 Antonella Verrienti1†
Antonella Verrienti1† Giorgio Grani1*†
Giorgio Grani1*† Marialuisa Sponziello1
Marialuisa Sponziello1 Valeria Pecce1
Valeria Pecce1 Giuseppe Damante2
Giuseppe Damante2 Cosimo Durante1
Cosimo Durante1 Diego Russo3
Diego Russo3 Sebastiano Filetti4
Sebastiano Filetti4- 1Department of Translational and Precision Medicine, Sapienza University of Rome, Rome, Italy
- 2Department of Medicine, University of Udine, Udine, Italy
- 3Department of Health Sciences, University “Magna Graecia” of Catanzaro, Catanzaro, Italy
- 4School of Health, UNITELMA Sapienza University of Rome, Rome, Italy
Aberrant activation of the RET proto-oncogene is implicated in a plethora of cancers. RET gain-of-function point mutations are driver events in multiple endocrine neoplasia 2 (MEN2) syndrome and in sporadic medullary thyroid cancer, while RET rearrangements are driver events in several non-medullary thyroid cancers. Drugs able to inhibit RET have been used to treat RET-mutated cancers. Multikinase inhibitors were initially used, though they showed modest efficacy and significant toxicity. However, new RET selective inhibitors, such as selpercatinib and pralsetinib, have recently been tested and have shown good efficacy and tolerability, even if no direct comparison is yet available between multikinase and selective inhibitors. The advent of high-throughput technology has identified cancers with rare RET alterations beyond point mutations and fusions, including RET deletions, raising questions about whether these alterations have a functional effect and can be targeted by RET inhibitors. In this mini review, we focus on tumors with RET deletions, including deletions/insertions (indels), and their response to RET inhibitors.
Introduction
The RET proto-oncogene encodes for a transmembrane glycoprotein receptor with tyrosine kinase activity. It is involved in several cell processes during embryogenesis, including proliferation, differentiation, motility, and survival (1). RET gene mutations and fusions are known to be gain-of-function driver events in many cancer types (Figure 1).
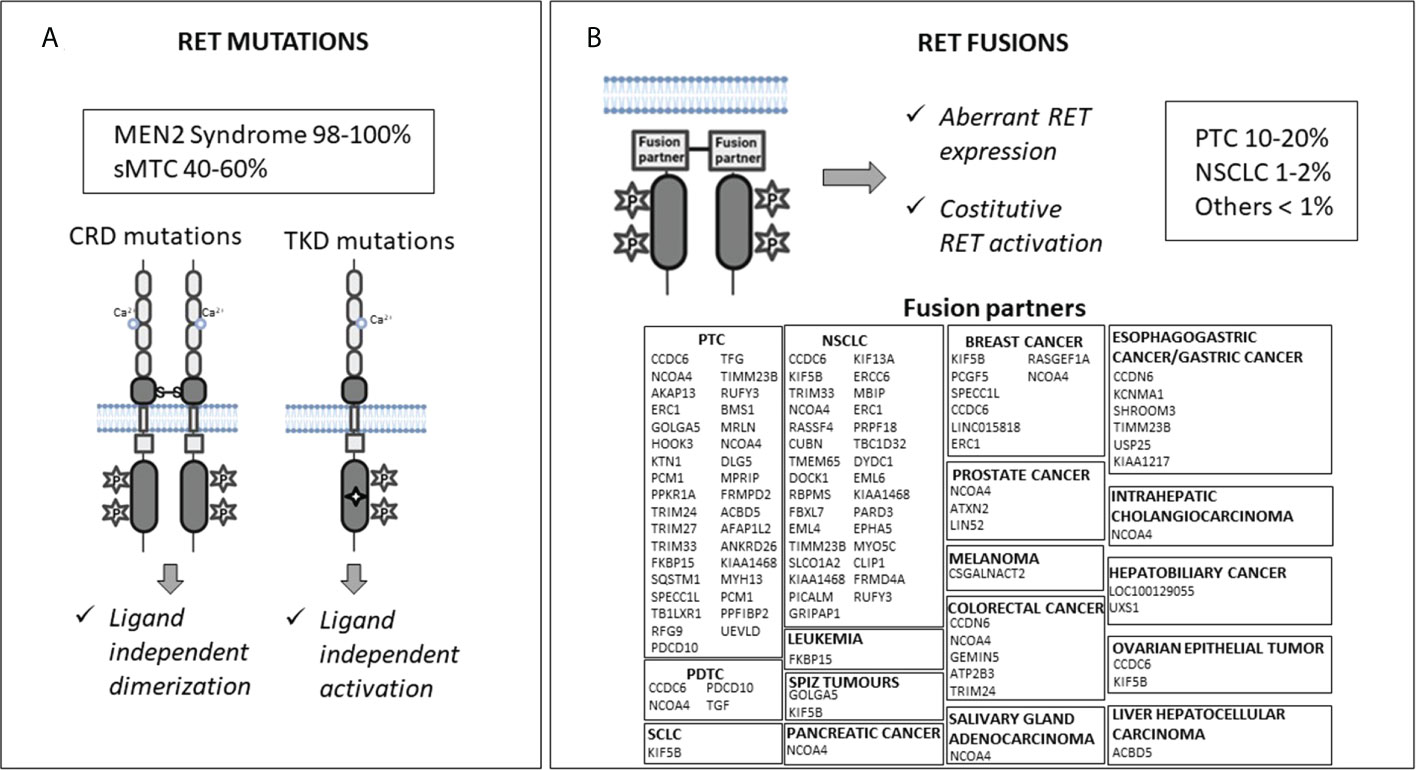
Figure 1 Molecular mechanisms of RET activation: mutations (Panel A) and fusions (Panel B). CRD, cysteine-rich domain; TKD, tyrosine kinase domain; sMTC, sporadic medullary thyroid cancer; PTC, papillary thyroid cancer; NSCLC, non-small cell lung cancer; PDTC, poorly differentiated thyroid cancer; SCLC, small cell lung cancer.
RET germline gain-of-function mutations cause predisposition to multiple endocrine neoplasia 2 (MEN2) syndrome, while somatic RET mutations have been found in 40-65% of sporadic medullary thyroid cancers (MTCs) (2–5). RET mutations carry out their oncogenic effect through two mechanisms. Mutations located in the extracellular cysteine-rich domain can lead to RET receptor constitutive dimerization and activation regardless of the presence of ligands, while mutations in the tyrosine kinase domains can cause a conformational change in the intracellular tyrosine kinase binding pocket, which allows constitutive kinase activation and altered substrate binding (1, 6–9) (Figure 1A).
Sanger sequencing is preferentially used for the detection of germline mutations in MEN2 syndrome; however, in evaluating the presence of somatic mutations, it may generate false negative results since it is not able to detect mutations below 15-20% of allele frequency. Alternatively, quantitative polymerase chain reaction (qPCR) or digital PCR can be used for the screening of somatic hotspot mutations, reaching a limit of detection of around 1% of allelic frequency or, in the case of digital PCR, less than 1% (10). The advent of high-throughput technologies, such as next-generation sequencing (NGS), has allowed hundreds to thousands of genes to be simultaneously analyzed, thus increasing the detection of novel or rare variants with a high sensitivity.
RET rearrangements occur in 10–20% of papillary thyroid carcinomas (PTCs), 1–2% of non-small cell lung cancers (NSCLCs), and in <1% of other cancers (e.g., colorectal cancer, breast cancer, chronic myelomonocytic leukemia, ovarian and salivary gland cancers, etc.) (11). RET rearrangements produce chimeras formed by the juxtaposition of an N-terminal partner to the RET C-terminal portion, including its catalytic domain. This leads to aberrant RET overexpression, ligand-independent dimerization, and kinase activation (Figure 1B). Notably, the RET fusion partner may influence the oncogenic potential of the chimeric RET protein affecting the intracellular location of the RET kinase (consequently the activated signaling pathways), and its expression levels (12). In addition, the altered function of the fusion partner may also have a role in the neoplastic transformation (13, 14).
Fluorescent in situ hybridization (FISH) is considered the standard used for the detection of RET rearrangements and has good sensitivity and specificity (10, 15). However, it may not be adequately informative regarding the specific RET fusion unless a specific fusion partner probe can be used (10, 15). Moreover, FISH results are difficult to interpret in many circumstances, such as in presence of pericentric fusions, deletions, and when possible partner genes are in close proximity to the RET gene (10). Reverse transcription-polymerase chain reaction (RT-PCR) can identify specific known RET fusion partners, but since it uses preselected primers, it is not able to detect novel fusion partners. Thus, it could underestimate the presence of RET rearrangements. Since this methodology is not ideal for the degraded and poor RNA quality isolated from formalin-fixed paraffin-embedded tissues, it is generally used together with other methodologies, such as immunohistochemistry (IHC) and FISH (10, 15). IHC is currently not indicated for the screening of RET alterations due to the high false positive and negative rates (15). Although DNA-based NGS can be designed for gene fusion detection, it doesn’t achieve high sensitivity and an RNA-based NGS is preferred (10).
The comprehensive genetic profiling of tumors made possible by novel detection technologies has resulted in the identification of multiple cancers with rare RET alterations beyond point mutations and fusions (16, 17), including a nonnegligible number of RET deletions. In this review, RET deletions also include deletions/insertions (indels).
Ret deletions in cancers
RET deletions are not frequently found in MTC. They have been reported in 5% of all RET-mutated sporadic MTCs, as reported in the Catalogue of Somatic Mutations in Cancer (COSMIC database: https://cancer.sanger.ac.uk/cosmic, accessed June 2022), and they represent around 3.5% of all germlines RET alterations found in MEN2 patients, as reported in the ARUP database (https://arup.utah.edu/database/MEN2/MEN2_display.php accessed June 2022). In non-MTC cancers, their frequency is very low, ranging from 0.03% (COSMIC database) to 0.2% (cBioPortal for Cancer Genomics public databases; https://www.cbioportal.org/, accessed June 2022).
Ret in-frame deletions
Overall, 37 RET deletions have been described in MTC patients and almost all of them (36/37, 97%) are in-frame. Seven are germline deletions and are mainly located in the cysteine-rich domain, at exons 11 and 10 (18–24), and two are in the cadherin-like coding regions, at exons 6 and 7 (25, 26). Most RET deletions have been found in the tumor tissue of sporadic MTCs, mainly at exons 11 and 15 (3, 4, 25, 27–49), and to a lesser extent at exons 10 and 8 (30, 45, 49–51). Although only a few deletions have been reported in non-”hotspot” exons (i.e., exons 6 and 7), we cannot exclude that their frequency may be higher since those exons are not routinely studied. The prognostic role of the RET deletions (including indels) has not been clearly proved due to the few available data. However, in a recent paper, Elisei R. et al. observed that MTC harboring RET indels, show a more aggressive phenotype with a high prevalence of advanced cases at diagnosis (45).
RET in-frame deletions have also been described in other cancer types, as reported in the COSMIC and cBioPortal databases (52, 53). Fifteen in-frame RET deletions have been found in 30 oncologic patients. Interestingly, 6/30 patients (20%) are affected by pheochromocytoma (PHEO) and carry deletions in common with MTCs, mapping at RET exons 11 and 15. This is not surprising since both MTC and PHEO can be induced by activating RET alterations. The remaining in-frame RET deletions have been observed in breast, large intestine, gastric, pancreatic, kidney, and lung cancers.
Ret frameshift deletions
Only one RET frameshift deletion, p. Gln681Argfs*50, has been reported in an MTC patient. However, it was found in copresence with the RET p.A680T point mutation and its functional effect has not yet been demonstrated (36). Conversely, a greater number of RET frameshift deletions has been described in other cancers, as reported in COSMIC and cBioPortal public databases (52, 53). These deletions are spread out along the gene, including the hotspot exons.
Frameshift deletions are commonly loss-of-function alterations since they result in a shift of the reading frame used for protein translation, leading to a completely different sequence of the polypeptide. They often introduce an early stop codon resulting in a truncated protein. However, the major mechanism explaining the loss of function is nonsense-mediated mRNA decay, by which mutated mRNA is degraded (54).
It has yet to be proven whether RET in-frame and frameshift deletions in non-MTC and non-PHEO cancers are pathogenic.
Ret-targeted therapies
The identification of key driver oncogenes as targetable activated kinases has allowed clinicians to explore new treatment options. Therefore, multikinase inhibitors (MKIs) that target multiple tyrosine kinase receptors, including RET and those involved in angiogenesis, such as VEGFRs and PDGFRs, were initially used to treat advanced RET-mutated MTC and subsequently other RET-altered cancers (55, 56). Given their multi-target inhibition, it is not clear whether their observed antitumor activity is due to RET inhibition or the inhibition of other kinase targets (57, 58) (Table 1).
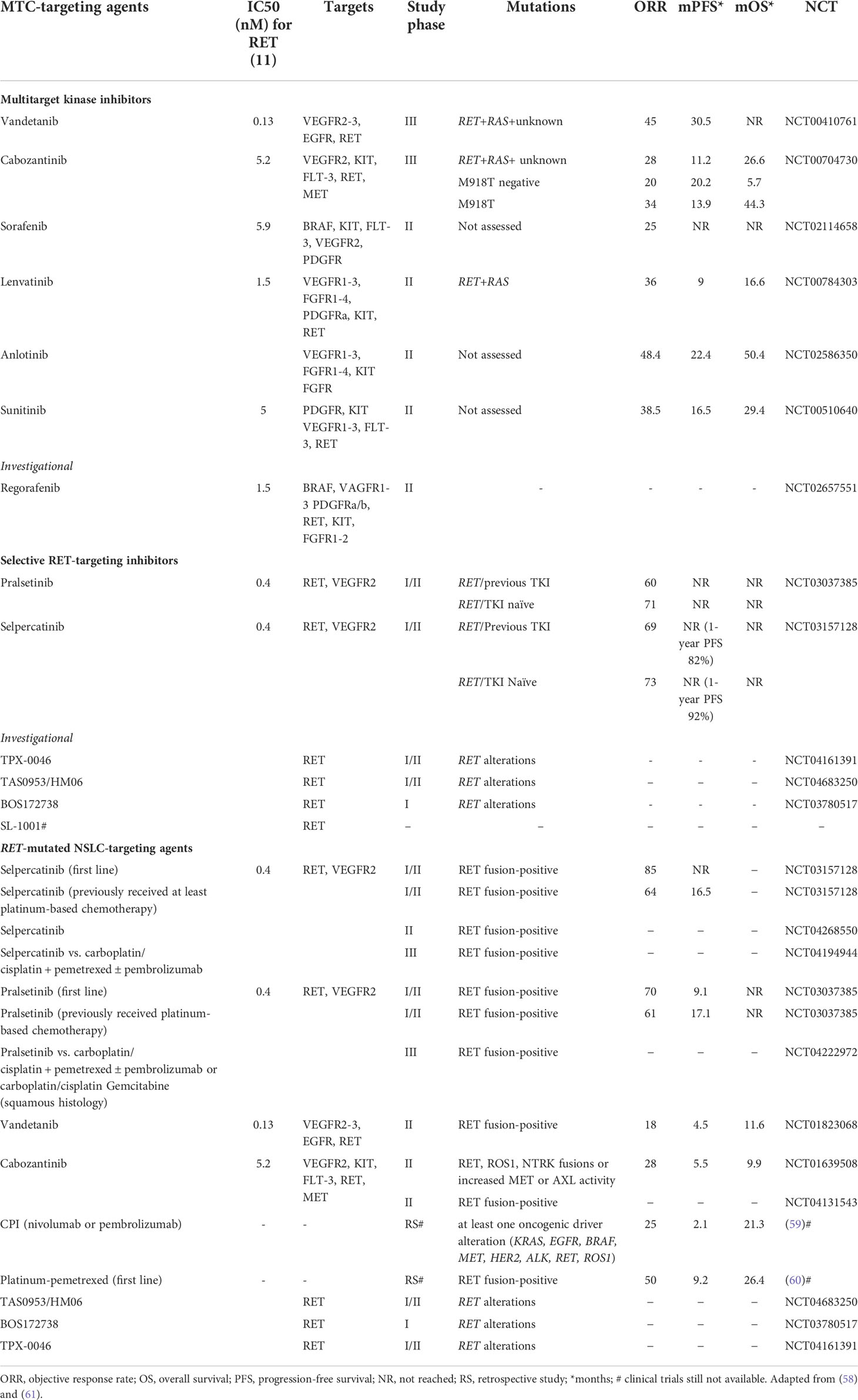
Table 1 Drugs targeting medullary thyroid cancers and RET-mutated NSCLC with relevant clinical trial data.
Cabozantinib and vandetanib have been approved for first-line treatment in MTC regardless of RET mutational status, even if the presence of RET mutations, particularly the RET p.M918T mutation, seems to be associated with a better response to cabozantinib in terms of overall response rate and progression-free survival (62, 63). Similarly, M918T mutation-positive patients also showed a higher response to vandetanib (64). Vandetanib showed a higher median progression-free survival (mPFS) than placebo (30.5 vs 19.3 months) in the ZETA trial (64), as had cabozantinib in the EXAM trial (11.2 vs 4.0 months) (65). The clinical effectiveness of vandetanib and cabozantinib in advanced MTC patients was also confirmed from real-world data, showing a mPFS up to 47 months for vandetanib (66–68) and up to 4 months for cabozantinib (66). The median overall survival (OS) for vandetanib and cabozantinib was 53 months and 24 months, respectively, in the German real-world multicenter cohort (66).
MKI treatment of RET-rearranged NSCLC showed a modest clinical benefit that was lower than that observed with EGFR, ALK, and ROS1 inhibitors (61) (Table 1). Moreover, MKI response can differ depending on the fusion partner. For example, vandetanib showed a greater effect in CCDC6-RET fusion tumors compared with KIF5B-RET (57). However, the adverse effects of non-selective RET inhibitors observed in all treated tumors due to their off-target side effects are responsible for high discontinuation and dose reduction rates (e.g., 12% and 35% for vandetanib and 16% and 79% for cabozantinib when used as thyroid cancer treatments, respectively) (57).
In the last years, small and highly selective RET inhibitors have been designed to overcome the treatment-related toxicities of non-selective RET inhibitors and acquired resistance to them (57). The new selective RET inhibitors pralsetinib (LOXO-292) and selpercatinib (BLU-667) have demonstrated both good efficacy and tolerability: in phase I/II trials, the mPFS was not reached, and the overall response rate was 71% and 73% (first line treatment), and 60% and 69% (second line treatment), respectively (Table 1). Currently, both drugs have been approved by the Food and Drug Administration (FDA) for the treatment of patients more than 12 years of age with: i) advanced or metastatic RET-mutant MTC; ii) RET fusion-positive metastatic NSCLC, and iii) advanced or metastatic RET fusion-positive thyroid cancer patients who require systemic therapy and who are radioactive iodine refractory. These drugs also show robust activity in other RET alteration-positive solid tumors (69). Interestingly, both drugs seem to be effective regardless of previous MKI or immune checkpoint therapies (57). Selpercatinib is also reported to be effective on CNS metastases (70) and uncommon metastatic sites, such as choroidal metastases (71). Common side effects of selpercatinib include dry mouth, hypertension, fatigue, increased aspartate aminotransferase level (AST), increased alanine aminotransferase level (ALT), increased glucose levels, and hypocalcemia, while pralsetinib additionally caused pain, constipation, and hematological toxicities such as decreased lymphocytes, neutrophils, and hemoglobin. Interstitial pneumonia is also reported for pralsetinib. The discontinuation and dose reduction rates in phase I/II trials were 2% and 30% for selpercatinib (72), and 4% and 44% for pralsetinib (73).
Given the availability of these drugs, the screening for and detection of RET driver alterations is now crucial in clinical practice since it provides more targeted treatment options. Unlike multitarget inhibitors, pralsetinib and selpercatinib have a selective nanomolar potency against RET and a diverse set of RET fusions and mutations. Head-to-head studies directly comparing efficacy and safety of selective RET inhibitors with MKI are currently ongoing (NCT04760288, NCT04211337); results are not yet available.
Specific mutations and acquired resistance
Some specific mutations are expected to cause acquired resistance to MKI treatments (12). Preclinical studies have shown that acquired gatekeeper mutation V804L is associated with MKI resistance (12). The emergence of a V804M mutation was reported in a patient with RET-mutant, sporadic MTC treated previously with multiple MKIs (74). Emergent V804L and S904F mutations were reported in patients with RET fusion-positive NSCLC during treatment with vandetanib (75, 76). The frequency, prognostic role, and clinical actionability of these mutations are not entirely clear (75). Some preclinical models identified other resistance mutations, including the V804E, G810A/S/R, I788N, 730I, E732K, V871I, V738A, A807V, F998V and Y806N (13, 77, 78).
Selpercatinib has a specific binding modality: both front and back pockets of RET are occupied without being affected by V804 mutations (unlike other tyrosine kinase inhibitors) (11). Selpercatinib was developed to be effective in RETV804L and RETV804M gatekeeper mutations and was found to be 60–1300 fold more effective than multitarget inhibitors against cell lines engineered with KIF5B-RETV804L/M gatekeeper mutations (74).
Conversely, RET mutations at the C-lobe solvent front (RET p.G810C/S/R), hinge (RETY p.806C/N), and β2 strand (RET p.V738A, only identified in cell lines) cause acquired resistance to selpercatinib (61, 79–81). Structural modeling showed that selpercatinib binding to the kinase ATP/selpercatinib binding site can be hindered if the glycine residue at position 810 in the RET solvent front is substituted with charged or polar residues (79, 80). In vitro experiments using BaF3/KIF5B-RET cells showed that pralsetinib and selpercatinib bind to RET in a similar mode and both are resistant to the same mutations (80), although some mutations (i.e., L730V/I) seem to be resistant only to pralsetinib (82).
New selective inhibitors in clinical development
New selective RET inhibitors are under development (Table 1). TPX-0046 is a dual RET/SRC kinase inhibitor, with activity in drug-resistant and naïve RET-driven cancer models. It is in phase I/II clinical trials for advanced solid tumors harboring RET fusions or mutations (NCT04161391). TAS0953 (HM06) is undergoing a phase I/II study in patients with advanced solid tumors with RET gene abnormalities (NCT04683250). SYHA1815 has an approximately 20-fold selectivity for RET over VEGFR2 and is being studied in a phase I trial in China (83). Other potential drug compounds, such as LOX-18228, LOX-19260, BOS172738 (DS-5010), and SL-1001 (84–86), are still in the preclinical stage. There are also research efforts to obtain mutant-selective inhibitors that may offer clinical advantages.
Clinical response in patients with ret deletions
Despite efforts to develop super-selective inhibitors, data available on the response of cancers harboring RET deletions to selective RET inhibitors are scarce and concern only MTCs. A RET p.D378_G385delinsE MTC was treated with selpercatinib and achieved partial response, with a maximum tumor reduction of 86% (87). The treatment of two RET p.L629_D631delinsH MTCs, one with cabozantinib and the other with a combination of sorafenib and tipifarnib, showed a partial response, with a tumor reduction of 48% and 46%, respectively (43, 44). Two MTCs with RET p.E632_636del and p.L633_A639del were treated with vandetanib and cabozantinib, respectively, showing stable disease (25, 43). In one case, disease progression was observed after seven months of treatment (22). Recently, two MTC patients with the p.E632_L633del and p.D631_L633delinsS RET deletion, respectively, who were previously treated with cabozantinib and/or vandetanib, experienced a treatment benefit with selpercatinib, with a rapid biochemical response. In particular, the first patient showed a partial response in the target lesions and stable disease in non-target lesions, while the second patient showed stable disease and a partial response in target and non-target lesions, respectively (45). An in vitro study provided evidence that the p.C630del RET alteration is sensitive to pralsetinib (23). A RET p.D898_E901del MTC was treated with cabozantinib, showing stable disease (43). Lastly, Zhao et al. used mutant-transformed Ba/F3 cells to demonstrate that p.D898_E901del is sensitive to selpercatinib and pralsetinib (88).
Discussion
The advancement of sequencing technologies has allowed comprehensive genetic profiling of tumors and the identification of new RET alterations, including deletions. Although the reported tumors with RET deletions are few, we cannot exclude that their real prevalence may be higher. Indeed, in clinical practice, RET deletions are usually not investigated through the gene.
Data on RET deletions as driver alterations in cancer are still scarce. In MTC and PHEO, only RET in-frame deletions have been reported, supporting their possible gain-of-function role. For a few of them, their oncogenic potential has been demonstrated through in vitro experiments (21, 23, 26, 49, 88–90). Conversely, frameshift deletions have been observed in a wide range of tumor types, except MTC and PHEO, though their functional role as driver alterations has not yet been demonstrated.
To date, limited information about the response of tumors with RET deletions to RET inhibitors is available and only concerns MTC patients. In those patients, treatment efficacy seems to be comparable to MTCs with RET point mutations. Considering the potential benefit of treating tumors with RET inhibitors, it is crucial to understand the real impact of these deletions in cancer development and progression and their response to RET targeted therapies.
Author contributions
Conceptualization, SF and CD. Writing—original draft preparation AV and GG. Writing—review and editing, MS, VP, DR, and GD. Supervision, SF and CD. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
The manuscript was edited by Melissa Kerr.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. De Groot JWB, Links TP, Plukker JTM, Lips CJM, Hofstra RMW. RET as a diagnostic and therapeutic target in sporadic and hereditary endocrine tumors. Endocrine Rev (2006) 27(5):535–60. doi: 10.1210/er.2006-0017
2. Ciampi R, Mian C, Fugazzola L, Cosci B, Romei C, Barollo S, et al. Evidence of a low prevalence of ras mutations in a large medullary thyroid cancer series. Thyroid (2013) 23(1):50–7. doi: 10.1089/thy.2012.0207
3. Boichard A, Croux L, Al Ghuzlan A, Broutin S, Dupuy C, Leboulleux S, et al. Somatic RAS mutations occur in a large proportion of sporadic RET-negative medullary thyroid carcinomas and extend to a previously unidentified exon. J Clin Endocrinol Metab (2012) 97(10):E2031–5. doi: 10.1210/jc.2012-2092
4. Moura MM, Cavaco BM, Pinto AE, Domingues R, Santos JR, Cid MO, et al. Correlation of RET somatic mutations with clinicopathological features in sporadic medullary thyroid carcinomas. Br J Cancer (2009) 100(11):1777–83. doi: 10.1038/sj.bjc.6605056
5. Verrienti A, Tallini G, Colato C, Boichard A, Checquolo S, Pecce V, et al. RET mutation and increased angiogenesis in medullary thyroid carcinomas. Endocr Relat Cancer (2016) 23(8):665–76. doi: 10.1530/ERC-16-0132. doi: 10.1210/jcem.81.8.8768845
6. Romei C, Ciampi R, Elisei R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat Rev Endocrinol (2016) 12(4):192–202. doi: 10.1038/nrendo.2016.11
7. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer (2014) 14(3):173–86. doi: 10.1038/nrc3680
8. D’Aloiso L, Carlomagno F, Bisceglia M, Anaganti S, Ferretti E, Verrienti A, et al. In vivo and in vitro characterization of a novel germline RET mutation associated with low-penetrant nonaggressive familial medullary thyroid carcinoma. J Clin Endocrinol Metab (2006) 91(3):754–9. doi: 10.1210/jc.2005-2338
9. Verrienti A, Carbone A, Bellitti P, Fabiano MC, De Rose RF, Maranghi M, et al. A novel double mutation VAL648ILE and VAL804LEU of ret proto-oncogene in multiple endocrine neoplasia type 2. Endocr Pract (2015) 21(11):1248–54. doi: 10.4158/EP15838.OR
10. Belli C, Penault-Llorca F, Ladanyi M, Normanno N, Scoazec JY, Lacroix L, et al. ESMO recommendations on the standard methods to detect RET fusions and mutations in daily practice and clinical research. Ann Oncol (2021) 32(3):337–50. doi: 10.1016/j.annonc.2020.11.021
11. Saha D, Ryan KR, Lakkaniga NR, Acharya B, Garcia NG, Smith EL, et al. Targeting rearranged during transfection in cancer: a perspective on small-molecule inhibitors and their clinical development. J Medicinal Chem (2021) 64(16):11747–73. doi: 10.1021/acs.jmedchem.0c02167
12. Román-Gil MS, Pozas J, Rosero-Rodríguez D, Chamorro-Pérez J, Ruiz-Granados Á, Caracuel IR, et al. Resistance to RET targeted therapy in thyroid cancer: Molecular basis and overcoming strategies. Cancer Treat Rev (2022) 105:102372. doi: 10.1016/j.ctrv.2022.102372
13. Subbiah V, Yang D, Velcheti V, Drilon A, Meric-Bernstam F. State-of-the-art strategies for targeting RET-dependent cancers. J Clin Oncol (2020) 38(11):1209–21. doi: 10.1200/JCO.19.02551
14. Santoro M, Moccia M, Federico G, Carlomagno F. Ret gene fusions in malignancies of the thyroid and other tissues. Genes (2020) 11(4):424. doi: 10.3390/genes11040424
15. Ferrara R, Auger N, Auclin E, Besse B. Clinical and translational implications of ret rearrangements in non–small cell lung cancer. J Thorac Oncol (2018) 13(1):27–45. doi: 10.1016/j.jtho.2017.10.021
16. Sponziello M, Benvenuti S, Gentile A, Pecce V, Rosignolo F, Virzì AR, et al. Whole exome sequencing identifies a germline MET mutation in two siblings with hereditary wild-type RET medullary thyroid cancer. Hum Mutat (2018) 39(3):371–7. doi: 10.1002/humu.23378
17. Pecce V, Sponziello M, Damante G, Rosignolo F, Durante C, Lamartina L, et al. A synonymous RET substitution enhances the oncogenic effect of an in-cis missense mutation by increasing constitutive splicing efficiency. PLoS Genet (2018) 14(10):e1007678. doi: 10.1371/journal.pgen.1007678
18. Ahmed SA, Snow-Bailey K, Highsmith WE, Sun W, Fenwick RG, Mao R. Nine novel germline gene variants in the RET proto-oncogene indentified in twelve unrelated cases. J Mol Diagnostics (2005) 7(2):283–8. doi: 10.1016/S1525-1578(10)60556-9
19. Giani C, Ramone T, Romei C, Ciampi R, Tacito A, Valerio L, et al. A new MEN2 syndrome with clinical features of both men2a and men2b associated with a new ret germline deletion. Case Rep Endocrinol (2020) 2020:4147097. doi: 10.1155/2020/4147097
20. Vandenbosch K, Renard M, Uyttebroeck A, Sciot R, Matthijs G, Legius E. Medullary thyroid carcinoma in a child with a new RET mutation and a RET polymorphism. Genet Couns (2005) 16(1):95–100.
21. Cordella D, Muzza M, Alberti L, Colombo P, Travaglini P, Beck-Peccoz P, et al. An in-frame complex germline mutation in the juxtamembrane intracellular domain causing RET activation in familial medullary thyroid carcinoma. Endocr Relat Cancer (2006) 13(3):945–53. doi: 10.1677/erc.1.01144
22. Yao B, Liu X, Liang H, Dong TT, Huang ZM, Chen X, et al. A novel mutation (D631del) of the RET gene was associated with MEN2A in a Chinese pedigree. Endocr J (2009) 56(1):99–104. doi: 10.1507/endocrj.K08E-066
23. Ma X, Ma X, Chin L, Zhu Z, Han H. A novel germline deletion of p.C630 in RET causes MTC and promotes cell proliferation and sensitivity to pralsetinib. J Clin Endocrinol Metab (2022), dgac352. doi: 10.1210/clinem/dgac352Epub ahead of print
24. Mulligan LM, Kwok JBJ, Healey CS, Elsdon MJ, Eng C, Gardner E, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature (1993) 363(6428):458–60. doi: 10.1038/363458a0
25. Vanden Borre P, Schrock AB, Anderson PM, Morris JC, Heilmann AM, Holmes O, et al. Pediatric, adolescent, and young adult thyroid carcinoma harbors frequent and diverse targetable genomic alterations, including kinase fusions. Oncologist (2017) 22(3):255–63. doi: 10.1634/theoncologist.2016-0279
26. Latteyer S, Klein-Hitpass L, Khandanpour C, Zwanziger D, Poeppel TD, Schmid KW, et al. A 6-base pair in frame germline deletion in exon 7 of RET leads to increased RET phosphorylation, ERK activation, and MEN2A. J Clin Endocrinol Metab (2016) 101(3):1016–22. doi: 10.1210/jc.2015-2948
27. Dvorakova S, Vaclavikova E, Sykorova V, Vcelak J, Novak Z, Duskova J, et al. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinomas. Mol Cell Endocrinol (2008) 284(1-2):21–7. doi: 10.1016/j.mce.2007.12.016
28. Jhiang SM, Fithian L, Weghorst CM, Clark OH, Falko JM, O’Dorisio TM, et al. RET mutation screening in MEN2 patients and discovery of a novel mutation in a sporadic medullary thyroid carcinoma. Thyroid (1996) 6(2):115–21. doi: 10.1089/thy.1996.6.115
29. Heilmann AM, Subbiah V, Wang K, Sun JX, Elvin JA, Chmielecki J, et al. Comprehensive genomic profiling of clinically advanced medullary thyroid carcinoma. Oncol (2016) 90(6):339–46. doi: 10.1159/000445978
30. Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab (2013) 98(2):E364–9. doi: 10.1210/jc.2012-2703
31. Romei C, Casella F, Tacito A, Bottici V, Valerio L, Viola D, et al. New insights in the molecular signature of advanced medullary thyroid cancer: Evidence of a bad outcome of cases with double RET mutations. J Med Genet (2016) 53(11):729–34. doi: 10.1136/jmedgenet-2016-103833
32. Hofstra RM, Stelwagen T, Stulp RP, de Jong D, Hulsbeek M, Kamsteeg EJ, et al. Extensive mutation scanning of RET in sporadic medullary thyroid carcinoma and of RET and VHL in sporadic pheochromocytoma reveals involvement of these genes in only a minority of cases. J Clin Endocrinol Metab (1996) 81(8):2881–4.
33. Kimura T, Yokogoshi Y, Saito S, Yoshimoto K. Mutations in the cysteine-rich region of the ret proto-oncogene in patients diagnosed as having sporadic medullary thyroid carcinoma. Endocr J (1995) 42(4):517–25. doi: 10.1507/endocrj.42.517
34. Alemi M, Lucas SD, Sällström JF, Bergholm U, Åkerström G, Wilander E. A complex nine base pair deletion in RET exon 11 common in sporadic medullary thyroid carcinoma. Oncogene (1997) 14(17):2041–5. doi: 10.1038/sj.onc.1201042
35. Marsh DJ, Andrew SD, Learoyd DL, Pojer R, Eng C, Robinson BG. Deletion-insertion mutation encompassing RET codon 634 is associated with medullary thyroid carcinoma. Hum Mutat (1998) Suppl 1:S3–4. doi: 10.1002/humu.1380110102
36. Jovanovic R, Kostadinova-Kunovska S, Janevska V, Bogoeva B, Spasevska L, Miladinova D, et al. Novel RET mutations in macedonian patients with medullary thyroid carcinoma: genotype-phenotype correlations. Pril (Makedonska Akad Na Nauk I Umet Oddelenie za Med Nauk (2015) 36(1):93–107.
37. Oriola J, Halperin I, Rivera-Fillat F, Donis-Keller H. The finding of a somatic deletion in RET exon 15 clarified the sporadic nature of a medullary thyroid carcinoma suspected to be familial. J Endocrinol Invest (2002) 25(1):25–31. doi: 10.1007/BF03343957
38. Pennelli G, Galuppini F, Barollo S, Cavedon E, Bertazza L, Fassan M, et al. The PDCD4/miR-21 pathway in medullary thyroid carcinoma. Hum Pathol (2015) 46(1):50–7. doi: 10.1016/j.humpath.2014.09.006
39. Uchino S, Noguchi S, Yamashita H, Sato M, Adachi M, Yamashita H, et al. Somatic mutations in RET exons 12 and 15 in sporadic medullary thyroid carcinomas: Different spectrum of mutations in sporadic type from hereditary type. Japanese J Cancer Res (1999) 90(11):1231–7. doi: 10.1111/j.1349-7006.1999.tb00701.x
40. Jozaghi Y, Zafereo M, Williams MD, Gule-Monroe MK, Wang J, Grubbs EG, et al. Neoadjuvant selpercatinib for advanced medullary thyroid cancer. Head Neck (2021) 43(1):E7–E12. doi: 10.1002/hed.26527
41. Ji JH, Oh YL, Hong M, Yun JW, Lee HW, Kim DG, et al. Identification of driving alk fusion genes and genomic landscape of medullary thyroid cancer. PLoS Genet (2015) 11(8):e1005467. doi: 10.1371/journal.pgen.1005467
42. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med (2017) 23(6):703–13. doi: 10.1038/nm.4333
43. Kurzrock R, Sherman SI, Ball DW, Forastiere AA, Cohen RB, Mehra R, et al. Activity of XL184 (cabozantinib), an oral tyrosine kinase inhibitor, in patients with medullary thyroid cancer. J Clin Oncol (2011) 29(19):2660–6. doi: 10.1200/JCO.2010.32.4145
44. Hong D, Ye L, Gagel R, Chintala L, El Naggar AK, Wright J, et al. Medullary thyroid cancer: Targeting the RET kinase pathway with sorafenib/tipifarnib. Mol Cancer Ther (2008) 7(5):1001–6. doi: 10.1158/1535-7163.MCT-07-2422
45. Elisei R, Ciampi R, Matrone A, Prete A, Gambale C, Ramone T, et al. Somatic RET indels in sporadic medullary thyroid Cancer : Prevalence and response to selpercatinib. J Clin Endocrinol Metab (2022), dgac325. doi: 10.1210/clinem/dgac325Online ahead of print
46. Simbolo M, Mian C, Barollo S, Fassan M, Mafficini A, Neves D, et al. High-throughput mutation profiling improves diagnostic stratification of sporadic medullary thyroid carcinomas. Virchows Arch (2014) 465(1):73–8. doi: 10.1007/s00428-014-1589-3
47. Musholt PB, Musholt TJ, Goodfellow PJ, Zehnbauer BA, Wells SA, Moley JF. “Cold” single-strand conformational variants for mutation analysis of the RET protooncogene. Surgery (1997) 122(2):363–71. doi: 10.1016/S0039-6060(97)90028-3
48. Moura MM, Cavaco BM, Pinto AE, Leite V. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J Clin Endocrinol Metab (2011) 96(5):E863–8. doi: 10.1210/jc.2010-1921
49. Ceccherini I, Pasini B, Pacini F, Gullo M, Bongarzone I, Romei C, et al. Somatic in frame deletions not involving juxtamembranous cysteine residues strongly activate the RET proto-oncogene. Oncogene (1997) 14(21):2609–12. doi: 10.1038/sj.onc.1201079
50. Pazaitou-Panayiotou K, Giatzakis C, Koutsodontis G, Vratimos A, Chrisoulidou A, Konstantinidis T, et al. Identification of two novel mutations in the RET proto-oncogene in the same family. Thyroid (2010) 20(4):401–6. doi: 10.1089/thy.2009.0262
51. Kalinin V, Frilling A. 27-bp deletion in the ret proto-oncogene as a somatic mutation associated with medullary thyroid carcinoma. J Mol Med (1998) 76:365–7. doi: 10.1007/s001090050228
52. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res (2019) 47(D1):D941–7. doi: 10.1093/nar/gky1015
53. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discovery (2012) 2(5):401–4. doi: 10.1158/2159-8290.CD-12-0095
54. Hentze MW, Kulozik AE. A perfect message: RNA surveillance and nonsense-mediated decay. Cell (1999) 96(3):307–10. doi: 10.1016/S0092-8674(00)80542-5
55. Durante C, Paciaroni A, Plasmati K, Trulli F, Filetti S. Vandetanib: Opening a new treatment practice in advanced medullary thyroid carcinoma. Endocrine (2013) 44(2):334–42. doi: 10.1007/s12020-013-9943-9
56. Durante C, Russo D, Verrienti A, Filetti S. XL184 (cabozantinib) for medullary thyroid carcinoma. Expert Opin Investig Drugs (2011) 20(3):407–13. doi: 10.1517/13543784.2011.559163
57. Belli C, Anand S, Gainor JF, Penault-Llorca F, Subbiah V, Drilon A, et al. Progresses toward precision medicine in RET-altered solid tumors. Clin Cancer Res (2020) 26(23):6102–11. doi: 10.1158/1078-0432.CCR-20-1587
58. Okafor C, Hogan J, Raygada M, Thomas BJ, Akshintala S, Glod JW, et al. Update on targeted therapy in medullary thyroid cancer. Front Endocrinology (2021) 12:708949. doi: 10.3389/fendo.2021.708949
59. Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann Oncol (2019) 30(8):1321–8. doi: 10.1093/annonc/mdz167
60. Shen T, Pu X, Wang L, Yu Z, Li J, Zhang Y, et al. Association between ret fusions and efficacy of pemetrexed-based chemotherapy for patients with advanced nsclc in china: a multicenter retrospective study. Clin Lung Cancer (2020) 21(5):e349–54. doi: 10.1016/j.cllc.2020.02.006
61. Drusbosky LM, Rodriguez E, Dawar R, Ikpeazu CV. Therapeutic strategies in RET gene rearranged non-small cell lung cancer. J Hematol Oncol (2021) 14(1):50. doi: 10.1186/s13045-021-01063-9
62. Schlumberger M, Elisei R, Müller S, Schöffski P, Brose M, Shah M, et al. Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann Oncol (2017) 28(11):2813–9. doi: 10.1093/annonc/mdx479
63. Sherman SI, Clary DO, Elisei R, Schlumberger MJ, Cohen EEW, Schöffski P, et al. Correlative analyses of RET and RAS mutations in a phase 3 trial of cabozantinib in patients with progressive, metastatic medullary thyroid cancer. Cancer (2016) 122(24):3856–64. doi: 10.1002/cncr.30252
64. Wells SA, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J Clin Oncol (2012) 30(2):134–41. doi: 10.1200/JCO.2011.35.5040
65. Elisei R, Schlumberger MJ, Müller SP, Schöffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol (2013) 31(29):3639–46. doi: 10.1200/JCO.2012.48.4659
66. Koehler VF, Adam P, Frank-Raue K, Raue F, Berg E, Hoster E, et al. Real-world efficacy and safety of cabozantinib and vandetanib in advanced medullary thyroid cancer. Thyroid (2021) 31(3):459–69. doi: 10.1089/thy.2020.0206
67. Ramos HE, Hecht F, Berdelou A, Borget I, Leboulleux S, Baudin E, et al. Long-term follow-up and safety of vandetanib for advanced medullary thyroid cancer. Endocrine (2021) 71(2):434–42. doi: 10.1007/s12020-020-02426-x
68. Valerio L, Bottici V, Matrone A, Piaggi P, Viola D, Cappagli V, et al. Medullary thyroid cancer treated with vandetanib: Predictors of a longer and durable response. Endocr Relat Cancer (2020) 27(2):97–110. doi: 10.1530/ERC-19-0259
69. Thein KZ, Velcheti V, Mooers BHM, Wu J, Subbiah V. Precision therapy for RET-altered cancers with RET inhibitors. Trends Cancer (2021) 7(12):1074–88. doi: 10.1016/j.trecan.2021.07.003
70. Andreev-Drakhlin A, Cabanillas M, Amini B, Subbiah V. Systemic and cns activity of selective ret inhibition with selpercatinib (loxo-292) in a patient with ret-mutant medullary thyroid cancer with extensive cns metastases. JCO Precis Oncol (2020) 4:PO.20.00096. doi: 10.1200/PO.20.00096
71. Matrone A, Prete A, Sartini MS, Elisei R. Significant response of medullary thyroid cancer choroidal metastases to highly selective RET inhibitor selpercatinib: a case report. Ann Oncol (2021) 32(11):1447–9. doi: 10.1016/j.annonc.2021.08.1987
72. Wirth LJ, Sherman E, Robinson B, Solomon B, Kang H, Lorch J, et al. Efficacy of selpercatinib in RET -altered thyroid cancers. N Engl J Med (2020) 383(9):825–35. doi: 10.1056/NEJMoa2005651
73. Subbiah V, Hu MI, Wirth LJ, Schuler M, Mansfield AS, Curigliano G, et al. Pralsetinib for patients with advanced or metastatic RET-altered thyroid cancer (ARROW): a multi-cohort, open-label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol (2021) 9(8):491–501. doi: 10.1016/S2213-8587(21)00120-0
74. Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, et al. Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol (2018) 29(8):1869–76. doi: 10.1093/annonc/mdy137
75. Wirth LJ, Kohno T, Udagawa H, Matsumoto S, Ishii G, Ebata K, et al. Emergence and targeting of acquired and hereditary resistance to multikinase RET inhibition in patients with RET-altered cancer. JCO Precis Oncol (2019) 3:PO.19.00189. doi: 10.1200/PO.19.00189
76. Nakaoku T, Kohno T, Araki M, Niho S, Chauhan R, Knowles PP, et al. A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat Commun (2018) 9(1):625. doi: 10.1038/s41467-018-02994-7
77. Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discovery (2018) 8(7):836–49. doi: 10.1158/2159-8290.CD-18-0338
78. Liu X, Shen T, Mooers BHM, Hilberg F, Wu J. Drug resistance profiles of mutations in the RET kinase domain. Br J Pharmacol (2018) 175(17):3504–15. doi: 10.1111/bph.14395
79. Solomon BJ, Tan L, Lin JJ, Wong SQ, Hollizeck S, Ebata K, et al. RET solvent front mutations mediate acquired resistance to selective ret inhibition in ret-driven malignancies. J Thorac Oncol (2020) 15(4):541–9. doi: 10.1016/j.jtho.2020.01.006
80. Subbiah V, Shen T, Terzyan SS, Liu X, Hu X, Patel KP, et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann Oncol (2021) 32(2):261–8. doi: 10.1016/j.annonc.2020.10.599
81. Lin JJ, Liu SV, McCoach CE, Zhu VW, Tan AC, Yoda S, et al. Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann Oncol (2020) 31(12):1725–33. doi: 10.1016/j.annonc.2020.09.015
82. Shen T, Hu X, Liu X, Subbiah V, Mooers BHM, Wu J. The L730V/I RET roof mutations display different activities toward pralsetinib and selpercatinib. NPJ Precis Oncol (2021) 5(1):48. doi: 10.1038/s41698-021-00188-x
83. Jiang Y, Peng X, Ji Y, Dai Y, Fang Y, Xiong B, et al. The novel RET inhibitor SYHA1815 inhibits RET-driven cancers and overcomes gatekeeper mutations by inducing G1 cell-cycle arrest through c-myc downregulation. Mol Cancer Ther (2021) 20(11):2198–206. doi: 10.1158/1535-7163.MCT-21-0127
84. Kolakowski GR, Anderson ED, Ballard JA, Brandhuber BJ, Condroski KR, Gomez EB, et al. Abstract 1464: Pre-clinical characterization of potent and selective next-generation RET inhibitors. Cancer Res (2021) 81(13_Supplement):1464. doi: 10.1158/1538-7445.AM2021-1464
85. Schoffski P, Aftimos PG, Massard C, Italiano A, Jungels C, Andreas K, et al. A phase I study of BOS172738 in patients with advanced solid tumors with RET gene alterations including non-small cell lung cancer and medullary thyroid cancer. J Clin Oncol (2019) 37(15S):TPS3162. doi: 10.1200/JCO.2019.37.15_suppl.TPS3162
86. Newton R, Waszkowycz B, Seewooruthun C, Burschowsky D, Richards M, Hitchin S, et al. Discovery and optimization of wt-ret/kdr-selective inhibitors of retv804m kinase. ACS Med Chem Lett (2020) 11(4):497–505. doi: 10.1021/acsmedchemlett.9b00615
87. Ortiz MV, Gerdemann U, Raju SG, Henry D, Smith S, Rothenberg SM, et al. Activity of the highly specific RET inhibitor selpercatinib (LOXO-292) in pediatric patients with tumors harboring RET gene alterations. JCO Precis Oncol (2020) 4:PO.19.00401. doi: 10.1200/PO.19.00401
88. Zhao Z, Fu T, Gao J, Xu Y, Wu X, Chen W, et al. Identifying novel oncogenic RET mutations and characterising their sensitivity to RET-specific inhibitors. J Med Genet (2021), jmedgenet–2019-106546. doi: 10.1136/jmedgenet-2019-106546Online ahead of print
89. Xing S. Characterization of ret oncogenic activation in MEN2 inherited cancer syndromes. Endocrinology (1996) 137(5):1512–9. doi: 10.1210/endo.137.5.8612479
Keywords: RET deletions, RET indels, acquired resistance, medullary thyroid cancer (MTC), RET-mutated cancers, pralsetinib, selpercatinib
Citation: Verrienti A, Grani G, Sponziello M, Pecce V, Damante G, Durante C, Russo D and Filetti S (2022) Precision oncology for RET-related tumors. Front. Oncol. 12:992636. doi: 10.3389/fonc.2022.992636
Received: 12 July 2022; Accepted: 29 July 2022;
Published: 24 August 2022.
Edited by:
Jill Kolesar, University of Kentucky, United StatesReviewed by:
Alessandro Prete, University of Pisa, ItalyCopyright © 2022 Verrienti, Grani, Sponziello, Pecce, Damante, Durante, Russo and Filetti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgio Grani, Z2lvcmdpby5ncmFuaUB1bmlyb21hMS5pdA==
†These authors have contributed equally to this work and share first authorship