




- Johnson & Johnson Pharmaceutical Research and Development, LLC, Spring House, PA, USA
Cannabinoids are known to be clinically beneficial for control of appetite disorders and nausea/vomiting, with emerging data that they can impact other GI disorders, such as inflammation. Post-inflammatory irritable bowel syndrome (PI-IBS) is a condition of perturbed intestinal function that occurs subsequent to earlier periods of intestinal inflammation. Cannabinoid 1 receptor (CB1R) and CB2R alterations in GI inflammation have been demonstrated in both animal models and clinically, but their continuing role in the post-inflammatory period has only been implicated to date. Therefore, to provide direct evidence for CBR involvement in altered GI functions in the absence of overt inflammation, we used a model of enhanced upper GI transit that persists for up to 4 weeks after a single insult by intracolonic 0.5% oil of mustard (OM) in mice. In mice administered OM, CB1R immunostaining in the myenteric plexus was reduced at day 7, when colonic inflammation is subsiding, and then increased at 28 days, compared to tissue from age-matched vehicle-treated mice. In the lamina propria CB2R immunostaining density was also increased at day 28. In mice tested 28 day after OM, either a CB1R-selective agonist, ACEA (1 and 3 mg/kg, s.c.) or a CB2R-selective agonist, JWH-133 (3 and 10 mg/kg, s.c.) reduced the enhanced small intestinal transit in a dose-related manner. Doses of ACEA and JWH-133 (1 mg/kg), alone or combined, reduced small intestinal transit of OM-treated mice to a greater extent than control mice. Thus, in this post-colonic inflammation model, both CBR subtypes are up-regulated and there is increased efficacy of both CB1R and CB2R agonists. We conclude that CBR remodeling occurs not only during GI inflammation but continues during the recovery phase. Thus, either CB1R- or CB2-selective agonists could be efficacious for modulating GI motility in individuals experiencing diarrhea-predominant PI-IBS.
Introduction
Debate about the medical benefits of cannabinoids in a wide array of diseases is based on anecdotal evidence from patients who have used medical marijuana, experimental animal models, and an increasing number of clinical studies. In gastrointestinal (GI) diseases there is overwhelming evidence that cannabinoids increase appetite and improve weight gain in patients during cancer chemotherapy or those suffering from AIDS wasting syndrome. However, as reviewed recently (Izzo and Sharkey, 2010), cannabinoids in the gut modulate gastric secretion and gastroprotection, GI motility, ion transport, visceral sensation, intestinal inflammation, and cell proliferation. In addition, evidence is emerging that exogenous and endogenous cannabinoids, have an important role in pathophysiology, such as in GI inflammation (D’Argenio et al., 2007; Izzo and Camilleri, 2009).
The primarily active constituent of marijuana, Δ9-tetrahydrocannabinol, acts on cannabinoid 1 (CB1R) and 2 (CB2R) receptors, which are G-protein coupled receptors (Glass and Northup, 1999; Bosier et al., 2010). There is a differential localization and distribution of CBR, where CB1R is located primarily on neuronal tissue and CB2R is located on peripheral blood leukocytes. In the GI tract, CB1R highly expressed in the submucosal and myenteric plexus neurons, as well as visceral afferent nerves (Kulkarni-Narla and Brown, 2000; Coutts et al., 2002; Pertwee and Ross, 2002; Casu et al., 2003). Immune cells in the intestinal lamina propria express CB2R, including plasma cells and activated macrophages (Wright et al., 2005).
In normal GI motility states, there is substantial experimental evidence that CB1R inhibits contractility and GI motility at multiple levels, reviewed in (Izzo and Sharkey, 2010). An enhanced role of CB1R has been reported in a number of rodent GI inflammatory models. For example, in mice with colitis induced by intracolonic dinitrobenzene sulfonic acid (Sibaev et al., 2006) and with ileitis-induced small intestinal croton oil (Izzo et al., 2001). Compared to CB1R, there is little evidence that CBR2 is involved in the control of normal GI motility, although CBR2 tonic inhibitory activity via endogenous cannabinoids has been reported in rat stomach (Storr et al., 2002), but not mouse stomach (Mule et al., 2007). However, in inflamed mice and rats models, there is now clearly an inhibitory role of CB2R in GI motility. For example, CB2R up-regulation and inhibition of GI transit have been reported in models of inflammation such as, croton oil-induced diarrhea (Izzo et al., 2000), trinitrobenzene sulfonic acid (TNBS)-induced colitis (Storr et al., 2009) and lipopolysaccharide (LPS)-induced GI transit in vivo and in isolated segments of ileum (Mathison et al., 2004; Duncan et al., 2008). Interestingly, in LPS-treated rat tissue, CB2R is localized in enteric neurons (Duncan et al., 2008) and in inflammatory bowel disease patients (Wright et al., 2005) CB2R is expressed on intestinal epithelium. Both of these are cell types that do not typically express CB2R in normal tissue and suggests extensive CBR remodeling in GI inflammation in diseased human tissue and experimental animal models.
There is also human genetic evidence of CBR in GI-related disorders. A frequent silent mutation is a common polymorphism (1359 G/A) of the CB1R gene (CNR1) in Caucasians (Gadzicki et al., 1999). This has been associated with metabolic syndrome in a Chinese population (Hu and Feng, 2010) and with irritable bowel syndrome (IBS) in Korean patients (Park et al., 2010). Although endocannabinoids and CBR have been linked to the underlying physiological processes of IBS (Storr et al., 2008), the association to the CNR1 gene is the first evidence linking CBR to IBS, to our knowledge. According to the Rome II and III criteria (Drossman, 2007), IBS is a spectrum of disorders characterized by abdominal discomfort and increased pain perception associated with altered bowel habits. Animal models of IBS have focused on the pain component and demonstrated that CBR agonists ameliorate visceral hyperalgesia during colorectal distention (Sanson et al., 2006; Brusberg et al., 2009) with increased efficacy after intracolonic TNBS (Kikuchi et al., 2008). However, there is little direct experimental evidence suggesting that cannabinoids may be beneficial for the dysmotility of IBS.
We have characterized a mouse model of accelerated transit (Kimball et al., 2005) that persists after the resolution of colonic inflammation (Kimball et al., 2006). In this mouse model, intracolonic oil of mustard (OM) produces an acute (3 day) colitis (Kimball et al., 2006) that is associated with an early increased mRNA expression of soluble inflammatory mediators and cytokine levels in the colon (Kimball et al., 2007). During the inflammatory stage, CB1R mRNA is rapidly up-regulated by 6 h but then down-regulated 2–3 days after OM (Kimball et al., 2007). After these inflammatory changes have resolved, mice exhibit increased small bowel transit 4 weeks after the initial insult (Kimball et al., 2005). Since this model may provide insight into CBR in the underlying pathophysiology of post-inflammatory IBS (PI-IBS), and we do not know the role of cannabinoids at this stage in the model, we used it to examine the time course of expression of CB1R and CB2R immunostaining in mouse small bowel following OM colitis induction, as well as compare the effects of cannabinoid agonists on intestinal transit after resolution of colonic inflammation.
Materials and Methods
Male CD-1 mice (Charles River Laboratories, Kingston, NC, USA), 10- to 12-weeks old, were used throughout these studies. All treatments were carried out in accordance with the Federal Animal Welfare Act and with methods approved by the Institutional Animal Care and Use Committee of Johnson and Johnson Pharmaceutical Research and Development, LLC.
Induction of Colitis
Freshly opened OM (95 or 98% pure allyl isothiocyanate, Sigma-Aldrich St. Louis, MO, USA), was used in each experiment. Mice (N = 12–15 per drug or vehicle treatment group) were briefly anesthetized with ketamine/xylasine (Sigma, St. Louis, MO, USA), and held vertically in a head down position so that 50 μl of a solution of 0.5% OM in 30% ethanol could be administered intracolonically. The OM administration occurred to a depth of 4 cm via a syringe equipped with a ball-tipped 22 G needle. The mice were allowed to recover from anesthesia under a warming light, and then were maintained with normal feed and water for 28 days at which time they were tested for small intestinal transit rate.
Drug Treatment
Cannabinoid agonists arachadonyl chloroethyl amide (ACEA), and JWH-133 (Tocris-Cookson, St. Louis, MO, USA) were administered subcutaneously, 30 min prior to carmine dye (cochineal powder; Sigma-Aldrich St. Louis, MO, USA) administration. In all experiments, CBR agonists were dissolved in a vehicle consisting of 5% Tween 80, 5% ethanol, and 5% dextrose in water. Drug-treated groups were compared against an OM control, which was administered vehicle only. The doses of agonists (ACEA and JWH-133) used in the present study were similar to that shown to be selectively acting at CB1R and CB2R in mice (Arevalo-Martin et al., 2003; Mathison et al., 2004).
Upper GI Transit
Mice received 250 μl of a 6% solution of carmine dye in 0.5% methylcellulose (w/v) by oral gavage. After 20 min, the mice were rapidly euthanized by cervical dislocation according to accepted procedures. The large and small intestines were resected, starting with the distal colon first, and working toward the pylorus until the entire small intestine, cecum, and colon were removed intact. Excess connective tissue was trimmed, the resected bowel was arranged lengthwise without being stretched, and the length of the entire small intestine was recorded. Percent transit was determined to be the distance that the carmine dye front traveled and then converted into a percentage of the length of the entire small intestine. For some comparisons values were normalized to their own respective non-drug-treated (i.e., vehicle) control group whose transit rate was set as 100% of control. Transit was compared in OM-treated mice vehicle-treated and naïve age-matched controls. Mice were not fasted prior to gavage and all were feeding normally at the start of the experiment. In pilot experiments with this model we determined that food deprivation has no effect on transit measured by carmine red. Potential inter-animal variability in the severity of colitis and small intestinal transit induced by OM were inconsequential for N = 12–15 per experimental treatment group, as previously reported (Kimball et al., 2005). When results were combined from multiple experiments each experimental treatment group contained data from multiples of N = 12–15. Throughout the text small intestinal transit is used interchangeably with upper GI transit though it is recognized that both gastric emptying and small bowel transit contribute to the distance traveled down the small intestine.
Immunohistochemistry
CB1R- and CB2R immunostaining was performed in 5 cm-long jejunoileal tissues, starting 5 cm proximal of the cecum. Tissues were collected from untreated mice, and from mice 7 and 28 days after OM-treatment. The tissues were formalin-fixed, and paraffin-embedded prior to immunohistochemical treatments. Briefly, 5 μm tissue sections were mounted on microscope slides and then routinely dewaxed and rehydrated. After a 5 min exposure in a microwave in Target Buffer (Dako, Carpinteria, CA, USA), slides were subsequently treated in 3% H2O2 for 5 min, followed by treatment with avidin–biotin reagent (Serotec, Raleigh, NC, USA) to eliminate endogenous peroxidase and biotin activity, respectively. The slides were then routinely processed for immunohistochemistry. All incubations were performed at room temperature for 30 min. After a 10 min blocking step with normal goat serum, the tissues were incubated with the primary antibodies. Rabbit anti-CB1R (1:20 dilution) and rabbit anti-CB2R (1:2 dilution; Chemicon, Temecula, CA, USA) polyclonal antibodies were used to identify CBR-immunoreactivity in tissue sections. Biotinylated goat anti-rabbit secondary antibody (Vector Labs, Burlingame, CA, USA), avidin–horseradish peroxidase (AbD Serotec, Raleigh, NC, USA) and 3,3′diaminobenzidine (Biomeda Foster City, CA, USA) used to detect immunostaining. Staining controls lacked primary antibodies, but included all other reagents. Since both primary antibodies were rabbit polyclonals the same staining controls could serve for both CB1R and CB2R, as reported in previous studies using these antibodies (Kimball et al., 2006).
Semi-quantitative analysis of CB1R- and CB2R staining was performed by a histologist who was blinded as to the treatment. For CB1R immunostaining myenteric ganglia were counted in the entire section (N = 8 sections) using a 40X objective, and assigned as weak-negative, moderate or intense staining. CB2R + ve cells in lamina propria were quantified as number of villi from eight high-powered fields showing either fewer or greater than five positively stained cells in the LP.
Statistics
Experimental groups were analyzed for significance of differences between the means of treatment groups and control groups by ANOVA with Bonferroni’s post-test using GraphPad Prism (GraphPad Software, San Diego, CA, USA). A P-value < 0.05 was considered statistically significant.
Results
CB1R and CB2R immunostaining was analyzed in jejunoileal tissues from normal mice and OM-treated mice taken 7 and 28 days after OM colitis induction. These times were based on our previous work, where at day 7 mice showed no alterations in intestinal transit with diminished inflammation in the large intestine and at day 28 mice had no inflammation but increased transit (Kimball et al., 2005). Representative photomicrographic images illustrate the marked changes observed in CB1R and CB2R immunostaining noted in myenteric ganglia and lamina propria (Figure 1). CB1R immunostaining in myenteric ganglia was moderately intense in untreated mouse tissue (Figure 1A) compared with little to no staining observed in tissues 7 days after OM administration (Figure 1C). However, by day 28 post-OM treatment (Figure 1E), more intense CB1R immunostaining was visualized in myenteric ganglia neurons, which appeared to exceed that seen in normal tissues. The myenteric ganglia also appeared to be larger in size, though size of ganglia was not quantified. CB2R immunostaining was most notably changed in the lamina propria. Lamina propria CB2R + ve cells were low to absent in untreated (Figure 1B) and day 7 post-OM administration tissues (Figure 1D), but were strongly positive and abundant 28 days post-OM administration (Figure 1F). Positively stained cells in the lamina propria appeared to be mononuclear (Figure 1F inset), but further work would be required to identify specific cell type.
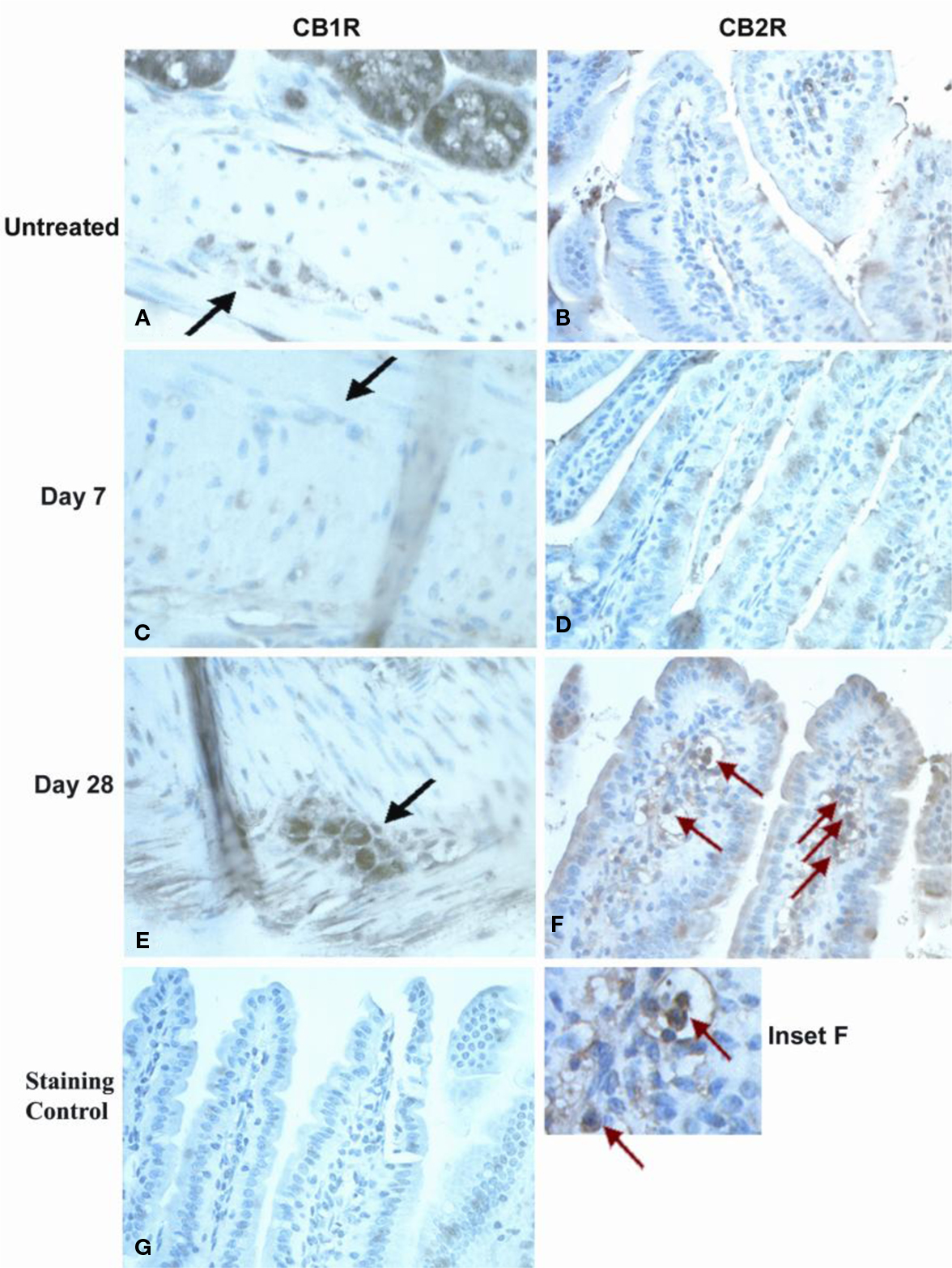
Figure 1. Immunohistochemical staining of small intestine tissues for CBR expression. Neuronal staining in myenteric plexus in normal controls (A), day 7 post-OM (C) and day 28 post-OM (E). Black arrows indicate myenteric ganglia. Moderate staining was observed in normal controls, deeply reduced at day 7, and restored to higher levels of expression at day 28. Immunohistochemical staining of small intestine tissues for CB2R expression is shown for lamina propria (LP) cells. Red arrows indicate stained cells in LP. LP cell staining is shown for normal controls (B), day 7 post-OM (D) and day 28 post-OM (F). Dense CB2R + staining was observed in numerous cells in day 28 tissues, but not in untreated or in day 7 tissues. Inset (F) was enlarged to highlight CB2R + LP cells with monocytic appearance. Non-specific staining in tissue where primary antiserum was omitted (G).
Semi-quantitative analysis of the changes in CB1R- and CB2R staining, by someone blinded as to treatment, illustrate these time dependent observations (Figure 2). Compared to untreated tissue, at day 7 post-OM tissues contained significantly fewer ganglia with moderate and intense CB1R + ve neuronal staining, whereas at day 28 post-OM these were significantly increased (Figure 2A). The number of villi with > 5 CB2R + ve cells in lamina propria cells was significantly increased at 28 days post-colitis (Figure 2B).
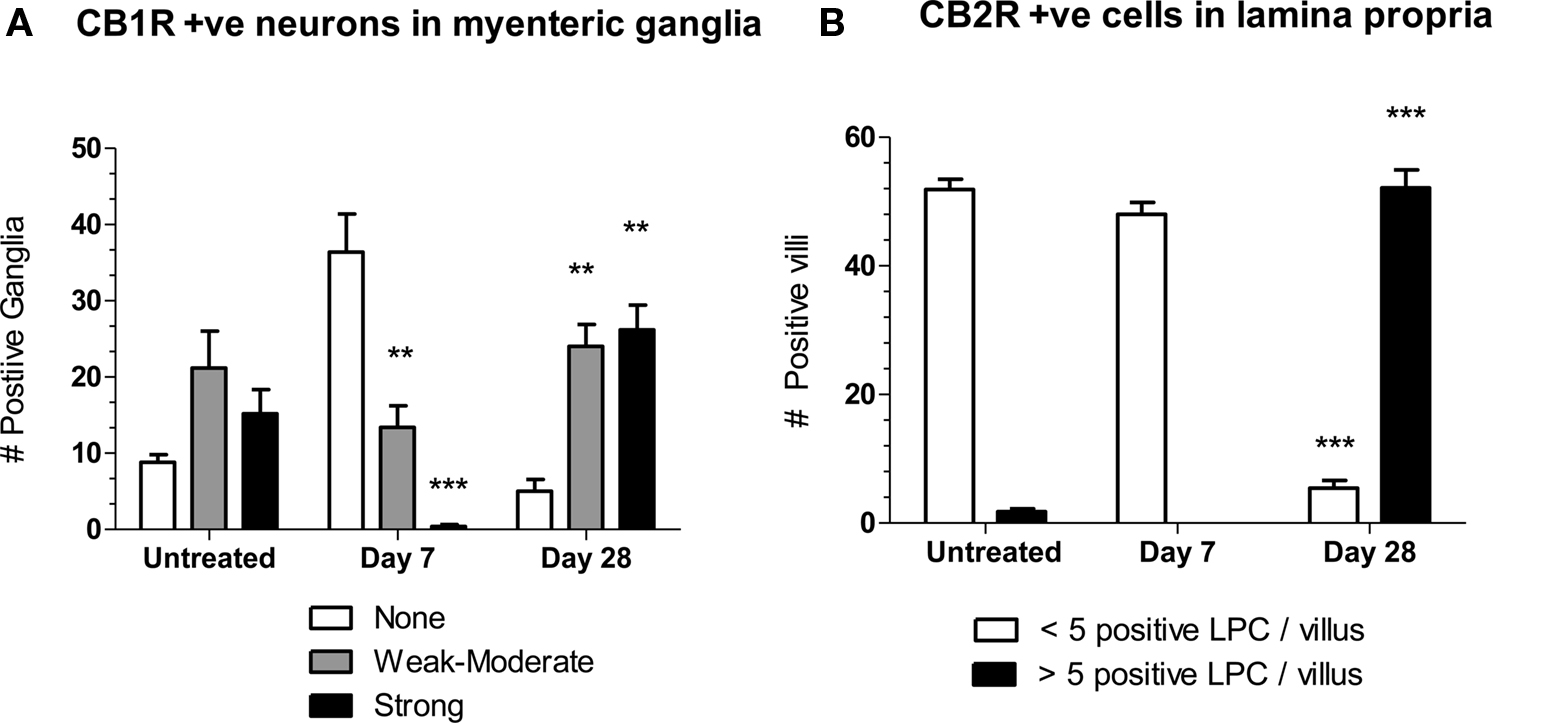
Figure 2. (A) The number of myenteric ganglia in jejunoileal tissue with moderate (gray bars) and intense (black bars) CB1R-stained neurons are decreased at day 7 and increased at day 28 compared to untreated tissue. Changes in the number of ganglia in which CB1R staining was absent (clear bars) were not significantly different in post-OM compared to untreated tissue. (B) The number of villi with < 5 CB2R + ve cells (clear bars) compared to > 5 CB2R + ve cells (black bars) in lamina. Differences two-way ANOVA and by Bonferroni’s post-test as a function of time were statistically significant and are shown as *P < 0.05, **P < 0.01, *** P < 0.001 compared to corresponding column for untreated.
In a separate group of animals small intestinal transit studies were performed with CBR agonists administered to mice 28 days after intracolonic vehicle (30% ethanol) or OM. As expected from our earlier study (Kimball et al., 2005), normal untreated mice upper GI transit (56 ± 2 and 56 ± 2% of small bowel length) was less than in mice 28 days after treatment with intracolonic OM (71 ± 2 and 68 ± 2% of small bowel length). Administration of the CB1R-selective agonist ACEA (1 and 3 mg/kg) to OM-treated mice effectively reduced the enhanced small intestinal transit to 59 ± 2 and 58 ± 4%, respectively, resulting in upper GI transit similar to that in normal mice (Figure 3A). Administration of the CB2R-selective agonist JWH-133 (3 and 10 mg/kg, s.c.) to OM-treated mice also reduced small intestinal transit to 49 ± 3 and 53 ± 4%, respectively. In this case, upper GI transit after CB2R agonist (3 mg/kg) administration in OM-treated mice appeared lower than that of normal mice but this was not reproduced at the highest dose (10 mg/kg; Figure 3B).
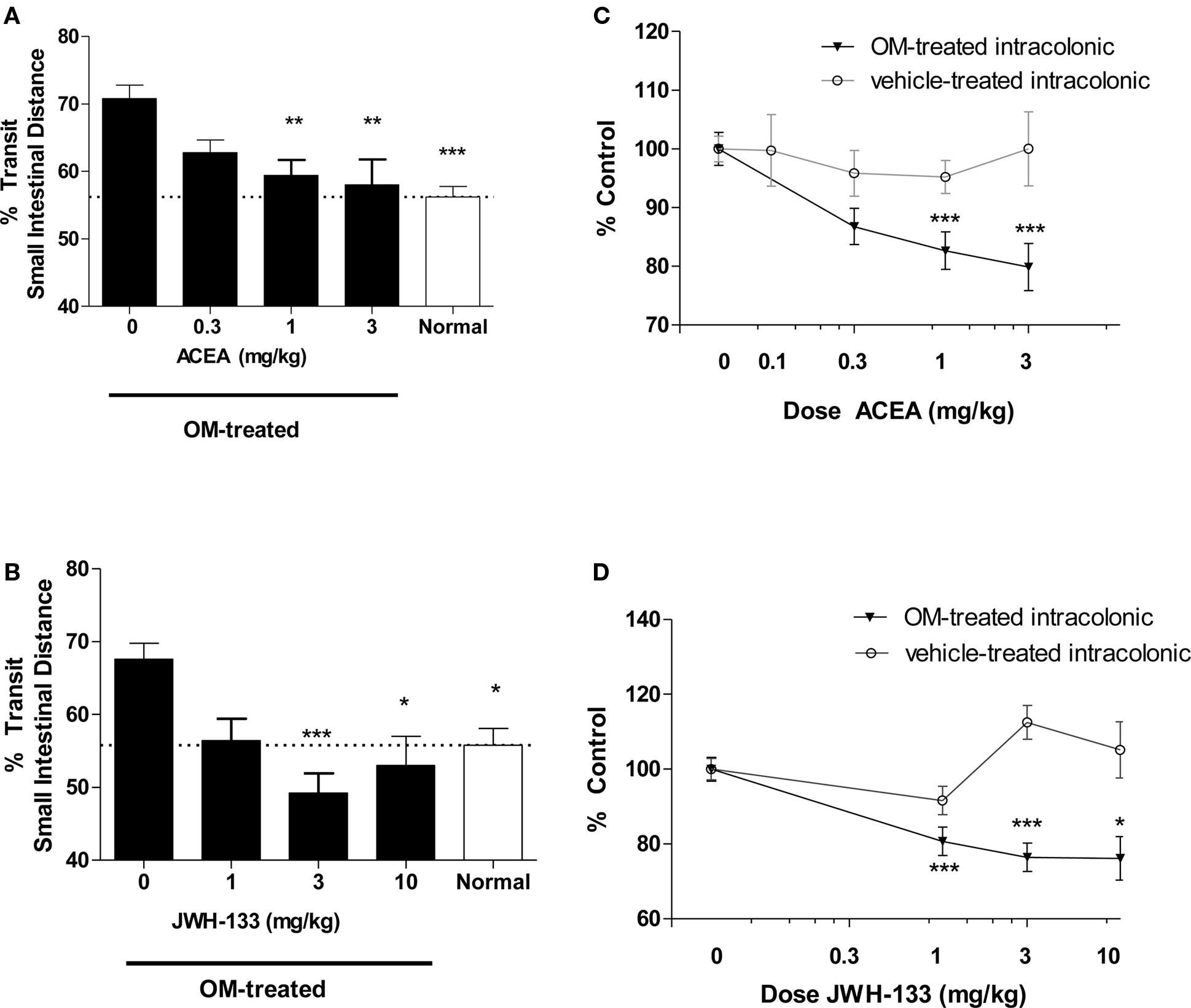
Figure 3. Effects on small intestinal transit of a range of doses of CB1R (A,C) and CB2R agonists (B,D) in mice 28 days after intracolonic 0.5% OM, vehicle (30% ethanol) and normal (untreated) age-matched mice. In (A) and (B) the % small intestinal distance traveled by carmine red is illustrated, with statistical comparisons to intracolonic vehicle-treated mice (i.e., first column in A, B). Effective doses (1 and 3 mg/kg) ACEA in OM-treated mice resulted in less upper GI transit than control (OM-treated only) mice and transit distance was similar to normal mice. Both ACEA (C) and JWH-133 (D) decrease transit in a dose-related fashion, when the values for CBR-treated groups are normalized to % of vehicle-treated control mice within each treatment. * P < 0.05, ** P < 0.01, ***P < 0.001.
In Figures 3C,D, all values for upper GI transit in CBR-treated mice were normalized to their respective CBR vehicle-treated control group, to enable comparison of CBR agonists in both intracolonic OM-and vehicle-treated mice. This illustrates a dose-related inhibition of accelerated transit in OM-treated mice by both CBR agonists. In intracolonic vehicle-treated control mice there was no effect of increasing doses of ACEA (Figure 3C). There was a trend for JWH-133 to increase upper GI transit in intracolonic vehicle-treated group (Figure 3D); however, the apparent increases did not attain the acceptable level of statistical significance.
Small intestinal transit is stable in normal (untreated) mice and those administered intracolonic vehicle (30% ethanol) after 28 days (Figure 4). Administration of the CB1R-selective agonist, ACEA, or the CB2R-selective agonist, JWH-133, individually at 1 mg/kg inhibited transit in OM-treated mice, but had no significant effect in normal mice (Figure 4). When the doses were combined they reduced the distance the marker traveled down the intestine to 65 ± 6 and 49 ± 7% of total length in normal and OM-treated mice, respectively (P < 0.001 compared to their respective controls). Although there was a trend to greater inhibition by the combination of CBR agonist in OM-treated mice this was not different than the same treatment in normal mice.
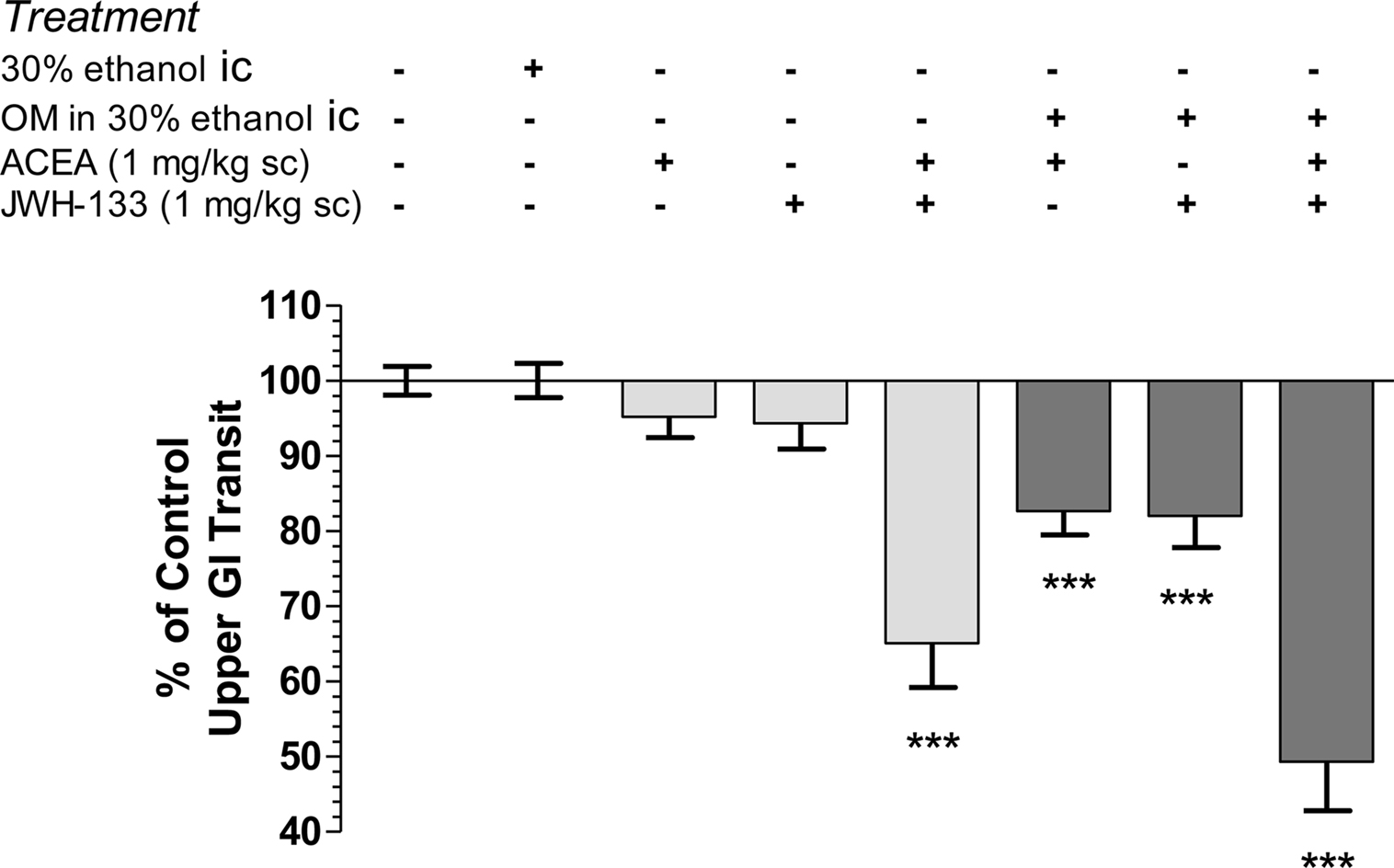
Figure 4. Intracolonic 30% ethanol had no impact on upper GI transit, measured 28 days later, compared to wild-type controls. CB1R (ACEA 1 mg/kg) and CB2R (JWH-133, 1 mg/kg) agonists alone inhibited small intestinal transit in mice 28 days post-OM administration, but not in control mice. A combination of both agonists inhibited transit in both groups. All values were normalized to the appropriate control group mice (i.e., columns 1–5 were normalized to mean transit in normal mice and columns 6–8 were normalized to mean transit in OM-treated treated mice) and are represented as % of control. ***P < 0.001 versus appropriate control (normal or 30% ethanol intracolonic).
Discussion
In the present study we have made the novel observations that CB1R in myenteric neurons and CB2R immunostaining in lamina propria are altered in the small intestine of mice up to 4 weeks after intracolonic OM. These changes are observed not only in the immediate post-colitis period (7 days) but also after all overt signs of inflammation have resolved (4 weeks) in mice that exhibit increased upper GI transit, which is a symptom consistent with clinical pathophysiology of PI-IBS (Kimball et al., 2005). Our results also demonstrate that both CB1R and CB2R agonists normalize upper GI transit in the OM-treated model and that the effect of these agonists is enhanced in the pathophysiological model compared to normal mice. The differences in efficacy by CBR agonists in the post-inflammatory compared to normal are consistent with cooperativity between CB1R and CB2R and amplification of signaling in a perturbed post-inflammatory system. Altogether these data suggest that altered CBR responsiveness is maintained long after an initial inflammatory period, and suggest a role in the underlying pathophysiology of PI-IBS.
We have previously characterized this model during acute inflammation (Kimball et al., 2006) and used it to determine the effects of CBR agonists in colitis (Kimball et al., 2006). We selected this model to study the CBR changes in post-inflammatory conditions because OM induces physiological perturbations similar to those associated with IBS, such as altered motility (Kimball et al., 2005) and visceral hyperalgesia (Laird et al., 2001). In addition, early changes in CBR mRNA expression was noted in OM-induced colitis (Kimball et al., 2007), but this had not been confirmed by protein staining for longer time periods after the acute colitis. Finally, CBR signaling is a consequence of OM neuronal stimulation (Bereiter et al., 2002) and seems to modulate the resulting hyperlagesia. Specifically, CBR interacts with TRPV1 channels on sensory afferents to ameliorate OM-induced hyperalgesia (Jordt et al., 2004; Akopian et al., 2008; Sawyer et al., 2009). Therefore this appears to be a useful model to study the role of CBR signaling in some of the pathophysiology associated with IBS.
We did not confirm selectivity of the CBR agonists in these experiments by using selective antagonists. This is because many studies have used CB1R and CB2R agonists, including ACEA and JWH-133, and already characterized their subtype selectivity using receptor antagonists or the CB1R inverse agonist in models of inflamed gut (Izzo et al., 1999, 2000, 2001, 2003; Landi et al., 2002; Mathison et al., 2004). The doses of agonists (ACEA and JWH-133) used in the present study were similar to that shown to be selectively acting at CB1R and CB2R to reduce rat LPS increased transit (Mathison et al., 2004). We recognize that without demonstrating competitive inhibition by using selective CB1R and CB2R antagonists in this study, we cannot completely exclude non-target related effects of the agonists such as TRPV1 receptors that bind endocannabinoids, and which ACEA structurally resembles. However, ACEA and JWH-133 has not been tested against TRPV1 or other TRPs, nor has the TRP-family receptor has been implicated in controlling small intestinal motility.
Twenty-eight days after OM administration we observed an up-regulation of CB2R in the small intestine and an inhibition of enhanced upper GI transit by CB2R agonists. This is similar to the situation that occurs in acute inflammation-induced hypermotility, reviewed in (Izzo, 2004; Wright et al., 2008; Izzo and Camilleri, 2009), where CB2R are increased in sensitivity compared to little or no effect in normal tissue. For example, upregulation of CB2R contributed to the increased efficacy of non-selective CBR agonists (Izzo et al., 2000, 2001) and a CB2R agonist reduced the electrically evoked ileal twitch responses in LPS-treated, but not normal, tissue (Duncan et al., 2008) and reduced motility in ileitis, but not in control mice (Capasso et al., 2008). Furthermore, a protective role of the CB2R in inflammation is demonstrated by a study in which a CB2R-selective antagonist exacerbated colitis (Storr et al., 2009). It is possible that CB2R is limiting the extent of enhanced transit in the post-inflammatory condition, but this has not been tested yet.
Up-regulation and enhanced anti-transit effects of CB1R agonists were also noted in the present study. Previously CB1R agonists inhibited inflammation in OM-induced acute colitis (Kimball et al., 2006) and dinitrobenzene sulfonic acid-induced colonic inflammation (Massa et al., 2004) with enhanced effects of CBR agonists reducing intestinal transit, fluid accumulation or the rate of colonic expulsion in rodents who were given an inflammatory stimulus (Izzo et al., 2000, 2001, 2003; Izzo, 2004; Mathison et al., 2004). Therefore our data support this enhanced role of CB1R in pathophysiology of the gut and show that it is maintained during the post-inflammatory period. In our study in normal mice, the CB1R-selective agonist (ACEA 1 mg/kg) did not inhibit transit in normal mice, in contrast to the situation in normal rats (Mathison et al., 2004). Two differences are apparent in these studies - species (rats versus mice) and the method of drug administration (i.p. versus s.c.). Discrepancies in CBR efficacy to inhibit gastric contractility has reported previously in rat (Storr et al., 2002) and mouse (Mule et al., 2007) stomach. Intraperitoneal drug administration in rats could introduce a higher local concentration of drug to the immediate vicinity of the intestinal tract than subcutaneous administration during a short term test (<30 min).
The increased sensitivity to either subtype of CBR agonists in OM-treated mice can be explained by the increased CB1R and CB2R expression in the small intestine in OM-treated mice tissues. Immunohistochemical data showed moderate CB1R expression in normal myenteric plexus that was reduced at 7 days post-OM colitis induction and which was restored, and appeared to be increased, by day 28. Likewise, striking changes for CB2R expression were seen in lamina propria, with dense staining in increased numbers of cells appearing at day 28, and appearing to occur mainly in monocytic cells.
Our results support a model for small intestinal motility dysfunction in which CB2R and CB1R agonists act in concert to affect transit. A review of cannabinoids and IBS concluded that the effects of the cannabinoid system on motility in humans are similar to those in rodents (Storr et al., 2008). Thus, although cannabinoids have not been studied in IBS patients to our knowledge, these data suggest that either CB1R- or CB2R-selective agonist could reduce GI motility in individuals experiencing diarrhea-predominant IBS. The utility of a CB2R agonist to reduce transit in mice with a post-inflammatory functional GI defect, coupled with a much lower CNS side effect liability, makes this an attractive concept.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The investigators are grateful to Brenda Hertzog and Meghan Towers (J&J PRD, Spring House, PA, USA) for performing histology and immunohistochemical preparations. We appreciate the discussions with Jeff Palmer, Steve Prouty, and Paul Wade (formerly Enterology Research Team at J&J PRD) during the development of this model.
References
Akopian, A. N., Ruparel, N. B., Patwardhan, A., and Hargreaves, K. M. (2008). Cannabinoids desensitize capsaicin and mustard oil responses in sensory neurons via TRPA1 activation. J. Neurosci. 28, 1064–1075.
Arevalo-Martin, A., Vela, J. M., Molina-Holgado, E., Borrell, J., and Guaza, C. (2003). Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J. Neurosci. 23, 2511–2516.
Bereiter, D. A., Bereiter, D. F., and Hirata, H. (2002). Topical cannabinoid agonist, WIN55,212-2, reduces cornea-evoked trigeminal brainstem activity in the rat. Pain 99, 547–556.
Bosier, B., Muccioli, G. G., Hermans, E., and Lambert, D. M. (2010). Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem. Pharmacol. 80, 1–12.
Brusberg, M., Arvidsson, S., Kang, D., Larsson, H., Lindstrom, E., and Martinez, V. (2009). CB1 receptors mediate the analgesic effects of cannabinoids on colorectal distension-induced visceral pain in rodents. J. Neurosci. 29, 1554–1564.
Capasso, R., Borrelli, F., Cascio, M. G., Aviello, G., Huben, K., Zjawiony, J. K., Marini, P., Romano, B., Di Marzo, V., Capasso, F., and Izzo, A. A. (2008). Inhibitory effect of salvinorin A, from Salvia divinorum, on ileitis-induced hypermotility: cross-talk between kappa-opioid and cannabinoid CB(1) receptors. Br. J. Pharmacol. 155, 681–689.
Casu, M. A., Porcella, A., Ruiu, S., Saba, P., Marchese, G., Carai, M. A., Reali, R., Gessa, G. L., and Pani, L. (2003). Differential distribution of functional cannabinoid CB1 receptors in the mouse gastroenteric tract. Eur. J. Pharmacol. 459, 97–105.
Coutts, A. A., Irving, A. J., Mackie, K., Pertwee, R. G., and Anavi-Goffer, S. (2002). Localisation of cannabinoid CB(1) receptor immunoreactivity in the guinea pig and rat myenteric plexus. J. Comp. Neurol. 448, 410–422.
D’Argenio, G., Petrosino, S., Gianfrani, C., Valenti, M., Scaglione, G., Grandone, I., Nigam, S., Sorrentini, I., Mazzarella, G., and Di Marzo, V. (2007). Overactivity of the intestinal endocannabinoid system in celiac disease and in methotrexate-treated rats. J. Mol. Med. 85, 523–530.
Drossman, D. A. (2007). Introduction. The Rome Foundation and Rome III. Neurogastroenterol. Motil. 19, 783–786.
Duncan, M., Mouihate, A., Mackie, K., Keenan, C. M., Buckley, N. E., Davison, J. S., Patel, K. D., Pittman, Q. J., and Sharkey, K. A. (2008). Cannabinoid CB2 receptors in the enteric nervous system modulate gastrointestinal contractility in lipopolysaccharide-treated rats. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G78–G87.
Gadzicki, D., Muller-Vahl, K., and Stuhrmann, M. (1999). A frequent polymorphism in the coding exon of the human cannabinoid receptor (CNR1) gene. Mol. Cell. Probes 13, 321–323.
Glass, M., and Northup, J. K. (1999). Agonist selective regulation of G proteins by cannabinoid CB(1) and CB(2) receptors. Mol. Pharmacol. 56, 1362–1369.
Hu, W. C., and Feng, P. (2010). G1359A polymorphism in the cannabinoid receptor-1 gene is associated with metabolic syndrome in the Chinese Han population. Arch. Med. Res. 41, 378–382.
Izzo, A. A. (2004). Cannabinoids and intestinal motility: welcome to CB2 receptors. Br. J. Pharmacol. 142, 1201–1202.
Izzo, A. A., and Camilleri, M. (2009). Cannabinoids in intestinal inflammation and cancer. Pharmacol. Res. 60, 117–125.
Izzo, A. A., Capasso, F., Costagliola, A., Bisogno, T., Marsicano, G., Ligresti, A., Matias, I., Capasso, R., Pinto, L., Borrelli, F., Cecio, A., Lutz, B., Mascolo, N., and Di Marzo, V. (2003). An endogenous cannabinoid tone attenuates cholera toxin-induced fluid accumulation in mice. Gastroenterology 125, 765–774.
Izzo, A. A., Fezza, F., Capasso, R., Bisogno, T., Pinto, L., Iuvone, T., Esposito, G., Mascolo, N., Di Marzo, V., and Capasso, F. (2001). Cannabinoid CB1-receptor mediated regulation of gastrointestinal motility in mice in a model of intestinal inflammation. Br. J. Pharmacol. 134, 563–570.
Izzo, A. A., Mascolo, N., Pinto, L., Capasso, R., and Capasso, F. (1999). The role of cannabinoid receptors in intestinal motility, defaecation and diarrhoea in rats. Eur. J. Pharmacol. 384, 37–42.
Izzo, A. A., Pinto, L., Borrelli, F., Capasso, R., Mascolo, N., and F., C. (2000). Central and peripheral cannabinoid modulation of gastrointestinal transit in physiological states or during the diarrhoea induced by croton oil. Br. J. Pharmacol. 129, 1627–1632.
Izzo, A. A., and Sharkey, K. A. (2010). Cannabinoids and the gut: new developments and emerging concepts. Pharmacol. Ther. 126, 21–38.
Jordt, S. E., Bautista, D. M., Chuang, H. H., McKemy, D. D., Zygmunt, P. M., Hogestatt, E. D., Meng, I. D., and Julius, D. (2004). Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265.
Kikuchi, A., Ohashi, K., Sugie, Y., Sugimoto, H., and Omura, H. (2008). Pharmacological evaluation of a novel cannabinoid 2 (CB2) ligand, PF-03550096, in vitro and in vivo by using a rat model of visceral hypersensitivity. J. Pharmacol. Sci. 106, 219–224.
Kimball, E. S., Palmer, J. M., D’Andrea, M. R., Hornby, P. J., and Wade, P. R. (2005). Acute colitis induction by oil of mustard results in later development of an IBS-like accelerated upper GI transit in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G1266–G1273.
Kimball, E. S., Prouty, S. P., Pavlick, K. P., Wallace, N. H., Schneider, C. R., and Hornby, P. J. (2007). Stimulation of neuronal receptors, neuropeptides and cytokines during experimental oil of mustard colitis. Neurogastroenterol. Motil. 19, 390–400.
Kimball, E. S., Schneider, C. R., Wallace, N. H., and Hornby, P. J. (2006). Agonists of cannabinoid receptor 1 and 2 inhibit experimental colitis induced by oil of mustard and by dextran sulfate sodium. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G364–G371.
Kulkarni-Narla, A., and Brown, D. R. (2000). Localization of CB1-cannabinoid receptor immunoreactivity in the porcine enteric nervous system. Cell Tissue Res. 302, 73–80.
Laird, J. M., Martinez-Caro, L., Garcia-Nicas, E., and Cervero, F. (2001). A new model of visceral pain and referred hyperalgesia in the mouse. Pain 92, 335–342.
Landi, M., Croci, T., Rinaldi-Carmona, M., Maffrand, J. P., Le Fur, G., and Manara, L. (2002). Modulation of gastric emptying and gastrointestinal transit in rats through intestinal cannabinoid CB(1) receptors. Eur. J. Pharmacol. 450, 77–83.
Massa, F., Marsicano, G., Hermann, H., Cannich, A., Monory, K., Cravatt, B. F., Ferri, G. L., Sibaev, A., Storr, M., and Lutz, B. (2004). The endogenous cannabinoid system protects against colonic inflammation. J. Clin. Invest. 113, 1202–1209.
Mathison, R., Ho, W., Pittman, Q. J., Davison, J. S., and Sharkey, K. A. (2004). Effects of cannabinoid receptor-2 activation on accelerated gastrointestinal transit in lipopolysaccharide-treated rats. Br. J. Pharmacol. 142, 1247–1254.
Mule, F., Amato, A., Baldassano, S., and Serio, R. (2007). Involvement of CB1 and CB2 receptors in the modulation of cholinergic neurotransmission in mouse gastric preparations. Pharmacol. Res. 56, 185–192.
Park, J. M., Choi, M. G., Cho, Y. K., Lee, I. S., Kim, S. W., Choi, K. Y., and Chung, I. S. (2010). Cannabinoid receptor 1 gene polymorphism and irritable bowel syndrome in the Korean population: a hypothesis-generating study. J. Clin. Gastroenterol. [Epub ahead of print].
Pertwee, R. G., and Ross, R. A. (2002). Cannabinoid receptors and their ligands. Prostaglandins Leukot. Essent. Fatty Acids 66, 101–121.
Sanson, M., Bueno, L., and Fioramonti, J. (2006). Involvement of cannabinoid receptors in inflammatory hypersensitivity to colonic distension in rats. Neurogastroenterol. Motil. 18, 949–956.
Sawyer, C. M., Carstens, M. I., and Carstens, E. (2009). Mustard oil enhances spinal neuronal responses to noxious heat but not cooling. Neurosci. Lett. 461, 271–274.
Sibaev, A., Massa, F., Yuce, B., Marsicano, G., Lehr, H. A., Lutz, B., Goke, B., Allescher, H. D., and Storr, M. (2006). CB1 and TRPV1 receptors mediate protective effects on colonic electrophysiological properties in mice. J. Mol. Med. 84, 513–520.
Storr, M., Gaffal, E., Saur, D., Schusdziarra, V., and Allescher, H. D. (2002). Effect of cannabinoids on neural transmission in rat gastric fundus. Can. J. Physiol. Pharmacol. 80, 67–76.
Storr, M. A., Keenan, C. M., Zhang, H., Patel, K. D., Makriyannis, A., and Sharkey, K. A. (2009). Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm. Bowel Dis. 15, 1678–1685.
Storr, M. A., Yuce, B., Andrews, C. N., and Sharkey, K. A. (2008). The role of the endocannabinoid system in the pathophysiology and treatment of irritable bowel syndrome. Neurogastroenterol. Motil. 20, 857–868.
Wright, K., Rooney, N., Feeney, M., Tate, J., Robertson, D., Welham, M., and Ward, S. (2005). Differential expression of cannabinoid receptors in the human colon: cannabinoids promote epithelial wound healing. Gastroenterology 129, 437–453.
Keywords: colitis, pathophysiology, motility, receptor up-regulation, endocannabinoids, inflammation, cannabinoid receptors, enteric nervous system
Citation: Kimball ES, Wallace NH, Schneider CR, D’Andrea MR and Hornby PJ (2010) Small intestinal cannabinoid receptor changes following a single colonic insult with oil of mustard in mice. Front. Pharmacol. 1:132. doi: 10.3389/fphar.2010.00132
Received: 21 June 2010;
Accepted: 20 October 2010;
Published online: 19 November 2010.
Edited by:
Martin Storr, University of Calgary, CanadaReviewed by:
Martin Storr, University of Calgary, CanadaMohammad Bashashati, Lorestan University of Medical Sciences, Iran
Marnie Duncan, Robert Gordon University, UK
Copyright: © 2010 Kimball, Wallace, Schneider, D’Andrea and Hornby. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Pamela J. Hornby, Cell Biology & Assay Technologies, J&J Biotechnology Center of Excellence, 145 King of Prussia Road, Radnor, PA 19087, USA. e-mail:cGhvcm5ieUBpdHMuam5qLmNvbQ==