 Carla Iacobini
Carla Iacobini Martina Vitale
Martina Vitale Giuseppe Pugliese
Giuseppe Pugliese Stefano Menini
Stefano Menini- Department of Clinical and Molecular Medicine, “La Sapienza” University, Rome, Italy
The hypoxia-inducible factor-1α (HIF-1α), a key player in the adaptive regulation of energy metabolism, and the M2 isoform of the glycolytic enzyme pyruvate kinase (PKM2), a critical regulator of glucose consumption, are the main drivers of the metabolic rewiring in cancer cells. The use of glycolysis rather than oxidative phosphorylation, even in the presence of oxygen (i.e., Warburg effect or aerobic glycolysis), is a major metabolic hallmark of cancer. Aerobic glycolysis is also important for the immune system, which is involved in both metabolic disorders development and tumorigenesis. More recently, metabolic changes resembling the Warburg effect have been described in diabetes mellitus (DM). Scientists from different disciplines are looking for ways to interfere with these cellular metabolic rearrangements and reverse the pathological processes underlying their disease of interest. As cancer is overtaking cardiovascular disease as the leading cause of excess death in DM, and biological links between DM and cancer are incompletely understood, cellular glucose metabolism may be a promising field to explore in search of connections between cardiometabolic and cancer diseases. In this mini-review, we present the state-of-the-art on the role of the Warburg effect, HIF-1α, and PKM2 in cancer, inflammation, and DM to encourage multidisciplinary research to advance fundamental understanding in biology and pathways implicated in the link between DM and cancer.
1 Introduction
Recent epidemiological studies have reported a transition from cardiovascular diseases to cancer as the leading cause of excess death associated with diabetes mellitus (DM) (1, 2). Cancer mortality among people with DM, especially type 2 (T2) DM, is approximately 30%-50% higher than in the general population, particularly for pancreatic, liver, colorectal, and endometrial cancers (3, 4). Clinical and preventive efforts must be directed at fighting DM-related risk factors for cancer to reduce the excess mortality risk in individuals with DM.
Possible mechanisms for a biological link between DM and cancer are hyperinsulinemia, inflammation, and hyperglycemia (5). Hyperglycemia is the distinctive feature of DM and the main cause of various life-threatening complications in both type 1 (T1) DM and T2DM (6, 7). A direct link between hyperglycemia and cancer comes from studies showing that, at high concentrations, glucose acts as DNA-damaging factor and impedes tumor suppressive functions, leading to genomic instability and eventually resulting in malignant transformation (8, 9). DM has been also associated with cancer promotion and progression (10–12); mechanisms involved in the regulation of cancer cell metabolism and the way cancer cells utilize glucose may mediate this association.
After briefly summarizing the molecular and biochemical characteristics of the Warburg effect in cancer cells, we will examine the updated evidence demonstrating similar metabolic and molecular changes in immune cells involved in inflammation and in target cells and tissues of chronic DM complications. In particular, the role of hypoxia inducible factor (HIF)-1α and M2 isoform of the glycolytic enzyme pyruvate kinase (PK) in driving the metabolic reprogramming of tumor, inflammatory, and diabetic cells will be discussed. Finally, to foster multidisciplinary investigation, the collected evidence will be illustrated in the context of a plausible hypothesis centered on changes in cellular glucose metabolism as mechanistic link between DM and cancer.
2 Warburg effect, HIF-1α, and PKM2 in cancer: when metabolism rhymes with opportunism
In cancer, a close relationship exists between the rate of glucose utilization and that of cell proliferation (13). In 1924, Otto Warburg identified the link between cancer and glucose by showing that tumor tissues consume and metabolize to lactate tremendous amounts of glucose relative to non-transformed tissues (14). While some cancer cells are oxidative and targeting mitochondrial oxidative phosphorylation (OXPHOS) may be a promising therapeutic target for oxidative carcinomas (15), most cancers cells exhibit suppressed mitochondrial respiration and a high rate of glucose uptake even in the presence of oxygen. This metabolic rewiring is known as both Warburg effect and aerobic glycolysis. Consistent with the importance of the Warburg effect for cancer cells, withdrawing glucose or inhibiting glycolysis is deleterious to tumorigenesis in experimental models of cancer (16, 17). How cancer cells take advantage from these metabolic changes and how glycolysis is related to cell proliferation is still not fully understood. Along with the proposal that the Warburg metabolism may be a way to produce ATP quickly (18), widely accepted hypothesis include: 1) expansion of the pool of glycolytic biosynthetic intermediates to support anabolic reactions and redox demand (19), 2) persistent NAD+ regeneration to sustain de novo lipogenesis (20), and 3) augmented lactate production to favor tumor growth and metastasis by affecting the tumor microenvironment (21). Along with changes in the tissue microenvironment, oncogenes and tumor suppressors that drives tumorigenesis contribute to the acquisition of the Warburg phenotype via activation of numerous transcription factors (including HIF-1α) regulating several genes encoding glycolytic proteins (including PKM2) (22).
HIF-1α is a master regulator of oxygen homeostasis playing a key role in the adaptive regulation of energy metabolism in mammalian tissues. By simultaneously increasing the expression of glycolytic enzymes and restraining mitochondrial function, HIF-1α can switch glucose metabolism from OXPHOS to glycolysis also in response to physiological and pathological stimuli other than hypoxia (23), including hyperglycemia-induced oxidative (24) and carbonyl (25) stress. In cancer cells, HIF-1α cooperates with the oncoprotein MYC to activate transcription of genes involved in glucose metabolism, including glucose transporters (e.g. GLUT1 and GLUT3) and glycolytic enzymes (e.g. lactate dehydrogenase A, hexokinase 2, PKM2, etc.) (26). In addition to stimulate glycolysis, HIF-1α actively represses mitochondrial respiration and biogenesis by inducing pyruvate dehydrogenase kinase 1 (27) and reducing peroxisome-proliferator-activated receptor γ co-activator-1α (28). Consistent with an important role in cancer cell biology, HIF-1α overexpression strongly correlates with poor prognosis for several solid cancers. Accordingly, pharmacological targeting of the HIF-1α signaling pathways has been recognized as a promising strategy for cancer therapy in the recent years (29).
The PKs are terminal enzymes of the glycolytic pathway that catalyze the conversion of phosphoenolpyruvate and ADP to pyruvate and ATP and are important modulators of cellular glucose metabolism. The PKM1/M2 isoforms are encoded by the same gene (PKM) and are generated by the alternative splicing of PKM mRNA (30). While PKM1 only exists as a stable and highly active tetrameric form and is expressed in most adult tissues (31), PKM2 is highly expressed during embryonic development and is reactivated in tissue regeneration and tumor development, suggesting that it is critical for actively proliferating cells (32). Unlike the constitutionally active PKM1, PKM2 is in equilibrium among the dimeric and monomer forms, which are catalytically inactive, and the active tetrameric form. Therefore, the glycolytic activity of PKM2 is subject to allosteric control (33). This implies that, at the same protein level, PKM2 is much less effective than PKM1 in catalyzing the last step within glycolysis (31). Accordingly, high ratios of PKM2/PKM1 lead to accumulation of all upstream glycolytic intermediates and diversion of metabolic flux towards the glycolytic biosynthetic branches, including the pentose-phosphate pathway, the hexosamine pathway, and the glycerol synthesis (34). This process is exploited by cancer cells to sustain their high biosynthetic and redox demand (35).
PKM2 and HIF-1α regulate each other. In fact, PKM2 is a transcriptional target of HIF-1α and a key player in the Warburg effect of glycolytic cancer cells (26). In turn, as a dimer, PKM2 translocates into the nucleus, interacts with, and promotes the transcriptional activity of HIF-1α (36). Therefore, HIF-1α and PKM2 are recognized as major drivers of cancer metabolism participating in a positive feedback loop that enhances the Warburg effect and feeds the glycolysis branch pathways (23, 30).
3 Warburg effect, HIF-1α, and PKM2 in inflammation: a matter of polarization
It has been almost 50 years since the first demonstration of aerobic glycolysis during lymphocyte proliferation (37). In the 2000s, some observations on metabolic reprogramming were extended to other cells of innate and adaptive immunity. Since then, a growing interest in the role of metabolism in immune regulation has bloomed. The exciting advances in the field of immunometabolism have been recently reviewed (38, 39). We summarize here the role of aerobic glycolysis, HIF-1α, and PKM2 in immune cells involved in chronic inflammation, which participates in all stages of tumorigenesis as well as DM development and progression to complications (40, 41).
In dendritic cells and macrophages, pro-inflammatory stimuli induce the shift to aerobic glycolysis and the production of inflammatory cytokines, such as interleukin (IL)-1β and tumor necrosis factor-α (TNF-α) (42). IL-1β and TNF-α are involved in insulin resistance (43, 44) and, together with other IL family members, promote tumorigenesis through complex mechanisms that involve direct growth stimulation and production of growth factors, recruitment of myeloid cells and immunosuppression, endothelial cell activation and promotion of angiogenesis (45, 46). In macrophages, the metabolic rewiring towards an enhanced glycolytic phenotype promotes polarization to the classically activated (or “M1”) phenotype and the production of many inflammatory mediators (44). Consistently, the glycolysis inhibitor 2-deoxy-D-glucose (2-DG) blocks (47), whereas GLUT1 overexpression enhances (48) M1 inflammatory functions. Conversely, OXPHOS is critical for the anti-inflammatory and tissue repair functions of alternatively activated (or “M2”) macrophages (49, 50). The balance between glycolysis and mitochondrial respiration also differentially regulates the phenotype and function of various subsets of T cells. For instance, T regulatory cells (Tregs) rely on glycolysis only during initial activation and proliferation, after that they switch toward oxidative metabolism for their regulatory functions. Consistently, GLUT1 expression increases the number of Tregs, but reduces their immunosuppressive capacity (51). Vice versa, T helper (Th)17 cells - a distinct subset of CD4+ T cells that produce the highly pro-inflammatory IL-17 – can be converted into Tregs by blocking glycolysis with 2-DG (52). Like the pro-inflammatory CD4+ Th17 cells, CD8+ lymphocytes require a Warburg-like metabolism not only for their proliferative capacity, but also for their effector functions (53, 54).
It is by now long-established that HIF-1α stabilization in immune cells can occur in an oxygen-independent manner. Bacteria and their cell membrane component lipopolysaccharide (LPS), inflammatory mediators, and endogenous molecules, such as the tricarboxylic-acid cycle intermediate succinate (47), can induce HIF-1α protein accumulation in macrophages through transcriptional and post-translational mechanisms under normoxic conditions (55–58). By cross talking with the nuclear factor-κB pathway, HIF-1α modulates essential inflammatory functions in myeloid cells (59). In keeping with a critical role for HIF-1α in the pro-inflammatory response in macrophage and T cells, HIF-1α deletion induces defective macrophage response to LPS and inhibits Th17 cell generation in mice (44, 60).
In addition to induce HIF-1α accumulation, pro-inflammatory stimuli trigger the expression of PKM2, which is now recognized as a critical determinant of the Warburg effect in macrophages. In fact, stabilizing PKM2 tetramerization with the allosteric activator TEPP-46, thus favoring PKM2 glycolytic activity and the glycolytic flux toward pyruvate, restores OXPHOS and reduces LPS-induced production of IL-1β, while promoting macrophage M2 polarization (61). In addition, PKM2 over expression induces, whereas downregulation inhibits, the activation of several toll-like receptor pathways (62). PKM2 tetramerization by TEPP-46 also blocks PKM2 nuclear translocation and restrains pro-inflammatory polarization in T cells by inhibiting the Warburg metabolism and favoring OXPHOS (63).
Overall, upregulation of aerobic glycolysis supports inflammatory immune functions. Hindering HIF-1α accumulation and PKM2 expression or favoring glycolytic activity over transcriptional activity of PKM2 by allosteric activation, blocks the Warburg metabolism and curbs inflammatory responses by supporting regulatory and anti-inflammatory immune phenotypes.
4 Warburg effect, HIF-1α, and PKM2 in diabetes: team members or individual runners on the road to complications?
While confirming previous findings of increased levels of glycolytic intermediates, recent omics studies in DM and related target organ damage have provided evidence of impaired mitochondrial metabolism and biogenesis, along with other features of a metabolic rewiring resembling the Warburg effect (64–66). Several metabolic intermediates and glycolytic enzymes, including PKM2, have been proposed as potential triggers of aerobic glycolysis and diversion of glycolytic intermediates into branch pathways (64).
Diabetic complications arise in tissues that exhibit insulin-independent glucose uptake (67, 68). In the cells of these tissues, activation of aerobic glycolysis may be a consequence of increased glucose uptake from systemic circulation and an attempt to quickly metabolize excess cellular glucose (69) (Figure 1). The drawback of this process is the cellular accumulation of toxic glucose metabolites (66, 70). In fact, at variance with cancer cells, normal cells are not actively proliferating. Accordingly, the enhanced glucose uptake, buildup of glycolytic intermediates, and flux through the glycolytic branch pathways results in an accumulation of sorbitol, diacylglycerol, and advanced glycation end products (AGEs) leading to the activation of pro-inflammatory and -oxidative pathways (71). To our knowledge, only one study has attempted to establish a relationship between mitochondrial dysfunction, the Warburg-like metabolism, and accumulation of toxic glucose metabolites in DM. By showing that high glucose induces HIF-1α activity and a switch from oxidative metabolism to glycolysis and its principal branches, this study suggests that aerobic glycolysis may play an initiating role in glucotoxicity and diabetic complications (25).
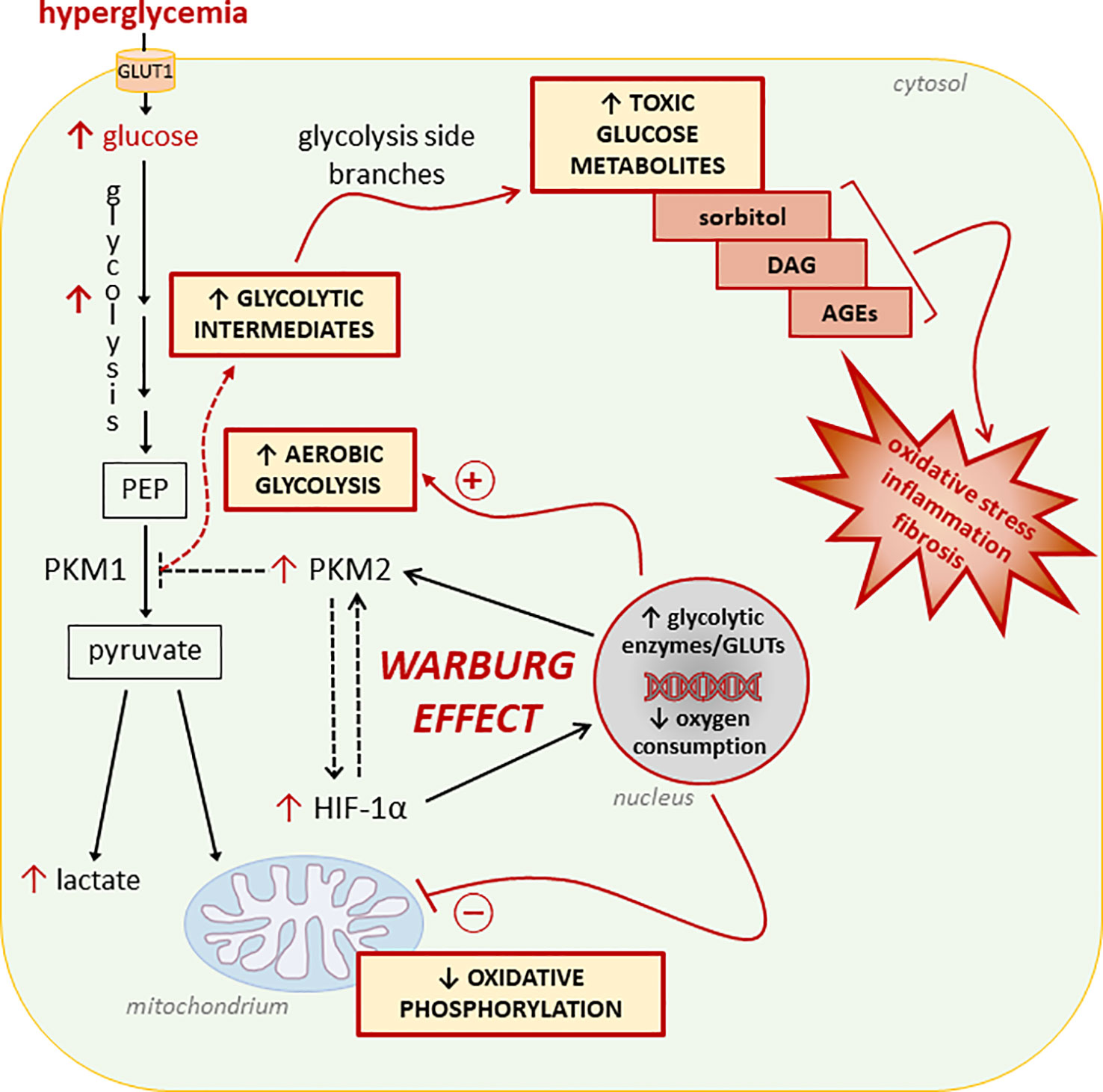
Figure 1 Glucose metabolic reprogramming induced by hyperglycemia in insulin-independent cells. Refer to the main text for detailed description and references. Dashed lines/arrows indicate well-established processes in cancer cells not yet confirmed in normal cells exposed to glucose concentrations in the diabetic range. In particular, the interaction between the hypoxia inducible factor 1-α (HIF-1α) and the M2 isoform of pyruvate kinase (PKM2), and the role of PKM2 in reducing the overall PK activity leading to accumulation of glycolytic intermediates need to be demonstrated. PEP, Phosphoenolpyruvate; GLUTs, glucose transporters; DAG, diacylglycerol; AGEs, advanced glycation end-products.
In addition to hypoxia, numerous metabolic stressors associated with DM, including hyperglycemia, affect HIF-1α stabilization and activity. Due to the heterogeneity of the findings, what is the net impact of DM-related factors on HIF-1α signaling is a matter of debate in literature (72). Concisely, hyperglycemia seems to inhibit HIF-1α expression induced by low-oxygen conditions (73, 74), suggesting a weaker HIF-1α-dependent response to hypoxia in DM. Conversely, high glucose concentrations promote HIF-1α nuclear translocation, transcriptional activity, and lactate accumulation in normoxic conditions (25, 72, 75–78). Ultimately, these discrepancies in the effect of DM on HIF-1α signaling are likely the result of different experimental conditions (e.g., hypoxia vs normoxia) used for modulating the levels and activity of HIF-1α. In this regard, it must be remembered that the Warburg effect occurs, by definition, in the presence of normal levels of oxygen.
Recently, proteomic studies conducted in T1DM patients have demonstrated increased circulating and renal levels of mitochondrial and glycolytic enzymes in those without diabetic nephropathy (DN). Curiously, elevated levels of the underactive glycolytic enzyme PKM2 were associated with reduced renal accumulation of toxic glucose metabolites and susceptibility to DN, suggesting a protective role of PKM2 by improving cellular glucose metabolism (66, 79). However, preclinical work by the same and other investigators has shown that TEPP-46 treatment reverses metabolic abnormalities, mitochondrial dysfunction, and kidney pathology of diabetic mice by enhancing PKM2 tetramer formation (i.e., glycolytic activity) and suppressing HIF-1α and lactate accumulation (66, 80, 81). These experimental findings suggest that also in DM, as in cancer and immune cells (61, 82), PKM2 overexpression may favor glucose metabolic reprogramming towards aerobic glycolysis, accumulation of glycolytic intermediates, and pro-inflammatory signaling. Collectively, these interesting findings could lead to greater understanding of DM complications if the functional complexity of PKM2 and its role as a key regulator of glucose metabolism is considered.
In general, the body of knowledge acquired in a century of research on cancer and immune metabolism has been overlooked in the interpretation of the data concerning several aspects of cellular glucose metabolism in DM, including aerobic glycolysis, HIF-1α induction, and PKM2 expression. For example, claims that HIF-1α activity should be enhanced in DM because HIF-1α signaling is submaximal for the degree of hypoxia in diabetic tissues (83, 84) overlook the role of this transcription factor in aerobic glycolysis and inflammation. Increased cellular glucose uptake, mitochondrial dysfunction, and pro-inflammatory signaling induced by HIF-1α activity (26–28, 44) would not be at all beneficial for tissues affected by DM complications (70). Rather, regarding the role of chronic hypoxia in DM complications, it might be more biologically sound to promote the activity of the HIF-2α isoform, which has different effects to HIF-1α on glucose metabolism and even opposite effects on redox state and inflammation (72, 85). Another naive conclusion, which is inconsistent with the mechanisms that regulate cellular glucose metabolism and drive hyperglycemia-induced cell damage, is that overall PKM2 expression should be increased to enhance PK activity and prevent DM complications (80, 86). Actually, to favor glycolytic flux to pyruvate and reduce the accumulation of toxic glucose metabolites, the expression of the constitutively active enzyme PKM1 should be preferred over that of PKM2. Favor the expression of the less active isoform M2 and then have to enhance PK activity with pharmacological agents does not seem the best strategy to improve cellular glucose metabolism and prevent DM-induced target organ damage. As in cancer (87), PKM2 activators are interesting drugs to consider for complications of DM, but the simplistic view that high PKM2 levels mean high levels of PK activity must be overcome. In fact, it is the exact opposite (31, 61).
Overall, more research is needed to fully characterize the changes in cellular glucose metabolism associated with DM complications and the role of HIF-1α and PKM2 as components of a molecular network that regulates metabolic reprograming towards the Warburg effect.
5 Discussion
Tight glucose control is important for reduction of cancer risk in T2DM (88). Substantial evidence supports a direct causal link between DM and carcinogenesis; hyperglycemia can in fact induce malignant transformation by causing DNA damage (9), oncogenic mutations (8), and loss of tumor suppressive functions (89). Besides this, cancer and DM share similar changes in some aspects of cellular energetics, particularly glucose metabolism. Hyperglycemia leads to excessive cellular glucose uptake and changes in glucose metabolism resembling the Warburg effect, including accumulation of glycolytic intermediates (64–66). Interestingly, among the molecular mechanisms associated with the anti-tumor activity of the anti-diabetic drug metformin, suppression of the Warburg effect has also been proposed (90). The question whether the Warburg effect, besides being a consequence, might also play a causal role in carcinogenesis has been raised in the past without receiving much attention, mainly because of the lack of plausible pathomechanisms (91). However, there are numerous clues that lead us to consider the metabolic reprogramming induced by hyperglycemia as a possible field of investigation to unravel the connections between diabetes and cancer.
Tumorigenesis comprises multiple steps of mutations subjected to a natural selection (Figure 2). Environmental forces and cellular adaption mechanisms that provide the mutated cell clone with survival and proliferative advantages over the neighboring cells govern this process (92). As tumor microenvironment factors influence cancer metabolism (93), hyperglycemia may promote the acquisition of a Warburg metabolism in transforming cells. In turn, hyperglycemia-mediated glycolytic reprogramming may contribute to shape the metabolic features of the evolving tumor cells by increasing the activity and fostering mutations of oncogenes regulating cell metabolism (8, 93), thus playing an active role in cancer promotion and progression. For example, high glucose was recently shown to stabilize and induce aberrant transcriptional activity of N-MYC - a member of the MYC family - even in normal cells, leading to increased proliferation and functional impairment (94). Overall, by inducing a Warburg-like metabolism, hyperglycemia might favor tumorigenesis by both contributing to the selection of more malignant phenotypes and directly inducing, in normal cells, the transcriptional activity of oncogenes that regulate multiple aspects of tumor metabolism, eventually increasing the chances of malignant transformation.
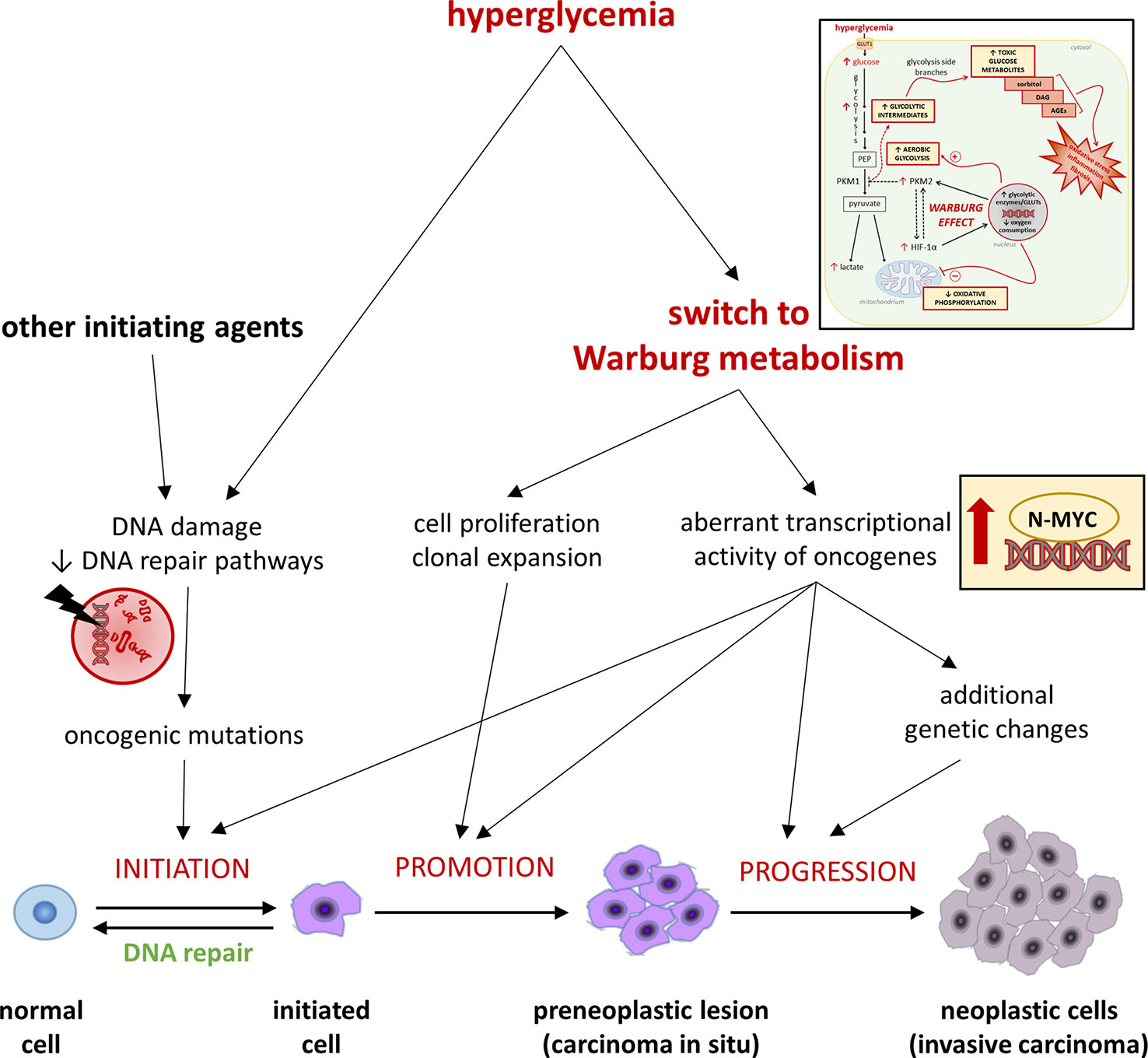
Figure 2 Hyperglycemia and the Warburg-like metabolism driven by excessive cellular glucose uptake in cancer initiation, promotion, and progression. Refer to the main text for detailed description and references.
The mechanisms by which aerobic glycolysis favors cell proliferation, regulates inflammatory immune functions, and induce cell damage in DM are not yet fully elucidated. The “anabolic” (34) and “energetic” (18) hypotheses explain how the Warburg metabolism provides building blocks and an increased rate of ATP to support the anabolic and energetic demand of proliferating cells. Regardless of the discussion of their validity (18), the current hypotheses do not address the question of the causal relationship and mechanistic link between aerobic glycolysis and cell proliferation in tumor and immune cells, or cell injury in DM. Studies in the fields of immunology and metabolism have identified interesting alternative (or complementary) mechanisms that may explain how glycolytic reprogramming benefits cancer and immune cells and promotes DM complications. These mechanisms rely on the signaling function of glycolytic intermediates and/or their spontaneous decomposition products, including the inevitable side-product of glycolysis methylglyoxal (MGO). This is a highly reactive dicarbonyl compound and major precursor of advanced glycation end-products (AGEs) (95). By acting at both transcriptional and post-translational levels, MGO plays important roles in the immune response to inflammatory stimuli (96–98) and, together with AGEs, is involved in the pathogenesis of DM complications (25, 95, 99–101) and in the onset and progression of many cancers (10, 11, 102–104).
In conclusion, oncology and immunology scientists have continued to build on the seminal work by biochemists to improve understanding of glucose metabolic rewiring in cancer and immune system biology and pathology. Researchers in endocrinology and metabolism have lagged behind in this process and are struggling to put the puzzle pieces together. A multidisciplinary approach could not only help to unravel the skein and effectively interpret data for a real progress in cardiometabolic research, it also may generate new knowledge on the mechanisms linking DM and cancer.
Author contributions
Writing—original draft preparation, CI and SM; writing—review and editing, MV and GP; visualization, SM; supervision, GP; funding acquisition, SM. All authors contributed to the article and approved the submitted version.
Funding
Some of the findings discussed stem from research funded by the EFSD and Sanofi European Research Programme in Macrovascular Complications of Diabetes 2019 and Sapienza University of Rome, Progetti di Ateneo 2017, 2018, 2019, 2020, and 2021 to SM.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Song M. Cancer overtakes vascular disease as leading cause of excess death associated with diabetes. Lancet Diabetes Endocrinol (2021) 9:131–3. doi: 10.1016/S2213-8587(21)00016-4
2. Pearson-Stuttard J, Bennett J, Cheng YJ, Vamos EP, Cross AJ, Ezzati M, et al. Trends in predominant causes of death in individuals with and without diabetes in England from 2001 to 2018: an epidemiological analysis of linked primary care records. Lancet Diabetes Endocrinol (2021) 9:165–73. doi: 10.1016/S2213-8587(20)30431-9
3. Harding JL, Andes LJ, Gregg EW, Cheng YJ, Weir HK, Bullard KM, et al. Trends in cancer mortality among people with vs without diabetes in the USA, 1988–2015. Diabetologia (2020) 63:75–84. doi: 10.1007/S00125-019-04991-X/TABLES/3
4. Ling S, Zaccardi F, Issa E, Davies MJ, Khunti K, Brown K. Inequalities in cancer mortality trends in people with type 2 diabetes: 20 year population-based study in England. Diabetologia (2023) 66(4):657–73. doi: 10.1007/S00125-022-05854-8/TABLES/4
5. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and CancerA consensus report. Diabetes Care (2010) 33:1674–85. doi: 10.2337/DC10-0666
6. Nathan DM, Genuth S, Lachin J, Cleary P, Crofford O, Davis M, et al. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med (1993) 329:977–86. doi: 10.1056/NEJM199309303291401
7. Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HAW. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med (2008) 359:1577–89. doi: 10.1056/NEJMOA0806470/SUPPL_FILE/NEJM_HOLMAN_1577SA1.PDF
8. Hu CM, Tien SC, Hsieh PK, Jeng YM, Chang MC, Chang YT, et al. High glucose triggers nucleotide imbalance through O-GlcNAcylation of key enzymes and induces KRAS mutation in pancreatic cells. Cell Metab (2019) 29:1334–1349.e10. doi: 10.1016/J.CMET.2019.02.005
9. Rahmoon MA, Elghaish RA, Ibrahim AA, Alaswad Z, Gad MZ, El-Khamisy SF, et al. High glucose increases DNA damage and elevates the expression of multiple DDR genes. Genes (Basel) (2023) 14:144. doi: 10.3390/GENES14010144/S1
10. Menini S, Iacobini C, Vitale M, Pesce C, Pugliese G. Diabetes and pancreatic cancer-a dangerous liaison relying on carbonyl stress. Cancers (Basel) (2021) 13:1–26. doi: 10.3390/CANCERS13020313
11. Menini S, Iacobini C, de Latouliere L, Manni I, Vitale M, Pilozzi E, et al. Diabetes promotes invasive pancreatic cancer by increasing systemic and tumour carbonyl stress in KrasG12D/+ mice. J Exp Clin Cancer Res (2020) 39(1):152. doi: 10.1186/S13046-020-01665-0
12. Wang W, Hapach LA, Griggs L, Smart K, Wu Y, Taufalele P v., et al. Diabetic hyperglycemia promotes primary tumor progression through glycation-induced tumor extracellular matrix stiffening. Sci Adv (2022) 8(46):eabo1673. doi: 10.1126/SCIADV.ABO1673
13. Lunt SY, vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol (2011) 27:441–64. doi: 10.1146/ANNUREV-CELLBIO-092910-154237
14. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi: 10.1126/SCIENCE.123.3191.309
15. Amoedo ND, Sarlak S, Obre E, Esteves P, Bégueret H, Kieffer Y, et al. Targeting the mitochondrial trifunctional protein restrains tumor growth in oxidative lung carcinomas. J Clin Invest (2021) 131(1):e133081. doi: 10.1172/JCI133081
16. Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-a expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell (2006) 9:425–34. doi: 10.1016/J.CCR.2006.04.023
17. Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell (2013) 24:213–28. doi: 10.1016/J.CCR.2013.06.014
18. Liberti MV, Locasale JW. The warburg effect: how does it benefit cancer cells? Trends Biochem Sci (2016) 41:211–8. doi: 10.1016/J.TIBS.2015.12.001
19. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2(5):e1600200. doi: 10.1126/SCIADV.1600200
20. Li Z, Ji BW, Dixit PD, Tchourine K, Lien EC, Hosios AM, et al. Cancer cells depend on environmental lipids for proliferation when electron acceptors are limited. Nat Metab (2022) 4:711–23. doi: 10.1038/s42255-022-00588-8
21. de la Cruz-López KG, Castro-Muñoz LJ, Reyes-Hernández DO, García-Carrancá A, Manzo-Merino J. Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol (2019) 9:1143/BIBTEX. doi: 10.3389/FONC.2019.01143/BIBTEX
22. Wiese EK, Hitosugi T. Tyrosine kinase signaling in cancer metabolism: PKM2 paradox in the warburg effect. Front Cell Dev Biol (2018) 6:79. doi: 10.3389/FCELL.2018.00079
23. Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, et al. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev (2007) 21:1037–49. doi: 10.1101/GAD.1529107
24. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/Glycolysis-Dependent axis. Cell Metab (2020) 32(3):437–446.e5. doi: 10.1016/j.cmet.2020.07.007
25. Iacobini C, Vitale M, Pugliese G, Menini S. Normalizing hif-1α signaling improves cellular glucose metabolism and blocks the pathological pathways of hyperglycemic damage. Biomedicines (2021) 9:1139. doi: 10.3390/BIOMEDICINES9091139/S1
26. Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab (2012) 23:560–6. doi: 10.1016/J.TEM.2012.06.010
27. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3:187–97. doi: 10.1016/J.CMET.2006.01.012
28. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of c-MYC activity. Cancer Cell (2007) 11:407–20. doi: 10.1016/J.CCR.2007.04.001
29. Ma Z, Xiang X, Li S, Xie P, Gong Q, Goh BC, et al. Targeting hypoxia-inducible factor-1, for cancer treatment: recent advances in developing small-molecule inhibitors from natural compounds. Semin Cancer Biol (2022) 80:379–90. doi: 10.1016/J.SEMCANCER.2020.09.011
30. Wong N, Ojo D, Yan J, Tang D. PKM2 contributes to cancer metabolism. Cancer Lett (2015) 356:184–91. doi: 10.1016/J.CANLET.2014.01.031
31. Christofk HR, vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature (2008) 452:230–3. doi: 10.1038/nature06734
32. Dayton TL, Jacks T, vander Heiden MG. PKM2, cancer metabolism, and the road ahead. EMBO Rep (2016) 17:1721–30. doi: 10.15252/EMBR.201643300
33. Zhang Z, Deng X, Liu Y, Liu Y, Sun L, Chen F. PKM2, function and expression and regulation. Cell Biosci (2019) 9:52. doi: 10.1186/s13578-019-0317-8
34. DeBerardinis RJ, Chandel NS. We need to talk about the warburg effect. Nat Metab (2020) 2:127–9. doi: 10.1038/s42255-020-0172-2
35. Macintyre AN, Rathmell JC. PKM2 and the tricky balance of growth and energy in cancer. Mol Cell (2011) 42:713–4. doi: 10.1016/J.MOLCEL.2011.06.003
36. Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell (2011) 145:732–44. doi: 10.1016/J.CELL.2011.03.054
37. Wang T, Marquardt C, Foker J. Aerobic glycolysis during lymphocyte proliferation. Nature (1976) 261:702–5. doi: 10.1038/261702a0
38. Kornberg MD. The immunologic warburg effect: evidence and therapeutic opportunities in autoimmunity. Wiley Interdiscip Rev Syst Biol Med (2020) 12:e1486. doi: 10.1002/WSBM.1486
39. Voss K, Hong HS, Bader JE, Sugiura A, Lyssiotis CA, Rathmell JC. A guide to interrogating immunometabolism. Nat Rev Immunol (2021) 21:637–52. doi: 10.1038/s41577-021-00529-8
40. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity (2019) 51:27–41. doi: 10.1016/J.IMMUNI.2019.06.025
41. Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes (2003) 52:1799–805. doi: 10.2337/DIABETES.52.7.1799
42. O’Neill LAJ, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med (2016) 213:15–23. doi: 10.1084/JEM.20151570
43. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356:1517–26. doi: 10.1056/NEJMOA065213
44. Wen H, Ting JPY, O’Neill LAJ. A role for the NLRP3 inflammasome in metabolic diseases – did warburg miss inflammation? Nat Immunol (2012) 13:352. doi: 10.1038/NI.2228
45. Garlanda C, Mantovani A. Interleukin-1 in tumor progression, therapy, and prevention. Cancer Cell (2021) 39:1023–7. doi: 10.1016/j.ccell.2021.04.011
46. Sethi G, Sung B, Aggarwal BB. TNF: a master switch for inflammation to cancer. Front Biosci (2008) 13:5094–107. doi: 10.2741/3066
47. Tannahill GM, Curtis AM, Adamik J, Palsson-Mcdermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature (2013) 496:238–42. doi: 10.1038/nature11986
48. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem (2014) 289:7884–96. doi: 10.1074/JBC.M113.522037
49. Rodríguez-Prados J-C, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, et al. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol (2010) 185:605–14. doi: 10.4049/JIMMUNOL.0901698
50. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab (2006) 4:13–24. doi: 10.1016/J.CMET.2006.05.011
51. Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and toll-like receptor signaling balance treg cell anabolic metabolism for suppression. Nat Immunol (2016) 17:1459–66. doi: 10.1038/NI.3577
52. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and treg cells. J Exp Med (2011) 208:1367–76. doi: 10.1084/JEM.20110278
53. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
54. Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol (2008) 38:2438–50. doi: 10.1002/EJI.200838289
55. Shatrov VA, Sumbayev V v., Zhou J, Brüne B. Oxidized low-density lipoprotein (oxLDL) triggers hypoxia-inducible factor-1α (HIF-1α) accumulation via redox-dependent mechanisms. Blood (2003) 101:4847–9. doi: 10.1182/BLOOD-2002-09-2711
56. Albina JE, Mastrofrancesco B, Vessella JA, Louis CA, Henry WL, Reichner JS. HIF-1 expression in healing wounds: HIF-1α induction in primary inflammatory cells by TNF-α. Am J Physiol Cell Physiol (2001) 281(6):C1971–7. doi: 10.1152/AJPCELL.2001.281.6.C1971/ASSET/IMAGES/LARGE/H01210786008.JPEG
57. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest (2005) 115:1806–15. doi: 10.1172/JCI23865
58. Blouin CC, Pageí EL, Soucy GM, Richard DE. Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1α. Blood (2004) 103:1124–30. doi: 10.1182/BLOOD-2003-07-2427
59. Palazon A, Goldrath AW, Nizet V, Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity (2014) 41:518–28. doi: 10.1016/J.IMMUNI.2014.09.008
60. McGettrick AF, O’Neill LAJ. The role of HIF in immunity and inflammation. Cell Metab (2020) 32:524–36. doi: 10.1016/J.CMET.2020.08.002
61. Palsson-Mcdermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab (2015) 21:65–80. doi: 10.1016/J.CMET.2014.12.005
62. Zhang X, Yang Y, Jing L, Zhai W, Zhang H, Ma Q, et al. Pyruvate kinase M2 contributes to TLR-mediated inflammation and autoimmunity by promoting Pyk2 activation. Front Immunol (2021) 12:680068/FULL. doi: 10.3389/FIMMU.2021.680068/FULL
63. Angiari S, Runtsch MC, Sutton CE, Palsson-McDermott EM, Kelly B, Rana N, et al. Pharmacological activation of pyruvate kinase M2 inhibits CD4+ T cell pathogenicity and suppresses autoimmunity. Cell Metab (2020) 31:391–405.e8. doi: 10.1016/J.CMET.2019.10.015
64. Sharma K, Karl B, Mathew A v., Gangoiti JA, Wassel CL, Saito R, et al. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol (2013) 24:1901–12. doi: 10.1681/ASN.2013020126
65. Sas KM, Kayampilly P, Byun J, Nair V, Hinder LM, Hur J, et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight (2016) 1(15):e86976. doi: 10.1172/JCI.INSIGHT.86976
66. Qi W, Keenan HA, Li Q, Ishikado A, Kannt A, Sadowski T, et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat Med (2017) 23:753. doi: 10.1038/NM.4328
67. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev (2013) 93:137–88. doi: 10.1152/PHYSREV.00045.2011
68. Iacobini C, Vitale M, Pesce C, Pugliese G, Menini S. Diabetic complications and oxidative stress: a 20-year voyage back in time and back to the future. Antioxidants (2021) 10:727. doi: 10.3390/ANTIOX10050727
69. Zhang G, Darshi M, Sharma K. The warburg effect in diabetic kidney disease. Semin Nephrol (2018) 38:111–20. doi: 10.1016/J.SEMNEPHROL.2018.01.002
70. Brownlee M. The pathobiology of diabetic complications - a unifying mechanism. Diabetes (2005) 54:1615–25. doi: 10.2337/DIABETES.54.6.1615
71. Nishikawa T, Edelstein D, Brownlee M. The missing link: a single unifying mechanism for diabetic complications. Kidney Int (2000) 58:S26–30. doi: 10.1046/J.1523-1755.2000.07705.X
72. Iacobini C, Vitale M, Haxhi J, Pesce C, Pugliese G, Menini S. Mutual regulation between redox and hypoxia-inducible factors in cardiovascular and renal complications of diabetes. Antioxidants (2022) 11:2183. doi: 10.3390/ANTIOX11112183
73. García-Pastor C, Benito-Martínez S, Moreno-Manzano V, Fernández-Martínez AB, Lucio-Cazaña FJ. Mechanism and consequences of the impaired hif-1α response to hypoxia in human proximal tubular HK-2 cells exposed to high glucose. Sci Rep (2019) 9(1):15868. doi: 10.1038/S41598-019-52310-6
74. Gao W, Ferguson G, Connell P, Walshe T, O’Brien C, Redmond EM, et al. Glucose attenuates hypoxia-induced changes in endothelial cell growth by inhibiting HIF-1α expression. Diabetes Vasc Dis Res (2014) 11:270–80. doi: 10.1177/1479164114533356/ASSET/IMAGES/LARGE/10.1177_1479164114533356-FIG2.JPEG
75. Isoe T, Makino Y, Mizumoto K, Sakagami H, Fujita Y, Honjo J, et al. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int (2010) 78:48–59. doi: 10.1038/KI.2010.99
76. Li R, Uttarwar L, Gao B, Charbonneau M, Shi Y, Chan JSD, et al. High glucose up-regulates ADAM17 through HIF-1α in mesangial cells. J Biol Chem (2015) 290:21603–14. doi: 10.1074/JBC.M115.651604
77. la Sala L, Pujadas G, de Nigris V, Canivell S, Novials A, Genovese S, et al. Oscillating glucose and constant high glucose induce endoglin expression in endothelial cells: the role of oxidative stress. Acta Diabetol (2015) 52:505–12. doi: 10.1007/S00592-014-0670-3/FIGURES/3
78. Lund J, Ouwens DM, Wettergreen M, Bakke SS, Thoresen GH, Aas V. Increased glycolysis and higher lactate production in hyperglycemic myotubes. Cells (2019) 8:1101. doi: 10.3390/CELLS8091101
79. Gordin D, Shah H, Shinjo T, St-Louis R, Qi W, Park K, et al. Characterization of glycolytic enzymes and pyruvate kinase M2 in type 1 and 2 diabetic nephropathy. Diabetes Care (2019) 42:1263–73. doi: 10.2337/DC18-2585
80. Fu J, Shinjo T, Li Q, St-Louis R, Park K, Yu MG, et al. Regeneration of glomerular metabolism and function by podocyte pyruvate kinase M2 in diabetic nephropathy. JCI Insight (2022) 7(5):e155260. doi: 10.1172/JCI.INSIGHT.155260
81. Liu H, Takagaki Y, Kumagai A, Kanasaki K, Koya D. The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1α accumulation. J Diabetes Investig (2021) 12:697–709. doi: 10.1111/JDI.13478
82. Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol (2012) 8:839. doi: 10.1038/NCHEMBIO.1060
83. Catrina SB, Zheng X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia (2021) 64:709–16. doi: 10.1007/S00125-021-05380-Z/FIGURES/2
84. Gunton JE. Hypoxia-inducible factors and diabetes. J Clin Invest (2020) 130:5063–73. doi: 10.1172/JCI137556
85. Packer M. Mutual antagonism of hypoxia-inducible factor isoforms in cardiac, vascular, and renal disorders. JACC Basic Transl Sci (2020) 5:961–8. doi: 10.1016/J.JACBTS.2020.05.006
86. Chen Z, Zhu Z, Liang W, Luo Z, Hu J, Feng J, et al. Reduction of anaerobic glycolysis contributes to angiotensin II-induced podocyte injury with foot process effacement. Kidney Int (2023) 103:735–48. doi: 10.1016/J.KINT.2023.01.007
87. Apostolidi M, Vathiotis IA, Muthusamy V, Gaule P, Gassaway BM, Rimm DL, et al. Targeting pyruvate kinase M2 phosphorylation reverses aggressive cancer phenotypes. Cancer Res (2021) 81:4346–59. doi: 10.1158/0008-5472.CAN-20-4190
88. Sjöholm K, Carlsson LMS, Svensson PA, Andersson-Assarsson JC, Kristensson F, Jacobson P, et al. Association of bariatric surgery with cancer incidence in patients with obesity and diabetes: long-term results from the Swedish obese subjects study. Diabetes Care (2022) 45:444–50. doi: 10.2337/DC21-1335
89. Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, et al. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature (2018) 559:637–41. doi: 10.1038/s41586-018-0350-5
90. Meng X, Lu Z, Lv Q, Jiang Y, Zhang L, Wang Z. Tumor metabolism destruction via metformin-based glycolysis inhibition and glucose oxidase-mediated glucose deprivation for enhanced cancer therapy. Acta Biomater (2022) 145:222–34. doi: 10.1016/j.actbio.2022.04.022
91. Devic S. Warburg effect - a consequence or the cause of carcinogenesis? J Cancer (2016) 7:817. doi: 10.7150/JCA.14274
92. Unterlass JE, Curtin NJ. Warburg and Krebs and related effects in cancer. Expert Rev Mol Med (2019) 21:e4. doi: 10.1017/ERM.2019.4
93. Anastasiou D. Tumour microenvironment factors shaping the cancer metabolism landscape. Br J Cancer (2017) 116:277–86. doi: 10.1038/bjc.2016.412
94. Choi S, Hong SP, Bae JH, Suh SH, Bae H, Kang P, et al. Hyperactivation of YAP/TAZ drives alterations in mesangial cells through stabilization of n-myc in diabetic nephropathy. J Am Soc Nephrol (2023) 34(5):809–28. doi: 10.1681/ASN.0000000000000075
95. Schalkwijk CG, Stehouwer CDA. Methylglyoxal, a highly reactive dicarbonyl compound, in diabetes, its vascular complications, and other age-related diseases. Physiol Rev (2020) 100:407–61. doi: 10.1152/PHYSREV.00001.2019/ASSET/IMAGES/LARGE/Z9J0012029260011.JPEG
96. Xiao W, Oldham WM, Priolo C, Pandey AK, Loscalzo J. Immunometabolic endothelial phenotypes: integrating inflammation and glucose metabolism. Circ Res (2021) 129:9–29. doi: 10.1161/CIRCRESAHA.120.318805
97. Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signaling. Nature (2018) 562:600. doi: 10.1038/S41586-018-0622-0
98. Galligan JJ, Wepy JA, Streeter MD, Kingsley PJ, Mitchener MM, Wauchope OR, et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc Natl Acad Sci USA (2018) 115:9228–33. doi: 10.1073/PNAS.1802901115/SUPPL_FILE/PNAS.1802901115.SD03.XLSX
99. Iacobini C, Menini S, Blasetti Fantauzzi C, Pesce CM, Giaccari A, Salomone E, et al. FL-926-16, a novel bioavailable carnosinase-resistant carnosine derivative, prevents onset and stops progression of diabetic nephropathy in db/db mice. Br J Pharmacol (2018) 175:53–66. doi: 10.1111/BPH.14070
100. Iacobini C, Vitale M, Haxhi J, Pesce C, Pugliese G, Menini S. Food-related carbonyl stress in cardiometabolic and cancer risk linked to unhealthy modern diet. Nutrients (2022) 14:1061. doi: 10.3390/NU14051061
101. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products. Circulation (2006) 114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854
102. Bellahcène A, Nokin MJ, Castronovo V, Schalkwijk C. Methylglyoxal-derived stress: an emerging biological factor involved in the onset and progression of cancer. Semin Cancer Biol (2018) 49:64–74. doi: 10.1016/J.SEMCANCER.2017.05.010
103. Menini S, Iacobini C, de Latouliere L, Manni I, Ionta V, Blasetti Fantauzzi C, et al. The advanced glycation end-product nϵ -carboxymethyllysine promotes progression of pancreatic cancer: implications for diabetes-associated risk and its prevention. J Pathol (2018) 245:197–208. doi: 10.1002/PATH.5072
Keywords: aerobic glycolysis, diabetes mellitus, hypoxia inducible factor (HIF)-1α, inflammation, methylglyoxal (MGO), oxidative phoshorylation, pyruvate kinase M isoform 2, Warburg effect
Citation: Iacobini C, Vitale M, Pugliese G and Menini S (2023) The “sweet” path to cancer: focus on cellular glucose metabolism. Front. Oncol. 13:1202093. doi: 10.3389/fonc.2023.1202093
Received: 07 April 2023; Accepted: 17 May 2023;
Published: 25 May 2023.
Edited by:
Thibaut Barnoud, Medical University of South Carolina, United StatesReviewed by:
Mary L. Taub, University at Buffalo, United StatesCristina Nuevo-Tapioles, New York University, United States
Mariana Renovato-Martins, Federal Fluminense University, Brazil
Copyright © 2023 Iacobini, Vitale, Pugliese and Menini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Menini, c3RlZmFuby5tZW5pbmlAdW5pcm9tYTEuaXQ=