis (the step-wise release of stored neutral lipids from lipid droplets) and lipophagy (a complete breakdown of lipid droplets) are distinct metabolic processes, it is not yet clear whether there is considerable crosstalk between the two processes and this may serve a distinct purpose in the cell (100, 101). It has been shown that perilipins (PLINs), lipid droplet surface proteins, act as gatekeepers to the lipophagy process and their degradation is a prerequisite for lipophagy. During starvation, perilipin 2 (PLIN2) and perilipin 3 (PLIN3) are proteolytically removed in parallel with the translocation of cytosolic ATGL and macroautophagy proteins onto lipid droplets (102). There is also evidence that PLIN2-mediated lipophagy regulation emerges as a key nodal point in modulating cellular sensitivity to ferroptosis. PLIN2 affects the proliferation of gastric carcinoma cells by modulating ferroptosis-related genes, including acyl-CoA synthetase long-chain family member 3 (ACL3), arachidonate 15-lipoxygenase (ALOXs), microtubule-associated protein 1 light chain 3 alpha (LC3), and the transcription factors pr/set domain 11 and importin 7 (IPO7) (103). Likewise, PLIN2 expression contributed to a decreased arachidonate 15-lipoxygenase (ALOX15) expression and arrested the occurrence of ferroptosis in gastric cancer. Conversely, PLIN2 knockdown facilitated a higher ALOX15 expression and accelerated ferroptosis (103). In contrast, increased lipid storage by tumor protein D52 (TPD52), a PLIN2 interactor protein, diminished lipid peroxidation to trigger ferroptosis (104). Recently, PLIN2 was also shown to play a key role in glioblastoma pathology, although its role in ferroptosis has not been directly investigated (105).
It has also been shown that the knockdown of RAB7A (member RAS oncogene family 7A) and autophagy-associated gene 5 (ATG5), a cargo receptor of lipid droplets, can prevent induced lipid peroxidation and subsequent ferroptosis (104, 106). Mechanistically, RAB7A is an indispensable factor for docking lysosomes to the lipid droplet surface during lipophagy under nutrient deprivation (107). Moreover, RAB7A has been demonstrated to act as a tumor suppressor in glioblastoma and prostate cancer (108), unlike other cancers.
Although emerging evidence reinforces the idea that the amount of lipid and the localization of lipids in lipid droplets potentially affect the ability to induce ferroptosis in cancer cells, studies on the effect of lipolysis and lipophagy on sensitizing glioblastoma cells for ferroptosis are limited. There is limited information on how microenvironment and disease progression affect the biogenesis and breakdown of lipid droplets (101). There is a need for a better understanding of specific trafficking itinerary of lipophagy which may lead to new anticancer approaches, particularly against “lipid addicted tumors” like glioblastoma. Finally, the molecular regulators of 1-O-acylceramide hydrolysis and whether this process impacts tumorigenesis are unknown.
Summary
In the field of cancer metabolism reprogramming and plasticity, we have learned some fascinating lessons. One of the classical examples of metabolic reprogramming is the Warburg effect (109) which was reported almost 100 years ago and demonstrated a high glucose uptake and an increase in the glycolysis rate in cancer cells in an aerobic environment. In addition to high glucose consumption, glioblastoma has increased glutamine uptake rate to fuel proliferation compared with healthy cells. Emerging data have also shown that lipid metabolism provides an important energy substrate and carbon source for glioblastoma cells, affecting cancer cell plasticity and persistence during therapy (110, 111). Multiple lines of evidence also suggest that lipid droplets act as a central hub for lipid trafficking in glioblastoma, allowing lipids to move in and out of lipid droplets.
However, developing cancer treatments by targeting altered lipid metabolism is in its infancy. It remains challenging, primarily due to an incomplete understanding of the mechanisms that regulate lipid synthesis, esterification, lipolysis and lipophagy in cancer cells (34, 36, 58, 112, 113). In addition, our understanding of the pathways downstream of peroxidized phospholipids that execute ferroptosis remains extremely limited (Box 1). For instance, it is unclear whether peroxidized phospholipids are cytotoxic by themselves or when metabolized into products (e.g., electrophilic intermediates) that function as the primary drivers of ferroptosis or other adaptive metabolic reprogramming. The extent to which a physiological niche (cell/tissue function) alters lipid droplets contributions to ferroptosis and the unique context-dependent vulnerabilities that can be targeted in combinatorial approaches have also not been well explored. Moreover, metabolic byproducts accumulate in the tumor and its microenvironment, increasing the need for cancer cells to engage waste management and recycling pathways, as shown in the case of other metabolic byproducts such as acetate, ammonia, and free fatty acids (73–76, 114–116). However, the existence of waste management and the recycling of peroxidized phospholipids are not yet clear and require further study.
What is beyond question is the continued lack of life-prolonging treatment for glioblastoma patients despite significant advances in genetic and molecular characterization and almost twenty years since the introduction of temozolomide as the standard of care. This necessitates the discovery of nuanced avenues to understand glioblastoma formation to overcome therapeutic resistance. Ferroptosis and lipid metabolism offer some very early promise in this direction and are worthy of further exploration in this lethal cancer.
BOX 1 Unanswered questions.
Although monitoring the balance of PUFAs and MUFAs is known to predict the sensitivity of cells to ferroptosis, cellular sensing and the feedback mechanism that balances PUFAs/MUFAs remain to be investigated.
There is imputes to define the relationship between lipid droplets, fatty acid metabolism, and ferroptosis. However, key protein mediators of these biological processes remain to be discovered.
The most important factors for ferroptosis-dependent cell death are lipid peroxides that self-propagate along the plasma membrane and result in the accumulation of oxidatively damaged lipids. Our understanding of the downstream pathways of peroxidized phospholipids that execute ferroptosis and potentially contribute to tumorigenesis remains at a nascent phase. The question remains whether there is a repair or antagonism mechanism of peroxide lipids at damage loci in the cell membrane. Are oxidatively damaged membranes prone to recycling? Can this metabolic plasticity contribute to glioblastoma therapeutic resistance? How do these factors cooperate their functions in cancer microenvironments?
Many cancers, including glioblastoma, consist of both stem and nonstem cells; however, the degree to which these cells are resistant to ferroptosis remains mostly unknown.
Author contributions
Conceptualization, AB-G. Writing-original draft preparation, AB-G, JD, and MW. Review and editing, AB-G, JD, and MW. Funding acquisition, AB-G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Brian Foundation and Rebecca L Cooper Medical Research Foundation for AB-G.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi: 10.1056/NEJMoa043330
2. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7
3. Park JK, Hodges T, Arko L, Shen M, Dello Iacono D, McNabb A, et al. Scale to predict survival after surgery for recurrent glioblastoma multiforme. J Clin Oncol (2010) 28(24):3838–43. doi: 10.1200/JCO.2010.30.0582
4. Ye X, Mahmud S, Skrabek P, Lix L, Johnston JB. Long-term time trends in incidence, survival and mortality of lymphomas by subtype among adults in Manitoba, Canada: A population-based study using cancer registry data. BMJ Open (2017) 7(7):e015106. doi: 10.1136/bmjopen-2016-015106
5. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70(1):7–30. doi: 10.3322/caac.21590
6. Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn MJ, et al. Five-year overall survival for patients with advanced NonSmall-cell lung cancer treated with pembrolizumab: Results from the phase I KEYNOTE-001 study. J Clin Oncol (2019) 37(28):2518–27. doi: 10.1200/JCO.19.00934
7. Krex D, Klink B, Hartmann C, von Deimling A, Pietsch T, Simon M, et al. Long-term survival with glioblastoma multiforme. Brain (2007) 130(Pt 10):2596–606. doi: 10.1093/brain/awm204
8. Stummer W, Reulen HJ, Meinel T, Pichlmeier U, Schumacher W, Tonn JC, et al. Extent of resection and survival in glioblastoma multiforme: Identification of and adjustment for bias. Neurosurgery (2008) 62(3):564–76. doi: 10.1227/01.neu.0000317304.31579.17
9. Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; m & b 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res (1987) 47(22):5846–52.
10. Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med (2017) 377(20):1954–63. doi: 10.1056/NEJMoa1707358
11. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med (2005) 352(10):997–1003. doi: 10.1056/NEJMoa043331
12. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol (2017) 18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X
13. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med (2014) 370(8):709–22. doi: 10.1056/NEJMoa1308345
14. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455(7216):1061–8. doi: 10.1038/nature07385
15. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell (2013) 155(2):462–77. doi: 10.1016/j.cell.2013.09.034
16. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell (2006) 9(3):157–73. doi: 10.1016/j.ccr.2006.02.019
17. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med (2015) 372(26):2499–508. doi: 10.1056/NEJMoa1407279
18. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi: 10.1016/j.ccr.2009.12.020
19. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell (2017) 32(1):42–56.e6. doi: 10.1016/j.ccell.2017.06.003
20. Teo WY, Sekar K, Seshachalam P, Shen J, Chow WY, Lau CC, et al. Relevance of a TCGA-derived glioblastoma subtype gene-classifier among patient populations. Sci Rep (2019) 9(1):7442. doi: 10.1038/s41598-019-43173-y
21. Park J, Shim JK, Yoon SJ, Kim SH, Chang JH, Kang SG, et al. Transcriptome profiling-based identification of prognostic subtypes and multi-omics signatures of glioblastoma. Sci Rep (2019) 9(1):10555. doi: 10.1038/s41598-019-47066-y
22. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A, et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol (2008) 116(6):597–602. doi: 10.1007/s00401-008-0455-2
23. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360(8):765–73. doi: 10.1056/NEJMoa0808710
24. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
25. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: Beyond IDO and tryptophan depletion. Cancer Res (2012) 72(21):5435–40. doi: 10.1158/0008-5472.CAN-12-0569
26. Ott M, Litzenburger UM, Rauschenbach KJ, Bunse L, Ochs K, Sahm F, et al. Suppression of TDO-mediated tryptophan catabolism in glioblastoma cells by a steroid-responsive FKBP52-dependent pathway. Glia (2015) 63(1):78–90. doi: 10.1002/glia.22734
27. Obara-Michlewska M, Szeliga M. Targeting glutamine addiction in gliomas. Cancers (Basel) (2020) 12(2):310. doi: 10.3390/cancers12020310
28. Badr CE, Silver DJ, Siebzehnrubl FA, Deleyrolle LP. Metabolic heterogeneity and adaptability in brain tumors. Cell Mol Life Sci (2020) 77(24):5101–19. doi: 10.1007/s00018-020-03569-w
29. Ekici S, Nye JA, Neill SG, Allen JW, Shu HK, Fleischer CC, et al. Glutamine imaging: A new avenue for glioma management. AJNR Am J Neuroradiol (2022) 43(1):11–8. doi: 10.3174/ajnr.A7333
30. Cohen AS, Grudzinski J, Smith GT, Peterson TE, Whisenant JG, Hickman TL, et al. First-in-human PET imaging and estimated radiation dosimetry of l-[5-(11)C]-glutamine in patients with metastatic colorectal cancer. J Nucl Med (2021) 63(1):36–43. doi: 10.2967/jnumed.120.261594
31. Subramani E, Radoul M, Najac C, Batsios G, Molloy AR, Hong D, et al. Glutamate is a noninvasive metabolic biomarker of IDH1-mutant glioma response to temozolomide treatment. Cancer Res (2020) 80(22):5098–108. doi: 10.1158/0008-5472.CAN-20-1314
32. Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C, et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med (2015) 7(274):274ra17. doi: 10.1126/scitranslmed.aaa1009
33. Guo D, Bell EH, Chakravarti A. Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol (2013) 2(3):289–99. doi: 10.2217/cns.13.20
34. Shakya S, Gromovsky AD, Hale JS, Knudsen AM, Prager B, Wallace LC, et al. Altered lipid metabolism marks glioblastoma stem and non-stem cells in separate tumor niches. Acta Neuropathol Commun (2021) 9(1):101. doi: 10.1186/s40478-021-01205-7
35. Tator CH, Evans JR, Olszewski J. Tracers for the detection of brain tumors. evaluation of radioiodinated human serum albumin and radioiodinated fatty acid. Neurology (1966) 16(7):650–61. doi: 10.1212/wnl.16.7.650
36. Bacci M, Lorito N, Smiriglia A, Morandi A. Fat and furious: Lipid metabolism in antitumoral therapy response and resistance. Trends Cancer (2021) 7(3):198–213. doi: 10.1016/j.trecan.2020.10.004
37. Ferraro GB, Ali A, Luengo A, Kodack DP, Deik A, Abbott KL, et al. Fatty acid synthesis is required for breast cancer brain metastasis. Nat Cancer (2021) 2(4):414–28. doi: 10.1038/s43018-021-00183-y
38. Spector AA, Steinberg D. Relationship between fatty acid and glucose utilization in Ehrlich ascites tumor cells. J Lipid Res (1966) 7(5):657–63. doi: 10.1016/S0022-2275(20)39247-6
39. Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc Natl Acad Sci U S A. (2019) 116(2):631–40. doi: 10.1073/pnas.1808834116
40. Wei J, Leit S, Kuai J, Therrien E, Rafi S, Harwood HJ Jr, et al. An allosteric mechanism for potent inhibition of human ATP-citrate lyase. Nature (2019) 568(7753):566–70. doi: 10.1038/s41586-019-1094-6
41. Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med (2019) 11(478):eaau5758. doi: 10.1126/scitranslmed.aau5758
42. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
43. Jiang X, Stockwell BR, Conrad M. Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
44. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
45. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer (2019) 19(7):405–14. doi: 10.1038/s41568-019-0149-1
46. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
47. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell (2003) 3(3):285–96. doi: 10.1016/S1535-6108(03)00050-3
48. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
49. Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep (2020) 30(10):3411–3423.e7. doi: 10.1016/j.celrep.2020.02.049
50. Turchi R, Faraonio R, Lettieri-Barbato D, Aquilano K. An overview of the ferroptosis hallmarks in friedreich's ataxia. Biomolecules (2020) 10(11):1489. doi: 10.3390/biom10111489
51. Mahoney-Sanchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC, et al. Ferroptosis and its potential role in the physiopathology of parkinson's disease. Prog Neurobiol (2021) 196:101890. doi: 10.1016/j.pneurobio.2020.101890
52. Gao X, Guo N, Xu H, Pan T, Lei H, Yan A, et al. Ibuprofen induces ferroptosis of glioblastoma cells via downregulation of nuclear factor erythroid 2-related factor 2 signaling pathway. Anticancer Drugs (2020) 31(1):27–34. doi: 10.1097/CAD.0000000000000825
53. Zhuo S, Chen Z, Yang Y, Zhang J, Tang J, Yang K, et al. Clinical and biological significances of a ferroptosis-related gene signature in glioma. Front Oncol (2020) 10:590861. doi: 10.3389/fonc.2020.590861
54. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol (2019) 20(3):137–55. doi: 10.1038/s41580-018-0085-z
55. Senkal CE, Salama MF, Snider AJ, Allopenna JJ, Rana NA, Koller A, et al. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab (2017) 25(3):686–97. doi: 10.1016/j.cmet.2017.02.010
56. Chapman KD, Aziz M, Dyer JM, Mullen RT. Mechanisms of lipid droplet biogenesis. Biochem J (2019) 476(13):1929–42. doi: 10.1042/BCJ20180021
57. Walther TC, Chung J, Farese RV Jr. Lipid droplet biogenesis. Annu Rev Cell Dev Biol (2017) 33:491–510. doi: 10.1146/annurev-cellbio-100616-060608
58. Petan T, Jarc E, Jusovic M. Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules (2018) 23(8):1941. doi: 10.3390/molecules23081941
59. Klemm RW, Ikonen E. The cell biology of lipid droplets: More than just a phase. Semin Cell Dev Biol (2020) 108:1–3. doi: 10.1016/j.semcdb.2020.06.016
60. Munir R, Lisec J, Swinnen JV, Zaidi N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer (2019) 120(12):1090–8. doi: 10.1038/s41416-019-0451-4
61. De Martino M, Daviaud C, Minns H, Attarwala N, Chen Q, Dephoure N, et al. Radiation therapy promotes unsaturated fatty acids to maintain survival of glioblastoma. bioRxiv (2022). doi: 10.1101/2022.06.01.494338
62. Jiang N, Xie B, Xiao W, Fan M, Xu S, Duan Y, et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat Commun (2022) 13(1):1511. doi: 10.1038/s41467-022-29137-3
63. Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep (2014) 9(1):349–65. doi: 10.1016/j.celrep.2014.08.056
64. Sperry J, Condro MC, Guo L, Braas D, Vanderveer-Harris N, Kim KKO, et al. Glioblastoma utilizes fatty acids and ketone bodies for growth allowing progression during ketogenic diet therapy. iScience (2020) 23(9):101453. doi: 10.1016/j.isci.2020.101453
65. Cruz ALS, Barreto EA, Fazolini NPB, Viola JPB, Bozza PT, et al. Lipid droplets: Platforms with multiple functions in cancer hallmarks. Cell Death Dis (2020) 11(2):105. doi: 10.1038/s41419-020-2297-3
66. Ookhtens M, Kannan R, Lyon I, Baker N. Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am J Physiol (1984) 247(1 Pt 2):R146–53. doi: 10.1152/ajpregu.1984.247.1.R146
67. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. (2007) 104(49):19345–50. doi: 10.1073/pnas.0709747104
68. Yoo H, Antoniewicz MR, Stephanopoulos G, Kelleher JK. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J Biol Chem (2008) 283(30):20621–7. doi: 10.1074/jbc.M706494200
69. Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature (2011) 481(7381):380–4.
70. Maher EA, Marin-Valencia I, Bachoo RM, Mashimo T, Raisanen J, Hatanpaa KJ, et al. Metabolism of [U-13 c]glucose in human brain tumors in vivo. NMR Biomed (2012) 25(11):1234–44. doi: 10.1002/nbm.2794
71. Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X, et al. Glucose-mediated n-glycosylation of SCAP is essential for SREBP-1 activation and tumor growth. Cancer Cell (2015) 28(5):569–81. doi: 10.1016/j.ccell.2015.09.021
72. Cheng C, Geng F, Li Z, Zhong Y, Wang H, Cheng X, et al. Ammonia stimulates SCAP/Insig dissociation and SREBP-1 activation to promote lipogenesis and tumour growth. Nat Metab (2022) 4(5):575–88. doi: 10.1038/s42255-022-00568-y
73. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell (2014) 159(7):1591–602. doi: 10.1016/j.cell.2014.11.020
74. Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell (2014) 159(7):1603–14. doi: 10.1016/j.cell.2014.11.025
75. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell (2015) 27(1):57–71. doi: 10.1016/j.ccell.2014.12.002
76. Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, et al. Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat Commun (2016) 7:11960. doi: 10.1038/ncomms11960
77. Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci U S A. (1998) 95(22):13018–23. doi: 10.1073/pnas.95.22.13018
78. Cases S, Stone SJ, Zhou P, Yen E, Tow B, Lardizabal KD, et al. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J Biol Chem (2001) 276(42):38870–6. doi: 10.1074/jbc.M106219200
79. Bayerle A, Marsching C, Rabionet M, Dworski S, Kamani MA, Chitraju C, et al. Endogenous levels of 1-o-acylceramides increase upon acidic ceramidase deficiency and decrease due to loss of Dgat1 in a tissue-dependent manner. Biochim Biophys Acta Mol Cell Biol Lipids (2020) 1865(9):158741. doi: 10.1016/j.bbalip.2020.158741
80. Bemlih S, Poirier MD, El Andaloussi A. Acyl-coenzyme a: Cholesterol acyltransferase inhibitor avasimibe affect survival and proliferation of glioma tumor cell lines. Cancer Biol Ther (2010) 9(12):1025–32. doi: 10.4161/cbt.9.12.11875
81. Nygren C, von Holst H, Mansson JE, Fredman P. Increased levels of cholesterol esters in glioma tissue and surrounding areas of human brain. Br J Neurosurg (1997) 11(3):216–20. doi: 10.1080/02688699746276
82. Ohmoto T, Nishitsuji K, Yoshitani N, Mizuguchi M, Yanagisawa Y, Saito H, et al. K604, a specific acylCoA: Cholesterol acyltransferase 1 inhibitor, suppresses proliferation of U251MG glioblastoma cells. Mol Med Rep (2015) 12(4):6037–42. doi: 10.3892/mmr.2015.4200
83. Liu JY, Fu WQ, Zheng XJ, Li W, Ren LW, Wang JH, et al. Avasimibe exerts anticancer effects on human glioblastoma cells via inducing cell apoptosis and cell cycle arrest. Acta Pharmacol Sin (2021) 42(1):97–107. doi: 10.1038/s41401-020-0404-8
84. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol (2019) 26(3):420–432.e9. doi: 10.1016/j.chembiol.2018.11.016
85. Dierge E, Debock E, Guilbaud C, Corbet C, Mignolet E, Mignard L, et al. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab (2021) 33(8):1701–1715.e5. doi: 10.1016/j.cmet.2021.05.016
86. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
87. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
88. Yuan H, Li X, Zhang X, Kang R, Tang D, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun (2016) 478(3):1338–43. doi: 10.1016/j.bbrc.2016.08.124
89. Cheng J, Fan YQ, Liu BH, Zhou H, Wang JM, Chen QX, et al. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep (2020) 43(1):147–58. doi: 10.3892/or.2019.7419
90. Bao C, Zhang J, Xian SY, Chen F. MicroRNA-670-3p suppresses ferroptosis of human glioblastoma cells through targeting ACSL4. Free Radic Res (2021) 55(7):853–64. doi: 10.1080/10715762.2021.1962009
91. Cheng X, Geng F, Pan M, Wu X, Zhong Y, Wang C, et al. Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab (2020) 32(2):229–242.e8. doi: 10.1016/j.cmet.2020.06.002
92. Wang H, Bell M, Sreenivasan U, Hu H, Liu J, et al. Unique regulation of adipose triglyceride lipase (ATGL) by perilipin 5, a lipid droplet-associated protein. J Biol Chem (2011) 286(18):15707–15. doi: 10.1074/jbc.M110.207779
93. Lass A, Zimmermann R, Oberer M, Zechner R. Lipolysis - a highly regulated multi-enzyme complex mediates the catabolism of cellular fat stores. Prog Lipid Res (2011) 50(1):14–27. doi: 10.1016/j.plipres.2010.10.004
94. Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science (2004) 306(5700):1383–6. doi: 10.1126/science.1100747
95. Taschler U, Radner FP, Heier C, Schreiber R, Schweiger M, Schoiswohl G, et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J Biol Chem (2011) 286(20):17467–77. doi: 10.1074/jbc.M110.215434
96. Haemmerle G, Zimmermann R, Strauss JG, Kratky D, Riederer M, Knipping G, et al. Hormone-sensitive lipase deficiency in mice changes the plasma lipid profile by affecting the tissue-specific expression pattern of lipoprotein lipase in adipose tissue and muscle. J Biol Chem (2002) 277(15):12946–52. doi: 10.1074/jbc.M108640200
97. Keenan SN, De Nardo W, Lou J, Schittenhelm RB, Montgomery MK, Granneman JG, et al. Perilipin 5 S155 phosphorylation by PKA is required for the control of hepatic lipid metabolism and glycemic control. J Lipid Res (2021) 62:100016. doi: 10.1194/jlr.RA120001126
98. Gimm T, Wiese M, Teschemacher B, Deggerich A, Schodel J, Knaup KX, et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J (2010) 24(11):4443–58. doi: 10.1096/fj.10-159806
99. Mao XG, Wang C, Liu DY, Zhang X, Wang L, Yan M, et al. Hypoxia upregulates HIG2 expression and contributes to bevacizumab resistance in glioblastoma. Oncotarget (2016) 7(30):47808–20. doi: 10.18632/oncotarget.10029
100. Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat Rev Mol Cell Biol (2017) 18(11):671–84. doi: 10.1038/nrm.2017.76
101. Ogasawara Y, Tsuji T, Fujimoto T. Multifarious roles of lipid droplets in autophagy - target, product, and what else? Semin Cell Dev Biol (2020) 108:47–54. doi: 10.1016/j.semcdb.2020.02.013
102. Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol (2015) 17(6):759–70. doi: 10.1038/ncb3166
103. Sun X, Yang S, Feng X, Zhou J, Wang H, et al. The modification of ferroptosis and abnormal lipometabolism through overexpression and knockdown of potential prognostic biomarker perilipin2 in gastric carcinoma. Gastric Cancer (2020) 23(2):241–59. doi: 10.1007/s10120-019-01004-z
104. Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun (2019) 508(4):997–1003. doi: 10.1016/j.bbrc.2018.12.039
105. Liu R, Lee JH, Li J, Yu R, Tan L, Xia Y, et al. Choline kinase alpha 2 acts as a protein kinase to promote lipolysis of lipid droplets. Mol Cell (2021) 81(13):2722–2735.e9. doi: 10.1016/j.molcel.2021.05.005
106. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy (2016) 12(8):1425–8. doi: 10.1080/15548627.2016.1187366
107. Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA, et al. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology (2015) 61(6):1896–907. doi: 10.1002/hep.27667
108. Wang W, Zhang H, Liu S, Kim CK, Xu Y, Hurley LA, et al. Internalized CD44s splice isoform attenuates EGFR degradation by targeting Rab7A. Proc Natl Acad Sci U S A. (2017) 114(31):8366–71. doi: 10.1073/pnas.1701289114
109. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol (1927) 8(6):519–30. doi: 10.1085/jgp.8.6.519
110. Cheng C, Geng F, Cheng X, Guo D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun (Lond) (2018) 38(1):27. doi: 10.1186/s40880-018-0301-4
111. Jones SF, Infante JR. Molecular pathways: Fatty acid synthase. Clin Cancer Res (2015) 21(24):5434–8. doi: 10.1158/1078-0432.CCR-15-0126
112. Wu X, Geng F, Cheng X, Guo Q, Zhong Y, Cloughesy TF, et al. Lipid droplets maintain energy homeostasis and glioblastoma growth via autophagic release of stored fatty acids. iScience (2020) 23(10):101569. doi: 10.1016/j.isci.2020.101569
113. Choi YK, Park KG. Targeting glutamine metabolism for cancer treatment. Biomol Ther (Seoul) (2018) 26(1):19–28. doi: 10.4062/biomolther.2017.178
114. Lyssiotis CA, Kimmelman AC. Metabolic interactions in the tumor microenvironment. Trends Cell Biol (2017) 27(11):863–75. doi: 10.1016/j.tcb.2017.06.003
115. Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC, et al. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science (2017) 358(6365):941–6. doi: 10.1038/s41556-018-0124-1
Keywords: brain cancer, metabolism, lipids, cell death, therapeutic vulnerabilities, glioma, lipid droplet (LD)
Citation: Bezawork-Geleta A, Dimou J and Watt MJ (2022) Lipid droplets and ferroptosis as new players in brain cancer glioblastoma progression and therapeutic resistance. Front. Oncol. 12:1085034. doi: 10.3389/fonc.2022.1085034
Received: 31 October 2022; Accepted: 18 November 2022;
Published: 15 December 2022.
Edited by:
Xingchun Gao, Xi’an Medical University, ChinaReviewed by:
Chunming Cheng, The Ohio State University, United StatesXiangjun Tang, Yale University, United States
Copyright © 2022 Bezawork-Geleta, Dimou and Watt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ayenachew Bezawork-Geleta, YmV6YXdvcmtnZWxldGEuYUB1bmltZWxiLmVkdS5hdQ==