 Caroline E. Nunes-Xavier1,2*
Caroline E. Nunes-Xavier1,2* Janire Mingo1Maite Emaldi1
Janire Mingo1Maite Emaldi1 Karine Flem-Karlsen2
Karine Flem-Karlsen2 Gunhild M. Mælandsmo2
Gunhild M. Mælandsmo2 Øystein Fodstad2Roberto Llarena3
Øystein Fodstad2Roberto Llarena3 José I. López1,4
José I. López1,4 Rafael Pulido1,5
Rafael Pulido1,5- 1Biomarkers in Cancer, Biocruces Bizkaia Health Research Institute, Barakaldo, Spain
- 2Department of Tumor Biology, Institute for Cancer Research, Oslo University Hospital Radiumhospitalet, Oslo, Norway
- 3Department of Urology, Cruces University Hospital, Barakaldo, Spain
- 4Department of Pathology, Cruces University Hospital, Barakaldo, Spain
- 5Ikerbasque, Basque Foundation for Science, Bilbao, Spain
Background: Pyruvate dehydrogenase (PDH) complex converts pyruvate into acetyl-CoA by pyruvate decarboxylation, which drives energy metabolism during cell growth, including prostate cancer (PCa) cell growth. The major catalytic subunit of PDH, PDHA1, is regulated by phosphorylation/dephosphorylation by pyruvate dehydrogenase kinases (PDKs) and pyruvate dehydrogenase phosphatases (PDPs). There are four kinases, PDK1, PDK2, PDK3 and PDK4, which can phosphorylate and inactivate PDH; and two phosphatases, PDP1 and PDP2, that dephosphorylate and activate PDH.
Methods: We have analyzed by immunohistochemistry the expression and clinicopathological correlations of PDHA1, PDP1, PDP2, PDK1, PDK2, PDK3, and PDK4, as well as of androgen receptor (AR), in a retrospective PCa cohort of patients. A total of 120 PCa samples of representative tumor areas from all patients were included in tissue microarray (TMA) blocks for analysis. In addition, we studied the subcellular localization of PDK2 and PDK3, and the effects of the PDK inhibitor dichloroacetate (DCA) in the growth, proliferation, and mitochondrial respiration of PCa cells.
Results: We found heterogeneous expression of the PDH complex components in PCa tumors. PDHA1, PDP1, PDK1, PDK2, and PDK4 expression correlated positively with AR expression. A significant correlation of PDK2 immunostaining with biochemical recurrence and disease-free survival was revealed. In PCa tissue specimens, PDK2 displayed cytoplasmic and nuclear immunostaining, whereas PDK1, PDK3 and PDK4 showed mostly cytoplasmic staining. In cells, ectopically expressed PDK2 and PDK3 were mainly localized in mitochondria compartments. An increase in maximal mitochondrial respiration was observed in PCa cells upon PDK inhibition by DCA, in parallel with less proliferative capacity.
Conclusion: Our findings support the notion that expression of specific PDH complex components is related with AR signaling in PCa tumors. Furthermore, PDK2 expression associated with poor PCa prognosis. This highlights a potential for PDH complex components as targets for intervention in PCa.
Introduction
PCa is a long-latency cancer, evolving from low malignancy early stages (prostatic intraepithelial neoplasia) to high-grade and metastatic adenocarcinomas, which frequently do not respond to anti-androgen hormone therapies (castrate-resistant prostate cancer, CRPC) (1–3). The androgen pathway is the central signaling pathway in PCa, together with the retinoblastoma (RB), PI3K/PTEN/AKT/mTOR, and RAS/RAF/MAPK pathways (4–7). Frequent alterations in PCa include gene amplification of MYC transcription factor and androgen receptor (AR), the gene deletion of NKX3.1 homeobox, RB1, and PTEN phosphatase, and the gene reorganization of the ETS family of transcription factors (8–12). Currently, the identification of early tumor markers, including metabolic biomarkers, and molecular targets for effective PCa treatment is a research priority (13–17).
PCa presents a high extent of metabolic modifications, mainly related with increase in aerobic glycolysis and protein and fatty acid synthesis (18, 19). As in other cancer types, this metabolic switch facilitates the synthesis of biomolecules required by the tumor cell to support its rapid growth and division (20). PCa cells display high levels of aerobic glycolysis in the more advanced tumor stages, while primary PCa cells show higher oxidative respiration than non-transformed prostatic cells. This is mainly due to a decrease in Zn accumulation in primary PCa cells, which allows citrate oxidation through the Krebs cycle. High de novo fatty acid synthesis is characteristic of PCa progression towards CRPC, which is facilitated by high expression of fatty acid synthase (FASN) and other lipogenic enzymes (21, 22). PCa progression and metastasis has been recently linked to glycolytic enzymes such as pyruvate kinase isoform M2 (PKM2) (23). Together, these observations suggest that specific interference with key metabolic reactions could be useful to improve the current therapies for advanced PCa.
The enzyme pyruvate dehydrogenase (PDH) is essential in the glycolytic and Krebs cycle metabolism, and play important roles in carcinogenesis, making this enzyme a feasible therapeutic target in cancer (24–27). PDH exists as a multi-enzyme complex formed by three catalytic (E1 [two genes: PDHA1-2], E2 [DLAT], and E3 [DLD]) and three regulatory subunits (E3BP [PDHX], PDKs [four genes: PDK1-4], and PDPs [two genes: PDP1-2). The mRNA expression patterns of these genes in prostate tissues and prostate tumors are distinct, as shown in databases GTEx (Genotype-Tissue Expression; https://gtexportal.org) and TCGA (The Cancer Genome Atlas; https://www.proteinatlas.org), but comprehensive comparative studies on the expression at the protein level of these enzymes in PCa are lacking. The association of PDKs expression with poor prognosis and resistance to anti-cancer therapies is widely documented, and PDKs inhibition (which results in PDH activation) constitutes a potential therapeutic possibility in several cancer types, including PCa (28–34). In addition, differing results have been reported on the association of other PDH components, such as PDHA1 and PDP1, with PCa prognosis (35, 36). Together, this makes relevant to investigate comparatively the individual expression and function of the distinct components of the PDH complex in relation with PCa progression and malignancy.
In this study, we have evaluated the expression and subcellular localization of components of the PDH complex in PCa, including PDHA1, PDP1, PDP2, PDK1, PDK2, PDK3 and PDK4. We have found specific correlations between the expression of some of these PDH complex components and AR expression in PCa tumors. Furthermore, a significant correlation of PDK2 PCa tumor immunostaining with patient biochemical recurrence and disease-free survival has been revealed. We discuss the potential of PDH complex components as targets for intervention in PCa.
Material and Methods
Cell Lines
Simian kidney COS-7 cells were cultured in DMEM (Dulbecco’s Modified Eagle’s Medium) (Lonza, Basel, Switzerland) medium supplemented with 5% FBS (Fetal Bovine Serum) (Sigma Aldrich, St. Louis, MO, USA). Human prostate carcinoma LNCaP cells were cultured in RPMI-1640 (Lonza) medium supplemented with 10% FBS. Human prostate carcinoma DU-145 cells were cultured in EMEM (Eagle’s Minimal Essential Medium) (Lonza) medium supplemented with 10% FBS. All media were supplemented with 1% L-Glutamine and 1% penicillin/streptomycin (Lonza). Cells were incubated at 37°C and 5% CO2.
Plasmids, Transfection, and Immunoblot
Human PDK2 (NM_002611.4) and PDK3 (NM_001142386) cDNAs, cloned in pcDNA3.1+/C-DYK mammalian expression plasmids (C-terminal Flag fusion), were purchased from GeneScript (Piscataway, NJ, USA). pRK5 Flag-PTEN was made by PCR incorporation of an N-terminal Flag sequence to human PTEN (NM_000314) from pRK5 PTEN (37). Cells were transiently transfected with empty vector, pRK5 Flag-PTEN, pCDNA3.1 PDK2-Flag, or pCDNA3.1 PDK3-Flag using GenJet reagent (SignaGen, Frederick, MD, USA). Cells were lysed in M-PER extraction reagent (ThermoFisher, Waltham, MA, USA) and processed for immunoblot as described (38). Primary antibody used was mouse anti-Flag (1:500, MAB3118, Sigma Aldrich). Secondary antibody was IRDye 680RD Goat anti-Mouse (LI-COR, Lincoln, NE, USA). Blots were processed with Odyssey CLx Imaging system (LI-COR).
Metabolism/Seahorse
Oxygen Consumption Rate Assay Kit (Cayman Chemical, Ann Arbor, MI, USA) was used to measure extracellular oxygen consumption levels according to manufacturer’s instructions. XF96 Mitochondrial stress test was performed using Seahorse Extracellular Flux Analyzer XF96e (Agilent Technologies, Santa Clara, CA, USA) to measure the oxygen consumption rate (OCR) of cells according to manufacturer’s instructions. Seahorse assays were performed in at least triplicate wells in three independent experiments for each condition.
Cell Proliferation and Confluence
Cell proliferation/viability of LNCaP and DU-145 cells was assessed as described (39). 5x103 cells/well were plated in 96-well culture plates. A day after plating the cells, different concentrations of dichloroacetate (DCA; Sigma Aldrich) or vehicle were added. Cell proliferation was measured with the CellTiter 96®AQueous One Solution Cell Proliferation Assay Kit (MTS Assay, Promega, Madison, WI, USA) in 96-well plates, and luminescence was measured at 490 nm using Victor3 microplate reader (PerkinElmer, Waltham MA, USA). To assess cell confluence, 5x103 cells/well were seeded on 96-well plates and the cell confluence was measured every three hours by the IncuCyte FLR imaging microscopes (Essen Biosciences, Ann Arbor, MI, USA), as described (40). The cells were treated with the indicated DCA concentrations 21 h post-plating and were scanned for 72 h after adding the drug.
Immunofluorescence
3x104 COS-7 cells per well were plated in 8-well chamber slides for immunofluorescence (Ibidi, Gräfelfing, Germany). Transient transfection was performed as described above. Cells were washed and mitochondria were stained with Mitotracker™ Red CMXRos following manufacturer’s instructions (250 nM, 20 min) (ThermoFisher). before they were fixed in methanol for 5 min at -20°C and blocked in blocking solution (Phosphate Buffered Saline (PBS) containing 3% Bovine Serum Albumin (BSA). Mouse anti-Flag primary antibody (1/100 in blocking solution) was incubated overnight at 4°C in a wet chamber. Subsequently, cells were washed three times with PBS-BSA for 10 min prior to incubation with anti-mouse FITC secondary antibody (1/100) for 1 h in a wet chamber and darkness at room temperature. Cells were washed and mounted in Mounting Medium with DAPI (4’6-diamidino-2-phenylindole) (Abcam) and visualized by standard [NIKON ECLIPSE TE2000 (Nikon, Tokyo, Japan)] or confocal microscopy [ZEISS LSM880 AIRYSCAN (Zeiss, Jena, Germany)].
Clinical Data and Tumor Samples
The PCa cohort has been previously described (41). Briefly, it consisted of 120 PCa patients treated with radical prostatectomy at Cruces University Hospital (Barakaldo, Spain) between 2000 and 2005. An experienced pathologist (JIL) selected tumor areas with well‐preserved tissue, representative of the whole tumor, from formalin‐fixed and paraffin‐embedded (FFPE) tumor tissue blocks, and TMA blocks were made from these areas. 4 μm sections were made from the TMA blocks, one of which was stained with hematoxylin and eosin (H&E) to verify the presence of tumor content. Biochemical recurrence (BR) was defined as a Prostate-Specific Antigen (PSA) measurement equal to or greater than 0.4 ng/ml after surgery. Follow‐up has been recorded until October 1, 2016. Cancer of the Prostate Risk Assessment Postsurgical (CAPRA‐S) score was calculated according to its definition (42), that is, by combining preoperative PSA, Gleason grade, surgical margins, extracapsular extension, seminal vesicle invasion, and lymph node invasion.
Immunohistochemistry and Scoring
Immunohistochemistry (IHC) was carried out using the following primary antibodies: PDHA1 (Sigma Aldrich, HPA047864, dilution: 1:10), PDP1 (Sigma Aldrich, HPA019081, dilution 1:10), PDP2 (Sigma Aldrich, HPA019950, dilution 1:65), PDK1 (Cell Signaling, Danvers, MA, USA, HPA027376, dilution: 1:120), PDK2 (Sigma Aldrich, HPA008287, dilution 1:25), PDK3 (Sigma Aldrich, HPA046583, dilution 1:50), PDK4 (Sigma Aldrich, HPA056731, dilution 1:100), and AR (SP107 ready to use, Ventana, Roche, Basel, Switzerland) antibodies. Antigen retrieval was performed at pH 6 and pH 9 using PT link system (Agilent Technologies). IHC immunostainings were performed in automated immunostainers (EnVision FLEX, Dako Autostainer Plus; Dako, Glostrup, Denmark and BenchMark Ultra, Ventana Medical Systems, Tucson, AZ, USA). Antibodies were incubated for 30 min, followed by secondary antibody incubation for 15 min using Goat Anti Mouse and Anti-rabbit Ig/HRP secondary antibodies (Dako), FLEX/HPR for 20 min, FLEX DAB/Sub Chromo for 10 min, and finally counterstaining with hematoxylin. Immunostainings were evaluated in tumor cells as negative (weak/no staining) or positive (medium/high staining). The analysis was performed using a Nikon Eclipse 80i microscope (Nikon, Tokyo, Japan).
Statistical Analysis
Error bars in results represent ± standard deviation (S.D.). Cell data was analyzed by GraphPad Prism t Test Calculator (San Diego, CA, USA), where significance was calculated using two-tailed student t-test. p values smaller than 0.05 were considered significant and are indicated with an asterisk (*). All experiments were performed at least twice, and results shown are from one representative experiment. The SPSS version 23 software (SPSS Inc., Chicago, IL, USA) was used for statistical calculations of the clinical material. For all the experiments, any p value below 0.05 was considered statistically significant.
Results
PDH complex components, including the negative regulators of PDH activity, PDKs, have been involved in PCa carcinogenesis (27). We analyzed the role of PDKs in the growth, proliferation, and mitochondrial respiration of PCa cells, using the PDK inhibitor dichloroacetate (DCA), which selectively shifts the cancer cell metabolism from glycolysis to oxidative phosphorylation (29). As expected, DCA treatment inhibited in a dose-response manner the growth/viability of LNCaP and DU-145 PCa cells, as shown by MTS assay and cell confluence measurements (Figures 1A–C). In addition, an increase in maximal oxygen consumption rate (OCR) was observed in LNCaP PCa cells upon PDK inhibition by DCA, in parallel with less proliferative capacity and cell viability (Figure 1D). These results suggest a role for PDKs in the regulation of cell growth and viability of PCa cells.
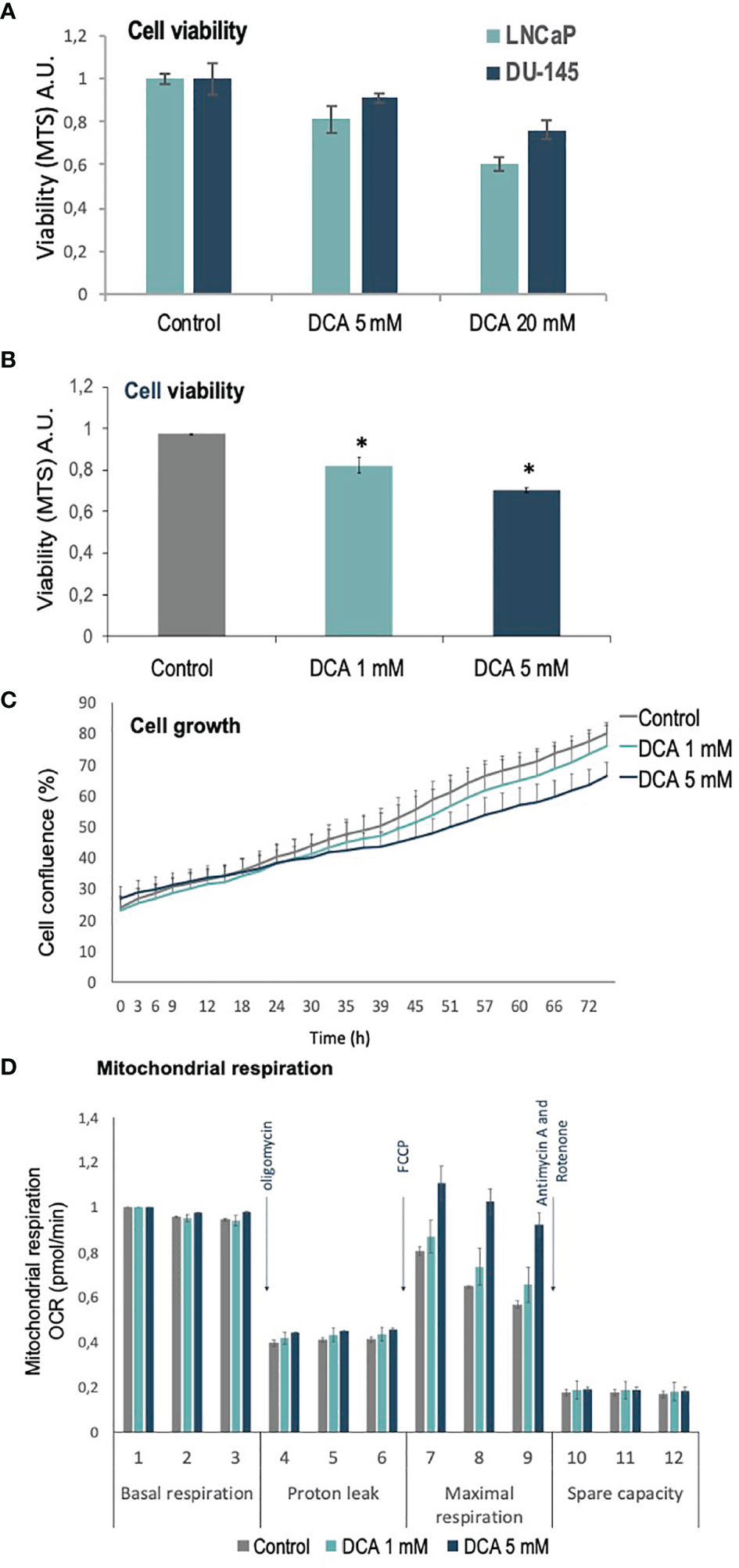
Figure 1 Viability, proliferation and mitochondrial function of PCa cells treated with DCA. (A) Cell viability is shown for LNCaP and DU-145 PCa cells, as determined by MTS analysis, after 72 h in the presence of DCA (5 mM and 20 mM). (B) Cell viability is shown for LNCaP cells, as determined by MTS analysis, after 72 h in the presence of DCA (1 mM and 5 mM). (C) Cell growth is shown for LNCaP cells, as determined by Incucyte live-cell analysis, after 72 h in the presence of DCA (1 mM and 5 mM). (D) Mitochondrial respiration is shown for LNCaP cells, as determined by Seahorse extracellular flux analysis, after 48 h in the presence of DCA (1 mM and 5 mM). p value below 0.05 are indicated with *.
This prompted us to investigate the expression of PDKs and other PHD complex components in PCa patient tumor samples. The expression of PDHA1, PDP1, PDP2, PDK1, PDK2, PDK3, and PDK4, as well as the expression of AR was evaluated by IHC in a retrospective cohort of 120 PCa patients (Tables 1–3). FFPE samples from representative tumor areas were included in TMAs for analysis, and expression was scored as negative or positive. We observed heterogeneous expression of PDHA1, PDP1, PDP2, PDK1, PDK2, PDK3, and PDK4 in PCa specimens, and examples of different patterns of staining for the different PDKs are shown in Figure 2. PDK2 expression in tumors displayed a nuclear/cytoplasmic pattern, whereas PDK1, PDK3, and PDK4 expression was mostly cytoplasmic (Figure 2). PDP1 expression positively correlated with stage (p = 0.037) and extracapsular extension (p = 0.027) (Table 1). Importantly, we found a significant positive correlation of PDK2 immunostaining with biochemical recurrence (p = 0.033), and negative correlation with disease-free survival (p = 0.045), suggesting a negative prognostic role for PDK2 expression in PCa (Table 2). Significant positive correlations were found with respect to AR expression for PDHA1 (p = 0.035), PDP1 (p = 0.046), PDK1 (p = 0.003), PDK2 (p = 0.001), and PDK4 (p = 0.031) expression (Table 3 and Figure 3). PDP2 and PDK3 did not show any significant correlation. Together, these findings show a heterogeneous expression pattern of PDH complex components in PCa related with AR and suggest an association between PDK2 expression and PCa progression.
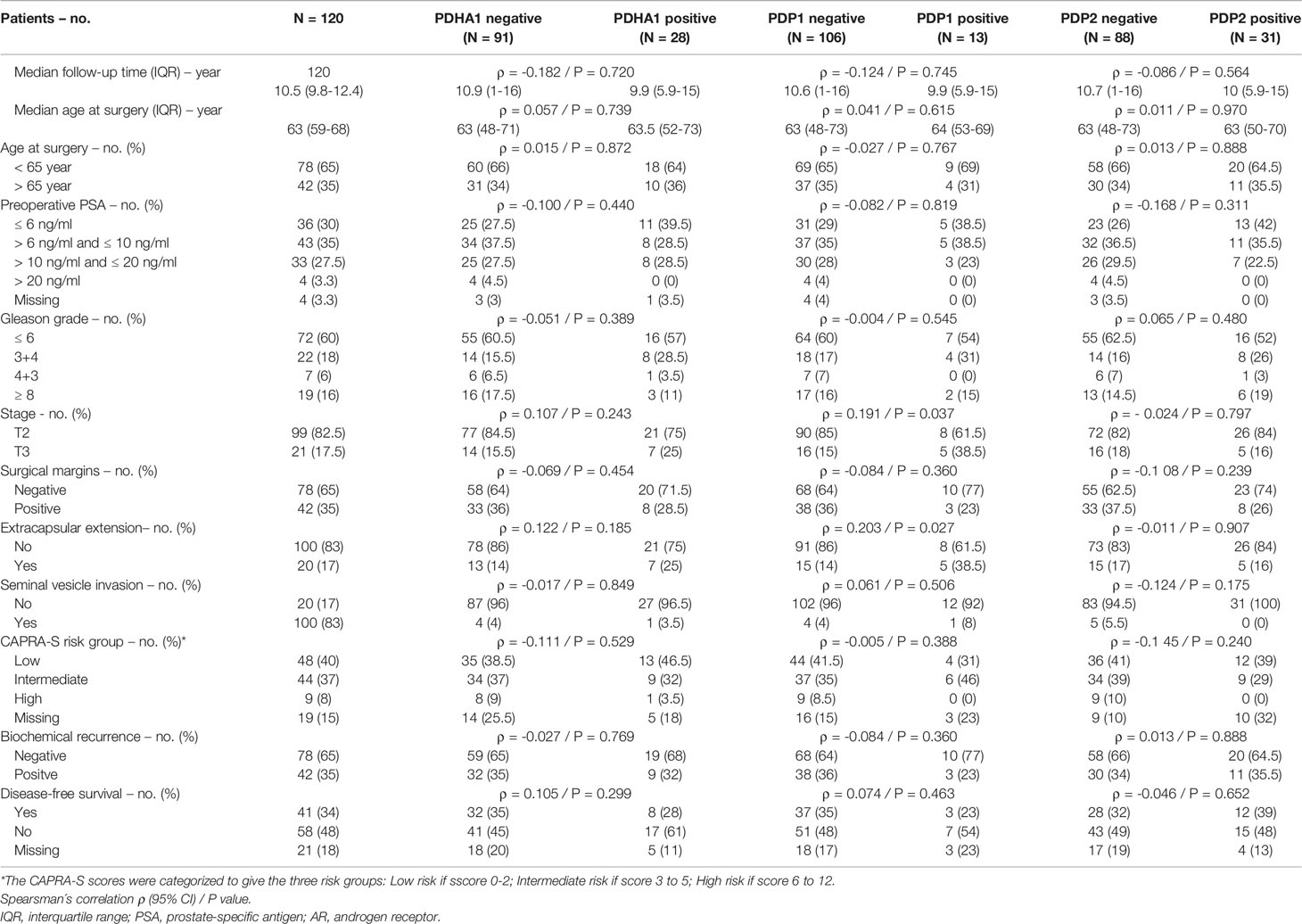
Table 1 Correlation between clinical and pathological variables and PDHA1, PDP1 and PDP2 protein expression in prostate cancer.
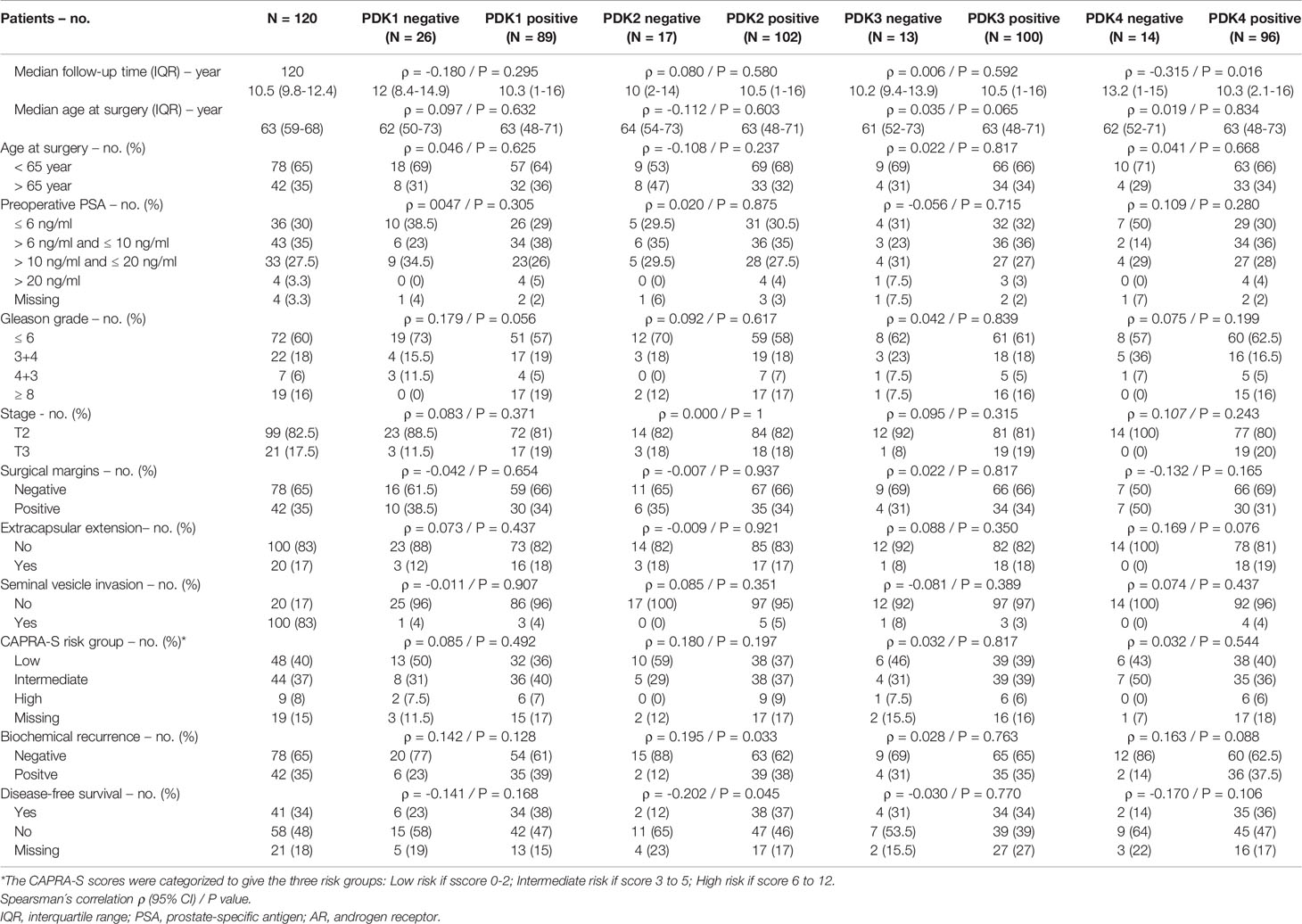
Table 2 Correlation between clinical and pathological variables and PDK1, PDK2, PDK3 and PDK4 protein expression in prostate cancer.

Table 3 Correlation between PDHA complex components and androgen receptor protein expression in prostate cancer.
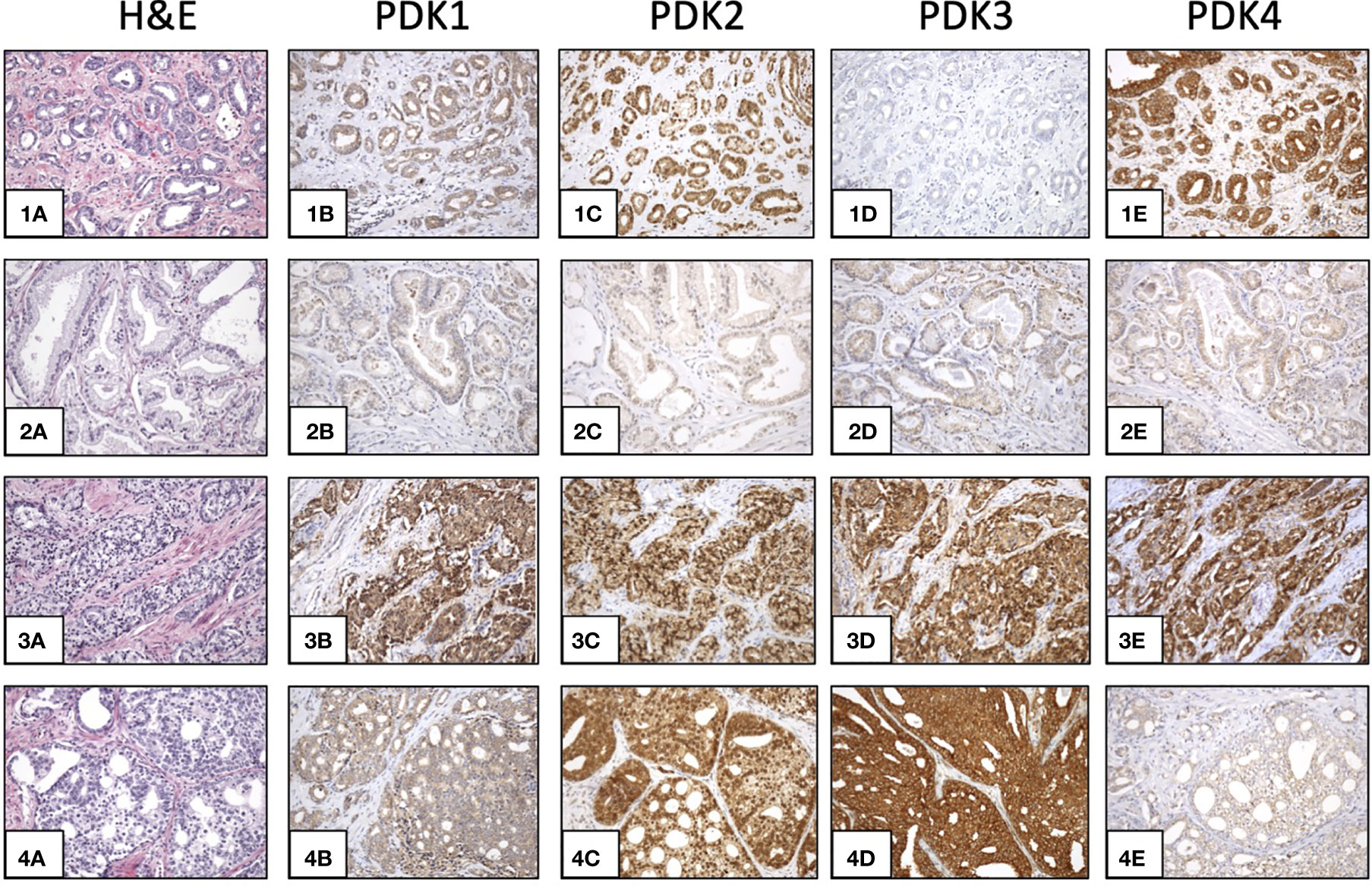
Figure 2 Expression of PDKs in PCa specimens. Immunohistochemical staining of expression of PDKs in four representative prostate carcinoma patient samples (1-4). Hematoxylin and eosin (H&E) staining (1A, 2A, 3A, 4A). High expression of all PDKs (case 3: 3B, 3C, 3D, 3E). Low expression of all PDKs (case 2: 2B, 2C, 2D, 2E). High expression of PDK2 and PDK4 (case 1: 1C, 1E), and low expression of PDK1 and PDK3 (case 1: 1B, 1D). High expression of PDK2 and PDK3 (case 4: 4C, 4D), and low expression of PDK1 and PDK4 (case 4: 4B, 4E). Magnification: X100.
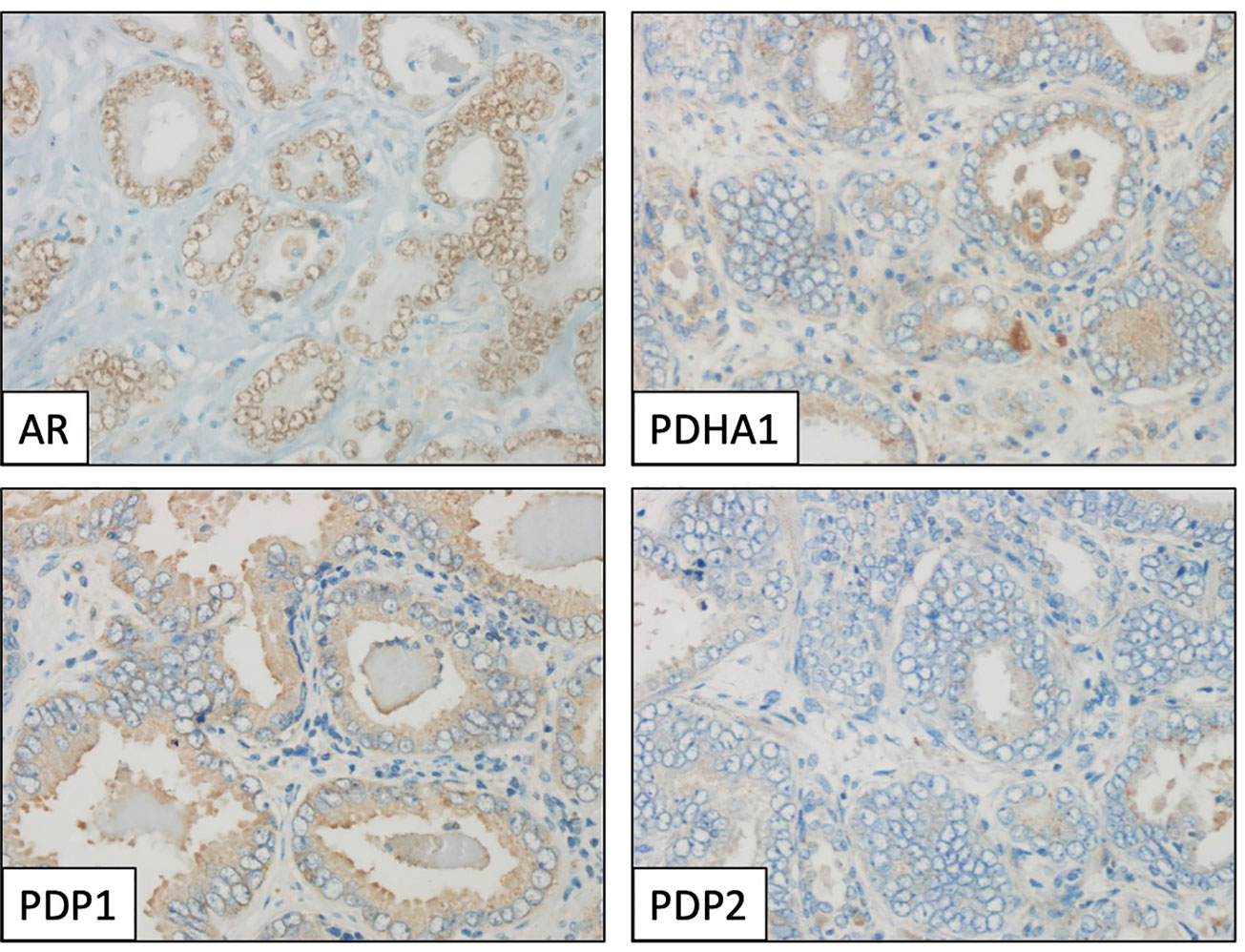
Figure 3 Immunohistochemical profile of a prostate adenocarcinoma specimen, showing positive staining for androgen receptor (AR, nuclear), PDHA1 and PDP1 (cytoplasmic), and negative for PDP2. Magnification: X400.
PDH complex components are found at the mitochondria, but they have also been found in the nucleus, which has been proposed to have clinical implications (36, 43). Next, we investigated by immunoblot and immunofluorescence the expression and subcellular localization of PDK2 and PDK3 (tagged with a Flag epitope at the C-terminus) ectopically expressed in COS-7 (as a suitable cell model for ectopic protein expression) and LNCaP PCa cells (Figures 4A–D). The expression of the phosphatase PTEN was monitored as a control. PDK2-Flag and PDK3-Flag proteins displayed a predominant punctate pattern of expression that overlapped with Mitotracker marker staining, indicating a major mitochondrial localization in cells (Figures 4B–D). This is in accordance with the mitochondrial subcellular localization reported for PDH components in other human cancer cell lines (43). In contrast, Flag-PTEN displayed cytoplasmic/nuclear localization (Figure 4B). Together, these results illustrate differential subcellular localization of PDKs in cells and in PCa tissues. In the case of PDK2, which showed predominant nuclear localization in PCa tissues, further studies are required to elucidate how its localization at specific subcellular compartments in PCa tumors may affect PCa progression.
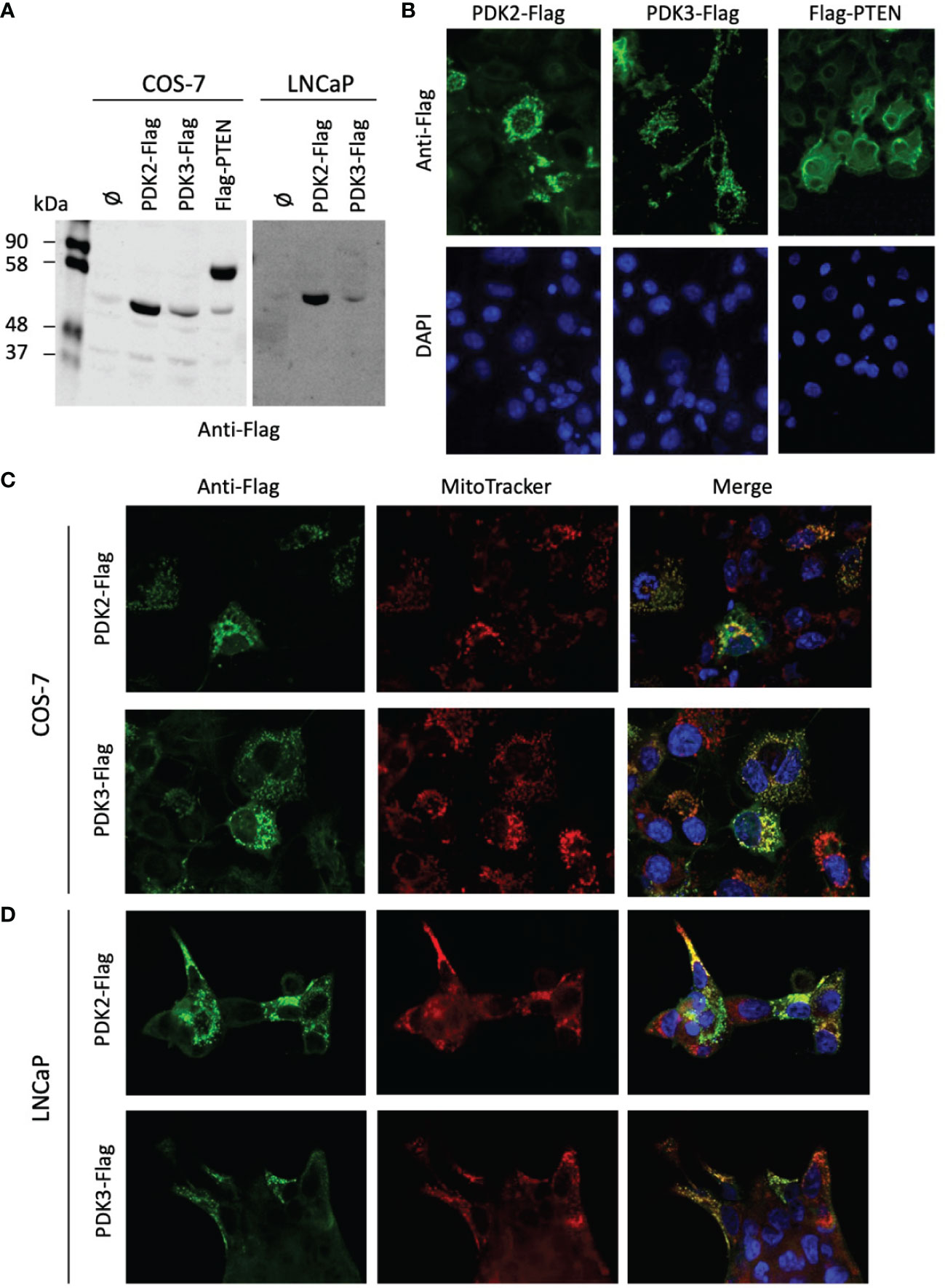
Figure 4 Expression and subcellular localization of PDK2 and PDK3 in PCa cells. (A) Immunoblot of ectopically expressed PDK2-Flag, PDK3-Flag, and Flag-PTEN (as a control) in COS-7 and LNCaP cells using anti-Flag antibody. (B) Immunofluorescence of PDK2-Flag, PDK3-Flag, and Flag-PTEN in COS-7 cells, using anti-Flag antibody (green). (C) Immunofluorescence of PDK2-Flag and PDK3-Flag (green) as in B, with Mitotracker as a mitochondria marker (red). (D) Immunofluorescence of PDK2-Flag and PDK3-Flag (green) in LNCaP cells, with Mitotracker as a mitochondria marker (red). In (B–D) nuclei were stained with DAPI (blue). Note the punctuated and mitochondrial localization of PDK2 and PDK3, as compared to the cytoplasmic PTEN localization.
Discussion
PCa cells show a unique metabolic reprogramming process during their progression towards malignancy, in which signaling through AR plays an essential role. Primary PCa tumor cells display unusual high oxidative respiration levels, which switch in CRPC cells to high aerobic glycolysis upon androgen-independent AR signaling (44). PDH enzymatic activity is a major universal driver of the energy metabolism in cells, coordinating the energy flux through the glycolytic and the mitochondrial TCA-oxidative pathways. Accordingly, PDH complex plays an important role in cancer-associated metabolic reprograming (27). Here, we have analyzed by IHC the expression of PDH components in PCa tumor samples. We have found a positive correlation of AR expression with PDHA1, PDP1, PDK1, PDK2, and PDK4 expression, which sustains the involvement of AR signaling in the control of PDH activity in PCa cells. In this regard, PDHA1 and PDK2 have been reported in a meta-analysis study as common androgen-regulated genes (45). In concordance, PDH/PDHA1 protein and its activator phosphatase PDP1 have been found to be overexpressed in PCa, in association with high Gleason score (36, 46), although low PDHA1 protein expression in PCa tumors has also been associated with poor prognosis (35). In our study, we found significant correlation of PDP1 expression, but not PDHA1, with stage and extracapsular extension. Prostate conditional Pten-null mice, knocked-out for PDHA1 expression in the prostate, displayed growth inhibition of prostate cells, and pharmacological inhibition of PDH activity in prostate Pten-null mice and in human PCa cells caused tumor and cell growth inhibition (36). Similarly, diminished cell growth was observed in PDHA1 knock-out LNCaP PCa cells (35, 47). Overall, these findings suggest a potential therapeutic benefit of PDH inhibition in advanced PCa tumors.
PDKs are physiologic negative regulators of PDH. In a variety of cancer types, PDK1-3 have been proposed to play oncogenic roles, whereas PDK4 has been proposed to play both oncogenic and tumor suppressive functions depending on the tumor type (33). In PCa, PDK1 has been found to be upregulated in correlation with disease progression, and PDK1 knock-down using siRNAs increased PCa cell migration and invasion, without significantly affecting cell proliferation (48). On the other hand, low PDK4 expression has been associated with biochemical recurrence in PCa datasets (49). PDK4 mRNA, followed by PDK2, are the more abundant PDK mRNAs detected in prostate and PCa (Supplementary Figure 1), and our IHC analysis revealed expression of all PDK proteins in PCa tumors. Notably, we detected correlation of PDK2 high expression with higher biochemical recurrence and lower disease-free survival, suggesting a pro-oncogenic role for PDK2 in PCa. This is in line with the proposed oncogenicity of PDK2 overexpression in other cancer types (50, 51). The tumor suppressor p53 negatively regulates PDK2 transcription (52), making of interest the analysis of the participation of p53 in the regulation of PDK2 expression in PCa cells. PDK2 showed a marked nuclear localization in PCa tumors, but not in PCa cell lines, which displayed PDK2 mitochondrial localization. Additional experiments are necessary to uncover the functional activities of nuclear PDK2 in PCa tissue.
The inhibition of PDKs by DCA, alone or in combination with other drugs, has been proposed as an alternative therapeutic anti-cancer approach, especially in chemoresistant tumors (34, 53–56). In our study, treatment of LNCaP and DU-145 PCa cells with DCA resulted in diminished cell proliferation, suggesting the feasibility of DCA, or DCA-related drugs, in the treatment of PCa. However, the clinical use of DCA in cancer therapy is limited, mainly due to undesired side effects, including peripheral neurotoxicity (32, 57). Kailavasan et al. reported metabolite ratios alterations in highly metastatic LNCaP-LN3 cells upon DCA treatment, which were not detected in poorly metastatic LNCaP cells (28). It has also been reported the sensitization to radiation of PCa cells by DCA (58), as well as PDK isozyme-specific effects of DCA on PCa cells (34). Interestingly, early studies on PDKs enzymatic activity revealed PDK2 as the PDK more efficiently inhibited by DCA (59). Whether DCA selectively targets PDK2 in PCa cells needs to be tested. It cannot be ruled out a DCA antiproliferative effect in PCa cells mediated by other PDKs. Dedicated studies are required to ascertain the involvement of inhibition of specific PDKs in the sensitivity to current anti-PCa therapies.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Comité Ético de Investigación Clínica, Hospital Universitario Cruces, Barakaldo, Spain. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
CN-X, JL, and RP contributed to conception and design of the study. CN-X, JM, ME, KF-K, JL, and RP performed experiments, collected and analyzed data. CN-X and KF-K performed the statistical analysis. RL and JL provided tumor samples, data acquisition and clinical details of patients. CN-X and RP wrote the first draft of the manuscript. CN-X, JM, KF-K, GM, ØF, RL, JL, and RP contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was funded by Instituto de Salud Carlos III (ISCIII; Spain and The European Social Fund+ ( "Investing in your future"; Grant number CP20/00008), and The Research Council of Norway (Grant number 239813) and Marie Skłodowska‐Curie Actions, UNIFOR‐FRIMED Legacy (2020, Norway) to CN-X; and by Ministerio de Economía y Competitividad (Spain and The European Regional Development Fund; Grant number SAF2016‐79847‐R) to RP and JL. CN-X is the recipient of a Miguel Servet Research Contract from ISCIII; Grant number CP20/00008).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Håkon Ramberg and Kristin A. Taskén (Oslo University Hospital) for providing LNCaP cells, and Arantza Perez Dobaran (University of the Basque Country, UPV/EHU, Leioa, Bizkaia, Spain) and Javier Diez García (Microscope Core facility at the Biocruces Bizkaia Health Research Institute) for expert technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2022.873516/full#supplementary-material
References
1. Knudsen BS, Vasioukhin V. Mechanisms of Prostate Cancer Initiation and Progression. Adv Cancer Res (2010) 109:1–50. doi: 10.1016/B978-0-12-380890-5.00001-6
2. Schrecengost R, Knudsen KE. Molecular Pathogenesis and Progression of Prostate Cancer. Semin Oncol (2013) 40:244–58. doi: 10.1053/j.seminoncol.2013.04.001
3. Attard G, Parker C, Eeles RA, Schroder F, Tomlins SA, Tannock I, et al. Prostate Cancer. Lancet (2016) 387:70–82. doi: 10.1016/S0140-6736(14)61947-4
4. Bluemn EG, Nelson PS. The Androgen/Androgen Receptor Axis in Prostate Cancer. Curr Opin Oncol (2012) 24:251–7. doi: 10.1097/CCO.0b013e32835105b3
5. Ramalingam S, Ramamurthy VP, Njar VCO. Dissecting Major Signaling Pathways in Prostate Cancer Development and Progression: Mechanisms and Novel Therapeutic Targets. J Steroid Biochem Mol Biol (2017) 166:16–27. doi: 10.1016/j.jsbmb.2016.07.006
6. Nunes-Xavier CE, Mingo J, Lopez JI, Pulido R. The Role of Protein Tyrosine Phosphatases in Prostate Cancer Biology. Biochim Biophys Acta Mol Cell Res (2019) 1866:102–13. doi: 10.1016/j.bbamcr.2018.06.016
7. Formaggio N, Rubin MA, Theurillat JP. Loss and Revival of Androgen Receptor Signaling in Advanced Prostate Cancer. Oncogene (2021) 40:1205–16. doi: 10.1038/s41388-020-01598-0
8. Baca SC, Garraway LA. The Genomic Landscape of Prostate Cancer. Front Endocrinol (Lausanne) (2012) 3:69. doi: 10.3389/fendo.2012.00069
9. Barbieri CE, Bangma CH, Bjartell A, Catto JW, Culig Z, Gronberg H, et al. The Mutational Landscape of Prostate Cancer. Eur Urol (2013) 64:567–76. doi: 10.1016/j.eururo.2013.05.029
10. Spans L, Clinckemalie L, Helsen C, Vanderschueren D, Boonen S, Lerut E, et al. The Genomic Landscape of Prostate Cancer. Int J Mol Sci (2013) 14:10822–51. doi: 10.3390/ijms140610822
11. Mitchell T, Neal DE. The Genomic Evolution of Human Prostate Cancer. Br J Cancer (2015) 113:193–8. doi: 10.1038/bjc.2015.234
12. Spratt DE, Zumsteg ZS, Feng FY, Tomlins SA. Translational and Clinical Implications of the Genetic Landscape of Prostate Cancer. Nat Rev Clin Oncol (2016) 13:597–610. doi: 10.1038/nrclinonc.2016.76
13. Crawford ED, Higano CS, Shore ND, Hussain M, Petrylak DP. Treating Patients With Metastatic Castration Resistant Prostate Cancer: A Comprehensive Review of Available Therapies. J Urol (2015) 194:1537–47. doi: 10.1016/j.juro.2015.06.106
14. Kelly RS, Vander Heiden MG, Giovannucci E, Mucci LA. Metabolomic Biomarkers of Prostate Cancer: Prediction, Diagnosis, Progression, Prognosis, and Recurrence. Cancer Epidemiol Biomark Prev (2016) 25:887–906. doi: 10.1158/1055-9965.EPI-15-1223
15. Baciarello G, Gizzi M, Fizazi K. Advancing Therapies in Metastatic Castration-Resistant Prostate Cancer. Expert Opin Pharmacother (2018) 19:1797–804. doi: 10.1080/14656566.2018.1527312
16. Lima AR, Pinto J, Bastos ML, Carvalho M, Guedes De Pinho P. NMR-Based Metabolomics Studies of Human Prostate Cancer Tissue. Metabolomics (2018) 14:88. doi: 10.1007/s11306-018-1384-2
17. Meehan J, Gray M, Martinez-Perez C, Kay C, Mclaren D, Turnbull AK. Tissue- and Liquid-Based Biomarkers in Prostate Cancer Precision Medicine. J Pers Med (2021) 11:664. doi: 10.3390/jpm11070664
18. Flavin R, Zadra G, Loda M. Metabolic Alterations and Targeted Therapies in Prostate Cancer. J Pathol (2011) 223:283–94. doi: 10.1002/path.2809
19. Giunchi F, Fiorentino M, Loda M. The Metabolic Landscape of Prostate Cancer. Eur Urol Oncol (2019) 2:28–36. doi: 10.1016/j.euo.2018.06.010
20. Ngo DC, Ververis K, Tortorella SM, Karagiannis TC. Introduction to the Molecular Basis of Cancer Metabolism and the Warburg Effect. Mol Biol Rep (2015) 42:819–23. doi: 10.1007/s11033-015-3857-y
21. Ciccarese C, Santoni M, Massari F, Modena A, Piva F, Conti A, et al. Metabolic Alterations in Renal and Prostate Cancer. Curr Drug Metab (2016) 17:150–5. doi: 10.2174/1389200216666151015112356
22. Zadra G, Loda M. Metabolic Vulnerabilities of Prostate Cancer: Diagnostic and Therapeutic Opportunities. Cold Spring Harb Perspect Med (2018) 8:a030569. doi: 10.1101/cshperspect.a030569
23. Guo W, Zhang Z, Li G, Lai X, Gu R, Xu W, et al. Pyruvate Kinase M2 Promotes Prostate Cancer Metastasis Through Regulating ERK1/2-COX-2 Signaling. Front Oncol (2020) 10:544288. doi: 10.3389/fonc.2020.544288
24. Stacpoole PW. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J Natl Cancer Inst (2017) 109:djx071. doi: 10.1093/jnci/djx071
25. Feng Y, Xiong Y, Qiao T, Li X, Jia L, Han Y. Lactate Dehydrogenase A: A Key Player in Carcinogenesis and Potential Target in Cancer Therapy. Cancer Med (2018) 7:6124–36. doi: 10.1002/cam4.1820
26. Sradhanjali S, Reddy MM. Inhibition of Pyruvate Dehydrogenase Kinase as a Therapeutic Strategy Against Cancer. Curr Top Med Chem (2018) 18:444–53. doi: 10.2174/1568026618666180523105756
27. Woolbright BL, Rajendran G, Harris RA, Taylor JA 3rd. Metabolic Flexibility in Cancer: Targeting the Pyruvate Dehydrogenase Kinase: Pyruvate Dehydrogenase Axis. Mol Cancer Ther (2019) 18:1673–81. doi: 10.1158/1535-7163.MCT-19-0079
28. Kailavasan M, Rehman I, Reynolds S, Bucur A, Tozer G, Paley M. NMR-Based Evaluation of the Metabolic Profile and Response to Dichloroacetate of Human Prostate Cancer Cells. NMR BioMed (2014) 27:610–6. doi: 10.1002/nbm.3101
29. Kankotia S, Stacpoole PW. Dichloroacetate and Cancer: New Home for an Orphan Drug? Biochim Biophys Acta (2014) 1846:617–29. doi: 10.1016/j.bbcan.2014.08.005
30. Harting T, Stubbendorff M, Willenbrock S, Wagner S, Schadzek P, Ngezahayo A, et al. The Effect of Dichloroacetate in Canine Prostate Adenocarcinomas and Transitional Cell Carcinomas In Vitro. Int J Oncol (2016) 49:2341–50. doi: 10.3892/ijo.2016.3720
31. Kinnaird A, Dromparis P, Saleme B, Gurtu V, Watson K, Paulin R, et al. Metabolic Modulation of Clear-Cell Renal Cell Carcinoma With Dichloroacetate, an Inhibitor of Pyruvate Dehydrogenase Kinase. Eur Urol (2016) 69:734–44. doi: 10.1016/j.eururo.2015.09.014
32. Tataranni T, Piccoli C. Dichloroacetate (DCA) and Cancer: An Overview Towards Clinical Applications. Oxid Med Cell Longev (2019) 2019:8201079. doi: 10.1155/2019/8201079
33. Atas E, Oberhuber M, Kenner L. The Implications of PDK1-4 on Tumor Energy Metabolism, Aggressiveness and Therapy Resistance. Front Oncol (2020) 10:583217. doi: 10.3389/fonc.2020.583217
34. Skorja Milic N, Dolinar K, Mis K, Matkovic U, Bizjak M, Pavlin M, et al. Suppression of Pyruvate Dehydrogenase Kinase by Dichloroacetate in Cancer and Skeletal Muscle Cells Is Isoform Specific and Partially Independent of HIF-1alpha. Int J Mol Sci (2021) 22:8610. doi: 10.3390/ijms22168610
35. Zhong Y, Li X, Ji Y, Li X, Li Y, Yu D, et al. Pyruvate Dehydrogenase Expression is Negatively Associated With Cell Stemness and Worse Clinical Outcome in Prostate Cancers. Oncotarget (2017) 8:13344–56. doi: 10.18632/oncotarget.14527
36. Chen J, Guccini I, Di Mitri D, Brina D, Revandkar A, Sarti M, et al. Compartmentalized Activities of the Pyruvate Dehydrogenase Complex Sustain Lipogenesis in Prostate Cancer. Nat Genet (2018) 50:219–28. doi: 10.1038/s41588-017-0026-3
37. Torres J, Navarro S, Rogla I, Ripoll F, Lluch A, Garcia-Conde J, et al. Heterogeneous Lack of Expression of the Tumour Suppressor PTEN Protein in Human Neoplastic Tissues. Eur J Cancer (2001) 37:114–21. doi: 10.1016/S0959-8049(00)00366-X
38. Mingo J, Luna S, Gaafar A, Nunes-Xavier CE, Torices L, Mosteiro L, et al. Precise Definition of PTEN C-Terminal Epitopes and Its Implications in Clinical Oncology. NPJ Precis Oncol (2019) 3:11. doi: 10.1038/s41698-019-0083-4
39. Nunes-Xavier CE, Aurtenetxe O, Zaldumbide L, Lopez-Almaraz R, Erramuzpe A, Cortes JM, et al. Protein Tyrosine Phosphatase PTPN1 Modulates Cell Growth and Associates With Poor Outcome in Human Neuroblastoma. Diagn Pathol (2019) 14:134. doi: 10.1186/s13000-019-0919-9
40. Flem-Karlsen K, Tekle C, Oyjord T, Florenes VA, Maelandsmo GM, Fodstad O, et al. P38 MAPK Activation Through B7-H3-Mediated DUSP10 Repression Promotes Chemoresistance. Sci Rep (2019) 9:5839. doi: 10.1038/s41598-019-42303-w
41. Nunes-Xavier CE, Kildal W, Kleppe A, Danielsen HE, Waehre H, Llarena R, et al. Immune Checkpoint B7-H3 Protein Expression Is Associated With Poor Outcome and Androgen Receptor Status in Prostate Cancer. Prostate (2021) 81:838–48. doi: 10.1002/pros.24180
42. Cooperberg MR, Hilton JF, Carroll PR. The CAPRA-S Score: A Straightforward Tool for Improved Prediction of Outcomes After Radical Prostatectomy. Cancer (2011) 117:5039–46. doi: 10.1002/cncr.26169
43. Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, et al. A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell (2014) 158:84–97. doi: 10.1016/j.cell.2014.04.046
44. Uo T, Sprenger CC, Plymate SR. Androgen Receptor Signaling and Metabolic and Cellular Plasticity During Progression to Castration Resistant Prostate Cancer. Front Oncol (2020) 10:580617. doi: 10.3389/fonc.2020.580617
45. Jin HJ, Kim J, Yu J. Androgen Receptor Genomic Regulation. Transl Androl Urol (2013) 2:157–77. doi: 10.3978/j.issn.2223-4683.2013.09.01
46. Lexander H, Palmberg C, Auer G, Hellstrom M, Franzen B, Jornvall H, et al. Proteomic Analysis of Protein Expression in Prostate Cancer. Anal Quant Cytol Histol (2005) 27:263–72.
47. Li Y, Li X, Li X, Zhong Y, Ji Y, Yu D, et al. PDHA1 Gene Knockout in Prostate Cancer Cells Results in Metabolic Reprogramming Towards Greater Glutamine Dependence. Oncotarget (2016) 7:53837–52. doi: 10.18632/oncotarget.10782
48. Subramaniam S, Jeet V, Gunter JH, Clements JA, Batra J. Allele-Specific MicroRNA-Mediated Regulation of a Glycolysis Gatekeeper PDK1 in Cancer Metabolism. Cancers (Basel) (2021) 13:3582. doi: 10.3390/cancers13143582
49. Oberhuber M, Pecoraro M, Rusz M, Oberhuber G, Wieselberg M, Haslinger P, et al. STAT3-Dependent Analysis Reveals PDK4 as Independent Predictor of Recurrence in Prostate Cancer. Mol Syst Biol (2020) 16:e9247. doi: 10.15252/msb.20199247
50. Cui L, Cheng Z, Liu Y, Dai Y, Pang Y, Jiao Y, et al. Overexpression of PDK2 and PDK3 Reflects Poor Prognosis in Acute Myeloid Leukemia. Cancer Gene Ther (2020) 27:15–21. doi: 10.1038/s41417-018-0071-9
51. Kitamura S, Yamaguchi K, Murakami R, Furutake Y, Higasa K, Abiko K, et al. PDK2 Leads to Cisplatin Resistance Through Suppression of Mitochondrial Function in Ovarian Clear Cell Carcinoma. Cancer Sci (2021) 112:4627–40. doi: 10.1111/cas.15125
52. Contractor T, Harris CR. P53 Negatively Regulates Transcription of the Pyruvate Dehydrogenase Kinase Pdk2. Cancer Res (2012) 72:560–7. doi: 10.1158/0008-5472.CAN-11-1215
53. Ferriero R, Iannuzzi C, Manco G, Brunetti-Pierri N. Differential Inhibition of PDKs by Phenylbutyrate and Enhancement of Pyruvate Dehydrogenase Complex Activity by Combination With Dichloroacetate. J Inherit Metab Dis (2015) 38:895–904. doi: 10.1007/s10545-014-9808-2
54. Roh JL, Park JY, Kim EH, Jang HJ, Kwon M. Activation of Mitochondrial Oxidation by PDK2 Inhibition Reverses Cisplatin Resistance in Head and Neck Cancer. Cancer Lett (2016) 371:20–9. doi: 10.1016/j.canlet.2015.11.023
55. Sun H, Zhu A, Zhou X, Wang F. Suppression of Pyruvate Dehydrogenase Kinase-2 Re-Sensitizes Paclitaxel-Resistant Human Lung Cancer Cells to Paclitaxel. Oncotarget (2017) 8:52642–50. doi: 10.18632/oncotarget.16991
56. Klose K, Packeiser EM, Muller P, Granados-Soler JL, Schille JT, Goericke-Pesch S, et al. Metformin and Sodium Dichloroacetate Effects on Proliferation, Apoptosis, and Metabolic Activity Tested Alone and in Combination in a Canine Prostate and a Bladder Cancer Cell Line. PloS One (2021) 16:e0257403. doi: 10.1371/journal.pone.0257403
57. Stacpoole PW, Martyniuk CJ, James MO, Calcutt NA. Dichloroacetate-Induced Peripheral Neuropathy. Int Rev Neurobiol (2019) 145:211–38. doi: 10.1016/bs.irn.2019.05.003
58. Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloroacetate (DCA) Sensitizes Both Wild-Type and Over Expressing Bcl-2 Prostate Cancer Cells In Vitro to Radiation. Prostate (2008) 68:1223–31. doi: 10.1002/pros.20788
Keywords: prostate cancer (PCa), pyruvate dehydrogenase (PDH), pyruvate dehydrogenase kinase (PDK), androgen receptor (AR), dichloroacetate (DCA)
Citation: Nunes-Xavier CE, Mingo J, Emaldi M, Flem-Karlsen K, Mælandsmo GM, Fodstad Ø, Llarena R, López JI and Pulido R (2022) Heterogeneous Expression and Subcellular Localization of Pyruvate Dehydrogenase Complex in Prostate Cancer. Front. Oncol. 12:873516. doi: 10.3389/fonc.2022.873516
Received: 10 February 2022; Accepted: 31 March 2022;
Published: 25 May 2022.
Edited by:
Alagarsamy Srinivasan, NanoBio Diagnostics, United StatesReviewed by:
Zhenbang Chen, Meharry Medical College, United StatesHumberto De Vitto, University of Minnesota Twin Cities, United States
Copyright © 2022 Nunes-Xavier, Mingo, Emaldi, Flem-Karlsen, Mælandsmo, Fodstad, Llarena, López and Pulido. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline E. Nunes-Xavier, Y2Fyb2xpbmVudW5lc3hhdmllckBnbWFpbC5jb20=