


- 1 Laboratory of Toxicology, University of Leuven, Leuven, Belgium
- 2 Institute for Clinical Immunology and Rheumatology, Group of Experimental Peptide Chemistry, Medical High School, Hannover, Germany
- 3 Center for Marine Bioproducts (CeBiMar), Havana, Cuba
Because of their prominent role in electro-excitability, voltage-gated sodium (NaV) channels have become the foremost important target of animal toxins. These toxins have developed the ability to discriminate between closely related NaV subtypes, making them powerful tools to study NaV channel function and structure. CgNa is a 47-amino acid residue type I toxin isolated from the venom of the Giant Caribbean Sea Anemone Condylactis gigantea. Previous studies showed that this toxin slows the fast inactivation of tetrodotoxin-sensitive NaV currents in rat dorsal root ganglion neurons. To illuminate the underlying NaV subtype-selectivity pattern, we have assayed the effects of CgNa on a broad range of mammalian isoforms (NaV1.2–NaV1.8) expressed in Xenopus oocytes. This study demonstrates that CgNa selectively slows the fast inactivation of rNaV1.3/β1, mNaV1.6/β1 and, to a lesser extent, hNaV1.5/β1, while the other mammalian isoforms remain unaffected. Importantly, CgNa was also examined on the insect sodium channel DmNaV1/tipE, revealing a clear phyla-selectivity in the efficacious actions of the toxin. CgNa strongly inhibits the inactivation of the insect NaV channel, resulting in a dramatic increase in peak current amplitude and complete removal of fast and steady-state inactivation. Together with the previously determined solution structure, the subtype-selective effects revealed in this study make of CgNa an interesting pharmacological probe to investigate the functional role of specific NaV channel subtypes. Moreover, further structural studies could provide important information on the molecular mechanism of NaV channel inactivation.
Introduction
Voltage-gated sodium (NaV) channels are the trademark of electro-excitable cells. These transmembrane proteins transiently open in response to membrane depolarizations and thereby provide the Na+ current pathway that underlies the initial phase of action potentials. To properly fulfill this crucial physiological role, NaV channels are bestowed with three key features: voltage-dependent activation, high selectivity for Na+ ions, and spontaneous fast inactivation (Hille, 2001). NaV channels are composed of a pore forming ∼260-kDa α-subunit associated with auxiliary β-subunits of ∼30 kDa. The α-subunit consists of four homologous, yet non-identical, repeats (DI–IV) connected by intracellular linkers, with each repeat containing six transmembrane segments (S1–6) (Catterall, 2000). The S4 segments in each repeat contain several positively charged Arg or Lys residues in every third position and are believed to act as voltage sensors, making the channel able to respond to voltage changes across the cell membrane (Stuhmer et al., 1989). Upon membrane depolarization, the positive charges move outward in the electrical field of the membrane, resulting in a conformational change of the protein structure that opens the ion conducting pore (Armstrong, 1981; Kontis et al., 1997). The intracellular loop that connects DIII and IV contains a highly conserved hydrophobic cluster of Ile, Phe, and Met residues, the so-called IFM-motif. This motif is proposed to be the inactivation gate, acting as a hinged lid that closes the ion conducting pore from the cytoplasmic side (West et al., 1992). Numerous studies have indicated that DIV, and more specifically segment IVS4, may play a unique role among the four homologous repeats in coupling activation to inactivation and it has been proposed that movement of this S4 segment facilitates closure of the inactivation gate (Chahine et al., 1994; Chen et al., 1996; Kontis and Goldin, 1997; Sheets et al., 1999). However, the precise molecular mechanism of coupling IVS4 movement to closure of the inactivation gate is still elusive today (Ulbricht, 2005). Several peptide toxins from the venom of scorpions, sea anemones and spiders have been shown to slow or inhibit the fast inactivation process of NaV channels by interacting with overlapping, yet non-identical binding sites, named receptor site 3 (Catterall, 2000). The molecular location of this receptor site is not entirely known but was shown to include several crucial amino acid residues in the extracellular S3–S4 loop in DIV (Rogers et al., 1996; Benzinger et al., 1998). It was proposed that by binding to this loop, site 3 toxins prevent the normal gating movement of the voltage sensor in DIV, thereby hindering the conformational changes associated with fast inactivation (Rogers et al., 1996).
In mammals, nine different genes that encode distinct NaV channel subtypes (NaV1.1–NaV1.9) have been identified until today (Goldin, 1999). These closely related subtypes (49–87% sequence identity among human subtypes) can have very different biophysical properties and are expressed in a tissue-specific manner. Evidently, this differential expression plays an important role in the diversity in electrical properties of excitable tissues and plasticity of nervous tissues. The importance of NaV subtype diversity is also reflected in the emerging roles that different NaV subtypes play in various channelopathies (for reviews, see Ashcroft, 2006; Catterall et al., 2008; Cannon, 2010). In contrast to the nine mammalian NaV channel genes, only one gene encoding NaV channels (para) has been identified in insects until today (Loughney et al., 1989). Functional diversity in insect NaV channels is very likely to be achieved by alternative splicing and RNA editing of the para transcript, rather than expression of distinct genes (Tan et al., 2002; Song et al., 2004). As a consequence, the insect NaV channel orthologs share much more sequence identity (typically 87–98%) than their mammalian counterparts (King et al., 2008). An important feature of animal toxins is that they can discriminate between closely related subtypes with high selectivity. However, for most of these toxins, the subtype-selectivity pattern is unknown. CgNa is a 47-amino acid residue type I toxin isolated from the venom of the Giant Caribbean Sea Anemone Condylactis gigantea. Previous studies showed that CgNa increases the action potential duration by slowing the inactivation of tetrodotoxin-sensitive (TTX-S) sodium currents in rat dorsal root ganglion (DRG) neurons (Standker et al., 2006; Salceda et al., 2007). In this study, we reveal the phyla- and subtype-selectivity of CgNa on NaV channels, using cloned NaV channel subtypes expressed in Xenopus oocytes.
Materials and Methods
Toxin Purification
CgNa was isolated and purified from the Giant Caribbean Sea Anemone Condylactis gigantea as described previously (Standker et al., 2006).
Expression of NaV Channels
For expression in Xenopus laevis oocytes, the cDNA encoding rNaV1.2 and mNaV1.6 was subcloned into pLCT1. The rNaV1.3, rNaV1.4, DmNaV1, and tipE cDNA was subcloned into vectors pNa3T, pUI-2, pGH19-13-5, and pGH19 respectively. For in vitro transcription, these plasmids were linearized with NotI. The rNaV1.7/pBSTA.rPN1 and hβ1/pGEM-HE were linearized with SacII and NheI respectively. Capped cRNAs were then synthesized from the linearized plasmid using the T7 mMESSAGE-mMACHINE transcription kit (Ambion, USA). The hNaV1.5/pSP64T, rβ1/pSP64T, and rNaV1.8/pSP64T vectors were linearized with XbaI, EcoRI, and XbaI respectively, and transcribed with the SP6 mMESSAGE-mMACHINE transcription kit (Ambion, USA). Female X. laevis frogs were anesthetized by submersion in ice water in the presence of 0.1% 3-aminobenzoic acid ethyl ester (tricaine mesylate). Stage V–VI oocytes were harvested from the ovarian lobes of anesthetized frogs as described previously (Liman et al., 1992). Care and use of X. laevis frogs in this study meet with the guidelines of the Catholic University Leuven (K.U. Leuven) and were approved by the ECD (Ethical Commission for Experiments on Animals, Belgian Federal Public Health Service). The oocytes were injected with up to 50 nl of cRNA at a concentration of 1 ng/nl using a microinjector (Drummond, USA). The oocyte incubation solution contained (in mM): NaCl 96, KCl 2, CaCl2 1.8, MgCl2 2, and HEPES-acid 5 (pH 7.4), supplemented with 50 mg/l gentamicin sulfate. Whole-cell currents from oocytes were recorded 2–5 days after injection.
Electrophysiological Study
Whole-cell currents were recorded in X. laevis oocytes using the two-electrode voltage-clamp technique as described by Liman et al. (1992). Experiments were performed at constant temperature 18–24°C using a GeneClamp 500 amplifier (Molecular Devises, USA) controlled by a pClamp data acquisition system (Molecular Devices, USA). Data were sampled at a frequency of 20 kHz and low-pass filtered at 2 kHz using a 4-pole low-pass Bessel filter. Leak subtraction was performed using a −P/4 protocol. The voltage-clamp data recorded in this study were discarded if oocytes exhibited large (>200 nA) or unstable (>10% deviation) leak currents throughout the experiment. To avoid overestimation of a potential toxin-induced shift in the current–voltage relationship as a result of inadequate voltage control when measuring large sodium currents in oocytes, only data obtained from cells with peak currents below 2 μA were considered for analysis. Voltage and current electrodes were filled with 3 M KCl and resistances were kept as low as possible (<1 MΩ). The bath solution contained (in nM): NaCl 96, KCl 2, CaCl2 1.8, MgCl2 2, and HEPES 5 (pH 7.4).
To examine the electrophysiological effects of CgNa on the cloned NaV channels, the following voltage protocols were applied. All protocols were applied from a holding potential of −90 mV and repeated with a start-to-start frequency of 0.2 Hz. (i) Test pulses were elicited by 100-ms depolarizations to the voltage corresponding to maximal activation in control conditions (between −15 and 10 mV, depending on the NaV channel subtype). (ii) Current–voltage relationships were determined by 100-ms step depolarizations ranging from −90 to 60 mV with 5-mV increments. The sodium conductance (gNa) was calculated according to Ohm’s law (Equation 1): gNa = INa/(V − Vrev), where INa represents the Na+ current peak amplitude at a given test potential V, and Vrev is the reversal potential. The values of gNa were plotted as a function of voltage and fitted using the Boltzmann equation (Equation 2): gNa/gmax = [1 + (exp(Vg − V)/k)]−1, where gmax represents maximal gNa, Vg is the voltage corresponding to half-maximal conductance, and k is the slope factor. (iii) To examine the toxin-induced effects on the steady-state inactivation process, a standard two-step voltage protocol was applied. In this protocol, 100-ms conditioning, 5-mV step prepulses ranging from −90 to 60 mV were immediately followed by a 50-ms test pulse to the voltage corresponding to maximal activation in control conditions. Data were normalized to the maximal Na+ current amplitude, plotted against prepulse potential, and fitted using the Boltzmann equation (Equation 3): INa/Imax = [(1 − C)/(1 + exp((V − Vh)/k))] + C, where Imax is the maximal INa, Vh is the voltage corresponding to half-maximal inactivation, V is the test voltage, k is the slope factor, and C is a constant representing a non-inactivating sustained fraction (close to 0 in control). (iv) The recovery from inactivation was assayed with a double-pulse protocol, where a 100-ms conditioning pulse to the potential corresponding to maximal activation in control was followed by a 50-ms test pulse to the same voltage. Both pulses were interspersed by a repolarization to −90 mV during a gradually increasing time interval (1–40 ms). The INa obtained in the test pulse was normalized to the INa obtained in the conditioning pulse and plotted against the corresponding time interval To assess the dose–response relationships, data were fitted according to the Hill equation (Equation 4): y = 100/[1 + (EC50/[toxin])h], where y is the amplitude of toxin-induced effect, EC50 is the toxin concentration at half-maximal efficacy, [toxin] is the toxin concentration and h is the Hill coefficient.
All data were analyzed using Clampfit 8.1 (Molecular Devices, USA), Excel 2003 (Microsoft, USA), and Origin 6.1 (OriginLab, USA) software. Statistical differences were determined using a Student’s t test. A test was considered to be significant when p < 0.05. All numerical data are presented as the mean ± SE for at least three experiments (n ≥ 3).
Results
CgNa was previously reported to slow the fast inactivation of TTX-S NaV currents in rat DRG neurons (Standker et al., 2006; Salceda et al., 2007). To illuminate the subtype-selectivity pattern behind these actions, we examined the effects of CgNa on a wide range of cloned mammalian NaV channels expressed in Xenopus oocytes. The toxin slowed the fast inactivation of specific NaV subtypes, resulting in an increase in INa peak amplitude and an incompletely inactivated or sustained current at the end of the 100-ms test depolarization (Figure 1, left-hand panels). The maximal degree of slowed inactivation was observed with subtypes rNaV1.3/β1 and mNaV1.6/β1, where addition of 10 μM CgNa to the bath medium produced a sustained current of 27.1 ± 2.7% (n = 7; p < 0.05) and 34.7 ± 1.5% (n = 6; p < 0.05) of INa peak amplitude, respectively. In parallel, their peak INa amplitudes increased by 36.6 ± 4.6% (n = 7; p < 0.05) and 28.5 ± 4.8% (n = 6; p < 0.05). In the case of hNaV1.5/β1, the peak INa amplitude was increased by 10.9 ± 3.2% (n = 3; p < 0.05), while the current was fully inactivated after 100 ms. The slowing of these mammalian NaV subtypes was not associated with a shift in current–voltage relationships nor a change in slope factor or midpoint potential of the activation curves. Neither was the reversal potential of any of the tested NaV subtypes changed, indicating that the ion selectivity of the channels was not altered by the toxin (Figure 1, middle panels). However, the observed effects were accompanied by a small but significant shift in steady-state inactivation of rNaV1.3/β1 and mNaV1.6/β1. The midpoint potential of steady-state inactivation (Vh) was shifted from −27.0 ± 0.2 to −37.0 ± 0.4 mV (n = 5; p < 0.05) and from −51.3 ± 0.3 to −54.4 ± 0.6 mV (n = 3; p < 0.05), respectively. Moreover, the steady-state inactivation of rNaV1.3/β1 and mNaV1.6/β1 became incomplete in presence of 10 μM CgNa, resulting in the appearance of a non-inactivating component of 28.8 ± 0.5 and 36.7 ± 0.7% (Figure 1, right-hand panels). The other mammalian NaV subtypes examined in this study remained unaffected by CgNa at concentrations up to 10 μM.
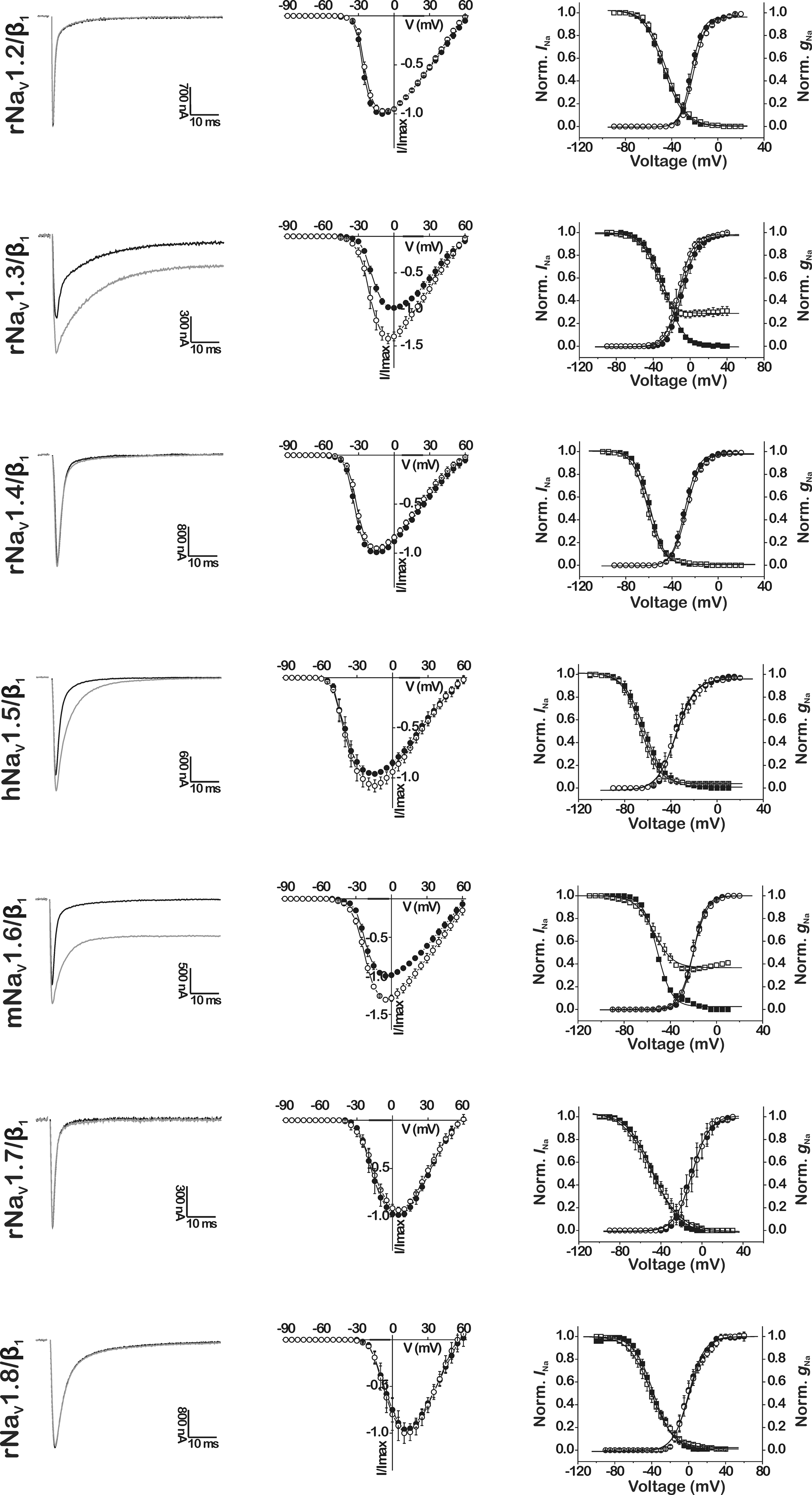
Figure 1. Effects of CgNa on cloned mammalian NaV channel subtypes NaV1.2–NaV1.8/β1 expressed in Xenopus oocytes. The tested mammalian isoforms originate from rat (r), human (h), or mouse (m). Left-hand panels show representative whole-cell current traces in control (black traces) and in presence of 10 μM CgNa (gray traces). Middle panels show normalized current–voltage relationships (n = 3–7) in control (l) and in presence of 10 μM CgNa (●). Right-hand panels show steady-state activation and inactivation curves (n = 3–7) in control (● and ■, respectively) and in presence of 10 μM CgNa (○ and □, respectively), fit with the Boltzmann equation.
In contrast to the clear effects, but modest potency, of the toxin on specific mammalian NaV channel subtypes, CgNa affected the inactivation of the insect sodium channel clone DmNaV1/tipE from Drosophila melanogaster much more profoundly. This was manifested by a dramatic increase in peak INa up to a maximum of 313.5 ± 19.2% (n = 6; p < 0.05) of control amplitude and a complete removal of real-time and steady-state inactivation in the presence of 10 μM CgNa (Figure 2B). At the more moderate concentration of 100 nM, the toxin increased the peak INa by 63.8 ± 19.2% (n = 4; p < 0.05) and induced a sustained current of 101.5 ± 20.3% (n = 4; p < 0.05) of INa peak amplitude (Figure 2A). The toxin-induced increase in INa was not associated with a shift in current–voltage relationships or reversal potential. However, 100 nM CgNa shifted the voltage dependence of steady-state inactivation (Vh from −41.6 ± 0.1 to −43.1 ± 0.3 mV; n = 6; p < 0.05) and caused the steady-state inactivation to become incomplete (47.8 ± 0.4% non-inactivating component; n = 6; p < 0.05). The toxin-induced increase in peak and sustained current amplitude was described by a relatively slow time course, reaching a steady-state after ∼12 min (Figure 2C; n = 6) and was not reversible by washing the bath medium. In addition, CgNa increased the rate of recovery from inactivation of the insect DmNaV1/tipE channel. The time constant of recovery significantly decreased from 1.89 ± 0.07 ms in control to 1.57 ± 0.11 ms in presence of 100 nM CgNa (Figure 2E; n = 7; p < 0.05).
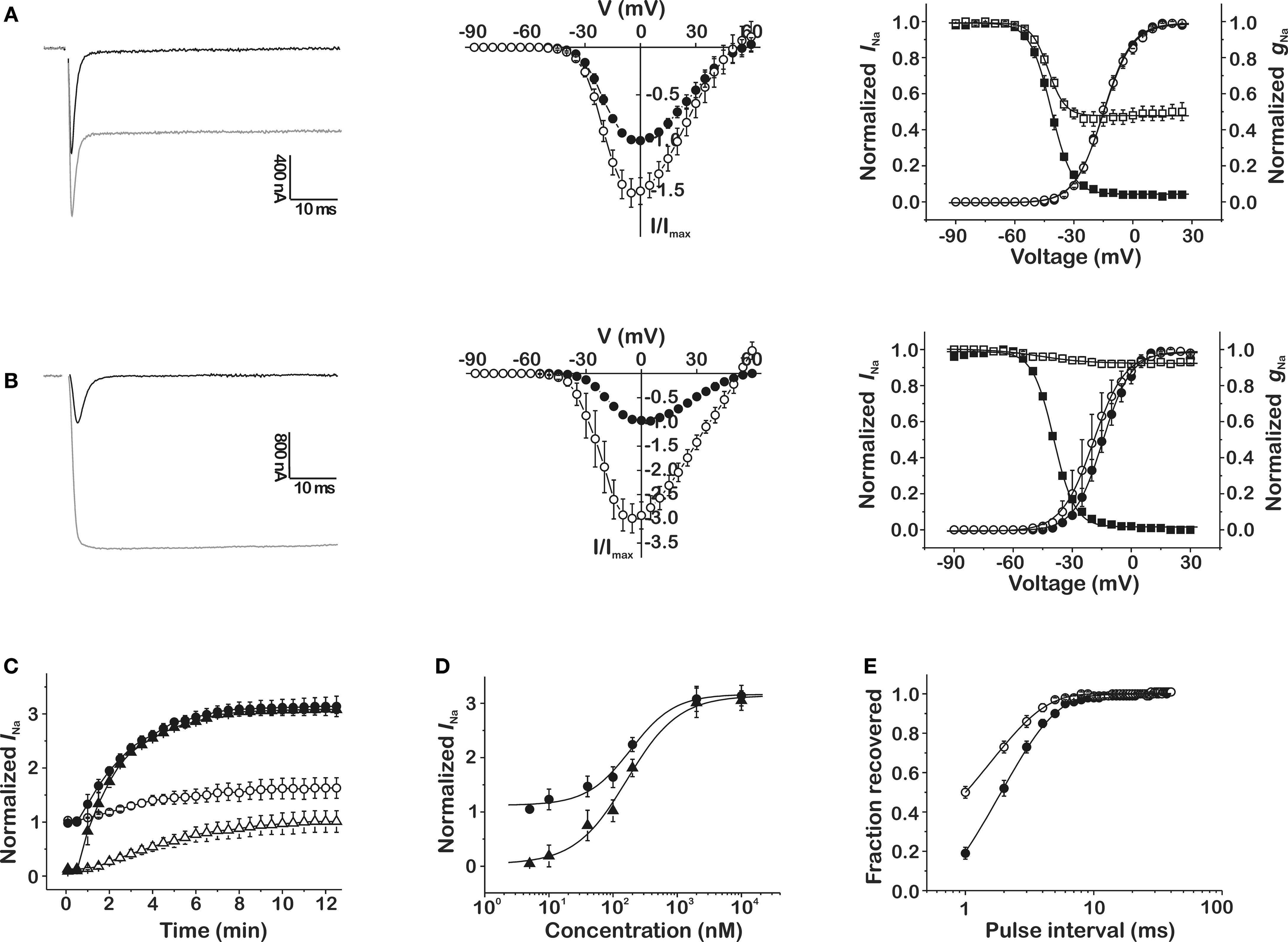
Figure 2. Effects of CgNa on the cloned insect NaV channel DmNaV1/tipE expressed in Xenopus oocytes. (A,B) Left-hand panels show representative whole-cell current traces in control (black traces) and in presence CgNa (gray traces) at a concentration of 100 nM (A) and 10 μM (B); middle panels show normalized current–voltage relationships (n = 3–6) in control (●) and in presence of CgNa (○) at a concentration of 100 nM (A) and 10 μM (B); right-hand panels display steady-state activation and inactivation curves (n = 3–6) in control (● and ■, respectively) and in presence of CgNa (○ and □, respectively) at a concentration of 100 nM (A) and 10 μM (B). (C) Time course of the increase (n = 6) in peak INa (Δ, 100 nM; ▲, 10 μM) and sustained INa measured after 100 ms (○, 100 nM; ●, 10 μM). (D) Dose–response curves displaying the concentration dependence of the CgNa-induced increase in INa peak (●) and sustained INa measured after 100 ms (▲). (E) Recovery from inactivation (n = 7) in control (●) and in the presence of 100 nM CgNa (○).
Given the differences in modulation of peak and steady-state current of specific NaV subtypes by CgNa, three different parameters were taken into account to properly quantify the subtype- and phylum-selectivity of the toxin. Therefore, dose–response curves were constructed using: (i) INa peak/INa peak, monitoring the toxin-induced increase in peak current amplitude; (ii) INa 5 ms/INa peak, showing the impairment of inactivation during the fast decay phase of the current; and (iii) INa 100 ms/INa peak, reflecting the fraction of remaining INa at steady-state, which displays the toxin-induced sustained current (Figures 3A–C) All dose–response data were fitted with the Hill equation (Eq. 4) and the resulting EC50 values can be found in Table 1. To facilitate comparison of the selectivity of the toxin for the tested subtypes, the normalized potency (inversed EC50 values with the highest set as 100%) and the normalized efficacy (relative amplitudes with the highest set as 100%) were plotted in bar diagrams (Figures 3D,E). Clearly, the selectivity of CgNa for DmNaV1/tipE is more pronounced in terms of efficacy (i.e., amplitude of the effect at saturating concentrations) than in terms of potency (i.e., the concentration required to produce an effect of a given amplitude). According to the potency, the following rank order was observed for CgNa: DmNaV1/tipE > rNaV1.3/β1 and mNaV1.6/β1 >>hNaV1.5/β1 >>> rNaV1.2/β1, rNaV1.4/β1, rNaV1.7/β1, and rNaV1.8/β1. The EC50 values observed on the insect channel were roughly two-fold lower than those on the most sensitive mammalian channels mNaV1.6/β1 and rNaV1.3/β1 (Table 1). A similar rank order was observed in terms of efficacy: DmNaV1/tipE >> rNaV1.3/β1 and mNaV1.6/β1 > hNaV1.5/β1 >>> rNaV1.2/β1, rNaV1.4/β1, rNaV1.7/β1, and rNaV1.8/β1. In this case, the efficacy of CgNa on the insect channel was roughly 10-fold higher than on rNaV1.3/β1 and mNaV1.6/β1. In general, no great differences were observed between the INa peak, INa 5 ms and INa 100 ms for each channel. The only great exception was seen with hNaV1.5/β1, were the INa 100 ms was not determinable, reflecting the lack of sustained current in this subtype.
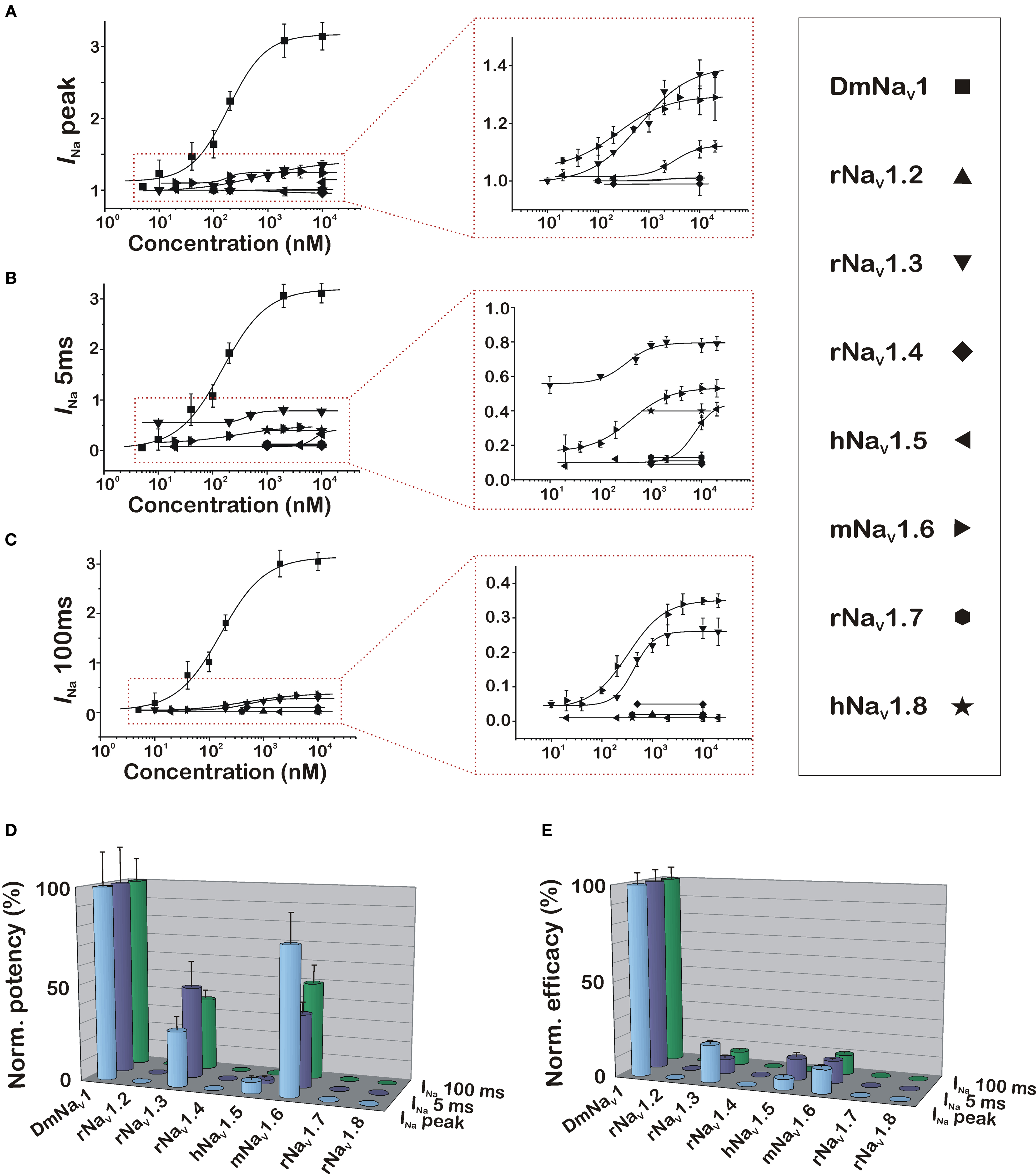
Figure 3. Dose–response relationships of the effects of CgNa on the insect and mammalian NaV channels. Dose–response curves are constructed using the following three parameters to quantify the effects induced by CgNa: increase in peak INa (A), increase in INa 5 ms (B), and increase in INa 100 ms (C). Left-hand graphs show phyla-selectivity between insect and mammalian NaV channels, while right-hand graphs zoom in to show the subtype-selectivity among the mammalian NaV subtypes. (D,E) Bar diagram illustrating the differences in potency and efficacy of CgNa on the variety of assayed insect and mammalian NaV channels.
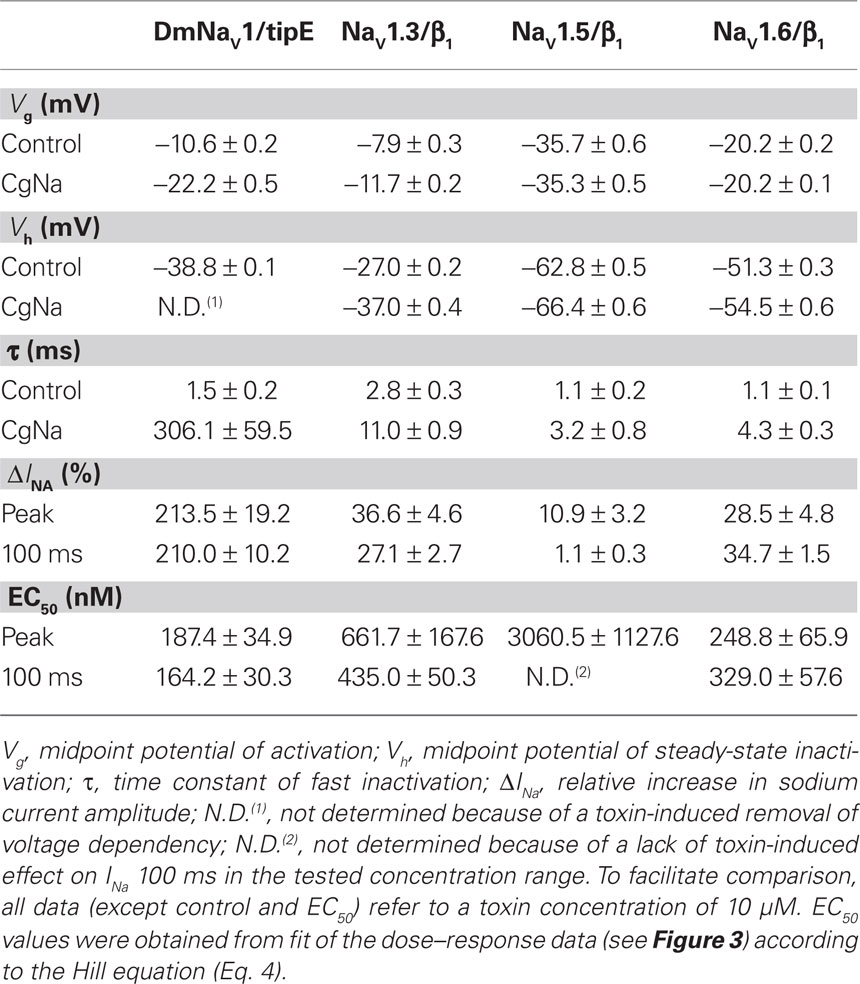
Table 1. Summary of the effects of CgNa on NaV channels.
Discussion
Because of their prominent role in electro-excitability, NaV channels have become one of the foremost important targets of venomous animals. Most of the presently known NaV channel toxins isolated from sea anemones modulate channel function by slowing or inhibiting the channel’s inactivation process and are able to discriminate between closely related NaV subtypes. Yet, for most known sea anemone toxins, the exact pattern of NaV subtypes they target is either unknown, or at best incomplete. In this work, we present the elaborate study of a sea anemone toxin on a broad range of mammalian NaV subtypes (NaV1.2–NaV1.9), and report its phyla-selectivity using the insect DmNaV1 channel from D. melanogaster.
This study demonstrates that CgNa causes a slowing of the fast inactivation of the neuronal NaV channel subtypes rNaV1.3/β1 and mNaV1.6/β1 and, to a lesser extent, of the cardiac isoform hNaV1.5/β1 (Figure 1). In contrast, CgNa failed to affect the other mammalian NaV channel subtypes rNaV1.2/β1, rNaV1.4/β1, rNaV1.7/β1, and rNaV1.8/β1, at concentrations up to 10 μM. Because of current difficulties in the expression of the rNaV1.1 clone in oocytes, this subtype was not included in the electrophysiological assay. The other mammalian NaV subtype that was not examined in this study, NaV1.9, fails to express in standard heterologous systems. However, because NaV1.9 channels are endogenously expressed in rat DRG neurons (Dib-Hajj et al., 1998) and CgNa was previously shown to be inactive in experiments with tetrodotoxin-resistant (TTX-R) NaV channels in rat DRG neurons (Salceda et al., 2007), we can exclude the possibility of CgNa affecting NaV1.9. The presently described NaV subtype-selectivity pattern accords well with previous studies which showed the activity of CgNa on TTX-S NaV channels in DRG neurons (Standker et al., 2006; Salceda et al., 2007). Rat DRG neurons express two types of NaV channels: TTX-S (carried mainly by NaV1.1, NaV1.3, NaV1.6, and NaV1.7) and TTX-R (NaV1.8 and NaV1.9) (Roy and Narahashi, 1992; Black et al., 1996; Dib-Hajj et al., 1998).
Beside these clear effects, but modest potency, observed on mammalian NaV channels, CgNa exhibits a marked phyla-selectivity in its actions. When tested at the same concentration as on the mammalian NaV channels (10 μM), CgNa drastically increases the peak current and causes complete removal of fast inactivation and steady-state inactivation of the insect DmNaV1/tipE channel (Figure 2B). At more moderate concentrations (100 nM), CgNa induces a sustained steady-state current and non-inactivating fraction of the steady-state inactivation of DmNaV1/tipE. Similar to the mammalian channels, the activation of the insect channel remains unaltered by the toxin (Figure 2A). In addition CgNa produced a significant increase in the repriming kinetics of the insect channel when channel returned to the resting state following activation (Figure 2E). Similar to previous reports in DRG neurons, these effects are not reversible by washing the bath medium (Salceda et al., 2007).
The apparent selectivity of CgNa for the insect DmNaV1/tipE channel over mammalian NaV channels is more pronounced in terms of efficacy (∼10-fold higher amplitude) than in terms of potency (only ∼2-fold lower EC50) (see Figures 3D,E). Although CgNa exhibits a selectivity for DmNaV1/tipE, it is not specific for insect NaV channels. While a few insect-selective sea anemone toxins are known, no truly insect-specific sea anemone toxin has been reported to this date. Nv1 from Nematostella vectensis was demonstrated to exert a high selectivity for insect channels (Moran et al., 2008). Although no dose–response data are available to quantify the reported selectivity and the tested mammalian channels in this study were limited to NaV1.2, 1.4, and 1.5, Nv1 was shown to affect DmNaV1/tipE profoundly (∼100% increase in peak current) at 1 μM while only small effects (∼20% increase in peak current) were observed on the mammalian NaV subtypes at 25 μM. Similarly, Av3 from Anemonia viridis (originally known as ATX III) showed selectivity for the insect channel over NaV1.2, 1.4, 1.5, and 1.6 (Moran et al., 2007). The toxins BgII and BgIII from Bunodosoma granulifera were also shown to target insect NaV channels with a remarkably high selectivity (Bosmans et al., 2002). The EC50 values of BgII and BgIII on the most sensitive mammalian channels were ∼50-fold and ∼5-fold higher than those described on the insect DmNaV1/tipE channel, respectively. Interestingly, this difference in selectivity was ascribed to a single N16D mutation, the sole difference in their amino acid sequences. This mutation seems to cause a great decrease in potency of BgIII for the DmNaV1/tipE channel, yet it slightly increased the efficacy of the toxin on the insect channel. This suggests that the introduction of a negatively charged residue at this position might be unfavorable for binding to the receptor site, but at the same time beneficial for the activity of the toxin. The only other type I sea anemone toxins reported until today that also exhibit this N16D substitution are CgTx II and III from Bunodosoma cangicum (Zaharenko et al., 2008; Wanke et al., 2009). Although these toxins were not assayed on insect NaV channels, this substitution did not seem to affect the activity of the toxins on mammalian NaV channels to the same extent as in the case of BgIII (Salceda et al., 2002). In CgNa, position 16 is occupied by the highly conserved Asn residue, but the role of a putative “unfavorable negative charge” might be taken over by neighboring acidic side chains, as the solution structure of CgNa revealed a significantly higher percentage of exposed negatively charged residues than is typical for type I sea anemone toxins (Salceda et al., 2007). The role of such a N16D substitution in the efficacy and potency of CgNa on insect channels certainly deserves further attention in future mutagenesis studies.
Because sea anemone toxins and scorpion α-toxins were shown to induce similar electrophysiological effects on NaV channels, they were examined in binding competition assays, which concluded that these toxins share a common binding site on NaV channels (Catterall and Beress, 1978). In fact, these toxins share an overlapping but non-identical binding site, later referred to as receptor site 3 (Catterall, 1979, 2000). Several mutagenesis studies indicated that the extracellular loop between S3 and S4 of DIV represents a substantial part of this receptor site (Thomsen and Catterall, 1989; Rogers et al., 1996; Benzinger et al., 1998; Sheets et al., 1999; Ulbricht, 2005). Moreover, a recent study using chimera channels indicated that scorpion and spider toxins that slow the inactivation of NaV channels target the voltage sensor paddle motif (S3b–S4) of DIV exclusively; additional interactions with paddle motifs from other domains will alter channel activation (Bosmans et al., 2008). Upon binding to the extracellular IVS3–S4 loop, site 3 toxins are thought to prevent normal voltage sensor movement in DIV, thereby affecting the coupling of activation and inactivation, and the conformational changes associated with fast inactivation of the channel (Rogers et al., 1996). Although CgNa has not been examined in competitive binding studies, it is not unreasonable to surmise, in the light of its typical electrophysiological behavior, that this toxin slows inactivation by interacting with receptor site 3 of the NaV channel.
Several extensive mutagenesis studies identified individual amino acid residues in IVS3–S4 as important determinants for binding of site 3 toxins (see Figure 4, IVS3, residues in bold). More specifically, the most extracellularly located negatively charged residue in the IVS3 segment was pointed out as a “hot spot” residue (Rogers et al., 1996; Benzinger et al., 1998). Interestingly, this negative charge is well conserved among mammalian and insect sodium channels, only being absent in rNaV1.8 and rNaV1.9. Neutralization or reversal of this charge in hNaV1.5 (D1612R or D1612N) caused a strong decrease in affinity for sea anemone toxin ApB (Benzinger et al., 1998). In contrast, when the corresponding Glu1613 residue in rNaV1.2 was mutated into a charge-conserving Asp, the binding affinity of sea anemone toxin ATX II increased significantly (Rogers et al., 1996). Markedly, CgNa appears to have a preference for the NaV subtypes that contain an Asp residue in this position over subtypes that possess a Glu residue (see Figure 4, gray background). Only rNaV1.4 deviates from this trend, as it contains an Asp residue, but is not targeted by CgNa. On the other hand, substitution of Glu1616 (shown in bold in Figure 4, IVS3–S4) into a Gln residue resulted in a strong decrease in affinity of ATX II for the rNaV1.2 subtype (Rogers et al., 1996). Similarly, the Gln residue in this position in rNaV1.4 might therefore account for the low affinity of CgNa for this NaV subtype. This E-Q substitution is also present in the CgNa-sensitive hNaV1.5, where it might be responsible for the lower affinity of CgNa compared to rNaV1.3 and mNaV1.6. Further comparison of the CgNa-insensitive rNaV1.4 with hNaV1.5 draws the attention to the V-F substitution in the IVS3–S4 loop in hNaV1.5. The otherwise highly similar loop regions of rNaV1.4 and hNaV1.5 suggests that this residue might also be important for the subtype-selectivity of CgNa. Mutation of this Val residue in rNaV1.2 into Ala reduced affinity of ATX II (Rogers et al., 1996), but substitution into Phe seems to be beneficial for CgNa binding in NaV1.5. Interestingly, CgNa slows the inactivation of hNaV1.5 without inducing a persistent steady-state current, as observed with rNaV1.3 and mNaV1.6 (see Figure 1 and Table 1). However, a sustained Na+ current component has long been recognized to be present in the plateau phase of the action potential of cardiomyocytes and Purkinje fibers (Zaza et al., 2008) and this sustained current was shown to be increased by site 3 toxins such as ATX II (Oliveira et al., 2004). It remains to be clarified why the steady-state inactivation of hNaV1.5 is not affected by CgNa. One possible explanation could be that this is the only human clone examined in this study. On the one hand, the mammalian orthologs (i.e., encoded by homologous genes in different species) hNaV1.5 and rNaV1.5 are extremely well conserved and their IVS3–S4 sequences are completely identical. On the other hand, there are several pathogenic mutations whose effect depends on the clone used, even if the amino acid sequence is very well conserved. Overall, the observations discussed above accord quite well with the previous studies that highlight the importance of individual amino acid residues in the IVS3–S4 loop, supporting the importance of this channel region in interaction of CgNa with NaV channels.

Figure 4. Sequence alignment of IVS3–S4 from insect and mammalian NaV subtypes. Amino acid alignment of the transmembrane segments S3 and S4 from the homologous repeat DIV connected by the extracellular loop. The two sequences on top are from insect channels (MdNaV1 and DmNaV1; Md, house fly; Dm, fruit fly), the other sequences are from mammalian channel subtypes (NaV1.1–NaV1.9; r, rat; m, mouse; h, human). The NaV channel subtypes affected by CgNa are shown on a gray background, residues that differ to those in the sequence of NaV1.1–NaV1.3 are colored red. Amino acid residues discussed in the text are printed in bold. Positions of the outermost N-terminal residues in the aligned sequence of each subtype are: MdNaV1, 1668 (Uniprot accession number Q94615); DmNaV1, 1680 (P35500); rNaV1.1, 1602 (P04774); rNaV1.2, 1592 (P04775); rNaV1.3, 1538 (P08104); rNaV1.4, 1407 (P15390); hNaV1.5, 1589 (Q14524); mNaV1.6, 1581 (Q9WTU3); rNaV1.7, 1574 (O08562); rNaV1.8, 1538 (Q62968); rNaV1.9, 1407 (O88457).
An interesting observation is that, though their IVS3–S4 sequences are completely identical, the rNaV1.3 subtype is clearly modulated by CgNa while rNaV1.2 remains unaffected. Although this does not rule out the presence of substantial contacts in IVS3–S4, it indicates that other critical residues outside this loop are responsible for the ability of CgNa to discriminate so thoroughly between these two subtypes. This finding strongly supports previous suggestions that sea anemone toxins interact with a discontinuous receptor site which may include channel regions outside the IVS3–S4 loop that contribute to the heterogeneity in NaV subtype selectivity (Rogers et al., 1996; Oliveira et al., 2004; Moran et al., 2009). Unfortunately, the presently available data do not give a quantitative idea concerning the extent of involvement of the IVS3–S4 loop in receptor site 3. Further structure–function studies analyzing the sensitivity of chimera or mutant channels will be necessary to explore the complete molecular identity of this receptor site. In the light of the observed preference of CgNa for insect NaV channels, the substitution of the outermost C-terminally aromatic Phe residue in the IVS3–S4 loop into a Leu residue definitely deserves further attention in these mutagenesis studies.
Another intriguing observation is that the TTX-R peripheral nervous system subtypes rNaV1.8 (and rNaV1.9) are in general quite resistant to site 3 toxins from sea anemones (Bosmans et al., 2002; Salceda et al., 2002, 2006), scorpions (Saab et al., 2002; Maertens et al., 2006), and spiders (Nicholson et al., 2004; Yamaji et al., 2009). This could, at least in part, be due to low conservation of important amino acid residues and longer size of the IVS3–S4 loop (see Figure 4). More specifically, in the IVS3–S4 loop of NaV1.8, a motif of four consecutive amino acid residues (Ser, Leu, Glu, and Asn or “SLEN”) was found to play a role in the resistance of this channel against venom from the scorpion Leiurus quinquestriatus (Saab et al., 2002). Transfer of this SLEN-motif from the venom-resistant NaV1.8 subtype to the analogous position in the venom-sensitive NaV1.4 subtype, rendered NaV1.4 resistant to the scorpion venom. This suggests an important contribution of either the length, or the specific residues of the SLEN-motif in toxin resistance of NaV1.8. Unfortunately, these experiments were not carried out in the context of sea anemone toxins. The search for potent and selective ligands for the neuronal TTX-R NaV subtypes is of particular interest, as these channels play an important role in neuropathic and inflammatory pain (Black et al., 2004; Dib-Hajj et al., 2010). A better understanding of the molecular basis of the resistance of NaV1.8 and NaV1.9 to CgNa could therefore significantly contribute in the development of novel pharmacological agents for the treatment of pain. Although toxins that slow NaV inactivation cause an increased Na+ influx, their actions result in a depolarization of the cell. This renders a fraction of the channels unavailable for a new activation by driving them into steady-state inactivation. In this way, the impairment of channel inactivation can ultimately lead to a decrease in NaV signaling. A good example of a toxin that exerts strong analgesic effects and induces persistent currents in NaV channels is Batrachotoxin from the poison Dart frog (Bosmans et al., 2004).
In summary, our data highlight that specific insect and mammalian NaV channel subtypes can be pharmacologically distinguished by their sensitivity to CgNa, as has only been partially described for other sea anemone toxins. Together with the previously determined three-dimensional structure (Salceda et al., 2007) and the presently described subtype-selectivity, future mutagenesis studies could give us more structural information on the interaction between CgNa and specific NaV channels. Because CgNa interacts with extracellular regions of the channel and most channel regions known to contribute to inactivation are located within membrane or at the intracellular side, this structural information might yield a better insight into the coupling of activation and inactivation in NaV channels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to A. L. Goldin (University of California, Irvine, USA) for sharing rNaV1.2, rNaV1.3, and mNaV1.6; G. Mandel (Stony Brook University, Stony Brook, USA) for sharing rNaV1.4; R. G. Kallen (University of Pennsylvania, Philadelphia, USA) for sharing hNaV1.5; P. Dietrich (Roche, Palo Alto, USA) for sharing rNaV1.7; J. N. Wood (University College, London, UK) for sharing rNaV1.8; S. H. Heinemann (Friedrich-Schiller-Universität, Jena, Germany) for sharing rβ1; S. C. Cannon (University of Texas, Southwestern Medical Center, Dallas, USA) for sharing hβ1; and M. S. Williamson (IACR Rothamsted, Harpenden, UK) for sharing DmNaV1 and tipE. This work was supported by grants G.0330.06 and G.0257.08 (F.W.O. Vlaanderen), OT-05–64 (K.U. Leuven) and UA P6/31 (Interuniversity Attraction Poles Program, Belgian State, Belgian Science Policy). The authors also thank Steve Peigneur, Marijke Stevens, and Annelies Van Der Haegen for their work in molecular biology and constructive discussions and Prof. Dr. Wolf-Georg Forssmann for supporting this work.
References
Benzinger, G. R., Kyle, J. W., Blumenthal, K. M., and Hanck, D. A. (1998). A specific interaction between the cardiac sodium channel and site-3 toxin anthopleurin B. J. Biol. Chem. 273, 80–84.
Black, J. A., Dib-Hajj, S., McNabola, K., Jeste, S., Rizzo, M. A., Kocsis, J. D., and Waxman, S. G. (1996). Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Brain Res. Mol. Brain Res. 43, 117–131.
Black, J. A., Liu, S., Tanaka, M., Cummins, T. R., and Waxman, S. G. (2004). Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108, 237–247.
Bosmans, F., Aneiros, A., and Tytgat, J. (2002). The sea anemone Bunodosoma granulifera contains surprisingly efficacious and potent insect-selective toxins. FEBS Lett. 532, 131–134.
Bosmans, F., Maertens, C., Verdonck, F., and Tytgat, J. (2004). The poison Dart frog’s batrachotoxin modulates Nav1.8. FEBS Lett. 577, 245–248.
Bosmans, F., Martin-Eauclaire, M. F., and Swartz, K. J. (2008). Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456, 202–208.
Cannon, S. C. (2010). Voltage-sensor mutations in channelopathies of skeletal muscle. J. Physiol. 588, 1887–1895.
Catterall, W. A. (1979). Neurotoxins as allosteric modifiers of voltage-sensitive sodium channels. Adv. Cytopharmacol. 3, 305–316.
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25.
Catterall, W. A., and Beress, L. (1978). Sea anemone toxin and scorpion toxin share a common receptor site associated with the action potential sodium ionophore. J. Biol. Chem. 253, 7393–7396.
Catterall, W. A., Dib-Hajj, S., Meisler, M. H., and Pietrobon, D. (2008). Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J. Neurosci. 28, 11768–11777.
Chahine, M., George, A. L. Jr., Zhou, M., Ji, S., Sun, W., Barchi, R. L., and Horn, R. (1994). Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron 12, 281–294.
Chen, L. Q., Santarelli, V., Horn, R., and Kallen, R. G. (1996). A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J. Gen. Physiol. 108, 549–556.
Dib-Hajj, S. D., Cummins, T. R., Black, J. A., and Waxman, S. G. (2010). Sodium channels in normal and pathological pain. Annu. Rev. Neurosci. 33, 325–347.
Dib-Hajj, S. D., Tyrrell, L., Black, J. A., and Waxman, S. G. (1998). NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc. Natl. Acad. Sci. U.S.A. 95, 8963–8968.
Goldin, A. L. (1999). Diversity of mammalian voltage-gated sodium channels. Ann. N.Y. Acad. Sci. 868, 38–50.
King, G. F., Escoubas, P., and Nicholson, G. M. (2008). Peptide toxins that selectively target insect NaV and CaV channels. Channels (Austin) 2, 100–116.
Kontis, K. J., and Goldin, A. L. (1997). Sodium channel inactivation is altered by substitution of voltage sensor positive charges. J. Gen. Physiol. 110, 403–413.
Kontis, K. J., Rounaghi, A., and Goldin, A. L. (1997). Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. J. Gen. Physiol. 110, 391–401.
Liman, E. R., Tytgat, J., and Hess, P. (1992). Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 9, 861–871.
Loughney, K., Kreber, R., and Ganetzky, B. (1989). Molecular analysis of the para locus, a sodium channel gene in Drosophila. Cell 58, 1143–1154.
Maertens, C., Cuypers, E., Amininasab, M., Jalali, A., Vatanpour, H., and Tytgat, J. (2006). Potent modulation of the voltage-gated sodium channel Nav1.7 by OD1, a toxin from the scorpion Odonthobuthus doriae. Mol. Pharmacol. 70, 405–414.
Moran, Y., Gordon, D., and Gurevitz, M. (2009). Sea anemone toxins affecting voltage-gated sodium channels – molecular and evolutionary features. Toxicon 54, 1089–1101.
Moran, Y., Kahn, R., Cohen, L., Gur, M., Karbat, I., Gordon, D., and Gurevitz, M. (2007). Molecular analysis of the sea anemone toxin Av3 reveals selectivity to insects and demonstrates the heterogeneity of receptor site-3 on voltage-gated Na+ channels. Biochem. J. 406, 41–48.
Moran, Y., Weinberger, H., Reitzel, A. M., Sullivan, J. C., Kahn, R., Gordon, D., Finnerty, J. R., and Gurevitz, M. (2008). Intron retention as a posttranscriptional regulatory mechanism of neurotoxin expression at early life stages of the starlet anemone Nematostella vectensis. J. Mol. Biol. 380, 437–443.
Nicholson, G. M., Little, M. J., and Birinyi-Strachan, L. C. (2004). Structure and function of delta-atracotoxins: lethal neurotoxins targeting the voltage-gated sodium channel. Toxicon 43, 587–599.
Oliveira, J. S., Redaelli, E., Zaharenko, A. J., Cassulini, R. R., Konno, K., Pimenta, D. C., Freitas, J. C., Clare, J. J., and Wanke, E. (2004). Binding specificity of sea anemone toxins to NaV1.1-1.6 sodium channels: unexpected contributions from differences in the IV/S3-S4 outer loop. J. Biol. Chem. 279, 33323–33335.
Rogers, J. C., Qu, Y., Tanada, T. N., Scheuer, T., and Catterall, W. A. (1996). Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 271, 15950–15962.
Roy, M. L., and Narahashi, T. (1992). Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J. Neurosci. 12, 2104–2111.
Saab, C. Y., Cummins, T. R., Dib-Hajj, S. D., and Waxman, S. G. (2002). Molecular determinant of NaV1.8 sodium channel resistance to the venom from the scorpion Leiurus quinquestriatus hebraeus. Neurosci. Lett. 331, 79–82.
Salceda, E., Garateix, A., Aneiros, A., Salazar, H., Lopez, O., and Soto, E. (2006). Effects of ApC, a sea anemone toxin, on sodium currents of mammalian neurons. Brain Res. 1110, 136–143.
Salceda, E., Garateix, A., and Soto, E. (2002). The sea anemone toxins BgII and BgIII prolong the inactivation time course of the tetrodotoxin-sensitive sodium current in rat dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 303, 1067–1074.
Salceda, E., Perez-Castells, J., Lopez-Mendez, B., Garateix, A., Salazar, H., Lopez, O., Aneiros, A., Standker, L., Beress, L., Forssmann, W. G., Soto, E., Jimenez-Barbero, J., and Gimenez-Gallego, G. (2007). CgNa, a type I toxin from the giant Caribbean sea anemone Condylactis gigantea shows structural similarities to both type I and II toxins, as well as distinctive structural and functional properties. Biochem. J. 406, 67–76.
Sheets, M. F., Kyle, J. W., Kallen, R. G., and Hanck, D. A. (1999). The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys. J. 77, 747–757.
Song, W., Liu, Z., Tan, J., Nomura, Y., and Dong, K. (2004). RNA editing generates tissue-specific sodium channels with distinct gating properties. J. Biol. Chem. 279, 32554–32561.
Standker, L., Beress, L., Garateix, A., Christ, T., Ravens, U., Salceda, E., Soto, E., John, H., Forssmann, W. G., and Aneiros, A. (2006). A new toxin from the sea anemone Condylactis gigantea with effect on sodium channel inactivation. Toxicon 48, 211–220.
Stuhmer, W., Conti, F., Suzuki, H., Wang, X. D., Noda, M., Yahagi, N., Kubo, H., and Numa, S. (1989). Structural parts involved in activation and inactivation of the sodium channel. Nature 339, 597–603.
Tan, J., Liu, Z., Nomura, Y., Goldin, A. L., and Dong, K. (2002). Alternative splicing of an insect sodium channel gene generates pharmacologically distinct sodium channels. J. Neurosci. 22, 5300–5309.
Thomsen, W. J., and Catterall, W. A. (1989). Localization of the receptor site for alpha-scorpion toxins by antibody mapping: implications for sodium channel topology. Proc. Natl. Acad. Sci. U.S.A. 86, 10161–10165.
Ulbricht, W. (2005). Sodium channel inactivation: molecular determinants and modulation. Physiol. Rev. 85, 1271–1301.
Wanke, E., Zaharenko, A. J., Redaelli, E., and Schiavon, E. (2009). Actions of sea anemone type 1 neurotoxins on voltage-gated sodium channel isoforms. Toxicon 54, 1102–1111.
West, J. W., Patton, D. E., Scheuer, T., Wang, Y., Goldin, A. L., and Catterall, W. A. (1992). A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc. Natl. Acad. Sci. U.S.A. 89, 10910–10914.
Yamaji, N., Little, M. J., Nishio, H., Billen, B., Villegas, E., Nishiuchi, Y., Tytgat, J., Nicholson, G. M., and Corzo, G. (2009). Synthesis, solution structure and phyla-selectivity of a spider {delta}-toxin that slows inactivation of specific voltage-gated sodium channel subtypes. J. Biol. Chem. 284, 24568–24582.
Zaharenko, A. J., Ferreira, W. A. Jr., de Oliveira, J. S., Konno, K., Richardson, M., Schiavon, E., Wanke, E., and de Freitas, J. C. (2008). Revisiting cangitoxin, a sea anemone peptide: purification and characterization of cangitoxins II and III from the venom of Bunodosoma cangicum. Toxicon 51, 1303–1307.
Keywords: sea anemone, toxin, inactivation, sodium channel, subtype, selectivity
Citation: Billen B, Debaveye S, Béress L, Garateix A and Tytgat J (2010) Phyla- and subtype-selectivity of CgNa, a Na+ channel toxin from the venom of the giant Caribbean sea anemone Condylactis gigantea. Front. Pharmacol. 1:133. doi: 10.3389/fphar.2010.00133
Received: 31 August 2010;
Accepted: 22 October 2010;
Published online: 23 November 2010.
Edited by:
Hugues Abriel, University of Bern, SwitzerlandReviewed by:
Stephan Kellenberger, University of Lausanne, SwitzerlandOscar Moran, National Research Council, Italy
Massimo Mantegazza, University of Nice Sophia Antipolis, France
Copyright: © 2010 Billen, Debaveye, Béress, Garateix and Tytgat. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Jan Tytgat, Laboratory of Toxicology, University of Leuven, Campus Gasthuisberg O&N2, Herestraat 49, P. O. Box 922, 3000 Leuven, Belgium. e-mail:amFuLnR5dGdhdEBwaGFybS5rdWxldXZlbi5iZQ==