 Yubao Lu
Yubao Lu Jiahe Zhang
Jiahe Zhang Yutong Chen2
Yutong Chen2 Cangyu Zhang
Cangyu Zhang- 1Department of Spine Surgery, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 2The Second Clinical Medical College, Lanzhou University, Lanzhou, China
- 3Department of Orthopaedics, Lanzhou University Second Hospital, Lanzhou, China
Osteosarcoma (OS) is the most common primary malignant bone sarcoma mainly affecting adolescents and young adults, which often progresses to pulmonary metastasis and leads to the death of OS patients. OS is characterized as a highly heterogeneous cancer type and the underlying pathologic mechanisms triggering tumor progress and metastasis are incompletely recognized. Surgery combined with neoadjuvant and postoperative chemotherapy has elevated 5-year survival to over 70% for patients with localized OS tumors, as opposed to only 20% of patients with recurrence and/or metastasis. Therefore, novel therapeutic strategies are needed to overcome the drawbacks of conventional treatments. Immunotherapy is gaining momentum for the treatment of OS with an increasing number of FDA-approved therapies for malignancies resistant to conventional therapies. Here, we review the OS tumor microenvironment and appraise the promising immunotherapies available in the management of OS.
Introduction
Osteosarcoma (OS), a malignant neoplasm with high morbidity in adolescents, basically stems from primitive mesenchymal cells, and is mostly seen in the metaphysis of femur, tibia, humerus, and other long bones (1). In light of incomplete statistics, the global incidence of osteosarcoma is about 4.8 per million (2, 3). Given the current clinical treatment, although surgery combined with neoadjuvant and postoperative chemotherapy has enhanced the 5-year survival rate by more than 70% in patients with localized OS, this conventional treatment failed in OS patients with recurrence and/or metastasis except for 20% of patients (4, 5). To make the matter worse, affected by the limitation in OS diagnosis, it is often the case that patients are tested for metastasis at the time of initial diagnosis (6). In terms of safety, mainstream chemotherapy (methotrexate, doxorubicin, cisplatin, and ifosfamide) appears with inescapable side effects like leukopenia, thrombocytopenia (7), mucositis (8), ototoxicity, and nephrotoxicity (9). Thus a novel less toxic therapy is needed. In a sense, OS is not only a disaster for patients and their families, but also a huge loss for social development. These losses do not simply come from the expensive medical services (for OS diagnosis and treatment) paid for by patients and the government. In fact, from a deeper perspective, it is because young patients are too unwell to enter society in order to fuel social development by that time. These losses are far more assessable and quantified. Therefore, applying new effective treatment methods to prolong the life span of patients and improve their quality of life is an essential problem that needs to be solved urgently.
The tumor microenvironment is a key problem in modern cancer research, which has a decisive function in the occurrence and progression of tumors (10). The normal tissue microenvironment of bone consists of bone marrow stroma and mineralized extracellular matrix. Bone marrow contains two different cell types: hematopoietic stem cells and bone marrow-derived mesenchyme stem cells with hematopoietic function, which differentiate into non-blood cell components of bone, such as mesenchymal stem cells (MSCs), osteoblasts, osteoclasts, osteocytes, fibroblasts, adipocytes etc. (11–13). Benefitting from the particularity of the bone tissue microenvironment, the porous mineralized extracellular matrix structure and abundant nutrition supply enable the bone tissue microenvironment to become ‘fertilized soil’ for tumor growth (14, 15). During OS genesis, changes occur in the tissue microenvironment of bone, the most important of which lies in the infiltration of immune cells (macrophages, neutrophils, dendritic cells, mast cells, natural killer cells, T lymphocytes, and B lymphocytes) (Figure 1) (16). The imbalance of these factors in the OS localized microenvironment is considered as the key to modulate the progression and metastasis of OS. As research into the OS microenvironment deepens, immunotherapy has undergone rapid development. The monotherapy or combination therapy of a tumor vaccine, immunomodulator, genetically modified T cell, cytokine, and immune checkpoint inhibitor brings hope to the dilemma of OS treatment. This significant progress is attributed to the immunotherapy that avoids drug resistance and immune escape to a large extent, reducing side effects paralleling therapeutic effect improvement (17).
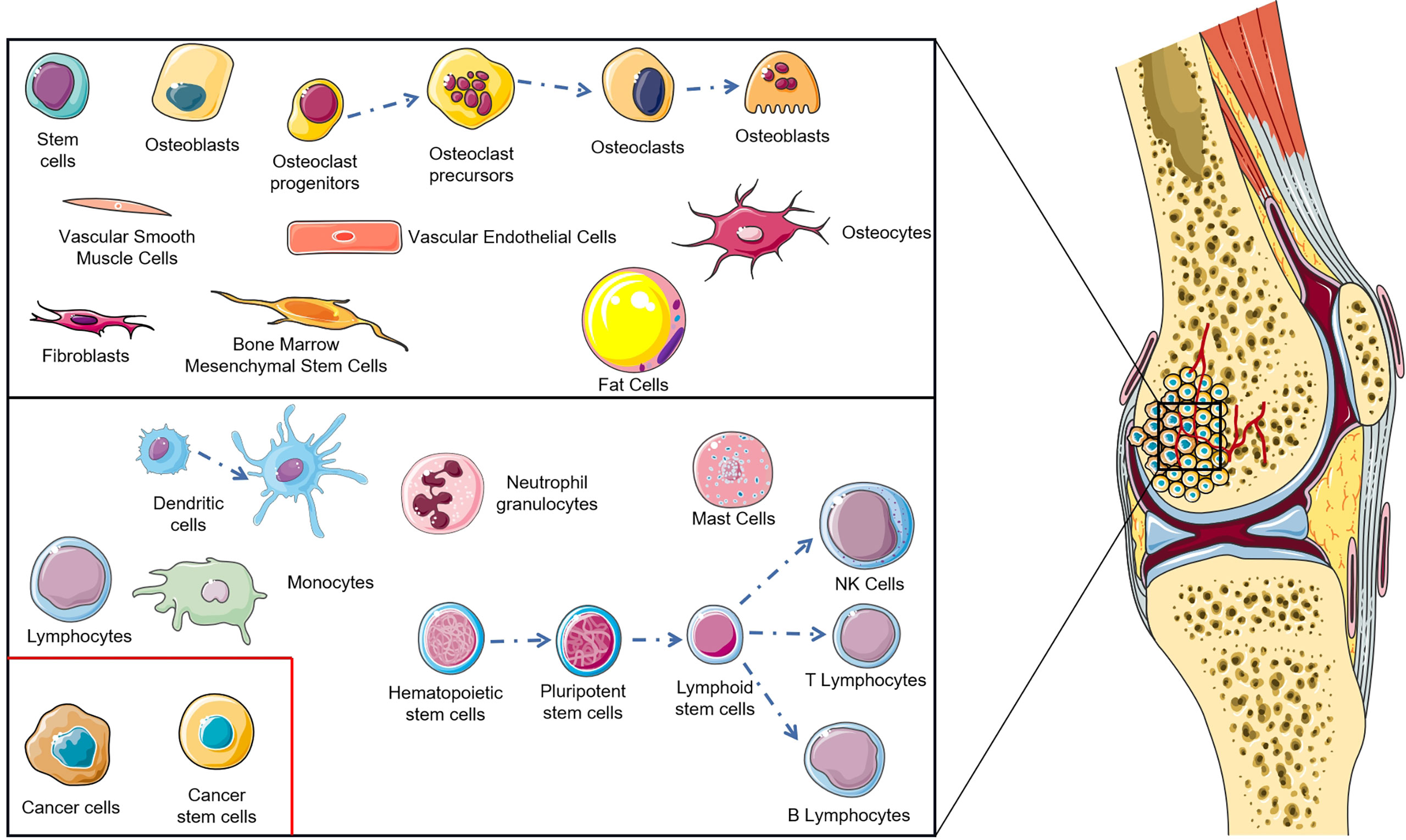
Figure 1 Cellular composition of osteosarcoma tumor microenvironment.
Hence, this review was carried out on immunotherapy in OS, aiming to provide systematic theoretical evidence for the development and application in the treatment, and give impetus to the progress of these kinds of studies.
Tumor Immune Microenvironment (TME)
The network of the OS immune microenvironment is composed of both innate and adaptive immune cells, infiltrating with macrophages, neutrophils, dendritic cells (DCs), mast cells, natural killer (NK) cells, T lymphocytes, and B lymphocytes (18). Specifically, macrophages and T lymphocytes are the dominating constitution therein (19). These immune cells can be located in the core and invasive margin of the tumor or in the adjacent tertiary lymphoid structures (13). Indeed, a normal immune system can recognize tumor cells and initiate a series of functional stepwise events to eliminate them. This process is termed the cancer-immunity cycle and consists of seven major steps: 1) DCs detect neoantigens and pro-inflammatory cytokines released by tumor cells, 2) the captured antigens are presented to T cells, 3) the tumor-specific immunity is initiated with priming and activation of T-cell response, 4) T cells traffic to tumors and 5) infiltrate herein, 6) T cells identify and combine with tumor cells, and 7) kill them. Killed malignant cells release neoantigens and start a subsequent circulation (20). However, OS cells are able to modulate the recruitment and differentiation of immune cells, and consequently sabotage the cancer-immunity cycle and induce an immune-tolerant microenvironment which is conducive to proliferating and metastasizing tumor cells (21). Notably, OS patients with higher immune scores with enhanced immune cell infiltration in the microenvironment have better prognosis (22, 23). Therefore, the focus on the tumor immune microenvironment, especially its crosstalk with tumor cells, may be indispensable to further understand the OS immune system for developing novel immunotherapies.
Tumor-Associated Macrophages in the TME
The immune component of OS TME predominantly consists of tumor-associated macrophages (TAMs), taking up a significantly high proportion compared with other immune cells (24), which play an essential role in inflammatory response and tissue homeostasis (25). Macrophages have high plasticity so that they present inverse phenotypes with different activating signals: the pro-inflammatory phenotype (M1) and the anti-inflammatory phenotype (M2) (26, 27). M1 macrophages serve as the key defense in tumor suppression by stimulating the immune system to express high levels of pro-inflammatory cytokines like IL-1, IL-6, and IL-12 (28), and both by inducting T helper type-1 (Th1) cell maturation and promoting inducible nitric-oxide synthase (iNOS) production (29). On the contrary, M2 macrophages are associated with immune suppression, matrix degrading, and tumor angiogenesis, and thus accelerate tumor progression and metastasis (27, 30). Many studies have proved that high infiltration of TAMs was correlated with worse prognosis in most solid cancers (31–33). Wolf-Dennen et al. and Li et al. revealed that M2-related cytokines, chemokines, and cell-markers showed an increased expression in lung metastasis of OS (34, 35). Similarly, Dhupkar et al. illustrated from another perspective that the shift from the M2 to M1 phenotype induced regression of lung metastasis in OS, strengthening the role of M2 macrophages in OS development (36). Nevertheless, it remains controversial to define whether TAMs are pro-tumor or anti-tumor in OS. Some previous studies showed that increased infiltration of TAMs was associated with reduced metastasis and a better survival outcome in high-grade OS (37, 38), whereas M2 macrophages indicated a bad prognosis (19, 38). Han et al. demonstrated that the number of M2 macrophages was negatively associated with TIM3+PD1+ T cells and relative pro-inflammatory cytokines (39). Through the CIBERSORT algorithm, M2 macrophages were verified as the major composition of TAMs. Buddingh et al. speculated that the constitutive of M2 macrophages may act in a metastatic suppression role rather than have a pro-metastatic effect in certain circumstances. M1 and M2 phenotypes are extremes of a continuum of macrophage polarization states (40). The extended transcriptional repertoire are detected from macrophages in chronic inflammation and tumors, which is mostly decided on the properties of diverse microenvironmental stimulation (41, 42). An intermediate phenotype M1-M2 found in primary OS with anti-metastatic activity suggested that the balance between M1 and M2 macrophages may play a decisive role in prognosis rather than the total number (38).
Therefore, M2-phenotype TAMs may work as promising targets to orchestrate OS progression. Novel treatment strategies focus on modulating TAM polarization to improve the ratio of M2 phenotype to M1 phenotype or inducing the shift from M2 phenotype to M1 phenotype to inhibit OS progression (Table 1). Mifamurtide, an immunomodulatory drug that triggers macrophages and increases the level of proinflammatory cytokines (56), has been approved by the European Medical Agency (EMA) to treat OS in combination with adjuvant chemotherapy (57, 58). Punzo et al. demonstrated mifamurtide’s anti-tumor effect from two aspects. Not only by shifting macrophage polarization towards the intermediate M1-M2 phenotype, it can also restrict M2 activation time via decreasing STAT3 and Akt phosphorylation to inhibit the STAT3 pathway and PI3K/Akt/mTOR pathway, both of which are activators of M2 polarization (43). In addition, all-trans retinoic acid (ATRA) has been proven to restrict the initiation of OS and reduce pulmonary OS metastasis (59, 60). ARTA could indirectly deviate macrophages from M2 polarization or disrupt the TAM-cancer stem cells (CSCs) pathway to limit CSC formation in OS (60). Naturally occurring compounds like dihydroxycoumarins (esculetin and fraxetin) were also expected to induce TAM polarization from M2 to M1 phenotypes in OS treatment (61). Dihydroxycoumarins act via inhibiting the production and growth of IL-10, MCP-1, and TGF-β1 as well as impeding the phosphorylation of Stat 3 during M2 phenotype differentiation to interfere with its activation (44, 61, 62). Zoledronic acid has been envisaged as a therapeutic drug because it is able to interfere with M2 phenotype polarization and cause TAM to polarize back to the M1 phenotype (45, 46). Although its function has been demonstrated in some bone metastasis tumors (63), a randomized study has suggested that zoledronate treatment resulted in higher risk than the placebo group (37). Gomez-Brouchet et al. found that zoledronate may induce deleterious polarization to CD68+/CD163+ bipotent macrophages (64). CD68 is stated to be an M1-polarized macrophage marker, while high level of CD163 staining is associated with high CMAF nuclear expression (a macrophage transcription factor correlated with M2 macrophage polarization) (65). Hence the effect of zoledronate needs more detailed research to clarify whether it is positive or negative for OS treatment.
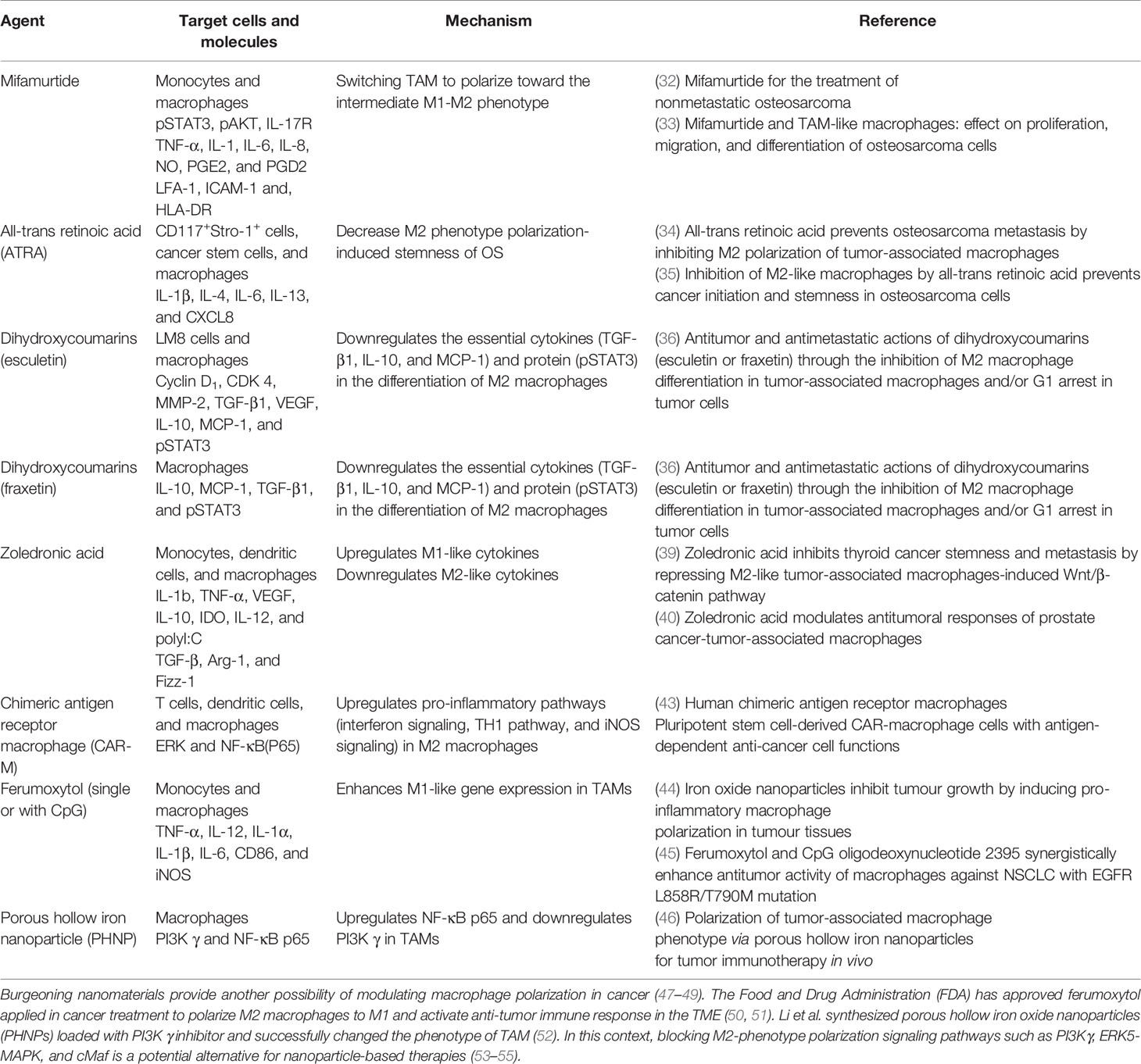
Table 1 Therapeutic TAM targeting agents in OS.
T Lymphocytes in the TME
T lymphocytes constitute the second most common infiltration cell type in OS. Particularly, tumor-infiltrating lymphocytes (TILs) are detected in 75% of OS with a peak around 86% in metastases, they include CD8+ T lymphocytes, CD4+ T lymphocytes, CD20+ B lymphocytes, and CD117+ mast cells (19). Helper and cytotoxic T cells can be activated by tumor antigen-triggered dendritic cells and directly attack tumors via cytotoxic cells (66). Besides, helper and cytotoxic T cells can also secret IFN-γ to inhibit tumor progression (67). In spite of a relatively low proportion of CD8+ T cell infiltration in OS, its ratio showed a significantly positive correlation with a lower rate of metastasis and better survival outcome (24). TILs are regarded as a selected population of T cells with higher specific immunological reactivity against tumors than normal lymphocytes. However, studies evaluated that CD8+ T cells were less abundant than myeloid cells in OS biopsies which suggests that OS has poorly immunogenic tumors with a lack of tumor neo-antigens (68). This would define OS as a “cold” tumor. Ligon et al. revealed that OS pulmonary metastasis was characterized with remarkable numbers of CD8+ T cells but the majority of them merely infiltrated on the edge of the metastasis because of immune resistance mechanisms (69). In addition, the expression of checkpoints TIM-3 and LAG-3 was detected on TILs at the interface of lung metastasis, indicating TIM-3 and LAG-3 are potential targets to enhanced TIL permeability and strengthen the cytotoxic effect (69).
On the basis of a data-driven mathematical model study, the population variation of cytotoxic T cells, helper T cells, and dendritic cells was in parallel with OS cell growth in the early stage and then decreased with time, and which was found to generally increase under the treatment of chemotherapy drugs as well (70). Whereas regulatory T cells were first reduced in the population and then raised (71). Fritzsching et al. showed that OS patients with a CD8+/FOXP3+ ratio above 3.08 possessed a notably improved survival, and the ratio of CD8+/FOXP3+ T cells to regulatory CD4+/FOXP3+ T cells in biopsies prior to chemotherapy allowed for the discrimination of OS patients with prolonged survival from non-survivors (68). On the other hand, the immune suppression molecule galectin-9 (Gal9) was primarily expressed on CD4+CD25+ Tregs in OS, with significantly higher frequencies than in non-cancer controls (72), as well as higher than other solid tumors (73). Furthermore, the Gal9 expressed by CD4+CD25+ Tregs could contribute to the development of M2 macrophages and lead to an increasingly suppressive anti-tumor CD8+ T cell response and inflammatory conditions (72). Another characteristic observed is the T cell exhaustion phenomenon that may be a vital mechanism contributing to impaired T-cell response against pathogens (74). The chronic inflammation in the TME can guide T cells to a dysfunctional or “exhausted” state, manifesting as an enhanced expression of multiple immune checkpoints of T cells (13). IL-21, mainly secreted from CD4+ T cells, has been proven to specifically enable homeostatic proliferation of cytotoxic CD8+ T cells by the effect of other cytokines such as IL-2, IL-7, and IL-15 (75, 76). Besides, it could also suppress the expression of FOXP3, a key transcription factor prompting Tregs, to relieve the inhibition on CD8+ T cell response (77). Gao et al. detected that CD4+ T cells in OS had decreased capacity to express IL-21 compared to healthy controls, especially follicular helper T (Tfh) cells which usually highly express PD-L1 with a severe reduction in this capacity as well as in proliferation capacity (78). Hence this evidence indicated that reverting the IL-21 decrease by blocking PD-L1 expression on Tfh cells or inhibiting naïve CD4+ T cell recruitment into tumors by interfering with PITPNM3 recognition of CCL18 may be an attractive strategy for immunotherapy in OS (24). IL-2 plays a crucial role in motivating expansion, differentiation, and function of effector T cells, and the loss or downward responsiveness of IL-2 are responsible for the exhausted phenotype in anti-tumor immunity (79). Tregs can be phenotypically identified by CD25 (IL-2 receptor-α subunit) (80). Remarkably, Solomon et al. developed Treg-depleting agents (RG6292) that targeted CD25, but did not interfere with IL-2 signaling on effector T cells (81). This breakthrough managed to reverse the suppressive states in the TME and preserved more available IL-2 to motivate effector T cells in the meanwhile.
Immune-Related Cells
There exist some non-immune cells capable of effecting tumor ontogeny by mediating immune responses. Mesenchymal stem cells (MSCs) derived from both normal tissue and tumor tissue are confirmed to facilitate the progression of OS. The tumor promotional effect of MSCs can be attributed to two main mechanisms. One is capacitating OS cells with stemness properties by IL-6 secreted from MSCs (82). OS extracellular vesicles can cause MSCs to shift to the pro-tumor phenotype distinguished by abundant IL-6 production (83). The other is immunosuppression of activated MSCs, including inhibiting the proliferation of T cells (84), B cells (85), and NK cells (86), leading to the differentiation of Tregs (87).
Bone matrix remodeling is a distinct characteristic of osteosarcoma which is basically mediated by osteoclasts. Osteoclasts derive from monocytic lineage with high heterogeneity. Just like other immune cells with monocytic lineages (monocytes, macrophages, and dendritic cells, etc.), osteoclasts are qualified to regulate T-cell activation (88). The role of osteoclasts is determined by their precursor cells and microenvironment. In malignancies, they suppress the T-cell-mediated cytotoxicity of CD4+ and CD8+ cells to shelter tumor cells from immune elimination (89). The role of osteoclasts in the pathogenesis of OS remains controversial. Based on previous research, a cogent hypothesis suggests that osteoclasts promote tumor cell spread in the early stage, yet destruct and reorganize bone niches in the later stage (19). Generally, it is indispensable that osteoclasts are crucial regulators of OS growth through their flexible immune function. As in neoadjuvant chemotherapy, the macrophage-phagocyte system phagocytizes drug-carrier particles and then recruits them into bone tissues. Hence the osteoclasts, normally stemmed from macrophages in the bone microenvironment, in the vicinity of OS can represent the viability of macrophages and drug-delivered volume into bone marrow (90).
Promising Immunotherapies
Immune Checkpoint Inhibitors (ICIs)
In most types of cancer, two immune checkpoints, programmed cell death protein 1 (PD-1) (91, 92) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) (93, 94), were observed with an upregulated expression on T cells in the TME. Their activation capacitates immune tolerance and treatment resistance through inhibiting T-cell activation (95). Whereas, OS cells specifically adopt this deregulation mechanism with highly expressed ligand proteins that activate those pathways (96, 97). Consequently, by prohibiting engagement between infiltrated T cells and tumor cells, ICIs are introduced to reverse immune tolerance and elicit anti-tumor immune response. The 2018 Nobel Prize in Physiology and Medicine was awarded to Allison and Honjo for elucidating the immunosuppression of CTLA-4 and PD-1, respectively, and suggesting potential immunotherapy targeted at specific immune checkpoints. As two of the earliest studied immune checkpoints, some molecular mechanisms of PD-1 and CTLA-4 blockade have been clarified in the published research, like eliciting CD8+ T-cell and ICOS+ Th1-like cell infiltration in the TME (98). Moreover, with the combined therapy of PD-1 blockade and radiotherapy, CD8+ T cells can be stimulated at a great extent and trafficked to the distant metastatic lesions which is identified as an abscopal effect (99). In view of their encouraging results in anti-tumor function, ICIs involved with CTLA-4 inhibitors (ipilimumab), PD-1 inhibitors (nivolumab, pembrolizumab, and cemiplimab), and PD-L1 inhibitors (atezolizumab, avelumab, and durvalumab) have been approved by the FDA to treat numerous types of solid tumors: Hodgkin and non-Hodgkin lymphomas, melanoma, Merkel cell carcinoma, and liver, kidney, cervical, head and neck, lung, gastric, colorectal, and bladder cancers, and cutaneous squamous cell carcinoma (100).
PD-1/PD-L1 Inhibitor and CTLA-4 Inhibitor
ICIs targeting CTLA-4, PD-1, and PD-L1 were used as initial generation ICIs in the treatment of OS (Table 2). CTLA-4 is a transmembrane glycoprotein receptor expressed on Tregs and memory T cells, which suppresses anti-tumor immunity by engaging CD80/86 on DCs. Similarly, PD-1 is a transmembrane immunoglobulin widely distrusted on activated T cells, which functions as a CTL inhibitor and Tregs activator (101). As for PD-L1, the expression is detected on OS cells, which symbolizes reduced immune cell infiltration, including T cells, NK cells, and dendritic cells, as well as increased T-cell apoptosis (102). These inhibitors are capable of reinvigorating the T-cell-mediated anti-tumor immunity.
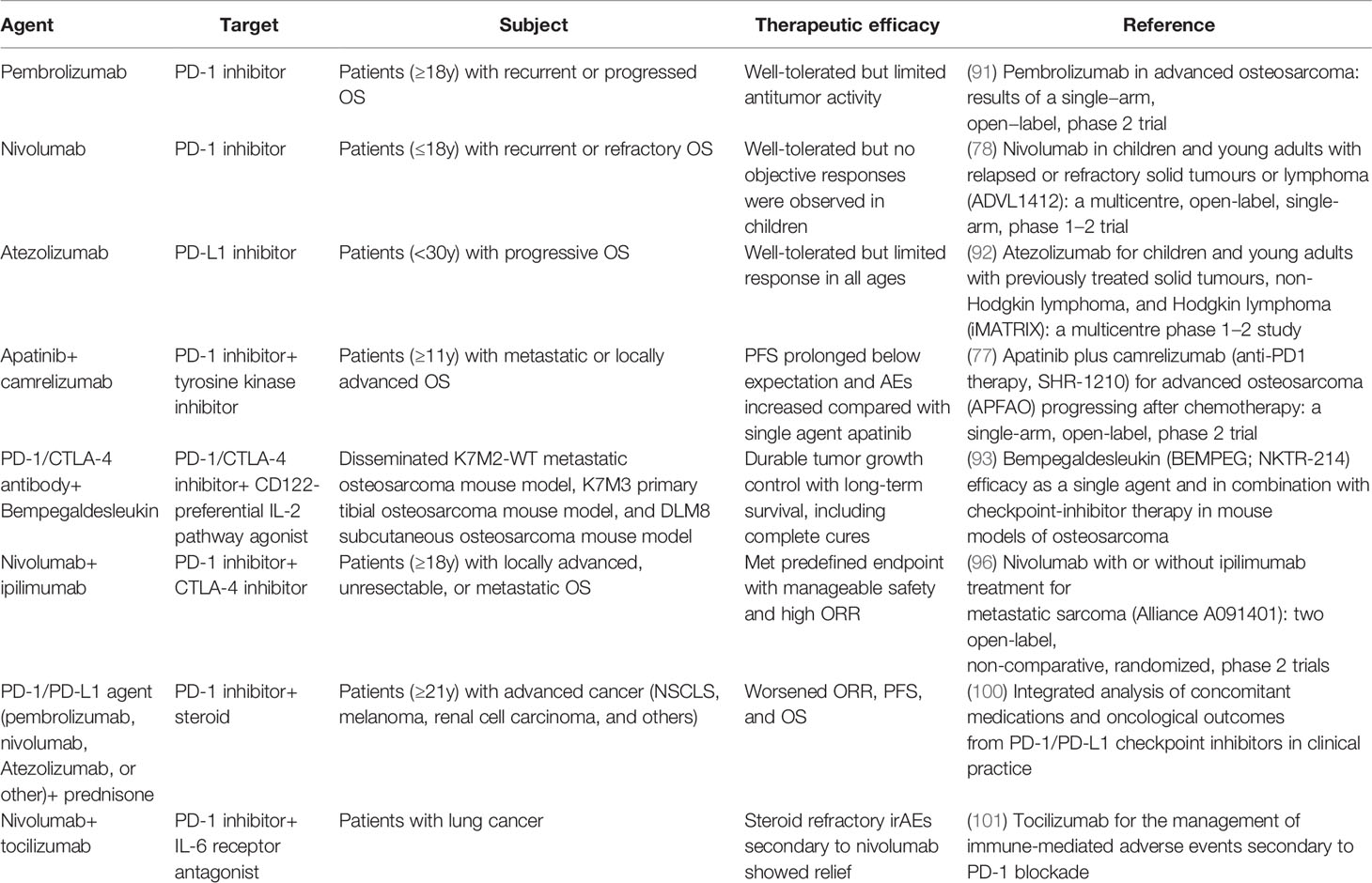
Table 2 Current PD-1/PD-L1 or CTLA-4-targeted ICI therapy.
However, prespecified results were not apparent in clinical trials by the use of single-agent PD-1/PD-L1 ICIs. The SARC028 trial evaluated the safety and efficacy of pembrolizumab and only 5% of patients with bone sarcoma (one OS and one chondrosarcoma) were observed to have an objective response (103). A novel phase 2 study (NCT03013127) validated the parallel result that pembrolizumab showed decent tolerance but insufficient clinical antitumor activity in adult patients with advanced OS (104). Further evidence on the lack of ICI activity in OS was also suggested with nivolumab (NCT02304458) and atezolizumab (NCT02541604) in pediatric trials. With 23 OS patients involved, neither study observed a positive response (90, 105). In addition, a trial (NCT03359018) of apatinib plus camrelizumab showed no extra survival benefits in the comparison of singe-agent apatinib (106). The low response in clinical trials with a single-agent ICI implied that OS was primarily resistant to the PD-1/PD-L1 blockade. The immune-genomic landscape of OS is characterized as a “cold” tumor, thus combination therapies of immune cell activation amplifiers synergized with ICIs were considered. In metastatic and orthotropic murine models of OS, bempegaldesleukin, a first-in-class CD122-preferred IL-2 pathway agonist, significantly enhanced activity of PD-1 and CTLA-4 ICIs through boosting the amassing of intratumoral effector T cells and NK cells (107). Another reason for the low objective response rate in PD-1/PD-L1 blockade therapy is that induced tumor-targeted CTLs are quickly exhausted in the tumor microenvironment so they cannot provide a sustainable and powerful anti-tumor effect (108). Consequently, it is apparent that single PD-1/PD-L1 ICI therapy may not be effective enough for treating OS. Lussier et al. revealed that T cells infiltrated in the OS tumor microenvironment unregulated additional inhibitory receptors like CTLA-4, which conspired to hinder tumor immunity. They combined CTLA-4 with a PD-1 ICI in the K7M2 murine model of metastatic OS and the tumors were completely under control in most of subjects as expected (109). Helm et al. found that OS mouse models treated with a combination of CTLA-4 and PD-1 ICIs detected a rise in CD8+ T cells. The Alliance A091401 trial aimed at evaluating nivolumab plus ipilimumab combination therapy for metastatic sarcoma and showed that in the combination group, 16% of patients reacted to the immunotherapy while only 5% of patients in the nivolumab group had a confirmed reaction (110). To date, although the therapeutic effect of ICI combination in OS has not been confirmed in clinical trials, several cases reported that immunotherapy with ipilimumab plus nivolumab displayed notable tumor manifestation remission and tumor mass stabilization with metastatic OS patients (111, 112). As is widely acknowledged, ICI-like anti-PD-1 inhibitors are antigen agnostic which directly points to a severe immune-related adverse event (irAE), immune hyperactivation. This cytokine release syndrome or cytokine storm can manifest as cardiovascular-associated and cerebrovascular-related events, coagulation disorders, encephalopathy, endocrinopathies, fever, gastro-hepatic-intestinal derangements, and hypoxia, and eventually progress to a stage of multiple organ failure. Steroids can be used to neutralize the toxicities, but they might impair the effectiveness of immunotherapy, and Cortellini et al. suggested that concomitant medication of a PD-1/PD-L1 ICI with prednisone can worsen patients’ ORR, PFS, and OS (113, 114). IL-6 blocker tocilizumab was tested to successfully temporize irAEs in a wide variety of models (115). It reminds us that a cytokine receptor antagonist plus ICI may be a therapeutic choice to manage a better prognosis in immunotherapy.
Interestingly, anti-PD-1 and anti-PD-L1 inhibitors have been found to mediate macrophages from M1 to M2 phenotypes in xenograft OS murine models (36). A characteristic of immune exclusion of OS pulmonary metastasis is the co-location of TAM and PD-L1-expressing tumor cells with CD8+ T cells on the pulmonary metastasis interface, posing a barrier to further TIL infiltration into pulmonary metastasis (69). Based on this, macrophage polarization-modulating drugs combined with a PD-1/PD-L1 ICI may help to break the barrier on the OS pulmonary metastasis interface and thus enhance the infiltration of TILs.
Novel Generation ICIs
It has been recognized that there are other checkpoint molecules co-expressed on TILs at the interface of tumors, conspiring against anti-tumor immunity. Among them, the checkpoint T-cell immunoglobulin and mucin-domain containing-3 (Tim-3) (72), lymphocyte-activation gene 3 (LAG-3) (116), indoleamine 2,3-dioxygenase (IDO) (69), and human epidermal growth factor receptor-2 (HER2) (117) have been examined in terms of OS.
Tim-3 regulates both the innate and adaptive immune pathways, and its expression has been confirmed in diverse immune cell types, involving CTLs, Tregs, macrophages, DCs, and NK cells (118, 119). The bond with its endogenous ligand GAL-9 leads to apoptosis and immune tolerance of T cells. A study indicated that Tim-3 was an independent predictor of survival, whose overexpression was a sign of poor prognosis in patients with OS (120). Furthermore, Tim-3 was proved to induce macrophages polarized to the M2 phenotype and promote lung metastasis in mouse models transplanted with OS-derived exosomes (121). In OS patients, both soluble Tim-3 and Tim-3-positive CD8+ and CD4+ T cells showed an elevated level in the peripheral circulation, which is negatively associated with the release of proinflammatory cytokines like IL-2, IFN-γ, and TNF-α (122). Similar to Tim-3, LAG-3 was broadly expressed on immune cells and inhibited anti-tumor immunity through interacting with ligand LSECtin in sarcoma (123). Pignon et al. suggested that cancerous patients with a higher percentage of CD8+PD-1+ T cells that are negative for Tim-3 and LAG-3 may better respond to anti-PD-1 immunotherapy (124). Assuming that there is a compensatory upregulation of Tim-3 and LAG-3 secondary to single-agent ICI therapy, resembling other diseases. Combinations of PD-1 ICI with Tim-3 or LAG-3 are rational candidates for further experiments in OS (116).
IDO is a tryptophan-degrading enzyme. The decrease in tryptophan and the rise in tryptophan metabolites restrain T-lymphocyte proliferation and induce immune tolerance. The activation of IDO can be induced by Bin1 gene losses and stimulation of IFN-γ in tumors. Especially, cytokines IL-12 and IL-18 also amplify IDO activity independently from the IFN-γ pathway in OS (125). Patients with higher IDO expression were associated with worse survival which may be attributable to the immune resistance induced by IDO (126). Toulmonde et al. revealed that TAM-expressing IDO1 is favorable to the M2 phenotype, indicating that a vital resistance mechanism to PD-1 ICI may be on account of preferential formation of these immune-suppressive phenotypic TAM (127). A clinical trial (NCT03414229) aimed at evaluating the combination of IDO inhibitor epacadostat with pembrolizumab in sarcoma is currently ongoing.
HER2 is highly expressed across a number of OS cell lines so is an ideal candidate for the combination of ICIs. Promisingly, a novel processing method that noncovalently binds TRA with nanomaterial grapheme oxide (GO) into stable TRA/GO complexes was achieved to eliminate OS cells, through inducing augmented oxidative stress and HER2 signaling (128). The clinical trial of TRA plus chemotherapy drug deruxtecan is ongoing (NCT04616560), and the cardiovascular safety in children and adolescent of this therapy has been proved in a previous study (129). We note that adjuvant high-dose chemotherapy is suggested to hinder tumor immune surveillance systems while killing tumor cells, and may increase the risk of a second malignancy as reported (130).
Adoptive T-Cell Therapy (ACT)
ICIs serve to revive a suppressed or suboptimal immune system. On the contrary, ACT is another great promising immunotherapy which directly “tells” T cells the characterization of tumors, then selectively recognizes, orients, and attacks tumors. Over a few decades of intense studies, three main ACTs have been distinguished: chimeric antigen receptor (CAR)-engineered T cells, T cell receptor (TCR)-engineered T cells, and tumor-infiltrating lymphocytes (TILs) (131). This treatment strategy represented impressive clinical responses with ex vivo-manufactured cellular therapies aiming at tumor antigens. After CAR T cell therapy obtained extraordinary clinical success in hematologic malignancies and lymphoma with FDA approval, the FDA has granted orphan drug designation to a CLDN18.2-specific CAR T cell agent in gastric/GEJ cancer, indicating a breakthrough in the ACT of solid tumors (132).
CAR T Therapy
CAR T cells are engineered to recognize tumor-associated antigens (TAAs). Briefly, the transduced CAR consists of (i) an ectodomain derived from a single chain variable fragment of an antibody that recognizes a neoantigen, (ii) the transmembrane domain, and (iii) an endodomain with intracellular signaling domains derived from the CD3 ζ chain and co-stimulatory molecules. This structure enables T cells to recognize TAAs and result in T-cell-mediated tumor cytotoxicity in a major histocompatibility complex (MHC)-independent manner (133). CAR T cells targeting CD19 have shown an unprecedented response against B cell hematologic malignancies, reaching 90% remission rates in clinical trials and becoming the first genetically modified cell-based treatment approved by the FDA (134, 135).
Three FDA-approved CAR T cell products and virtually all the current clinical trials use γ-retroviral or lentiviral to modify gene ex vivo. Nevertheless, viral vectors come with some noteworthy safety concerns and production challenges. Due to the broad distribution of proviral insertion sites of retroviral vectors, oncogenic transformation may be secondary to potential insertional mutagenesis (131). Ruella et al. reported that a mis-insertion of the CAR gene into a single leukemic B cell mediated by lentiviral vector during T cell manufacturing can lead to overexpansion of CAR-transduced B cell leukemia (CARB) cells and irreversible death in a B cell acute lymphoblastic leukemia patient. Moreover, these CAR B cells integrate with CD19 on the surface of leukemic cells, disguising it from recognition by and conferring resistance to CAR T cells. Despite no exact mechanism being identified, leukemic blasts were detected to have lentiviral vector insertions in two sites: one was in chromosome 13 in intron 18 of the propionyl-CoA carboxylase-A gene9, and the other was in chromosome 11, 62.5 kb downstream of the neuropilin-1 gene. Although it is a specific case, the mis-insertion resulted in oncogenesis and resistance to CAR T cells. This study highlighted that we should be more vigilant, and that better manufacturing technologies are needed when using lentiviral vectors (136). In addition, the limitation of the long-lasting transgene expression induced by viral vectors may lead to persistent B cell aplasia and cytokine release syndrome (137, 138). In terms of production, lot-to-lot variation in viral transduction efficiencies as well as the high cost and time-consuming nature of the T cell manufacturing procedure also remain noteworthy challenges. Based on the defects above, novel non-viral transfection methods were designed and classified as membrane permeabilization-based means and carrier-based means, which are featured with an added bonus of high flexibility in effector molecules and cell types (131).
Due to the heterogeneity of tumor antigens, limited targets are identified in OS. Exhausted CAR T cells over-expressed inhibitive receptors such as PD-1, with an upregulation of PD-L1 on tumor cells at the same time. Combination therapy with PD-1 ICI or genetically engineered CAR T cells expressing tumor-limited PD-1 blockade can rescue the immunosuppression of single CAR T therapy partially (139). In addition, a clinical trial (NCT04433221) that combined CAR T cells with low dose chemotherapy which was demonstrated to modulate surface PD-L1 level is undergoing. Furthermore, multi-target CAR T cells are constructed to improve antigen recognition and avert tumor recurrence caused by the overgrowth of certain antigen-negative or low-expressed cells (140). In the view that CTLs co-expressing TIM-3-PD-1 function more exhaustedly than those expressing PD-1 only, homogenously inhibiting TIM-3 and PD-1 may better prime T cells and strengthen their anti-tumor cytotoxicity (141, 142). For instance, CD19 CAR T cells exhibiting considerable co-expression of inhibitory receptors TIM-3+/PD-1+ showed a lower CD4/CD8 ratio in TexMACS medium, indicating an increased immunity (143).
Another factor of failure in combating solid tumors with CAR T cells accounts for restricted infiltration and poor persistence caused by a stiff osteoid bone tumor matrix and immunosuppressive components in the TME (133). Hence the incorporation of costimulatory molecules and cytokines are designed to enhance CAR T-cell activation and strengthen its function. In fact, in order to improve costimulatory signaling, CD28-based and CD28-CD3ζ-OX40 CAR T cells are being tested in clinical trials respectively on patients with sarcoma (NCT00902044 and NCT01953900). The transgenic cytokine expression of IL-2, IL-15, or IL-23 on CAR T cells also improved proliferative activity (144). Simultaneously, T cells redirected for universal cytokine killing were developed with a nuclear factor of activated T cell (NFAT)-responsive promoter that only initiated cytokine release when CAR recognized a tumor antigen, making for minimizing systemic toxicity and elevating cytokine concentration at the tumor site (140). As to facilitating migration to the tumor site, CAR T cells were modified with co-expression of CCL5 and CXCL9 which created a loop to magnify lymphocyte engraftment through effective CD8+ recruitment (145).
TCR T Therapy
Different from CAR T cells, receptors expressed on the TCR T cells are produced from high affinity and high acidity tumor antigen-specific T-cell clones, which allow TCR T cells’ specificity and sensitivity for targeting cell-surface human leukocyte antigens (HLA) (146). Superior to antibodies and CARs in targeting efficiency, TCRs are able to penetrate tumors and engage with both tumor intracellular and surface antigenic peptides presented by HLA (147, 148). This feature allows for more latent targets and potential for further application in solid tumors. Cancer germline antigens express in a restricted manner in testis tissues and tumor tissues from different histological origins, while germ cells lack MHC molecule expression and are protected from TCR T cell-mediated immune attack (149). A cancer germline antigen NY-ESO-1 is an ideal target for TCR T cell therapy. In a phase I/II trial of NY-ESO-1-specific TCR-T treatment, 61% of synovial cell sarcoma patients benefited from the treatment without severe side effects (150). Moreover, NY-ESO-1 expression is found in 31.3% of OS tumors and TCR that targets NY-ESO-1 is being tested currently in patients with OS (NCT03462316). Most previous studies engineered CD8+ T cells that encode MHC-I-restricted TCRs for treatment, however, Lu et al. managed to treat patients with OS by CD4+ T cells transduced with MHC-II-restricted TCRs and MAGE-A3 and all experimented patients showed objective partial responses on metastatic lung lesions (151).
Papillomavirus binding factor (PBF) is a kind of DNA-binding transcription factor whose expression reaches up to 92% in OS. PBF A24.2 peptides are determined to activate CTLs from HLA-A24-positive patients with OS and therefore trigger immunity to eliminate tumor cells (152). A PBF TCR-multimer has been successfully created to recognize the naturally presented PBF peptide on HLA-A24+PBF+ OS cells (153). Although data about the usage of PBF-modified T cells in treating OS are not available, studies of this issue are critical, as suggested by the encouraging results with PBF’s role in oncogenicity and immunogenicity.
On the contrary, limitations of TCR T therapy come from the fact that tumors are likely to escape immunity by decreasing the expression of their MHCs and the usage is restricted in different patients with various HLA haplotypes because of HLA restriction (133). Baeuerle et al. solved this problem through developing TCR fusion constructs (TRuCs) with five fused TCR subunits, which activate anti-tumor immune response in a HLA-independent way. Of note, TRuC T cells showed better safety in well-controlled cytokine release compared with CAR T cell therapy (154). Another challenge is mispairing of the introduced α/β TCR chains with endogenous α/β TCR chains, that not only weakens the expression and efficacy of TCR T cells, but can also result in recognition of unintended antigens and may go further with autoimmune response and toxicities (101). A substitute of invariant natural killer T TCR T cells engineered and later differentiated from hematopoietic stem cells has been explored and potently protected a melanoma mouse model from lung metastasis (155). A relevant clinical trial in patients with OS is ongoing, testing the safety of transplanting TCRαβ+/CD19+-depleted haploidentical hematopoietic stem cells for treatment (NCT02508038).
TIL T Therapy
OS is characterized with a high proportion of TILs which play a vital role in regulating the progression of tumors. They were proposed with several advantages compared to non-infiltrating lymphocytes. First of all, TILs are organized from the TME with requisite chemokine receptors and thus they have highly specific directionality into tumors (156). Secondly, most TILs target mutated tumor-specific antigens instead of self-antigens, lowering the risk of autoimmunity subsequent to TIL T therapy (157). What is more, T cells recognizing mutated antigens is independent of central tolerance which allows for the expression of higher affinity TCR (158). Given those benefits, TIL T therapy was pioneered by Rosenberg in patients with melanoma, as the first adopted form of effective T cell therapy for solid tumors (159).
TILs are obtained from resected tumors followed by ex vivo expansion, and subsequently are transferred to patients in enormous quantity after lymphdepletion. Co-cultured with IL-2, TILs are exposed to rapid expansion protocols (REPs), under the restimulation of monoclonal anti-CD3 antibodies in the presence of allogeneic irradiated peripheral mononuclear cells and IL-2 (160). However, the isolation and expansion of TILs from OS tissue are considered to be uncertain, because the level of obtained TILs is far from sufficient regarding the requirement for immunotherapy (161). An additional obstacle in OS is low immunomodulatory molecules and suffusion of suppressive mediators on OS cells that may hinder activation and proliferation of TILs (162). The novel generation of TIL T cells showed satisfactory persistence, consistent with memory phenotypes of the majority of T cells. Sarnaik et al. reported a one-time cellular therapy named lifileucel (LN-144) with durable responses and an 80% disease control rate in advanced melanoma patients following failure of ICI therapy (163). The promising clinical trial about applying TIL LN-145 in treating patients with OS is ongoing (NCT03449108). Furthermore, combining ICI with TIL T cells may also represent a valid treatment option for OS patients progressing to individual therapies. Anti-CTLA-4 inhibitors were validated to boost HLA binding affinity of TIL T cells in melanoma as well as promoting CD8+ TILs expansion in Lewis lung carcinoma (164). The most recent study conducted by Wang et al. suggested that TILs plus anti-PD1 therapy showed remarkable clinical outcomes in metastatic OS patients, with a nearly quintuple objective response rate than single anti-PD1 therapy with a significantly prolonged medium progression-free survival time and medium overall survival time (165).
Cancer Vaccines
Cancer vaccines are another novel immunotherapy that induce anti-tumor effects through stimulating patient’s endogenous immune response. Tumor antigens of whole cells, lysates, DNA, RNA, peptides, or proteins are presented or exposed to initiate the immune system (21, 166). In the 1970s, Marcove et al. pioneered the use of autologous tumor lysates as cancer vaccine therapy and resulted in increased overall survival in patients with OS (167). For cross-century researching, the usage of cancer vaccines has gained momentum and is mainly classified into three types: immune cell vaccines, autologous tumor cell vaccines, and non-cell-based vaccines. Immune cell vaccines were developed to exploit innate immunocyte (DCs, γδ T cells, and macrophages) advantages of activating effector T cells to the full. However, at the same time, limited by the immunosuppressive molecules in the TME and quality/quantity of compromised immune effector cells in patients, the availability of migration and activation is a main concern (66). On the other side, autologous tumor cell vaccines and non-cell-based vaccines bypass this obstacle. The recognition mechanism of autologous tumor cell vaccines is HLA-I-independent, and patient immune systems function to specifically select the most immunogenic antigen (164). Non-cell-based vaccines are characterized with higher efficiency and safety because these certain peptides or viruses can avoid an off-target effect (168).
Immune Cell Vaccines
DC vaccines are the most widely used vaccination approaches in tumors. DCs are professional antigen-presenting cells with robust stimulation function. They endocytose and present antigens to naïve T cells which are subsequently stimulated to differentiation into tumor-killing cells (66). DC vaccines are developed to reverse the tumor-induced inhibition of APC antigen presentation activity and remove immunosuppression (169, 170). The latest progress in DC vaccines is FDA-granted orphan drug designation for ilixadencel against hepatocellular carcinoma considering the great success obtained in a phase 1 trial implemented with ilixadencel in Sweden (171). The general manufacturing procedure of DC vaccines is as follows: DCs are isolated from peripheral blood mononuclear cells, matured and pulsed with tumor antigen ex vivo, and ultimately injected back into the patient (66). The classification based on the various sources of pulsed antigens is divided into three major types: 1) DCs co-cultured with tumor-specific peptides or proteins; 2) DCs transfected with DNA or RNA encoding for tumor antigens; and 3) DCs co-cultured with tumor lysates or fused with devitalized tumor cells. The first two approaches are particularly effective for identified antigenic targets, whereas the third one bypasses the necessity for identified antigens and automatically forces the patient’s immune system to target the most antineoplastic antigen (164). Krishnadas et al. showed a decent clinical response with DCs pulsed with peptides stemmed from cancer germline antigens (MAGE-A1, MAGE-A3, and NY-ESO-1) against neuroblastoma, Ewing sarcoma, osteosarcoma, and rhabdomyosarcoma (172). As for some kinds of OS with cancer germline antigen genes silenced, combining demethylating treatment can be introduced to raising their expression (173). Besides, combination therapy of DC vaccines and targeted drugs such as anti-transforming growth factor-β/glucocorticoid-induced tumor necrosis factor receptor antibodies, has been elucidated to efficiently suppress the progression of primary and metastatic tumors by remodeling the TME to be more immunized (174). Another notable improvement came from the source of DCs. The major source of DCs in clinical trials is human CD14+ monocytes or CD34+ progenitors, but lately Zhou et al. proved that type 1 conventional dendritic cells can also elicit systemic and sustaining tumor-specific T cell-mediated cytotoxicity in a mice model loaded with OS (175). This finding further expanded the usage and development of DC vaccines.
However, tumor-associated suppression results in some unsatisfactory outcomes in clinical trials of DC vaccines (172, 176). The combinations are being evaluated now with gemcitabine which inhibits myeloid-derived suppressor cells to improve effects of tumor-associated suppression (NCT01803152). To eliminate deregulation from immune checkpoints, ICIs are adopted together. For instance, Nagaoka et al. demonstrated that anti-PD-1 treatment enhanced the infiltration and function of TILs induced by DC vaccines in melanoma-loaded mice models (177). Blockade of CLTA-4 has also been reported to augment T cell priming capacity of the DC vaccine and showed reduced angiogenesis and metastasis progression in colon and breast tumor-bearing mice (178). Hence, using ICIs with DC vaccines plays a complementary role in elevating antitumor efficacy and warrants further evaluation in clinical trials.
Apart from DCs, another two types of immune cells, γδ T cells, and macrophages have been put forward for tumor vaccines in the form of peptide-pulsed γδ T vaccines and chimeric antigen receptor macrophages (CAR-Ms). γδ T cells have a powerful ability of priming CD8+ T cells (179). In addition, γδ T cells are superior to traditional DC vaccines because the way they activate cytotoxic activity against cancer cells is HLA-independent (66). In the initial study, the researchers reported that γδ T cells can directly recognize and attack OS cells, despite the fact that targeted cell lines were merely moderately susceptible to γδ T cell cytotoxicity (180). FDA-approved rapamycin as an mTOR inhibitor can enlarge the elicited γδ T cell response through boosting proinflammatory factor release and enhancing tumor core infiltration (181, 182). Given the intrinsic function to penetrate tumors and activate T cell-mediated anti-tumor immunity, researchers recently engineered novel CAR-Ms to attain three aims: 1) presenting tumor antigens to naïve T cells and instigating them into CTLs, 2) fostering a pro-inflammatory TME by converting M2 macrophages to the M1 phenotype, and 3) phagocytizing tumor cells directly. CAR-Ms can be endowed with stable M1 phenotype after being genetically engineered by the chimeric adenoviral vector (Ad5f35). Klichinsky et al. developed an anti-HER2 CAR-M which displayed a notable reduction in tumor burden and a prolonged overall survival in mice models with metastatic lung cancer (183). To optimize the producing process and elude the risk of viral vectors’ oncogenicity with ex vivo CAR-M production, Kang et al. fabricated polymer nanocarriers which delivered genes encoding CAR and interferon-γ to macrophages in vivo and managed to in situ code to tumor-specific CAR-expressing and M1 phenotype macrophages (184). Thus far, these previous studies may pave the way for applying γδ T cell- and CAR-M-based vaccines in treating OS.
Autologous Tumor Cell Vaccines
Autologous tumor cell vaccines bypass DC isolation and culture ex vivo, and directly initiate DC response in vivo. Tumor cells are isolated from the patient, expanded if necessary, and then receive irradiation before being re-fused into the patient (185). In patients with EWS, an autologous tumor cell vaccine pulsed with GM-CSF and a furin convertase-targeted snRNA has been demonstrated to trigger a valid immune response in half of the patients (186). In the field of OS, a recent study combined an autologous tumor cell vaccine, ACT, and IL-2 together in dogs with OS and obtained remarkably prolonged survival compared to traditional amputation therapy (187). Apparently an autologous tumor cell vaccine has great potential in single or combination therapy, but its safety and efficacy in human beings with OS requires further study to elucidate.
Non-Cell-Based Vaccine
New approaches are breathing new life to tumor vaccine strategies. Peptide- and viral-based vaccines have a similar mechanism of presenting the antigen directly to DCs in vivo. The tumor–associated antigen PBF-derived peptide vaccination has long been explored for HLA-A2/A24+ patients with OS, Li et al. performed a breakthrough study in creating a peptide-specific tetramer targeting QVT and LSA peptides and showed cytotoxicity against HLA-A11+PBF+ OS cells (188). Other vaccinations based on HER2 have been underscored by benefits in canine models with OS, including a HER2-targeted recombinant listeria vaccine and an epidermal growth factor receptor (homology to HER2)-targeted peptide vaccine, which reduced metastasis and improved prognosis compared to the control group (189, 190). The peptide-based vaccine has to function in the context of HLA-I, and immunologic adjuvant is needed to gain a sufficient T cell response. Apart from polarization-inducing ability on macrophages mentioned above, mifamurtide can stimulate TLR4 to upregulate the expression of type 1 interferon, so it may have the potential as a substitute for INF-α (164). Furthermore, two recently completed phase I/II clinical trials with oncolytic HSV1716 (NCT00931931) and unmodified oncolytic reovirus REOLYSIN® (NCT00503295) explored their effectiveness in treating OS, which may become novel approaches for oncolytic viral vaccines.
Conclusion
Based on deepening understanding of the biological characteristics of OS, treatment has broadly developed across the centuries. Immunotherapy has revolutionized the management of OS since it was introduced, which can change non-responders to responders and strengthen the responses that do occur. In this review, we explored the specific TME and current application of immunotherapy in OS, classified as ICIs, ACT, and cancer vaccines. OS induces an immune-negative microenvironment characterized by high densities of M2 macrophages and low densities of TILs, which leads to drug resistance and inferior overall survival of OS patients. Particularly, immunotherapy breaks bottlenecks in tumor immune escape and chemotherapeutic resistance; ICIs target those protein molecules or their ligands that downregulate autoimmune function and thus re-activate the autoimmune system. ACT produces engineered T lymphocytes with high precision and efficiency which are equipped with specific neoplastic antigen-targeted receptors in vitro. Tumor vaccines are a range of biological ornaments loaded with TAA to provoke the immune system into recognizing and attacking those extrinsic antigens. Immunotherapy in melanoma, prostate cancer, renal cell carcinoma, and non-small-cell lung cancer has become a reality, and is now approved by the FDA.
However, to date, novel immunotherapies remain limited for OS. The main obstacles focus on two fields: finite T-cell infiltration and secondary immune toxicity. It is hard to permeate into targeted tissues because of immunosuppressive TME and dense fibrous tissue around solid tumors that impede T-cell infiltration. Oncolytic virus, which can infect and lyse tumor cells, has been identified to reverse the primary resistance to PD-1 blockade therapy through increasing intratumoral infiltration by CD8+ T cells and elevating PD-L1 expression (191, 192). At present, oncolytic virus for OS is in preclinical exploration and it is hoped that it can break the defense of solid surfaces in OS (193). Moreover, angiotensin inhibitors have been suggested to lessen extracellular matrix sclerosis in solid tumors, involving pancreatic tumors, breast tumors, and colorectal adenocarcinoma (194, 195). In addition, nanotechnology has become widespread in medication. These biodegradable nanoparticles are used as adjuvants to guide and control molecules that come into play at targeted sites (196). They can enter certain cells through phagocytosis, specific endocytosis, or by penetrating cells directly and thus induce a series of inflammatory chemokines to be released in the TME, and eventually attract T-cell infiltration (197). Beyond enhancing delivery, biomaterials can be applied in prompting the expansion of T cells ex vivo, such as constructing an APC-mimetic scaffold to precisely reproduce expansion stimulus signals in vivo (198). Multitarget approaches, providing a simultaneous inhibition of TME components, are being considered to offer a more efficient way to treat cancer. At present, the “immune cocktail therapy” of combining multiple immunotherapeutic strategies holds great promise, which can modulate the cancer-immunity cycle including but not limited to increasing immune cell infiltration and augmenting the cytotoxicity of T cells. We summarize the ongoing or planned clinical experiments that were mentioned above in Table 3.

Table 3 Some ongoing or planned clinical trials of novel immunotherapy.
With regard to toxicity, immune agents can cause systematic cytokine release syndrome, capillary leak syndrome, and a sepsis-like syndrome, and irAEs can be involved in any organ or tissue. It has been reported that combination of ICIs brought about more severe and broader AEs and worse even, irAEs not only appear in the early stage of treatment but also may develop in a prolonged period (199). To lower the risk of side effects to the maximum extent, more personalized therapeutic approaches are in urgent need of development. Patients should be stratified by the susceptibility to develop irAEs before starting, with the identification of biomarkers that predict patient responses and control the progress of treatment. In addition, agents need to be altered on the basis of targets recognized in patient biopsy samples and usability immune-related grading in clinical trials. In general, immunotherapy poses great prospects for the treatment of OS and further research is needed on molecular mechanisms to realize more accurate curative effects.
Author Contributions
The co-first author is responsible for paper writing and literature search. The co-corresponding author is responsible for proofreading the article. The other authors are all members of this research group, and they jointly undertake the drawing and revision work of the paper. All authors contributed to the article and approved the submitted version.
Funding
Science and Technology Project of Gansu Province (20JR5RA325) and Lanzhou Science and Technology Bureau Project (2017-4-60).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
OS, osteosarcoma; MSCs, mesenchymal stem cells; DCs, dendritic cells; NK, natural killer; TAMs, tumor-associated macrophages; iNOS, inducible nitric-oxide synthase; EMA, European Medical Agency; ATRA, all-trans retinoic acid; CSCs, cancer stem cells; CAR-Ms, chimeric antigen receptor macrophages; FDA, Food and Drug Administration; PHNPs, porous hollow iron oxide nanoparticles; TILs, tumor-infiltrating lymphocytes; Tfh, follicular helper T; PD-1, programmed cell death protein 1; CTLA-4, cytotoxic T-lymphocyte associated protein 4; irAE, immune-related adverse event; Tim-3, T-cell immunoglobulin and mucin-domain containing-3; LAG-3, lymphocyte-activation gene 3; IDO, indoleamine 2,3-dioxygenase; HER2, human epidermal growth factor receptor-2; TRA, trastuzumab; GO, grapheme oxide; ACT, adoptive T-cell therapy; CAR, chimeric antigen receptor; TCR, T cell receptor; TILs, tumor-infiltrating lymphocytes; TAAs, tumor-associated antigens; MHC, major histocompatibility complex; CARB, CAR-transduced B cell leukemia; NFAT, nuclear factor of activated T cells; HLA, human leukocyte antigens; PBF, papillomavirus binding factor; TRuCs, TCR fusion constructs; REPs, rapid expansion protocols.
References
1. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: Current Treatment and a Collaborative Pathway to Success. J Clin Oncol (2015) 33(27):3029–35. doi: 10.1200/jco.2014.59.4895
2. Pingping B, Yuhong Z, Weiqi L, Chunxiao W, Chunfang W, Yuanjue S, et al. Incidence and Mortality of Sarcomas in Shanghai, China, During 2002-2014. Front Oncol (2019) 9:662. doi: 10.3389/fonc.2019.00662
3. Zhou Y, Yang D, Yang Q, Lv X, Huang W, Zhou Z, et al. Single-Cell RNA Landscape of Intratumoral Heterogeneity and Immunosuppressive Microenvironment in Advanced Osteosarcoma. Nat Commun (2020) 11(1):6322. doi: 10.1038/s41467-020-20059-6
4. Kansara M, Teng MW, Smyth MJ, Thomas DM. Translational Biology of Osteosarcoma. Nat Rev Cancer (2014) 14(11):722–35. doi: 10.1038/nrc3838
5. Gaspar N, Occean BV, Pacquement H, Bompas E, Bouvier C, Brisse HJ, et al. Results of Methotrexate-Etoposide-Ifosfamide Based Regimen (M-EI) in Osteosarcoma Patients Included in the French OS2006/sarcome-09 Study. Eur J Cancer (Oxford England: 1990) (2018) 88:57–66. doi: 10.1016/j.ejca.2017.09.036
6. Zhao J, Dean DC, Hornicek FJ, Yu X, Duan Z. Emerging Next-Generation Sequencing-Based Discoveries for Targeted Osteosarcoma Therapy. Cancer Lett (2020) 474:158–67. doi: 10.1016/j.canlet.2020.01.020
7. Zhang B, Zhang Y, Li R, Li J, Lu X, Zhang Y. The Efficacy and Safety Comparison of First-Line Chemotherapeutic Agents (High-Dose Methotrexate, Doxorubicin, Cisplatin, and Ifosfamide) for Osteosarcoma: A Network Meta-Analysis. J Orthop Surg Res (2020) 15(1):51. doi: 10.1186/s13018-020-1576-0
8. Smrke A, Anderson PM, Gulia A, Gennatas S, Huang PH, Jones RL. Future Directions in the Treatment of Osteosarcoma. Cells (2021) 10(1):172. doi: 10.3390/cells10010172
9. Meyers PA, Healey JH, Chou AJ, Wexler LH, Merola PR, Morris CD, et al. Addition of Pamidronate to Chemotherapy for the Treatment of Osteosarcoma. Cancer (2011) 117(8):1736–44. doi: 10.1002/cncr.25744
10. Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res (2019) 79(18):4557–66. doi: 10.1158/0008-5472.Can-18-3962
11. Zheng Y, Zhou H, Dunstan CR, Sutherland RL, Seibel MJ. The Role of the Bone Microenvironment in Skeletal Metastasis. J Bone Oncol (2013) 2(1):47–57. doi: 10.1016/j.jbo.2012.11.002
12. Junttila MR, de Sauvage FJ. Influence of Tumour Micro-Environment Heterogeneity on Therapeutic Response. Nature (2013) 501(7467):346–54. doi: 10.1038/nature12626
13. Belli C, Trapani D, Viale G, D’Amico P, Duso BA, Della Vigna P, et al. Targeting the Microenvironment in Solid Tumors. Cancer Treat Rev (2018) 65:22–32. doi: 10.1016/j.ctrv.2018.02.004
14. Yang C, Tian Y, Zhao F, Chen Z, Su P, Li Y, et al. Bone Microenvironment and Osteosarcoma Metastasis. Int J Mol Sci (2020) 21(19):6985. doi: 10.3390/ijms21196985
15. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun Signal (2020) 18(1):59. doi: 10.1186/s12964-020-0530-4
16. Chen C, Xie L, Ren T, Huang Y, Xu J, Guo W. Immunotherapy for Osteosarcoma: Fundamental Mechanism, Rationale, and Recent Breakthroughs. Cancer Lett (2021) 500:1–10. doi: 10.1016/j.canlet.2020.12.024
17. Burgess M, Tawbi H. Immunotherapeutic Approaches to Sarcoma. Curr Treat Options Oncol (2015) 16(6):26. doi: 10.1007/s11864-015-0345-5
18. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-Related Inflammation. Nature (2008) 454(7203):436–44. doi: 10.1038/nature07205
19. Heymann MF, Lézot F, Heymann D. The Contribution of Immune Infiltrates and the Local Microenvironment in the Pathogenesis of Osteosarcoma. Cell Immunol (2019) 343:103711. doi: 10.1016/j.cellimm.2017.10.011
20. Kodet O, Němejcova K, Strnadová K, Havlínová A, Dundr P, Krajsová I, et al. The Abscopal Effect in the Era of Checkpoint Inhibitors. Int J Mol Sci (2021) 22(13):7204. doi: 10.3390/ijms22137204
21. Anwar MA, El-Baba C, Elnaggar MH, Elkholy YO, Mottawea M, Johar D, et al. Novel Therapeutic Strategies for Spinal Osteosarcomas. Semin Cancer Biol (2020) 64:83–92. doi: 10.1016/j.semcancer.2019.05.018
22. Hu C, Liu C, Tian S, Wang Y, Shen R, Rao H, et al. Comprehensive Analysis of Prognostic Tumor Microenvironment-Related Genes in Osteosarcoma Patients. BMC Cancer (2020) 20(1):814. doi: 10.1186/s12885-020-07216-2
23. Zhang C, Zheng JH, Lin ZH, Lv HY, Ye ZM, Chen YP, et al. Profiles of Immune Cell Infiltration and Immune-Related Genes in the Tumor Microenvironment of Osteosarcoma. Aging (2020) 12(4):3486–501. doi: 10.18632/aging.102824
24. Yu Y, Zhang H, Ren T, Huang Y, Liang X, Wang W, et al. Development of a Prognostic Gene Signature Based on an Immunogenomic Infiltration Analysis of Osteosarcoma. J Cell Mol Med (2020) 24(19):11230–42. doi: 10.1111/jcmm.15687
25. Murray PJ, Wynn TA. Protective and Pathogenic Functions of Macrophage Subsets. Nat Rev Immunol (2011) 11(11):723–37. doi: 10.1038/nri3073
26. Sica A, Mantovani A. Macrophage Plasticity and Polarization: In Vivo Veritas. J Clin Invest (2012) 122(3):787–95. doi: 10.1172/jci59643
27. Mosser DM, Edwards JP. Exploring the Full Spectrum of Macrophage Activation. Nat Rev Immunol (2008) 8(12):958–69. doi: 10.1038/nri2448
28. Mirabello L, Zhu B, Koster R, Karlins E, Dean M, Yeager M, et al. Frequency of Pathogenic Germline Variants in Cancer-Susceptibility Genes in Patients With Osteosarcoma. JAMA Oncol (2020) 6(5):724–34. doi: 10.1001/jamaoncol.2020.0197
29. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
30. Yang L, Zhang Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J Hematol Oncol (2017) 10(1):58. doi: 10.1186/s13045-017-0430-2
31. Lee CH, Espinosa I, Vrijaldenhoven S, Subramanian S, Montgomery KD, Zhu S, et al. Prognostic Significance of Macrophage Infiltration in Leiomyosarcomas. Clin Cancer Res (2008) 14(5):1423–30. doi: 10.1158/1078-0432.Ccr-07-1712
32. Jensen TO, Schmidt H, Møller HJ, Høyer M, Maniecki MB, Sjoegren P, et al. Macrophage Markers in Serum and Tumor Have Prognostic Impact in American Joint Committee on Cancer Stage I/II Melanoma. J Clin Oncol (2009) 27(20):3330–7. doi: 10.1200/jco.2008.19.9919
33. van Dongen M, Savage ND, Jordanova ES, Briaire-de Bruijn IH, Walburg KV, Ottenhoff TH, et al. Anti-Inflammatory M2 Type Macrophages Characterize Metastasized and Tyrosine Kinase Inhibitor-Treated Gastrointestinal Stromal Tumors. Int J Cancer (2010) 127(4):899–909. doi: 10.1002/ijc.25113
34. Han Y, Guo W, Ren T, Huang Y, Wang S, Liu K, et al. Tumor-Associated Macrophages Promote Lung Metastasis and Induce Epithelial-Mesenchymal Transition in Osteosarcoma by Activating the COX-2/STAT3 Axis. Cancer Lett (2019) 440-441:116–25. doi: 10.1016/j.canlet.2018.10.011
35. Wolf-Dennen K, Gordon N, Kleinerman ES. Exosomal Communication by Metastatic Osteosarcoma Cells Modulates Alveolar Macrophages to an M2 Tumor-Promoting Phenotype and Inhibits Tumoricidal Functions. Oncoimmunology (2020) 9(1):1747677. doi: 10.1080/2162402x.2020.1747677
36. Dhupkar P, Gordon N, Stewart J, Kleinerman ES. Anti-PD-1 Therapy Redirects Macrophages From an M2 to an M1 Phenotype Inducing Regression of OS Lung Metastases. Cancer Med (2018) 7(6):2654–64. doi: 10.1002/cam4.1518
37. Gomez-Brouchet A, Illac C, Gilhodes J, Bouvier C, Aubert S, Guinebretiere JM, et al. CD163-Positive Tumor-Associated Macrophages and CD8-Positive Cytotoxic Lymphocytes are Powerful Diagnostic Markers for the Therapeutic Stratification of Osteosarcoma Patients: An Immunohistochemical Analysis of the Biopsies Fromthe French OS2006 Phase 3 Trial. Oncoimmunology (2017) 6(9):e1331193. doi: 10.1080/2162402x.2017.1331193
38. Buddingh EP, Kuijjer ML, Duim RA, Bürger H, Agelopoulos K, Myklebost O, et al. Tumor-Infiltrating Macrophages are Associated With Metastasis Suppression in High-Grade Osteosarcoma: A Rationale for Treatment With Macrophage Activating Agents. Clin Cancer Res (2011) 17(8):2110–9. doi: 10.1158/1078-0432.Ccr-10-2047
39. Han Q, Shi H, Liu F. CD163(+) M2-Type Tumor-Associated Macrophage Support the Suppression of Tumor-Infiltrating T Cells in Osteosarcoma. Int Immunopharmacol (2016) 34:101–6. doi: 10.1016/j.intimp.2016.01.023
40. Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
41. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity (2014) 40(2):274–88. doi: 10.1016/j.immuni.2014.01.006
42. Atri C, Guerfali FZ, Laouini D. Role of Human Macrophage Polarization in Inflammation During Infectious Diseases. Int J Mol Sci (2018) 19(6):1801. doi: 10.3390/ijms19061801
43. Punzo F, Bellini G, Tortora C, Pinto DD, Argenziano M, Pota E, et al. Mifamurtide and TAM-Like Macrophages: Effect on Proliferation, Migration and Differentiation of Osteosarcoma Cells. Oncotarget (2020) 11(7):687–98. doi: 10.18632/oncotarget.27479
44. Fukuda H, Nakamura S, Chisaki Y, Takada T, Toda Y, Murata H, et al. Daphnetin Inhibits Invasion and Migration of LM8 Murine Osteosarcoma Cells by Decreasing RhoA and Cdc42 Expression. Biochem Biophys Res Commun (2016) 471(1):63–7. doi: 10.1016/j.bbrc.2016.01.179
45. Lv J, Chen FK, Liu C, Liu PJ, Feng ZP, Jia L, et al. Zoledronic Acid Inhibits Thyroid Cancer Stemness and Metastasis by Repressing M2-Like Tumor-Associated Macrophages Induced Wnt/β-Catenin Pathway. Life Sci (2020) 256:117925. doi: 10.1016/j.lfs.2020.117925
46. Tsagozis P, Eriksson F, Pisa P. Zoledronic Acid Modulates Antitumoral Responses of Prostate Cancer-Tumor Associated Macrophages. Cancer Immunol Immunother (2008) 57(10):1451–9. doi: 10.1007/s00262-008-0482-9
47. Li Z, Liu Y, Fang X, Shu Z. Nanomaterials Enhance the Immunomodulatory Effect of Molecular Targeted Therapy. Int J Nanomedicine (2021) 16:1631–61. doi: 10.2147/ijn.S290346
48. Liu J, Liang H, Li M, Luo Z, Zhang J, Guo X, et al. Tumor Acidity Activating Multifunctional Nanoplatform for NIR-Mediated Multiple Enhanced Photodynamic and Photothermal Tumor Therapy. Biomaterials (2018) 157:107–24. doi: 10.1016/j.biomaterials.2017.12.003
49. Dai L, Li X, Duan X, Li M, Niu P, Xu H, et al. A Ph/ROS Cascade-Responsive Charge-Reversal Nanosystem With Self-Amplified Drug Release for Synergistic Oxidation-Chemotherapy. Adv Sci (Weinheim Baden-Wurttemberg Germany) (2019) 6(4):1801807. doi: 10.1002/advs.201801807
50. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron Oxide Nanoparticles Inhibit Tumour Growth by Inducing Pro-Inflammatory Macrophage Polarization in Tumour Tissues. Nat Nanotechnol (2016) 11(11):986–94. doi: 10.1038/nnano.2016.168
51. Wang G, Zhao J, Zhang M, Wang Q, Chen B, Hou Y, et al. Ferumoxytol and CpG Oligodeoxynucleotide 2395 Synergistically Enhance Antitumor Activity of Macrophages Against NSCLC With EGFR(L858R/T790M) Mutation. Int J Nanomedicine (2019) 14:4503–15. doi: 10.2147/ijn.S193583
52. Li K, Lu L, Xue C, Liu J, He Y, Zhou J, et al. Polarization of Tumor-Associated Macrophage Phenotype via Porous Hollow Iron Nanoparticles for Tumor Immunotherapy In Vivo. Nanoscale (2020) 12(1):130–44. doi: 10.1039/c9nr06505a
53. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. Pi3kγ is a Molecular Switch That Controls Immune Suppression. Nature (2016) 539(7629):437–42. doi: 10.1038/nature19834
54. Liu M, Tong Z, Ding C, Luo F, Wu S, Wu C, et al. Transcription Factor C-Maf is a Checkpoint That Programs Macrophages in Lung Cancer. J Clin Invest (2020) 130(4):2081–96. doi: 10.1172/jci131335
55. Medvedeva GF, Kuzmina DO, Nuzhina J, Shtil AA, Dukhinova MS. How Macrophages Become Transcriptionally Dysregulated: A Hidden Impact of Antitumor Therapy. Int J Mol Sci (2021) 22(5):2662. doi: 10.3390/ijms22052662
56. Múdry P, Kýr M, Rohleder O, Mahdal M, Staniczková Zambo I, Ježová M, et al. Improved Osteosarcoma Survival With Addition of Mifamurtide to Conventional Chemotherapy - Observational Prospective Single Institution Analysis. J Bone Oncol (2021) 28:100362. doi: 10.1016/j.jbo.2021.100362
57. Groisberg R, Hong DS, Behrang A, Hess K, Janku F, Piha-Paul S, et al. Characteristics and Outcomes of Patients With Advanced Sarcoma Enrolled in Early Phase Immunotherapy Trials. J Immunother Cancer (2017) 5(1):100. doi: 10.1186/s40425-017-0301-y
58. Ando K, Mori K, Corradini N, Redini F, Heymann D. Mifamurtide for the Treatment of Nonmetastatic Osteosarcoma. Expert Opin Pharmacother (2011) 12(2):285–92. doi: 10.1517/14656566.2011.543129
59. Zhou Q, Xian M, Xiang S, Xiang D, Shao X, Wang J, et al. All-Trans Retinoic Acid Prevents Osteosarcoma Metastasis by Inhibiting M2 Polarization of Tumor-Associated Macrophages. Cancer Immunol Res (2017) 5(7):547–59. doi: 10.1158/2326-6066.Cir-16-0259
60. Shao XJ, Xiang SF, Chen YQ, Zhang N, Cao J, Zhu H, et al. Inhibition of M2-Like Macrophages by All-Trans Retinoic Acid Prevents Cancer Initiation and Stemness in Osteosarcoma Cells. Acta Pharmacol Sin (2019) 40(10):1343–50. doi: 10.1038/s41401-019-0262-4
61. Kimura Y, Sumiyoshi M. Antitumor and Antimetastatic Actions of Dihydroxycoumarins (Esculetin or Fraxetin) Through the Inhibition of M2 Macrophage Differentiation in Tumor-Associated Macrophages and/or G1 Arrest in Tumor Cells. Eur J Pharmacol (2015) 746:115–25. doi: 10.1016/j.ejphar.2014.10.048
62. Sica A, Bronte V. Altered Macrophage Differentiation and Immune Dysfunction in Tumor Development. J Clin Invest (2007) 117(5):1155–66. doi: 10.1172/jci31422
63. Kähkönen TE, Halleen JM, Bernoulli J. Osteoimmuno-Oncology: Therapeutic Opportunities for Targeting Immune Cells in Bone Metastasis. Cells (2021) 10(6):1529. doi: 10.3390/cells10061529
64. Gomez-Brouchet A, Gilhodes J, Acker NV, Brion R, Bouvier C, Assemat P, et al. Characterization of Macrophages and Osteoclasts in the Osteosarcoma Tumor Microenvironment at Diagnosis: New Perspective for Osteosarcoma Treatment? Cancers (2021) 13(3):423. doi: 10.3390/cancers13030423
65. Barros MH, Hauck F, Dreyer JH, Kempkes B, Niedobitek G. Macrophage Polarisation: An Immunohistochemical Approach for Identifying M1 and M2 Macrophages. PloS One (2013) 8(11):e80908. doi: 10.1371/journal.pone.0080908
66. Wang Z, Wang Z, Li B, Wang S, Chen T, Ye Z. Innate Immune Cells: A Potential and Promising Cell Population for Treating Osteosarcoma. Front Immunol (2019) 10:1114. doi: 10.3389/fimmu.2019.01114
67. Castro F, Cardoso AP, Gonçalves RM, Serre K, Oliveira MJ. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front Immunol (2018) 9:847. doi: 10.3389/fimmu.2018.00847
68. Fritzsching B, Fellenberg J, Moskovszky L, Sápi Z, Krenacs T, Machado I, et al. CD8(+)/FOXP3(+)-Ratio in Osteosarcoma Microenvironment Separates Survivors From non-Survivors: A Multicenter Validated Retrospective Study. Oncoimmunology (2015) 4(3):e990800. doi: 10.4161/2162402x.2014.990800
69. Ligon JA, Choi W, Cojocaru G, Fu W, Hsiue EH, Oke TF, et al. Pathways of Immune Exclusion in Metastatic Osteosarcoma are Associated With Inferior Patient Outcomes. J Immunother Cancer (2021) 9(5):e001772. doi: 10.1136/jitc-2020-001772
70. Le T, Su S, Shahriyari L. Investigating Optimal Chemotherapy Options for Osteosarcoma Patients Through a Mathematical Model. Cells (2021) 10(8):2009. doi: 10.3390/cells10082009
71. Le T, Su S, Kirshtein A, Shahriyari L. Data-Driven Mathematical Model of Osteosarcoma. Cancers (2021) 13(10):2367. doi: 10.3390/cancers13102367
72. Li X, Chen Y, Liu X, Zhang J, He X, Teng G, et al. Tim3/Gal9 Interactions Between T Cells and Monocytes Result in an Immunosuppressive Feedback Loop That Inhibits Th1 Responses in Osteosarcoma Patients. Int Immunopharmacol (2017) 44:153–9. doi: 10.1016/j.intimp.2017.01.006
73. Mochizuki K, Kawana S, Yamada S, Muramatsu M, Sano H, Kobayashi S, et al. Various Checkpoint Molecules, and Tumor-Infiltrating Lymphocytes in Common Pediatric Solid Tumors: Possibilities for Novel Immunotherapy. Pediatr Hematol Oncol (2019) 36(1):17–27. doi: 10.1080/08880018.2019.1578843
74. Wherry EJ, Kurachi M. Molecular and Cellular Insights Into T Cell Exhaustion. Nat Rev Immunol (2015) 15(8):486–99. doi: 10.1038/nri3862
75. Topchyan P, Xin G, Chen Y, Zheng S, Burns R, Shen J, et al. Harnessing the IL-21-BATF Pathway in the CD8(+) T Cell Anti-Tumor Response. Cancers (2021) 13(6):1236. doi: 10.3390/cancers13061263
76. Liu S, Lizée G, Lou Y, Liu C, Overwijk WW, Wang G, et al. IL-21 Synergizes With IL-7 to Augment Expansion and Anti-Tumor Function of Cytotoxic T Cells. Int Immunol (2007) 19(10):1213–21. doi: 10.1093/intimm/dxm093
77. Li Y, Yee C. IL-21 Mediated Foxp3 Suppression Leads to Enhanced Generation of Antigen-Specific CD8+ Cytotoxic T Lymphocytes. Blood (2008) 111(1):229–35. doi: 10.1182/blood-2007-05-089375
78. Gao W, Zhou J, Ji B. Evidence of Interleukin 21 Reduction in Osteosarcoma Patients Due to PD-1/PD-L1-Mediated Suppression of Follicular Helper T Cell Functionality. DNA Cell Biol (2017) 36(9):794–800. doi: 10.1089/dna.2017.3669
79. Spolski R, Li P, Leonard WJ. Biology and Regulation of IL-2: From Molecular Mechanisms to Human Therapy. Nat Rev Immunol (2018) 18(10):648–59. doi: 10.1038/s41577-018-0046-y
80. Walker LS. CD4+ CD25+ Treg: Divide and Rule? Immunology (2004) 111(2):129–37. doi: 10.1111/j.0019-2805.2003.01788.x
81. Solomon I, Amann M, Goubier A, Vargas FA, Zervas D, Qing C, et al. CD25-T(reg)-Depleting Antibodies Preserving IL-2 Signaling on Effector T Cells Enhance Effector Activation and Antitumor Immunity. Nat Cancer (2020) 1(12):1153–66. doi: 10.1038/s43018-020-00133-0
82. Chang X, Ma Z, Zhu G, Lu Y, Yang J. New Perspective Into Mesenchymal Stem Cells: Molecular Mechanisms Regulating Osteosarcoma. J Bone Oncol (2021) 29:100372. doi: 10.1016/j.jbo.2021.100372
83. Sarhadi VK, Daddali R, Seppänen-Kaijansinkko R. Mesenchymal Stem Cells and Extracellular Vesicles in Osteosarcoma Pathogenesis and Therapy. Int J Mol Sci (2021) 22(20):11035. doi: 10.3390/ijms222011035
84. Duffy MM, Ritter T, Ceredig R, Griffin MD. Mesenchymal Stem Cell Effects on T-Cell Effector Pathways. Stem Cell Res Ther (2011) 2(4):34. doi: 10.1186/scrt75
85. Franquesa M, Hoogduijn MJ, Bestard O, Grinyó JM. Immunomodulatory Effect of Mesenchymal Stem Cells on B Cells. Front Immunol (2012) 3:212. doi: 10.3389/fimmu.2012.00212
86. Casado JG, Tarazona R, Sanchez-Margallo FM. NK and MSCs Crosstalk: The Sense of Immunomodulation and Their Sensitivity. Stem Cell Rev Rep (2013) 9(2):184–9. doi: 10.1007/s12015-013-9430-y
87. Song N, Scholtemeijer M, Shah K. Mesenchymal Stem Cell Immunomodulation: Mechanisms and Therapeutic Potential. Trends Pharmacol Sci (2020) 41(9):653–64. doi: 10.1016/j.tips.2020.06.009
88. Kelleher FC, O’Sullivan H. Monocytes, Macrophages, and Osteoclasts in Osteosarcoma. J Adolesc Young Adult Oncol (2017) 6(3):396–405. doi: 10.1089/jayao.2016.0078
89. Li H, Hong S, Qian J, Zheng Y, Yang J, Yi Q. Cross Talk Between the Bone and Immune Systems: Osteoclasts Function as Antigen-Presenting Cells and Activate CD4+ and CD8+ T Cells. Blood (2010) 116(2):210–7. doi: 10.1182/blood-2009-11-255026
90. Davis KL, Fox E, Merchant MS, Reid JM, Kudgus RA, Liu X, et al. Nivolumab in Children and Young Adults With Relapsed or Refractory Solid Tumours or Lymphoma (ADVL1412): A Multicentre, Open-Label, Single-Arm, Phase 1-2 Trial. Lancet Oncol (2020) 21(4):541–50. doi: 10.1016/s1470-2045(20)30023-1
91. Xin Yu J, Hodge JP, Oliva C, Neftelinov ST, Hubbard-Lucey VM, Tang J. Trends in Clinical Development for PD-1/PD-L1 Inhibitors. Nat Rev Drug Discov (2020) 19(3):163–4. doi: 10.1038/d41573-019-00182-w
92. Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol (2017) 8:561. doi: 10.3389/fphar.2017.00561
93. Kraehenbuehl L, Weng CH, Eghbali S, Wolchok JD, Merghoub T. Enhancing Immunotherapy in Cancer by Targeting Emerging Immunomodulatory Pathways. Nat Rev Clin Oncol (2021) 19(1):37–50. doi: 10.1038/s41571-021-00552-7
94. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: A Moving Target in Immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
95. Jia D, Li S, Li D, Xue H, Yang D, Liu Y. Mining TCGA Database for Genes of Prognostic Value in Glioblastoma Microenvironment. Aging (2018) 10(4):592–605. doi: 10.18632/aging.101415
96. Huang W, Ran R, Shao B, Li H. Prognostic and Clinicopathological Value of PD-L1 Expression in Primary Breast Cancer: A Meta-Analysis. Breast Cancer Res Treat (2019) 178(1):17–33. doi: 10.1007/s10549-019-05371-0
97. Pollack SM, He Q, Yearley JH, Emerson R, Vignali M, Zhang Y, et al. T-Cell Infiltration and Clonality Correlate With Programmed Cell Death Protein 1 and Programmed Death-Ligand 1 Expression in Patients With Soft Tissue Sarcomas. Cancer (2017) 123(17):3291–304. doi: 10.1002/cncr.30726
98. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell (2017) 170(6):1120–33.e17. doi: 10.1016/j.cell.2017.07.024
99. Chen DS, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity (2013) 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012
100. Poto R, Marone G, Pirozzi F, Galdiero MR, Cuomo A, Formisano L, et al. How can We Manage the Cardiac Toxicity of Immune Checkpoint Inhibitors? Expert Opin Drug Saf (2021) 20(6):685–94. doi: 10.1080/14740338.2021.1906860
101. Wang Z, Li B, Ren Y, Ye Z. T-Cell-Based Immunotherapy for Osteosarcoma: Challenges and Opportunities. Front Immunol (2016) 7:353. doi: 10.3389/fimmu.2016.00353
102. Koirala P, Roth ME, Gill J, Piperdi S, Chinai JM, Geller DS, et al. Immune Infiltration and PD-L1 Expression in the Tumor Microenvironment are Prognostic in Osteosarcoma. Sci Rep (2016) 6:30093. doi: 10.1038/srep30093
103. Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in Advanced Soft-Tissue Sarcoma and Bone Sarcoma (SARC028): A Multicentre, Two-Cohort, Single-Arm, Open-Label, Phase 2 Trial. Lancet Oncol (2017) 18(11):1493–501. doi: 10.1016/s1470-2045(17)30624-1
104. Boye K, Longhi A, Guren T, Lorenz S, Næss S, Pierini M, et al. Pembrolizumab in Advanced Osteosarcoma: Results of a Single-Arm, Open-Label, Phase 2 Trial. Cancer Immunol Immunother (2021) 70(9):2617–24. doi: 10.1007/s00262-021-02876-w
105. Geoerger B, Zwaan CM, Marshall LV, Michon J, Bourdeaut F, Casanova M, et al. Atezolizumab for Children and Young Adults With Previously Treated Solid Tumours, non-Hodgkin Lymphoma, and Hodgkin Lymphoma (iMATRIX): A Multicentre Phase 1-2 Study. Lancet Oncol (2020) 21(1):134–44. doi: 10.1016/s1470-2045(19)30693-x
106. Xie L, Xu J, Sun X, Guo W, Gu J, Liu K, et al. Apatinib Plus Camrelizumab (Anti-PD1 Therapy, SHR-1210) for Advanced Osteosarcoma (APFAO) Progressing After Chemotherapy: A Single-Arm, Open-Label, Phase 2 Trial. J Immunother Cancer (2020) 8(1):e000798. doi: 10.1136/jitc-2020-000798
107. Hennessy M, Wahba A, Felix K, Cabrera M, Segura MG, Kundra V, et al. Bempegaldesleukin (BEMPEG; NKTR-214) Efficacy as a Single Agent and in Combination With Checkpoint-Inhibitor Therapy in Mouse Models of Osteosarcoma. Int J Cancer (2021) 148(8):1928–37. doi: 10.1002/ijc.33382
108. Lussier DM, O’Neill L, Nieves LM, McAfee MS, Holechek SA, Collins AW, et al. Enhanced T-Cell Immunity to Osteosarcoma Through Antibody Blockade of PD-1/PD-L1 Interactions. J Immunother (Hagerstown Md: 1997) (2015) 38(3):96–106. doi: 10.1097/cji.0000000000000065
109. Lussier DM, Johnson JL, Hingorani P, Blattman JN. Combination Immunotherapy With α-CTLA-4 and α-PD-L1 Antibody Blockade Prevents Immune Escape and Leads to Complete Control of Metastatic Osteosarcoma. J Immunother Cancer (2015) 3:21. doi: 10.1186/s40425-015-0067-z
110. D’Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN, et al. Nivolumab With or Without Ipilimumab Treatment for Metastatic Sarcoma (Alliance A091401): Two Open-Label, non-Comparative, Randomised, Phase 2 Trials. Lancet Oncol (2018) 19(3):416–26. doi: 10.1016/s1470-2045(18)30006-8
111. Sterz U, Grube M, Herr W, Menhart K, Wendl C, Vogelhuber M. Case Report: Dual Checkpoint Inhibition in Advanced Metastatic Osteosarcoma Results in Remission of All Tumor Manifestations-A Report of a Stunning Success in a 37-Year-Old Patient. Front Oncol (2021) 11:684733. doi: 10.3389/fonc.2021.684733
112. Nuytemans L, Sys G, Creytens D, Lapeire L. NGS-Analysis to the Rescue: Dual Checkpoint Inhibition in Metastatic Osteosarcoma - a Case Report and Review of the Literature. Acta Clin Belg (2021) 76(2):162–7. doi: 10.1080/17843286.2019.1683129
113. Haanen J, Carbonnel F, Robert C, Kerr KM, Peters S, Larkin J, et al. Management of Toxicities From Immunotherapy: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann Oncol (2017) 28(suppl_4):iv119–iv42. doi: 10.1093/annonc/mdx225
114. Cortellini A, Tucci M, Adamo V, Stucci LS, Russo A, Tanda ET, et al. Integrated Analysis of Concomitant Medications and Oncological Outcomes From PD-1/PD-L1 Checkpoint Inhibitors in Clinical Practice. J Immunother Cancer (2020) 8(2):e001361. doi: 10.1136/jitc-2020-001361
115. Stroud CR, Hegde A, Cherry C, Naqash AR, Sharma N, Addepalli S, et al. Tocilizumab for the Management of Immune Mediated Adverse Events Secondary to PD-1 Blockade. J Oncol Pharm Pract (2019) 25(3):551–7. doi: 10.1177/1078155217745144
116. Dancsok AR, Setsu N, Gao D, Blay JY, Thomas D, Maki RG, et al. Expression of Lymphocyte Immunoregulatory Biomarkers in Bone and Soft-Tissue Sarcomas. Mod Pathol (2019) 32(12):1772–85. doi: 10.1038/s41379-019-0312-y
117. Gill J, Gorlick R. Advancing Therapy for Osteosarcoma. Nat Rev Clin Oncol (2021) 18(10):609–24. doi: 10.1038/s41571-021-00519-8
118. Wing JB, Tay C, Sakaguchi S. Control of Regulatory T Cells by Co-Signal Molecules. Adv Exp Med Biol (2019) 1189:179–210. doi: 10.1007/978-981-32-9717-3_7
119. Li X, Lu H, Gu Y, Zhang X, Zhang G, Shi T, et al. Tim-3 Suppresses the Killing Effect of Vγ9vδ2 T Cells on Colon Cancer Cells by Reducing Perforin and Granzyme B Expression. Exp Cell Res (2020) 386(1):111719. doi: 10.1016/j.yexcr.2019.111719
120. Pu F, Chen F, Zhang Z, Qing X, Lin H, Zhao L, et al. TIM-3 Expression and its Association With Overall Survival in Primary Osteosarcoma. Oncol Lett (2019) 18(5):5294–300. doi: 10.3892/ol.2019.10855
121. Cheng Z, Wang L, Wu C, Huang L, Ruan Y, Xue W. Tumor-Derived Exosomes Induced M2 Macrophage Polarization and Promoted the Metastasis of Osteosarcoma Cells Through Tim-3. Arch Med Res (2021) 52(2):200–10. doi: 10.1016/j.arcmed.2020.10.018
122. Liu H, Zhi L, Duan N, Su P. Abnormal Expression of Tim-3 Antigen on Peripheral Blood T Cells is Associated With Progressive Disease in Osteosarcoma Patients. FEBS Open Bio (2016) 6(8):807–15. doi: 10.1002/2211-5463.12079
123. Que Y, Fang Z, Guan Y, Xiao W, Xu B, Zhao J, et al. LAG-3 Expression on Tumor-Infiltrating T Cells in Soft Tissue Sarcoma Correlates With Poor Survival. Cancer Biol Med (2019) 16(2):331–40. doi: 10.20892/j.issn.2095-3941.2018.0306
124. Pignon JC, Jegede O, Shukla SA, Braun DA, Horak CE, Wind-Rotolo M, et al. irRECIST for the Evaluation of Candidate Biomarkers of Response to Nivolumab in Metastatic Clear Cell Renal Cell Carcinoma: Analysis of a Phase II Prospective Clinical Trial. Clin Cancer Res (2019) 25(7):2174–84. doi: 10.1158/1078-0432.Ccr-18-3206
125. Liebau C, Baltzer AW, Schmidt S, Roesel C, Karreman C, Prisack JB, et al. Interleukin-12 and Interleukin-18 Induce Indoleamine 2,3-Dioxygenase (IDO) Activity in Human Osteosarcoma Cell Lines Independently From Interferon-Gamma. Anticancer Res (2002) 22(2a):931–6.
126. Urakawa H, Nishida Y, Nakashima H, Shimoyama Y, Nakamura S, Ishiguro N. Prognostic Value of Indoleamine 2,3-Dioxygenase Expression in High Grade Osteosarcoma. Clin Exp Metastasis (2009) 26(8):1005–12. doi: 10.1007/s10585-009-9290-7
127. Toulmonde M, Penel N, Adam J, Chevreau C, Blay JY, Le Cesne A, et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas: A Phase 2 Clinical Trial. JAMA Oncol (2018) 4(1):93–7. doi: 10.1001/jamaoncol.2017.1617
128. Xiao H, Jensen PE, Chen X. Elimination of Osteosarcoma by Necroptosis With Graphene Oxide-Associated Anti-HER2 Antibodies. Int J Mol Sci (2019) 20(18):4360. doi: 10.3390/ijms20184360
129. Kopp LM, Womer RB, Schwartz CL, Ebb DH, Franco VI, Hall D, et al. Effects of Dexrazoxane on Doxorubicin-Related Cardiotoxicity and Second Malignant Neoplasms in Children With Osteosarcoma: A Report From the Children’s Oncology Group. Cardiooncology (London England) (2019) 5:15. doi: 10.1186/s40959-019-0050-9
130. Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM, et al. The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol Res (2017) 5(9):767–77. doi: 10.1158/2326-6066.Cir-16-0365
131. Raes L, De Smedt SC, Raemdonck K, Braeckmans K. Non-Viral Transfection Technologies for Next-Generation Therapeutic T Cell Engineering. Biotechnol Adv (2021) 49:107760. doi: 10.1016/j.biotechadv.2021.107760
132. Jiang H, Shi Z, Wang P, Wang C, Yang L, Du G, et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J Natl Cancer Inst (2019) 111(4):409–18. doi: 10.1093/jnci/djy134
133. Balta E, Wabnitz GH, Samstag Y. Hijacked Immune Cells in the Tumor Microenvironment: Molecular Mechanisms of Immunosuppression and Cues to Improve T Cell-Based Immunotherapy of Solid Tumors. Int J Mol Sci (2021) 22(11):5736. doi: 10.3390/ijms22115736
134. Geyer MB, Brentjens RJ. Review: Current Clinical Applications of Chimeric Antigen Receptor (CAR) Modified T Cells. Cytotherapy (2016) 18(11):1393–409. doi: 10.1016/j.jcyt.2016.07.003
135. Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell (2020) 38(4):473–88. doi: 10.1016/j.ccell.2020.07.005
136. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of Resistance to Chimeric Antigen Receptor T Cell Therapy by Transduction of a Single Leukemic B Cell. Nat Med (2018) 24(10):1499–503. doi: 10.1038/s41591-018-0201-9
137. Tomuleasa C, Fuji S, Berce C, Onaciu A, Chira S, Petrushev B, et al. Chimeric Antigen Receptor T-Cells for the Treatment of B-Cell Acute Lymphoblastic Leukemia. Front Immunol (2018) 9:239. doi: 10.3389/fimmu.2018.00239
138. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood (2014) 124(2):188–95. doi: 10.1182/blood-2014-05-552729
139. Chen N, Morello A, Tano Z, Adusumilli PS. CAR T-Cell Intrinsic PD-1 Checkpoint Blockade: A Two-in-One Approach for Solid Tumor Immunotherapy. Oncoimmunology (2017) 6(2):e1273302. doi: 10.1080/2162402x.2016.1273302
140. Tian Y, Li Y, Shao Y, Zhang Y. Gene Modification Strategies for Next-Generation CAR T Cells Against Solid Cancers. J Hematol Oncol (2020) 13(1):54. doi: 10.1186/s13045-020-00890-6
141. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J Exp Med (2010) 207(10):2187–94. doi: 10.1084/jem.20100643
142. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-Cell Exhaustion During Chronic Viral Infection. Proc Natl Acad Sci U S A (2010) 107(33):14733–8. doi: 10.1073/pnas.1009731107
143. Alnabhan R, Gaballa A, Mörk LM, Mattsson J, Uhlin M, Magalhaes I. Media Evaluation for Production and Expansion of Anti-CD19 Chimeric Antigen Receptor T Cells. Cytotherapy (2018) 20(7):941–51. doi: 10.1016/j.jcyt.2018.04.007
144. Ma X, Shou P, Smith C, Chen Y, Du H, Sun C, et al. Interleukin-23 Engineering Improves CAR T Cell Function in Solid Tumors. Nat Biotechnol (2020) 38(4):448–59. doi: 10.1038/s41587-019-0398-2
145. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation Between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell (2019) 35(6):885–900.e10. doi: 10.1016/j.ccell.2019.05.004
146. Zhao Q, Jiang Y, Xiang S, Kaboli PJ, Shen J, Zhao Y, et al. Engineered TCR-T Cell Immunotherapy in Anticancer Precision Medicine: Pros and Cons. Front Immunol (2021) 12:658753. doi: 10.3389/fimmu.2021.658753
147. Brameshuber M, Kellner F, Rossboth BK, Ta H, Alge K, Sevcsik E, et al. Monomeric TCRs Drive T Cell Antigen Recognition. Nat Immunol (2018) 19(5):487–96. doi: 10.1038/s41590-018-0092-4
148. Harper J, Adams KJ, Bossi G, Wright DE, Stacey AR, Bedke N, et al. An Approved In Vitro Approach to Preclinical Safety and Efficacy Evaluation of Engineered T Cell Receptor Anti-CD3 Bispecific (ImmTAC) Molecules. PloS One (2018) 13(10):e0205491. doi: 10.1371/journal.pone.0205491
149. Iura K, Kohashi K, Ishii T, Maekawa A, Bekki H, Otsuka H, et al. MAGEA4 Expression in Bone and Soft Tissue Tumors: Its Utility as a Target for Immunotherapy and Diagnostic Marker Combined With NY-ESO-1. Virchows Arch (2017) 471(3):383–92. doi: 10.1007/s00428-017-2206-z
150. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. J Clin Oncol (2011) 29(7):917–24. doi: 10.1200/jco.2010.32.2537
151. Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol (2017) 35(29):3322–9. doi: 10.1200/jco.2017.74.5463
152. Tsukahara T, Kawaguchi S, Torigoe T, Kimura S, Murase M, Ichimiya S, et al. Prognostic Impact and Immunogenicity of a Novel Osteosarcoma Antigen, Papillomavirus Binding Factor, in Patients With Osteosarcoma. Cancer Sci (2008) 99(2):368–75. doi: 10.1111/j.1349-7006.2008.00695.x
153. Watanabe K, Tsukahara T, Toji S, Saitoh S, Hirohashi Y, Nakatsugawa M, et al. Development of a T-Cell Receptor Multimer With High Avidity for Detecting a Naturally Presented Tumor-Associated Antigen on Osteosarcoma Cells. Cancer Sci (2019) 110(1):40–51. doi: 10.1111/cas.13854
154. Baeuerle PA, Ding J, Patel E, Thorausch N, Horton H, Gierut J, et al. Synthetic TRuC Receptors Engaging the Complete T Cell Receptor for Potent Anti-Tumor Response. Nat Commun (2019) 10(1):2087. doi: 10.1038/s41467-019-10097-0
155. Smith DJ, Liu S, Ji S, Li B, McLaughlin J, Cheng D, et al. Genetic Engineering of Hematopoietic Stem Cells to Generate Invariant Natural Killer T Cells. Proc Natl Acad Sci USA (2015) 112(5):1523–8. doi: 10.1073/pnas.1424877112
156. Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of Tumor-Specific T Cells With CXCR2 Chemokine Receptor Improves Migration to Tumor and Antitumor Immune Responses. Clin Cancer Res (2010) 16(22):5458–68. doi: 10.1158/1078-0432.Ccr-10-0712
157. Junker N, Kvistborg P, Køllgaard T, Straten P, Andersen MH, Svane IM. Tumor Associated Antigen Specific T-Cell Populations Identified in Ex Vivo Expanded TIL Cultures. Cell Immunol (2012) 273(1):1–9. doi: 10.1016/j.cellimm.2011.12.004
158. van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor Exome Analysis Reveals Neoantigen-Specific T-Cell Reactivity in an Ipilimumab-Responsive Melanoma. J Clin Oncol (2013) 31(32):e439–42. doi: 10.1200/jco.2012.47.7521
159. Spiess PJ, Yang JC, Rosenberg SA. In Vivo Antitumor Activity of Tumor-Infiltrating Lymphocytes Expanded in Recombinant Interleukin-2. J Natl Cancer Inst (1987) 79(5):1067–75.
160. Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J Clin Oncol (2016) 34(20):2389–97. doi: 10.1200/jco.2016.66.7220
161. Hinrichs CS, Rosenberg SA. Exploiting the Curative Potential of Adoptive T-Cell Therapy for Cancer. Immunol Rev (2014) 257(1):56–71. doi: 10.1111/imr.12132
162. Muthana M, Rodrigues S, Chen YY, Welford A, Hughes R, Tazzyman S, et al. Macrophage Delivery of an Oncolytic Virus Abolishes Tumor Regrowth and Metastasis After Chemotherapy or Irradiation. Cancer Res (2013) 73(2):490–5. doi: 10.1158/0008-5472.Can-12-3056
163. Sarnaik AA, Hamid O, Khushalani NI, Lewis KD, Medina T, Kluger HM, et al. Lifileucel, a Tumor-Infiltrating Lymphocyte Therapy, in Metastatic Melanoma. J Clin Oncol (2021) 39(24):2656–66. doi: 10.1200/jco.21.00612
164. Tsukahara T, Emori M, Murata K, Mizushima E, Shibayama Y, Kubo T, et al. The Future of Immunotherapy for Sarcoma. Expert Opin Biol Ther (2016) 16(8):1049–57. doi: 10.1080/14712598.2016.1188075
165. Wang C, Li M, Wei R, Wu J. Adoptive Transfer of TILs Plus Anti-PD1 Therapy: An Alternative Combination Therapy for Treating Metastatic Osteosarcoma. J Bone Oncol (2020) 25:100332. doi: 10.1016/j.jbo.2020.100332
166. Pollack SM, Ingham M, Spraker MB, Schwartz GK. Emerging Targeted and Immune-Based Therapies in Sarcoma. J Clin Oncol (2018) 36(2):125–35. doi: 10.1200/jco.2017.75.1610
167. Marcove RC, Miké V, Huvos AG, Southam CM, Levin AG. Vaccine Trials for Osteogenic Sarcoma. A Preliminary Report. CA Cancer J Clin (1973) 23(2):74–80. doi: 10.3322/canjclin.23.2.74
168. Shemesh CS, Hsu JC, Hosseini I, Shen BQ, Rotte A, Twomey P, et al. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol Ther (2021) 29(2):555–70. doi: 10.1016/j.ymthe.2020.09.038
169. Suryawanshi A, Manicassamy S. Tumors Induce Immune Tolerance Through Activation of β-Catenin/TCF4 Signaling in Dendritic Cells: A Novel Therapeutic Target for Cancer Immunotherapy. Oncoimmunology (2015) 4(12):e1052932. doi: 10.1080/2162402x.2015.1052932
170. Cornwall SM, Wikstrom M, Musk AW, Alvarez J, Nowak AK, Nelson DJ. Human Mesothelioma Induces Defects in Dendritic Cell Numbers and Antigen-Processing Function Which Predict Survival Outcomes. Oncoimmunology (2016) 5(2):e1082028. doi: 10.1080/2162402x.2015.1082028
171. Rizell M, Sternby Eilard M, Andersson M, Andersson B, Karlsson-Parra A, Suenaert P. Phase 1 Trial With the Cell-Based Immune Primer Ilixadencel, Alone, and Combined With Sorafenib, in Advanced Hepatocellular Carcinoma. Front Oncol (2019) 9:19. doi: 10.3389/fonc.2019.00019
172. Krishnadas DK, Shusterman S, Bai F, Diller L, Sullivan JE, Cheerva AC, et al. A Phase I Trial Combining Decitabine/Dendritic Cell Vaccine Targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for Children With Relapsed or Therapy-Refractory Neuroblastoma and Sarcoma. Cancer Immunol Immunother (2015) 64(10):1251–60. doi: 10.1007/s00262-015-1731-3
173. Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis Antigens, Gametogenesis and Cancer. Nat Rev Cancer (2005) 5(8):615–25. doi: 10.1038/nrc1669
174. Kawano M, Tanaka K, Itonaga I, Iwasaki T, Miyazaki M, Ikeda S, et al. Dendritic Cells Combined With Anti-GITR Antibody Produce Antitumor Effects in Osteosarcoma. Oncol Rep (2015) 34(4):1995–2001. doi: 10.3892/or.2015.4161
175. Zhou Y, Slone N, Chrisikos TT, Kyrysyuk O, Babcock RL, Medik YB, et al. Vaccine Efficacy Against Primary and Metastatic Cancer With In Vitro-Generated CD103(+) Conventional Dendritic Cells. J Immunother Cancer (2020) 8(1):e000474. doi: 10.1136/jitc-2019-000474
176. Miwa S, Nishida H, Tanzawa Y, Takeuchi A, Hayashi K, Yamamoto N, et al. Phase 1/2 Study of Immunotherapy With Dendritic Cells Pulsed With Autologous Tumor Lysate in Patients With Refractory Bone and Soft Tissue Sarcoma. Cancer (2017) 123(9):1576–84. doi: 10.1002/cncr.30606
177. Nagaoka K, Hosoi A, Iino T, Morishita Y, Matsushita H, Kakimi K. Dendritic Cell Vaccine Induces Antigen-Specific CD8(+) T Cells That are Metabolically Distinct From Those of Peptide Vaccine and is Well-Combined With PD-1 Checkpoint Blockade. Oncoimmunology (2018) 7(3):e1395124. doi: 10.1080/2162402x.2017.1395124
178. Esmaily M, Masjedi A, Hallaj S, Nabi Afjadi M, Malakotikhah F, Ghani S, et al. Blockade of CTLA-4 Increases Anti-Tumor Response Inducing Potential of Dendritic Cell Vaccine. J Control Release (2020) 326:63–74. doi: 10.1016/j.jconrel.2020.06.017
179. Altvater B, Pscherer S, Landmeier S, Kailayangiri S, Savoldo B, Juergens H, et al. Activated Human γδ T Cells Induce Peptide-Specific CD8+ T-Cell Responses to Tumor-Associated Self-Antigens. Cancer Immunol Immunother (2012) 61(3):385–96. doi: 10.1007/s00262-011-1111-6
180. Kato Y, Tanaka Y, Miyagawa F, Yamashita S, Minato N. Targeting of Tumor Cells for Human Gammadelta T Cells by Nonpeptide Antigens. J Immunol (Baltimore Md: 1950) (2001) 167(9):5092–8. doi: 10.4049/jimmunol.167.9.5092
181. Dao V, Liu Y, Pandeswara S, Svatek RS, Gelfond JA, Liu A, et al. Immune-Stimulatory Effects of Rapamycin Are Mediated by Stimulation of Antitumor γδ T Cells. Cancer Res (2016) 76(20):5970–82. doi: 10.1158/0008-5472.Can-16-0091
182. Cao G, Wang Q, Li G, Meng Z, Liu H, Tong J, et al. mTOR Inhibition Potentiates Cytotoxicity of Vγ4 γδ T Cells via Up-Regulating NKG2D and TNF-α. J Leukoc Biol (2016) 100(5):1181–9. doi: 10.1189/jlb.5A0116-053RR
183. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human Chimeric Antigen Receptor Macrophages for Cancer Immunotherapy. Nat Biotechnol (2020) 38(8):947–53. doi: 10.1038/s41587-020-0462-y
184. Kang M, Lee SH, Kwon M, Byun J, Kim D, Kim C, et al. Nanocomplex-Mediated In Vivo Programming to Chimeric Antigen Receptor-M1 Macrophages for Cancer Therapy. Adv Mater (Deerfield Beach Fla) (2021) 33(43):e2103258. doi: 10.1002/adma.202103258
185. Dyson KA, Stover BD, Grippin A, Mendez-Gomez HR, Lagmay J, Mitchell DA, et al. Emerging Trends in Immunotherapy for Pediatric Sarcomas. J Hematol Oncol (2019) 12(1):78. doi: 10.1186/s13045-019-0756-z
186. Senzer N, Barve M, Kuhn J, Melnyk A, Beitsch P, Lazar M, et al. Phase I Trial of “bi-shRNAi(Furin)/GMCSF DNA/autologous Tumor Cell” Vaccine (FANG) in Advanced Cancer. Mol Ther (2012) 20(3):679–86. doi: 10.1038/mt.2011.269
187. Flesner BK, Wood GW, Gayheart-Walsten P, Sonderegger FL, Henry CJ, Tate DJ, et al. Autologous Cancer Cell Vaccination, Adoptive T-Cell Transfer, and Interleukin-2 Administration Results in Long-Term Survival for Companion Dogs With Osteosarcoma. J Vet Intern Med (2020) 34(5):2056–67. doi: 10.1111/jvim.15852
188. Li D, Toji S, Watanabe K, Torigoe T, Tsukahara T. Identification of Novel Human Leukocyte Antigen-A*11:01-Restricted Cytotoxic T-Lymphocyte Epitopes Derived From Osteosarcoma Antigen Papillomavirus Binding Factor. Cancer Sci (2019) 110(4):1156–68. doi: 10.1111/cas.13973
189. Mason NJ, Gnanandarajah JS, Engiles JB, Gray F, Laughlin D, Gaurnier-Hausser A, et al. Immunotherapy With a HER2-Targeting Listeria Induces HER2-Specific Immunity and Demonstrates Potential Therapeutic Effects in a Phase I Trial in Canine Osteosarcoma. Clin Cancer Res (2016) 22(17):4380–90. doi: 10.1158/1078-0432.Ccr-16-0088
190. Doyle HA, Gee RJ, Masters TD, Gee CR, Booth CJ, Peterson-Roth E, et al. Vaccine-Induced ErbB (EGFR/HER2)-Specific Immunity in Spontaneous Canine Cancer. Trans Oncol (2021) 14(11):101205. doi: 10.1016/j.tranon.2021.101205
191. Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell (2017) 170(6):1109–19.e10. doi: 10.1016/j.cell.2017.08.027
192. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight Into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov (2016) 6(8):827–37. doi: 10.1158/2159-8290.Cd-15-1545
193. Wedekind MF, Cripe TP. Oncolytic Viruses and Their Potential as a Therapeutic Opportunity in Osteosarcoma. Adv Exp Med Biol (2020) 1258:77–89. doi: 10.1007/978-3-030-43085-6_5
194. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin Inhibition Enhances Drug Delivery and Potentiates Chemotherapy by Decompressing Tumour Blood Vessels. Nat Commun (2013) 4:2516. doi: 10.1038/ncomms3516
195. Xie G, Cheng T, Lin J, Zhang L, Zheng J, Liu Y, et al. Local Angiotensin II Contributes to Tumor Resistance to Checkpoint Immunotherapy. J Immunother Cancer (2018) 6(1):88. doi: 10.1186/s40425-018-0401-3
196. Di Gioacchino M, Petrarca C, Gatta A, Scarano G, Farinelli A, Della Valle L, et al. Nanoparticle-Based Immunotherapy: State of the Art and Future Perspectives. Expert Rev Clin Immunol (2020) 16(5):513–25. doi: 10.1080/1744666x.2020.1762572
197. Ma Q, Zhou D, DeLyria ES, Wen X, Lu W, Thapa P, et al. Synthetic Poly(L-Glutamic Acid)-Conjugated CpG Exhibits Antitumor Efficacy With Increased Retention in Tumor and Draining Lymph Nodes After Intratumoral Injection in a Mouse Model of Melanoma. J Immunother (Hagerstown Md: 1997) (2017) 40(1):11–20. doi: 10.1097/cji.0000000000000145
198. Cheung AS, Zhang DKY, Koshy ST, Mooney DJ. Scaffolds That Mimic Antigen-Presenting Cells Enable Ex Vivo Expansion of Primary T Cells. Nat Biotechnol (2018) 36(2):160–9. doi: 10.1038/nbt.4047
Keywords: immunotherapies, osteosarcoma, immune microenvironment, PD-1, CAR T, TCR T, TIL T, cancer vaccines
Citation: Lu Y, Zhang J, Chen Y, Kang Y, Liao Z, He Y and Zhang C (2022) Novel Immunotherapies for Osteosarcoma. Front. Oncol. 12:830546. doi: 10.3389/fonc.2022.830546
Received: 07 December 2021; Accepted: 28 February 2022;
Published: 01 April 2022.
Edited by:
Alessandro Poggi, San Martino Hospital (IRCCS), ItalyReviewed by:
Pawel Muranski, Columbia University, United StatesPeter Anderson, Cleveland Clinic, United States
Copyright © 2022 Lu, Zhang, Chen, Kang, Liao, He and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cangyu Zhang, Ym9zdXllMTJAMTI2LmNvbQ==
†ORCID: Yubao Lu, orcid.org/0000-0003-4375-8316
Jiahe Zhang, orcid.org/0000-0003-4147-394X
‡These authors share first authorship