

- Consiglio Nazionale delle Ricerche, Istituto di Biofisica, Genova, Italy
CLC-K channels belong to the CLC gene family, which comprises both Cl− channels and Cl−/H+ antiporters. They form homodimers which additionally co-assemble with the small protein barttin. In the kidney, they are involved in NaCl reabsorption; in the inner ear they are important for endolymph production. Mutations in CLC-Kb lead to renal salt loss (Bartter’s syndrome); mutations in barttin lead additionally to deafness. CLC-K channels are interesting potential drug targets. CLC-K channel blockers have potential as alternative diuretics, whereas CLC-K activators could be used for the treatment of patients with Bartter’s syndrome. Several small organic acids inhibit CLC-K channels from the outside by binding to a site in the external vestibule of the ion conducting pore. Benzofuran derivatives with affinities better than 10 μM have been discovered. Niflumic acid (NFA) exhibits a complex interaction with CLC-K channels. Below ∼1 mM, NFA activates CLC-Ka, whereas at higher concentrations NFA inhibits channel activity. The co-planarity of the rings of the NFA molecule is essential for its activating action. Mutagenesis has led to the identification of potential regions of the channel that interact with NFA. CLC-K channels are also modulated by pH and [Ca2+]ext. The inhibition at low pH has been shown to be mediated by a His-residue at the beginning of helix Q, the penultimate transmembrane helix. Two acidic residues from opposite subunits form two symmetrically related intersubunit Ca2+ binding sites, whose occupation increases channel activity. The relatively high affinity CLC-K blockers may already serve as leads for the development of useful drugs. On the other hand, the CLC-K potentiator NFA has a quite low affinity, and, being a non-steroidal anti-inflammatory drug, can be expected to exert significant side effects. More specific and more potent activators will be needed and it will be important to understand the molecular mechanisms that underlie NFA activation.
Introduction
CLC proteins form a family of voltage-gated Cl− channels and Cl−/H+-exchangers involved in important physiological processes, including transepithelial transport, membrane excitability, cell volume regulation, and luminal acidification of the endosomal–lysosomal system (Zifarelli and Pusch, 2007). Knockout mouse models and human diseases have provided insights into the physiologic role of CLC proteins. Mutations in four of the nine human CLC genes underlie inherited diseases. CLC-1 mutations cause myotonia; mutations in CLC-Kb and in BSDN, the gene codifying barttin, determine different forms of Bartter’s syndrome with renal salt loss; CLC-5 mutations are responsible for Dent’s disease; mutations of CLC-7 lead to osteopetrosis (Zifarelli and Pusch, 2007; Jentsch, 2008).
Kidney and inner ear specific CLC-K channels are particularly interesting potential drug targets. In the present review, after providing a brief overview over CLC proteins in general and CLC-K channels in particular, we will review in detail the current knowledge on the interaction of CLC-K channels with various organic and inorganic ligands.
All CLC proteins share several basic features. Structurally, they share the same homodimeric architecture (Ludewig et al., 1996; Middleton et al., 1996; Weinreich and Jentsch, 2001), in which each of two identical subunits contains an independent anion permeation pathway (“double-barreled channel”) (see Figure 3A); even though some CLC proteins require small β-subunits (barttin and ostm1) for proper function (Estévez et al., 2001; Waldegger et al., 2002; Lange et al., 2006), the basic mechanisms of ion conduction are mostly dependent of the CLC α-subunits. So far all CLC proteins that have been functionally investigated, appear to be impermeable to cations (except protons, see below). The open probability (Po) of the CLC channels is regulated by two distinct gating mechanisms: a “protopore gate” or “fast gate” (in CLC-0) closes individual pores and an additional, slow (or common) gate closes both pores simultaneously. It is still unclear whether similar gating mechanisms are present also in CLC transporters, like the human CLC-5 Cl−/H+ antiporter or the plant CLC-a NO3−/H+ antiporter (De Angeli et al., 2006, 2009; Zdebik et al., 2008; Alekov and Fahlke, 2009; Zifarelli and Pusch, 2009b; Picollo et al., 2010).
Important structural and functional information was provided by the crystal structures of bacterial CLCs (Dutzler et al., 2002, 2003). Each monomer consists of 18 intramembrane α-helices (labeled A-R) and exhibits a complex topology. Each subunit presents an internal pseudo-two-fold symmetry with the two halves spanning the membrane in opposite directions. Each subunit bears three distinct Cl− binding sites which can be occupied simultaneously, called Sint, Scen, Sext (Dutzler et al., 2002, 2003; Lobet and Dutzler, 2006; Picollo et al., 2009). In the crystal structure of CLC-ec1, Sext was found to be occupied by the side chain of the conserved glutamate 148. When E148 was mutated to alanine or glutamine the same site was occupied by a Cl− ion and the ion conduction pathway was open (Dutzler et al., 2003). In fact, in most CLC proteins, selective conduction and gating are intimately coupled: the permeant ions directly affect gating (Pusch et al., 1995) and the critical E148 is the main determinant of the “fast gate” (Dutzler et al., 2003). Interestingly, in contrast to most CLC channels and CLC transporters, CLC-K channels have a hydrophobic residue (valine) in place of the critical glutamate (Waldegger and Jentsch, 2000; Zifarelli and Pusch, 2007; Jentsch, 2008).
The common characteristics of CLC proteins initially led to the implicit assumption that the family is composed of only Cl− channels. Surprisingly, Accardi and Miller (2004) discovered that CLC-ec1 is a Cl−/H+ exchanger. Subsequent studies have shown that also several eukaryotic CLC proteins are anion/proton antiporters (Picollo and Pusch, 2005; Scheel et al., 2005; De Angeli et al., 2006; Neagoe et al., 2010), demonstrating that two different transport mechanisms can be based on the same molecular architecture.
In contrast to the bacterial CLC-ec1, all eukaryotic and some prokaryotic CLC proteins have large C-terminal intracellular domains which bear two so-called CBS domains (Estévez and Jentsch, 2002). These domains have been found to bind adenine nucleotides in isolated CLC-5 and CLC-2 C-terminals fragments (Scott et al., 2004; Wellhauser et al., 2006; Meyer et al., 2007) and adenine nucleotides affect the function of CLC-1 (Bennetts et al., 2005, 2007; Tseng et al., 2007), CLC-5 (Zifarelli and Pusch, 2009b), and the plant atCLC-a (De Angeli et al., 2009). However, the CBS domains of CLC-Ka do not appear to bind nucleotides (Markovic and Dutzler, 2007), and no consistent effects of intracellular nucleotides on CLC-K function have been described so far.
CLC proteins are expressed in many organs, yet the kidney is the preferential expression site for several CLC channels and exchangers. In fact the physiological functions of this organ require a matching system of transporters targeted specifically to the apical or the basolateral membranes. In particular, a large fraction of NaCl and cation reabsorption happens at the level of Henle’s loop of the nephron. In the thick ascending limb (TAL) the concerted action of the Na/K-ATPase, an apical Na–K–2Cl co-transporter, an apical K+ recycling channel, and basolateral CLC-Kb/barttin Cl− channels mediates NaCl reabsorption (Figure 1A).
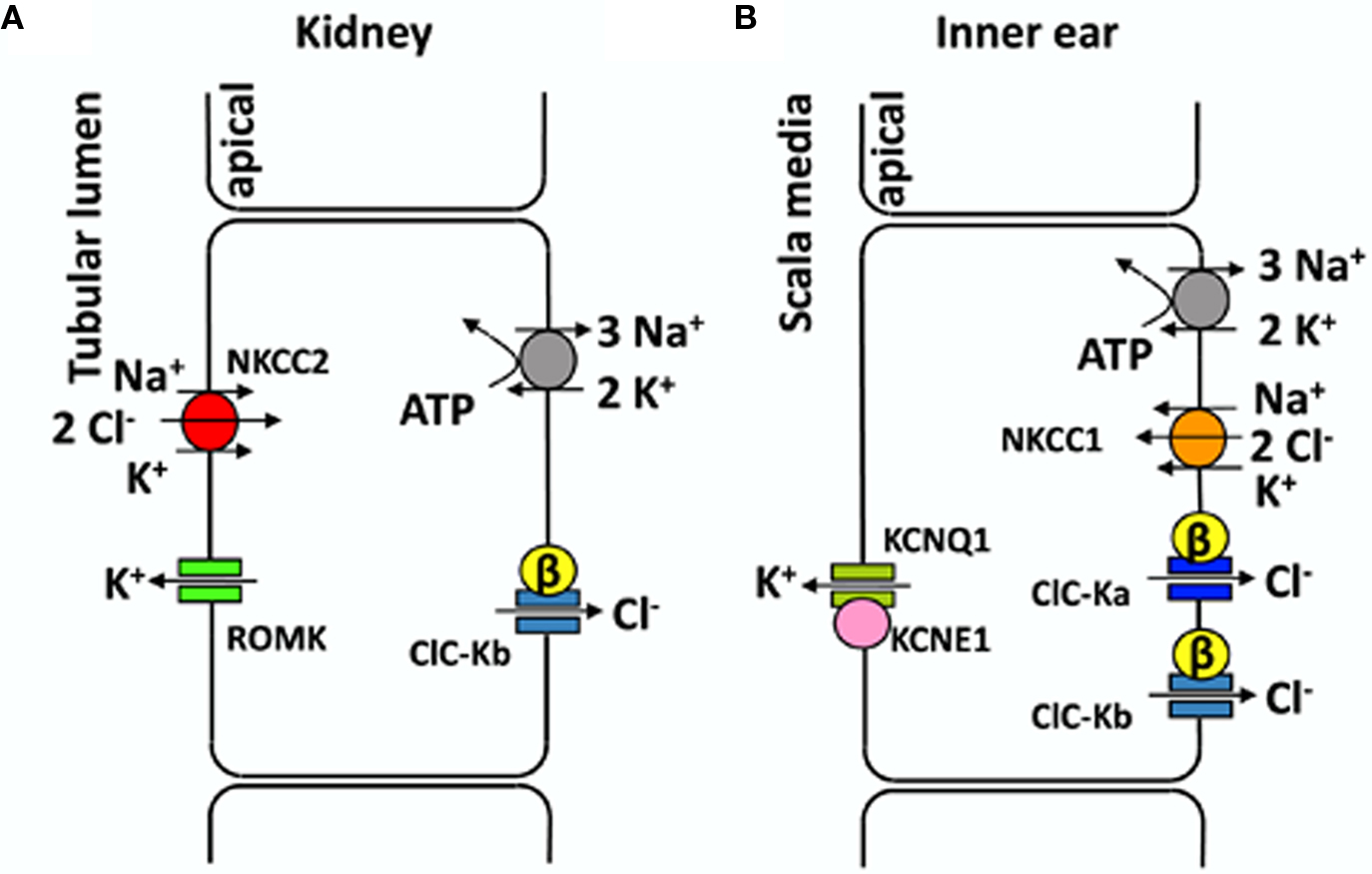
Figure 1. Transport models for NaCl reabsorption in the TAL (A) and for transepithelial K+ transport in the inner ear (B). (A) Schematic representation of an epithelial cell from the thick ascending limb of Henle’s loop. All transporters involved in NaCl reabsorption are drawn in different colors. (B) Schematic representation of marginal cell from the stria vascularis (for simplicity other epithelial cell layers are not shown). See text for details.
Human CLC-Ka and CLC-Kb share 90% of identity whereas they are 80% identical to rat CLC-K1 and CLC-K2 (Uchida et al., 1993; Kieferle et al., 1994). CLC-K channels are expressed in the kidney and in the inner ear and co-assemble with the β-subunit barttin (Birkenhäger et al., 2001; Estévez et al., 2001). CLC-K1 is the only CLC-K channel that is functional without barttin upon heterologous expression (Uchida et al., 1993; Waldegger and Jentsch, 2000; Estévez et al., 2001). Barttin not only enhances trafficking from the endoplasmic reticulum to the plasma membrane but also affects the processes of permeation and gating of CLC-K channels (Scholl et al., 2006; Fischer et al., 2010; Lang, 2010).
CLC-K2 (CLC-Kb) is expressed in basolateral membranes of several nephron segments (TAL, connecting tubule, distal convoluted tubule, and intercalated cells), whereas CLC-K1 is expressed in both apical and basolateral membranes of the thin ascending limb of Henle’s loop (Uchida et al., 1995; Vandewalle et al., 1997; Matsumura et al., 1999; Estévez et al., 2001; Kobayashi et al., 2001, 2002). The fact that CLC-K1 is also found on apical membranes might be related to its ability to function without barttin. CLC-K channels are found in the inner ear also, in the basolateral membranes of marginal cells of the stria vascularis and in dark cells of the vestibular organ, where they are involved in the endolymph production (Estévez et al., 2001; Rickheit et al., 2008; Zdebik et al., 2009). Several transporters contribute to maintain a high K+ concentration of the endolymph and its positive potential of ∼80 mV with respect to normal extracellular space. Both factors (high [K+], and high potential) are essential for the stimulation of hair cells and the sound transmission in the ear (Figure 1B). A recent study (Rickheit et al., 2008), using mice in which barttin was deleted in the inner ear but not in the kidney, showed that while [K+] and fluid secretion were maintained, the endocochlear potential was reduced. This drastic decrease reduces the driving force for K+ entry into hair cells and causes deafness in Bartter’s syndrome type IV (Rickheit et al., 2008).
The renal diseases associated with mutations in the genes coding for CLC-Ks and barttin underscore the physiological role of these channels. Mutations in CLC-Kb and barttin cause Bartter’s syndrome (type III and type IV, respectively), a kidney disease characterized by renal salt wasting due to a reduced NaCl reabsorption in the TAL (Simon et al., 1997). Additional deafness is present in Bartter’s syndrome type IV (Birkenhäger et al., 2001). Diseases associated with CLC-Ka mutations are not known, but simultaneous mutations in CLC-Ka and CLC-Kb lead to Bartter’s syndrome (type IV) with deafness (Schlingmann et al., 2004). Moreover, mice lacking CLC-K1 exhibit nephrogenic diabetes insipidus (Matsumura et al., 1999), suggesting a role for CLC-K1 and, possibly CLC-Ka, in the urine concentrating mechanism (Matsumura et al., 1999). CLC-Kb polymorphisms have been involved in high blood pressure (Jeck et al., 2004). However, subsequent studies were in conflict with these results (Kokubo et al., 2005; Speirs et al., 2005).
Specific modulators of CLC-K channels could be useful in the treatment of several pathological conditions. For example CLC-Ka block, increasing water diuresis, might reduce cardiac load after heart failure (Fong, 2004; Picollo et al., 2004). On the other hand, CLC-Ka potentiators may be useful in patients with Bartter’s syndrome with residual activity of CLC-K channels (Zifarelli et al., 2010). Because there is no information on the functional properties of the rat CLC-K2 channel, we limit our review on the data that have been obtained for rat CLC-K1 and the two human isoforms.
The Discovery of Bis-Phenoxy Derivatives of CPP as Blockers of CLC-K Channels
Before the discovery of barttin as an essential β subunit of CLC-K channels (Estévez et al., 2001), only the rat CLC-K1 could be functionally expressed in heterologous systems (Uchida et al., 1993). However, the expression level was too low to allow a detailed pharmacological characterization. To overcome this limitation and to explore the functional properties of the human CLC-K homologs, Waldegger and Jentsch (2000) constructed chimeras between rat CLC-K1 and human CLC-Kb. One of these chimeras was used by Liantonio et al. (2002) in an early pharmacological investigation. These authors tested various derivatives of CPP (p-chlorophenoxy-propionic acid) (Figure 2A) as potential CLC-K inhibitors. CPP, a blocker of the skeletal muscle Cl− conductance (De Luca et al., 1992), had previously been shown to block CLC-0 and CLC-1 from the intracellular side (Aromataris et al., 1999; Pusch et al., 2000). While CPP itself was ineffective (from the outside), bis-phenoxy derivatives of CPP (e.g., GF-100, Figure 2B) were shown to be relatively potent inhibitors of the CLC-K chimera when applied from the outside (KD ∼100 μM) (Liantonio et al., 2002). GF-100 was also effective on wild-type CLC-K1 co-expressed with barttin with a similar apparent KD of ∼100 μM (Liantonio et al., 2002). Interestingly, GF-100 was highly effective also on CLC-1, but only when applied from the inside, whereas it had almost no effect on CLC-K1 from the intracellular side (Liantonio et al., 2002).
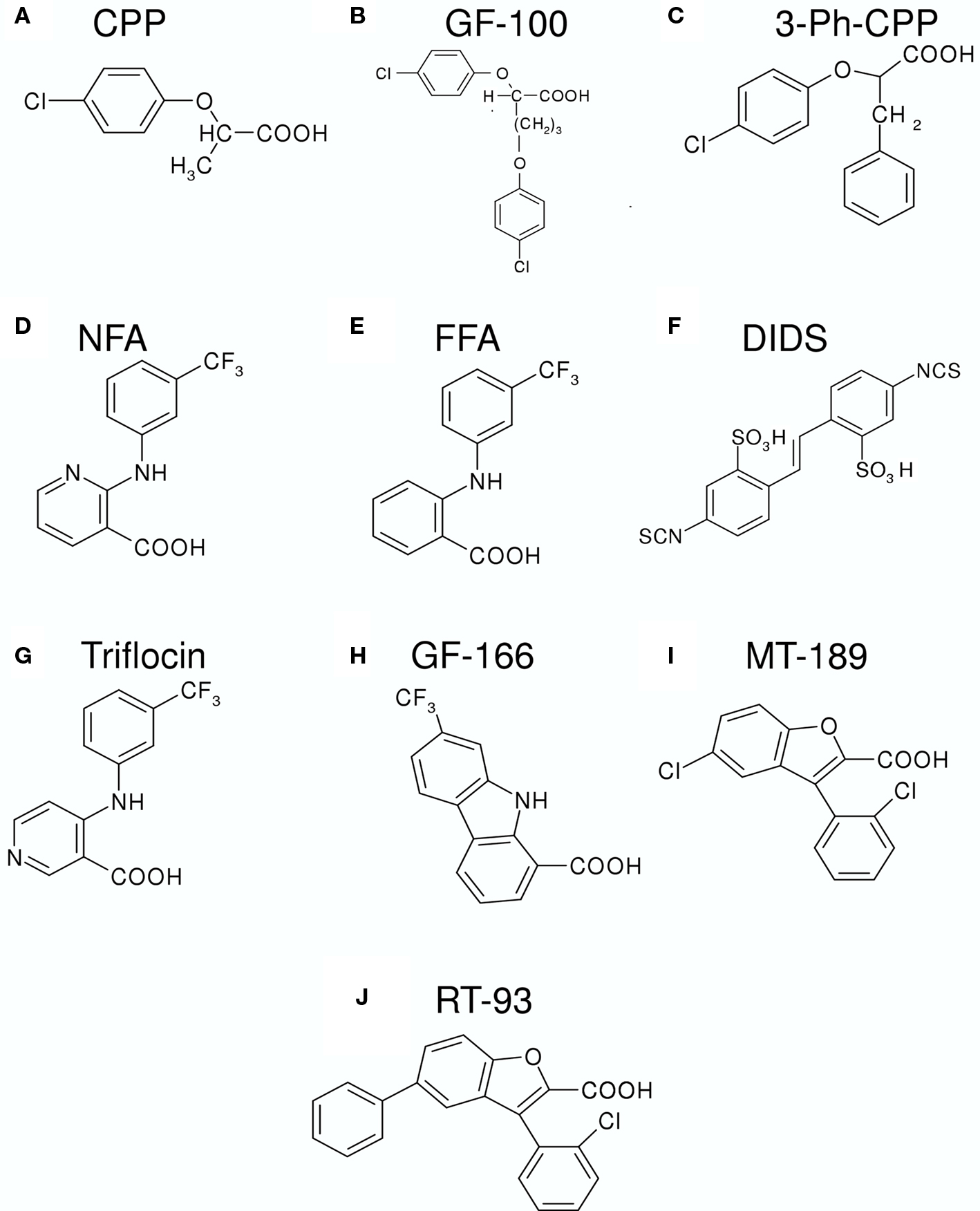
Figure 2. Chemical structures of various substances that inhibit or activate CLC-K channels. (A) CPP (p-chlorophenoxy-propionic acid); (B) GF-100 (proper name); (C) 3-Ph-CPP (3-phenyl-CPP); (D) NFA (niflumic acid); (E) FFA (flufenamic acid); (F) DIDS (4,4′-Diisothiocyanato-2,2′-stilbenedisulfonic acid); (G) Triflocin; (H) GF-166 (proper name); (I) MT-189 (proper name); (J) RT-93 (proper name).
Following this pioneering work, Liantonio et al. (2004) performed a structure–activity relationship (SAR) analysis based on the GF-100 molecule, using rat CLC-K1 co-expressed with barttin. Among several molecules, the simplest structure compatible with an elevated blocking potency (KD ∼100 μM) was 3-phenyl-CPP (Figure 2C) (Liantonio et al., 2004). Furthermore, it was found that the block had a rapid onset and was quickly reversible, demonstrating that the binding site must be exposed to the extracellular side of the channel. The block by 3-phenyl-CPP was slightly voltage-dependent and was significantly increased in low [Cl]ext (Liantonio et al., 2004). This finding suggested that the binding site is located in or close to the Cl− ion conducting pathway. Among several “classical” Cl− channel blockers, CLC-K1 was found to be quite sensitive to extracellular application of 5-nitro-2-(3-phenylpropylamino)benzoic acid (NPPB, KD ∼230 μM), niflumic acid (NFA, KD ∼250 μM), and 4,4′-Diisothiocyanato-2,2′-stilbenedisulfonic acid (DIDS, KD ∼150 μM) (Liantonio et al., 2004). However, data on DIDS inhibition have to be interpreted with caution because the DIDS molecule is unstable in aqueous solution (see below).
A Pore-Blocking 3-Phenyl, Dids, and Benzofuran Binding Site
The initial pharmacological studies employed the rat CLC-K1 homolog co-injected with barttin which manifests an elevated functional expression level in Xenopus oocytes. In following studies it was found that the human homolog CLC-Ka, co-expressed with barttin, showed a similar sensitivity to 3-phenyl-CPP (Figure 2C) and DIDS (Figure 2F) as the rat CLC-K1 with apparent KD values of 80 and 90 μM, respectively (Picollo et al., 2004). Surprisingly, human CLC-Kb, despite a more than 90% sequence identity with CLC-Ka, was significantly less sensitive to both compounds (apparent KD about five-fold larger, Figure 4A) (Picollo et al., 2004). By comparing the sequences of CLC-K1, CLC-Ka, and CLC-Kb, and based on the structure of bacterial CLC homologs (Dutzler et al., 2002, 2003) four residues of helix B (Figure 4B) were selected as candidates that are responsible for the differential block of CLC-Ka and CLC-Kb by 3-phenyl-CPP and DIDS (Picollo et al., 2004). In fact, the amino acid at position 68, a negatively charged Asp in the less sensitive CLC-Kb and a neutral Asn in the sensitive CLC-Ka, was found to be the major determinant of 3-phenyl-CPP sensitivity (Figures 4B,C) (Picollo et al., 2004). The amino acid at position 72, a negatively charged Glu in the less DIDS sensitive CLC-Kb and a neutral Gly in the DIDS sensitive CLC-Ka, was found to be additionally important for DIDS sensitivity (Picollo et al., 2004). Residue D/N68 is localized in the putative extracellular vestibule of the channel, in agreement with the notion of a pore-blocking mechanism (colored red in Figure 3). However, details of this blocking mechanism remain to be investigated. Most CLC proteins carry a negatively charged amino acid at the equivalent position and mutation of the respective residue rather drastically alters functional properties (Fahlke et al., 1995; Ludewig et al., 1997; Picollo et al., 2004) and can lead to myotonia in the case of CLC-1 (Heine et al., 1994).
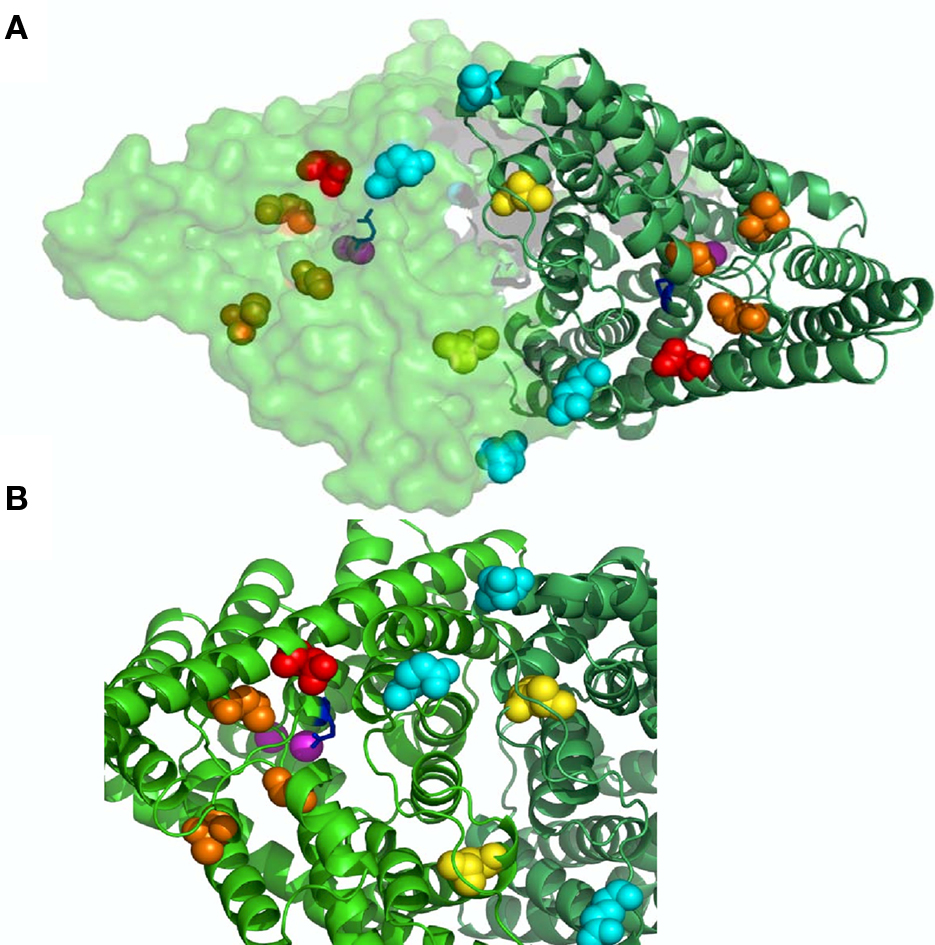
Figure 3. Binding sites on CLC-K channels. In (A) a view from the outside of the bacterial CLC-ec1 (pdb code 1OTS) homodimer is shown with one subunit in cartoon and the other in surface representation. In (B) a zoom of a selected region highlighting the residues involved in ligand binding. Cl− ions in pink; gating glutamate in blue; N68 implicated in block by CPP in red (D54 in CLC-ec1); H497 mediating H+ block in yellow (L421 in CLC-ec1); E261 and D278 forming the Ca binding site in cyan (E235 and N250 in CLC-ec1); L155, G345, and A349 which are important for NFA potentiation in orange (L139, T312, and G316 in CLC-ec1).
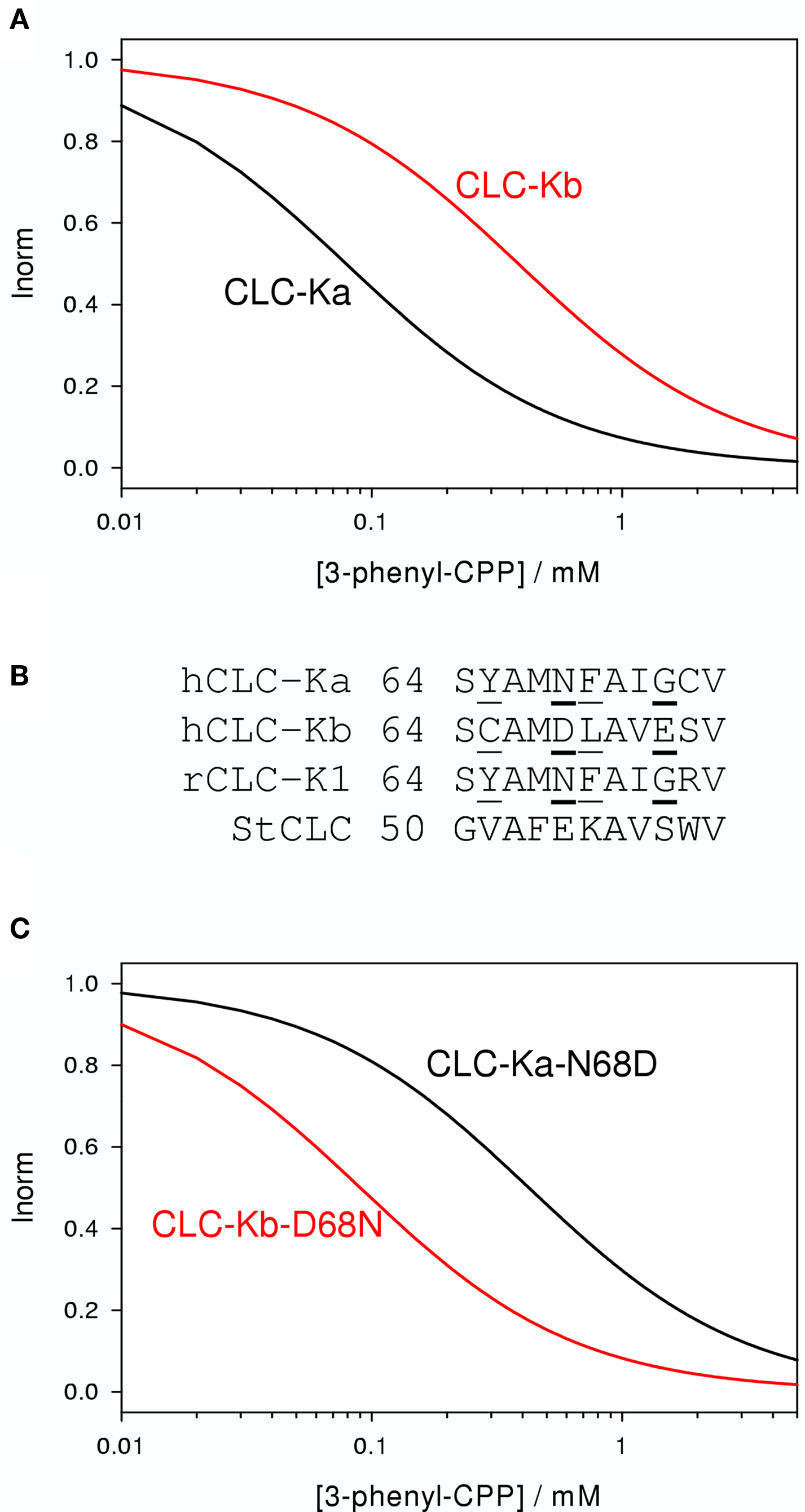
Figure 4. Effect of 3-phenyl-CPP on CLC-Ka, CLC-Kb, and mutants. (A) The blocking effect of 3-phenyl-CPP is schematically indicated for CLC-Ka and CLC-Kb. (B) A sequence alignment of a short stretch of residues of helix B of CLC-Ka, CLC-Kb, CLC-K1, and StCLC, a bacterial homolog (Dutzler et al., 2002) is shown. Residues which are identical in CLC-Ka and CLC-K1, and different in CLC-Kb, are underlined. CLC-K residues at position 68 drastically affect block by 3-phenyl-CPP. DIDS block is additionally dependent on the residue at position 72. Sequence of the bacterial StCLC is shown because the mutagenesis work was guided by the crystal structure of this protein. Residues at positions 68 and 72 are mostly responsible for the difference between CLC-Ka and CLC-Kb and are shown in bold. (C) Schematically the blocking effect of 3-phenyl-CPP of mutants CLC-Ka-N68D and CLC-Kb-D68N. Plots were generated based on data from Picollo et al. (2004).
Regarding the effect of DIDS on CLC-K channels it has to be noted that Matulef et al. (2008) reported that the DIDS molecule itself (Figure 2F) is rather unstable in aqueous solutions and that by virtue of the reactivity of DIDS, higher order “DIDS oligomers” can form. Some of these oligomers have a highly increased potency of block of CLC-Ka with KD values of the order of a few micromolar (Matulef et al., 2008). Block was drastically reduced by the N68D mutation suggesting that the DIDS oligomers bind to the same site as the DIDS monomer. The high affinity of “DIDS tetramers” or “DIDS pentamers” is consistent with the hypothesis that the two ends of these long molecules bind each in one of the pores of the homodimeric channel (Matulef et al., 2008). These oligomeric DIDS molecules are so far the most potent CLC-K blockers. However, even though these molecules are highly interesting as tools to probe the channel function and structure, they are probably rather useless from a pharmacological point of view, because of their chemical reactivity.
Recently, benzofuran derivatives of 3-phenyl-CPP were found to block CLC-K channels with greatly improved potency compared to 3-phenyl-CPP (Liantonio et al., 2008). The two most potent molecules were MT-189 (Figure 2I) and RT-93 (Figure 2J) with apparent KD values of 7 μM. Both compounds had a significantly reduced potency on the CLC-Ka-N68D mutant, suggesting that they bind to the same site as 3-phenyl-CPP and DIDS (the residue corresponding to N68 is colored in red in Figure 3). In line with this finding, compound MT-189 had also a reduced affinity for CLC-Kb (apparent KD ∼20 μM). Interestingly, the more hydrophobic compound RT-93 was equally effective on CLC-Kb (apparent KD ∼6 μM) as on CLC-Ka (Liantonio et al., 2008). This compound represents thus the most potent blocker of CLC-Kb described so far. In addition, this result suggests that it is probably difficult to develop drugs that are highly specific for one of the human CLC-K homologs.
Activation and Block by Fenamates
Niflumic acid (Figure 2D) and flufenamic acid (FFA) (Figure 2E) belong to a class of fenamates usually used as non-steroidal anti-inflammatory drugs. However, in particular NFA, is also known as a classical Cl− channel inhibitor. In fact, CLC-K1 was found to be blocked by NFA with an apparent KD of ∼100 μM (Liantonio et al., 2004; Picollo et al., 2007) (Figure 5A). It was therefore rather surprising that the human CLC-K homologs are activated rather than blocked by sub-millimolar concentrations of NFA (Liantonio et al., 2006). CLC-Ka is blocked by higher concentrations (above ∼1 mM) of NFA leading to a biphasic concentration dependence (Figure 5B). In contrast, CLC-Kb is potentiated by NFA at all concentrations tested (Liantonio et al., 2006) (Figure 5C). Currents start to increase within a few seconds after application of NFA strongly suggesting that the respective binding site is located on the extracellular face of the protein (Liantonio et al., 2006; Picollo et al., 2007). Interestingly, flufenamic acid (FFA, Figure 2E), whose structure seems to be very similar to that of NFA (see Figures 2D,E), does not activate CLC-Ka, but rather induces channel block. The FFA block is probably mediated by the same pore binding site as 3-phenyl-CPP block (see above) because the mutant CLC-Ka-N68 was much less sensitive to FFA (Liantonio et al., 2006). However, the block of CLC-Ka by NFA at higher concentrations (Figure 5B) and the block of CLC-K1 by NFA (Figure 5A) is probably not mediated by the same binding site, because it is unaltered by the N68D mutation (Picollo et al., 2007). CLC-Kb appears somewhat more susceptible to the potentiating effect of fenamates in that also FFA enhances currents (Liantonio et al., 2006) (Figure 5C).
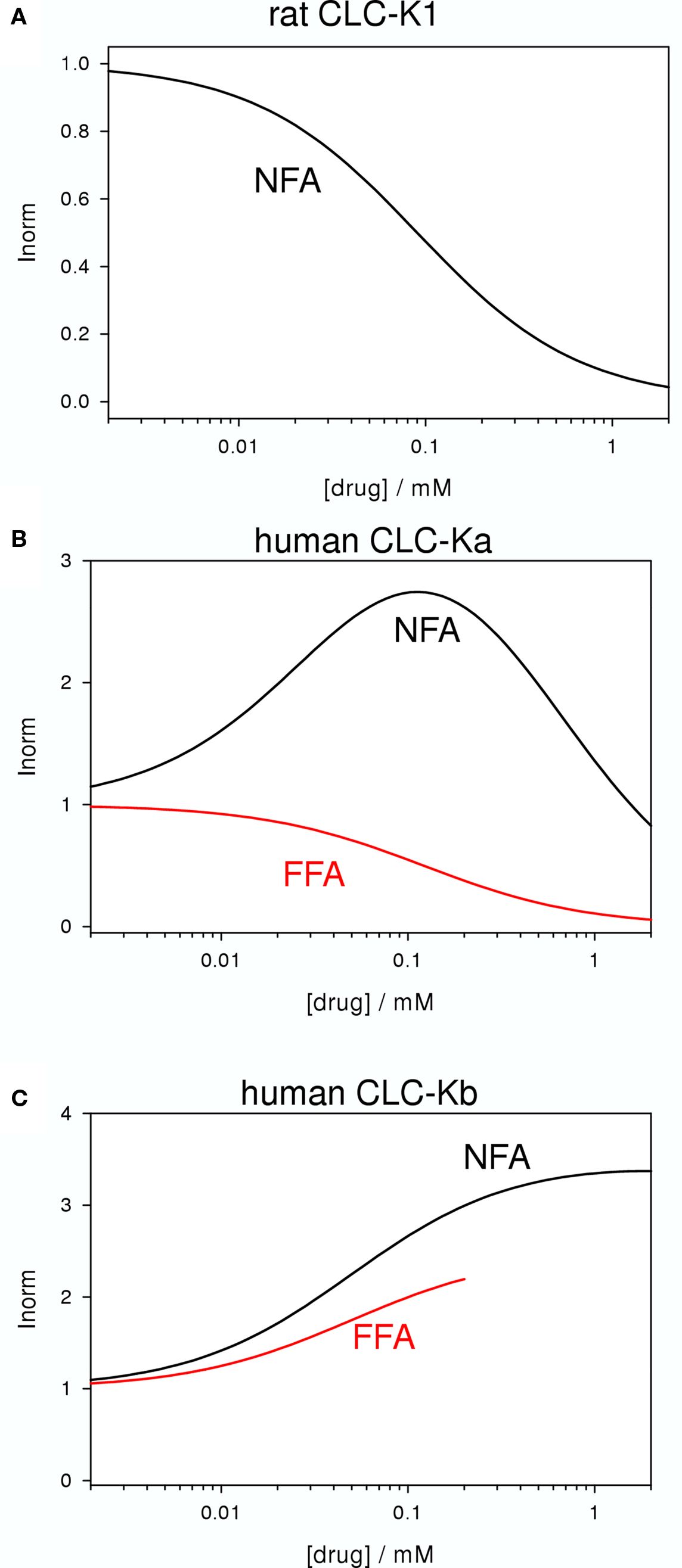
Figure 5. Blocking and potentiating effects of NFA and FFA on various CLC-K homologs (A: CLC-K1, B: CLC-Ka, C: CLC-Kb). The effect of NFA is schematically drawn in black, whereas the effect of FFA is shown in red. No measurements with FFA on CLC-K1 have been reported.
The striking difference between the effects of NFA (activating) and FFA (blocking) on CLC-Ka could be explained by a fundamental difference between these two molecules: NFA has a rather rigid structure in which the aromatic rings are strictly co-planar. In contrast, in FFA, lacking the N-atom in the COOH-bearing ring, the H-atom of the C corresponding to the N of NFA induces a steric hindrance that disturbs the co-planarity (Liantonio et al., 2006). Similarly, also 3-phenyl-CPP has a flexible, non-co-planar conformation. This hypothesis (Liantonio et al., 2006) was nicely confirmed by designing a “flexible, non-co-planar version” of NFA, and similarly by designing a “rigid, co-planar version” of FFA (Liantonio et al., 2008). Triflocin differs from NFA only in the position of the pyridine N (Figure 2G) but the H-atom at the ortho position now induces the same steric hindrance as in FFA. In agreement with the above mentioned hypothesis, triflocin induces block of CLC-Ka at all concentrations tested (Liantonio et al., 2008). On the other hand, the “co-planar FFA”, in which the two phenyl groups are forced to assume a co-planar conformation by a cyclization (molecule GF-166, Figure 2H), showed an activating effect on CLC-Ka at concentrations below 200 μM (Liantonio et al., 2008).
Niflumic acid activates CLC-Ka and CLC-Kb by increasing the probability of the channel to be in the open state. In fact, the basal Po of CLC-Ka and CLC-Kb is small, i.e., at least smaller than about 0.1 (Picollo et al., 2007; Gradogna et al., 2010; Zifarelli et al., 2010). This small basal Po provides a large margin for the activation by various ligands. It is not clear, however, which is the molecular mechanism that underlies the regulation of the open-probability of CLC-K channels. In a first attempt to obtain more insight into the molecular mechanism of NFA effects on CLC-Ka, Picollo et al. (2007) separated the blocking effect of NFA from the potentiating effect and concluded that probably two NFA molecules have to bind in order to induce channel closure, whereas binding of one NFA molecule suffices to induce potentiation.
In an attempt to identify the NFA binding site(s), Zifarelli et al. (2010) functionally screened about 80 mutations of CLC-Ka, testing for the activating effect of 200 μM. They could identify two regions of the protein which are distant on the primary sequence but relatively close in the 3D structure. The three amino acids which abolished the activating effect of NFA were L155, G345, and A349, and are colored orange in Figure 3. However, in that work, it could not be excluded that these residues are not involved in NFA binding but rather in transmitting the binding event to the resulting change in Po. Further work is clearly necessary in order to understand better the complex interaction of NFA with CLC-K channels.
Regulation by Ca and pH
Previous studies demonstrated in several models that CLC-K channels are enhanced by increasing of extracellular Ca2+ (Uchida et al., 1995; Sauvé et al., 2000; Waldegger and Jentsch, 2000; Estévez et al., 2001; Waldegger et al., 2002) (Figure 6B). Such a regulation by divalent cations has not been found in other members of the CLC family. Moreover, CLC-K channels are blocked by acid pH (Uchida et al., 1995; Sauvé et al., 2000; Waldegger and Jentsch, 2000; Estévez et al., 2001; Waldegger et al., 2002) (Figure 6A). Proton modulation is common in CLC proteins both in Cl−/H+ antiporters as well as in most of the channels (Friedrich et al., 1999; Accardi and Miller, 2004; Picollo and Pusch, 2005; Zifarelli and Pusch, 2009a). Interestingly, CLC-K channels are lacking the “critical glutamate” whose protonation/deprotonation is involved in the opening of most CLC channels (Hanke and Miller, 1983; Rychkov et al., 1996; Jordt and Jentsch, 1997; Chen and Chen, 2001; Arreola et al., 2002; Dutzler et al., 2003; Traverso et al., 2006; Zifarelli et al., 2008; Niemeyer et al., 2009; Zifarelli and Pusch, 2010). This suggested that a different mechanism of pH regulation is present in CLC-K channels. Since the kidney is involved in calcium reabsorption and the maintenance of acid-base balance (Frick and Bushinsky, 2003; Jeck et al., 2005), Ca2+ and proton regulation of CLC-K channels is potentially of physiological relevance.
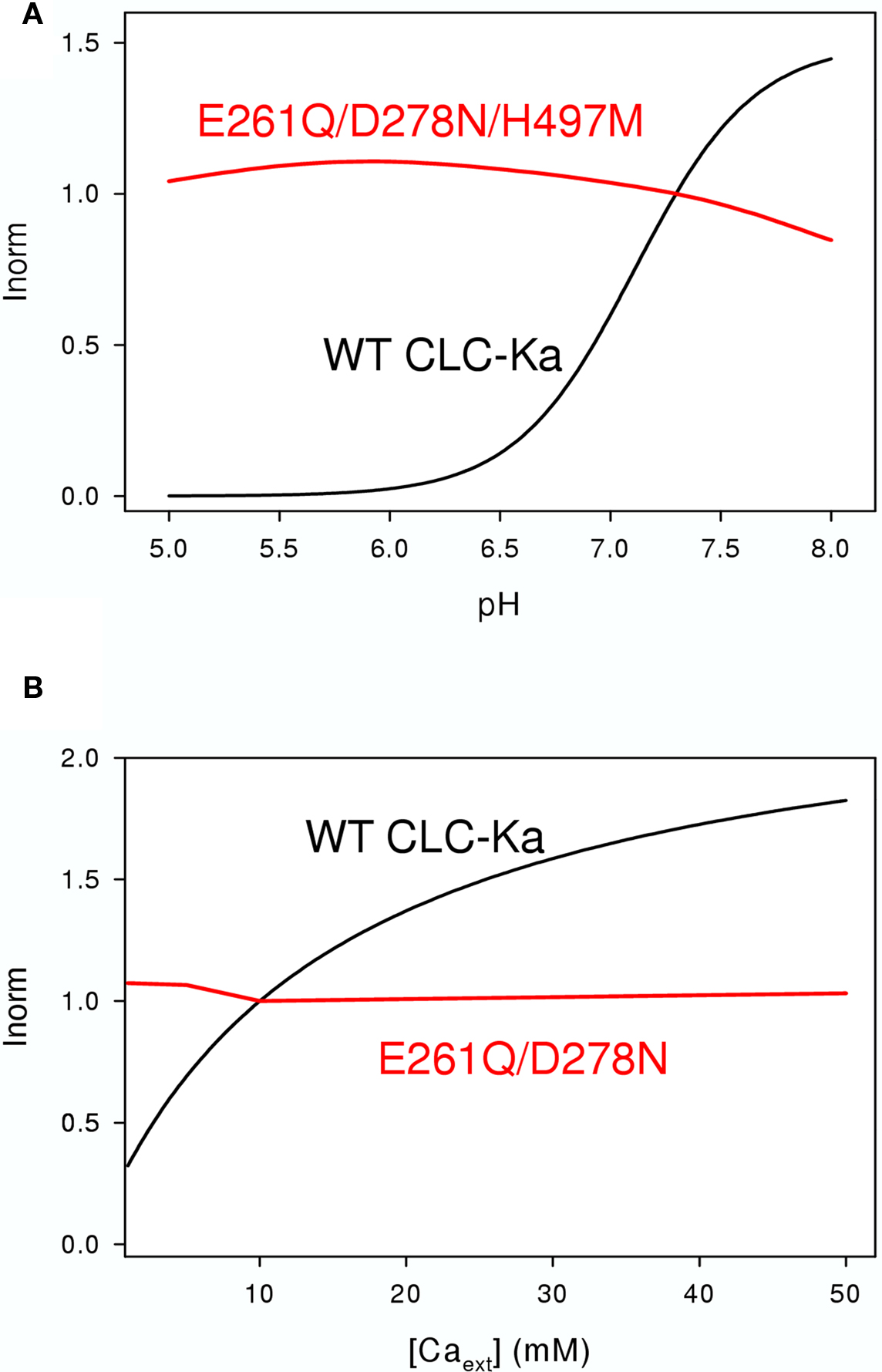
Figure 6. Effects of extracellular pH (A) and [Ca2+] (B) on WT CLC-Ka (black line) and the mutants (red lines) E261Q/D278N/H497M (A) and E261Q/D278N (B) schematically drawn according to the results of Gradogna et al. (2010).
The CLC-Kb mutation, R351W, found in patients with Bartter’s syndrome, was reported to abolish Ca2+ activation and to reduce the pH effect in CLC-Kb (Yu et al., 2009). However, the currents mediated by the mutant CLC-Kb construct were barely above background, and similar mutations in CLC-Ka had no effect on Ca2+ or H+ sensitivity (Gradogna et al., 2010). Similarly, a mutation in the C-terminus altered Ca2+ sensitivity of CLC-Kb (but not CLC-Ka) probably by an indirect effect (Martinez and Maduke, 2008).
Gradogna et al. (2010) performed a biophysical analysis of Ca2+ and proton modulation of human CLC-K channels expressed in Xenopus oocytes. Kinetic experiments, performed by patch recordings and fast solution exchange, showed an allosteric modulation of CLC-Ka for both, Ca2+ and protons (Gradogna et al., 2010). It was demonstrated that Ca2+ and protons act via independent mechanisms and binding sites (Gradogna et al., 2010). Modeling predicted a two state (blocked/unblocked) mechanism with a pK = 7.1 for proton modulation and it was suggested two protons are necessary to block the channel. Because Ca2+ is not strictly essential for opening, a four-state mechanism was used for modeling the Ca2+-dependence, with Ca2+ binding constants of KO = 1.5 mM for the open state and KC = 19.6 mM for the closed state (Gradogna et al., 2010).
To identify the Ca2+ binding site, all charged and titratable residues accessible from the extracellular side were mutated. This extensive mutagenesis led to the identification of two mutations, E261Q and D278N, that reduced calcium sensitivity of CLC-Ka. E261 and D278 from different subunit are close to each other and likely form an intersubunit Ca2+ binding site; in fact the double mutant E261Q/D278N completely abolished modulation by calcium (Gradogna et al., 2010). The corresponding residues are colored cyan in Figure 3.
Recently Niemeyer et al. (2009) identified an off-pore histidine as being responsible for the proton induced block of the CLC-2 channel. Based on this discovery, the corresponding residue of CLC-Ka, H497, was mutated in several amino acids (Gradogna et al., 2010). The only functional mutant H497M showed very small currents. Interestingly, the triple mutant E261Q/D278N/H497M yielded bigger currents than H497M and completely abolished proton block. This result suggested that the same mechanism controls proton block of CLC-2 and CLC-K channels (Gradogna et al., 2010). The residue His-497, at the beginning of helix Q is colored yellow in Figure 3. Both Ca2+ and H+ act by altering the Po of the channel. However, it is unclear if they affect the same or different gating processes.
Possible Applications of CLC-K Channel Activators and Inhibitors and General Outlook
CLC-K channels are highly promising drug targets. Firstly, blockers of CLC-K channels are promising alternative diuretics (Fong, 2004). Secondly, pharmacological tools can be used in cellular or animal models in order to explore the physiological role of CLC-Ks. Third, activators of CLC-K channels may be useful for patients with Bartter’s syndrome by activating the available CLC-Ka channels or by increasing residual CLC-Kb channel activity.
However, since CLC-K channels are also necessary for the generation of a positive endocochlear potential (Rickheit et al., 2008), side effects of any CLC-K specific drug have to be carefully evaluated. In this respect it is interesting to note that recessive loss of function of CLC-Kb leads to Bartter’s syndrome without deafness, presumably because of sufficient CLC-Ka function in the inner ear. Thus, drugs that specifically block CLC-Kb, but not CLC-Ka, might avoid such side effects. The same may be true for CLC-Ka specific drugs. If CLC-Ka plays a similar role in humans as CLC-K1 in mice (Matsumura et al., 1999), CLC-Ka specific blockers may induce water diuresis, a condition that may be advantageous in certain pathological conditions (Fong, 2004).
Regarding the blockers, the relatively high affinity compounds RT-93 and MT-189 might be tested as potential diuretic and for unspecific effects in animal experiments, and eventually these compounds may serve as leads for the development of useful drugs.
Regarding the CLC-K potentiators, NFA is the most potent compound which is able to increase CLC-Ka and CLC-Kb currents. However, on the one hand its affinity is rather low, and on the other hand NFA, being a non-steroidal anti-inflammatory drug, can be expected to exert significant side effects. More specific and more potent activators will be needed before approaching in vivo tests. Additionally, murine models are possibly not well suited to test CLC-K activators because rat CLC-K1 is blocked by NFA and nothing is known about the effect of NFA on CLC-K2. In the meantime, it will also be important to understand the molecular mechanisms that underlie the activating effects of NFA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The financial support by Telethon Italy (GGP08064), the Italian “Ministero dell’Istruzione, dell’Università e della Ricerca” (MIUR PRIN 20078ZZMZW_002), the Compagnia San Paolo, and the Italian Institute of Technology (“progetto seed”) is gratefully acknowledged.
References
Accardi, A., and Miller, C. (2004). Secondary active transport mediated by a prokaryotic homologue of ClC Cl− channels. Nature 427, 803–807.
Alekov, A. K., and Fahlke, C. (2009). Channel-like slippage modes in the human anion/proton exchanger ClC-4. J. Gen. Physiol. 133, 485–496.
Aromataris, E. C., Astill, D. S., Rychkov, G. Y., Bryant, S. H., Bretag, A. H., and Roberts, M. L. (1999). Modulation of the gating of ClC-1 by S-(−) 2-(4-chlorophenoxy) propionic acid. Br. J. Pharmacol. 126, 1375–1382.
Arreola, J., Begenisich, T., and Melvin, J. E. (2002). Conformation-dependent regulation of inward rectifier chloride channel gating by extracellular protons. J. Physiol. 541, 103–112.
Bennetts, B., Parker, M. W., and Cromer, B. A. (2007). Inhibition of skeletal muscle CLC-1 chloride channels by low intracellular pH and ATP. J. Biol. Chem. 282, 32780–32791.
Bennetts, B., Rychkov, G. Y., Ng, H.-L., Morton, C. J., Stapleton, D., Parker, M. W., and Cromer, B. A. (2005). Cytoplasmic ATP-sensing domains regulate gating of skeletal muscle ClC-1 chloride channels. J. Biol. Chem. 280, 32452–32458.
Birkenhäger, R., Otto, E., Schurmann, M. J., Vollmer, M., Ruf, E. M., Maier-Lutz, I., Beekmann, F., Fekete, A., Omran, H., Feldmann, D., Milford, D. V., Jeck, N., Konrad, M., Landau, D., Knoers, N. V., Antignac, C., Sudbrak, R., Kispert, A., and Hildebrandt, F. (2001). Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 29, 310–314.
Chen, M. F., and Chen, T. Y. (2001). Different fast-gate regulation by external Cl(−) and H(+) of the muscle- type ClC chloride channels. J. Gen. Physiol. 118, 23–32.
De Angeli, A., Monachello, D., Ephritikhine, G., Frachisse, J. M., Thomine, S., Gambale, F., and Barbier-Brygoo, H. (2006). The nitrate/proton antiporter AtCLCa mediates nitrate accumulation in plant vacuoles. Nature 442, 939–942.
De Angeli, A., Moran, O., Wege, S., Filleur, S., Ephritikhine, G., Thomine, S., Barbier-Brygoo, H., and Gambale, F. (2009). ATP binding to the C terminus of the Arabidopsis thaliana nitrate/proton antiporter, AtCLCa, regulates nitrate transport into plant vacuoles. J. Biol. Chem. 284, 26526–26532.
De Luca, A., Tricarico, D., Wagner, R., Bryant, S. H., Tortorella, V., and Conte Camerino, D. (1992). Opposite effects of enantiomers of clofibric acid derivative on rat skeletal muscle chloride conductance: antagonism studies and theoretical modeling of two different receptor site interactions. J. Pharmacol. Exp. Ther. 260, 364–368.
Dutzler, R., Campbell, E. B., Cadene, M., Chait, B. T., and MacKinnon, R. (2002). X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature 415, 287–294.
Dutzler, R., Campbell, E. B., and MacKinnon, R. (2003). Gating the selectivity filter in ClC chloride channels. Science 300, 108–112.
Estévez, R., Boettger, T., Stein, V., Birkenhäger, R., Otto, E., Hildebrandt, F., and Jentsch, T. J. (2001). Barttin is a Cl− channel beta-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 414, 558–561.
Estévez, R., and Jentsch, T. J. (2002). CLC chloride channels: correlating structure with function. Curr. Opin. Struct. Biol. 12, 531–539.
Fahlke, C., Rüdel, R., Mitrovic, N., Zhou, M., and George, A. L. Jr. (1995). An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron 15, 463–472.
Fischer, M., Janssen, A. G., and Fahlke, C. (2010). Barttin activates ClC-K channel function by modulating gating. J. Am. Soc. Nephrol. 21, 1281–1289.
Frick, K. K., and Bushinsky, D. A. (2003). Molecular mechanisms of primary hypercalciuria. J. Am. Soc. Nephrol. 14, 1082–1095.
Friedrich, T., Breiderhoff, T., and Jentsch, T. J. (1999). Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J. Biol. Chem. 274, 896–902.
Gradogna, A., Babini, E., Picollo, A., and Pusch, M. (2010). A regulatory calcium-binding site at the subunit interface of CLC-K kidney chloride channels. J. Gen. Physiol. 136, 311–323.
Hanke, W., and Miller, C. (1983). Single chloride channels from Torpedo electroplax. Activation by protons. J. Gen. Physiol. 82, 25–45.
Heine, R., George, A. L. Jr., Pika, U., Deymeer, F., Rüdel, R., and Lehmann-Horn, F. (1994). Proof of a non-functional muscle chloride channel in recessive myotonia congenita (Becker) by detection of a 4 base pair deletion. Hum. Mol. Genet. 3, 1123–1128.
Jeck, N., Schlingmann, K. P., Reinalter, S. C., Komhoff, M., Peters, M., Waldegger, S., and Seyberth, H. W. (2005). Salt handling in the distal nephron: lessons learned from inherited human disorders. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R782–R795.
Jeck, N., Waldegger, S., Lampert, A., Boehmer, C., Waldegger, P., Lang, P. A., Wissinger, B., Friedrich, B., Risler, T., Moehle, R., Lang, U. E., Zill, P., Bondy, B., Schaeffeler, E., Asante-Poku, S., Seyberth, H., Schwab, M., and Lang, F. (2004). Activating mutation of the renal epithelial chloride channel ClC-Kb predisposing to hypertension. Hypertension 43, 1175–1181.
Jentsch, T. J. (2008). CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit. Rev. Biochem. Mol. Biol. 43, 3–36.
Jordt, S. E., and Jentsch, T. J. (1997). Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 16, 1582–1592.
Kieferle, S., Fong, P., Bens, M., Vandewalle, A., and Jentsch, T. J. (1994). Two highly homologous members of the ClC chloride channel family in both rat and human kidney. Proc. Natl. Acad. Sci. U. S. A. 91, 6943–6947.
Kobayashi, K., Uchida, S., Mizutani, S., Sasaki, S., and Marumo, F. (2001). Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J. Am. Soc. Nephrol. 12, 1327–1334.
Kobayashi, K., Uchida, S., Okamura, H. O., Marumo, F., and Sasaki, S. (2002). Human CLC-KB gene promoter drives the EGFP expression in the specific distal nephron segments and inner ear. J. Am. Soc. Nephrol. 13, 1992–1998.
Kokubo, Y., Iwai, N., Tago, N., Inamoto, N., Okayama, A., Yamawaki, H., Naraba, H., and Tomoike, H. (2005). Association analysis between hypertension and CYBA, CLCNKB, and KCNMB1 functional polymorphisms in the Japanese population–the Suita Study. Circ. J. 69, 138–142.
Lang, F. (2010). Modulation of ClC-K channel function by the accessory subunit barttin. J. Am. Soc. Nephrol. 21, 1238–1239.
Lange, P. F., Wartosch, L., Jentsch, T. J., and Fuhrmann, J. C. (2006). ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature 440, 220–223.
Liantonio, A., Accardi, A., Carbonara, G., Fracchiolla, G., Loiodice, F., Tortorella, P., Traverso, S., Guida, P., Pierno, S., De Luca, A., Camerino, D. C., and Pusch, M. (2002). Molecular requisites for drug binding to muscle CLC-1 and renal CLC-K channel revealed by the use of phenoxy-alkyl derivatives of 2-(p-chlorophenoxy)propionic acid. Mol. Pharmacol. 62, 265–271.
Liantonio, A., Picollo, A., Babini, E., Carbonara, G., Fracchiolla, G., Loiodice, F., Tortorella, V., Pusch, M., and Camerino, D. C. (2006). Activation and inhibition of kidney CLC-K chloride channels by fenamates. Mol. Pharmacol. 69, 165–173.
Liantonio, A., Picollo, A., Carbonara, G., Fracchiolla, G., Tortorella, P., Loiodice, F., Laghezza, A., Babini, E., Zifarelli, G., Pusch, M., and Camerino, D. C. (2008). Molecular switch for CLC-K Cl− channel block/activation: Optimal pharmacophoric requirements towards high-affinity ligands. Proc. Natl. Acad. Sci. U.S.A. 105, 1369–1373.
Liantonio, A., Pusch, M., Picollo, A., Guida, P., De Luca, A., Pierno, S., Fracchiolla, G., Loiodice, F., Tortorella, P., and Conte Camerino, D. (2004). Investigations of pharmacologic properties of the renal CLC-K1 chloride channel co-expressed with barttin by the use of 2-(p-Chlorophenoxy)propionic acid derivatives and other structurally unrelated chloride channels blockers. J. Am. Soc. Nephrol. 15, 13–20.
Lobet, S., and Dutzler, R. (2006). Ion-binding properties of the ClC chloride selectivity filter. EMBO J. 25, 24–33.
Ludewig, U., Jentsch, T. J., and Pusch, M. (1997). Inward rectification in ClC-0 chloride channels caused by mutations in several protein regions. J. Gen. Physiol. 110, 165–171.
Ludewig, U., Pusch, M., and Jentsch, T. J. (1996). Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature 383, 340–343.
Markovic, S., and Dutzler, R. (2007). The structure of the cytoplasmic domain of the chloride channel ClC-Ka reveals a conserved interaction interface. Structure 15, 715–725.
Martinez, G. Q., and Maduke, M. (2008). A cytoplasmic domain mutation in ClC-Kb affects long-distance communication across the membrane. PLoS One 3, e2746. doi: 10.1371/journal.pone.0002746.
Matsumura, Y., Uchida, S., Kondo, Y., Miyazaki, H., Ko, S. B., Hayama, A., Morimoto, T., Liu, W., Arisawa, M., Sasaki, S., and Marumo, F. (1999). Overt nephrogenic diabetes insipidus in mice lacking the CLC-K1 chloride channel. Nat. Genet. 21, 95–98.
Matulef, K., Howery, A. E., Tan, L., Kobertz, W. R., Du Bois, J., and Maduke, M. (2008). Discovery of potent CLC chloride channel inhibitors. ACS Chem. Biol. 3, 419–428.
Meyer, S., Savaresi, S., Forster, I. C., and Dutzler, R. (2007). Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 14, 60–67.
Middleton, R. E., Pheasant, D. J., and Miller, C. (1996). Homodimeric architecture of a ClC-type chloride ion channel. Nature 383, 337–340.
Neagoe, I., Stauber, T., Fidzinski, P., Bergsdorf, E. Y., and Jentsch, T. J. (2010). The late endosomal ClC-6 mediates proton/chloride countertransport in heterologous plasma membrane expression. J. Biol. Chem. 285, 21689–21697.
Niemeyer, M. I., Cid, L. P., Yusef, Y. R., Briones, R., and Sepúlveda, F. V. (2009). Voltage-dependent and -independent titration of specific residues accounts for complex gating of a ClC chloride channel by extracellular protons. J. Physiol. 587, 1387–1400.
Picollo, A., Liantonio, A., Babini, E., Camerino, D. C., and Pusch, M. (2007). Mechanism of interaction of niflumic acid with heterologously expressed kidney CLC-K chloride channels. J. Membr. Biol. 216, 73–82.
Picollo, A., Liantonio, A., Didonna, M. P., Elia, L., Camerino, D. C., and Pusch, M. (2004). Molecular determinants of differential pore blocking of kidney CLC-K chloride channels. EMBO Rep. 5, 584–589.
Picollo, A., Malvezzi, M., and Accardi, A. (2010). Proton block of the CLC-5 Cl−/H+ exchanger. J. Gen. Physiol. 135, 653–659.
Picollo, A., Malvezzi, M., Houtman, J. C., and Accardi, A. (2009). Basis of substrate binding and conservation of selectivity in the CLC family of channels and transporters. Nat. Struct. Mol. Biol. 16, 1294–1301.
Picollo, A., and Pusch, M. (2005). Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature 436, 420–423.
Pusch, M., Liantonio, A., Bertorello, L., Accardi, A., De Luca, A., Pierno, S., Tortorella, V., and Camerino, D. C. (2000). Pharmacological characterization of chloride channels belonging to the ClC family by the use of chiral clofibric acid derivatives. Mol. Pharmacol. 58, 498–507.
Pusch, M., Ludewig, U., Rehfeldt, A., and Jentsch, T. J. (1995). Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature 373, 527–531.
Rickheit, G., Maier, H., Strenzke, N., Andreescu, C. E., De Zeeuw, C. I., Muenscher, A., Zdebik, A. A., and Jentsch, T. J. (2008). Endocochlear potential depends on Cl(−) channels: mechanism underlying deafness in Bartter syndrome IV. EMBO J. 2, 2.
Rychkov, G. Y., Pusch, M., Astill, D. S., Roberts, M. L., Jentsch, T. J., and Bretag, A. H. (1996). Concentration and pH dependence of skeletal muscle chloride channel ClC- 1. J. Physiol. 497, 423–435.
Sauvé, R., Cai, S., Garneau, L., Klein, H., and Parent, L. (2000). pH and external Ca(2+) regulation of a small conductance Cl(−) channel in kidney distal tubule. Biochim. Biophys. Acta. 1509, 73–85.
Scheel, O., Zdebik, A. A., Lourdel, S., and Jentsch, T. J. (2005). Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature 436, 424–427.
Schlingmann, K. P., Konrad, M., Jeck, N., Waldegger, P., Reinalter, S. C., Holder, M., Seyberth, H. W., and Waldegger, S. (2004). Salt wasting and deafness resulting from mutations in two chloride channels. N. Engl. J. Med. 350, 1314–1319.
Scholl, U., Hebeisen, S., Janssen, A. G., Müller-Newen, G., Alekov, A., and Fahlke, C. (2006). Barttin modulates trafficking and function of ClC-K channels. Proc. Natl. Acad. Sci. U. S. A. 103, 11411–11416.
Scott, J. W., Hawley, S. A., Green, K. A., Anis, M., Stewart, G., Scullion, G. A., Norman, D. G., and Hardie, D. G. (2004). CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 113, 274–284.
Simon, D. B., Bindra, R. S., Mansfield, T. A., Nelson-Williams, C., Mendonca, E., Stone, R., Schurman, S., Nayir, A., Alpay, H., Bakkaloglu, A., Rodriguez-Soriano, J., Morales, J. M., Sanjad, S. A., Taylor, C. M., Pilz, D., Brem, A., Trachtman, H., Griswold, W., Richard, G. A., John, E., and Lifton, R. P. (1997). Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 17, 171–178.
Speirs, H. J., Wang, W. Y., Benjafield, A. V., and Morris, B. J. (2005). No association with hypertension of CLCNKB and TNFRSF1B polymorphisms at a hypertension locus on chromosome 1p36. J. Hypertens. 23, 1491–1496.
Traverso, S., Zifarelli, G., Aiello, R., and Pusch, M. (2006). Proton sensing of CLC-0 mutant E166D. J. Gen. Physiol. 127, 51–66.
Tseng, P.-Y., Bennetts, B., and Chen, T.-Y. (2007). Cytoplasmic ATP inhibition of CLC-1 is enhanced by low pH. J. Gen. Physiol. 130, 217–221.
Uchida, S., Sasaki, S., Furukawa, T., Hiraoka, M., Imai, T., Hirata, Y., and Marumo, F. (1993). Molecular cloning of a chloride channel that is regulated by dehydration and expressed predominantly in kidney medulla. J. Biol. Chem. 268, 3821–3824.
Uchida, S., Sasaki, S., Nitta, K., Uchida, K., Horita, S., Nihei, H., and Marumo, F. (1995). Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J. Clin. Invest. 95, 104–113.
Vandewalle, A., Cluzeaud, F., Bens, M., Kieferle, S., Steinmeyer, K., and Jentsch, T. J. (1997). Localization and induction by dehydration of ClC-K chloride channels in the rat kidney. Am. J. Physiol. 272, F678–F688.
Waldegger, S., Jeck, N., Barth, P., Peters, M., Vitzthum, H., Wolf, K., Kurtz, A., Konrad, M., and Seyberth, H. W. (2002). Barttin increases surface expression and changes current properties of ClC-K channels. Pflügers Arch. 444, 411–418.
Waldegger, S., and Jentsch, T. J. (2000). Functional and structural analysis of ClC-K chloride channels involved in renal disease. J. Biol. Chem. 275, 24527–24533.
Weinreich, F., and Jentsch, T. J. (2001). Pores formed by single subunits in mixed dimers of different CLC chloride channels. J. Biol. Chem. 276, 2347–2353.
Wellhauser, L., Kuo, H. H., Stratford, F. L., Ramjeesingh, M., Huan, L. J., Luong, W., Li, C., Deber, C. M., and Bear, C. E. (2006). Nucleotides bind to the C-terminus of ClC-5. Biochem. J. 398, 289–294.
Yu, Y., Xu, C., Pan, X., Ren, H., Wang, W., Meng, X., Huang, F., and Chen, N. (2009). Identification and functional analysis of novel mutations of the CLCNKB gene in Chinese patients with classic Bartter syndrome. Clin. Genet. 77, 155–162.
Zdebik, A. A., Wangemann, P., and Jentsch, T. J. (2009). Potassium ion movement in the inner ear: insights from genetic disease and mouse models. Physiology (Bethesda) 24, 307–316.
Zdebik, A. A., Zifarelli, G., Bergsdorf, E. Y., Soliani, P., Scheel, O., Jentsch, T. J., and Pusch, M. (2008). Determinants of anion-proton coupling in mammalian endosomal CLC proteins. J. Biol. Chem. 283, 4219–4227.
Zifarelli, G., Liantonio, A., Gradogna, A., Picollo, A., Gramegna, G., De Bellis, M., Murgia, A. R., Babini, E., Camerino, D. C., and Pusch, M. (2010). Identification of sites responsible for the potentiating effect of niflumic acid on ClC-Ka kidney chloride channels. Br. J. Pharmacol. 160, 1652–1661.
Zifarelli, G., Murgia, A. R., Soliani, P., and Pusch, M. (2008). Intracellular proton regulation of ClC-0. J. Gen. Physiol. 132, 185–198.
Zifarelli, G., and Pusch, M. (2007). CLC chloride channels and transporters: a biophysical and physiological perspective. Rev. Physiol. Biochem. Pharmacol. 158, 23–76.
Zifarelli, G., and Pusch, M. (2009a). Conversion of the 2 Cl(−)/1 H(+) antiporter ClC-5 in a NO(3)(−)/H(+) antiporter by a single point mutation. EMBO J. 28, 175–182.
Zifarelli, G., and Pusch, M. (2009b). Intracellular regulation of human ClC-5 by adenine nucleotides. EMBO Rep. 10, 1111–1116.
Keywords: chloride channel, CLC, kidney, inner ear, chloride transport, diuretic, fenamates, calcium
Citation: Pusch M and Gradogna A (2010) Molecular pharmacology of kidney and inner ear CLC-K chloride channels. Front. Pharmacol. 1:130. doi: 10.3389/fphar.2010.00130
Received: 11 August 2010;
Paper pending published: 01 September 2010;
Accepted: 30 September 2010;
Published online: 25 October 2010.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Anselm Zdebik, University College London, UK;Peying Fong, Kansas State University, USA;
Antonella Liantonio, University of Bari, Italy
Copyright: © 2010 Pusch and Gradogna. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Michael Pusch, Consiglio Nazionale delle Ricerche, Istituto di Biofisica, Via De Marini, 6, 16149 Genova, Italy. e-mail:cHVzY2hAZ2UuaWJmLmNuci5pdA==