 Jingyu Zhao
Jingyu Zhao Yaqi Wang
Yaqi Wang Lei Tao1,2
Lei Tao1,2 Ligong Chen
Ligong Chen- 1School of Pharmaceutical Sciences, Key Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology (Ministry of Education), Tsinghua University, Beijing, China
- 2Collaborative Innovation Center for Biotherapy, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, West China Medical School, Sichuan University, Chengdu, China
- 3Advanced Innovation Center for Human Brain Protection, Beijing Tiantan Hospital, Capital Medical University, Beijing, China
Malignant brain tumors represent approximately 1.5% of all malignant tumors. The survival rate among patients is relatively low and the mortality rate of pediatric brain tumors ranks first among all childhood malignant tumors. At present malignant brain tumors remain incurable. Although some tumors can be treated with surgery and chemotherapy, new treatment strategies are urgent owing to the poor clinical prognosis. Iron is an essential trace element in many biological processes of the human body. Iron transporters play a crucial role in iron absorption and transport. Ferroptosis, an iron-dependent form of nonapoptotic cell death, is characterized by the accumulation of lipid peroxidation products and lethal reactive oxygen species (ROS) derived from iron metabolism. Recently, compelling evidence has shown that inducing ferroptosis of tumor cells is a potential therapeutic strategy. In this review, we will briefly describe the significant regulatory factors of ferroptosis, iron, its absorption and transport under physiological conditions, especially the function of iron transporters. Then we will summarize the relevant mechanisms of ferroptosis and its role in malignant brain tumors, wherein the role of transporters is not to be ignored. Finally, we will introduce the current research progress in the treatment of malignant brain tumors by inducing ferroptosis in order to explain the current biological principles of potential treatment targets and treatment strategies for malignant brain tumors.
1 Introduction
Brain tumors can be categorized as primary malignant types and secondary forms from metastasis (1). Of these, roughly 40% will be malignant and the incidence rate of malignant brain tumors is higher in males (2, 3). Primary brain tumors are the first common tumor and the first cause of tumor death in children (3). Brain tumors can be classified based on origin, such as glioblastoma (GBM), neuroblastoma and meningioma (4). GBM is the most common and aggressive malignant primary brain tumor, with a limited response to the current standard of treatment. Most GBM patients can only live up to 15-20 months (5).
Malignant brain tumors are commonly intratumoral heterogenic, which likely explains their poor clinical prognosis of malignant brain tumors poor and easy to relapse (6). Despite current multimodality treatment efforts, combining in surgical resection when feasible, with radiotherapy, chemotherapy and symptomatic treatment, the median survival remains short (7).
Iron is necessary for life (8). Iron plays an extremely significant role in brain development and function, and is involve in many biological processes such as embryonic neuronal development, myelin formation, neurotransmitter synthesis and oxidative phosphorylation (9, 10). Iron deficiency impairs the function of iron-requiring enzymes in all tissues, however, excessive iron accumulation leads to toxicity through oxidative stress activation of cell death signaling pathways (11). To maintain adequate and safe amounts of iron levels, cells express a the coordination of a wide variety of proteins, which tightly control both intracellular and systemic iron metabolism (12). Iron transporters participate in the regulation of iron uptake, storage and distribution, wherein help maintain iron homeostasis (13).
Ferroptosis is an iron-dependent form of regulated cell death (14). The intracellular iron homeostasis and balance between the oxidation and reduction of phospholipids is tightly associated with ferroptosis. Ferroptosis occurs when iron overload induces lipid peroxidation (11). Recent studies showed that ferroptosis is involved in the death of pathological cells in malignant brain tumors, which may have a therapeutic potential towards malignant brain tumors (15, 16). The specific way of ferroptosis inhibiting cancer may be to induce oxidative stress and resist treatment antagonism of cancer cells, in which iron transporters may has a stronger role. Although great progress has been made in the study of the biological function and disease correlation of ferroptosis, its biological signal pathway and underlying mechanism remain to be elucidated.
Starting from the iron transport in the body under physiological conditions, we further summarize the specific mechanism of iron metabolism disorder and ferroptosis in the pathological condition of malignant brain tumors, in particular, the crucial role of transporters. Finally, we summarized the specific mechanisms and targets for inducing ferroptosis in the treatment of malignant brain tumors and introduced potentially related drugs.
2 Iron Physiology
2.1 Iron and Iron Transporters
2.1.1 Iron Function
Iron is a vital micronutrient for nearly all living organisms due to its significant role in many biological processes such as catalyzing redox reactions and transporting oxygen. In addition, iron is essential for the functions of many enzymes and prosthetic groups (17, 18).
2.1.2 Dietary Sources of Iron
Iron is required across all human life stages, from embryological development, to infancy or old age. Estimated daily average iron requirements are the highest in pregnancy 3rd trimester (19). Despite having an efficient iron recycling mechanism, humans need to absorb about 10% of our total iron needs from regular dietary to maintain normal health. Dietary iron exists as either heme iron or non-heme iron. Heme iron is derived from hemoglobin, myoglobin and neuroglobin found in animal foods, and its absorption is not affected by diet; meanwhile non-heme iron is found mainly in plant foods, and its absorption is influenced by inhibitors and enhancers found in the diet. Nonetheless both are affected by iron storage levels in the body (19, 20).
2.1.3 Iron Absorption
The absorption site of iron is mainly in the mucosa of duodenum and upper jejunum. In a nutshell, iron absorption can be divided into two steps; first iron in food enters intestinal mucosal cells, second iron in intestinal mucosal cells crosses the cell membrane into capillaries and is transported systemically to the whole body in bloodstream (19).
2.1.4 Iron Transport
In humans, a number of proteins have evolved which tightly regulate iron homeostasis since we cannot rapidly excrete iron in the urine and iron must be transported and stored intracellular on a protein carrier due to extremely low free iron levels both systemically and intracellular (21). These includes the proteins that are involved in iron transport, both in the circulation and intracellularly, the reductases and oxidases that facilitate the movement of iron across cell membranes, and other proteins that regulate these processes (22). Iron transporters are vital role to maintain iron homeostasis in the body, and a total of 22 iron transporters have been identified (Table 1). The functions of several iron transporters are introduced below.
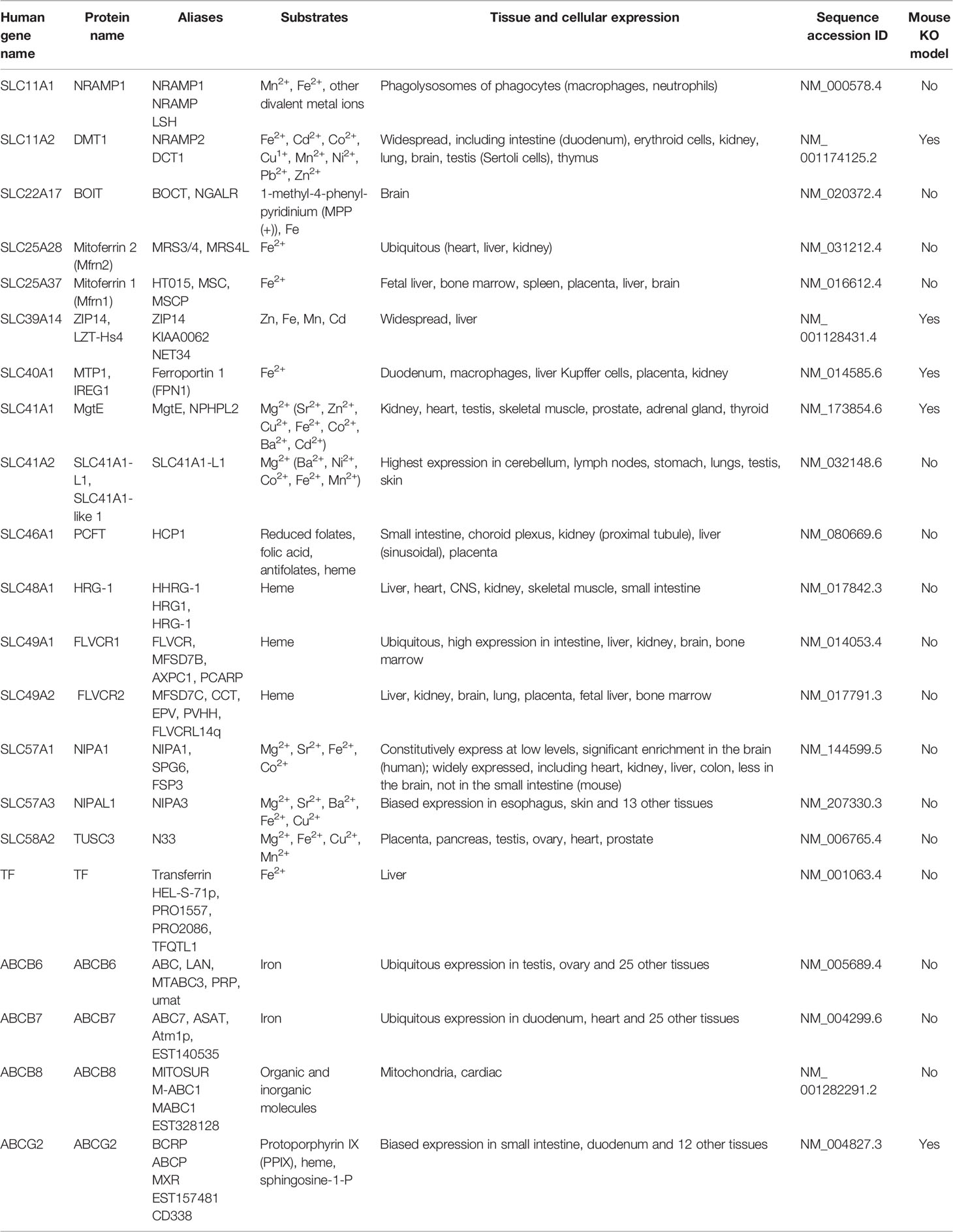
Table 1 Iron Transporters. For detailed information about the gene tables, please visit: http://www.bioparadigms and http://www.org.genecards.org.
Transferrin (TF) is regulator of free iron levels in body fluids, binding, sequestering, and transporting Fe3+ ions. This iron carrier protein helps maintain iron availability systemically and prevents tissue oxidative damage caused by excessive free radical accumulation (23).
SLC25A37 (Mitoferrin 1, Mfrn1) is a solute carrier localized in the mitochondrial inner membrane. When iron enters cells, Mfrn1 transport iron into mitochondria, which is used to synthesize mitochondrial heme and iron sulfur clusters. Mitoferrin-1 is necessary for neuronal energy metabolism and influences brain function (24).
SLC11A2 (Divalent metal cation transporter 1, DMT1), is a proton-dependent iron importer of Fe2+, is involved in systemic iron recycling and cellular iron absorption. DMT1 is located on the parietal membrane of duodenal intestinal epithelial cells, where it brings dietary free iron into cells and promotes iron absorption (25). DMT1 is also involved in transferrin/transferrin receptor 1 (TF/TFR1) pathway, wherein transports iron absorbed by this pathway from the endosome into the cytosol (26).
SLC40A1 (Ferroportin 1, Fpn1), is a major iron export protein, is expressed in many cells, such as placental syncytiotrophoblasts, wherein plays a role in transferring maternal iron to the fetus and releasing iron from tissue into the blood. It should be noted that inactivating the murine Fpn1 gene globally is embryonic lethal (27).
2.2 Brain Iron Transport
2.2.1 Brain Iron Function
Iron in the brain plays a crucial role in maintaining normal physiological function through its participation in many cellular activities such as mitochondrial respiration, myelin synthesis, neurotransmitter synthesis and metabolism (10). Iron is also essential in enzymes involved in the production of monoamines (dopamine, epinephrine, norepinephrine and serotonin), which are involved in social emotional development, executive function and memory processes. Therefore, maintaining iron homeostasis is essential for normal physiological activity of the brain (28).
Blood-brain barrier (BBB) and blood cerebrospinal fluid barrier (BCSFB) are of great significance to maintain the relative stability of physical and chemical factors in the internal environment of brain tissue and prevent harmful substances in blood from entering brain tissue (29). The BBB and BCSFB also controls iron transport from the bloodstream to the brain parenchyma, allowing for some independence of brain iron levels from the total body iron and providing some resistance to systemic iron toxicity (30, 31). Different cells types in the brain acquire iron through different pathways, which involving a myriad iron transporters (Table 2) (29).
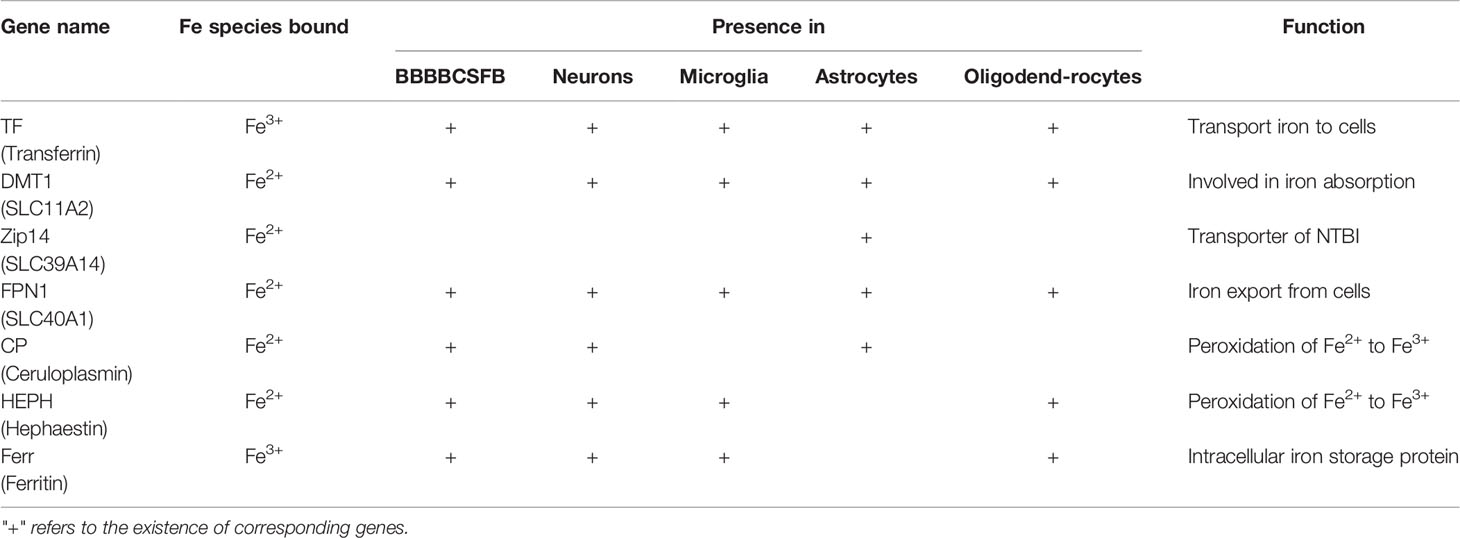
Table 2 Proteins Involved in Brain Iron Transport.
Herein we provide a summary of recent literature unveiling the mechanism of iron transport and regulation across the BBB and BCSFB, as well as the characteristics of iron transport and metabolism in different cell types of the central nervous system (CNS) such as neurons, microglia, astrocytes, and oligodendrocytes.
2.2.2 Iron and Iron Transporters in BBB and BCSFB
CNS is tightly sealed from the changeable milieu of blood by the BBB and the BCSFB (31). BBB is an heterogenous multicellular complex system. This system includes tightly connected endothelial cells and a unique basement membrane. In addition to the parenchymal basement membrane, the basement membrane also contains an ensheathment of astrocytic end-feet, pericytes and perivascular antigen-presenting cells (32). BCSFB lies at the choroid plexuses in the lateral, third and fourth ventricles of the brain where the choroid plexus epithelial cells of the nonporous capillary wall contain a special carrier system for transporting various substances. This system is responsible for the exchange of substances between cerebrospinal fluid (CSF) and blood, and transport across BBB and BCSFB is important for the entry of iron into brain (33, 34).
TF/TFR1 pathway may be the main route of iron transporter across the luminal (apical) membrane of the BBB. Additionally, non-transferrin-bound iron (NTBI) uptake from the blood through luminal DMT1 and H-ferritin uptake may be partly responsible for iron transport across the BBB. Iron transport across the abluminal (basal) membrane is a Fpn1/hephaestin (Fpn1/Heph) and/or Fpn1/ceruloplasmin (CP)-mediated process (35, 36).
TF/TFR1/DMT1 pathway is an important pathway for iron transport across the BCSFB. Furthermore, iron export from the choroid epithelium to the CSF is mediated by the Fpn1/CP or Fpn1/Heph pathways. Beyond restriction of the access of substances from the blood to the CSF, it is possible that the BCSFB has a bigger impact on iron removal from the brain than iron uptake into the brain (35–37).
2.2.3 Iron and Iron Transporters in Neurons
Iron is essential for neuron development and function (38). First iron is an essential cofactor for enzymes involved in energy metabolism and amino acid biosynthesis. Iron also plays a significant role for division of embryonic neurons as it is a cofactor for the enzyme ribonucleotide reductase. In addition, during early embryonic development, the dysfunction of yolk sac cells caused by excessive iron uptake leads to the necrotic degeneration of neuroectodermal cells (39, 40).
The neuronal expression levels of the TFR1 reflects their need for iron (41). DMT1 is also expressed in neurons, suggesting that after transferrin binding, iron is transported to the cytoplasm through DMT1 (42). DMT1 is involved in hippocampal neuronal iron uptake during development and memory formation (43). The presence of NTBI in brain extracellular fluids suggests that neurons can also take up iron as transferrin-free iron (44). Fpn1 and Heph are involved in the output of iron from the neuron (45, 46).
2.2.4 Iron and Iron Transporters in Microglia
Microglia have vital roles in brain development and CNS homeostasis, including programmed cell death, clearance of apoptotic newborn neurons, as well as pruning developing axons and synapses (47, 48). Microglia are immune cells of the CNS, which are implicated in brain inflammation and can modulate the transport and metabolism of essential metal iron according to the anti-inflammatory and pro-inflammatory environment (49).
The mechanism of iron transport in microglia has been addressed in cell culture. The different sources of cells include primary adult mouse microglia (49), primary 2-day-old Sprague-Dawley microglia, primary newborn Wistar rat microglia (50), primary C57BL/6 mice microglial (51) and BV-2 microglial cells (52). Microglial cells interact with both TF bound-iron (TBI) and NTBI. TBI is taken up via the TFR1/DMT1 pathway, and after the release of iron in the acidic milieu of the endosome, this is translocated into the cytosol by DMT1 or other transporters (53). For NTBI uptake, an endogenous cell surface ferrireductase reduces Fe3+ to Fe2+ for uptake by DMT1 in a pH-dependent manner at the cell surface (54).
2.2.5 Iron and Iron Transporters in Astrocytes
Astrocytes are the most abundant glial cells in the brain (55). In healthy CNS tissue, astrocytes maintain homeostasis of extracellular fluids, provide energy substrates to neurons, modulate local blood flow, and play essential roles in synapse development and plasticity (56). In addition, astrocytic end-feet form intimate contacts with the abluminal side of brain capillary endothelial cells (BCECs) in all brain regions. This close relationship makes it denotes an important role in nutrient capture from the circulating blood such as iron (57). Astrocytes theoretically can transport iron directly from BCECs to neurons and oligodendrocytes through intracellular transport (58).
The TF cycle is probably not the main process by which astrocytes obtain iron from endothelial cells (59). It is more likely that DMT1 mediates some of this uptake, since this transporter is strongly expressed in the astrocyte end-feet contacting with BCECs directly. This suggests that astrocytes can potentially uptake NTBI directly from BCECs (57).
In addition, the zinc transporter Zip14 and resident transient receptor potential channels have been suggested to be involved in the uptake of NTBI by astrocytes (60). Fpn1 and CP are highly expressed on astrocytic cell membranes, and both proteins may be essential in iron mobilization from these cells into the extracellular brain space (61, 62).
2.2.6 Iron and Iron Transporters in Oligodendrocytes
Oligodendrocytes create myelin sheaths for CNS axons, assist in the jumping and efficient transmission of bioelectric signals, maintain and protect the normal function of neurons (63, 64). Oligodendrocytes are the cells with the highest iron levels in the brain. Oligodendroglia cells require iron as a cofactor for several enzymes involved in the proliferation and differentiation of oligodendrocyte precursor cells (OPCs), as well as enzymes required for the production of cholesterol and phospholipids, which are essential myelin components (65, 66).
In oligodendrocytes, TF/TFR1/DMT1 pathway plays a significant role in iron transport in immature oligodendrocytes, however the proportion of iron transported by this pathway may decrease with the beginning of myelination (36). DMT1 is essential for OPC maturation and normal myelination in mouse brain, which is considered to be a crucial pathway for many cells to uptake NTBI (67). Extensive literature suggests that H-ferritin is the main source of iron in oligodendrocytes, conferring high buffering capacity for iron (68). Heph is expressed by mature oligodendrocytes and plays a role in iron efflux from these cells, but white and gray matter oligodendrocytes can regulate iron efflux differently; while white matter oligodendrocytes upregulate the expression of Cp in the absence of Heph, likely as a fail-safe mechanism, gray matter oligodendrocytes lacks such compensatory pathway (69).
3 Ferroptosis and Transporters in Malignant Brain Tumors
3.1 The Transport Mechanisms in Ferroptosis
Ferroptosis is a form of iron-dependent regulatory cell death distinguished from necrosis, apoptosis and autophagy (70), which can be triggered by the small-molecule compound erastin and RSL3 (71, 72). Iron and polyunsaturated fatty acids (PUFAs) act as raw materials for lipid peroxidation to promote the occurrence of ferroptosis (73, 74). While glutathione peroxidase 4 (GPX4) using glutathione (GSH) as the substrate effectively removes excess ROS through antioxidant mechanism and inhibits ferroptosis (75). The increase of intracellular iron content, the accumulation of ROS and excessive lipid peroxidation are crucial to induce ferroptosis (76). Ferroptosis is closely related to iron metabolism, amino acid metabolism and lipid metabolism in cells. Therefore, iron transporters and amino acid transporters involved in metabolism have a marked effect on the cell sensitivity to ferroptosis (70, 77).
3.1.1 Iron Transporters in Ferroptosis
DMT1 and TfR1 are involved in the absorption of intracellular iron (78, 79), while Fpn1 transports iron from the cell to the blood (27). They are both ubiquitous and crucial proteins that regulate the iron content in cells and are essential for the maintenance of iron homeostasis (Table 3). Iron is essential for cell growth, but it can promote the formation of toxic ROS during ferroptosis. In the case of excessive iron in cells, Fe2+ and H2O2 can generate hydroxyl radicals (OH-) through Fenton reaction, promoting the oxidation of PUFAs on the cell membrane, greatly accelerating lipid peroxidation and ultimately causing cell damage or death (80). Therefore, increasing the expression of TFR1 or decreasing the expression of Fpn1 will increase the accumulation of iron in the cell and result in ferroptosis. DMT1 located on the lysosomal membrane mediates iron transfer and the inhibitors of DMT1 can kill cells by accelerating lysosomal iron overload and an increase of ROS production (81).
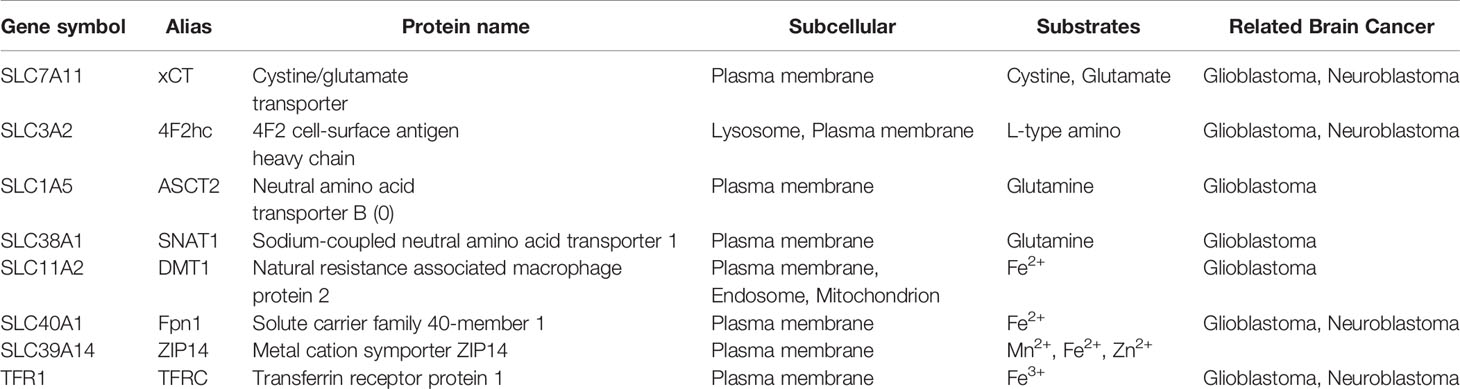
Table 3 The characteristics of ferroptosis-related transport protein associated with malignant brain tumors.
Recently identified ferroptosis-related iron transporters ZIP14 (SLC39A14) can transport manganese, iron and zinc (Table 3). However, its main function is to transport manganese ions, while iron ions are not the main transport substrate of ZIP14 under normal physiological conditions (82, 83). Only in the state of iron overload, ZIP14 exhibits the function of transporting iron ions and mediating ferroptosis (84).
3.1.2 Amino Acid Transporters in Ferroptosis
The amino acid transporter system Xc− on the cell membrane is composed of two core components, SLC7A11 (Solute Carrier Family 7 Member 11, xCT) and SLC3A2 (Solute Carrier Family 3 Member 2, 4F2hc), involved in the exchange of extracellular cystine (Cys2) by transporting intracellular glutamate (Glu) (Table 3) (70). Intracellularly, Cys2 will be reduced to cysteine (Cys), thereby promoting the synthesis of GSH, the cofactor of GPX4. As a central regulatory protein for ferroptosis, GPX4 can convert GSH to oxidized glutathione (GSSG) whilst also reducing lipid hydroperoxides (L-OOH) to lipid alcohols (L-OH), which is the main mechanism to prevent lipid peroxidation and inhibit ferroptosis (85). In fact, knockout and inactivation of GPX4 both contribute to ferroptosis (86). Ferroptosis inducer erastin can result in GSH depletion and GPX4 inactivation by inhibiting system Xc− transport of cystine (71), while RSL3 directly induces ferroptosis by inhibiting the activity of GPX4 (72). Cys is the crucial limiting amino acid for intracellular GSH synthesis and GSH depletion directly affects the function of GPX4. Therefore, system Xc− that participates in the uptake of Cys2 is considered to be one of the most critical regulators of ferroptosis. Recent studies suggest that regulation of TP53 (87), Nrf2 (15), ATF4 (88), BECN1 (89) or interferon γ(IFNγ) released by CD8+ T cells (90) significantly inhibits the system Xc−, leading to a decrease in GSH synthesis and ferroptosis.
The transmembrane transport of glutamine (Gln) is dependent on SLC1A5 (Solute Carrier Family 1 Member 5) and SLC38A1 (Solute Carrier Family 38 Member 1) (Table 3). After entering the cell, Gln is catalyzed by glutaminase (GLS) and broken down into Glu and ammonia in the mitochondria (91). Subsequently, Glu can be converted to α-ketoglutarate (α-KG) that is involved in the oxidative energy supply as an important intermediate for the tricarboxylic acid (TCA) cycle (92). Glu is an indispensable molecule for generating GSH, which can effectively scavenge intracellular ROS. In cancer cells, inhibition of ferroptosis has been shown to be associated with high levels of Gln (93). Although glutaminolysis promotes cancer cell growth, this metabolic process can also induce ferroptosis toward cell death (94). The pivotal role of dihydrolipoamide dehydrogenase (DLD) in prompting ferroptosis induced by cystine deprivation or cystine import inhibition has been recently confirmed. Apart from stimulating DLD to produce hydrogen peroxides, α-KG can be further converted into acetyl-CoA, facilitating fatty acid synthesis and lipid peroxidation-dependent ferroptosis (95). MIR137 (microRNA137) has also been recently identified as a negative regulator of erastin or RSL3-induced ferroptosis through down-regulation of SLC1A5 in melanoma cells (96).
3.2 Ferroptosis and Malignant Brain Tumors
In 2021, the World Health Organization (WHO) released the fifth edition of the Classification of Tumors of the Central Nervous System (CNS) (WHO CNS5). Among various brain tumors, childhood brain tumors, adult gliomas and meningiomas are currently the most common brain neoplasia. Neuroglioma is one of the common primary central nervous system tumors that originate from glial cells. GBM is the most malignant and deadliest type of neuroglioma (97). Neuroblastoma is the most common extracranial tumor in children and nearly half of neuroblastoma occurs in infants and young children under 2 years of age (98). Meningiomas are tumors originating from arachnoid cap cells, most of which are benign. However, about 3% meningiomas are malignant, including invasive meningiomas (99). The current treatment methods for malignant brain tumors mainly include surgical resection, radiotherapy and chemotherapy.
Recently increasing numbers of studies have shown that ferroptosis is associated with the pathological process of a variety of neurological diseases, including neurodegenerative diseases, neurotrauma and brain tumors (100). Nevertheless, there has been less research on brain tumors compared to the other types of tumors so far. It is undeniable that ferroptosis, a new form of non-apoptotic cell death, will open up new therapeutic avenues for eliminating brain tumor cells (101).
Soon after ferroptosis was defined, researchers injected iron-containing water into the rats transplanted with glioma-35 cells and then focused on treating the tumor site with radiotherapy (102). They found that the tumor volume in the experimental group was significantly smaller than that in the control group. Mechanistically, in a separate report, it is suggested that iron-containing water treatment before radiation induces glioma cell death through the combination of apoptosis and ferroptosis (103). Furthermore, ferroptosis is proved to be involved in the GBM cell death which can be induced by neutrophils. It appears that this process requires activation signals given by the tumor microenvironment. When mature neutrophils infiltrating into the tumors are activated, they will trigger lipid peroxidation by transferring myeloperoxidase into GBM cells and increase cellular ROS, finally causing tumor cell ferroptosis (104).
Although most ferroptosis-related studies have concentrated on gliomas, neuroblastoma, another malignant brain tumor, is gradually coming into focus. Research suggests that overexpression of Mitochondrial ferritin (FtMt) in dopaminergic neuroblastoma cell line SH-SY5Y cells can significantly inhibit erastin-induced ferroptosis (105). This is mainly due to FtMt-mediated inhibition of cellular labile iron pool (LIP) and the accumulation of cytoplasmic ROS which protects against effects of ferroptosis. In another study with SH-SY5Y, the ferroptosis inhibitor Ferrostatin-1 (Fer-1) was found to have a neuroprotective effect under Rotenone-induced oxidative stress conditions (106).
In a recently published study, researchers evaluated the expression of Merlin/Neurofibromin2 (NF2) and the ferroptosis regulator GPX4 in patients with primary meningioma and found a positive correlation between them. They speculated that the inactivation of NF2 in meningiomas may be more likely to cause ferroptosis. Furthermore, it has been determined that inhibition of NF2 and E-Cadherin can promote ferroptosis-related cytotoxicity and lipid peroxidation in meningioma cell lines. The transcription factor MEF2C has been shown to regulate the transcription of NF2 and E-cadherin genes. Silencing MEF2C, the expression levels of NF2 and E-cadherin in meningiomas decreased, which inhibited the growth of meningiomas mediated by ferroptosis (Figure 1). Therefore, MEF2C can be used as a potential molecular target for the treatment of aggressive meningiomas through modulating ferroptosis (107).
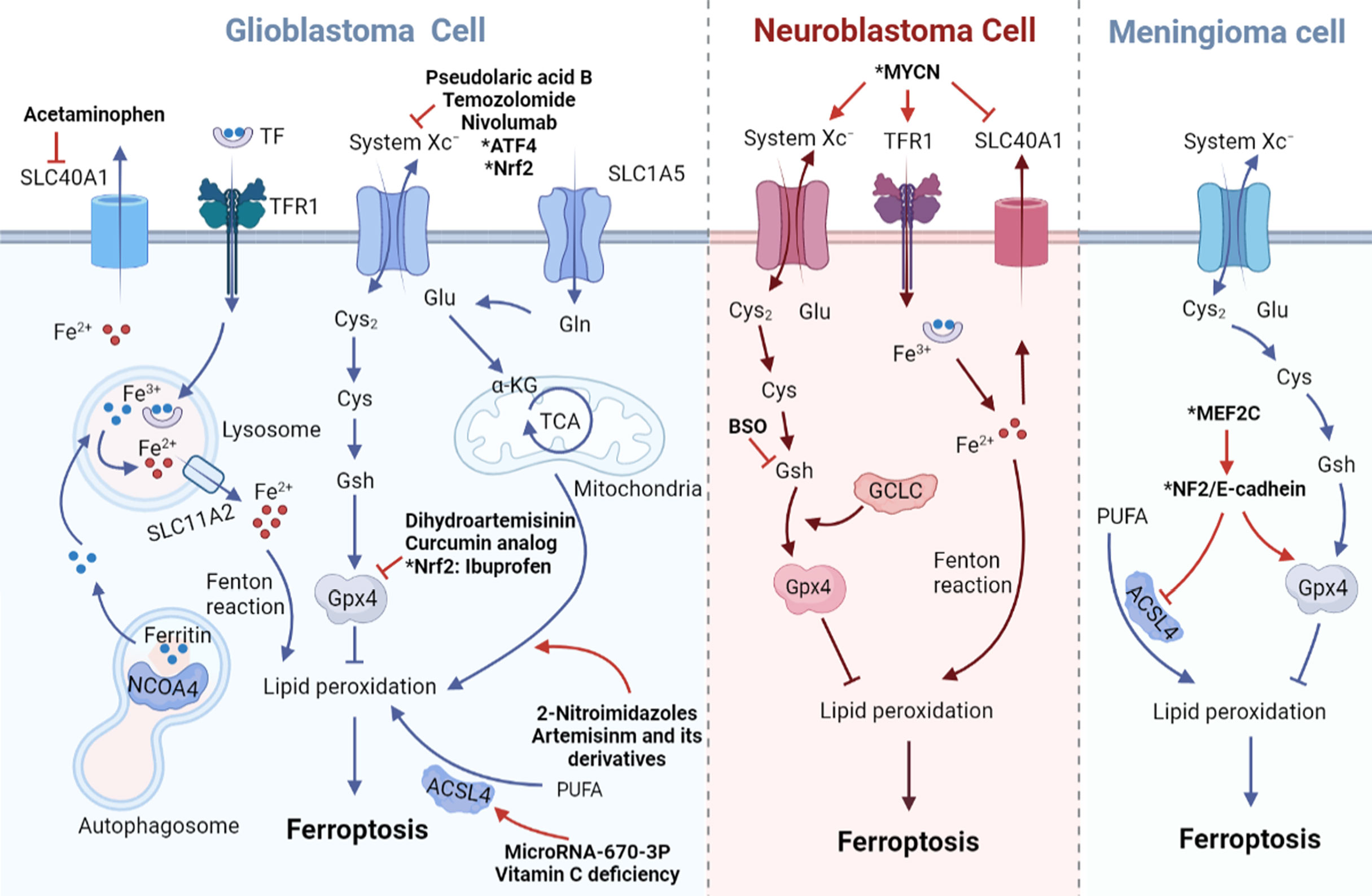
Figure 1 Impacts of ferroptosis-related transport proteins in three malignant brain tumor cells. In GBM cell, iron transport-related proteins DMT1 (SLC11A2), Fpn1 (SLC40A1), TFR1 and amino acid transporters system Xc– (SLC7A11/SLC3A2), ASCT2 (SLC1A5) regulate the occurrence of ferroptosis all together. In MYCN-amplified neuroblastoma cell, lipid peroxidation and cell death are promoted due to increased expression of TFR1 and System Xc– and lower expression of Fpn1. In meningioma cell, MEF2C mediated upregulation of NF2 and E-cadherin inhibits Erastin-induced ferroptosis. Arrows indicate promotion and blunt-ended lines indicate inhibition. Cys, cysteine; Cys2, cystine; GSH, glutathione; GPX4, glutathione peroxidase 4; Glu, glutamate; Gln, glutamine; TF, transferrin; TFR1, transferrin receptor 1; PUFA, polyunsaturated fatty acid; ACSL4, acyl-CoA synthetase long-chain family member 4; TCA, tricarboxylic acid cycle; α-KG, α-ketoglutarate; NCOA4, nuclear receptor coactivator 4; ATF4,activating transcription factor 4; Nrf2, nuclear factor erythroid-2-related factor; MYCN, BHLH Transcription Factor; MEF2C, Myocyte Enhancer Factor 2C; NF2, neurofibromatosis type 2; BSO, buthionine sulphoximine.
3.3 The Role of Transporters Associated With Ferroptosis in Malignant Brain Tumors
Ferroptosis plays a key role in the development of malignant brain tumors. As an important part of ferroptosis, relevant transporters can regulate amino acid metabolism and iron metabolism and are essential for the maintenance of iron homeostasis. Disorders of iron homeostasis in the brain will increase the risk of tumors, which may be one of the factors leading to the increased incidence of brain tumors (108). In addition, a group of ferroptosis-related genes have been discovered that may predict the prognosis of glioma patients based on clinical databases (109). In terms of iron metabolism, CDGSH iron-sulfur domain-containing protein 1 (CISD1) (110), poly r(C) binding protein 1 (PCBP1) (111) and transferrin (TF) (94) have a marked impact on ferroptosis by regulating the cellular content of iron. Here we compared the survival curve of brain tumor patients with the expression of ferroptosis-related genes and the results showed that the decrease in survival rate was related to the high-level expression of the protein required for iron intake (Figure 2). These data indicate that a better understanding of the role of ferroptosis-related transporters in malignant brain tumors may help provide more options for the treatment and prevention of brain tumors.
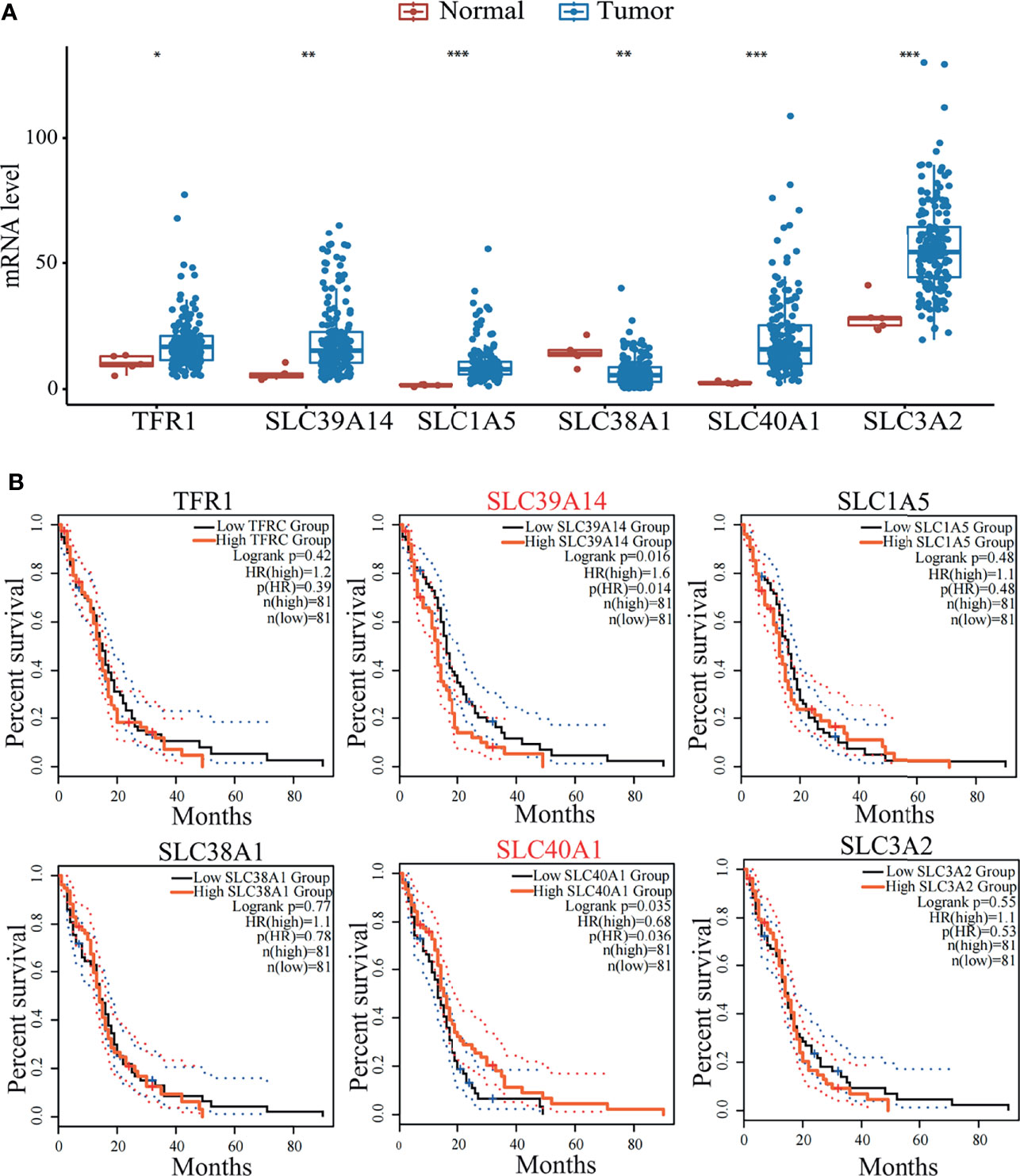
Figure 2 (A) Expression level of transporters (TFR1, SLC39A14, SLC1A5, SLC38A1, SLC40A1, SLC3A2) in tumor patients and normal people. Data mined from TCGA (https://cancergenome.nih.gov/). *p < 0.05, **p < 0.01, ***p < 0.001, compared with tumor patient group. (B) Survival curves of GBM patients mined from GEPIA2 (http://gepia2.cancer-pku.cn/). GBM patients were stratified into high or low expression groups based on the expression level of transporters (TFR1, SLC39A14, SLC1A5, SLC38A1, SLC40A1, SLC3A2) of patients. p<0.05 in Log‐rank test. OS, overall survival in months.
The obvious increase of lipid and cytoplasmic ROS is an important feature of ferroptosis and part of its regulatory factors have been used as small molecule drug targets to induce the death of cancer cells. Fpn1 can inhibit ferroptosis by reducing the accumulation of iron-dependent lipid ROS. Studies have found that in neuroblastoma cells, erastin induces the accumulation of iron and the low expression of Fpn1 involved in iron outflow (112). Furthermore, hepcidin, an amino acid peptide hormone (113) that binds with Fpn1 and stimulates Fpn1 degradation, increases antitumor activity of Erastin. This suggests that Fpn1 can be used as a potential therapeutic target for neuroblastoma in the future and Fpn1 inhibitors may provide a new approach for the treatment of neuroblastoma.
In neuroblastoma, gene amplification of the oncogenic transcription factor MYCN makes tumor cells more malignant and difficult to eliminate. Increased TFR1 expression and decreased Fpn1 expression in MYCN-amplified neuroblastoma cells results in high intracellular iron content. Overexpression of MYCN activates Xc−/GPX4 pathway, resulting in increased intracellular cystine and enhanced antioxidant protection (114). Therefore, the use of system Xc− selective inhibitors or TFR1 agonists to treat MYCN-amplified neuroblastoma will increase the level of lipid peroxidation and eventually lead to ferroptosis of tumor cells (Figure 1).
In addition to neuroblastoma, GSH depletion caused by system Xc− inhibition is associated with other malignant brain tumors (115). Nuclear factor (erythoid-derived)-like 2(Nrf2) overexpression or Kelch-like ECH associated protein 1(Keap1) knockdown can accelerate the growth of glioblastoma and promote the development of glioma cells (15). Similarly, xCT is positively regulated by Nrf2 and plays a crucial role in the inhibition of ROS accumulation during the ferroptosis process of glioma cells. Drug inhibitors targeting system Xc− can rescue ROS generation, thereby increasing the sensitivity of glioma cells to ferroptosis and achieving the goal of treating malignant gliomas (Figure 1) (15).
The first-line treatment anti-tumor drug Temozolomide can inhibit the growth of glioblastoma. In order to explore the role of ferroptosis in this process, researchers treated human glioblastoma cell line TG905 cells with siRNA and found that knockdown of DMT1 reduced the level of ROS and iron production induced by Temozolomide (116). In addition, down-regulation of DMT1 also increased the expression of GPX4, Nrf2 and HO-1, thereby preventing the occurrence of ferroptosis. Temozolomide induces ferroptosis of some glioblastoma cells by increasing the expression of DMT1, so the divalent metal transporter DMT1 can be used as a drug target in glioblastoma.
4 Therapeutic Strategy
Mounting evidence suggests ferroptosis plays a beneficial role in tumors treatment. With the need for new treatments for malignant brain tumors, increased attention has been paid to drugs inducing ferroptosis that designed based on the regulatory pathways of ferroptosis. The main types of malignant brain tumors targeted by the novel Ferroptosis-based include GBM (117), fibrosarcoma (118), head and neck carcinoma (119).
Ferroptosis can be induced by increasing intracellular iron or ROS level (11). Inhibition of the glutathione peroxidase GPx4 or glutamate/cystine antiporter system Xc− through the drugs is beneficial, promoting ferroptosis though increased ROS accumulation. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis (15). Although some studies have reported that inhibiting ferroptosis by activating Nrf2 pathway can play a neuroprotective role, for example, astrocytes protect neurons from ferroptosis by activating the Nrf2 pathway to supply neurons with GSTM2 and other antioxidants, inhibiting Nrf2 pathway in tumor cells to promote ferroptosis plays a therapeutic effect (120). ATF4 and Pseudolaric acid B promotes ferroptosis in a xCT-dependent manner (89, 121). Dihydroartemisinin initiates ferroptosis through GPx4 inhibition (122). Ibuprofen induces ferroptosis via downregulation of Nrf2-Keap1 signaling pathway (123).
Other mechanisms of promoting ferroptosis have also been reported, including activating the transcription factor BACH1 (BTB domain and CNC homology 1) (124) or Nox4 (121) to promote oxidative stress, inhibition of autophagy (125), vitamin C deficiency to reduce proliferation (126) and targeting ACSL4 which suppresses proliferation (127). Based on these mechanisms, related drugs have been found, such as 2-Nitroimidazoles, temozolomide, artemisinin and its derivatives.
Ferroptosis inducers may expand our arsenal of frontline therapeutic agents for combinatory approaches. Temozolomide toxicity operates is boost by ferroptosis (128). Androgen receptor (AR) ubiquitination is induced by the curcumin analog which suppresses growth of temozolomide-resistant GBM through disruption of GPX4-mediated redox homeostasis (129). Furthermore, T cell-promoted tumor ferroptosis is an anti-tumor mechanism, and targeting this pathway in combination with immunotherapy is another potential therapeutic approach (91, 130, 131). Nivolumab therapy revealed that clinical benefits correlate with reduced expression of SLC3A2 and increased IFNγ and CD8 (91).
Although many anticancer compounds that promote ferroptosis have been found, there are still many treasures to be discovered. Drugs targeting other mechanisms of ferroptosis need to be explored, such as targeted iron accumulation. A systematic assessment of the relationship between ferroptosis related genes (FRGs) expression profiles and the occurrence and development of tumors based on the Cancer Genome Atlas (TCGA), Chinese Glioma Genome Atlas (CGGA) datasets and FerrDb datasets may unveil new targets (77, 132). In fact, the potential impact of Acetaminophen in ferroptosis through interaction with CD44, HSPB1, and SLC40A1 was found this way (132).
To find out the potential correlation of GBM with transporters involved in ferroptosis. Here we compared the expression of several ferroptosis related transporters (SLC7A11, SLC3A2, SLC1A5, SLC38A1, SLC11A2, SLC40A1, SLC39A14, TFR1, TF) in normal people and GBM patients based on TCGA data. It is worth mentioning that the differentially expressed genes (DEGs) covered the majority of the transporters that we screened related to ferroptosis. Box-plot shows the expressions of SLC3A2, SLC1A5, SLC40A1, SLC39A14 and TFR1 increased significantly, the expression of SLC38A1 decreased significantly (Figure 2A). The effect of DEGs on the survival curve of GBM patients was further explored based on TCGA data (Figure 2B). As shown in the Kaplan‐Meier survival curve, median survival of GBM patients changed significantly according to the expression of SLC39A14 (p = 0.016) and SLC40A1 (p = 0.035), but the mechanism behind it remains to be explored. The above analysis further proves the potential of targeting these transporters and ferroptosis in the treatment of GBM.
Ferroptosis induction may prove as an effective therapeutic strategy against malignant brain tumors, yet a wide range of ferroptosis inducers are prone to off-target effects and may cause significant damage to normal cells. Therefore, it is urgent to develop tumor targeting delivery strategies of ferroptosis inducers. At present, many research are focusing on this aspect. Class I histone deacetylase (HDAC) inhibitors can selectively inhibit ferroptosis in neurons, but promote ferroptosis in tumor cells, which may be due to its different epigenetic regulation on the two cells. The combination of HDAC inhibitors and ferroptosis inducers can not only reduce the dosage of ferroptosis inducers to reduce toxicity, but also protect neurons (133, 134). Nano-targeting of WA allows systemic application and suppressed tumor growth due to an enhanced accumulation at the tumor site (135, 136).
At present, the treatment strategy targeting ferroptosis has been widely studied in various tumors, among which the advanced treatment strategy can potentially use for malignant brain tumors as well. Some new therapeutic mechanisms are worth learning. For example, gene interference by transferring genes with adeno-associated virus and iron nanoparticles enhance ferroptosis and inhibit tumor growth (137); ferroptosis inducer erastin or rsl3 is used independently or in combination with standard-of-care second-generation for the treatment of advanced prostate cancer (138); and activating ferroptosis by sequestering iron in lysosomes kills cancer stem cells (139). Studies have showed that targeted ferroptosis can used to overcome drug resistance of tumors. For example, Vorinostat promotes ferroptosis to overcome the resistance to epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) (140); and artesunate inhibits growth of therapy-resistant renal cell carcinoma through induction of ferroptosis (141). Some advanced strategies are for targeted therapy. For example, Photodynamic therapy site-specifically produces reactive oxygen species for the Fenton reaction, which promotes ferroptosis and suppresses tumors (142); and catalytic nanomedicine that contains natural glucose oxidase and ultrasmall Fe3O4 nanoparticles selectively and effectively strengthens ferroptosis of tumor cells (143). In short, the essence can be drawn from the treatment of other tumors and used in the treatment of malignant brain tumors.
Inducing ferroptosis of tumor cells is a newly discovered strategy for the treatment of malignant brain tumors, but many problems remain to be solved, including elucidating the mechanism of ferroptosis in different malignant brain tumors, discovering new therapeutic targets for inducing ferroptosis of tumor cells, and increasing the tumor cell targeting of ferroptosis inducers. It is worth noting that the regulation of iron transport in tumor cells and the expression of transporters related to ferroptosis may have good therapeutic potential. Many transporters have become drug targets in recent years (144, 145). At the same time, clarifying iron transport under physiological conditions also provides an important research basis for targeted therapy of tumor cells, crucial to avoid the damage of normal tissues through off target effects.
Author Contributions
LC proposed the research. JZ and YW both reviewed the literature and collected references. JZ, YW, and LT wrote the manuscript and finalized the paper. All authors contributed to the article and approved the submitted manuscript.
Funding
This work was supported by National Natural Science Foundation of China (32130048, 92157301, 31971085 and 91857108 to LC), the Ministry of Science and Technology of China National Key R&D Programs (2018YFA0506903 to LC), Nation Science and Technology Major Projects for Major New Drugs Innovation and Development (2018ZX09711003-004-002 to LC), Tsinghua University Spring Breeze Fund (2021Z99CFY012 to LC), Tsinghua-Foshan Innovation Special Fund (2020THFS0133 to LC).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013-2017. Neuro Oncol (2020) 22:iv1–96. doi: 10.1093/neuonc/noaa200
2. Meyer MA. Malignant Gliomas in Adults. N Engl J Med (2008) 359:1850–0. doi: 10.1016/S0140-6736(18)30990-5
3. Ostrom QT, Adel Fahmideh M, Cote DJ, Muskens IS, Schraw JM, Scheurer ME, et al. Risk Factors for Childhood and Adult Primary Brain Tumors. Neuro Oncol (2019) 21:1357–75. doi: 10.1093/neuonc/noz123
5. Constantinos A, Dimitrios TT. Glioblastoma Multiforme: Pathogenesis and Treatment. Pharmacol Ther (2015) 152:63–82. doi: 10.1016/j.pharmthera.2015.05.005
6. Klekner L, Szivos L, Virga J, Rkosy P, Nagy B. Significance of Liquid Biopsy in Glioblastoma-A Review. J Biotechnol (2019) 298:82–7. doi: 10.1016/j.jbiotec.2019.04.011
7. Sathornsumetee S, Rich JN. New Approaches to Primary Brain Tumor Treatment. Anticancer Drugs (2006) 17:1003–16. doi: 10.1097/01.cad.0000231473.00030.1f
8. Qiu Y. The Relation Between Necessary Trace Element Iron and Various Diseases. Biol Trace Elem Res (1997) 4:19–22. doi: 10.16755/j.cnki.issn.1006-446x.1997.10.006
10. Mccann S, Amadó M, Moore SE. The Role of Iron in Brain Development: A Systematic Review. Nutrients (2020) 12:2001–23. doi: 10.3390/nu12072001
11. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: Process and Function. Cell Death Differ (2016) 23:369–79. doi: 10.1038/cdd.2015.158
12. Rouault TA. The Role of Iron Regulatory Proteins in Mammalian Iron Homeostasis and Disease. Nat Chem Biol (2006) 2:406–14. doi: 10.1038/nchembio807
13. Montalbetti N, Simonin A, Kovacs G, Hediger MA. Mammalian Iron Transporters: Families SLC11 and SLC40. Mol Aspects Med (2013) 34:270–87. doi: 10.1016/j.mam.2013.01.002
14. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
15. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 Pathway Promotes Cell Proliferation and Diminishes Ferroptosis. Oncogenesis (2017) 6:e371. doi: 10.1038/oncsis.2017.65
16. Zhu J, Xiong Y, Zhang Y, Wen J, Zhang W. The Molecular Mechanisms of Regulating Oxidative Stress-Induced Ferroptosis and Therapeutic Strategy in Tumors. Oxid Med Cell Longev (2020) 2020:1–14. doi: 10.1155/2020/8810785
18. Srole DN, Ganz T. Erythroferrone Structure, Function, and Physiology: Iron Homeostasis and Beyond. J Cell Physiol (2020) 236:4888–901. doi: 10.1155/2020/8810785
19. Anderson GJ, Mclaren GD. Iron Physiology and Pathophysiology in Humans. New York: Humana Press (2012). doi: 10.1007/978-1-60327-485-2
20. Monsen ER, Hallberg L, Layrisse M, Hegsted DM, Cook JD, Mertz W, et al. Estimation of Available Dietary Iron. Am J Clin Nutr (1978) 31:134–41. doi: 10.1093/ajcn/31.1.134
21. Eisenstein RS, Blemings KP. Iron Regulatory Proteins, Iron Responsive Elements and Iron Homeostasis. J Nutr (1998) 128:2295. doi: 10.1002/jcp.30247
22. Crichton RR, Wilmet S, Legssyer R, Ward RJ. Molecular and Cellular Mechanisms of Iron Homeostasis and Toxicity in Mammalian Cells. J Inorg Biochem (2002) 91:9–18. doi: 10.1016/s0162-0134(02)00461-0
23. Gomme PT, Mccann KB, Bertolini J. Transferrin: Structure, Function and Potential Therapeutic Actions. Drug Discov Today (2005) 10:267–73. doi: 10.1016/S1359-6446(04)03333-1
24. Baldauf L, Endres T, Scholz J, Kirches E, Ward DM, Lessmann V, et al. Mitoferrin-1 Is Required for Brain Energy Metabolism and Hippocampus-Dependent Memory. Neurosci Lett (2019) 713:134521–41. doi: 10.1016/j.neulet.2019.134521
25. Gunshin H, Fujiwara Y, Custodio AO, Direnzo C, Andrews NC. Slc11a2 Is Required for Intestinal Iron Absorption and Erythropoiesis But Dispensable in Placenta and Liver. J Clin Investig (2005) 115:1258–66. doi: 10.1172/JCI24356
26. Touret N, Furuya W, Forbes J, Gros P, Grinstein S. Dynamic Traffic Through the Recycling Compartment Couples the Metal Transporter Nramp2 (DMT1) With the Transferrin Receptor. J Biol Chem (2003) 278:25548–57. doi: 10.1074/jbc.M212374200
27. Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The Iron Exporter Ferroportin/Slc40a1 Is Essential for Iron Homeostasis. Cell Metab (2005) 1:191–200. doi: 10.1016/j.cmet.2005.01.003
28. Thirupathi A, Chang YZ. Brain Iron Metabolism and CNS Diseases. Adv Exp Med Biol (2019) 1173:1–19. doi: 10.1007/978-981-13-9589-5_1
29. Hu CL, Tao L, Cao XZ, Chen LG. The Solute Carrier Transporters and the Brain:Physiological and Pharmacological Implications. Asian J Pharm Sci (2020) 15:131–44. doi: 10.1016/j.ajps.2019.09.002
30. Rouault TA, Cooperman S. Brain Iron Metabolism. Semin Pediatr Neurol (2006) 13:142–8. doi: 10.1016/j.spen.2006.08.002
31. Engelhardt B, Sorokin L. The Blood-Brain and the Blood-Cerebrospinal Fluid Barriers: Function and Dysfunction. Semin Immunopathol (2009) 31:497–511. doi: 10.1007/s00281-009-0177-0
32. Abbott NJ, Patabendige A, Dolman D, Yusof SR, Begley DJ. Structure and Function of the Blood-Brain Barrier. Neurobiol Dis (2010) 37:13–25. doi: 10.1016/j.nbd.2009.07.030
33. Tina S, Birk ML, Torben M. Impairment of Interrelated Iron- and Copper Homeostatic Mechanisms in Brain Contributes to the Pathogenesis of Neurodegenerative Disorders. Front Pharmacol (2012) 3:169. doi: 10.3389/fphar.2012.00169
34. Li GJ, Choi BS, Wang X, Jie L, Wei Z. Molecular Mechanism of Distorted Iron Regulation in the Blood–CSF Barrier and Regional Blood–Brain Barrier Following In Vivo Subchronic Manganese Exposure. Neurotoxicology (2006) 27:737–44. doi: 10.1016/j.neuro.2006.02.003
35. Wei Z, Monnot AD. Regulation of Brain Iron and Copper Homeostasis by Brain Barrier Systems: Implication in Neurodegenerative Diseases. Pharmacol Ther (2012) 133:177–88. doi: 10.1016/j.pharmthera.2011.10.006
37. Wang X, Li GJ, Zheng W. Efflux of Iron From the Cerebrospinal Fluid to the Blood at the Blood-CSF Barrier: Effect of Manganese Exposure. Exp Biol Med (Maywood) (2008) 233:1561–71. doi: 10.3181/0803-RM-104
38. Beard JL. Iron Biology in Immune Function, Muscle Metabolism and Neuronal Functioning. J Nutr (2001) 131:568S–79S. doi: 10.1093/jn/131.2.568S
39. Moos T, Morgan EH. The Metabolism of Neuronal Iron and Its Pathogenic Role in Neurological Disease: Review. Ann NY Acad Sci (2004) 1012:14–26. doi: 10.1196/annals.1306.002
40. Kuchta B. Experiments and Ultrastructural Investigations on the Mouse Embryo During Early Teratogen-Sensitive Stages. Acta Anat (1982) 113:218–25. doi: 10.1159/000145558
41. Giometto B, Bozza F, Argentiero V, Gallo P, Pagni S, Piccinno MG, et al. Transferrin Receptors in Rat Central Nervous System. An Immunocytochemical Study. J Neurol Sci (1990) 98:81–90. doi: 10.1016/0022-510x(90)90183-n
42. Burdo JR, Menzies SL, Simpson IA, Garrick LM, Connor JR. Distribution of Divalent Metal Transporter 1 and Metal Transport Protein 1 in the Normal and Belgrade Rat. J Neurosci Res (2010) 66:1198–207. doi: 10.1002/jnr.1256
43. Carlson ES, Tkac I, Magid R, O'Connor MB, Andrews NC, Schallert T, et al. Iron Is Essential for Neuron Development and Memory Function in Mouse Hippocampus. J Nutr (2009) 139:672–9. doi: 10.3945/jn.108.096354
44. Williamson DL. Normalizing a Hyperactive mTOR Initiates Muscle Growth During Obesity. Aging (2011) 3:83–4. doi: 10.18632/aging.100290
45. Vulpe CD, Kuo YM, Murphy TL, Cowley L, Anderson GJ. Hephaestin, a Ceruloplasmin Homologue Implicated in Intestinal Iron Transport, Is Defective in the Sla Mouse. Nat Genet (1999) 21:195–9. doi: 10.1038/5979
46. Wong BX, Tsatsanis A, Lim LQ, Adlard PA, Bush AI, Duce JA. β-Amyloid Precursor Protein Does Not Possess Ferroxidase Activity but Does Stabilize the Cell Surface Ferrous Iron Exporter Ferroportin. PLoS One (2014) 9:e114174. doi: 10.1371/journal.pone.0114174
47. Block ML, Zecca L, Hong JS. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat Rev Neurosci (2007) 8:57–69. doi: 10.1038/nrn2038
48. Kreutzberg GW. Microglia: A Sensor for Pathological Events in the CNS. Trends Neurosci (1996) 19:312–8. doi: 10.1016/0166-2236(96)10049-7
49. Mccarthy RC, Sosa JC, Gardeck AM, Baez AS, Lee CH, Wessling-Resnick M. Inflammation-Induced Iron Transport and Metabolism by Brain Microglia. J Biol Chem (2018) 293(20):7853–63. doi: 10.1074/jbc.RA118.001949
50. Carden T, Correale J, Pasquini JM, Pérez M. Transferrin Enhances Microglial Phagocytic Capacity. Mol Neurobiol (2019) 56:6324–40. doi: 10.1007/s12035-019-1519-0
51. Rathore KI, Redensek A, David S. Iron Homeostasis in Astrocytes and Microglia Is Differentially Regulated by TNF-α and TGF-β1. Glia (2012) 60:738–50. doi: 10.1002/glia.22303
52. Xu YX, Du F, Jiang LR, Gong J, Zhou YF, Luo QQ, et al. Effects of Aspirin on Expression of Iron Transport and Storage Proteins in BV-2 Microglial Cells. Neurochem Int (2015) 91:72–7. doi: 10.1016/j.neuint.2015.10.014
53. Aral LA, Ergn MA, Engn AB, Brcek AZ, Belen HB. Iron Homeostasis Is Altered in Response to Hypoxia and Hypothermic Preconditioning in Brain Glial Cells. Turk J Med Sci (2020) 50:2005–16. doi: 10.3906/sag-2003-41
54. Bishop GM, Dang TN, Dringen R, Robinson SR. Accumulation of Non-Transferrin-Bound Iron by Neurons, Astrocytes, and Microglia. Neurotox Res (2011) 19:443–51. doi: 10.1007/s12640-010-9195-x
55. Zhang HY, Wang ND, Ning S, Xu HM, Shi LM, Jiang H, et al. 6-Hydroxydopamine Promotes Iron Traffic in Primary Cultured Astrocytes. Biometals (2013) 26:705–14. doi: 10.1007/s10534-013-9647-x
56. Sofroniew MV. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol (2020) 41:758–70. doi: 10.1016/j.it.2020.07.004
57. Lane D, Robinson SR, Czerwinska H, Bishop GM, Lawen A. Two Routes of Iron Accumulation in Astrocytes: Ascorbate-Dependent Ferrous Iron Uptake via the Divalent Metal Transporter (DMT1) Plus an Independent Route for Ferric Iron. Biochem J (2010) 432:123–32. doi: 10.1042/BJ20101317
58. Franca C, Ilaria P, Daniele Z, Fabio G. Iron Entry in Neurons and Astrocytes: A Link With Synaptic Activity. Front Mol Neurosci (2015) 8:18. doi: 10.3389/fnmol.2015.00018
59. Pelizzoni I, Zacchetti D, Campanella A, Grohovaz F, Codazzi F. Iron Uptake in Quiescent and Inflammation-Activated Astrocytes: A Potentially Neuroprotective Control of Iron Burden. Biochim Biophys Acta Mol Basis Dis (2013) 1832:1326–33. doi: 10.1016/j.bbadis.2013.04.007
60. Bishop GM, Scheiber IF D, Ringen R, Robinson SR. Synergistic Accumulation of Iron and Zinc by Cultured Astrocytes. J Neural Transm (2010) 117:809–17. doi: 10.1007/s00702-010-0420-9
62. Gaasch JA, Lockman PR, Geldenhuys WJ, Allen DD, Schyf C. Brain Iron Toxicity: Differential Responses of Astrocytes, Neurons, and Endothelial Cells. Neurochem Res (2007) 32:1196–208. doi: 10.1007/s11064-007-9290-4
63. Kipp M. Oligodendrocyte Physiology and Pathology Function. Cells (2020) 9:2078. doi: 10.3390/cells9092078
64. Bradl M, Lassmann H. Oligodendrocytes: Biology and Pathology. Acta Neuropathol (2010) 119:37–53. doi: 10.1007/s00401-009-0601-5
65. Connor JR, Menzies SL. Relationship of Iron to Oligodendrocytes and Myelination. Glia (1996) 17:83–93. doi: 10.1002/(SICI)1098-1136(199606)17:2<83::AID-GLIA1>3.0.CO;2-7
66. Gerber MR, Connor JR. Do Oligodendrocytes Mediate Iron Regulation in the Human Brain? Ann Neurol (1989) 26:95–8. doi: 10.1002/ana.410260115
67. Cheli VT, González D, Marziali LN, Zamora NN, Guitart ME, Spreuer V, et al. The Divalent Metal Transporter 1 (DMT1) Is Required for Iron Uptake and Normal Development of Oligodendrocyte Progenitor Cells. J Neurosci (2018) 38:9142–59. doi: 10.1523/JNEUROSCI.1447-18.2018
68. Qi Y, Jamindar TM, Dawson G. Hypoxia Alters Iron Homeostasis and Induces Ferritin Synthesis in Oligodendrocytes. J Neurochem (2010) 64:2458–64. doi: 10.1046/j.1471-4159.1995.64062458.x
69. Schulz K, Vulpe CD, Harris LZ, David S. Iron Efflux From Oligodendrocytes Is Differentially Regulated in Gray and White Matter. J Neurosci (2011) 31:13301–11. doi: 10.1523/JNEUROSCI.2838-11.2011
70. Cao JY, Dixon SJ. Mechanisms of Ferroptosis. Cell Mol Life Sci (2016) 73:2195–209. doi: 10.1007/s00018-016-2194-1
71. Zhao Y, Li Y, Zhang R, Wang F, Wang T, Jiao Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther (2020) 13:5429–41. doi: 10.2147/OTT.S254995
72. Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front Pharmacol (2018) 9:1371. doi: 10.3389/fphar.2018.01371
73. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
74. Zhang HL, Hu BX, Li ZL, Du T, Shan JL, Ye ZP, et al. PKCβII Phosphorylates ACSL4 to Amplify Lipid Peroxidation to Induce Ferroptosis. Nat Cell Biol (2022) 24:88–98. doi: 10.1038/s41556-021-00818-3
75. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: Past, Present and Future. Cell Death Dis (2020) 11:88. doi: 10.1038/s41419-020-2298-2
76. Chen X, Kang R, Kroemer G, Tang D. Broadening Horizons: The Role of Ferroptosis in Cancer. Nat Rev Clin Oncol (2021) 18:280–96. doi: 10.1038/s41571-020-00462-0
77. Zhu X, Zhou Y, Ou Y, Cheng Z, Han D, Chu Z, et al. Characterization of Ferroptosis Signature to Evaluate the Predict Prognosis and Immunotherapy in Glioblastoma. Aging (Albany NY) (2021) 13:17655–72. doi: 10.18632/aging.203257
78. Yanatori I, Kishi F. DMT1 and Iron Transport. Free Radic Biol Med (2019) 133:55–63. doi: 10.1016/j.freeradbiomed.2018.07.020
79. Gammella E, Buratti P, Cairo G, Recalcati S. The Transferrin Receptor: The Cellular Iron Gate. Metallomics (2017) 9:1367–75. doi: 10.1039/c7mt00143f
80. Kajarabille N, Latunde-Dada GO. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int J Mol Sci (2019) 20:4968. doi: 10.3390/ijms20194968
81. Turcu AL, Versini A, Khene N, Gaillet C, Cañeque T, Müller S, et al. DMT1 Inhibitors Kill Cancer Stem Cells by Blocking Lysosomal Iron Translocation. Chemistry (2020) 26:7369–73. doi: 10.1002/chem.202000159
82. Liuzzi JP, Aydemir F, Nam H, Knutson MD, Cousins RJ. Zip14 (Slc39a14) Mediates Non-Transferrin-Bound Iron Uptake Into Cells. Proc Natl Acad Sci USA (2006) 103:13612–7. doi: 10.1073/pnas.0606424103
83. Aydemir TB, Cousins RJ. The Multiple Faces of the Metal Transporter ZIP14 (SLC39A14). J Nutr (2018) 148:174–84. doi: 10.1093/jn/nxx041
84. Yu Y, Jiang L, Wang H, Shen Z, Cheng Q, Zhang P, et al. Hepatic Transferrin Plays a Role in Systemic Iron Homeostasis and Liver Ferroptosis. Blood (2020) 136:726–39. doi: 10.1182/blood.2019002907
85. Forcina GC, Dixon SJ. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics (2019) 19:e1800311. doi: 10.1002/pmic.201800311
86. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr Top Microbiol Immunol (2017) 403:143–70. doi: 10.1007/82_2016_508
87. Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The Tumor Suppressor P53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep (2017) 20:1692–704. doi: 10.1016/j.celrep.2017.07.055
88. Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 Promotes Angiogenesis and Neuronal Cell Death and Confers Ferroptosis in a xCT-Dependent Manner. Oncogene (2017) 36:5593–608. doi: 10.1038/onc.2017.146
89. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc- Activity. Curr Biol (2018) 28:2388–99.e5. doi: 10.1016/j.cub.2018.05.094
90. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T Cells Regulate Tumour Ferroptosis During Cancer Immunotherapy. Nature (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
91. Bröer A, Rahimi F, Bröer S. Deletion of Amino Acid Transporter ASCT2 (SLC1A5) Reveals an Essential Role for Transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) to Sustain Glutaminolysis in Cancer Cells. J Biol Chem (2016) 291:13194–205. doi: 10.1074/jbc.M115.700534
92. Dong W, Keibler MA, Stephanopoulos G. Review of Metabolic Pathways Activated in Cancer Cells as Determined Through Isotopic Labeling and Network Analysis. Metab Eng (2017) 43:113–24. doi: 10.1016/j.ymben.2017.02.002
93. Jin L, Alesi GN, Kang S. Glutaminolysis as a Target for Cancer Therapy. Oncogene (2016) 35:3619–25. doi: 10.1038/onc.2015.447
94. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
95. Shin D, Lee J, You JH, Kim D, Roh JL. Dihydrolipoamide Dehydrogenase Regulates Cystine Deprivation-Induced Ferroptosis in Head and Neck Cancer. Redox Biol (2020) 30:101418. doi: 10.1016/j.redox.2019.101418
96. Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, et al. miR-137 Regulates Ferroptosis by Targeting Glutamine Transporter SLC1A5 in Melanoma. Cell Death Differ (2018) 25:1457–72. doi: 10.1038/s41418-017-0053-8
97. Wirsching HG, Galanis E, Weller M. Glioblastoma. Handb Clin Neurol (2016) 134:381–97. doi: 10.1016/B978-0-12-802997-8.00023-2
98. Nakagawara A, Li Y, Izumi H, Muramori K, Inada H, Nishi M. Neuroblastoma. Jpn J Clin Oncol (2018) 48:214–41. doi: 10.1093/jjco/hyx176
99. Fountain DM, Young AMH, Santarius T. Malignant Meningiomas. Handb Clin Neurol (2020) 170:245–50. doi: 10.1016/B978-0-12-822198-3.00044-6
100. Weiland A, Wang Y, Wu W, Lan X, Han X, Li Q, et al. Ferroptosis and Its Role in Diverse Brain Diseases. Mol Neurobiol (2019) 56:4880–93. doi: 10.1007/s12035-018-1403-3
101. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
102. Ivanov SD, Semenov AL, Kovan'ko EG, Yamshanov VA. Effects of Iron Ions and Iron Chelation on the Efficiency of Experimental Radiotherapy of Animals With Gliomas. Bull Exp Biol Med (2015) 158:800–3. doi: 10.1007/s10517-015-2865-1
103. Ivanov SD, Semenov AL, Mikhelson VM, Kovan'ko EG, Iamshanov VA. Effects of Iron Ion Additional Introduction in Radiation Therapy of Tumor-Bearing Animals. Radiats Biol Radioecol (2013) 53:296–303. doi: 10.7868/s0869803113030065
104. Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, et al. Neutrophil-Induced Ferroptosis Promotes Tumor Necrosis in Glioblastoma Progression. Nat Commun (2020) 11:5424. doi: 10.1038/s41467-020-19193-y
105. Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM, et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front Aging Neurosci (2016) 8:308. doi: 10.3389/fnagi.2016.00308
106. Kabiraj P, Valenzuela CA, Marin JE, Ramirez DA, Mendez L, Hwang MS, et al. The Neuroprotective Role of Ferrostatin-1 Under Rotenone-Induced Oxidative Stress in Dopaminergic Neuroblastoma Cells. Protein J (2015) 34:349–58. doi: 10.1007/s10930-015-9629-7
107. Bao Z, Hua L, Ye Y, Wang D, Li C, Xie Q, et al. MEF2C Silencing Downregulates NF2 and E-Cadherin and Enhances Erastin-Induced Ferroptosis in Meningioma. Neuro Oncol (2021) 23:2014–27. doi: 10.1093/neuonc/noab114
108. Wang Y, Yu L, Ding J, Chen Y. Iron Metabolism in Cancer. Int J Mol Sci (2018) 20:95. doi: 10.3390/ijms20010095
109. Zhuo S, Chen Z, Yang Y, Zhang J, Tang J, Yang K. Clinical and Biological Significances of a Ferroptosis-Related Gene Signature in Glioma. Front Oncol (2020) 10:590861. doi: 10.3389/fonc.2020.590861
110. Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 Inhibits Ferroptosis by Protection Against Mitochondrial Lipid Peroxidation. Biochem Biophys Res Commun (2016) 478:838–44. doi: 10.1016/j.bbrc.2016.08.034
111. Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O, et al. Iron Chaperone Poly rC Binding Protein 1 Protects Mouse Liver From Lipid Peroxidation and Steatosis. Hepatology (2021) 73:1176–93. doi: 10.1002/hep.31328
112. Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of Ferroportin Accelerates Erastin-Induced Ferroptosis in Neuroblastoma Cells. Eur Rev Med Pharmacol Sci (2018) 22:3826–36. doi: 10.26355/eurrev_201806_15267
113. Ganz T, Nemeth E. Hepcidin and Iron Homeostasis. Biochim Biophys Acta (2012) 1823:1434–43. doi: 10.1016/j.bbamcr.2012.01.014
114. Floros KV, Cai J, Jacob S, Kurupi R, Fairchild CK, Shende M, et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res (2021) 81:1896–908. doi: 10.1158/0008-5472.CAN-20-1641
115. Sehm T, Fan Z, Ghoochani A, Rauh M, Engelhorn T, Minakaki G, et al. Sulfasalazine Impacts on Ferroptotic Cell Death and Alleviates the Tumor Microenvironment and Glioma-Induced Brain Edema. Oncotarget (2016) 7:36021–33. doi: 10.18632/oncotarget.8651
116. Song Q, Peng S, Sun Z, Heng X, Zhu X. Temozolomide Drives Ferroptosis via a DMT1-Dependent Pathway in Glioblastoma Cells. Yonsei Med J (2021) 62:843–9. doi: 10.3349/ymj.2021.62.9.843
117. Zhang WL, Kong FH, Nan JI, LI HW, Qi LI, Lu HE, et al. Study of Correlation Between Ferroptosis and Glioblastoma. Labeled Immunoassays Clin Med (2018) 25:275–88. doi: 10.11748/bjmy.issn.1006-1703.2018.02.031
118. Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An Essential Role for Functional Lysosomes in Ferroptosis of Cancer Cells. Biochem J (2016) 473:769–77. doi: 10.1042/BJ20150658
119. Lin R, Zhang Z, Chen L, Zhou Y, Zou P, Feng C, et al. Dihydroartemisinin (DHA) Induces Ferroptosis and Causes Cell Cycle Arrest in Head and Neck Carcinoma Cells. Cancer Lett (2016) 381:165–75. doi: 10.1016/j.canlet.2016.07.033
120. Tetsuro I, Eiji W, Mann GE. Circadian Control of BDNF-Mediated Nrf2 Activation in Astrocytes Protects Dopaminergic Neurons From Ferroptosis. Free Radic Biol Med (2019) 133:169–78. doi: 10.1016/j.freeradbiomed.2018.09.002
121. Wang Z, Ding Y, Wang X, Lu S, Wang C, He C, et al. Pseudolaric Acid B Triggers Ferroptosis in Glioma Cells via Activation of Nox4 and Inhibition of xCT. Cancer Lett (2018) 428:21–33. doi: 10.1016/j.canlet.2018.04.021
122. Yi R, Wang H, Deng C, Wang X, Yao L, Niu W, et al. Dihydroartemisinin Initiates Ferroptosis in Glioblastoma Through GPX4 Inhibition. Biosci Rep (2020) 40:BSR20193314. doi: 10.1042/BSR20193314
123. Gao X, Guo N, Xu H, Pan T, Xu L. Ibuprofen Induces Ferroptosis of Glioblastoma Cells via Downregulation of Nuclear Factor Erythroid 2-Related Factor 2 Signaling Pathway. Anticancer Drugs (2019) 31:27–34. doi: 10.1097/CAD.0000000000000825
124. Igarashi K, Nishizawa H, Saiki Y, Matsumoto M. The Transcription Factor BACH1 at the Crossroads of Cancer Biology: From Epithelial-Mesenchymal Transition to Ferroptosis. J Biol Chem (2021) 297:101032. doi: 10.1016/j.jbc.2021.101032
125. Mariachiara B, Matteo M, Simone P, Ivana DP, Giorgio DQ, Maurizio M, et al. Inhibition of Autophagy Increases Susceptibility of Glioblastoma Stem Cells to Temozolomide by Igniting Ferroptosis. Cell Death Dis (2018) 9:841. doi: 10.1038/s41419-018-0864-7
126. Jara N, Ramirez E, Ferrada L, Salazar K, Espinoza F, González-Chavarría I, et al. Vitamin C Deficient Reduces Proliferation in a Human Periventricular Tumor Stem Cell-Derived Glioblastoma Model. J Cell Physiol (2021) 236:5801–17. doi: 10.1002/jcp.30264
127. Cheng J, Fan YQ, Liu BH, Zhou H, Chen QX. ACSL4 Suppresses Glioma Cells Proliferation via Activating Ferroptosis. Oncol Rep (2020) 43:147–58. doi: 10.3892/or.2019.7419
128. Sehm T, Rauh M, Wiendieck K, Buchfelder M, Eyüpoglu I, Savaskan NE. Temozolomide Toxicity Operates in a xCT/SLC7a11 Dependent Manner and Is Fostered by Ferroptosis. Oncotarget (2016) 7:74630–47. doi: 10.18632/oncotarget.11858
129. Chen TC, Chuang JY, Ko CY, Kao TJ, Yang PY, Yu CH, et al. AR Ubiquitination Induced by the Curcumin Analog Suppresses Growth of Temozolomide-Resistant Glioblastoma Through Disrupting GPX4-Mediated Redox Homeostasis. Redox Biol (2020) 30:101413. doi: 10.1016/j.redox.2019.101413
130. Chen R, Chen L. Solute Carrier Transporters: Emerging Central Players in Tumour Immunotherapy. Trends Cell Biol (2022) 32:186–201. doi: 10.1016/j.tcb.2021.08.002
131. Ji L, Zhao X, Zhang B, Kang L, Hu X. Slc6a8-Mediated Creatine Uptake and Accumulation Reprogram Macrophage Polarization via Regulating Cytokine Responses. Immunity (2019) 51:272–84.e7. doi: 10.1016/j.immuni.2019.06.007
132. Deng S, Zhenga Y, Mo Y, Xu X, Ke Y. Ferroptosis Suppressive Genes Correlate With Immunosuppression in Glioblastoma. World Neurosurg (2021) 152:e436–48. doi: 10.1016/j.wneu.2021.05.098
133. Rroji O, Kumar A, Karuppagounder SS, Ratan RR. Epigenetic Regulators of Neuronal Ferroptosis Identify Novel Therapeutics for Neurological Diseases: HDACs, Transglutaminases, and HIF Prolyl Hydroxylases. Neurobiol Dis (2021) 147:105145. doi: 10.1016/j.nbd.2020.105145
134. Zille M, Kumar A, Kundu N, Bourassa MW, Wong VSC, Willis D, et al. Ferroptosis in Neurons and Cancer Cells Is Similar But Differentially Regulated by Histone Deacetylase Inhibitors. Eneuro (2019) 6:ENEURO.0263–18. doi: 10.1523/ENEURO.0263-18.2019
135. Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W, et al. Fenton-Reaction-Accelerable Magnetic Nanoparticles for Ferroptosis Therapy of Orthotopic Brain Tumors. ACS Nano (2018) 12:11355–65. doi: 10.1021/acsnano.8b06201
136. Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J Clin Invest (2018) 128:3341–55. doi: 10.1172/JCI99032
137. Gao JL, Luo T, Wang JK. Gene Interfered-Ferroptosis Therapy for Cancers. Nat Commun (2021) 12:5311–27. doi: 10.1038/s41467-021-25632-1
138. Ghoochani A, Hsu EC, Aslan M, Rice MA, Nguyen HM, Brooks JD, et al. Ferroptosis Inducers Are a Novel Therapeutic Approach for Advanced Prostate Cancer. Cancer Res (2021) 81:1583–94. doi: 10.1158/0008-5472.CAN-20-3477
139. Mai TT, Hama A, Hienzsch A, Cañeque T, Müller S, Wicinski J, et al. Salinomycin Kills Cancer Stem Cells by Sequestering Iron in Lysosomes. Nat Chem (2017) 9:1025–33. doi: 10.1038/nchem.2778
140. Zhang T, Sun BB, Zhong CX, Xu K, Wang ZX, Hofman P, et al. Targeting Histone Deacetylase Enhances the Therapeutic Effect of Erastin-Induced Ferroptosis in EGFR-Activating Mutant Lung Adenocarcinoma. Transl Lung Cancer Res (2021) 10:1857–72. doi: 10.21037/tlcr-21-303
141. Markowitsch SD, Schupp P, Lauckner J, Vakhrusheva O, Slade KS, Mager R, et al. Artesunate Inhibits Growth of Sunitinib-Resistant Renal Cell Carcinoma Cells Through Cell Cycle Arrest and Induction of Ferroptosis. Cancers (2020) 12:3150–73. doi: 10.3390/cancers12113150
142. Sakharnova TA, Balalaeva IV, Vedunova MV, Krysko D. Ferroptosis and Photodynamic Therapy Synergism: Enhancing Anticancer Treatment. Trends Cancer (2021) 7(6):484–7. doi: 10.1016/j.trecan.2021.01.013
143. Huo MF, Wang LY, Chen Y, Shi JL. Tumor-Selective Catalytic Nanomedicine by Nanocatalyst Delivery. Nat Commun (2017) 8:357–68. doi: 10.1038/s41467-017-00424-8
144. Song W, Li D, Tao L, Luo Q, Chen L. Solute Carrier Transporters: The Metabolic Gatekeepers of Immune Cells. Acta Pharm Sin B (2020) 10:61–78. doi: 10.1016/j.apsb.2019.12.006
Keywords: iron transport, transporters, ferroptosis, malignant brain tumors, therapeutic strategy
Citation: Zhao J, Wang Y, Tao L and Chen L (2022) Iron Transporters and Ferroptosis in Malignant Brain Tumors. Front. Oncol. 12:861834. doi: 10.3389/fonc.2022.861834
Received: 25 January 2022; Accepted: 21 March 2022;
Published: 21 April 2022.
Edited by:
Ali Ghoochani, Stanford University, United StatesReviewed by:
Wentao Dong, Stanford University, United StatesMarta Garcia-Contreras, Johns Hopkins Medicine, United States
Copyright © 2022 Zhao, Wang, Tao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ligong Chen, bGlnb25nY2hlbkB0c2luZ2h1YS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship