 Seema Rai
Seema Rai Gunja Roy
Gunja Roy Younis Ahmad Hajam
Younis Ahmad Hajam- 1Department of Zoology Guru Ghasidas Vishwavidyalaya, Bilaspur, India
- 2Department of Life Sciences and Allied Health Sciences, Sant Bhag Singh University, Jalandhar, India
Melatonin, (N-acetyl-5-methoxytryptamine) an indoleamine exerts multifaced effects and regulates numerous cellular pathways and molecular targets associated with circadian rhythm, immune modulation, and seasonal reproduction including metabolic rewiring during T cell malignancy. T-cell malignancies encompass a group of hematological cancers characterized by the uncontrolled growth and proliferation of malignant T-cells. These cancer cells exhibit a distinct metabolic adaptation, a hallmark of cancer in general, as they rewire their metabolic pathways to meet the heightened energy requirements and biosynthesis necessary for malignancies is the Warburg effect, characterized by a shift towards glycolysis, even when oxygen is available. In addition, T-cell malignancies cause metabolic shift by inhibiting the enzyme pyruvate Dehydrogenase Kinase (PDK) which in turn results in increased acetyl CoA enzyme production and cellular glycolytic activity. Further, melatonin plays a modulatory role in the expression of essential transporters (Glut1, Glut2) responsible for nutrient uptake and metabolic rewiring, such as glucose and amino acid transporters in T-cells. This modulation significantly impacts the metabolic profile of T-cells, consequently affecting their differentiation. Furthermore, melatonin has been found to regulate the expression of critical signaling molecules involved in T-cell activations, such as CD38, and CD69. These molecules are integral to T-cell adhesion, signaling, and activation. This review aims to provide insights into the mechanism of melatonin’s anticancer properties concerning metabolic rewiring during T-cell malignancy. The present review encompasses the involvement of oncogenic factors, the tumor microenvironment and metabolic alteration, hallmarks, metabolic reprogramming, and the anti-oncogenic/oncostatic impact of melatonin on various cancer cells.
1 Introduction
The human body’s homeostasis depends on the blood and immune system derived from the hematopoietic stem cells (HSCs), which are multipotent stem cells that can generate various types of blood cells. HSCs produce erythrocytes, which transport life-sustaining oxygen; Thrombocytes, which help with blood clotting; and leukocytes primarily white blood cells, which fight infections and disease. These are derived from versatile sources of stem cells. HSCs coordinate a key transition within this milieu of white blood cells and produce T lymphocytes also known as T-cells, which act as guardians of the immune system (1).
T cells play a crucial role in immune response by generating various cytokines and interleukins, while B cells produce antibodies. These cell types exhibit different cell surface characteristics, functions, and developmental pathways. Cell surface markers are protein molecules expressed on the surface of a cell that particular antibodies may recognize. They may be used as an indicator for cell identification, functions, and conditions. Neoplastic cells are those that have grown abnormally and uncontrollably, resulting in the creation of tumors or malignancies. By analyzing the cell surface markers on neoplastic cells, their origin (which type of normal cell they are derived from), nature (whether they are benign or malignant, and what subtype of cancer they belong to), and involvement in the development of the lymphoproliferative disease can be recognized (2). Dysregulation in the functioning of immune cells can lead to serious disorders, and immune dysfunction may contribute to the development and progression of malignancies or cancer (3). Cancer cells are aberrant cells that have undergone abnormal development and uncontrolled proliferation. Different cells of the immune system, including T cells and natural killer (NK) cells, play a crucial role in shriveling the growth and proliferation of cancer cells, as well as eliminating malignant cells. Therefore, targeting NK cells and T-cell immunotherapy hold significant promise as potential tools for cancer treatment (4). Several factors, such as genetic predisposition (5), human T-cell leukemia virus (HTLV-1), Epstein-Barr virus, and autoimmune disorders, have been implicated in the pathogenesis of T-cell malignancies (6).
T-cell malignancies are a diverse set of illnesses that each result from the clonal evolution of damaged T cells at different phases of life. Nonetheless, acute lymphoblastic leukemia (T-ALL), the most frequent kind of T cell malignancy found in children has been reported to account for 15% to 25% (7). A non-Hodgkin lymphoma with a biology comparable to T-ALL is T-lymphoblastic lymphoma (T-LLy). The human T cell lymphocytic virus type 1 (HTLV1) is the primary cause of the exceedingly aggressive blood malignancy known as adult T cell leukemia/lymphoma (ATLL) (8, 9).T cell large granular lymphocytic leukemia (T-LGL) and T prolymphocytic leukemia (T-PLL) are two further uncommon types of T cell leukemia (10). The two main types of T cell lymphomas are cutaneous T cell lymphoma (CTCL) and peripheral T cell lymphoma (PTCL). The two most prevalent subtypes of CTCL are mycosis fungoids (MF) and Sezary syndrome (SS), which together account for the majority of cases. Angioimmunoblastic T cell lymphoma (AITL), extranodal natural killer (NK)-T cell lymphoma (ENKTL), enteropathy-associated T cell lymphoma (EATL), hepatosplenic T cell lymphoma (HSTCL), and PTCL-not otherwise specified (PTCL-NOS), the most prevalent of the group, are among the subtypes of PTCL that can be distinguished (11). According to previous reports, the incidence of cancer is expected to increase significantly by 2050, with about 19.3 million new cases annually, making it the second leading cause of death worldwide (12). The report by the World Health Organization (WHO) confirms that as of 2018, there were 9.6 million cancer-related deaths worldwide, with morbidity and mortality rates increasing significantly over time. Cancer has become a major health issue globally, and it requires immediate attention from researchers and clinicians. The Global Cancer Observatory (GLOBOCAN) of the International Agency for Research on Cancer of the World Health Organisation predicts that by 2040, there will be 27.5 million new cases of various cancers annually. This represents an increase of 61.7% when compared to recent statistics (13).
Chemotherapy, radiation therapy, and surgery represent the conventional approaches recommended and employed by doctors for treating cancer patients. Regrettably, these methods often come with a host of adverse effects and provide no guarantee against cancer recurrence. Consequently, there exists a pressing demand for novel therapeutic agents capable of enhancing post-treatment survival and quality of life for cancer patients. These innovative agents might take the form of biological molecules designed to modulate the immune system and possess anti-cancer properties. One illustrative instance of such a biological molecule is melatonin, a naturally occurring hormone renowned for its relatively minimal side effects when compared to synthetic drugs. Melatonin also exhibits the ability to target specific genes and DNA associated with the development and progression of cancer. In recent years, numerous studies have been conducted to delve into the multifaceted functions and role of melatonin in human health and various disease states (14).
In recent years, extensive study has been conducted to discover the various functions, uses, and properties of melatonin in human physiology and pathology. Melatonin is an indolamine and neurohormone secreted rhythmically by the pineal gland. Aaron B Lerner in 1958, isolated melatonin from the bovine pineal extract and identified it chemically as 5-Methoxy-N-Acetyl tryptamine. The molecular mechanism for the physiological effects of melatonin has been explained which provides evidence regarding its involvement in the regulation of the sleep-wake cycle, circadian rhythms, reproduction, immunological response, apoptosis, proliferation, metastasis, angiogenesis, and oxidative stress via receptor-mediated (Nuclear and membrane receptors) and nonreceptor activities carried out through non-receptor-dependent pathways (15–20). There are numerous cytoplasmic and mitochondrial binding sites for melatonin in addition to membrane and nuclear receptors (15, 21–28). It is a lipophilic molecule and hence virtue makes it a potent antioxidative agent, thereby it reduces the free radicals like- reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation and maintains a balance between the anti-oxidant and ROS/RNS (14) (Figure 1).
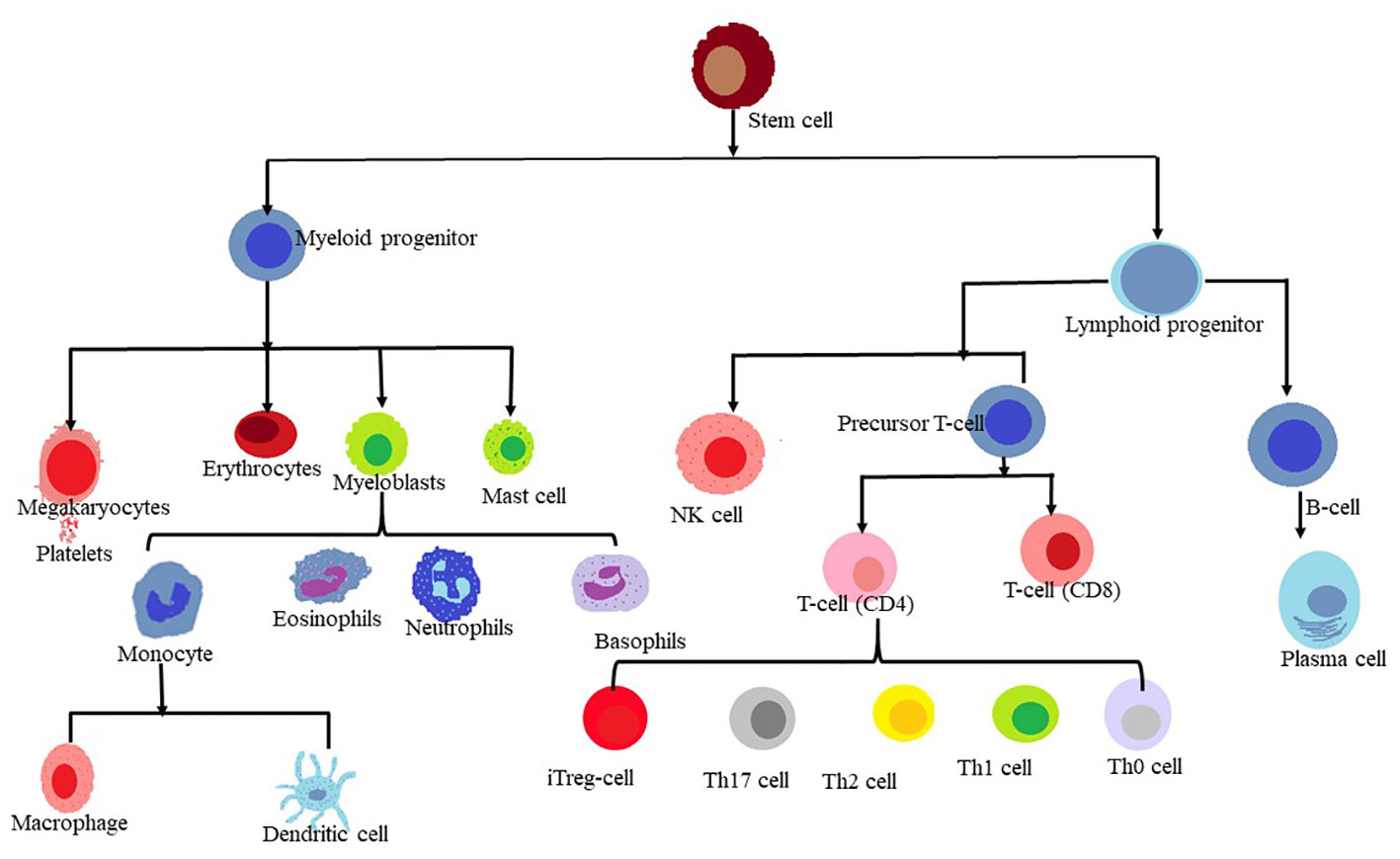
Figure 1 Lineage of myeloid and lymphoid cells.
In addition, melatonin is a well-known molecule that can rewire metabolic pathways. Melatonin has also been found to have a significant role in the regulation of various types of cancer. It exhibits anti-angiogenic and anti-metastatic properties and has the potential to prevent cell necrosis, as observed in some studies on breast cancer cell lines. Moreover, melatonin regulates transcription factors such as NF-kB and influences the production of cytokines and interleukins (29, 30). A recent study using a nude mice model demonstrated that melatonin treatment (10 mg/kg) via intraperitoneal administration for three weeks resulted in a reduction in tumor size (31). It regulates factors such as vascular endothelial growth factor (VEGF), hypoxia-inducible factor-1α (HIF-1α), and human epidermal growth factor receptor 2 (HER-2) (32). In patients with lung cancer caused by smoking cigarettes, a significant increase in the levels of pro-inflammatory cytokines, which are signaling molecules involved in promoting inflammation, has been observed (32). However, it has been studied that the administration of melatonin can counteract these effects by reducing neutrophil counts and inhibiting the production of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), interleukin-8 (IL-8), and interleukin-6 (IL-6) (33).
Numerous investigations also supported the anticancer and oncostatic properties of melatonin, which are mediated by various modes of action (34, 35). Melatonin was further employed in conjunction with other anticancer therapies to supplement standard treatments and lessen negative effects (34–36).
2 Cancer and oncogenic factors
Cancer is a multifaceted disease characterized by uncontrolled cell growth and proliferation, leading to the formation of malignant tumors. It arises from a combination of genetic and environmental factors, which interact to initiate and promote oncogenesis. Understanding the intricate interplay of these oncogenic factors is crucial for elucidating the underlying mechanisms of cancer development and identifying potential targets for therapeutic interventions. In recent years, significant advancements in cancer research have shed light on the diverse array of oncogenic factors involved in this complex disease. Genetic alterations play a fundamental role in the development of cancer (34). Mutations in key genes, such as oncogenes and tumor suppressor genes, can disrupt the delicate balance of cellular processes, leading to uncontrolled cell growth. Oncogenes are genes that, when mutated or overexpressed, promote cell proliferation and survival, while tumor suppressor genes act as guardians of the genome, regulating cell cycle progression and preventing the formation of tumors. Research has uncovered numerous oncogenes and tumor suppressor genes, including well-known examples like TP53, KRAS, and BRCA1/2, which have provided critical insights into the molecular basis of cancer. In addition to genetic alterations, Carcinogens present in the environment, such as tobacco smoke, ultraviolet radiation, and certain chemicals, can induce DNA damage and initiate oncogenic processes, and lifestyle factors, such as diet, physical activity, and exposure to toxins, can influence cancer risk by modulating various signaling pathways and epigenetic modifications contribute significantly to cancer development (Figure 2).
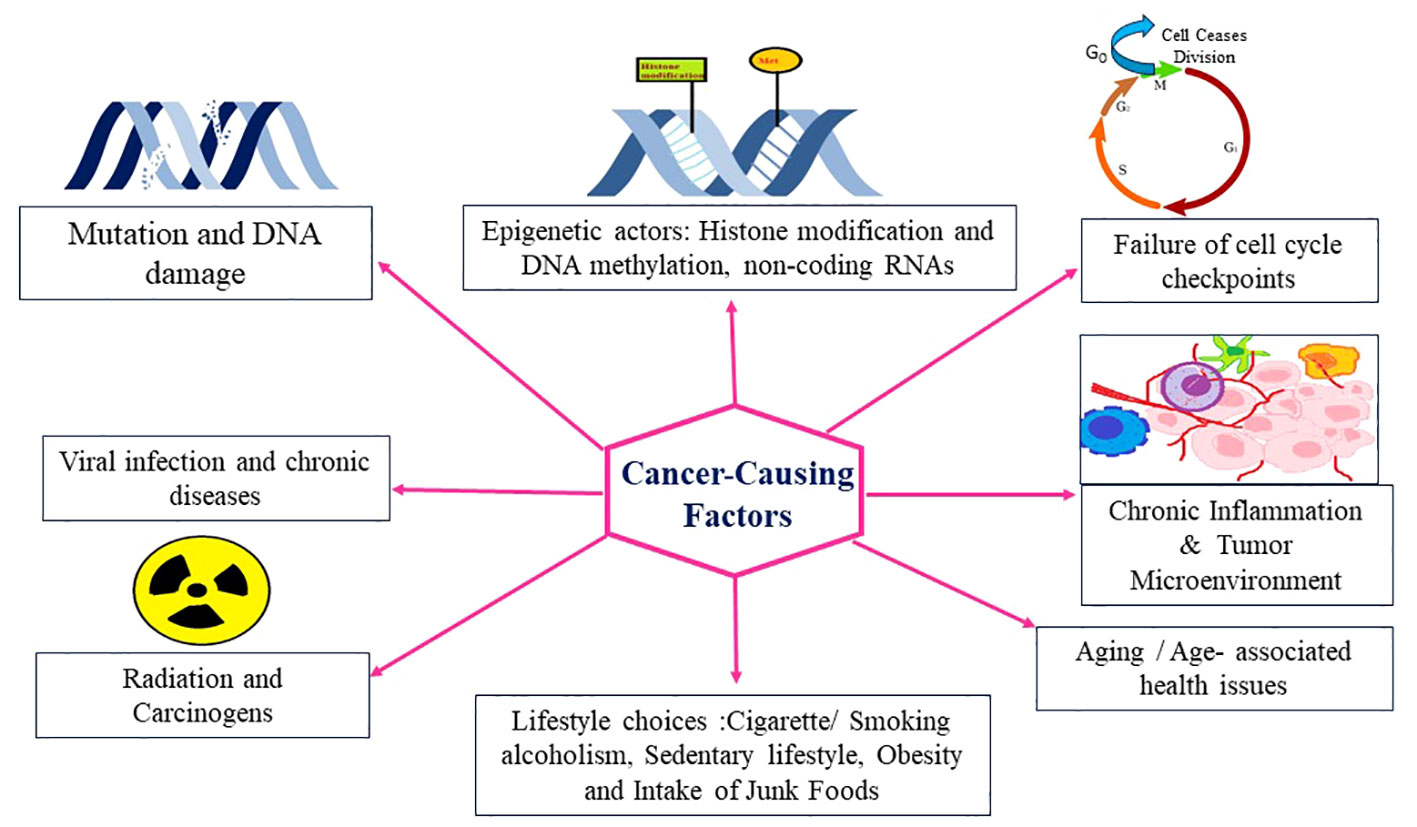
Figure 2 Different factors responsible for cancer.
2.1 Tumor microenvironment and metabolic alteration in malignant cells
Cells require macromolecules that support them in growth and division similarly malignant cells also require macromolecules for their development and these macromolecules are synthesized from scratch via rewiring the metabolic pathways inside the cells or may be obtained from other sources, to meet the metabolic demands required for their development and proliferation (37). The demand for malignant cells is aided by the tumor microenvironment (TEM). This is a complex dynamic network of host components that are recruited and reprogrammed by the tumor to support its proliferation and survival. The TEM is composed of various cellular and noon-cellular elements that interact with each other and with the malignant cells through physical interaction and molecular signals. The cellular components comprise immune cells –macrophage, dendritic cell, T-cells, B-cells, NK-cells, and mast cells, fibroblasts these are the main source of extracellular matrix (EMC) proteins and growth factors; endothelial cells, which form blood vessels that supply oxygen nutrients to the growing tumors and other cell types such as adipocytes, pericytes, and neutral cells (38, 39). The TEM plays a crucial role in the development, proliferation, metastasis, and response to therapy of tumors by affecting the biological nature and phenotype of tumor cells. The TEM can promote tumor cell proliferation, survival, migration, angiogenesis, stemness, epithelial-mesenchymal transition (EMT), drug resistance, and immune evasion. However, the TEM is not a static or homogenous entity, but rather a dynamic and heterogenous that modulates itself over time. And these modifications also alter the metabolic reprogramming in tumor cells (38, 40, 41). Previously it was believed that the TEM are consequence of the metabolic by-products of tumor cells, such as lactate, ammonia, and ROS. However, studies have shown that TEM can also initiate and sustain the metabolic rewiring of tumor cells. These tumor cells show some hallmarks in terms of metabolic reprogramming which favors their survival, development, and proliferation (41, 42).
The hallmarks of cancer cell metabolic rewiring involve aerobic glycolysis (Warburg effect), a hypoxic condition, that contributes to the cancer cell’s transformation by providing a tumor microenvironment (TEM) (43). Aerobic glycolysis or Warburg effect is the phenomenon of glucose fermentation into lactate or lactic acid in the presence of oxygen i.e., in aerobic conditions. This process provides anabolic carbons that can be used in biosynthetic pathways. Glucose uptake in normal cells is strictly regulated but, malignant cells have been reprogrammed to fuel unconstrained proliferation (44) and many malignant cell types have been reported to take up glucose at unusually elevated rates. Glucose could be transformed into a variety of cell-building components like lipids, amino acids, and, nucleotides, necessary for the development and proliferation of malignant cells. Aerobic glycolysis promotes the generation of electron carriers, which are required as cofactors in the redox process in cells. Elevated glucose uptake allows for increased synthesis of the reducing equivalent NADPH via the oxidative branch of the pentose phosphate pathways, whereas fermentation entails regeneration of the oxidizing NAD+ via the activity of lactate dehydrogenase (LDH) (45). These electron carriers are required for cellular redox balance and biosynthesis. Under hypoxic circumstances, aerobic glycolysis optimizes ATP generation (45). Hypoxia, or low oxygen level, is prevalent in solid tumors due to insufficient blood flow. Hypoxia can activate hypoxia-inducible factor-1α (HIF-1α), a transcription factor that controls numerous glycolysis-related genes. HIF-1α is a key regulator of cellular response to hypoxia condition and it also regulates the expression of genes that are responsible for angiogenesis, erythropoiesis, and acidosis regulation (45). HIF-1α induces genes like vascular endothelial growth factor (VEGF) (46). Angiogenesis generation of new blood vessels in developing malignant cells in an uncontrolled manner to meet the demand for oxygen and nutrients. On the contrary, it results in some disorganized structure with branching and splitting forming a chaotic and leaky network (47). Such a clumsy structure leads to inadequate blood flow, anaerobic conditions (hypoxic environment), and the generation of free radicals. Aerobic glycolysis leads to the generation of lactate which makes the tumor environment acidic and increases the free radical load. “Earlier” lactate is considered a by-product of enhanced aerobic glycolysis and accumulates in malignant cells, raising the pH of the tumor microenvironment but a recent investigation by Brandon et al. suggests that it fuels the tricarboxylic acid cycle (TCA) (48). Lactate promotes HIF-1α activity by blocking the prolyl hydroxylases 2 (PDH 2). However, the specific mechanism is still unclear (49). When PDH 2 gets inhibited HIF-1α localized into the nucleus and binds with its other subunit HIF-1β. Further, this complex binds with hypoxia-responsive elements (HREs) and activates the gene expression for the NOX genes (NOX1, NOX 2, NOX 4, and NOX 5) NOX enzymes also contribute to the production of free radicals Superoxide and hydrogen peroxide (50) by transferring the electrons from NADPH to oxygen (51). All these factors contribute to the establishment of a suitable condition for the development and proliferation of malignant cells known as a tumor microenvironment (TEM). TEM favors the abnormal development and proliferation of malignant cells and is associated with various pathways such as the PI3K/Akt/mTOR pathways, insulin-like growth factor-1 (IGF-1), hypoxia-inducible factor-1 (HIF-1), and NF-KB. These pathways play a direct role in promoting tumor development in the tumor microenvironment (TME), a condition characterized by low oxygen levels known as hypoxia (52).
The PI3K/Akt pathway is one of the most commonly altered pathways in human cancer. This pathway, along with mTOR, regulates the intake and use of various nutrients such as glucose, amino acids, nucleotides, and lipids. The PI3K/Akt pathway is downstream of several receptors such as G-protein coupled receptors (GPCR), receptor tyrosine kinase (RTK), integrins, and cytokine receptors. It is critical for maintaining regular cellular functions and enhancing cellular proliferation. While normal cells require external growth factors to activate these signaling pathways, malignant cells acquire activating mutations in PI3K subunits such as p110a (PI3CA) or enduring inactivating mutations in PTEN, a phosphatase that controls PI3K/Akt signaling (53, 54). MYC, a transcription factor, is connected to the PI3K/Akt/mTOR signaling pathway and is pivotal in regulating cellular nutrient utilization. It governs the expression of key metabolic genes such as HK2 and ENO1, that facilitate glucose breakdown, and glucose transporters that lead glucose into cells. The stability of MYC is maintained by PI3K/Akt suppression of FOXO and GSK3, which otherwise target MYC for degradation (53, 55) Figure 3.
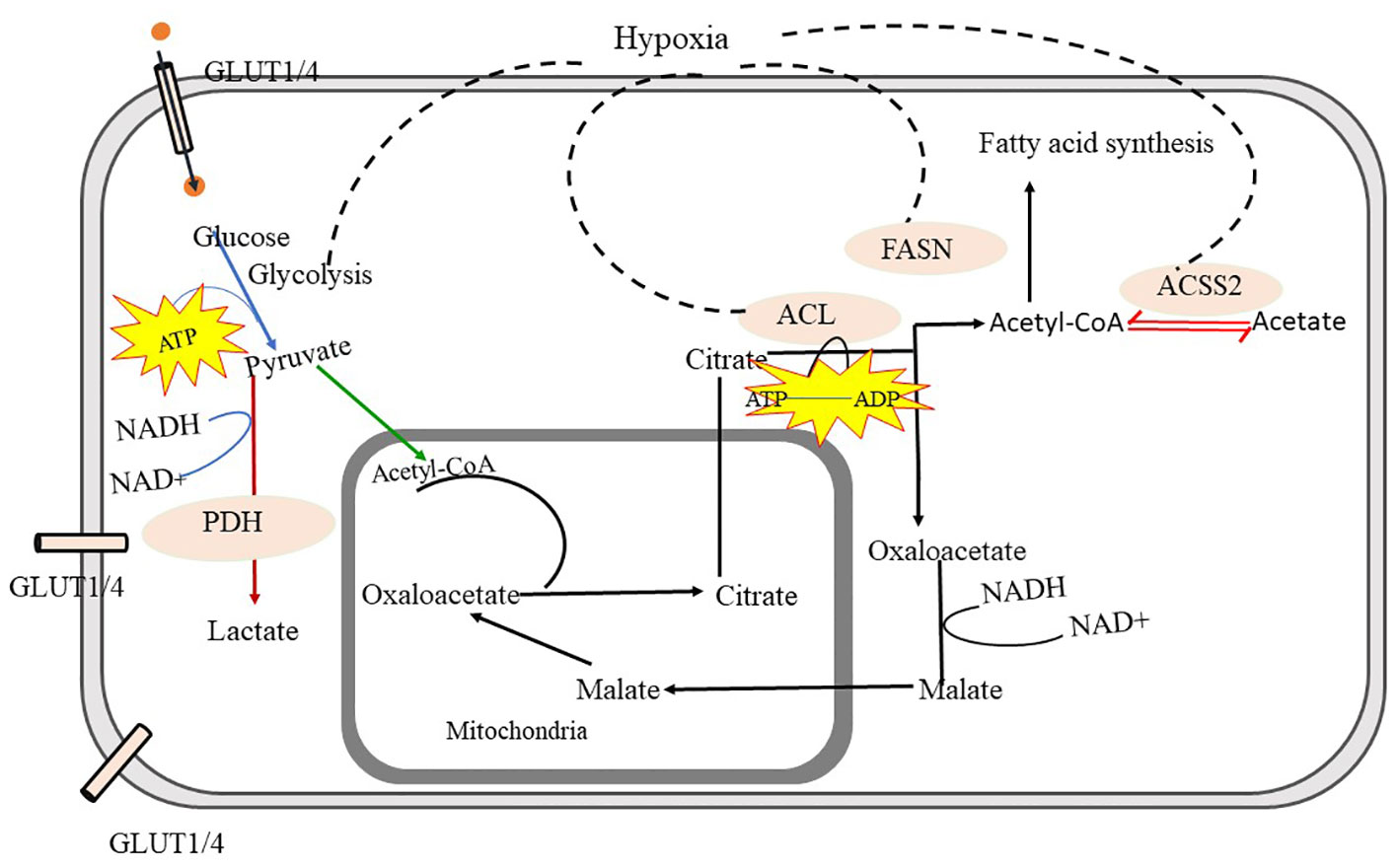
Figure 3 Metabolic Pathways in Malignant Form- Glycolysis is the crucial source to obtain energy through which glucose is converted into pyruvate. Pyruvate is further converted into acetyl-CoA and gets transferred into mitochondria, through the mediation of pyruvate dehydrogenase (PDH) and converted into lactate via fermentation. The tricarboxylic acid cycle (TCA) in mitochondria provides Citrate. Citrate is exported to cytosol and converted into acetyl-CoA and oxaloacetate with the help of ATP citrate lyase (ACL). Cytosolic acetyl-CoA is utilized in fatty-acid synthesis and oxaloacetate is further converted to malate and again malate is imported back into mitochondria and converted into oxaloacetate in malignant cells. cytosolic acetyl-CoA synthetase (ACSS2) facilitates the interconversion between acetyl-CoA and acetate, which helps to maintain the microenvironment in malignant cells. further fatty acid synthase (FASN) facilitates fatty acid synthesis using acetyl-CoA. In malignant cells, glycolysis, fatty acid synthesis via FACN, ACL, and ACSS, and import of oxaloacetate into mitochondria is highly increased. ATP citrate lyase -ACL, Acetyl-CoA synthetase- (ACSS2), Fatty acid synthesis (FACN).
3 Hallmarks of T-cell malignancies
T-cell malignancy, also known as T-cell leukemia or lymphoma, represents an aggressive form of T-cell cancer characterized by the uncontrolled proliferation of T lymphocytes, a type of white blood cells responsible for immune responses. Various factors contribute to the development of T-cell malignancies, including genetic abnormalities and chromosomal rearrangements that disrupt the normal cell cycle and apoptotic mechanisms (56). These abnormalities lead to rapid and uncontrolled proliferation of malignant T-cells, enabling their survival and accumulation in the body. Glucose and glutamine serve as essential fuel sources for this aberrant proliferation (57). The tumor microenvironment may also play a role in triggering the formation of T-cell malignancies, as the presence of a small population of T-cells within the tumor microenvironment can be negatively regulated by PD-L1 (programmed cell death ligand 1) (58).
T-cell non-Hodgkin lymphomas (NHLs) are a subtype of NHLs and represent rare cancers, accounting for approximately 12% of all lymphomas (56). The prevalence of T-cell NHLs varies across different geographic regions, ranging from 18.3% in Hong Kong to 1.5% in Vancouver, British Columbia, Canada. This disparity may be attributed to varying exposure levels to pathogenic agents such as HTLV-1 and Epstein-Barr virus (EBV) in Asian countries. T-cell NHLs often manifest as extranodal diseases, making the distinction between reactive processes and lymphoma challenging due to varying rates of necrosis and apoptosis observed in biopsy specimens. However, advancements in immunophenotypic, cytogenetic, and molecular studies have significantly improved diagnostic capabilities, classification, and prognosis for T-cell NHLs (59).
T-cell malignancy is a complex and diverse disorder that is still under investigation to understand the relationship between aberrant T-cell behavior and the development of malignant cells (60). It can be categorized into T-cell lymphomas and T-cell leukemia, both of which exhibit dysfunctional T-cell characteristics. These hematologic malignancies often present with aggressive features, such as high tumor burden, elevated white blood cell counts, and the presence of large thymic masses and pleural effusions. The rapid tumor growth observed in T-cell malignancies may be attributed to the ability of immature thymocytes to undergo high rates of division during critical stages of early T-cell development. This accelerated growth is fueled by the activation of oncogenic pathways that regulate cell growth and proliferation in early T-cell progenitors (60). T-cell lymphoid malignancies exhibit heterogeneity and display thymus-specific cell surface markers. The most common T-cell cancers in children (10-15%) and young adults (25%) are T-ALL and T-LLy (61). T-ALL arises from the dysfunction of T-cells due to translocations involving specific genes associated with T-cell receptors (43). T-cell lymphoblastic leukemia, on the other hand, originates from the malignant transformation of T-cell progenitors. Adult T-cell leukemia/lymphoma (ATL) was first identified as an aggressive leukemia/lymphoma of mature T cells in Kyoto, Japan, in 1977 by Uchyama and colleagues. It was later discovered that the pathogen responsible for ATL is the human T-cell lymphotropic virus type-1 (HTLV-1), which was found in a patient with an aggressive form of mycosis fungoides three years later. HTLV-1 is endemic in the Caribbean Islands, central Africa, and certain regions of Japan, particularly Kyushu and Okinawa. Transmission of HTLV-1 occurs through breast milk from carrier mothers, sexual contact (mainly between males and females), blood transfusions, and the sharing of contaminated intravenous needles. After HTLV-1 infection, only a subset of carriers (6% male and 2% female patients) develop ATL after a long latent period. HTLV-1 is not very dominant and known in India, but there has been a recent increase in ATL cases in a tertiary healthcare center in northern Kerala. ATL can manifest in various forms, including acute, chronic, lymphomatous, and smoldering types, with skin lesions often appearing initially (44). Other forms of T-cell malignancy include T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) arising from T-cell precursors, such as thymocytes, as well as T-cell large granular lymphocyte (LGL) leukemia, adult T-cell leukemia/lymphoma (ATL), and T-cell prolymphocytic leukemia (T-PLL) originating from mature T cells (62). T-cell lymphomas are rare and aggressive cancers with a poor prognosis, often due to chemotherapy resistance and patient intolerance to existing toxic chemotherapy regimens (45). T-cell lymphoblastic leukemia is considered a model for studying cancer genesis and development. Despite extensive research on the genetic rewiring of T-cell lymphoblastic leukemia, the precise molecular mechanisms underlying the transition from precursor T-cells to a malignant form remain unclear (46).
The development of T-cell malignancy involves complex interactions between environmental factors and genetics. Whole-genome sequencing studies have revealed distinct epigenetic signatures in exhausted T cells. Abnormal chromatin landscapes and altered epigenetic states have been observed in exhausted or dysfunctional T cells, closely linked to their functional state (58). Specific markers exhibit upregulation or downregulation in malignancy. Following repeated antigenic stimulation, high expression of PD-1 and other co-inhibitory molecules has been identified as an indicator of T-cell exhaustion and dysfunction, particularly in cells isolated from the tumor microenvironment (63). Epigenetic factors such as DNA methylation and histone modification control PD-1 expression and T-cell dysfunction. DNA methylation enzymes DNMT1 and DNMT3B are increased in dysfunctional or exhausted T cells, and inhibition of DNA methylation can restore normal T-cell function (64). DNA methylation plays a significant role in the expression of many genes involved in cell growth, differentiation, and genomic integrity during lineage commitment of lymphoid precursor cells, contributing to the production of mature lymphocytes with effective antigen receptors (65). Decreased expression of DNMT1 leads to gene hypomethylation, resulting in increased production of IL-2 and IFN-γ in T lymphocytes. IL-2 and IFN-γ play crucial roles in T-cell differentiation (66). Increased DNMT expression and subsequent hypermethylation of the cyclin-dependent kinase inhibitor p15INK4B have been associated with acute myeloid leukemia (AML) and the acute phase of chronic myeloid leukemia (CML) (67).
Malignant or dysfunctional T cells exhibit alterations in the expression levels of various markers. The expression of CD30 marker is increased in T-cell lymphomas. CD30 is a glycoprotein belonging to the tumor necrosis factor receptor superfamily and serves as a marker of T-cell activation and proliferation. CD30 receptor (CD30R) interaction with CD30 ligand enhances T-cell survival through the stimulation of NF-kB and MAP kinase pathways. Previous studies suggest that CD30 promotes the survival of malignant T cells by activating NF-kB and anti-apoptotic factors (68). This marker is specifically involved in the differentiation of CD4+ cells into Th2 cells and stimulates the synthesis and secretion of Th2-associated cytokines interleukin-13 (IL-13) and interleukin-4 (IL-4) while inhibiting the expression of Treg cells (69). CD30 also promotes the production of proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), contributing to inflammation and the development of malignancy. Interleukin-2 receptor (IL-2R), also known as CD25, exhibits upregulation in malignant T cells. IL-2R plays a role in T-cell proliferation, activation, and survival. CD25 upregulation is associated with the expression of regulatory T cells (Treg), which play a crucial role in immunological tolerance by suppressing immune response generation and activating CD8+ cells and NK cells, thus aiding in immune escape (70, 71). Increased IL-2 production promotes T-cell proliferation (72). CD52 receptor, also known as Campath 1 antigen, is also upregulated in malignant T cells and is associated with immune tolerance (73). The upregulation of these receptors contributes to immune evasion. On the other hand, some markers show downregulation in malignant T cells. The T-cell receptor complex (TCR), an essential component of the normal T-cell immune response, consists of TCR proteins responsible for recognizing specific antigens presented by major histocompatibility complex (MHC) molecules on antigen-presenting cells (APCs). The expression of TCR complex markers is reduced or diminished in malignant T cells, leading to functional consequences. Studies suggest the presence of dysfunctional assembly of the beta chain of the TCR complex in malignant T cells (74, 75). CD3 marker expression is also decreased in malignant T cells. CD3 promotes T-cell differentiation into various T-cell subtypes, and its reduced expression level restricts T-cell differentiation (76).
3.1 Metabolic reprogramming in T-cell malignancy
Malignant T-cells show various alterations in terms of their metabolic reprogramming to fuel their survival and vigorous proliferation and to meet these demands rewire their several signaling pathways like Glycolysis and glutaminolysis pathways to meet the demands of energy,
3.1.1 Glucose metabolism in malignant T-cells
Aerobic glycolysis or Warburg effect is the phenomenon of glucose fermentation into lactate or lactic acid in the presence of oxygen i.e., in aerobic conditions. This process provides anabolic carbons that can be used in biosynthetic pathways. Glucose uptake in normal cells is strictly regulated but, malignant cells have been reprogrammed to fuel unconstrained proliferation (77) and many malignant cell types have been reported to take up glucose at unusually elevated rates. Glucose could be transformed into a variety of cell-building components like lipids, amino acids, and, nucleotides, necessary for the development and proliferation of malignant cells. Aerobic glycolysis promotes the generation of electron carriers, which are required as cofactors in the redox process in cells. Elevated glucose uptake allows for increased synthesis of the reducing equivalent NADPH via the oxidative branch of the pentose phosphate pathways, whereas fermentation entails regeneration of the oxidizing NAD+ via the activity of lactate dehydrogenase (LDH) (78). These electron carriers are required for cellular redox balance and biosynthesis. Under hypoxic circumstances, aerobic glycolysis optimizes ATP generation. Hypoxia, or low oxygen level, is prevalent in solid tumors due to insufficient blood flow. Hypoxia can activate hypoxia-inducible factor-1α (HIF-1α), a transcription factor that controls numerous glycolysis-related genes. HIF-1α is a key regulator of cellular response to hypoxia condition and it also regulates the expression of genes that are responsible for angiogenesis, erythropoiesis, and acidosis regulation (79). HIF-1α induces genes like vascular endothelial growth factor (VEGF) (80). Angiogenesis generation of new blood vessels in developing malignant cells in an uncontrolled manner to meet the demand for oxygen and nutrients. On the contrary, it results in some disorganized structure with branching and splitting forming a chaotic and leaky network (81). Such a clumsy structure leads to inadequate blood flow, anaerobic conditions (hypoxic environment), and the generation of free radicals. Anaerobic glycolysis leads to the generation of lactate which makes the tumor environment acidic and increases the free radical load. “Earlier” lactate is considered a by-product of enhanced aerobic glycolysis and accumulated in malignant cells, raising the pH of the tumor microenvironment but a recent investigation by Brandon et al. suggests that it fuels the tricarboxylic acid cycle (TCA) (48). Lactate promotes HIF-1α activity by blocking the prolyl hydroxylases 2 (PDH 2). However, the specific mechanism is still unclear (49). When PDH 2 gets inhibited HIF-1α localized into the nucleus and binds with its other subunit HIF-1β. Further, this complex binds with hypoxia-responsive elements (HREs) and activates the gene expression for the NOX genes (NOX1, NOX 2, NOX 4, and NOX 5) NOX enzymes also contribute to the production of free radicals Superoxide and hydrogen peroxide (50) by transferring the electrons from NADPH to oxygen (51).
3.1.2 Amino acid metabolism in malignant T-cells
Amino acids are one of the crucial nutrients for T cells, and they have various roles in T cell metabolism, such as building blocks for protein synthesis, sources of energy, and regulators of signaling pathways. A different subset of T-cells has a distinct metabolic profile and preferences for amino acids. Essential and non-essential amino acids also uptake get rewired in malignant T-cells. Glutamate is the essential amino acid that provides energy to a malignant form of T-cells along with enhanced glycolysis. Glutaminolysis is the conversion of glutamine an amino acid, into glutamate and then into α-ketoglutrate, which can enter the TCA cycle to generate energy and biosynthetic substrate. It fuels the TCA cycles and supports anabolic processes, such as nucleotide synthesis. It also modulates the redox balance and the epigenetic regulation of T-cells by producing NADPH and donating methyl group. Glutaminolysis pathways regulate the mTOR pathway to control T-cell proliferation (82). Various factors impact the survival and proliferation of malignant T-cells like TEM which provide stimulation via cytokines environment, antigen stimulation environments, and crucial factors.
Glutamines (Glu) are transported into T-cells via amino acid transporters (AATs), which are sodium (Na+)-dependent transporters. These transporters use concentration gradients to allow Glu and Na+ ions into the cell and expel any excess concentration of Na+ ions. The transporters that perform this function belong to the solute carrier protein 1 member 5 (SLC1A5) and SLC38A1 families, with ASCT2 being the most important. Subsequently, Glutamine (Glu) enters the mitochondria via the SLC1A5 variant (SLC1A5_var), which is an AAT located on the mitochondrial membrane through its N-terminal targeting signal. Here, Gln is decomposed into glutamate (Glu) and ammonia by the action of mitochondrial glutaminase. Alternatively, Gln can be transformed into α-ketoglutaric acid via glutamate dehydrogenase 1 (GLUD1) (83).
Arginine metabolism is the conversion of arginine to ornithine and urea using arginase, or into citrulline and nitric oxide (NO) through nitric oxide synthase (NOS). Arginine metabolism is crucial for nucleotide synthesis, NO production, urea cycle, and immune regulation. Different subsets of T-cells, and other immune cells in the tumor microenvironment modulating arginine metabolism.
In summary, metabolic reprogramming in cancer cells, including T-cell malignancies, is associated with altered nutrient utilization and adaptations to the TME. Understanding the specific metabolic pathways involved in T-cell malignancies can pave the way for developing targeted therapies. The PI3K/Akt/mTOR pathway is among the key metabolic features observed in malignant T cells.
4 Melatonin
Melatonin (N-acetyl-5-methoxytryptamine), is a naturally occurring indole amine produced by the human body through various mechanisms. Its primary production occurs in the pineal gland, particularly in response to darkness. In addition to the pineal gland, other organs such as the gastrointestinal tract, bone marrow, lymphocytes, retina, and skin are known to synthesize melatonin (84). The suprachiasmatic nucleus (SCN) of the hypothalamus serves as the biological clock governing the 24-hour rhythm of melatonin production and secretion (85). Melatonin levels rise during the night and tend to rise during the day and night, gradually declining in the morning and throughout the day. Elevated melatonin levels at night play a vital role in orchestrating optimal homeostatic metabolic rhythms in target organs, contributing to the prevention of numerous diseases. Exposure to the light at night can disrupt the circadian rhythm and hamper melatonin generation. Melatonin is a pleiotropic hormone with diverse functions, including anti-oxidants, oncostatic, antiaging, and immune modulation activities (86–88). It functions by inhibiting the production of pro-oxidants and promoting the synthesis of antioxidants such as glutathione peroxidase and superoxide dismutase, thereby offering protection against reactive oxygen and nitrogen species (ROS/RNS) (89). Additionally, melatonin has demonstrated cardioprotective effects. Due to its lipophilic nature, melatonin can easily traverse various morphological and physiological barriers and reach the subcellular compartments of heart cells, mitigating oxidative stress generation (90). This productive action helps prevent DNA damage, which is essential for maintaining genomic stability (86, 90, 91). It exerts its effects through two receptors, namely MT1R and MT2R, which belong to the family of G-protein coupled receptors (GPCRs) and regulate various physiological processes (88) (Figure 4).
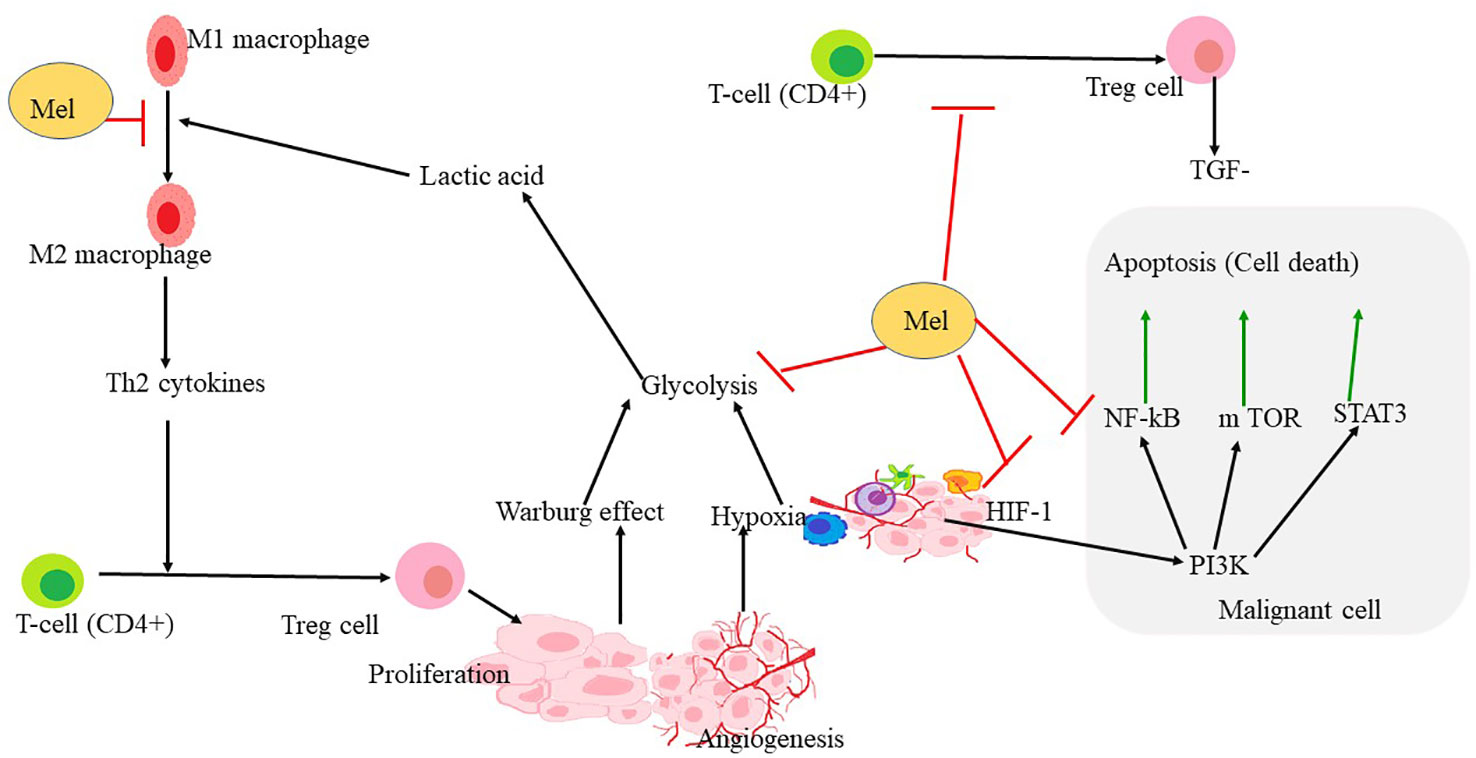
Figure 4 Mechanism through which melatonin rewires the pathways involved in hypoxia and angiogenesis formation. HIF1α is the key regulator of hypoxia and angiogenesis. Polarization of macrophage to M2 also triggers the proliferation of Treg-cells, leading to the upregulation of some apoptotic factors in malignant cells. PI3K, NF- κB, STAT3 leading to apoptosis that may be upregulated in hypoxia conditions or in the tumor microenvironment. Melatonin reverses the polarization of macrophages and inhibits Treg-cell proliferation and angiogenesis. Melatonin further inhibits HIF-1α, NF-κ B, and also STAT 3 and PI3K pathways leading to inhibition of glycolysis, ultimately leading to apoptosis.
4.1 Anti-oncogenic/oncostatic impact of melatonin on various cancer cells
The concentration of melatonin in cerebrospinal fluid (CSF) and blood reaches its peak between 2:00 a.m. and 4:00 a.m. and then gradually decreases as the day progresses. During night time, the melatonin concentration in the blood typically ranges from 80 to 120 pg/ml, but it goes down to 10-20pg/ml during the daytime (92). The melatonin concentration is usually deficient, typically ranging from 0.1 to 0.6 pg/ml. This is significantly lower than the standard level of melatonin found in healthy humans, which can reach up to 120 pg/ml at night. This indicates that cancer cells may have difficulties in producing melatonin or maybe breaking it down more quickly, which could be contributing to their malignant behavior (Table 1).
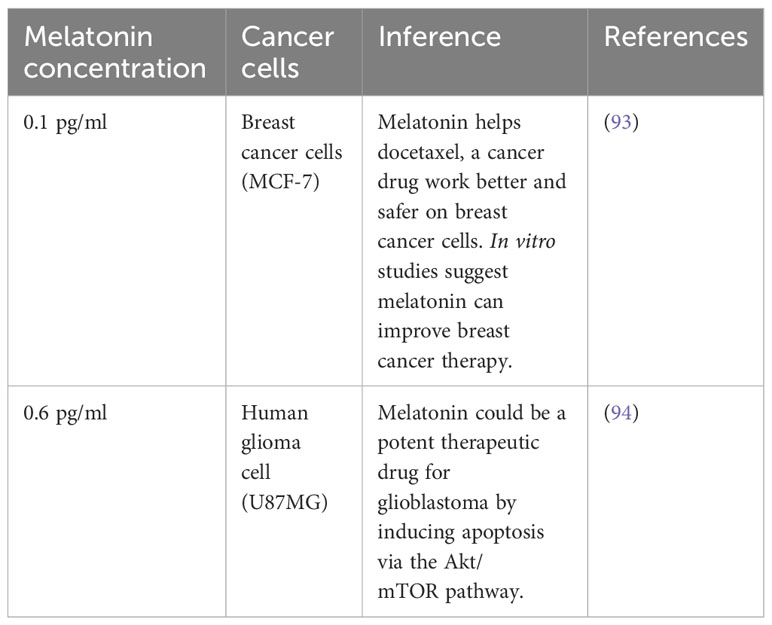
Table 1 Melatonin levels in some malignant cell lines.
Numerous in vivo and in vitro studies have demonstrated that melatonin can suppress cancer development. It interacts with several metabolic pathways and plays a modulatory role in estrogen-controlled pathways in breast cancer, potentially exerting an oncostatic effect (95, 96). Different pharmaceutical doses of melatonin are given by Meharzadi et al. (13) (Table 2).
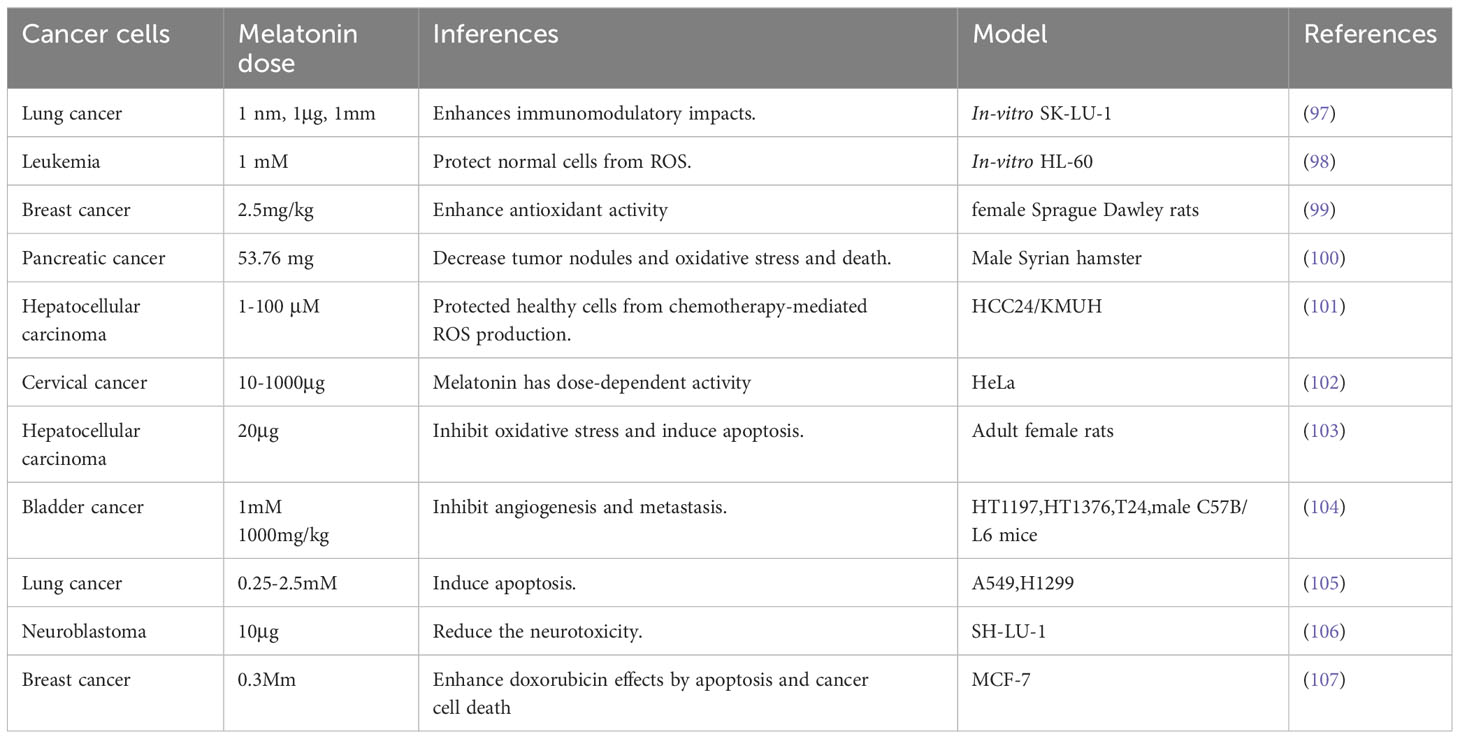
Table 2 Different pharmaceutical doses of melatonin.
4.2 Action mechanism of melatonin in cancer cell
Melatonin exhibits the ability to attach to distinct types of receptors, situated both along the cell membrane and within the cellular nucleus. The membrane receptors are MT1R and MT2R, a type of G-Protein coupled receptor (GPCR). These receptors are prevalent in humans and other mammals, affecting diverse processes like circadian rhythm, immune modulation, and blood pressure regulation (22). Within the cell nucleus, the binding locations referred to as orphan receptor retinoic-acid receptor (ROR) ROR-α and orphan receptor retinoid Z receptor (RZR) RZR-β orphan receptors engaged in gene regulation. Some studies suggest that these receptors regulate the gene expression which is involved in inflammation, metabolism, and bone formation (21). Furthermore, melatonin can also attach to other membranes with the ROR family, including RZR-β AND ROR-γ. Previous reports suggest that human CD4+ cells which are helper T-cells and CD8+ cells which are cytotoxic T-cells have a higher affinity for melatonin and may respond to blood serum melatonin levels 150-200pg/mL, as they express both membrane receptors MTIR and nuclear receptors RZR-α, ROR-α1, ROR-α2, for melatonin. T-cells also express the enzymes which are crucial for the biosynthesis of melatonin like tryptophan-5-hydroxylase (TPH), L-aromatic amino acid decarboxylase (AADC), N-acetyltransferase (NAT), and Hydroxyindole-O-methyltransferase (HIOMT) (25). This suggests that melatonin has an important role in regulating immune cells as other Peripheral blood mononuclear cells PBMCs also express these receptors and enzymes giving a strong viewpoint on melatonin as an immune modulation and impacting the T-cell functions. Melatonin also influences the survival of T-memory cells (25). Most active T-cells undergo apoptosis but a few subsets of memory T-cells remain alive. T memory cells can recall and react to previous encounters with pathogens. These memory T-cells (Tm) are sub-categorized into effector T-cell (Tem), memory stem T-cell (Tscm), Follicular helper T-cells, and resident memory T-cells (Trm). Melatonin declines the apoptosis of human and mouse T-cells via TCR/CD3 and suppresses the CD3-driven expression of CD95L. CD95L is a protein that belongs to the Tumor necrosis factor (TNF) family. It can bind with the CD95 marker expressed on T-lymphocytes and other cells. CD95L triggers several pathways leading to cell death or survival depending on the context. Inhibiting the CD95L expression increases the survival of Tm cells by preventing apoptosis (108). Melatonin influences the production of IL-2 a cytokine crucial for immune response. Melatonin stimulates the Th1 cells to produce IL-2 and CGP 52608 which operates similarly to melatonin (a ligand for RZR/ROR) (109). Whereas in Jurkat cells CGP52608 an antagonist to the melatonin nuclear receptor inhibits the secretion and production of IL-2. Suggests that melatonin modulates the T-cells via nuclear receptors (110). Th17 cells, a category of T-lymphocytes that generate IL-17, cytokines causing inflammation and immune harm, are influenced by melatonin (111). Melatonin restrains the development of Th17 cells, a characteristic that could prove beneficial for autoimmune conditions. Melatonin stimulates ROR-α to translocate from the nucleus to the cytoplasm where it loses the capacity to activate its target genes. Consequently, this decreases the expression of RORγt another nuclear receptor essential for TH17 development. Melatonin binding with MT1R a GPCR, sets off a signaling process involving ERK1/2, C/EBPα, REV-ERBα, and NFIL3. These proteins control the expression of genes related to Th17 maturation by binding to their promotor regions. Melatonin’s action extended to decrease the expression of RORα and RORγt, achieved by inhibiting REV-ERB-α and NFIL3, respectively. Binding with MT1R phosphorylation of Erk1/2 occurs which further stimulates CAAT/enhancer binding protein α (C/EBPAα) which suppresses the REV-ERBα (expressed by nr1d1 gene) which further suppresses the expression of NFIL3 (transcription factor inhibits the RORα and RORγt (112, 113).
Malignancy is the abnormal development and proliferation of malignant cells, which is associated with various pathways such as the PI3K/Akt/mTOR pathways, insulin-like growth factor-1 (IGF-1), hypoxia-inducible factor-1 (HIF-1), and NF-KB. These pathways play a direct role in promoting tumor development in the tumor microenvironment (TME), a condition characterized by low oxygen levels known as hypoxia (52). Melatonin, a hormone produced by the pineal gland, regulates a variety of physiological activities. Numerous studies have demonstrated its significant impact on various cancer cell lines and their programmed cell death, also known as apoptosis. However, the effects of melatonin may vary depending on specific carcinogenic molecules and the growth medium conditions (114, 115).
Melatonin inhibits tumor growth by suppressing the actions of prolactin-insulin-like growth factor-1 (IGF-1), growth hormone-dependent growth factors (GHFs), epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF), hepatocyte growth factor (HGF), transforming growth factor (TGF), and platelet-derived growth factors (PDGF). By blocking these growth factors, melatonin indirectly hampers the malignant transformation of healthy cells (32). Moreover, melatonin induces mitochondrial-dependent activities and promotes apoptosis in tumor cells. Cancer cells in the tumor microenvironment undergo altered metabolic rewiring, converting glucose to lactate even in the presence of oxygen. Lactate creates an acidic environment that favors cell proliferation, but melatonin counteracts this effect by inhibiting the mitochondrial enzyme pyruvate dehydrogenase kinase (PDK) and stimulating the production of acetyl-CoA in the tumor microenvironment (TME) (116).
4.3 Melatonin influencing the metabolic rewiring in T-cell malignancy
Metabolic rewiring is a powerful strategy employed by malignant cells to adapt to different environmental conditions and physiological demands. Through metabolic rewiring, the metabolic pathways are modified and preferences, ultimately influencing crucial cellular functions such as energy production, signaling mechanism, biosynthesis, and survival (117). Metabolic reprogramming has been implicated in cancer progression (118). Malignant cell metabolism differs significantly from that of normal cells to meet the biosynthetic and bioenergetic demands of rapid proliferation and adaptation to the tumor microenvironment (TME) (119). Cancer cell metabolic reprogramming and adaptability play crucial roles in therapy resistance. The characteristics and activity of immune cells within the TME are closely associated with cancer growth, and the metabolic activity of cancer cells influences the TME (120). This metabolic interplay alters nutrient availability in the microenvironment as cancer cells and the immune system compete for essential nutrients like glucose, glutamine, lipids, and amino acids (121).
T-cell malignancies are characterized by dysregulated metabolic pathways that sustain the increased energy and metabolic demands of malignant cells Identifying these metabolic pathways is crucial for developing novel therapeutics targeting the metabolic vulnerabilities of T-cell malignancies (43). The development and progression of T-cell malignancies rely on various pathways involved in metabolic regulation (120). Tumor cells exhibit enhanced glycolysis rates even in the presence of adequate oxygen, a phenomenon known as the Warburg effect (122). This metabolic shift enables cancer cells to generate the energy and biosynthetic precursors necessary for growth and survival. Malignant T-cells undergo a significant metabolic shift. This entails an increased dependency on glycolysis, and glutaminolysis as well as a modification in lipid metabolism. The Warburg effect describes cancer cells’ preference for anaerobic glycolysis over aerobic glycolysis, even under normal aerobic conditions (Warburg, 1956). Anaerobic glycolysis facilitates rapid ATP production (123). All these metabolic reprogramming increases the free radical loads. Nicotinamide adenine dinucleotide (NAD) plays a vital role in redox reactions within various metabolic pathways, DNA repair, and post-translational modifications (124). Malignant cells require elevated levels of NAD for their survival and proliferation (124). Nicotinamide adenine dinucleotide phosphate (NADPH) protects tumor cells from reactive oxygen species (ROS) and reactive nitrogen species (RNS). Maintaining a balanced level of ROS is necessary for cancer cells’ signal transduction, but excessive ROS can trigger cytotoxicity, DNA damage, and cellular apoptosis. To overcome this, tumor cells develop a defense system that regulates the level of antioxidants required for their survival, and this defense system depends on NADPH production (125). Lipid metabolism rewiring is the major aspect of metabolic rewiring in malignant T-cells. Lipids are present in the form of droplets, and rafts, act as signaling molecules and as a precursor for the synthesis of macromolecules. Lipid rafts activate the expression of fatty acid synthase (FASN) in malignant cells (126). FASN is an enzyme that catalyzes fatty acid synthesis through acetyl-CoA. Fatty acids are utilized by mitochondria to generate ATP (117) (Figure 5).
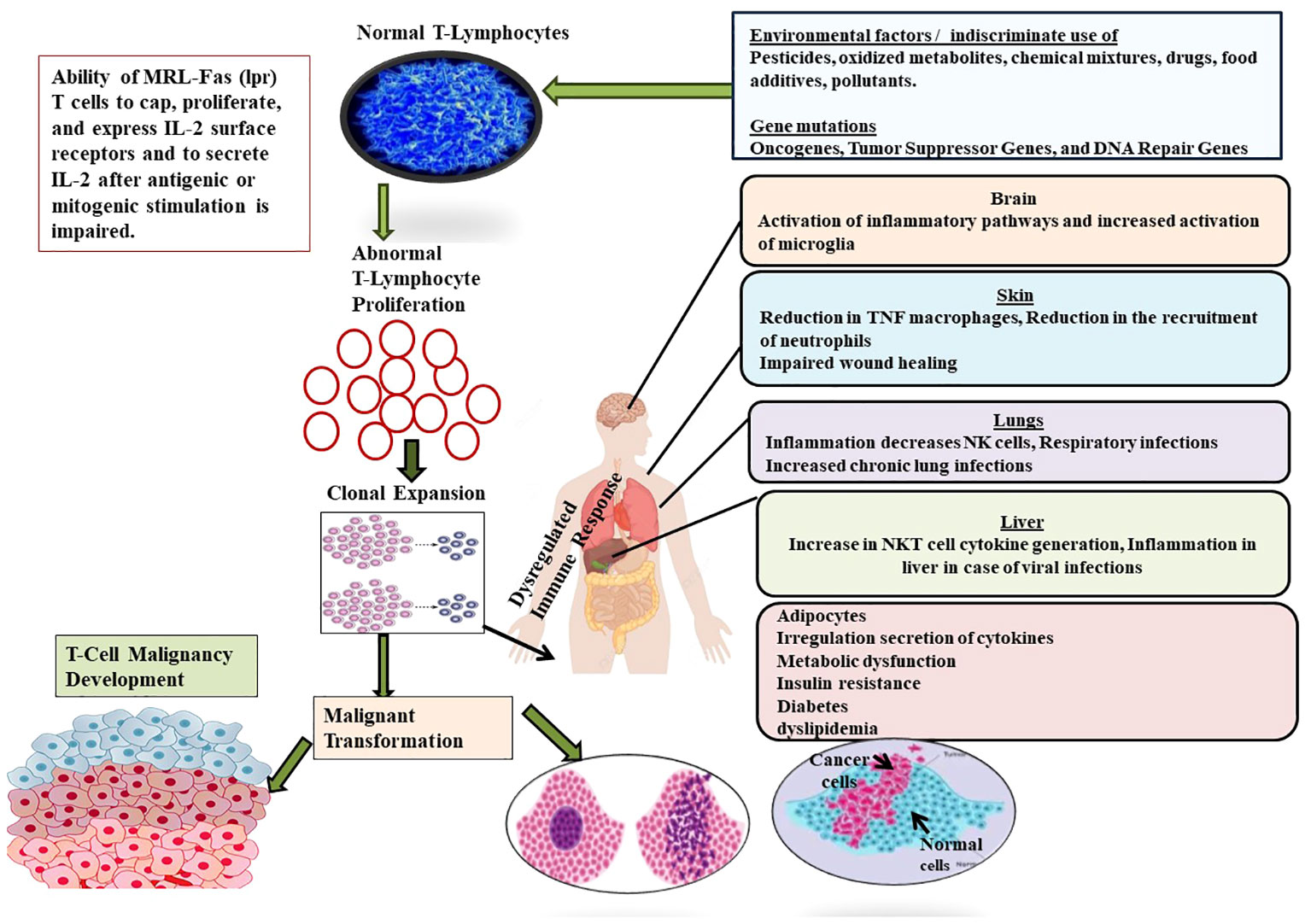
Figure 5 Showing the role of different factors such as environmental factors (Pesticides, oxidized metabolites, chemical mixtures, drugs, food additives, pollutants) and genetic factors (Oncogenes, Tumor Suppressor Genes, and DNA Repair Genes) in the development of T-cell malignancy. Malignancy in T-cell induces complications in different organs such as brain, liver, skin, lungs and finally suppresses the formation and development of normal T-cells.
Previous research has demonstrated that melatonin directly influences T/B cell activation. During T-cell activation, the expression of CD38 and CD69 on the cell surface is often observed. CD38 is an NAD+ glycosyl hydrolase/ADP ribose cyclase involved in cell adhesion and signaling. It is present in early activated T cells as well as various immunological cells, including CD4+ T cells. CD38 expression increases during infections. Additionally, CD69 serves as a well-known marker of early lymphocyte and T cell activation, primarily expressed by CD8+, and CD45RO+ activated T cells (Table 3).
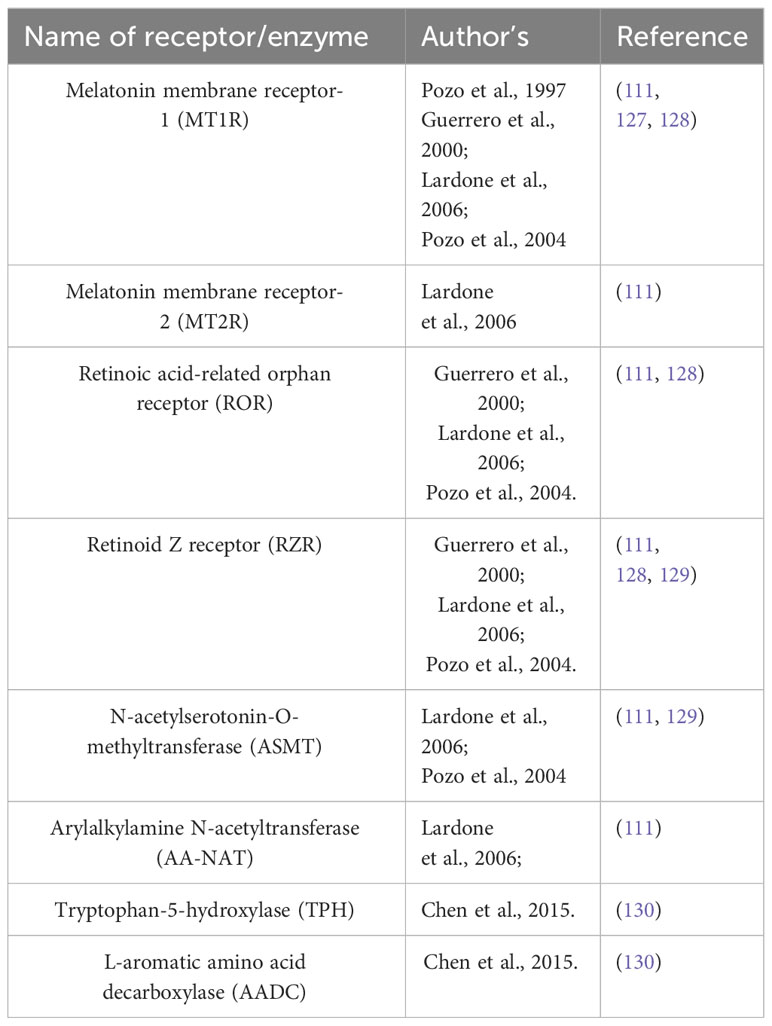
Table 3 Receptors/enzymes reported in T-cells.
4.3.1 Melatonin inhibiting glycolysis in T-cell malignancy
Glycolysis is a metabolic mechanism that transforms glucose into pyruvate and generates ATP, the primary source of energy for cells. Even in the presence of oxygen, cancer cells frequently rely on glycolysis more than normal cells. The Warburg effect is a phenomenon that permits malignant cells to survive and proliferate in hypoxia and nutrient-depleted environments. Glycolysis also provides cancer cells with biosynthetic intermediates while decreasing oxidative stress by redirecting glucose away from mitochondrial oxidative phosphorylation (OXPHOS). One of the mechanisms by which melatonin exerts its anti-cancer effect is by inhibiting glycolysis (13).
4.3.1.1 Melatonin inhibiting the Hexokinase II
Hexokinase II plays a pivotal role in the glycolytic pathway, which is a series of chemical reactions that convert glucose into energy in the form of adenosine triphosphate (ATP). In the first step of glycolysis, HKII phosphorylates glucose, which is essential for trapping glucose inside the cell and preparing it for further processing in the glycolytic pathway. In other words, HKII acts as a gatekeeper, ensuring that glucose is efficiently utilized for energy production within the cells (131, 132).
Melatonin has been proven to suppress the action of HKII by binding to the enzyme directly, modifying its structure and activity. This interaction stops HKII from phosphorylating glucose and inhibiting glycolysis efficiently. Because of this glucose trapping is reduced and glycolysis cannot continue as normal leading to a decrease in the level of glycolysis and hence the energy supply to malignant cells declines (133).
4.3.1.2 Melatonin activating AMP-activated protein kinase and reducing the Glut1 expression
Melatonin stimulates the enzyme AMP-activated protein kinase (AMPPK). AMPK is an important cellular energy sensor that detects variations in the Adenosine monophosphate (AMP) to adenosine triphosphate (ATP) ratio. AMPK is activated when cellular energy levels are low and the AMP/ATP ratio rises (indicating an energy deficit). This activation has numerous consequences for glycolysis inhibition (134). AMPK controls the mTOR signaling pathway. mTOR is a key regulator of cell growth and metabolism. It combines signals from many sources, including growth factors, nutrient availability, and energy status. Melatonin activation of AMPK inhibits mTOR signaling (135).
Glut 1 expression is reduced when is inhibited by AMPK. Glut1 is a protein that aids in the transport of glucose into cells. Melatonin indirectly decreases the amount of glucose available for glycolysis by lowering the expression of Glut1 (134, 136).
4.4 Melatonin inducing apoptotic proteins in malignant T-cells
Metabolic reprogramming is necessary for T-cell activation and differentiation to fulfill new demands, including increased synthesis of various inflammatory mediators. Upon T-cell receptor (TCR) activation, there is an upregulation of glucose, amino acid, and iron uptake transporters (137). These transporters are essential for the differentiation of effector T cells. Consequently, the nutritional microenvironment surrounding T cells has the potential to modulate T-cell immunological responses (137). Melatonin induces apoptosis of malignant cells through intrinsic and extrinsic pathways. The intrinsic pathways of apoptosis are activated by melatonin by impacting the membrane potential of mitochondria and cytochrome c release via increasing the expression of pro-apoptotic genes belonging to Bcl-2 family like Bax, Bak, Bid these proteins from pores in the mitochondrial membrane and facilitating the release of cytochrome c (138). Cytochrome c is released from the mitochondrial permeability transition pore (MPTP) due to damage or stress in mitochondria (138). Now further cytochrome c activates other proteolytic activity via caspases. Caspase activity leads to apoptosis (138).
4.5 Melatonin and p53 targeting malignant T-cells
The p53 encoded by the TP53 gene, a tumor suppressor protein, regulates the cell death/cell cycle arrest and acts as a transcription factor. DNA damage in cells is sensed by p53 leading to cell cycle arrest or cell death (139). The role of p53 in tumor suppression has been identified as an intricate combination of several biological mechanisms and rewiring the metabolic pathways thereby, suppressing the tumor progression (140). Under these, the control of cellular metabolism and autophagy, as well as inhibiting the ferroptosis process, have a substantial influence on how well cells can respond and adapt to disturbance, limiting the build-up of damage that can lead to cancer. By restricting glycolysis, p53 promotes mitochondrial respiration. The glucose transporters GLUT1, GLUT3, and GLUT4 are repressed (141). Furthermore, p53 inhibits the expression of TIGAR (TP53-inducible glucolysis and apoptosis regulator), a regulator of glucose breakdown. TIGAR expression decreases the fructose-2,6 bisphosphate expression in cells inhibiting glycolysis (142) as well as PDK2 (pyruvate dehydrogenase kinase 2), which inactivates the pyruvate dehydrogenase complex controlling pyruvate access to the Kreb cycle (131).
Melatonin inhibits the STAT 3 signaling which enhances the p53 expression. p53 also regulates the immune system. It controls the inflammatory response via STAT3, which is triggered by the inflammatory cytokine IL-6 (143) In studies, it was reported that p53 population declines in pancreatic cancer lead to an increase in STAT3 activation. STAT3 is proinflammatory and makes the environment suitable for the progression of tumors (144).
5 Summary and conclusion
The emerging evidence highlights melatonin as a significant modulator in the metabolic rewiring processes observed in T-cell malignancies through its pleiotropic effects. on various cellular pathways and molecular targets, melatonin exerts regulatory control over key metabolic alterations seen in these malignancies. It may be suggested that melatonin by promoting mitochondrial-dependent activities and stimulating the production of acetyl-CoA, participates in the suppression of tumor growth and proliferation and hence disrupts the metabolic rewiring. Moreover, melatonin’s influence on nutrient uptake and metabolism in T cells, including the modulation of glucose and amino acid transporters, highlights its ability to shape the metabolic profile of malignant T cells. In addition, such modulation has implications for T-cell differentiation, function, and overall immunological responses in T-cell malignancies. The regulation of critical signaling molecules involved in T-cell activation, such as CD38 and CD69, further underscores melatonin’s role in modulating T-cell behavior and immune responses within the tumor microenvironment.
Understandings of the research evidence present the picture more clearly that harnessing the therapeutic potential of melatonin in T-cell malignancies may open new avenues for targeted interventions by exploiting its effects on metabolic rewiring and T-cell activation. Therefore, melatonin-based strategies may be offered as novel therapeutic approaches to combat these aggressive hematological cancers. However, further research is needed to unravel the precise molecular mechanisms underlying melatonin’s modulatory effects on metabolic rewiring in T-cell malignancies and to establish an effective pharmacological dose. Additionally, well-designed clinical studies are warranted to validate its efficacy and safety as an adjunctive therapy or standalone treatment option to disrupt the metabolic rewiring processes driving T-cell malignancies, thereby offering potential improvements in patient outcomes and survival rates.
6 Recommendations
6.1 Mechanistic studies
Perform extensive mechanistic studies to uncover the precise signaling pathways and molecular targets involved in melatonin’s impact on metabolic reprogramming in T-cell cancers. This will offer a valuable understanding of the underlying mechanisms and aid in pinpointing prospective therapeutic targets.
6.2 In vivo models
To comprehensively examine the effects of melatonin on tumor metabolism in a complex microenvironment, it is imperative to employ in vivo models such as mouse models of T-cell malignancies. This will facilitate a more physiologically relevant setting to verify the observations made in vitro and further investigate the impact of melatonin on metabolic rewiring.
6.3 Combination therapies
It is recommended to explore the potential synergies between melatonin and current treatment methods such as chemotherapy or immunotherapy. Preclinical investigations and clinical trials should be conducted to evaluate the advantages of combining melatonin with conventional therapies. The ultimate goal is to improve treatment effectiveness and overcome resistance mechanisms in T-cell cancers.
6.4 Immunological responses
Explore the immunomodulatory effects of melatonin in the context of T-cell malignancies. Investigate its impact on T-cell differentiation, activation, and anti-tumor immune responses. This will help elucidate the role of melatonin in shaping the immune microenvironment and its potential for combination with immunotherapies.
6.5 Clinical trials
Design and conduct well-controlled clinical trials to evaluate the efficacy and safety of melatonin-based interventions in patients with T-cell malignancies. Consider patient selection criteria, optimal dosing regimens, and combination strategies to determine the clinical utility of melatonin in improving patient outcomes.
6.6 Biomarker development
Identify and validate biomarkers associated with melatonin response in T-cell malignancies. Establish robust biomarker panels to predict patient response to melatonin-based therapies and monitor treatment effectiveness.
7 Future perspective
The future perspectives of this review article involve unraveling the underlying mechanisms, exploring synergistic effects with existing therapies, investigating melatonin as an adjuvant to immunotherapies, developing targeted analogs, and conducting rigorous clinical trials. By advancing our understanding and harnessing the therapeutic potential of melatonin, we can pave the way for innovative approaches to tackle metabolic rewiring in T-cell malignancies and improve patient outcomes in the future.
Author contributions
Corresponding and joint first authors from Guru Ghasidas Vishwavidyalaya wrote and edited the manuscript. All other co-authors contributed intellectually to the designing, editing, and proofreading of the manuscript.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The author acknowledges the untiring efforts of all the co-authors (GR, YH) in finalizing this review article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The views expressed in this article are those of the authors and do not necessarily represent the official views of their affiliated institutions.
References
1. Haas S. Hematopoietic stem cells in health and disease—insights from single-cell multi-omic approaches. Curr Stem Cell Rep (2020) 6:67–76. doi: 10.1007/s40778-020-00174-2
2. Barrena S, Almeida J, Yunta M, López A, Fernandez-Mosteirin N, Giralt M, et al. Aberrant expression of tetraspanin molecules in B-cell chronic lymphoproliferative disorders and its correlation with normal B-cell maturation. Leukemia (2005) 19(8):1376–83. doi: 10.1038/sj.leu.2403822
3. Prestwich RJ, Errington F, Hatfield P, Merrick AE, Ilett EJ, Selby PJ, et al. The immune system–is it relevant to cancer development, progression, and treatment? Clin Oncol (R Coll Radiol) (2008) 20(2):101–12. doi: 10.1016/j.clon.2007.10.011
4. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol (2012) 12(4):239–52. doi: 10.1038/nri3174
5. Nagasawa T, Zhang Q, Raghunath PN, Wong HY, El-Salem M, Szallasi A, et al. Multi-gene epigenetic silencing of tumor suppressor genes in T-cell lymphoma cells; delayed expression of the p16 protein upon reversal of the silencing. Leuk Res (2006) 30(3):303–12. doi: 10.1016/j.leukres.2005.08.012
6. Uchiyama T, Yodoi J, Sagawa K, Takatsuki K, Uchino H. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood (1977) 50(3):481–92. doi: 10.1182/blood.V50.3.481.481
7. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med (2015) 373(16):1541–52. doi: 10.1056/NEJMra1400972
8. Utsunomiya A, Choi I, Chihara D, Seto M. Recent advances in the treatment of adult T-cell leukemia-lymphomas. Cancer Sci (2015) 106(4):344–51. doi: 10.1111/cas.12617
9. Matutes E. Adult T-cell leukemia/lymphoma. J Clin Pathol (2007) 60(12):1373–7. doi: 10.1136/jcp.2007.052456
10. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood (2016) 127(20):2375–90. doi: 10.1182/blood-2016-01-643569
11. Laribi K, Alani M, Truong C, Baugier de Materre A. Recent advances in the treatment of peripheral T-cell lymphoma. Oncologist (2018) 23(9):1039–53. doi: 10.1634/theoncologist.2017-0524
12. Hulvat MC. Cancer incidence and trends. Surg Clinics (2020) 100(3):469–81. doi: 10.1016/j.suc.2020.01.002
13. Mehrzadi S, Pourhanifeh MH, Mirzaei A, Moradian F, Hosseinzadeh A. An updated review of mechanistic potentials of melatonin against cancer: pivotal roles in angiogenesis, apoptosis, autophagy, endoplasmic reticulum stress and oxidative stress. Cancer Cell Int (2021) 21(1):1–28. doi: 10.1186/s12935-021-01892-1
14. Davoodvandi A, Nikfar B, Reiter RJ, Asemi Z. Melatonin and cancer suppression: insights into its effects on DNA methylation. Cell Mol Biol Letters (2022) 27(1):1–11. doi: 10.1186/s11658-022-00375-z
15. Reiter RJ, Tan D-X, Manchester L, Terron MP, Flores LJ, Koppisepi S. Medical implications of melatonin: receptor-mediated and receptor-independent actions. Adv Med Sci (De Gruyter Open) (2007) 52:11–28.
16. Slominski RM, Reiter RJ, Schlabritz-Loutsevitch N, Ostrom RS, Slominski AT. Melatonin membrane receptors in peripheral tissues: distribution and functions. Mol Cell Endocrinol (2012) 351(2):152–66. doi: 10.1016/j.mce.2012.01.004
17. He B, Zhao Y, Xu L, Gao L, Su Y, Lin N, et al. The nuclear melatonin receptor ROR α is a novel endogenous defender against myocardial ischemia/reperfusion injury. J Pineal Res (2016) 60(3):313–26. doi: 10.1111/jpi.12312
18. Tan DX, Hardeland R, Manchester LC, Paredes SD, Korkmaz A, Sainz RM, et al. The changing biological roles of melatonin during evolution: from an antioxidant to signals of darkness, sexual selection and fitness. Biol Rev (2010) 85(3):607–23. doi: 10.1111/j.1469-185X.2009.00118.x
19. Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G, et al. Update on melatonin receptors: IUPHAR Review 20. Br J Pharmacol (2016) 173(18):2702–25. doi: 10.1111/bph.13536
20. Reiter RJ, Tan DX, Galano A. Melatonin: exceeding expectations. Physiology (2014) 29(5):325–33. doi: 10.1152/physiol.00011.2014
21. Dragojevic Dikic S, Jovanovic AM, Dikic S, Jovanovic T, Jurisic A, Dobrosavljevic A. Melatonin: a “Higgs boson” in human reproduction. Gynecol Endocrinol (2015) 31(2):92–101. doi: 10.3109/09513590.2014.978851
22. Miller E, Morel A, Saso L, Saluk J. Melatonin redox activity. Its potential clinical applications in neurodegenerative disorders. Curr Topics Medicinal Chem (2015) 15(2):163–9.
23. Vriend J, Reiter RJ. Melatonin feedback on clock genes: a theory involving the proteasome. J Pineal Res (2015) 58(1):1–11. doi: 10.1111/jpi.12189
24. Manchester LC, Coto-Montes A, Boga JA, Andersen LPH, Zhou Z, Galano A, et al. Melatonin: an ancient molecule that makes oxygen metabolically tolerable. J Pineal Res (2015) 59(4):403–19. doi: 10.1111/jpi.12267
25. Sanchez-Barcelo EJ, Mediavilla MD, Vriend J, Reiter RJ. Constitutive photomorphogenesis protein 1 (COP 1) and COP 9 signalosome, evolutionarily conserved photomorphogenic proteins as possible targets of melatonin. J Pineal Res (2016) 61(1):41–51. doi: 10.1111/jpi.12340
26. Golombek DA, Pandi-Perumal SR, Brown GM, Cardinali DP. Some implications of melatonin use in chronopharmacology of insomnia. Eur J Pharmacol (2015) 762:42–8. doi: 10.1016/j.ejphar.2015.05.032
27. Opie LH, Lecour S. Melatonin has multiorgan effects. Eur Heart Journal–Cardiovascular Pharmacotherapy (2016) 2(4):258–65. doi: 10.1093/ehjcvp/pvv037
28. Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L. Melatonin as an antioxidant: under promises but over delivers. J Pineal Res (2016) 61(3):253–78. doi: 10.1111/jpi.12360
29. Gelaleti GB, Borin TF, Maschio-Signorini LB, Moschetta MG, Jardim-Perassi BV, Calvinho GB, et al. Efficacy of melatonin, IL-25 and siIL-17B in tumorigenesis-associated properties of breast cancer cell lines. Life Sci (2017) 183:98–109. doi: 10.1016/j.lfs.2017.06.013
30. Papazisis KT, Kouretas D, Geromichalos GD, Sivridis E, Tsekreli OK, Dimitriadis KA, et al. Effects of melatonin on proliferation of cancer cell lines. J Pineal Res (1998) 25(4):211–8. doi: 10.1111/j.1600-079X.1998.tb00390.x
31. Huang J, Shan W, Li N, Zhou B, Guo E, Xia M, et al. Melatonin provides protection against cisplatin-induced ovarian damage and loss of fertility in mice. Reprod BioMedicine Online (2021) 42(3):505–19. doi: 10.1016/j.rbmo.2020.10.001
32. Chuffa L, RJ R, Lupi LA. Melatonin as a promising agent to treat ovarian cancer: molecular mechanisms. Carcinogenesis (2017) 38(10):945–52. doi: 10.1093/carcin/bgx054
33. Shin IS, Shin NR, Park JW, Jeon CM, Hong JM, Kwon OK, et al. Melatonin attenuates neutrophil inflammation and mucus secretion in cigarette smoke-induced chronic obstructive pulmonary diseases via the suppression of Erk-Sp1 signaling. J Pineal Res (2015) 58(1):50–60. doi: 10.1111/jpi.12192
34. Sanchez-Barcelo EJ, Mediavilla MD, Alonso-Gonzalez C, Reiter RJ. Melatonin uses in oncology: breast cancer prevention and reduction of the side effects of chemotherapy and radiation. Expert Opin Investigational Drugs (2012) 21(6):819–31. doi: 10.1517/13543784.2012.681045
35. Talib WH, Alsayed AR, Abuawad A, Daoud S, Mahmod AI. Melatonin in cancer treatment: current knowledge and future opportunities. Molecules (2021) 26(9):2506. doi: 10.3390/molecules26092506
36. Odeh LH, Talib WH, Basheti IA. Synergistic effect of thymoquinone and melatonin against breast cancer implanted in mice. J Cancer Res Ther (2018) 14(Suppl 2):S324–S30. doi: 10.4103/0973-1482.235349
37. Martínez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer (2021) 21(10):669–80. doi: 10.1038/s41568-021-00378-6
38. Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Communication Signaling (2020) 18:1–19. doi: 10.1186/s12964-020-0530-4
39. Tiwari A, Trivedi R, Lin S-Y. Tumor microenvironment: barrier or opportunity towards effective cancer therapy. J Biomed Sci (2022) 29(1):1–27. doi: 10.1186/s12929-022-00866-3
40. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: a milieu hindering and obstructing antitumor immune responses. Front Immunol (2020) 11:940. doi: 10.3389/fimmu.2020.00940
41. Tiwari S, Siddiqui B, Singh S, Praveen A. Tumor microenvironment: a therapeutic aid in cancer. Indian J Surg (2023) 52:11–28. doi: 10.1007/s12262-023-03828-7
42. Liu J, Gao M, Yang Z, Zhao Y, Guo K, Sun B, et al. Macrophages and metabolic reprograming in the tumor microenvironment. Front Oncol (2022) 12:795159. doi: 10.3389/fonc.2022.795159
43. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell (2002) 1(1):75–87. doi: 10.1016/S1535-6108(02)00018-1
44. Khader A, Shaan M, Balakrishnan S, Ambooken B, Muhammed K, Rajan U. Multifaceted adult T-cell leukemia/lymphoma in India: A case series. Indian J Dermatol (2015) 60(1):103. doi: 10.4103/0019-5154.147846
45. Ma H, Abdul-Hay M. T-cell lymphomas, a challenging disease: types, treatments, and future. Int J Clin Oncol (2017) 22:18–51. doi: 10.1007/s10147-016-1045-2
46. Durinck K, Goossens S, Peirs S, Wallaert A, Van Loocke W, Matthijssens F, et al. Novel biological insights in T-cell acute lymphoblastic leukemia. Exp Haematol (2015) 43(8):625–39. doi: 10.1016/j.exphem.2015.05.017
47. Zhang Z, Liu S, Zhang B, Qiao L, Zhang Y, Zhang Y. T cell dysfunction and exhaustion in cancer. Front Cell Dev Biol (2020) 8:17. doi: 10.3389/fcell.2020.00017
48. Singh L, Nair L, Kumar D, Arora MK, Bajaj S, Gadewar M, et al. Hypoxia induced lactate acidosis modulates tumor microenvironment and lipid reprogramming to sustain the cancer cell survival. Front Oncol (2023) 13:1034205. doi: 10.3389/fonc.2023.1034205
49. Nalbandian M, Takeda M. Lactate as a signaling molecule that regulates exercise-induced adaptations. Biology (2016) 5(4):38. doi: 10.3390/biology5040038
50. Smolková K, Ježek P. The role of mitochondrial NADPH-dependent isocitrate dehydrogenase in cancer cells. Int J Cell Biol (2012) 2012:273947–12. doi: 10.1155/2012/273947
51. Gong S, Wang S, Shao M. NADPH oxidase 4: a potential therapeutic target of Malignancy. Front Cell Dev Biol (2022) 10:884412. doi: 10.3389/fcell.2022.884412
52. Albini A, Tosetti F, Li VW, Noonan DM, Li WW. Cancer prevention by targeting angiogenesis. Nat Rev Clin Oncol (2012) 9(9):498–509. doi: 10.1038/nrclinonc.2012.120
53. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell (2017) 169(3):381–405. doi: 10.1016/j.cell.2017.04.001
54. Dey P, Kimmelman AC, DePinho RA. Metabolic codependencies in the tumor microenvironment. Cancer Discov (2021) 11(5):1067–81. doi: 10.1158/2159-8290.CD-20-1211
56. Rizvi MA, Evens AM, Tallman MS, Nelson BP, Rosen ST. T-cell Non-Hodgkin Lymphoma Blood. Blood (2006) 107(4):1255–64. doi: 10.1182/blood-2005-03-0306
57. Gabriel S, Kallies A. Glucose-and glutamine-fueled stabilization of C-Myc is required for T-cell proliferation and Malignant transformation. Cell Death Discov (2016) 2(1):1–2. doi: 10.1038/cddiscovery.2016.47
58. Zhang H, Dai Z, Wu W, Wang Z, Zhang N, Zhang L, et al. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J Exp Clin Cancer Res (2021) 40(1):1–22. doi: 10.1186/s13046-021-01987-7
59. Rüdiger T, Weisenburger D, Anderson J, Armitage J, Diebold J, MacLennan K, et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkin’s Lymphoma Classification Project. Ann Oncol (2002) 13(1):140–9. doi: 10.1093/annonc/mdf033
60. Sehn LH, Soulier J. Introduction to the review series on T-cell Malignancies. Am Soc Haematol Washington DC; (2017) p:1059–60. doi: 10.1182/blood-2017-01-741389
61. Smith SD, Morgan R, Link MP, McFall P, Hecht F. Cytogenetic and immunophenotypic analysis of cell lines established from patients with T cell leukemia/lymphoma. Blood Journal (1986) 67(3):650–656. doi: 10.1182/blood.V67.3.650.650
62. Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood J Am Soc Haematol (2017) 129(9):1124–33. doi: 10.1182/blood-2016-09-692582
63. Haymaker C, Wu RC, Bernatchez C, Radvanyi LG. PD-1 and BTLA and CD8+ T-cell “exhaustion” in cancer: “Exercising” an alternative viewpoint. Oncoimmunology (2012) 1(5):735–8. doi: 10.4161/onci.20823
64. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell (2017) 170(1):142–57. e19. doi: 10.1016/j.cell.2017.06.007
65. Rahmani T, Azad M, Chahardouli B, Nasiri H, Vatanmakanian M, Kaviani S. Patterns of DNMT1 promoter methylation in patients with acute lymphoblastic leukemia. Int J Haematol-Oncol Stem Cell Res (2017) 11(3):172. doi: 10.1182/blood.v97.5.1172
66. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12(3):180–90. doi: 10.1038/nri3156
67. Mizuno S-i, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood J Am Soc Haematol (2001) 97(5):1172–9.
68. Jones D, Fletcher CD, Pulford K, Shahsafaei A, Dorfman DM. The T-cell activation markers CD30 and OX40/CD134 are expressed in nonoverlapping subsets of peripheral T-cell lymphoma. Blood J Am Soc Haematol (1999) 93(10):3487–93. doi: 10.1182/blood.V93.10.3487.410k39_3487_3493
69. Takemoto S, Iwanaga M, Sagara Y, Watanabe T. Plasma soluble CD30 as a possible marker of adult T-cell leukemia in HTLV-1 carriers: a nested case-control study. Asian Pac J Cancer Prev (2015) 16(18):8253–8. doi: 10.7314/apjcp.2015.16.8253
70. Li Q, Zou J, Wang M, Ding X, Chepelev I, Zhou X, et al. Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T-cell differentiation. Nat Commun (2014) 5(1):5780. doi: 10.1038/ncomms6780
71. Li D, Liang J, Yang W, Guo W, Song W, Zhang W, et al. A distinct lipid metabolism signature of acute myeloid leukemia with prognostic value. Front Oncol (2022) 12:876981. doi: 10.3389/fonc.2022.876981
72. Duprez V, Lenoir G, Dautry-Varsat A. Autocrine growth stimulation of a human T-cell lymphoma line by interleukin 2. Proc Natl Acad Sci (1985) 82(20):6932–6. doi: 10.1073/pnas.82.20.6932
73. Geissinger E, Bonzheim I, Roth S, Rosenwald A, Müller-Hermelink HK, Rüdiger T. CD52 expression in peripheral T-cell lymphomas determined by combined immunophenotyping using tumor cell specific T-cell receptor antibodies. Leukemia Lymphoma (2009) 50(6):1010–6. doi: 10.1080/10428190902926981
74. Hinz T, Weidmann E, Kabelitz D. Dual TCR-expressing T lymphocytes in health and disease. Int Arch Allergy Immunol (2001) 125(1):16–20. doi: 10.1159/000053792
75. Fischer MB, Hauber I, Födinger M, Wolf HM, Thon V, Donath P, et al. Defective TCR surface expression associated with impaired TCR beta-chain assembly in a patient with cutaneous T-cell lymphoma. J Invest Dermatol (1995) 104(4):537–40. doi: 10.1111/1523-1747.ep12606046
76. Zelle-Rieser C, Thangavadivel S, Biedermann R, Brunner A, Stoitzner P, Willenbacher E, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Haematol Oncol (2016) 9:1–12. doi: 10.1186/s13045-016-0345-3
77. Dang CV. Role of aerobic glycolysis in genetically engineered mouse models of cancer. BMC Biol (2013) 11(1):1–4. doi: 10.1186/1741-7007-11-3
78. Ganapathy-Kanniappan S, Geschwind J-FH. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer (2013) 12(1):1–11. doi: 10.1186/1476-4598-12-152
79. Al Tameemi W, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol (2019) 7:4. doi: 10.3389/fcell.2019.00004
80. Sebestyén A, Kopper L, Dankó T, Tímár J. Hypoxia signaling in cancer: from basics to clinical practice. Pathol Oncol Res (2021) 27:1609802. doi: 10.3389/pore.2021.1609802
81. Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev (2007) 26:281–90. doi: 10.1007/s10555-007-9066-y
82. Feng X, Li X, Liu N, Hou N, Sun X, Liu Y. Glutaminolysis and CD4+ T-cell metabolism in autoimmunity: From pathogenesis to therapy prospects. Front Immunol (2022) 13:986847. doi: 10.3389/fimmu.2022.986847
83. Fotiadis D, Kanai Y, Palacín M. The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med (2013) 34(2-3):139–58. doi: 10.1016/j.mam.2012.10.007
84. Talib WH. Melatonin and cancer hallmarks. Molecules (2018) 23(3):518. doi: 10.3390/molecules23030518
85. Li Y, Li S, Zhou Y, Meng X, Zhang J-J, Xu D-P, et al. Melatonin for the prevention and treatment of cancer. Oncotarget (2017) 8(24):39896. doi: 10.18632/oncotarget.16379
86. Galano A, Tan D-X, Reiter RJ. Melatonin: a versatile protector against oxidative DNA damage. Molecules (2018) 23(3):530. doi: 10.3390/molecules23030530
87. Dubocovich ML, Delagrange P, Krause DN, Sugden D, Cardinali DP, Olcese J. International Union of Basic and Clinical Pharmacology. LXXV. Nomenclature, classification, and pharmacology of G protein-coupled melatonin receptors. Pharmacol Rev (2010) 62(3):343–80. doi: 10.1124/pr.110.002832
88. Liu J, Clough SJ, Hutchinson AJ, Adamah-Biassi EB, Popovska-Gorevski M, Dubocovich ML. MT1 and MT2 melatonin receptors: a therapeutic perspective. Annu Rev Pharmacol Toxicol (2016) 56:361–83. doi: 10.1146/annurev-pharmtox-010814-124742
89. Rodriguez C, Mayo JC, Sainz RM, Antolín I, Herrera F, Martín V, et al. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res (2004) 36(1):1–9. doi: 10.1046/j.1600-079X.2003.00092.x
90. Reppert SM, Weaver DR, Ebisawa T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron (1994) 13(5):1177–85. doi: 10.1016/0896-6273(94)90055-8
91. Lindahl T. Instability and decay of the primary structure of DNA. Nature (1993) 362(6422):709–15. doi: 10.1038/362709a0
92. Zetner D, Andersen LPK, Alder R, Jessen ML, Tolstrup A, Rosenberg J. Pharmacokinetics and safety of intravenous, intravesical, rectal, transdermal, and vaginal melatonin in healthy female volunteers: A cross-over study. Pharmacology (2021) 106(3-4):169–76. doi: 10.1159/000510252
93. Alonso-González C, Menéndez-Menéndez J, González-González A, González A, Cos S, Martínez-Campa C. Melatonin enhances the apoptotic effects and modulates the changes in gene expression induced by docetaxel in MCF−7 human breast cancer cells. Int J Oncol (2018) 52(2):560–70. doi: 10.3892/ijo.2017.4213
94. Bao Z, Chen K, Krepel S, Tang P, Gong W, Zhang M, et al. High glucose promotes human glioblastoma cell growth by increasing the expression and function of chemoattractant and growth factor receptors. Trans Oncol (2019) 12(9):1155–63. doi: 10.1016/j.tranon.2019.04.016
95. Carbajo-Pescador S, Ordoñez R, Benet M, Jover R, García-Palomo A, Mauriz J, et al. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br J Cancer (2013) 109(1):83–91. doi: 10.1038/bjc.2013.285
96. Basler M, Jetter A, Fink D, Seifert B, Kullak-Ublick GA, Trojan A. Urinary excretion of melatonin and association with breast cancer: meta-analysis and review of the literature. Breast Care (2014) 9(3):182–7. doi: 10.1159/000363426
97. Plaimee P, Weerapreeyakul N, Thumanu K, Tanthanuch W, Barusrux S. Melatonin induces apoptosis through biomolecular changes, in SK-LU-1 human lung adenocarcinoma cells. Cell Prolif (2014) 47(6):564–77. doi: 10.1111/cpr.12140
98. Bejarano I, Espino J, Marchena AM, Barriga C, Paredes SD, Rodríguez AB, et al. Melatonin enhances hydrogen peroxide-induced apoptosis in human promyelocytic leukaemia HL-60 cells. Mol Cell Biochem (2011) 353:167–76. doi: 10.1007/s11010-011-0783-8
99. Moselhy SS, Al Mslmani MA. Chemopreventive effect of lycopene alone or with melatonin against the genesis of oxidative stress and mammary tumors induced by 7, 12 dimethyl (a) benzanthracene in sprague dawely female rats. Mol Cell Biochem (2008) 319:175–80. doi: 10.1007/s11010-008-9890-6
100. Padillo FJ, Ruiz-Rabelo JF, Cruz A, Perea MD, Tasset I, Montilla P, et al. Melatonin and celecoxib improve the outcomes in hamsters with experimental pancreatic cancer. J Pineal Res (2010) 49(3):264–70. doi: 10.1111/j.1600-079X.2010.00791.x
101. Lin Z-Y, Chuang W-L. Pharmacologic concentrations of melatonin have diverse influence on differential expressions of angiogenic chemokine genes in different hepatocellular carcinoma cell lines. Biomed Pharmacother (2010) 64(10):659–62. doi: 10.1016/j.biopha.2010.09.006
102. Zhu H, Chen Y, Bai L-c, Cao X-r, Xu R. Different effects of melatonin on X-rays-irradiated cancer cells in a dose-dependent manner. Dose-Response (2019) 17(3):1559325819877271. doi: 10.1177/1559325819877271
103. El-Magd MA, Mohamed Y, El-Shetry ES, Elsayed SA, Gazia MA, Abdel-Aleem GA, et al. Melatonin maximizes the therapeutic potential of non-preconditioned MSCs in a DEN-induced rat model of HCC. Biomed Pharmacother (2019) 114:108732. doi: 10.1016/j.biopha.2019.108732
104. Chen YT, Yang CC, Shao PL, Huang CR, Yip HK. Melatonin-mediated downregulation of ZNF746 suppresses bladder tumorigenesis mainly through inhibiting the AKT-MMP-9 signaling pathway. J Pineal Res (2019) 66(1):e12536. doi: 10.1111/jpi.12536
105. Gurunathan S, Jeyaraj M, Kang M-H, Kim J-H. Melatonin enhances palladium-nanoparticle-induced cytotoxicity and apoptosis in human lung epithelial adenocarcinoma cells A549 and H1229. Antioxidants (2020) 9(4):357. doi: 10.3390/antiox9040357
106. Waseem M, Sahu U, Salman M, Choudhury A, Kar S, Tabassum H, et al. Melatonin pre-treatment mitigates SHSY-5Y cells against oxaliplatin induced mitochondrial stress and apoptotic cell death. PloS One (2017) 12(7):e0180953. doi: 10.1371/journal.pone.0180953
107. Koşar PA, Nazıroğlu M, Övey İS, Çiğ B. Synergic effects of doxorubicin and melatonin on apoptosis and mitochondrial oxidative stress in MCF-7 breast cancer cells: involvement of TRPV1 channels. J Membrane Biol (2016) 249:129–40. doi: 10.1007/s00232-015-9855-0
108. Levoin N, Jean M, Legembre P. CD95 structure, aggregation and cell signaling. Front Cell Dev Biol (2020) 8:314. doi: 10.3389/fcell.2020.00314
109. Reppert SM. Melatonin receptors: molecular biology of a new family of G protein-coupled receptors. J Biol Rhythms (1997) 12(6):528–31. doi: 10.1177/074873049701200606
110. Garcia-Maurino S, Gonzalez-Haba MG, Calvo JR, Rafii-El-Idrissi M, Sanchez-Margalet V, Goberna R, et al. Melatonin enhances IL-2, IL-6, and IFN-gamma production by human circulating CD4+ cells: a possible nuclear receptor-mediated mechanism involving T helper type 1 lymphocytes and monocytes. J Immunol (Baltimore Md: 1950) (1997) 159(2):574–81. doi: 10.4049/jimmunol.159.2.574
111. Lardone PJ, Carrillo-Vico A, Naranjo MC, De Felipe B, Vallejo A, Karasek M, et al. Melatonin synthesized by Jurkat human leukemic T cell line is implicated in IL-2 production. J Cell Physiol (2006) 206(1):273–9. doi: 10.1002/jcp.20461
112. Farez MF, Mascanfroni ID, Méndez-Huergo SP, Yeste A, Murugaiyan G, Garo LP, et al. Melatonin contributes to the seasonality of multiple sclerosis relapses. Cell (2015) 162(6):1338–52. doi: 10.1016/j.cell.2015.08.025
113. Yu X, Rollins D, Ruhn KA, Stubblefield JJ, Green CB, Kashiwada M, et al. TH17 cell differentiation is regulated by the circadian clock. Science (2013) 342(6159):727–30. doi: 10.1126/science.1243884
114. Shellard S, Whelan R, Hill B. Growth inhibitory and cytotoxic effects of melatonin and its metabolites on human tumour cell lines in vitro. Br J Cancer (1989) 60(3):288. doi: 10.1038/bjc.1989.272
115. Di Bella G, Mascia F, Gualano L, Di Bella L. Melatonin anticancer effects. Int J Mol Sci (2013) 14(2):2410–30. doi: 10.3390/ijms14022410
116. Reiter R, Sharma R, Ma Q, Rosales Corral S, Manucha WAF. Circadian and non-circadian melatonin: Influence on glucose metabolism in cancer cells. J Curr Sci Technol JCST (2020) 10(1):85–98.
117. Mehta A, Ratre YK, Soni VK, Shukla D, Sonkar SC, Kumar A, et al. Orchestral role of lipid metabolic reprogramming in T-cell Malignancy. Front Oncol (2023) 13:1122789. doi: 10.3389/fonc.2023.1122789
118. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi: 10.1016/j.cell.2015.08.016
119. Viale A, Draetta GF. Metabolic features of cancer treatment resistance. Metab Cancer (2016) 207:35–56. doi: 10.1007/978-3-319-42118-6_6
120. Chang C-H, Pearce EL. Emerging concepts of T cell metabolism as a target of immunotherapy. Nat Immunol (2016) 17(4):364–8. doi: 10.1038/ni.3415
121. Cairns RA, Mak TW. Fire and water: Tumor cell adaptation to metabolic conditions. Exp Cell Res (2017) 356(2):204–8. doi: 10.1016/j.yexcr.2017.04.029
122. Koppenol WH, Bounds PL. The Warburg effect and metabolic efficiency: re-crunching the numbers. Science (2009) 324(5930):1029–33. doi: 10.1126/science.1160809
123. Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett (2015) 356(2):156–64. doi: 10.1016/j.canlet.2014.04.001
124. Yaku K, Okabe K, Nakagawa T. NAD metabolism: Implications in aging and longevity. Ageing Res Rev (2018) 47:1–17. doi: 10.1016/j.arr.2018.05.006
125. Ju H-Q, Lin J-F, Tian T, Xie D, Xu R-H. NADPH homeostasis in cancer: functions, mechanisms and therapeutic implications. Signal Transduct Targeted Ther (2020) 5(1):231. doi: 10.1038/s41392-020-00326-0
126. Menendez JA, Vellon L, Lupu R. Targeting fatty acid synthase-driven lipid rafts: a novel strategy to overcome trastuzumab resistance in breast cancer cells. Med Hypotheses (2005) 64(5):997–1001. doi: 10.1016/j.mehy.2004.09.027
127. Pozo D, Delgado M, Fernandez-Santos JM, Calvo JR, Gomariz RP, Martin-Lacave I, et al. Expression of the Mel1a-melatonin receptor mRNA in T and B subsets of lymphocytes from rat thymus and spleen. FASEB J (1997) 11(6):466–73. doi: 10.1096/fasebj.11.6.9194527
128. Guerrero JM, Pozo D, García-Mauriño S, Osuna C, Molinero P, Calvo JR. Involvement of nuclear receptors in the enhanced IL-2 production by melatonin in Jurkat cells. Ann NY Acad Sci (2000) 917(1):397–403. doi: 10.1111/j.1749-6632.2000.tb05404.x
129. Pozo D, García-Mauriño S, Guerrero JM, Calvo JR. mRNA expression of nuclear receptor RZR/RORα, melatonin membrane receptor MT1, and hydroxindole-O-methyltransferase in different populations of human immune cells. J Pineal Res (2004) 37(1):48–54. doi: 10.1111/j.1600-079X.2004.00135.x
130. Chen Y, Leon-Ponte M, Pingle S, O’Connell P, Ahern G. T lymphocytes possess the machinery for 5-HT synthesis, storage, degradation and release. Acta Physiologica (2015) 213(4):860–7. doi: 10.1111/apha.12470
131. Xiao C, Tian H, Zheng Y, Yang Z, Li S, Fan T, et al. Glycolysis in tumor microenvironment as a target to improve cancer immunotherapy. Front Cell Dev Biol (2022) 10:1013885. doi: 10.3389/fcell.2022.1013885
132. Duan S-l, Wu M, Zhang Z-J, Chang S. The potential role of reprogrammed glucose metabolism: an emerging actionable codependent target in thyroid cancer. J Trans Med (2023) 21(1):735. doi: 10.1186/s12967-023-04617-2
133. Pourhanifeh MH, Hosseinzadeh A, Juybari KB, Mehrzadi S. Melatonin and urological cancers: A new therapeutic approach. Cancer Cell Int (2020) 20(1):1–11. doi: 10.1186/s12935-020-01564-6
134. de Lima Mota A, Jardim-Perassi BV, de Castro TB, Colombo J, Sonehara NM, Nishiyama VKG, et al. Melatonin modifies tumor hypoxia and metabolism by inhibiting HIF-1α and energy metabolic pathway in the in vitro and in vivo models of breast cancer. Melatonin Res (2019) 2(4):83–98. doi: 10.32794/mr11250042
135. Pokhrel RH, Acharya S, Ahn J-H, Gu Y, Pandit M, Kim J-O, et al. AMPK promotes antitumor immunity by downregulating PD-1 in regulatory T cells via the HMGCR/p38 signaling pathway. Mol Cancer (2021) 20:1–15. doi: 10.1186/s12943-021-01420-9
136. Hevia D, González-Menéndez P, Quiros-González I, Miar A, Rodríguez-García A, Tan DX, et al. Melatonin uptake through glucose transporters: a new target for melatonin inhibition of cancer. J Pineal Res (2015) 58(2):234–50. doi: 10.1111/jpi.12210
137. Sinclair LV, Rolf J, Emslie E, Shi Y-B, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol (2013) 14(5):500–8. doi: 10.1038/ni.2556
138. Ren W, Liu G, Chen S, Yin J, Wang J, Tan B, et al. Melatonin signaling in T cells: Functions and applications. J Pineal Res (2017) 62(3):e12394. doi: 10.1111/jpi.12394
139. Panatta E, Zampieri C, Melino G, Amelio I. Understanding p53 tumour suppressor network. Biol Direct (2021) 16(1):1–7. doi: 10.1186/s13062-021-00298-3
140. Li X, Guo M, Cai L, Du T, Liu Y, Ding H-F, et al. Competitive ubiquitination activates the tumor suppressor p53. Cell Death Differentiation (2020) 27(6):1807–18. doi: 10.1038/s41418-019-0463-x
141. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res (2004) 64(7):2627–33. doi: 10.1158/0008-5472.CAN-03-0846
142. Bensaad K, Tsuruta A, Selak MA, Vidal MNC, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell (2006) 126(1):107–20. doi: 10.1016/j.cell.2006.05.036
143. Wörmann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Görgülü K, et al. Loss of P53 function activates JAK2–STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology (2016) 151(1):180–93.e12. doi: 10.1053/j.gastro.2016.03.010
144. Niu G, Wright KL, Ma Y, Wright GM, Huang M, Irby R, et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol (2005) 25(17):7432–40. doi: 10.1128/MCB.25.17.7432-7440.2005
Glossary
Keywords: T-cell malignancy, melatonin, metabolic rewiring, tumor microenvironment, T-cell exhaustion, cancer
Citation: Rai S, Roy G and Hajam YA (2024) Melatonin: a modulator in metabolic rewiring in T-cell malignancies. Front. Oncol. 13:1248339. doi: 10.3389/fonc.2023.1248339
Received: 27 June 2023; Accepted: 04 December 2023;
Published: 08 January 2024.
Edited by:
Santosh Kumar, National Institute of Technology Rourkela, IndiaReviewed by:
Sukh Mahendra Singh, Banaras Hindu University, IndiaPedro Gonzalez-Menendez, University of Oviedo, Spain
Copyright © 2024 Rai, Roy and Hajam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seema Rai, ZHJzZWVtYWthbWxlc2hAZ21haWwuY29t