
- Divisão de Farmacologia, Coordenação de Pesquisa, Instituto Nacional de Câncer, Rio de Janeiro, Brazil
Brazil is the fifth largest country in the world and its present population, in excess of 190 million, is highly heterogeneous, as a result of centuries of admixture between Amerindians, Europeans, and Sub-Saharan Africans. The estimated individual proportions of biogeographical ancestry vary widely and continuously among Brazilians: most individuals, irrespective of self-identification as White, Brown or Black – the major categories of the Brazilian Census “race/color” system – have significant degrees of European and African ancestry, while a sizeable number display also Amerindian ancestry. These features have important pharmacogenetic (PGx) implications: first, extrapolation of PGx data from relatively well-defined ethnic groups is clearly not applicable to the majority of Brazilians; second, the frequency distribution of polymorphisms in pharmacogenes (e.g., CYP3A5, CYP2C9, GSTM1, ABCB1, GSTM3, VKORC, etc) varies continuously among Brazilians and is not captured by race/color self-identification; third, the intrinsic heterogeneity of the Brazilian population must be acknowledged in the design and interpretation of PGx studies in order to avoid spurious conclusions based on improper matching of study cohorts. The peculiarities of PGx in Brazilians are illustrated with data for different therapeutic groups, such as anticoagulants, HIV protease inhibitors and non-steroidal antinflammatory drugs, and the challenges and advantages created by population admixture for the study and implementation of PGx are discussed. PGx data for Amerindian groups and Brazilian-born, first-generation Japanese are presented to illustrate the rich diversity of the Brazilian population. Finally, I introduce the reader to the Brazilian Pharmacogenetic Network or Refargen1, a nation-wide consortium of research groups, with the mission to provide leadership in PGx research and education in Brazil, with a population health impact.
Introduction
“Pharmacogenetics deals with pharmacological responses and their modification by hereditary influences”. This definition, offered by Werner Kalow in the first book dedicated to pharmacogenetics (Kalow, 1962), highlights the three pillars of this discipline: pharmacology, genetics, and human diversity. Pharmacogenetics has evolved greatly over the 50 years elapsed since Kalow’s book was published, was re-christened as pharmacogenomics in the fashion of the “omics” revolution, but its conceptual development and praxis remain contingent upon a better understanding of human genomic diversity and its impact on drug pharmacokinetics and pharmacodynamics. In a recent review of the evolution and structure of human genetic diversity, Pena (2007) discussed three models: The first, essentially typological, is based on the partition of humanity into races, visualized as being very different from each other, but internally homogeneous; the second, proposes a division into (continental) populations rather than races; the third, labeled the variable mosaic genome – VMG – paradigm, stresses individuality rather than membership in population categories based on race, ethnicity or geographical origin. I would suggest that most pharmacogenetics/-genomics (PGx) studies continue to be performed, and the data analyzed, reported, and fed into databases, in consonance with either the first or the second models described by Pena (2007). However, both models are poor descriptors of admixed populations, since genetic admixture is best modeled as a continuous variable, consistent with the VMG paradigm (Suarez-Kurtz and Pena, 2006, 2007; Pena, 2007). The individual uniqueness that is central to this paradigm implies that “each person must be treated as an individual… rather than as an exemplar of a race” (Patrinos, 2004) or in the words of McLeod (2007) “data from ethnic groups will not be as useful as analysis of individuals patients. While knowledge of ethnic differences may be relevant to much of the world’s population, its usefulness is significantly limited in situations of extensive genetic mixing”. With increasing global migration, admixture gains relevance as an additional challenge to the successful worldwide implementation of PGx in clinical practice. From this perspective, the Brazilian population, with tri-hybrid ancestral roots in Amerindian, European and African groups, and five centuries of extensive interethnic mating, provides a valuable model for studying the impact of admixture on the conceptual development and clinical implementation of PGx-informed prescription. In this review, I will initially offer a brief overview of the genetic diversity of present-day Brazilians and then move to the PGx arena to discuss data from our laboratory for different therapeutic groups and the PGx implications of the Brazilian population heterogeneity and admixture.
PGx and Population Diversity: Brazil as a Model Case
Brazil is the fifth largest country in the world and occupies an area of 8.5 million km2. Its present population exceeds 190 million people, who speak Portuguese, in contrast to all other Latin American nations in which Spanish is the official language. The language reflects the colonization of Brazil by the Portuguese, initiated in the year 1500. At that time, the indigenous population living in the area of what is now Brazil was estimated at ∼2.5 million (Ribeiro, 1995). The Portuguese–Amerindian admixture started soon after the arrival of the first colonizers. The slave trade of Sub-Saharan Africans, from the middle of the sixteenth century until the late 1800s, provided the third major ancestral root of the Brazilian population. Centuries of admixture of Europeans – mainly Portuguese, but also Spaniards, Italians and Germans – Amerindians and Africans account for the heterogeneity and diversity of the present-day population of Brazil. The extent of admixture is well documented in a wealth of population genetic studies in Brazilians (Salzano and Bortolini, 2002; Parra et al., 2003; Pena et al. 2009), and is illustrated in Figure 1 with data from a cohort of 300 healthy, unrelated individuals living in the city of Rio de Janeiro, in the Southeast region of Brazil (Suarez-Kurtz et al., 2007a). Individual DNA was genotyped with a panel of autosomal insertion/deletion (indel) polymorphisms, validated as ancestry-informative markers (Bastos-Rodrigues et al., 2006) and the individual proportions of European, Amerindian and African biogeographical ancestry were estimated using the software STRUCTURE (Pritchard et al., 2000). Each individual self-identified as white, brown, or black, according to the “race/color” classification adopted by the Brazilian Census, which relies on self-perception of skin Color. The term Color is capitalized to call attention to its special meaning in the context of the Brazilian Census classification, Color (in Portuguese, “cor”) denoting the Brazilian equivalent of the English term “race”.
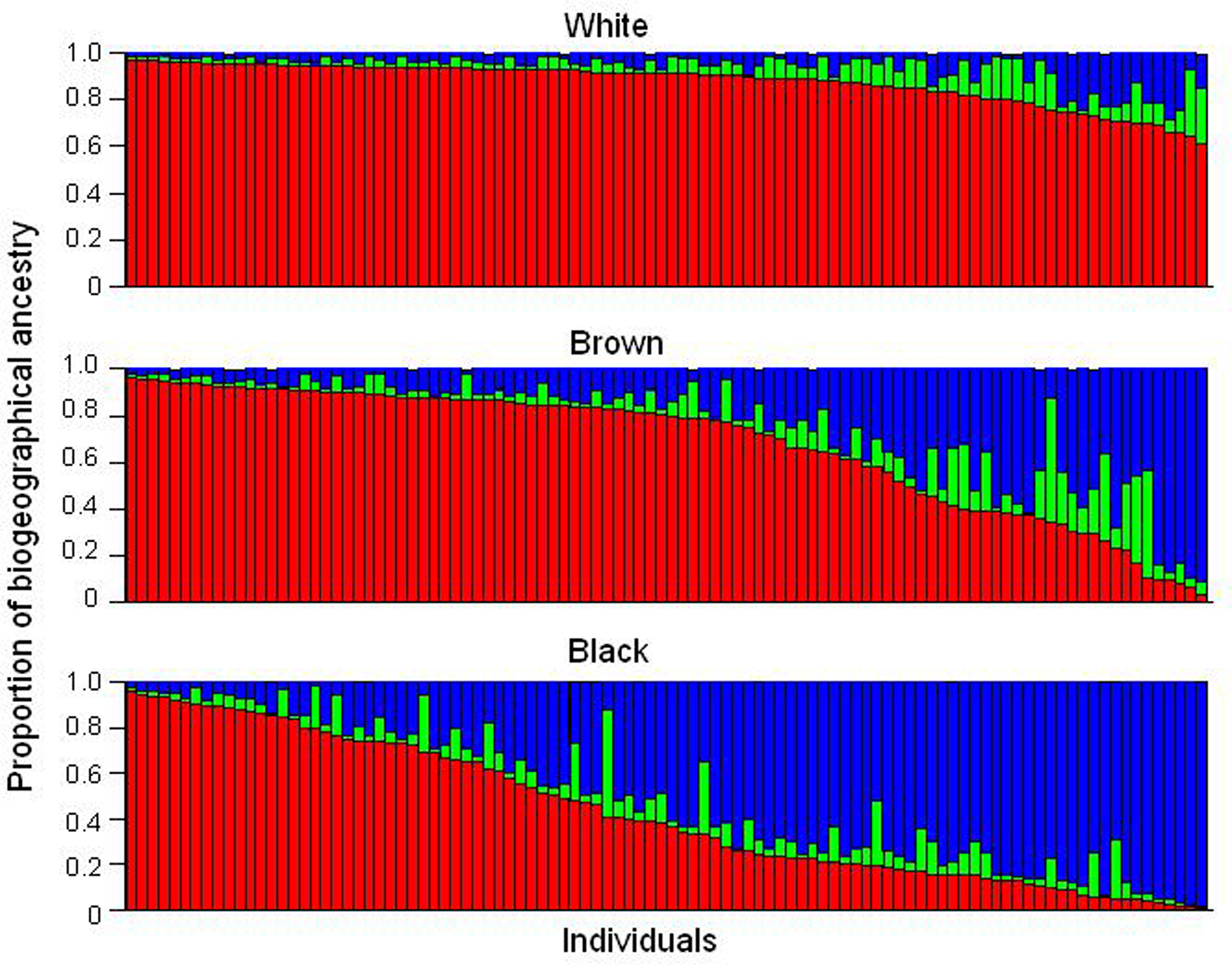
Figure 1. Assignment of Brazilian samples to genetic clusters inferred using a panel of ancestry-informative markers and the program STRUCTURE. Data from 300 individuals, resident in Rio de Janeiro, self-identified as White, Brown, or Black, according to the Color classification adopted by the Brazilian Census. Each individual is represented by a vertical line, which is partitioned in three colored segments corresponding to the individual’s estimated membership proportion in the African (blue), Amerindian (green), and European (red) clusters. Data from Suarez-Kurtz et al. (2007a).
It is clear from Figure 1 that the individual proportions of Amerindian, European, and African ancestry, vary widely among Brazilians, and that most individuals have significant degrees of European and African ancestry, while a sizeable number display also Amerindian ancestry. The average proportions of European ancestry decreased progressively from self-reported White (0.86, n = 100), to Brown (0.68, n = 100) and then to Black individuals (0.43, n = 100), whereas the opposite trend was observed with respect to African ancestry, which averaged 0.07, 0.24, and 0.50 in White, Brown, and Black persons, respectively. Amerindian ancestry was relatively constant across the three Color groups, ranging from 0.07 and 0.09 (Suarez-Kurtz et al., 2007a). Further analysis of these data is beyond the scope of this review, and herein I will focus on two features that are evident in Figure 1 and have important implications for PGx: First, European and African components together account for 0.93 (SD 0.08) of the genetic diversity in this cohort, as compared to 0.07 (0.08) for Amerindian ancestry. Consequently, European and African ancestry will have a considerably greater impact on the frequency of polymorphisms of PGx relevance, as compared to Amerindian ancestry. Second, the individual proportions of European and African ancestry vary over wide ranges, and most importantly, as a continuum across the Color categories of the Brazilian Census. Based on these two features, it is reasonable to anticipate that (i) the greater the difference in frequency of a given polymorphism between European and sub-Saharan African ancestral populations, the more likely the allele frequency will vary within the African–European admixed Brazilian population, and (ii) polymorphism frequency will vary continuously as a function of the individual proportions of European and/or African ancestry.
These predictions were verified for polymorphisms in several pharmacogenes (CYP3A5, CYP2C8, CYP2C9, CYP2C19, GSTM1, GSTT1 GSTM3, ABCB1, GNB3, and VKORC1), initially in individuals from Rio de Janeiro (Suarez-Kurtz et al., 2007a,b; Estrela et al., 2008a; Vargens et al., 2008) and more recently in a representative cohort of the overall Brazilian population, which included 1,100 self-identified White, Brown, and Black adults recruited at four different geographical regions (Suarez-Kurtz et al., 2010a,b). Figure 2 presents data for CYP2C8 to illustrate the procedure used to infer the statistical association between PGx polymorphisms and biogeographical ancestry. The plot shows that the individual odds of having the CYP2C8*2 allele increases, while the odds of having CYP2C8*3 and CYP2C8*4 decrease, as the proportion of African ancestry increases. Notably, the changes in allele probability are modeled as a continuum across the entire cohort, irrespective of self-identity with a Census Color category, consistent with the distribution of African ancestry among Brazilians shown in Figure 1. For comparison, the right column in Figure 2 shows the frequency of the CYP2C8 polymorphisms in a sample of 70 Mozambicans genotyped in our lab. Mozambicans may be considered as proxies of the African ancestors of Brazilians, since Mozambique, a former Portuguese colony in the southeast coast of Africa, was a source of slaves brought to Brazil in the nineteenth century. The agreement between the model-estimated frequency of each CYP2C8 polymorphism for Brazilians with 100% African ancestry and the observed frequency in Mozambicans is striking. However, we must caution that this result was not systematically observed for other pharmacogenes that we have studied, e.g., VKORC1 (Suarez-Kurtz et al., 2010a).
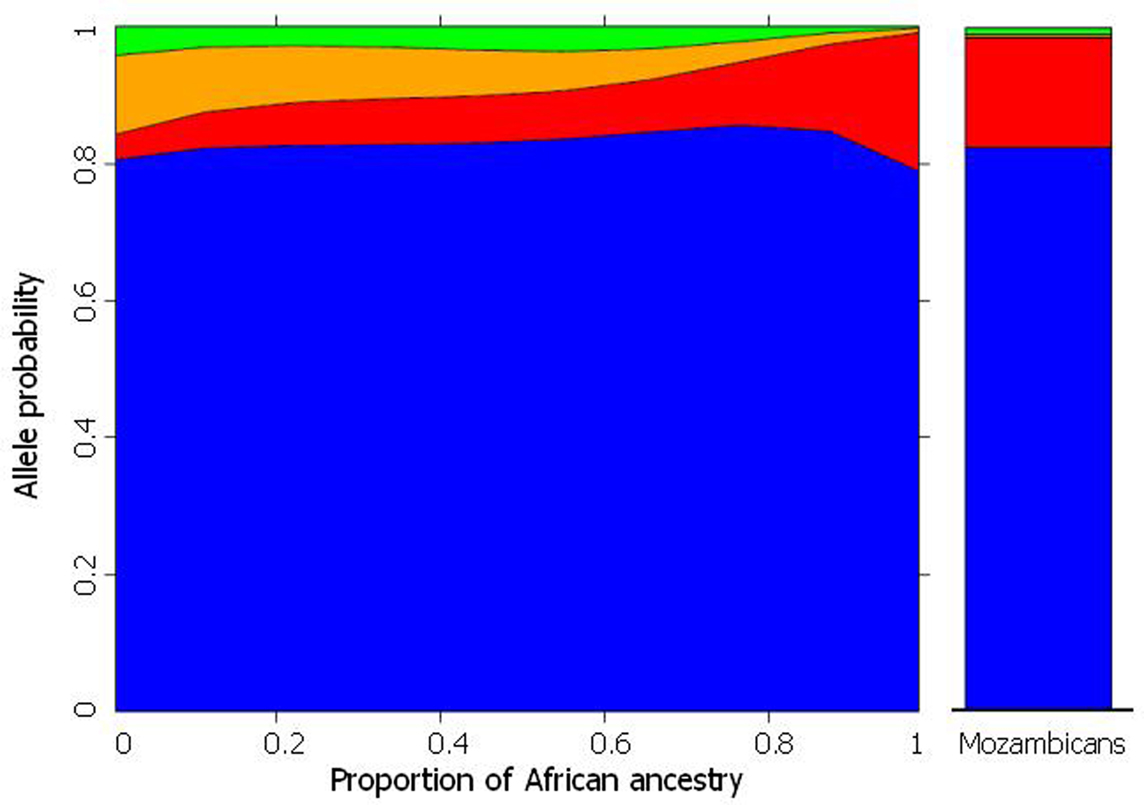
Figure 2. The plot on the left shows the frequency distribution of CYP2C8*1 (default, blue) *2 (red), *3 (orange), and *4 (red) alleles according to the estimated individual proportion of African ancestry in 958 healthy, unrelated Brazilians. Non-linear logistic regression was applied to describe the association between biogeographical ancestry and frequency of CYP2C8 alleles. Data from Suarez-Kurtz et al. (2010b). The plot on the right shows the frequency of the CYP2C8 alleles in 70 healthy, unrelated Mozambicans (Suarez-Kurtz and Damasceno, unpublished).
Assessment of the distribution of PGx polymorphisms according to biogeographical ancestry is a novel approach, which I consider more appropriate for Brazilians, and most likely, other admixed populations of the Americas. Nevertheless, most PGx studies in Brazilians are designed, analyzed, and reported according to racial/ethnic categories defined by different criteria and designated by a variety of labels. For example, the terms white, Caucasian, Caucasoid, European derived, Euro Brazilians or “of European descent” have been used to designate individuals who would, for the most part, self-identify as White according to the Brazilian Census. A similar situation is observed with respect to the Brown (reported as interethnic-admixed, mullato, intermediate, of mixed-ancestry, etc.) and Black (labeled as black, African-derived, Afro- or African–Brazilians, etc) Color categories. The poor correlation between Color and ancestry in Brazilians (see above) casts a shadow of uncertainty over studies that have used only self-reported or investigator assessment of race/color without genomic ancestry analysis. We are fully aware of this caveat, and by adopting the Census categorization in our studies do not expect to circumvent it. Quite on the contrary, we have recently shown that the frequency distribution of CYP2C8 and CYP2C9 polymorphisms among self-identified White, Brown, and Black Brazilians varies significantly across different geographical regions (Suarez-Kurtz et al., 2010b). Thus, it makes little sense to extrapolate data on CYP2C8 and CYP2C9 polymorphisms from one or more Color strata recruited at a given geographical region (or city) to the ensemble of Brazilians. This is consistent with the notion that “people who are White, Brown, or Black in the northern part of Brazil are ancestrally quite unlike people who are White, Brown, or Black in the southern part of the Country” (Pena et al., 2009). Brazilian Census Color categories are, at best, poor proxies of biogeographical ancestry, a conclusion that has important PGx implications, since Brazilians may not fit into “race” or continental categories used to stratify participants of multinational PGx studies. For example, Brazilians self-identified as Pardo (Brown) could not be allocated into any of the four sub-groups (White, Asian, Black, or Mixed) of the International Warfarin Pharmacogenetic Consortium, a very important initiative that enrolled over 5,000 patients from nine countries and four continents (IWPC, 2009). Also, Black Brazilians, with an estimated average proportion of African ancestry of 0.51 (Suarez-Kurtz et al., 2007), would not properly fit into the IWPC Black category, which included Africans and also African–Americans, the latter having average proportions of African ancestry around 0.80 (Parra, 2007). As a corollary, warfarin dosing algorithms that have a “race” term defined by criteria prevalent in a given region or country (e.g., the United States OMB) are unlikely to be applicable worldwide, especially in extensively admixed populations, such as Brazilians. On a broader perspective, one may ask whether the global population diversity may be captured by inserting “race” terms in PGx algorithms that are sufficiently “friendly” to be adopted by the practicing prescriber?
PGx Studies in Brazilians
The population diversity of Brazil implies that extrapolation of data derived from relatively well-defined ethnic groups is clearly not applicable to the majority of Brazilians (Suarez-Kurtz, 2008a,b). Only recently, recognition of this fact translated into PGx research on the clinical response to prescribed drugs. By comparison, much more information has accumulated over the last 15 years on genetic variation in metabolic pathways for environmental pro-carcinogens and its impact on cancer risk in Brazilians. Furthermore, various PGx targets such as alpha- and beta-adrenergic receptors, dopamine and 5HT receptors, components of the renin–angiotensin system, vascular endothelial growth factor and methylenetetrahydrofolate reductase have been the object of studies of disease susceptibility and phenotypes, rather than drug response in a clinical setting (Suarez-Kurtz and Pena, 2007). Nevertheless, a few academic groups have conducted important PGx research on different therapeutic classes in the Brazilian population. Space limitations do not allow for a comprehensive review (and referencing) of all published PGx clinical trials in Brazilians, but I would like to highlight the contribution of a few distinct groups in this area, before presenting an overview of our own studies. Mara Hutz et al. (Universidade Federal do Rio Grande do Sul) investigated the impact of genetic polymorphisms on the efficacy and toxicity of HMG-CoA reductase inhibitors (“statins”) in hypercholesterolemia (Hutz and Fiegenbaum, 2008), on the effects of methylphenidate in attention-deficit/hyperactivity disorders (Kieling et al., 2010), and on the efficacy of clozapine in schizophrenic patients (Kohlrausch et al., 2008). Rosario Hirata et al. (Universidade de São Paulo, USP) explored genetic determinants of the lipid lowering effect of atorvastatin (Rodrigues et al., 2007) and the impact of statins on the expression of ABC drug transporters and CYP3A drug metabolizing enzymes (Rebecchi et al., 2009). José-Eduardo Tanus-Santos (USP) examined the influence of polymorphisms in the endothelial nitric oxide synthase gene on the therapeutic drug response in gestational hypertension and preeclampsia (Sandrim et al., 2010) and on the atorvastatin-induced changes in blood nitrite levels and erythrocyte membrane fluidity (Nagassaki et al., 2009).
Pharmacokinetics of Antiretrovirals
The Brazilian public health system pioneered, in the early 1990's, the universal access to HIV medications free of charge to all citizens who need it2. Ritonavir-boosted lopinavir formulations are frequently included in highly active antiretroviral therapy (HAART) regimens for HIV infection. Both drugs are HIV protease inhibitors, but ritonavir at the doses used in co-formulations with lopinavir, serves the purpose of inhibiting CYP3A drug metabolizing enzymes, thereby increasing (“boosting”) the exposure to lopinavir, a CYP3A substrate. We studied the impact of polymorphisms in the ABCB1, CYP3A5, and SLCO1B1 genes on the pharmacokinetics of lopinavir and ritonavir in HIV-infected men under stable treatment with Kaletra® at the Hospital Universitário Clementino Fraga Filho, Universidade Federal do Rio de Janeiro. With respect to ABCB1 the study cohort was genotyped for the 1236C > T, 2677G > T/A, and 3435C > T SNPs. Multivariate regression analysis was applied to assess the influence of ABCB1 genotypes and haplotypes on the trough (pre-dose) concentrations of lopinavir and ritonavir in blood plasma, semen, and saliva (Estrela et al., 2009). We observed marked inter-individual variability in the concentrations of both protease inhibitors in the three matrices (e.g., a 27.5-fold range for lopinavir in plasma, Figure 3), but no association with ABCB1 genotypes or haplotypes.
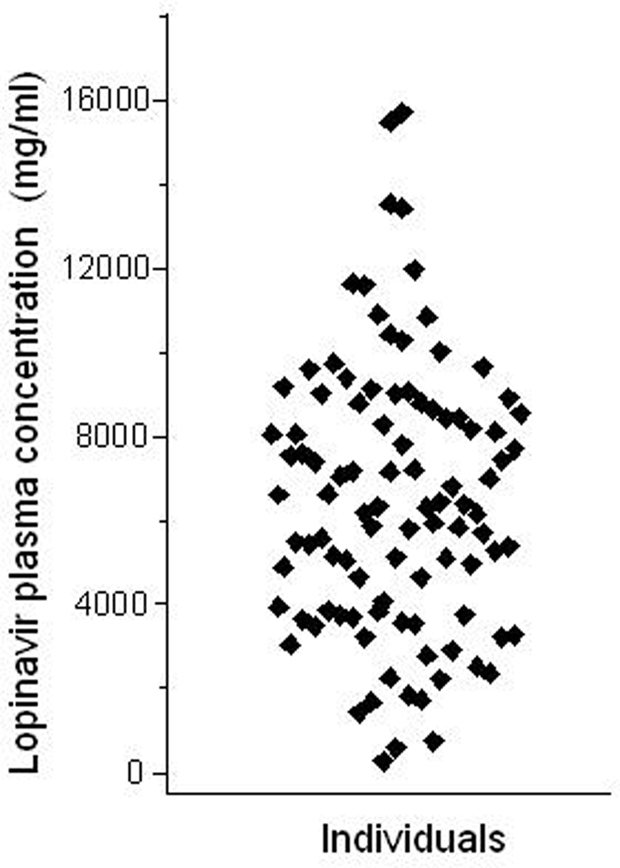
Figure 3. Intra-individual variability of lopinavir pharmacokinetics. The plot shows the trough (pre-dose) concentration of lopinavir in the plasma of 99 HIV-infected males, under stable HAART treatment including two daily doses of lopinavir (400 mg) and ritonavir (100 mg). Each data point corresponds to one individual. Data from Estrela et al. (2009).
The goal of the CYP3A5 study in HIV-infected men (Estrela et al., 2008b) was to examine whether the reported impact of the CYP3A5 genotype on the disposition of the HIV protease inhibitor, saquinavir, administered as a single dose and with no ritonavir boosting to Tanzanian healthy individuals (Josephson et al., 2007) prevailed under a natural clinical setting. Our data revealed no influence of the CYP3A5 genotype on the trough plasma concentration of lopinavir and ritonavir in HIV-infected males on a stable HAART regimen. We ascribed the apparent discrepancy between our results (Estrela et al., 2008b) and those of Josephson et al. (2007) to ritonavir boosting, which was thought to occlude any pharmacokinetic consequences of functional polymorphisms in the CYP3A5 gene. Together, the two studies describe complementary steps towards the implementation of PGx in clinical practice, namely the discovery of association(s) between genetic polymorphisms and pharmacological traits under specific experimental conditions, followed by examination of the impact of the association(s) in the targeted patient populations, studied in the relevant clinical setting.
Human immunodeficiency virus protease inhibitors have been recently reported to be substrates of the SLCO1B1/OATP1 drug transporter, and a SNP (521T > C) in the SLCO1B1 gene was associated with plasma levels of lopinavir in HIV-infected individuals (Hartkoorn et al., 2010). The availability of plasma concentration data for lopinavir and ritonavir from our cohort of HIV-infected men under stable HAART, provided the opportunity for seeking independent confirmation of these results and for extending these observations in two directions: first, by examining two other SLCO1B1 SNPs, (388A > G, 463C > A) and second, by exploring the association of the SLCO1B1 polymorphisms with ritonavir plasma concentrations (Kohlrausch et al., 2010). Our results confirmed that carriers of the 521C allele display significantly higher lopinavir (but not ritonavir) plasma concentrations relative to the wild-type TT genotype. There was no significant effect of either 388A > G or 463C > A SNPs on lopinavir or ritonavir plasma concentrations. Reduced OATP-mediated uptake of lopinavir by the hepatocytes in carriers of the SLCO1B1 521C allele was thought to account for the increased plasma levels of lopinavir. The clinical usefulness of this observation is uncertain at present, in view of both the extensive overlap of the trough concentrations of lopinavir in the plasma across the three 521T > C genotypes and the low prevalence (<5%) of the homozygous variant genotype (521CC) in most populations. Further studies to confirm the importance of SLCO1B1 polymorphisms in lopinavir pharmacokinetics are warranted.
Pharmacokinetics and Pharmacodynamics of NSAIDs
The implementation of regulatory legislation for generic drugs in Brazil, in the late 1990s provided an impetus for bioequivalence studies. The relatively large number of samples collected from healthy subjects under the rigorously controlled conditions of bioequivalence trials offer a favorable experimental setting for assessing the influence of genetic polymorphisms on pharmacokinetic parameters. Our group explored this opportunity with respect to the impact of CYP2C9 polymorphisms on the pharmacokinetics and pharmacodynamics of the non-steroidal anti-inflammatory drugs (NSAIDs), tenoxicam, and piroxicam (Vianna-Jorge et al., 2004; Perini et al., 2005). These were “gene-candidate” studies, the choice of CYP2C9 being based on previous knowledge that the encoded cytochrome P-450 (CYP) enzyme, CYP2C9, provides the major metabolic pathway for the inactivation of both piroxicam and tenoxicam. The polymorphisms investigated, CYP2C9*2 and CYP2C9*3, encode CYP2C9 isoenzymes with reduced metabolic activity towards these NSAIDs. The results of the pharmacokinetic analyses are shown in Table 1.

Table 1. Impact of CYP2C9 polymorphisms on pharmacokinetic parameters for single doses of piroxicam and tenoxicam in healthy Brazilian individuals.
The CYP2C9 genotypes had no influence on the peak concentration of either drug in plasma (Cmax). By contrast, the plasma concentration–time curve from zero to infinity (AUCinf), a measure of drug exposure, was significantly greater in CYP2C9*1/*2 or *1/*3 heterozygous, as compared to wild-type homozygous subjects (CYP2C9*1/*1). This effect was associated with, and explained by the significantly lower oral clearance (corrected for body weight, CL/Fcor) of either NSAID in heterozygous carriers of the CYP2C9*2 or *3 alleles, compared to wild-type homozygous subjects.
The piroxicam study was extended to include a pharmacodynamic parameter, namely the ex vivo production of thromboxane B2 (TxB2), catalyzed by cyclo-oxygenase 1 (COX-1), a distinct target of NSAIDs. Our work hypothesis was that inhibition of COX-1 by piroxicam would be greater in carriers of the defective CYP2C9*2 and CYP2C9*3 alleles, because of the increased exposure to piroxicam in these individuals, as compared to wild-type homozygous (Perini et al., 2005). The results verified our hypothesis by showing significant differences in TxB2 production between either CYP2C9*1/*2 or CYP2C9*1/*3 genotypes and CYP2C9*1/*1. The ANOVA R2 (coefficient of variation) among the three genotypes, indicated that 78% of the inter-individual variability may be explained by the CYP2C9 polymorphisms examined, namely CYP2C9*2 and CYP2C9*3. This is a remarkable example of PGx modulation, as there are not many instances of such a large contribution of genetic polymorphisms to the inter-individual variability of pharmacodynamic phenotypes.
The piroxicam bioequivalence trial disclosed one individual who displayed a unique pharmacokinetic profile, such that his plasma piroxicam concentration 13 days after the administration of a single drug dose remained at 70% of the peak concentration measured at 2 h (estimated half-life of elimination, 500 h). Nevertheless, no adverse effects were observed or reported by the individual. After a 6-month washout, when piroxicam was no longer detected in his plasma, this individual was re-exposed to a single piroxicam dose, and blood samples were collected for 120 days for CYP2C9 genotyping, measurements of piroxicam plasma concentrations and assessment of the activity of COX-1 and COX-2 – the pharmacodynamic targets of NSAIDs – through the ex vivo formation of TBX2 and prostaglandin E2, respectively (Perini and Suarez-Kurtz, 2006). The individual was genotyped as CYP2C9*3/*3, an extremely rare genotype in Brazilians, which encodes a non-functional CYP2C9 isoform. The impact of this genotype was remarkable: the effects on pharmacokinetic parameters observed with the first dose were reproduced and the pharmacodynamic analyses showed that COX-2 activity was maintained below 50% of its control value for more than one month and that COX-1 was below 20% of its control value for more than 2 months after the single piroxicam dose! Again, no adverse effects were observed. We speculated on the potential advantages conferred by the PGx polymorphisms associated with impaired drug metabolism for long-term strategies in disease prevention and treatment (Perini and Suarez-Kurtz, 2006).
Warfarin Dosing Algorithms for Brazilians
Warfarin combines several characteristics that make it a model target for individualized drug therapy: (i) it is the most commonly prescribed oral anticoagulant in Brazil and worldwide, (ii) there is large inter-individual variation in the required dosage, (iii) its therapeutic index is narrow and incorrect dosage, especially during the initial phase of treatment, carries a high risk of bleeding or failure to prevent thromboembolism, and (iv) a reliable biomarker, the international normalized ratio (INR), is available for quantifying warfarin’s anticoagulant effect. Polymorphisms in two genes, namely CYP2C9 and VKORC1, have been repeatedly found to associate with the clinical response to warfarin. CYP2C9 encodes CYP2C9, the primary metabolic route for the disposition of S-warfarin, the most active isomer of clinically used racemic warfarin. The polymorphic VKORC1 gene codifies VKORC1 the molecular target of coumarin anticoagulants. In collaboration with the Instituto Nacional de Cardiologia Laranjeiras (INCL), a reference cardiology hospital of the Brazilian Public Health System located in Rio de Janeiro, we assessed the contribution of CYP2C9 and VKORC1 polymorphisms, and of demographic and clinical variables to the inter-individual variability in warfarin dose requirement for stable anticoagulation, defined as three successive INR readings in the 2–3.5 range (Perini et al., 2008a). The warfarin dose required for stable anticoagulation varied from 3 to 75 mg/week, i.e., a 25-fold inter-individual variability! Multivariate regression modeling was applied to develop a dosing algorithm for prediction of the appropriate warfarin dose for each patient. As shown in Figure 4, the VKORC1 3673G > A genotype proved to be the most important predictor in our model, with a partial R2 value of 23.8%, followed by the number of copies of variant CYP2C9 alleles (R2 = 6.9%) and by co-treatment with amiodarone (R2 = 5.6%). Body weight, therapeutic indication for the use of warfarin, age, and co-treatment with simvastatin were the other covariates associated with warfarin dose requirement. A dosing algorithm including all these covariates explained 51% of the variance in warfarin dose requirement in our study cohort.
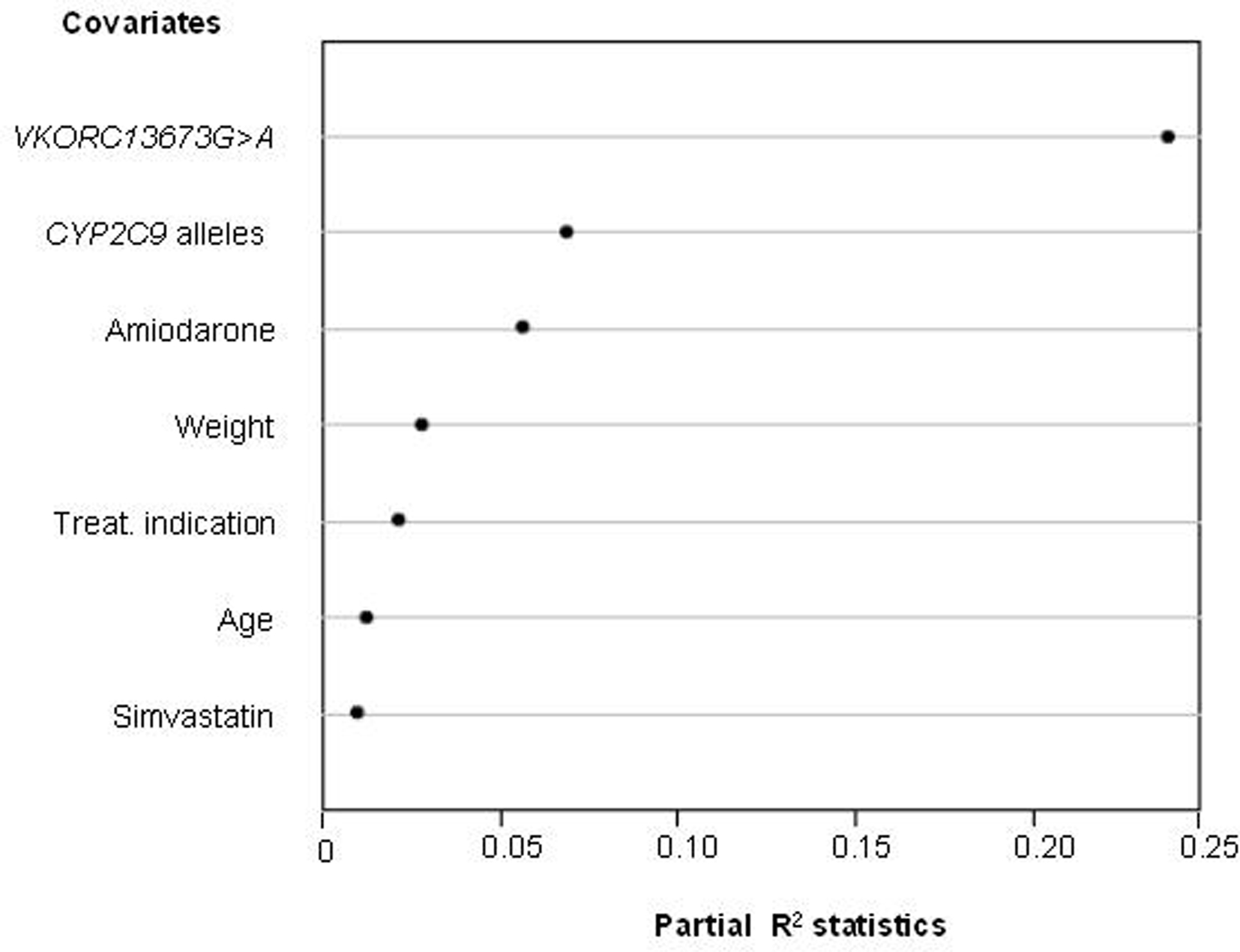
Figure 4. Influence of genetic and non-genetic factors on the warfarin dose requirement for stable anticoagulation in 390 patients from the INCL, Rio de Janeiro. Multivariate regression modeling was applied to identify the factors (covariates) that associate significantly with the individual warfarin weekly dose requirement. The relative contribution of each covariate to the final model is expressed by the partial R2 statistics (abscissa), which measures the degree of association between two random variables, with the effect of a set of controlling random variables removed. The higher the R2 value, the greater the contribution of the covariate to the model. Data from Perini et al. (2008).
We extended our studies on the INCL cohort to explore the association of other variables with warfarin dose requirements (Suarez-Kurtz et al., 2009), and reported that the inclusion of INR/dose as a covariate in regression modeling of the stable warfarin dose leads to a novel algorithm with greater predictive power (R2 = 60%; Figure 5). The most informative model retained the same covariates previously identified as associated with stable warfarin weekly dose in this cohort (age, weight, treatment indication, co-medication with amiodarone, or simvastatin, VKORC1 and CYP2C9 genotypes) but included also an INR/dose term. A distinct feature of the novel algorithm is that the individual INR/dose term does not represent a fixed time point after starting warfarin therapy, but rather the first measurement taken after admission of the patients in the anticoagulant unit. This feature is potentially useful for patients under continuous warfarin treatment, who had not reached stable dosing despite repeated dose adjustments.
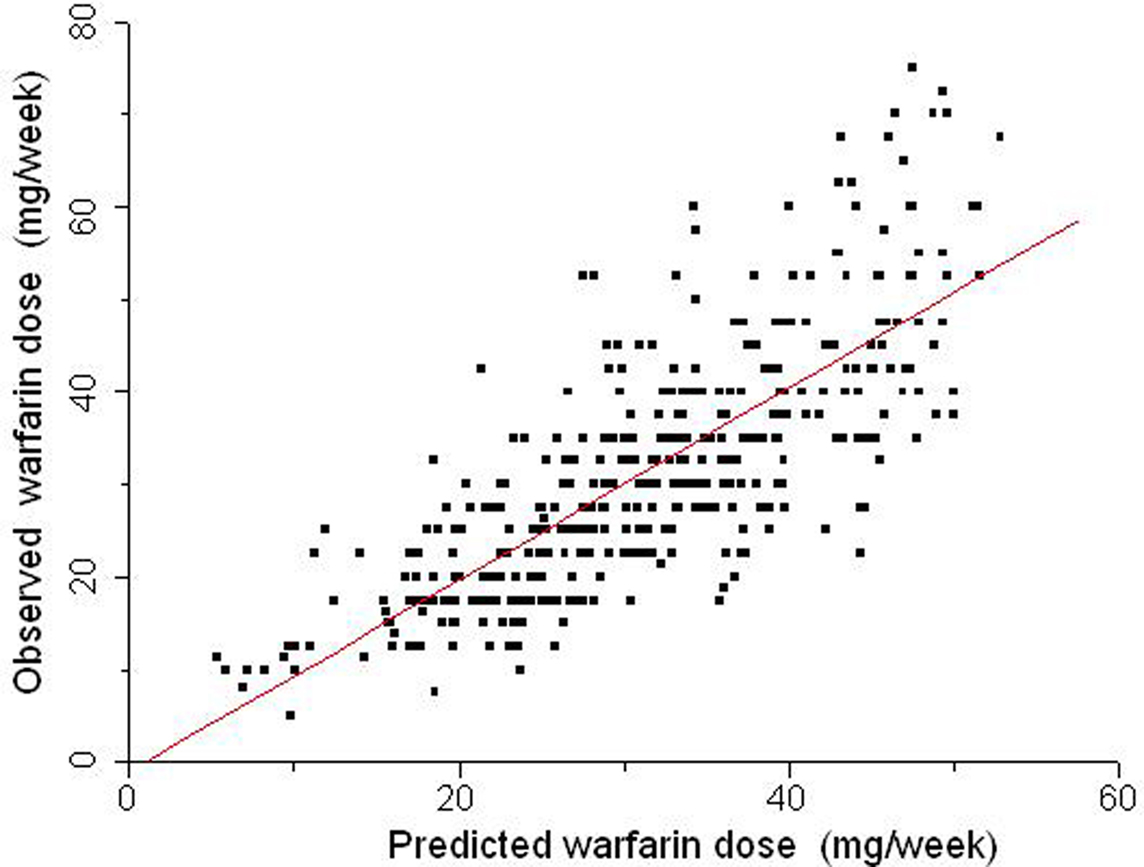
Figure 5. Relationship between warfarin weekly doses predicted by a multiple regression model (abscissa) and the stable doses actually taken by 390 patients (ordinate) from INCL, Rio de Janeiro. The covariates in the dosing algorithm were age, weight, treatment indication, co-medication with amiodarone or simvastatin, VKORC1, and CYP2C9 genotypes and the first available INR reading for each patient. The identity line is traced. Data from Suarez-Kurtz et al. (2009).
In a recently published study (Perini et al., 2010) we investigated the association between warfarin dose requirement and a polymorphism in CYP4F2, namely, rs2108622 C > T (V33M). Biological plausibility for this association was provided by the evidence that CYP4F2 is a vitamin K1 oxidase and carriers of the CYP4F2 33M allele are likely to have elevated hepatic levels of vitamin K1, requiring higher warfarin doses (McDonald et al., 2009). Previous studies had reported controversial results regarding the contribution of the CYP4F2 rs2108622 to the warfarin dose requirement for stable anticoagulation, but had one common feature: the enrolled patients were exclusively or predominantly (>90%) white, of European descent. The allele frequency of rs2108622 differs markedly among populations, being lower in sub-Saharan Africans and African–Americans (0–9%) as compared with Europeans and white North Americans (17–33%)3. From a population perspective, it might be anticipated that rs2108622 will have a smaller influence, if any, on the warfarin dose requirements in black individuals. We explored this hypothesis in the INCL cohort, which included self-identified White, Brown, and Black individuals, and observed that addition of the rs2108622 genotype as a variable had only a marginal effect on the predictive power of the warfarin dosing algorithm previously derived from this patient cohort. We concluded that prospective CYP4F2 genotyping is not justified in Brazilians who are potential candidates for warfarin therapy (Perini et al., 2010).
Collectively, out studies on the PGx of warfarin in Brazilians have distinct strengths. First, they reflect real-life community prescribing and dispensing of warfarin in the context of a public hospital in a developing country. Second, the recruitment of patients from the notoriously admixed and heterogeneous Brazilian population allowed for the investigation of PGx associations in individuals with heterogeneous genetic ancestry, under the same environmental conditions. A distinct feature of a PGx algorithms derived from our patient cohort is the ability to predict equally well the warfarin dose requirement in White and Black patients, in marked contrast with the poorer outcome of several warfarin dosing algorithms in blacks, notably African–Americans, as compared to white patients (Gage et al., 2008). Third, the non-genetic covariates of our dosing algorithms are readily available in the patient medical record, whereas the genetic covariates, namely, the CYP2C9 and VKORC1 3673G > A genotypes, can be determined in a time frame compatible with the urgency of starting warfarin administration to many patients. However, we also recognize that our studies have limitations, most of which are common to studies of similar design performed in other populations. First, by being retrospective, the associations and dosing algorithm described in our studies must be prospectively tested in an appropriately large cohort. Second, we did not assess the impact of the algorithms on the clinical response to warfarin but instead used the INR as a surrogate for it. Third, by explaining 51–60% of the variance in warfarin dosing, our algorithms leave 40–50% of this variance to be accounted for by other variables, whether genetic or non-genetic. Finally, our algorithms, developed for a heterogeneous and admixed population of a large Brazilian city, may not be applicable to other population groups.
Studies in Amerindian Groups Living in Brazil
Native American (Amerindian) populations are poorly represented in PGx research and PGx databases (reviewed by Jaja et al., 2008). The extant Amerindian population of Brazil is very diverse, consisting of more than 227 ethnic groups speaking more than 180 languages4. This diversity is reflected in the frequency of polymorphisms in genes encoding drug metabolizing enzymes and drug targets (reviewed in Suarez-Kurtz and Pena, 2007). In collaboration with Maria Luiza Petzl-Erler et al. from Universidade Federal do Paraná we assessed the distribution of four SNPs in VKORC1 in three endogamous Amerindian populations, namely Guarani-Kaiowá, Guarani-Ñandeva, and Kaingang, living in indigenous reservation areas in the South and Center-West regions of Brazil (Perini et al., 2008b). Despite living side by side for centuries, the Guarani (Tupi-Guarani linguistic family) and the Kaingang (Gê-speaking people) maintain their cultural and linguistic distinctiveness, which is paralleled by substantial genetic distance between them. Our results indicated that the VKORC1 5808T > G SNP was absent or rare (<3%), whereas 3673G > A, 6853G > C and 9041G > A were frequent (34–63%) in the three Amerindian populations. No difference was detected in VKORC1 allele or genotype frequency between the two Guarani populations, whereas significant differences were observed between Kaingang and Guarani. The data disclosed a uniqueness of the frequency distribution of the VKORC1 SNPs in the Guarani and Kaingang, compared to Asian, African, and European populations. However, in view of the vast inter-populational diversity among Amerindians, these results should not be interpreted as representative of other extant Amerindian peoples. Our observation that 40% of Kaingang and 60% of Guarani have VKORC1 haplotypes that include the variant 3673A allele associated with increased susceptibility to warfarin, suggests that these two Amerindian populations comprise high proportions of individuals requiring reduced warfarin doses for appropriate anticoagulation in the prevention or treatment of thromboembolism.
Studies in Brazilians with Japanese Ancestry
Brazil hosts the largest Japanese community outside Japan, estimated at 1.5 million individuals, one third of whom are first-generation, Brazilian-born with native Japanese parents. This large community provided a unique opportunity for comparative studies of the distribution of PGx polymorphisms in native Japanese versus their Brazilian-born descendants. In collaboration with ândrea Ribeiro-dos-Santos et al. (Universidade Federal do Pará) and Emilio Moriguchi (Universidade Federal do Rio Grande do Sul) we explored this opportunity with respect to functional polymorphisms in genes that modulate drug disposition (CYP2C9, CYP2C19, and GSTM3) or response (VKORC1), and that differ significantly in frequency in native Japanese versus Brazilians with no Japanese ancestry (Perini et al., 2009). Our study detected no difference in the frequency of the CYP2C9*2, CYP2C9*3, CYP2C19*2, CYP2C19*3, GSTM3*A/*B, or VKORC1 3673G > A, 5808T > G, 6853G > C, and 9041G > A polymorphisms between native Japanese and first-generation Japanese descendants. In contrast, significant differences in the frequency of each polymorphism were observed between native or first-generation Japanese and Brazilians with no Japanese ancestry. The striking similarity in the frequency of clinically relevant PGx polymorphisms between Brazilian-born Japanese descendants and native Japanese led us to suggest that the former may be recruited for clinical trials designed to generate bridging data for the Japanese population in the context of the International Conference on Harmonization for drug development.
The Brazilian Pharmacogenetic Network (Refargen)
To deal with the specificity of PGx in Brazilians, a nation-wide network named Rede Nacional de Farmacogenética (Brazilian National Pharmacogenetics Network, Refargen) was established in 2004 (Suarez-Kurtz, 2004). Presently, the network congregates 18 research groups from various Brazilian institutions, and its mission is to provide leadership in PGx research and education in Brazil, with a population health impact. Information on Refargen’s members and activities, as well as an updated database of allele, genotype and haplotype frequencies of PGx polymorphisms in the Brazilian population may be accessed at the network internet site1.
Concluding Remarks and Perspectives
The kaleidoscopic diversity of the admixed Brazilian population, with tri-hybrid biogeographical ancestry in Europe, Africa, and America adds complexity to, but also creates advantages for PGx research. Advantages include the opportunity to explore PGx associations in individuals with heterogeneous genetic ancestry, under similar environmental and socio-economical conditions, and to gather information on peoples that are excluded or under-represented in clinical drug trials. For example, studies in Brazilians could fill in PGx information gaps, pertinent to Africans and Native Americans. A major challenge to pharmacogeneticists studying the Brazilian population is population stratification, which if not controlled for, will confound the outcomes of PGx association studies. Our studies have provided examples of how to control for this caveat, by combining ancestry-informative markers and appropriate statistical approaches. A distinct message that emerges from these studies is that race/color categorization does not capture the distribution of PGx polymorphisms among Brazilians, which is best modeled by continuous functions of the individual proportions of European and African ancestry, irrespective of self-identified Color. Recognition of this fact is important in the design and interpretation of PGx clinical trials in Brazilians but does not imply that PGx-informed drug prescription requires investigation of individual ancestry. Rather, individual genotyping should be directed to PGx polymorphisms of proven clinical utility for the specific medical condition being treated, irrespective of biogeographical ancestry. Because of their tri-ancestral roots and extensive admixture, the vast majority of Brazilians will require PGx tests for polymorphisms that have been clinically validated in European, sub-Saharan African and, to a lesser extent, Amerindian populations. The CYP2C9 gene may be used as an example, in the context of warfarin dose requirement: variant alleles CYP2C9*5, CYP2C9*6, CYP2C9*8, and CYP2C9*11 occur in Africans but are rare or absent in Europeans, whereas the opposite is observed with respect to CYP2C9*2 and CYP2C9*3. Genotyping of the six polymorphisms could be justified in Brazilians and, most likely, African–Americans, but not Europeans, in whom only CYP2C9*2 and CYP2C9*3 genotyping might be adequate for predicting the CYP2C9 phenotype.
Personalized drug therapy, the promise of pharmacogenomics, must be based on the recognition of the inherent genetic individuality. This notion is particularly relevant to admixed populations, in which substructure increases further the fluidity of racial/ethnic labels (Suarez-Kurtz, 2005). Because interethnic admixture is either common or increasing at a fast pace in many, if not most populations, extrapolation on a global scale of PGx data from well-defined ethnic groups is plagued with uncertainty. To impact positively on global health, PGx must broaden its scope, with respect to both target and population diversity, and be inclusive of admixed populations with their perceived challenges and advantages. From this perspective, PGx studies in Brazilians have the potential to contribute relevant information towards personalized drug prescription worldwide.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in the author’s laboratory is sponsored by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior (CAPES), Financiadora de Estudos e Projetos (Finep) and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (Faperj).
Footnotes
References
Bastos-Rodrigues, L., Pimenta, J. R., and Pena, S. D. J. (2006). The genetic structure of human populations studied through short insertion-deletion polymorphisms. Ann. Hum. Genet. 70, 658–665.
Estrela, R. C. E., Ribeiro, F. S., Carvalho, R. S., Gregorio, S. P., Dias-Netto, E., Struchiner, C. J., and Suarez-Kurtz, G. (2008a).The distribution of ABCB1 polymorphisms among Brazilians: Impact of population admixture. Pharmacogenomics 9, 267–276.
Estrela, R. C. E., Santoro, A. B., Barroso, P. F., Tuyama, M., and Suarez-Kurtz, G. (2008b). CYP3A5 genotype has no impact on plasma trough concentrations of lopinavir and ritonavir in HIV-infected subjects. Clin. Pharmacol. Ther. 84, 205–207.
Estrela, R. C. E., Ribeiro, F. S., Barroso, P. F., Tuyama, M., Gregório, S. P., Dias-Neto, E., Struchiner, C. J., and Suarez-Kurtz. G. (2009). ABCB1 polymorphisms have no impact on the concentrations of lopinavir and ritonavir in blood, semen and saliva of HIV-infected men under stable antiretroviral therapy. Pharmacogenomics 10, 311–318.
Gage, B. F., Eby, C., Johnson, J. A., Deych, E., Rieder, M. J., Ridker, P. M., Milligan, P. E., Grice, G., Lenzini, P., Rettie, A. E., Aquilante, C. L., Grosso, L., Marsh, S., Langaee, T., Farnett, L. E., Voora, D., Veenstra, D. L., Glynn, R. J., Barrett, A., and McLeod, H. L. (2008). Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin. Pharmacol. Ther. 84, 326–331.
Hartkoorn, R. C., Kwan, W. S., Shallcross, V., Chaikan, A., Liptrott, N., Egan, D., Sora, E. S., James, C. E., Gibbons, S., Bray, P. G., Back, D. J., Khoo, S. H., and Owen, A. (2010). HIV protease inhibitors are substrates for OATP1A2, OATP1B1 and OATP1B3 and lopinavir plasma concentrations are influenced by SLCO1B1 polymorphisms. Pharmacogenet. Genomics 20, 112–120.
Hutz, M. H., and Fiegenbaum, M. (2008). Impact of genetic polymorphisms on the efficacy of HMG-CoA reductase inhibitors. Am. J. Cardiovasc. Drugs 8, 161–170.
IWPC, The International Warfarin Pharmacogenetic Consortium, Klein, T. E., Altman, R. B., Eriksson, N., Gage, B. F., Kimmel, S. E., Lee, M. T., Limdi, N. A., Page, D., Roden, D. M., Wagner, M. J., Caldwell, M. D., and Johnson, J. A. (2009). Estimation of the warfarin dose with clinical and pharmacogenetic data. New Engl. J. Med. 360, 753–764.
Jaja, C., Burke, W., Thummel, K., Edwards, K., and Veenstra, D. L. (2008). Cytochrome p450 enzyme polymorphism frequency in indigenous and native american populations, a systematic review. Community Genet. 11, 141–149.
Josephson, F., Allqvist, A., Janabi, M., Sayi, J., Aklillu, E., Jande, M., Mahindi, M., Burhenne, J. Bottiger, Y., Gustafsson, L. L., Haefeli, W. E., and Bertilsson, L. (2007). CYP3A5 genotype has an impact on the metabolism of the HIV protease inhibitor saquinavir. Clin. Pharmacol. Ther. 81, 708–712.
Kalow, W. (1962). Pharmacogenetics, Heredity and the Response to Drugs. Philadelphia: WB Saunders Co.
Kieling, C., Genro, J. P., Hutz, M. H., and Rohde, L. A. (2010). A current update on ADHD pharmacogenomics. Pharmacogenomics 11, 407–419.
Kohlrausch, F. B., Estrela, R. C. E., Barroso, P. F., and Suarez-Kurtz, G. (2010). The impact of SLCO1B1 polymorphisms on the plasma concentration of lopinavir and ritonavir in HIV-infected men. Br. J. Clin. Pharmacol. 69, 95–98.
Kohlrausch, F. B., Salatino-Oliveira, A., Gama, C. S., Lobato, M. I., Belmonte-de-Abreu, P., and Hutz, M. H. (2008). G-protein gene 825C > T polymorphism is associated with response to clozapine in Brazilian schizophrenics. Pharmacogenomics 9, 1429–1436.
McDonald, M. G., Rieder, M. J., Nakano, M., Hsia, C. K., and Rettie, A. E. (2009). CYP4F2 is a vitamin K1 oxidase, An explanation for altered warfarin dose in carriers of the V433M variant. Mol. Pharmacol. 75, 1337–1346.
McLeod, H. L. (2007). “Pharmacogenetics in the African American population,” In Pharmacogenomics in Admixed Populations, ed. G. Suarez-Kurtz (Austin, TX: Landes Bioscience). 47–59.
Nagassaki, S., Herculano, R. D., Graeff, C. F., and Tanus-Santos, J. E. (2009). eNOS T-786C polymorphism affects atorvastatin-induced changes in erythrocyte membrane fluidity. Eur. J. Clin. Pharmacol. 65, 385–392.
Parra, E. J. (2007). “Admixture in north America,” in Pharmacogenomics in Admixed Populations, ed. G. Suarez-Kurtz (Austin, TX: Landes Bioscience), 28–46.
Parra, F. C., Amado, R. C., Lambertucci, J. R., Rocha, J., Antunes, C. M., and Pena, S. D. J. (2003). Color and genomic ancestry in Brazilians. Proc. Natl. Acad. Sci. USA 100, 177–182.
Pena, S. D. J. (2007). “The evolution and structure of human genetic diversity,” in Pharmacogenomics in Admixed Populations, ed. G. Suarez-Kurtz (Austin, TX: Landes Bioscience), 1–11.
Pena, S. D., Bastos-Rodrigues, L., Pimenta, J. R., and Bydlowski, S. P. (2009). DNA tests probe the genomic ancestry of Brazilians. Braz. J. Med. Biol. Res. 42, 870–876.
Perini, J. A., Struchiner, C. J., Silva-Assunção, E., Santana, I. S. C., Rangel, F., Ojopi, E. B., Dias-Neto, E., and Suarez-Kurtz, G. (2008a). Pharmacogenetics of warfarin: development of a dosing algorithm for Brazilian patients. Clin. Pharmacol. Ther. 84, 722–728.
Perini, J. A., Petzl-Erler, M. L., Tsuneto, L. T., and Suarez-Kurtz, G. (2008b). VKORC1 polymorphisms in Amerindian populations of Brazil. Pharmacogenomics 9, 1623–1629.
Perini, J. A., Struchiner, C. J., Silva-Assunção, E., and Suarez-Kurtz, G. (2010). Impact of CYP4F2 rs2108622 on the stable warfarin dose in an admixed patient cohort. Clin. Pharmacol. Ther. 87, 417–420.
Perini, J. A., and Suarez-Kurtz, G. (2006). Impact of CYP2C9*3/*3 genotype on the pharmacokinetics and pharmacodynamics of piroxicam. Clin. Pharmacol. Ther. 80, 549–551.
Perini, J. A., Vargens, D. D., Santana, I. S., Moriguchi, E. H., Ribeiro-Dos-Santos, A. K., Tsutsumi, M., and Suarez-Kurtz, G. (2009). Pharmacogenetic polymorphisms in Brazilian-born, first-generation Japanese descendants. Braz. J. Med. Biol. Res. 42, 1179–1184.
Perini, J. A., Vianna-Jorge, R., Brogliato, A. R., and Suarez-Kurtz, G. (2005). Influence of CYP2C9 genotypes on the pharmacokinetics and pharmacodynamics of piroxicam. Clin. Pharmacol. Ther. 78, 362–369.
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Rebecchi, I. M., Rodrigues, A. C., Arazi, S. S., Genvigir, F. D., Willrich, M. A., Hirata, M. H., Soares, S. A., Bertolami, M. C., Faludi, A. A., Bernik, M. M., Dorea, E. L., Dagli, M. L., Avanzo, J. L., and Hirata, R. D. (2009). ABCB1 and ABCC1 expression in peripheral mononuclear cells is influenced by gene polymorphisms and atorvastatin treatment. Biochem. Pharmacol. 77, 66–75.
Ribeiro, D. (1995). O Povo Brasileiro, a Formação e o Sentido do Brasil. São Paulo: Companhia das Letras.
Rodrigues, A. C., Hirata, M. H., and Hirata, R. D. (2007). The genetic determinants of atorvastatin response. Curr. Opin. Mol. Ther. 9, 545–553.
Salzano, F. M., and Bortolini, M. C. (2002). A Study of Brazilian Populations. Detroit: Wayne State University Press.
Sandrim, V. C., Palei, A. C., Luizon, M. R., Izidoro-Toledo, T. C., Cavalli, R. C., and Tanus-Santos, J. E. (2010). eNOS haplotypes affect the responsiveness to antihypertensive therapy in preeclampsia but not in gestational hypertension. Pharmacogenomics J. 10, 40–45.
Suarez-Kurtz, G. (2004). Pharmacogenomics in admixed populations, the Brazilian pharmacogenetics/pharmacogenomics network – REFARGEN. Pharmacogenomics J. 4, 347–438.
Suarez-Kurtz, G. (2005). Pharmacogenetics in admixed populations. Trends Pharmacol. Sci. 26, 196–201.
Suarez-Kurtz, G. (2008a). The implications of population admixture in race-based therapy. Clin. Pharmacol. Ther. 3, 399–400.
Suarez-Kurtz, G. (2008b). Ethnic differences in drug therapy, a pharmacogenomics perspective. Expert Rev. Clin. Pharmacol. 1, 337–339.
Suarez-Kurtz, G., Amorim, A., Damasceno, A., Hutz, M. H., Moraes, M. O., Ojopi, E. B., Pena, S. D. J., Perini, J. A., Prata, M. J., Ribeiro-dos-Santos, A., Romano-Silva, M. A., Teixeira, D., and Struchiner, C. J. (2010a). VKORC1 polymorphisms in Brazilians, comparison with the Portuguese and Portuguese-speaking Africans and pharmacogenetic implications. Pharmacogenomics 11, 1257–1267.
Suarez-Kurtz, G., Genro, J. P., Moraes, M. O., Ojopi, E. B., Pena, S. D. J., Perini, J. A., Ribeiro-dos-Santos, A., Romano-Silva, M. A., Santana, I., and Struchiner, C. J. (2010b). Global pharmacogenomics: Impact of population diversity on the distribution of polymorphisms in the CYP2C cluster among Brazilians. in preparation.
Suarez-Kurtz, G., and Pena, S. D. J. (2006). Pharmacogenomics in the Americas, Impact of genetic admixture. Curr. Drug Targets 7, 1649–1658.
Suarez-Kurtz, G., and Pena, S. D. J. (2007). “Pharmacogenetic studies in the Brazilian population” in Pharmacogenomics in Admixed Populations, ed. G. Suarez-Kurtz (Austin, TX: Landes Bioscience), 75–98.
Suarez-Kurtz, G., Vargens, D. D., Struchiner, C. J., Bastos-Rodrigues, L., and Pena, S. D. J. (2007a). Self-reported skin color, genomic ancestry and the distribution of GST polymorphisms. Pharmacogenet. Genomics 17, 765–771.
Suarez-Kurtz, G., Perini, J. A., Bastos-Rodrigues, L., Pena, S. D. J., and Struchiner, C. J. (2007b). Impact of population admixture on the distribution of the CYP3A5*3 polymorphism. Pharmacogenomics 8, 1299–1306.
Suarez-Kurtz, G., Perini, J. A., Silva-Assunção, E., and Struchiner, C. J. (2009). Relative contribution of VKORC1, CYP2C9 and INR response to warfarin stable dose. Blood 113, 4125–4126.
Vargens, D. D., Almendra, L., Struchiner, C. J., and Suarez-Kurtz, G. (2008). Distribution of the GNB3 825C > T polymorphism among Brazilians, impact of population structure. Eur. J. Clin. Pharmacol. 3, 253–256.
Keywords: biogeographical ancestry, Brazilian pharmacogenetic network, population admixture, warfarin, HIV protease inhibitors, non-steroidal anti-inflammatory drugs
Citation: Suarez-Kurtz G (2010) Pharmacogenetics in the Brazilian population. Front. Pharmacol. 1:118. doi: 10.3389/fphar.2010.00118
Received: 29 July 2010;
Paper pending published: 20 August 2010;
Accepted: 27 August 2010;
Published online: 04 October 2010.
Edited by:
Andrea Gaedigk, The Children’s Mercy Hospital and Clinics, USAReviewed by:
Liewei Wang, Mayo Clinic, USAMara H. Hutz, Universidade Federal do Rio Grande do Sul, Brazil
Copyright: © 2010 Suarez-Kurtz. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Guilherme Suarez-Kurtz, Instituto Nacional de Câncer, Coordenação de Pesquisa, Divisão de Farmacologia, Rua André Cavalcanti 37, Rio de Janeiro, RJ 22290-290, Brazil. e-mail:a3VydHpAaW5jYS5nb3YuYnI=