


- 1 School of Pharmaceutical Sciences, University of Geneva, University of Lausanne, Geneva, Switzerland
- 2 Department of Clinical Research, University of Bern, Bern, Switzerland
A growing number of drugs have been shown to prolong cardiac repolarization, predisposing individuals to life-threatening ventricular arrhythmias known as Torsades de Pointes. Most of these drugs are known to interfere with the human ether à-gogo related gene 1 (hERG1) channel, whose current is one of the main determinants of action potential duration. Prolonged repolarization is reflected by lengthening of the QT interval of the electrocardiogram, as seen in the suitably named drug-induced long QT syndrome. Chirality (presence of an asymmetric atom) is a common feature of marketed drugs, which can therefore exist in at least two enantiomers with distinct three-dimensional structures and possibly distinct biological fates. Both the pharmacokinetic and pharmacodynamic properties can differ between enantiomers, as well as also between individuals who take the drug due to metabolic polymorphisms. Despite the large number of reports about drugs reducing the hERG1 current, potential stereoselective contributions have only been scarcely investigated. In this review, we present a non-exhaustive list of clinically important molecules which display chiral toxicity that may be related to hERG1-blocking properties. We particularly focus on methadone cardiotoxicity, which illustrates the importance of the stereoselective effect of drug chirality as well as individual variations resulting from pharmacogenetics. Furthermore, it seems likely that, during drug development, consideration of chirality in lead optimization and systematic assessment of the hERG1 current block with all enantiomers could contribute to the reduction of the risk of drug-induced LQTS.
Introduction
In cardiac cells, the action potential (AP) is elicited by the orchestrated activity of several types of ion channels. Among them, the voltage-gated potassium channel is crucial for normal human cardiac electrical activity. The human ether à-gogo related gene (hERG1) channel, responsible for the rapid component of the delayed rectifier potassium current (IKr), is one of the main determinants of action potential duration (APD). Functional hERG1 channels are formed by the assembly of four α-subunits encoded by the KCNH2 gene. Each α-subunit of hERG1 consists of six transmembrane domains (S1–S6) and intracellular amino- and carboxy-termini. The voltage sensor resides in the first four transmembrane domains, while the S5 and S6 helices, the selectivity filter, and the pore-helix compose the pore-forming units (Figure 1).
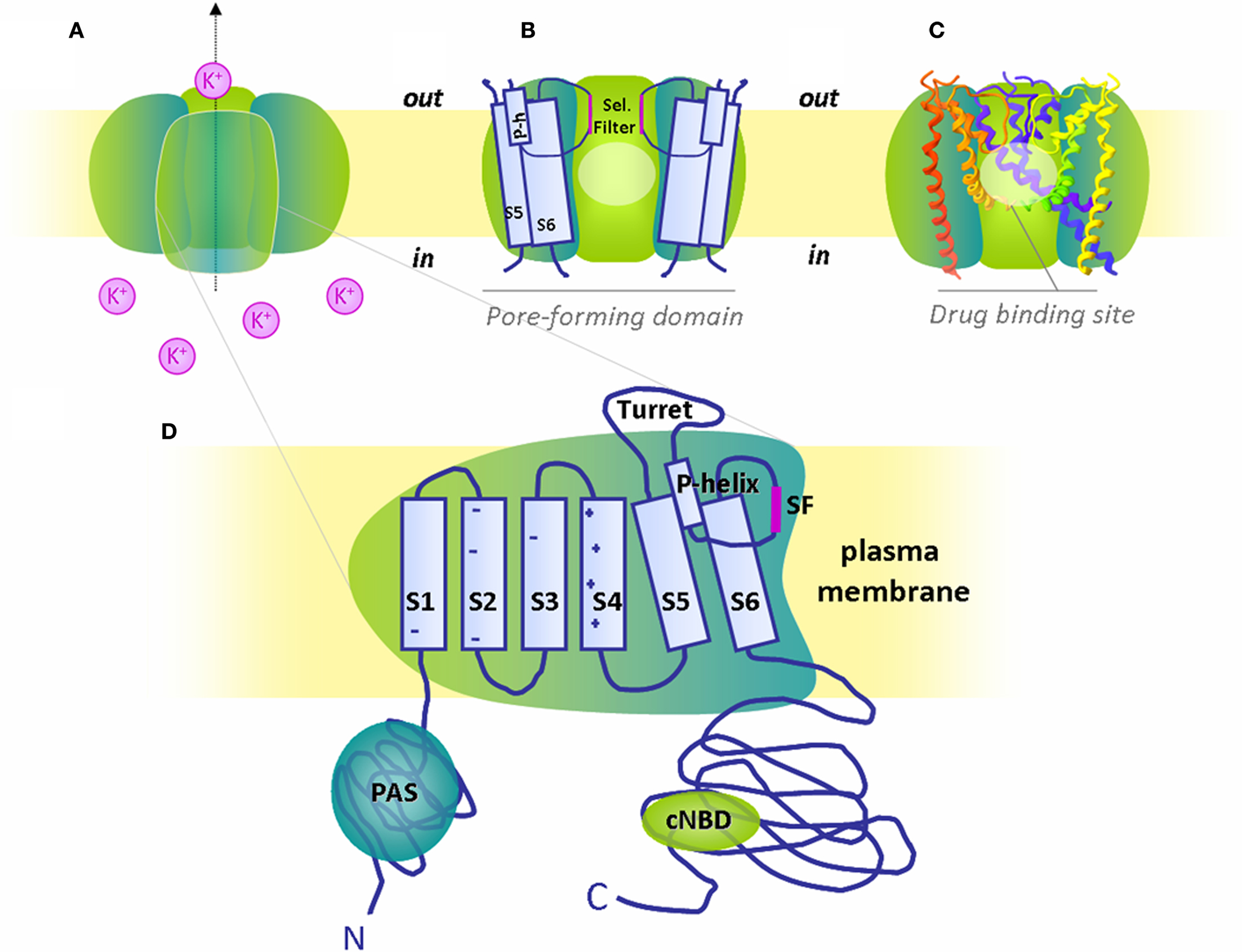
Figure 1. The hERG1 channel structure and putative binding region for blocking drugs. (A) Schematized hERG1 tetrameric channel at the plasma membrane and (B) pore-forming region in schematic and (C) ribbon representation. The pale circles locate the general drug binding site in the cavity created by the pore-forming domain, i.e., transmembrane domains (S5, S6), pore-helix (P-h) and selectivity filter (Sel. Filter) of the four α-subunits. For clarity, only three subunits are depicted. (D) Magnification of one α-subunit with six transmembrane domains (S1-S6), S5-P linker or turret loop, pore-helix (P-helix) and selectivity filter (SF). Both N- and C-termini are intracellular with respectively identified Per-Arnt-Sim (PAS) domain and cyclic nucelotide binding domain (cNBD).
Many loss-of-function mutations in the KCNH2 gene can prolong APD, reflected by an increase of the QT interval on the electrocardiogram (ECG), due to the reduction of repolarizing currents as presented in Figure 2. Lengthening of the QT interval when corrected to the heart rate (QTc) is a well-known risk factor for arrhythmias that can cause fainting and/or sudden death, named Torsades de Pointes (TdP) (Figure 2). Such genetic disorders are referred to as congenital long QT syndromes (LQTS). Another hERG1-related LQTS has been shown to be drug-induced (Abriel et al., 2004). A variety of medications have the undesired ability to trigger life-threatening arrhythmias and sudden cardiac death due to their inhibition of the hERG1 channel. This adverse effect raises important concern in the pharmaceutical area (Brown, 2004). Since structures and therapeutic classes involved are so diverse, hERG1 liability must be assessed for all new drugs, sometimes leading to considerable economic loss if, after years of development, the drug reveals to block this potassium channel.
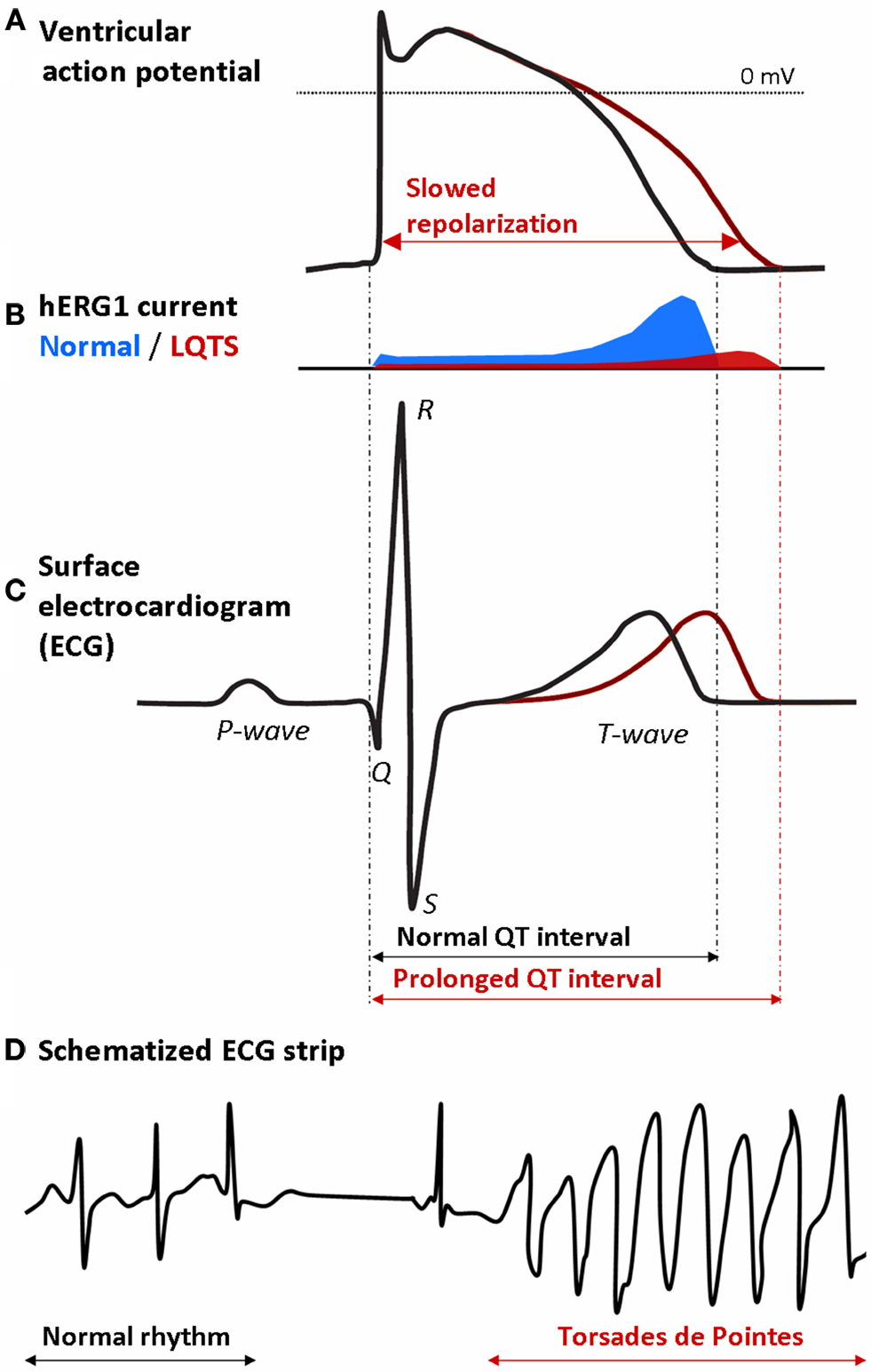
Figure 2. Ventricular action potentials and related ECG signals: prolonged QT interval is a risk factor for Torsades de Pointes. (A) Normal and prolonged ventricular action potential due to (B) reduction of hERG1 currents in LQTS are reflected (C) on the ECG recording with lengthening of the QT interval. (D) Schematized ECG recording presenting the onset of Torsades de Pointes in a patient with long QT syndrome.
Recently, stereoselectivity in both pharmacokinetics (PK) and pharmacodynamics (PD) disclosed the chiral toxicity of methadone on the hERG1 channel (Eap et al., 2007). The main goal of this review is to summarize the most relevant literature regarding potential stereoselective inhibition of the cardiac hERG1 channel and to discuss the advantages of focusing on single enantiomers for the development of drugs devoid of hERG1-blocking properties.
Drug Block of the hERG1 Channel
The first described case of drug-induced LQTS was attributable to quinidine, an antiarrhythmic class Ia drug intended to act on Na+ channels (Roden et al., 1986). Syncopal episodes following initiation of quinidine therapy were recognized shortly after the introduction of the drug in the 1920s, well before the description of LQTS and the understanding of the molecular mechanisms (reviewed in Roden, 2006). Later in the development of antiarrhythmic therapy, molecules were specifically designed to block K+ currents (class III antiarrhythmic agents), such as dofetilide and ibutilide. Unfortunately, such drugs predictably prolong the QTc interval, resulting in potentially lethal ventricular arrhythmias in 5–7% of recipients (Marban, 2002). These drugs, along with other molecules of the methanesulfonanilide group, have been shown to block the hERG1 channel (Spector et al., 1996; Yang et al., 1997). It is now recognized that many structurally unrelated drugs, designed to act on non-cardiac targets, unintentionally block the repolarizing current IKr. Although disruption of hERG1 protein trafficking has also been reported, blockage of the hERG1 current or IhERG1 accounts for more than 95% of drug-induced delayed repolarization.
In the past 15 years, the most common reason for withdrawal or restriction of marketed drugs has been the prolongation of the QT interval associated with polymorphic ventricular tachycardia or TdP (Roden, 2004). The rare incidence of these life-threatening arrhythmias explains why they are not readily observed during clinical trials or post-marketing surveillance. Many drugs have been removed from the market after years of use or had their availability severely restricted because of this rare and potentially fatal toxicity. Although not an exhaustive list of hERG1 channel blockers, some examples include the antimuscarinic drug terodiline (withdrawal in 1991), the antianginal agent mibefradil (1998), the anti-histaminics terfenadine and astemizole (withdrawal in 1998 and 1999, respectively), the antibiotic grepafloxacin (1999), the gastrointestinal prokinetic cisapride (2000), the analgesic levo-α-acetylmethadol (2003), and the neuroleptics sertindole (since 1998) and thioridazine (2005). A growing number of non-class III antiarrhythmic agents have been shown to prolong cardiac repolarization, predisposing an individual to sudden cardiac death. Drugs proven to lengthen the QT interval and/or drugs associated with TdP were estimated to comprise ∼2–3% of all drug prescriptions (De Ponti et al., 2000). Many of these drugs remain on the market1 because their benefits are still considered to outweigh their risks. Regulatory authorities now demand systematic assessment of the torsadogenic risk (ICH S7B, 2005) of any newly developed drug.
Molecular Determinants of the hERG1 Channel Block
Their are a wide variety of drugs of different classes and structures that can inhibit the hERG1 channel. The first insights into the reasons for this widespread block came about a decade ago. Based upon the assumption that the block of hERG1 occurred in the cavity of the channel (Figure 1), similar to the Na+ and Ca2+ channels, Mitcheson et al. (2000a) used alanine-scanning mutagenesis of the hERG1 pore-helix and S6 domain to determine the binding sites for MK-499, a potent methanesulfonanilide blocker of the K+ channel. Channels with alanine mutations in the S6 domain (G648A, Y652A, and F656A) were minimally inhibited by MK-499 as compared to wild-type channels (Mitcheson et al., 2000a). Based on a homology model of the hERG1 channel, these aromatic residues were predicted to line the pore cavity. In addition, three channels with a mutation located in the base of the pore-helix (T623A, S624A, and V625A) and one located in the S6 domain (V659A) were also less sensitive to MK-499 inhibition (Mitcheson et al., 2000a). Similar reductions in block potency were obtained with Y652A and F656A mutants for terfenadine and cisapride, as well as many other compounds as reviewed in Stansfeld et al. (2007). Mitcheson’s group provided further evidence to the intra-cavity block of the hERG1 channel by rather large molecules, such as methanesulfonanilide drugs. Using the hERG1-D540K mutant that has the unusual property of opening in response to hyperpolarization, they confirmed that MK-499 blocked the hERG1 channel in the inner vestibule, as MK-499 was trapped by closure of the activation gate (Mitcheson et al., 2000b). The ability of HERG1 (wild-type and D540K) channels to trap MK-499 suggests that its vestibule is larger than other well studied Kv1 (Shaker family) channels. Unlike other voltage-gated K+ channels (e.g., Kv1–Kv4), hERG1 lacks the Pro-x-Pro (x for any amino acid) sequence in the S6 domain, which is thought to be responsible for the sharp bend in the S6 helices, thereby reducing the volume of the channel cavity (Mitcheson et al., 2000a; Labro et al., 2003). Altogether, these results show that the residues lining the pore (especially Tyr652 and Phe656) are major structural determinants for hERG1 current block.
Chiral Drugs and Stereoselectivity
A chiral molecule – from the Greek word χείρ for hand – is one whose structure lacks an internal plane of symmetry and has a non-superimposable mirror image (Figure 3). Although chiral molecules share the same molecular formula, molecular weight, atom-to-atom linkages, and bonding distances, their three-dimensional arrangements differ. Chirality most often results from the presence of an asymmetrical atom, usually a carbon atom with four different substituents. A chiral molecule and its mirror image are called enantiomers, and different methods are used to distinguish them. Chiral nomenclature can either describe how each enantiomer affects the rotation of polarized light or its absolute chemical configuration, as presented in Figure 3. An equimolar mixture of a pair of enantiomers is known as racemate and does not have any optical activity (reviewed in Smith, 2009). When a compound has multiple chiral centers, two molecules that are not perfect mirror images of each other are named diastereomers.
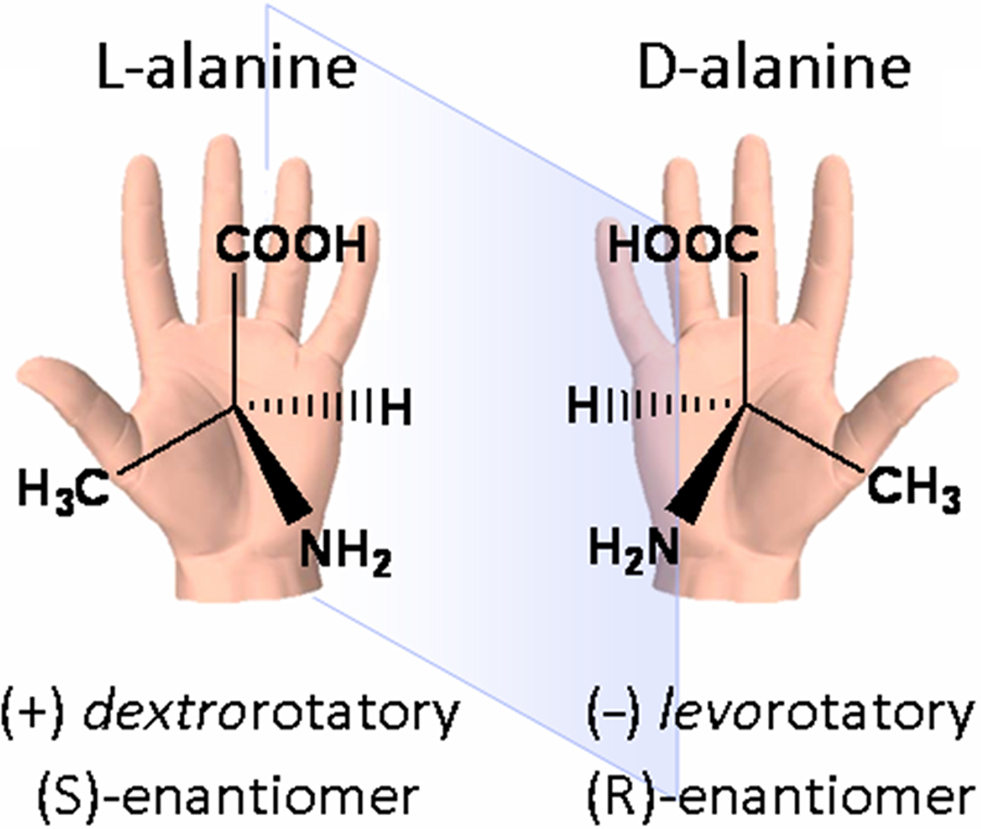
Figure 3. A chiral molecule and its different names: the amino acid alanine. The left-handed molecule (L-alanine or (S)-alanine) is presented with its mirror image, the right-handed molecule (D-alanine or (R)-alanine). Note that the optical activity (dextro- or levorotatory) cannot be deduced from the structure.
Except for their ability to rotate polarized light in opposite directions, enantiomers have identical chemical and physical properties. However, in the presence of other chiral molecules or environments, one enantiomer can behave differently from another. The enantiomers of a compound may taste and smell differently, or they may exert different activities when used as therapeutics. These effects reflect the chirality inherent in biological systems, as the main “building blocks” of life (i.e., amino acids and sugars) are chiral. Chirality can introduce marked selectivity, and often specificity, in drug action (Tucker, 2000). Consequently, stereoselectivity is the preferential interaction of one enantiomer molecule with a biological target. Since many commonly used drugs are racemates, differentiation of the relative contribution of each enantiomer to the overall drug action, including stereoselective PK and PD, is essential. While one enantiomer is likely responsible for the activity of interest, its paired enantiomer could be partially or equally active, inactive or may have an antagonist effect. One enantiomer may also have a separate activity that could be desirable or not (Caner et al., 2004).
Pharmacogenetics and Stereoselective Metabolism
A number of genetic variants, either “silent” mutations or polymorphisms, have been found to be associated with an increased vulnerability for drug-induced LQTS (e.g., Yang et al., 2002; Paavonen et al., 2003; Paulussen et al., 2004; Sun et al., 2004). There is also evidence that differences in the pharmacokinetic properties of drugs may be risk factors for drug-induced QT interval prolongation. Pharmacogenetic studies assess the genetic causes of inter-individual differences in drug response. It is well known that genetic variability of drug metabolism can result in functional polymorphisms of metabolizing enzyme genes, and among them cytochromes P450 (CYP) isoenzymes – essential for the catalysis of oxidation of endogenous molecules or xenobiotics during phase I – have been extensively studied. Many different human CYP genes exist, the most important being CYP2C9, CYP2D6, and CYP3D4, since their gene products account for 60–70% of the phase I metabolism of clinically important drugs (Ingelman-Sundberg, 2001). Mutations in the CYP genes can generate enzymes with abolished, reduced, altered, or increased activity. The phenotype of a CYP enzyme can be tested by administering a specific substrate, e.g., debrisoquine for CYP2D6, and measuring the metabolite-to-parent drug ratio in the urine. Individuals with a low ratio have reduced enzyme activity and are termed slow metabolizers (SMs) in order to differentiate them from extensive metabolizers (EMs). Perhexiline and thioridazine are examples of drugs that depend on CYP2D6 activity and both act on the hERG1 channel (reviewed in Roden and Viswanathan, 2005). Walker et al. (1999) studied the block of IhERG1 by perhexiline, an antianginal agent known to cause QT interval prolongation and TdP. Interestingly, half-maximal inhibitory concentrations (IC50) were above the clinical plasma concentrations usually observed. Nevertheless, since 8–10% of Caucasians and Black individuals are CYP2D6 SMs, the authors proposed that these patients may be more likely to have high plasma concentrations of perhexiline with a consequent increased susceptibility to adverse effects (Walker et al., 1999). Some drugs are able to convert extensive metabolizers into slow metabolizers by interfering with the cytochrome pathway, and may cause drug–drug interactions. The gastrointestinal prokinetic agent cisapride is a good example of a high-affinity blocker of the hERG1 channel being substrate of the CYP3A4. Prolongation of the QT interval along with severe cardiac outcomes have been reported after concomitant administration of cisapride with macrolide antibiotics, such as erythromycin (Tierney et al., 1997), or with azole antifungal agents like ketoconazole (Michalets and Williams, 2000). These drugs alone may prolong the QT interval by blocking IhERG1, but they are mainly responsible for the drastic increase in the plasma concentration of cisapride due to CYP3A4 inhibition.
Drug-metabolizing enzymes are constituted of chiral entities, thus their tertiary structure creates unique three-dimensional binding and/or catalytic domains that enable the identification of one enantiomer from another (Smith, 2009). Stereoselective metabolism has long been recognized as a source of variable disposition between enantiomers. Fluoxetine is an antidepressant of the selective serotonin reuptake inhibitor class (Figure 4). In a study by Margolis et al. (2000), N-demethylation of this molecule was assessed by heterogeneously expressed human cytochrome P450 enzymes. Their results suggested that CYP2D6, CYP2C9, and CYP3A4 greatly contribute to the hepatic metabolism of this chiral antidepressant drug. In a contemporaneous study, stereoselective biotransformation of racemic fluoxetine was assessed in CYP2D6 sparteine slow and extensive metabolizers (Fjordside et al., 1999). In SMs, both oral clearance (3 versus 17 L/h) and half-life (9.5 versus 6.1 days) revealed a significantly slower metabolism of (R)-fluoxetine compared to the (S)-form, respectively; while no difference between enantiomers was observed in EMs (oral clearance: 36 and 40 L/h, half-life: 2.1 and 1.1 days). Because (R)-fluoxetine has a shorter half-life – it yields an inactive metabolite, whereas (S)-norfluoxetine is as active as (R)- or (S)-fluoxetine in selective serotonin reuptake inhibition – the clinical availability of this enantiomer could prove beneficial in elderly individuals. Results from early clinical trials, however, raised concerns over its potential to prolong the QTc interval at the highest doses administered. This unexpected finding led to the termination of the clinical development of this enantiomer (reviewed in Shah, 2002). Interestingly, fluoxetine and its major metabolite norfluoxetine, have been shown to inhibit the hERG1 current in vitro (Thomas et al., 2002; Rajamani et al., 2006), and the estimated IC50s of direct hERG1 current inhibition in mammalian cells are in the range of plasma concentrations reached in humans (reviewed in Thomas et al., 2002). Unfortunately, none of these studies assessed a potential stereoselective effect of fluoxetine enantiomers. It remains unknown whether the CYP2D6 SM status and consequent reduced biotransformation of (R)-fluoxetine played a role in the significantly longer QTc intervals measured in the clinical trial. Nevertheless, since this antidepressant is widely prescribed and the frequency of the sparteine SM phenotype ranges from 2.3 to 9% according to populations (Eichelbaum et al., 1986; Niewinski et al., 2002), it would be instructive to know if this metabolic condition is an additional risk factor for polymorphic arrhythmias for individuals taking fluoxetine.
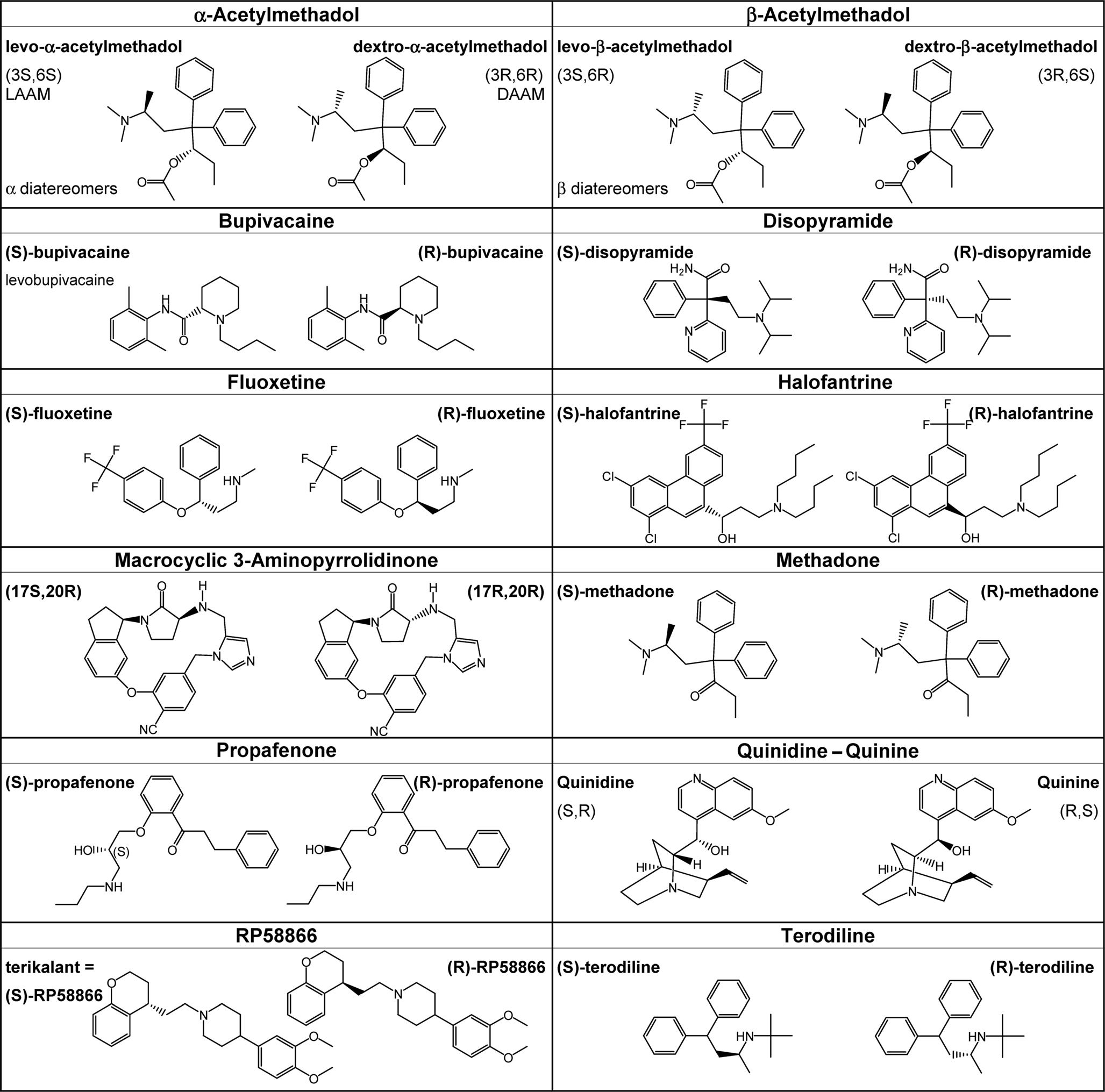
Figure 4. Chemical structures of selected chiral compounds blocking the hERG1 channel.
Halofantrine is an antimalarial drug effective for the treatment of uncomplicated chloroquine-resistant Plasmodium falciparum. Regrettably, this drug has been associated with QT interval prolongation and arrhythmias in patients without any known underlying cardiac abnormalities (Mbai et al., 2002). Halofantrine enantiomers (Figure 4) extensively accumulate upon daily administration and have high intersubject PK variability. In the clinical study of Abernethy et al. (2001), the steady-state plasma concentration of (R)-halofantrine exceeded the concentration of (S)-halofantrine by approximately two-fold. The correlation between the plasma concentration and QT interval appeared to be stronger for the (R)- versus the (S)-halofantrine in all but one patient, which may be consistent with the increased cardiotoxic potential of the (R)-isomer (Abernethy et al., 2001). Abernethy et al. (2001) did, however, observe a high variability in the plasma concentrations of halofantrine and QTc intervals, but they did not search for any ethnical correlations (38% Caucasians versus 62% African-Americans) in their cohort of essentially young males. Inter-ethnical variations in genes involved in the biotransformation of halofantrine into its metabolite may, in part, account for these variations. In addition to the PK difference, halofantrine produced a stereoselective block of the delayed rectifier potassium current in isolated feline myocytes, with the (R)-enantiomer being more potent (Wesche et al., 2000). Mbai et al. (2002) confirmed that halofantrine and its major liver metabolite block the hERG1 channel (IC50s at the nM range), but the authors did not separately assess the enantiomers. Again, further investigation of the pharmacokinetics and cardiac toxicity of each chiral entity would be valuable to ensure safer drug treatment.
Stereoselectivity and ION Channels
Membrane receptors, such as β-adrenergic receptors, often display marked stereoselectivity in drug pharmacodynamics, in either agonist or antagonist activity (Popp et al., 2004). Different stereoselective blockers have also been identified for ion channels. The levorotatory chromanol 293B was demonstrated to be nearly seven-fold more potent than its dextro-form in the block of the cardiac KCNQ1/KCNE1 current (Yang et al., 2000). Verapamil was the first L-type calcium channel blocker available for the treatment of angina pectoris, hypertension and supraventricular arrhythmias (Sandler et al., 1968), and its racemic mixture is still widely used in the management of these conditions. Although the two enantiomers of verapamil possess similar spectra of pharmacological effects, they differ in their pharmacokinetic and pharmacodynamic properties. (S)-verapamil is more potent than the (R)-form in the block of the L-type Ca2+ current, but it is also preferentially metabolized during hepatic first-passage and has a greater volume of distribution (Busse et al., 2006). Dihydropyridine molecules, another class of calcium channel modulators, also present marked stereoselectivity. Whereas the enantiomer (S)-Bay K 8644 is a potent activator of L-type Ca2+ channels, the (R)-Bay K 8644 acts as an antagonist (Triggle, 1996). Moreover, the stereoselective effect may be state-dependent, which is a feature unique to ion channels. This phenomenon is clearly seen with (S)-Bay K 8644, which has activating properties at polarized membrane potentials and antagonistic properties at depolarized potentials (reviewed in Triggle, 1996).
Bupivacaine (Figure 4), a local anesthetic drug belonging to the amino amide group, also displays stereoselective effects on its main targets, the voltage-gated sodium channels (Nav). Although some studies demonstrated that the potency and duration of local anesthesia in vivo were the same for (S)- and (R)-bupivacaine (Kanai et al., 1999), in vitro observations indicated that the (R)-enantiomer more potently blocks neuronal sodium channels (Lee-Son et al., 1992). Accidental intravascular injection or the use of high concentrations of local anesthetics can produce profound systemic effects, especially in the central nervous and cardiovascular systems. Valenzuela and co-workers studied the potential block of cardiac channels and the putative differences between enantiomers. Both cardiac Nav1.5 and Kv1.5 were inhibited by bupivacaine, with the (R)-form being in both cases a more potent blocker (Valenzuela et al., 1995a,b). Altogether, these results spoke in favor of the use of (S)-bupivacaine, commonly known as levobupivacaine, for an increased cardiovascular safety.
Stereoselective Effects on Cardiac Repolarization
Although many reports of drugs blocking the hERG1 current are found in the literature (e.g., Snyders et al., 1991; Roy et al., 1996; Snyders and Chaudhary, 1996; Mohammad et al., 1997; Monserrat and Bianchini, 2001; Zhang et al., 2001; Thomas et al., 2003; Perry et al., 2004), the potential stereoselective contribution of the distinct enantiomers has only been scarcely investigated. Among the few publications existing, it is worth mentioning the work of Yang et al. (2000) in which enantiomers of chromanol 293B showed no differences on the hERG1 current, but demonstrated a seven-fold stereoselectivity on the KCNQ1/KCNE1 current block. Likewise, verapamil enantiomers that blocked the L-type Ca2+ channels also blocked the K+ hERG1 channels, but they had equal potencies of inhibition when tested in Xenopus oocytes (Waldegger et al., 1999). The first demonstration of the stereoselective block of hERG1 was by Gonzalez et al. (2002). The authors, using a heterologous expression system, revealed that (S)-bupivacaine was more potent at blocking the IhERG1 than (R)-bupivacaine, which is remarkably the inverse stereoselectivity to that observed in other cardiac channels (Gonzalez et al., 2002). The same observation was made for ropivacaine, a structurally related anesthetic differing only in the size of the N-substituent (Siebrands et al., 2005).
Another historical example is quinidine and its dextrorotatory diastereomer quinine (Figure 4). As previously mentioned, quinidine has been mainly used as a class I antiarrhythmic drug, even though both diastereomers can block the cardiac Nav1.5 (reviewed in White, 2007). Despite quinidine has a greater anti-plasmodium activity, quinine is still preferentially used to treat malaria, because, in contrast to quinidine, no significant cardiotoxicity has been reported in large prospective studies of quinine in uncomplicated and severe malaria (reviewed in White, 2007). In a study by Sanchez-Chapula et al. (2003), both diastereomers where tested for their ability to block hERG1 channels expressed in Xenopus oocytes. Results showed a ∼12-fold difference in the IC50 of block and mutagenesis experiments also unveiled the intracellular and open-state block of the channel by the two diastereomers. In addition to the stereoselective effect on the K+ channel, pharmacokinetic differences were observed (in terminal half-life, volume of distribution at steady-state and plasma protein binding) resulting in plasma concentrations of unbound quinidine being twice that of quinine in a series of subjects receiving the drug intravenously (Karbwang et al., 1993). In the same study by Karbwang et al. (1993), although both alkaloids prolonged the QTc interval compared to baseline, the maximum change after quinine infusion was approximately half that after quinidine. According to +White’s review (2007), the large difference in hERG1 channel inhibition may explain why quinine has very little effect on ventricular repolarization in vivo. In this case, the stereoselective pharmacodynamic effects, associated with different therapeutic concentrations, determined the clinical indications of these two stereoisomers.
Other enantiomers have been reported to prolong AP or QT interval durations, although to the best of our knowledge, the stereoselective involvement of hERG1 channel block has not yet been demonstrated. Jurkiewicz et al. (1996) studied the effects of the class III antiarrhythmic agent RP58866 and its (S)-enantiomer, terikalant (Figure 4), on outward K+ currents in guinea pig ventricular myocytes. RP58866 and terikalant potently blocked the IKr, with IC50s of 22 and 31 nM, respectively (Jurkiewicz et al., 1996). The difference reported is small and potentially irrelevant, but only studies of hERG1 current block by the (R)-enantiomer may validate the possible stereoselectivity of block. The (R)-enantiomer was not tested because terikalant is considered to be the active enantiomer of RP58866. Escande et al. (1992) demonstrated that RP58866 and terikalant lengthened the AP in a comparable manner, while the (R)-isomer produced a three-fold lower effect at the same concentration. Since the racemate is a 1:1 mixture of enantiomers, the pure active enantiomer would be expected to exert a larger effect. The fact that equal concentrations of racemic and pure (S)-form have similar effects on APD provides additional evidence supporting the diverse modulation by RP58866 enantiomers on various cardiac channels that determine the APD.
Disopyramide is another antiarrhythmic drug (class Ia) marketed as a racemic mixture since 1977 (Figure 4). In a study by Vanhoutte et al. (1991), both the (R)- and (S)-enantiomers were tested on guinea pig papillary muscle. At physiological K+ concentrations and pacing, 20 μM of (S)-disopyramide significantly increased APD by ∼20%, while the same concentration (R)-disopyramide diminished APD by ∼5%. Controversy exists concerning the enantioselective effects on the sodium channel, the intended clinical target of disopyramide, for which both the absence and increased block by the (R)-form have been reported (reviewed in Vanhoutte et al., 1991). Nevertheless, even at lower concentrations (2 μM), APD was still significantly increased by (S)-disopyramide and not by the (R)-form, thus excluding it from contributing to sodium current inhibition (Vanhoutte et al., 1991). Importantly, this drug has been associated with drug-induced LQTS and TdP events (Kimura et al., 1994; Hirose et al., 2008). Meanwhile, disopyramide was demonstrated to block IhERG1 at clinically relevant concentrations (IC50 < 10 μM), and this action was proposed to constitute the molecular basis of its proarrhythmic effect (Paul et al., 2001). Investigation of the preferential block of IhERG1/IKr by disopyramide enantiomers would be very instructive and might lead to the reconsideration of the safety of the racemic administration.
Similar observations have been reported for terodiline, a drug used to treat incontinence. This molecule (Figure 4) was first marketed in 1965 as an antianginal drug before it was re-introduced in the mid-1980s for urinary incontinence (reviewed in Shah, 2002). Terodiline acts as a mixed antimuscarinic (effect of the (R)-terodiline) and Ca2+ channel blocker (effect of (S)-terodiline), and both enantiomers are considered to contribute to the beneficial effects on the urinary bladder (Martin et al., 2006). This antispasmodic drug was withdrawn from the market after it was linked to serious tachyarrhythmias and QT interval prolongation, as well as to bradycardia and atrioventricular-dissociation (Hartigan-Go et al., 1996). Taken together, the description of the adverse effects likely result from excessive delay of ventricular repolarization. In 1996, Hartigan-Go et al. studied the cardiovascular and electrocardiographic effects of both (R)- and (S)-terodiline on healthy volunteers. Interestingly, both racemic (single dose of 200 mg) and (R)-terodiline (100 mg) significantly increased QTc interval and QRS duration, while the pure (S)-enantiomer (100 mg) did not affect QTc values (Hartigan-Go et al., 1996). Although (S)-terodiline plasma concentrations were higher than those for the (R)-form, the authors demonstrated that the pharmacokinetics were not sufficient to account for the differences in ECGs. Hartigan-Go et al. (1996) concluded that QT prolongation with the racemic terodiline administration was exclusively imputable to the (R)-enantiomer. Later, racemic terodiline was demonstrated to block IKr and IhERG1 at clinically relevant concentrations (Jones et al., 1998; Martin et al., 2006). To our knowledge, no publication has addressed the stereoselective effect of terodiline on hERG1.
Propafenone (Figure 4) is another class I antiarrhythmic drug, delivered as a racemate, that deserves to be mentioned. In rare clinical cases, propafenone has been associated with QT interval prolongation and TdP (e.g., Rosengarten and Brooks, 1987; Hii et al., 1991). In the early 2000s, racemic propafenone was demonstrated to block overexpressed hERG1 channels in cellular models (Mergenthaler et al., 2001; Paul et al., 2002; Witchel et al., 2004) and native IKr current from isolated rabbit ventricular myocytes (Cahill and Gross, 2004) at IC50 values comparable with those of quinidine 0.4–0.8 μM in mammals) and within therapeutic plasma concentrations. Both enantiomers have comparable depressant effects on Na+ current (QRS widening) in dog Purkinje fibers (Kroemer et al., 1989), but, in contrast, only (S)-propafenone exerts a β-blocker activity (Kroemer et al., 1989). A possible stereoselective interaction with the hERG1 channel remains to be evaluated. Interestingly, Kroemer and co-workers evidenced the stereoselective disposition of propafenone in humans. In a small study population patients receiving the racemate (150 mg), clearance of (S)-propafenone was significantly lower (approximately −55%) than when receiving the same dose of the (S)-enantiomer alone, and this resulted in higher plasma concentrations (Kroemer et al., 1994). The authors concluded that when given as a racemic mixture (R)-propafenone impaired the disposition of (S)-propafenone due to an enantiomer–enantiomer interaction along the metabolic pathway. In addition, propafenone undergoes extensive hepatic transformation, mainly through the CYP2D6, which activity is genetically determined. In another study by Kroemer et al. (1989), patients with the SM genotype had plasma concentrations significantly elevated compared with the EM group, although S/R ratios were similar. Clinical relevance of this stereoselective disposition in hERG1-related cardiotoxicity is difficult to estimate, since the major metabolites of propafenone also inhibit IKr (Cahill and Gross, 2004), and the β-blocker and Ca2+ channel antagonism of propafenone (Dukes and Williams, 1984) may somehow counterbalance repolarization defects.
The Particular Case of Methadone as a Herg1 Blocker
Methadone is a synthetic analgesic drug, primarily a μ-agonist with pharmacological properties similar to morphine, though lacking some of the euphoric actions of the opium poppy alkaloid (Mattick et al., 2009). First synthesized in the 1930s, methadone is currently widely used in maintenance treatments (or MMTs) in order to reduce illicit opioid misuse, but also for the management of chronic pain, generally when other opioids are ineffective or poorly tolerated. Methadone is a chiral drug with an asymmetrical carbon (Figure 4). Although it is commonly administered as a racemic mixture of (R)- and (S)-methadone, the (R)-enantiomer accounts for most if not all of the opioid effects (Eap et al., 2002). In vitro competitive binding experiments (versus labeled naloxone in rat brain homogenates) have shown that the necessary concentration for the binding of the (R)-enantiomer was 10 times lower than that of the (S)-methadone. Similarly, a 10-fold difference of affinity was found between the two enantiomers for purified μ1 and μ2 opioid receptors. Finally, in human analgesia, (R)-methadone was ∼50 times as potent as the (S)-form (reviewed in Eap et al., 2002). Despite these results, the racemate is still widely used in MMTs, namely because only racemic methadone is available in the international market and because of its lower cost when compared to the pure (R)-enantiomer (Gaertner et al., 2008).
Recent studies have demonstrated that pharmacokinetics of methadone, mainly the metabolism step, are highly variable owing to genetic and environmental influences (Eap et al., 2002; Ferrari et al., 2004). In humans, methadone is extensively metabolized in the body, predominantly by the liver and probably also the intestinal cytochrome P450 isoenzymes. More precisely, CYP3A4 and CYP2B6 are the major isoforms involved in vivo, with CYP2D6 contributing to a minor extent (Crettol et al., 2006). It is worth mentioning that the stereoselective metabolism of methadone by CYP2B6 has also been reported in vitro, with an estimated 1.8-fold preference for (S)-methadone. Crettol and co-workers confirmed this in vivo by assessing the influence of the CYP2B6 *6/*6 genotype, a slow metabolizer status representing ∼6% of Caucasians and Africans. These authors confirmed that CYP2B6 contributed to (S)-methadone metabolism and, to a lesser extent, (R)-methadone metabolism, with the homozygous carriers of allele *6 showing higher (S)- and (R,S)-methadone plasma levels (Crettol et al., 2005; Crettol et al., 2006).
Importantly, a disproportionate number of unexpected sudden deaths is seen in patients receiving methadone. Although many of these deaths are likely to be a result of respiratory depression, Torsades de Pointes is also a contributing factor (Wilcock and Beattie, 2009). The number of reports of cardiac events under methadone treatment continuously increased over the last decade (Valko et al., 2001; Sticherling et al., 2005; Ehret et al., 2006), thus raising the suspicion about block of the hERG1 channel. It was without surprise that methadone racemate was demonstrated to block IhERG1 in transfected HEK293 cells with an IC50 of ∼10 μM (reviewed in Eap et al., 2002).
However, given the marked differences in the pharmacokinetics and pharmacodynamics of the two enantiomers, neither stereoselectivity of hERG1 block nor metabolic implications of the CYP2B6 *6/*6 genotype were assessed in regards to methadone cardiac safety until the study by Eap et al. (2007). In this study, whole-cell patch-clamp experiments using HEK293 cells expressing hERG1 showed that (S)-methadone blocks IhERG1 3.5-fold more potently than (R)-methadone (IC50s at 37°C: 2 and 7 μM, respectively). As CYP2B6 slow metabolizer status results in a reduced ability to metabolize (S)-methadone, ECGs, CYP2B6 genotypes, and (R)- and (S)-methadone plasma concentrations were obtained for a cohort of 179 MMT patients. Individuals identified with CYP2B6 *6/*6 genotype showed anticipated higher plasma concentrations of (R,S)- and (S)-methadone, but also higher frequencies of prolonged and borderline QTc intervals than extensive metabolizers. The CYP2B6 SM status was associated with an increased risk of prolonged QTc (odds ratio = 4.5, P = 0.03; Eap et al., 2007). Since the proportion of CYP2B6 SMs in Caucasian and African populations is non-negligible (∼6%), carriers of such polymorphism are at a potentially higher risk for severe cardiac arrhythmias and sudden death while receiving racemic methadone treatment. Since the (S)-enantiomer is thought to be more cardiotoxic and that (R)-methadone is widely accepted as the eutomer, the active enantiomer for the desired effects, the authors suggested that the safety of the commercialized racemate toward pure (R)-methadone be reconsidered (Eap et al., 2007). Indeed, the administration of the (R)-form would decrease plasma concentrations of methadone by two-fold, thereby producing a safer cardiac profile, while not affecting the activity on the μopioid receptors. Prescription of (R)-methadone would also greatly diminish the clinical concern of CYP2B6 slow metabolizer status for cardiotoxic effects. In addition, no major changes would be expected on the other beneficial activities of methadone, i.e., on the N-methyl-D-aspartate receptor, or serotonin and norepinephrine reuptake, since in all these cases the (R)- is more potent than the (S)-enantiomer (reviewed in Eap et al., 2007). Chiral switching is the term used for the development and/or commercialization of single enantiomers in place of a previous racemic mixture. Reasons for marketing a pure isomer may be purely economic, as substitution by the single enantiomer may be a good way to extend patent franchise, or may result from the demonstration of a new safety profile, as is the case for (R)-methadone.
Eap and colleagues recently investigated in a small cohort of 39 MMT patients the effects of the substitution of a dose of racemate by half-dose of (R)-methadone. They demonstrated significant reduction in the QTc interval values (Ansermot et al., 2010). Opioid adverse effects and withdrawal symptoms were either absent or of low intensity and remained unchanged during the study, which confirmed that the half-dose of (R)-methadone was therapeutically equivalent to the racemic mixture (Ansermot et al., 2010). These encouraging results, in agreement with in vitro and in vivo pharmacogenetic data, should be confirmed by larger and prospective studies that can assess changes in mortality. It seems clear that as a preventive measure pure (R)-methadone, that is currently only available in Germany, should be considered for all patients (treated for pain or under MMT) in order to reduce the risk of cardiac toxic effects and sudden death. Early studies dating back to the 1960s pointed out disadvantageous side-effects of racemic methadone when compared to the (R)-form, but they did not receive the deserved attention (reviewed in Gaertner et al., 2008). Since the article revealing the chiral cardiotoxicity of methadone was published (Eap et al., 2007), increasing number of health professionals have demanded a better assessment of the risks and pure (R)-methadone prescription (Gaertner et al., 2008; Lin et al., 2009; Wilcock and Beattie, 2009; Ansermot et al., 2010).
It is worth mentioning that the acetylated analog of methadone, acetylmethadol, has been implicated in several cases of severe arrhythmias and TdP, thus prompting its withdrawal from the market (Deamer et al., 2001). Acetylation of methadone creates an additional asymmetric center and consequently four enantiomers of acetylmethadol, or two pairs of diastereomers, α and β (Figure 4). However, only the levo-α-acetylmethadol (LAAM), derived from (S)-methadone, has been commercialized. Since the dextrorotary α-enantiomer (DAAM) derives from the active (R)-methadone, DAAM is expected to be more potent than LAAM (reviewed in Newcombe, 2006). Indeed, LAAM has proven to be a less potent analgesic than its diastereomer, but it appeared to be the least toxic of all isomers in early animal studies (lower half-maximal lethal dose). In later studies, the α-acetyl derivative of (S)-methadone was demonstrated to inhibit the hERG1 current (IC50,LAAM: ∼2 μM), but information about DAAM or other enantiomers is lacking (Katchman et al., 2002). A study of IhERG1 block evaluating all four enantiomers of acetylmethadol could certainly provide useful information about the mechanisms and/or determinants of the stereoselectivity toward the K+ channel.
Conclusion and Perspectives
About 20 years ago, Ariens and Wuis estimated that ∼57% of marketed drugs were chiral and of these, ∼55% were used clinically in the racemic form (reviewed in Mannhold et al., 2006). For newly approved drugs, the tendency is toward the chiral switching or the development of the pure enantiomer with ∼60% of single enantiomers and only 5–10% of racemic mixtures accepted in the early 2000s (Miller, 2006; Shimazawa et al., 2008). This observation reveals that researchers are increasingly aware of the complex effects of enantiomers. Although the pharmacokinetics and PD of chiral compounds are routinely investigated, thus far the pharmacogenetic risks and the stereoselective bock of the hERG1 channel have only been scarcely studied. In this review, we presented a non-exhaustive list of the compounds displaying chiral toxicity that is most likely related to hERG1-blocking properties. The particular case of methadone illustrates the need to consider the overall stereoselective effect of a chiral drug while taking into account any known pharmacogenetic information. Even though methadone is not a high-affinity blocker of hERG1 (IC50s in the μM range), and that a 3.5-fold difference between enantiomers might not be clinically relevant, the difference in hERG1 inhibition is accompanied by additional stereoselective PK and PD properties influencing (S)-methadone toxicity. Careful use of hERG1 blockers with respect to the patient’s genetic background may be possible in the future if rapid and affordable genetic screening tools become available. The inter-ethnic distribution of polymorphisms is known to be significant for drug response and should be taken into account when dealing with drugs that possibly affect cardiac repolarization.
The cavity of the hERG1 channel is large and promiscuous, and there might not be a therapeutic compound that is able to exert an “all or nothing” blocking effect. Nevertheless, there is clear evidence that some chiral molecules have a marked stereoselective effect on the hERG1 channel, and that these molecules could be highly informative with respect to the molecular requirements for drug binding and for safer enantiomeric drug development. Jamieson et al. (2006) reviewed the case of macrocyclic 3-aminopyrrolidinone farnesyltransferase inhibitors and structural modifications to design out hERG1 affinity. Within this macrocyclic series, hERG1 binding proved to be highly sensitive to chiral modifications, as one (R,R) compound (Figure 4) presented high affinity toward hERG1 (in binding assays, IC50 = 0.15 μM), whereas its (S,R)-diastereomer did so to a lesser extent (IC50 = 9.1 μM). In addition, both presented similar potencies in the block of the pharmacological target (Bell et al., 2002). It is likely that the chemical optimization of lead compounds with the introduction of chiral centers as well as the systematic assessment of hERG1 current block with all enantiomers will contribute to the reduction of the risk of drug-induced LQTS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The group of HA is supported by a grant of the Swiss National Science Foundation (310030_120707). We are grateful to Dr. Allison Felley Jacquemont for her thorough reading of the manuscript and helpful comments and to the reviewers for their constructive comments.
Footnote
- ^see www.QTdrugs.org
References
Abernethy, D. R., Wesche, D. L., Barbey, J. T., Ohrt, C., Mohanty, S., Pezzullo, J. C., and Schuster, B. G. (2001). Stereoselective halofantrine disposition and effect: concentration-related QTc prolongation. Br. J. Clin. Pharmacol. 51, 231–237.
Abriel, H., Schlapfer, J., Keller, D. I., Gavillet, B., Buclin, T., Biollaz, J., Stoller, R., and Kappenberger, L. (2004). Molecular and clinical determinants of drug-induced long QT syndrome: an iatrogenic channelopathy. Swiss. Med. Wkly. 134, 685–694.
Ansermot, N., Albayrak, O., Schlapfer, J., Crettol, S., Croquette-Krokar, M., Bourquin, M., Deglon, J. J., Faouzi, M., Scherbaum, N., and Eap, C. B. (2010). Substitution of (R,S)-methadone by (R)-methadone: Impact on QTc interval. Arch. Intern. Med. 170, 529–536.
Bell, I. M., Gallicchio, S. N., Abrams, M., Beese, L. S., Beshore, D. C., Bhimnathwala, H., Bogusky, M. J., Buser, C. A., Culberson, J. C., Davide, J., Ellis-Hutchings, M., Fernandes, C., Gibbs, J. B., Graham, S. L., Hamilton, K. A., Hartman, G. D., Heimbrook, D. C., Homnick, C. F., Huber, H. E., Huff, J. R., Kassahun, K., Koblan, K. S., Kohl, N. E., Lobell, R. B., Lynch, J. J. Jr., Robinson, R., Rodrigues, A. D., Taylor, J. S., Walsh, E. S., Williams, T. M., and Zartman, C. B. (2002). 3-Aminopyrrolidinone farnesyltransferase inhibitors: design of macrocyclic compounds with improved pharmacokinetics and excellent cell potency. J. Med. Chem. 45, 2388–2409.
Busse, D., Templin, S., Mikus, G., Schwab, M., Hofmann, U., Eichelbaum, M., and Kivisto, K. T. (2006). Cardiovascular effects of (R)- and (S)-verapamil and racemic verapamil in humans: a placebo-controlled study. Eur. J. Clin. Pharmacol. 62, 613–619.
Cahill, S. A., and Gross, G. J. (2004). Propafenone and its metabolites preferentially inhibit IKr in rabbit ventricular myocytes. J. Pharmacol. Exp. Ther. 308, 59–65.
Caner, H., Groner, E., Levy, L., and Agranat, I. (2004). Trends in the development of chiral drugs. Drug Discov. Today 9, 105–110.
Crettol, S., Deglon, J. J., Besson, J., Croquette-Krokkar, M., Gothuey, I., Hammig, R., Monnat, M., Huttemann, H., Baumann, P., and Eap, C. B. (2005). Methadone enantiomer plasma levels, CYP2B6, CYP2C19, and CYP2C9 genotypes, and response to treatment. Clin. Pharmacol. Ther. 78, 593–604.
Crettol, S., Deglon, J. J., Besson, J., Croquette-Krokar, M., Hammig, R., Gothuey, I., Monnat, M., and Eap, C. B. (2006). ABCB1 and cytochrome P450 genotypes and phenotypes: influence on methadone plasma levels and response to treatment. Clin. Pharmacol. Ther. 80, 668–681.
De Ponti, F., Poluzzi, E., Montanaro, N., and Ferguson, J. (2000). QTc and psychotropic drugs. Lancet 356, 75–76.
Deamer, R. L., Wilson, D. R., Clark, D. S., and Prichard, J. G. (2001). Torsades de pointes associated with high dose levomethadyl acetate (ORLAAM). J. Addict. Dis. 20, 7–14.
Dukes, I. D., and Williams, E. M. V. (1984). The Multiple-Modes of Action of Propafenone. Eur. Heart J. 5, 115–125.
Eap, C. B., Buclin, T., and Baumann, P. (2002). Interindividual variability of the clinical pharmacokinetics of methadone: implications for the treatment of opioid dependence. Clin. Pharmacokinet. 41, 1153–1193.
Eap, C. B., Crettol, S., Rougier, J. S., Schläpfer, J., Sintra Grilo, L., Déglon, J. J., Besson, J., Croquette-Krokar, M., Carrupt, P. A., and Abriel, H. (2007). Stereoselective block of hERG channel by (S)-methadone and QT interval prolongation in CYP2B6 slow metabolizers. Clin. Pharmacol. Ther. 81, 719–728.
Ehret, G. B., Voide, C., Gex-Fabry, M., Chabert, J., Shah, D., Broers, B., Piguet, V., Musset, T., Gaspoz, J. M., Perrier, A., Dayer, P., and Desmeules, J. A. (2006). Drug-induced long QT syndrome in injection drug users receiving methadone: high frequency in hospitalized patients and risk factors. Arch. Intern. Med. 166, 1280–1287.
Eichelbaum, M., Reetz, K. P., Schmidt, E. K., and Zekorn, C. (1986). The genetic polymorphism of sparteine metabolism. Xenobiotica 16, 465–481.
Escande, D., Mestre, M., Cavero, I., Brugada, J., and Kirchhof, C. (1992). RP 58866 and its active enantiomer RP 62719 (terikalant): blockers of the inward rectifier K + current acting as pure class III antiarrhythmic agents. J. Cardiovasc. Pharmacol. 20 (Suppl. 2), S106–S113.
Ferrari, A., Coccia, C. P., Bertolini, A., and Sternieri, E. (2004). Methadone – metabolism, pharmacokinetics and interactions. Pharmacol. Res. 50, 551–559.
Fjordside, L., Jeppesen, U., Eap, C. B., Powell, K., Baumann, P., and Brosen, K. (1999). The stereoselective metabolism of fluoxetine in poor and extensive metabolizers of sparteine. Pharmacogenetics 9, 55–60.
Gaertner, J., Voltz, R., and Ostgathe, C. (2008). Methadone: a closer look at the controversy. J. Pain Symptom. Manage. 36, e4–e7.
Gonzalez, T., Arias, C., Caballero, R., Moreno, I., Delpon, E., Tamargo, J., and Valenzuela, C. (2002). Effects of levobupivacaine, ropivacaine and bupivacaine on HERG channels: stereoselective bupivacaine block. Brit. J. Pharmacol. 137, 1269–1279.
Hartigan-Go, K., Bateman, D. N., Daly, A. K., and Thomas, S. H. L. (1996). Stereoselective cardiotoxic effects of terodiline. Clin. Pharmacol. Ther. 60, 89–98.
Hii, J. T., Wyse, D. G., Gillis, A. M., Cohen, J. M., and Mitchell, L. B. (1991). Propafenone-induced torsade de pointes: cross-reactivity with quinidine. Pacing Clin. Electrophysiol. 14, 1568–1570.
Hirose, K., Yamaguchi, H., Oshima, Y., Choraku, M., Hirono, A., Takamori, N., and Tamura, K. (2008). Severe respiratory failure and torsades de pointes induced by disopyramide in a patient with myasthenia gravis. Intern. Med. 47, 1703–1708.
ICH S7B. (2005). International Conference on Harmonisation; guidance on S7B nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals. Fed. Regist. 70, 61133–61134.
Ingelman-Sundberg, M. (2001). Pharmacogenetics: an opportunity for a safer and more efficient pharmacotherapy. J. Intern. Med. 250, 186–200.
Jamieson, C., Moir, E. M., Rankovic, Z., and Wishart, G. (2006). Medicinal chemistry of hERG optimizations: highlights and hang-ups. J. Med. Chem. 49, 5029–5046.
Jones, S. E., Ogura, T., Shuba, L. M., and McDonald, T. F. (1998). Inhibition of the rapid component of the delayed-rectifier K+ current by therapeutic concentrations of the antispasmodic agent terodiline. Br. J. Pharmacol. 125, 1138–1143.
Jurkiewicz, N. K., Wang, J., Fermini, B., Sanguinetti, M. C., and Salata, J. J. (1996). Mechanism of action potential prolongation by RP 58866 and its active enantiomer, terikalant. Block of the rapidly activating delayed rectifier K+ current, IKr. Circulation 94, 2938–2946.
Kanai, Y., Tateyama, S., Nakamura, T., Kasaba, T., and Takasaki, M. (1999). Effects of levobupivacaine, bupivacaine, and ropivacaine on tail-flick response and motor function in rats following epidural or intrathecal administration. Reg. Anesth. Pain Med. 24, 444–452.
Karbwang, J., Davis, T. M., Looareesuwan, S., Molunto, P., Bunnag, D., and White, N. J. (1993). A comparison of the pharmacokinetic and pharmacodynamic properties of quinine and quinidine in healthy Thai males. Br. J. Clin. Pharmacol. 35, 265–271.
Katchman, A. N., McGroary, K. A., Kilborn, M. J., Kornick, C. A., Manfredi, P. L., Woosley, R. L., and Ebert, S. N. (2002). Influence of opioid agonists on cardiac human ether-a-go-go-related gene K(+) currents. J. Pharmacol. Exp. Ther. 303, 688–694.
Kimura, Y., Takayanagi, K., Sakai, Y., Satoh, T., Fujito, T., Inoue, T., Hayashi, T., Morooka, S., and Takabatake, Y. (1994). Torsades de pointes in paced patients with sick sinus syndrome after disopyramide administration. Jpn. Heart J. 35, 153–161.
Kroemer, H. K., Fromm, M. F., Buhl, K., Terefe, H., Blaschke, G., and Eichelbaum, M. (1994). An enantiomer-enantiomer interaction of (S)-and (R)-propafenone modifies the effect of racemic drug therapy. Circulation 89, 2396–2400.
Kroemer, H. K., Funck-Brentano, C., Silberstein, D. J., Wood, A. J., Eichelbaum, M., Woosley, R. L., and Roden, D. M. (1989). Stereoselective disposition and pharmacologic activity of propafenone enantiomers. Circulation 79, 1068–1076.
Labro, A. J., Raes, A. L., Bellens, I., Ottschytsch, N., and Snyders, D. J. (2003). Gating of shaker-type channels requires the flexibility of S6 caused by prolines. J. Biol. Chem. 278, 50724–50731.
Lee-Son, S., Wang, G. K., Concus, A., Crill, E., and Strichartz, G. (1992). Stereoselective inhibition of neuronal sodium channels by local anesthetics. Evidence for two sites of action? Anesthesiology 77, 324–335.
Lin, C. R., Somberg, T., Molnar, J., and Somberg, J. (2009). The effects of chiral isolates of methadone on the cardiac potassium channel IKr. Cardiology 113, 59–65.
Margolis, J. M., O’Donnell, J. P., Mankowski, D. C., Ekins, S., and Obach, R. S. (2000). (R)-, (S)-, and racemic fluoxetine N-demethylation by human cytochrome P450 enzymes. Drug Metab. Dispos. 28, 1187–1191.
Martin, R. L., Su, Z., Limberis, J. T., Palmatier, J. D., Cowart, M. D., Cox, B. F., and Gintant, G. A. (2006). In vitro preclinical cardiac assessment of tolterodine and terodiline: multiple factors predict the clinical experience. J. Cardiovasc. Pharmacol. 48, 199–206.
Mattick, R. P., Breen, C., Kimber, J., and Davoli, M. (2009). Methadone maintenance therapy versus no opioid replacement therapy for opioid dependence. Cochrane Database Syst. Rev. CD002209.
Mbai, M., Rajamani, S., and January, C. T. (2002). The anti-malarial drug halofantrine and its metabolite N-desbutylhalofantrine block HERG potassium channels. Cardiovasc. Res. 55, 799–805.
Mergenthaler, J., Haverkamp, W., Huttenhofer, A., Skryabin, B. V., Musshoff, U., Borggrefe, M., Speckmann, E. J., Breithardt, G., and Madeja, M. (2001). Blocking effects of the antiarrhythmic drug propafenone on the HERG potassium channel. Naunyn Schmiedebergs Arch. Pharmacol. 363, 472–480.
Michalets, E. L., and Williams, C. R. (2000). Drug interactions with cisapride: clinical implications. Clin. Pharmacokinet. 39, 49–75.
Miller, S. (2006). Scientific and Regulatory Aspects of Quality Control for Chiral Drugs. Chirality International Symposium, Busan, South Korea; www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ucm103532.pdf.
Mitcheson, J. S., Chen, J., Lin, M., Culberson, C., and Sanguinetti, M. C. (2000a). A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U.S.A. 97, 12329–12333.
Mitcheson, J. S., Chen, J., and Sanguinetti, M. C. (2000b). Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J. Gen. Physiol. 115, 229–239.
Mohammad, S., Zhou, Z. F., Gong, Q. M., and January, C. T. (1997). Blockage of the HERG human cardiac K + channel by the gastrointestinal prokinetic agent cisapride. Am. J. Physio. Heart Circ. Physiol. 273, H2534–H2538.
Monserrat, J. M., and Bianchini, A. (2001). Anticholinesterase effect of eserine (physostigmine) in fish and crustacean species. Braz. Arch. Biol. Techn. 44, 63–68.
Newcombe, D. A. L. (2006). An Examination of the Pharmacodynamics and Pharmacokinetics of Levo-alpha-actelymethadol (LAAM), Compared to Methadone, in Opioid Maintenance Patients. Ph.D. Thesis. University of Adelaide, Australia.
Niewinski, P., Orzechowska-Juzwenko, K., Hurkacz, M., Rzemislawska, Z., Jazwinska-Tarnawska, E., Milejski, P., and Forkasiewicz, Z. (2002). CYP2D6 extensive, intermediate, and poor phenotypes and genotypes in a Polish population. Eur. J. Clin. Pharmacol. 58, 533–535.
Paavonen, K. J., Chapman, H., Laitinen, P. J., Fodstad, H., Piippo, K., Swan, H., Toivonen, L., Viitasalo, M., Kontula, K., and Pasternack, M. (2003). Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG). Cardiovasc. Res. 59, 603–611.
Paul, A. A., Witchel, H. J., and Hancox, J. C. (2002). Inhibition of the current of heterologously expressed HERG potassium channels by flecainide and comparison with quinidine, propafenone and lignocaine. Br. J. Pharmacol. 136, 717–729.
Paul, A. A., Witchel, H. J., and Hancox, J. C. (2001). Inhibition of HERG potassium channel current by the class 1a antiarrhythmic agent disopyramide. Biochem. Biophys. Res. Commun. 280, 1243–1250.
Paulussen, A. D., Gilissen, R. A., Armstrong, M., Doevendans, P. A., Verhasselt, P., Smeets, H. J., Schulze-Bahr, E., Haverkamp, W., Breithardt, G., Cohen, N., and Aerssens, J. (2004). Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. J. Mol. Med. 82, 182–188.
Perry, M., de Groot, M. J., Helliwell, R., Leishman, D., Tristani-Firouzi, M., Sanguinetti, M. C., and Mitcheson, J. (2004). Structural determinants of HERG channel block by clofilium and ibutilide. Mol. Pharmacol. 66, 240–249.
Popp, B. D., Hutchinson, D. S., Evans, B. A., and Summers, R. J. (2004). Stereoselectivity for interactions of agonists and antagonists at mouse, rat and human beta3-adrenoceptors. Eur. J. Pharmacol. 484, 323–331.
Rajamani, S., Eckhardt, L. L., Valdivia, C. R., Klemens, C. A., Gillman, B. M., Anderson, C. L., Holzem, K. M., Delisle, B. P., Anson, B. D., Makielski, J. C., and January, C. T. (2006). Drug-induced long QT syndrome: hERG K + channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br. J. Pharmacol. 149, 481–489.
Roden, D. M. (2006). Long QT syndrome: reduced repolarization reserve and the genetic link. J. Intern. Med. 259, 59–69.
Roden, D. M., and Viswanathan, P. C. (2005). Genetics of acquired long QT syndrome. J. Clin. Invest. 115, 2025–2032.
Roden, D. M., Woosley, R. L., and Primm, R. K. (1986). Incidence and clinical features of the quinidine-associated long QT syndrome: implications for patient care. Am. Heart J. 111, 1088–1093.
Rosengarten, M., and Brooks, R. (1987). Torsade de pointes ventricular tachycardia in a hypothyroid patient treated with propafenone. Can. J. Cardiol. 3, 234–239.
Roy, M. L., Dumaine, R., and Brown, A. M. (1996). HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation 94, 817–823.
Sanchez-Chapula, J. A., Ferrer, T., Navarro-Polanco, R. A., and Sanguinetti, M. C. (2003). Voltage-dependent profile of human ether-a-go-go-related gene channel block is influenced by a single residue in the S6 transmembrane domain. Mol. Pharmacol. 63, 1051–1058.
Sandler, G., Clayton, G. A., and Thornicroft, S. G. (1968). Clinical evaluation of verapamil in angina pectoris. Br. Med. J. 3, 224–227.
Shah, R. R. (2002). The significance of QT interval in drug development. Br. J. Clin. Pharmacol. 54, 188–202.
Shimazawa, R., Nagai, N., Toyoshima, S., and Okuda, H. (2008). Present state of new chiral drug development and review in Japan. J. Health Sci. 54, 23–29.
Siebrands, C. C., Schmitt, N., and Friederich, P. (2005). Local anesthetic interaction with human ether-a-go-go-related gene (HERG) channels. Anesthesiology 103, 102–112.
Smith, S. W. (2009). Chiral toxicology: it’s the same thing…only different. Toxicol. Sci. 110, 4–30.
Snyders, D. J., and Chaudhary, A. (1996). High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol. Pharmacol. 49, 949–955.
Snyders, D. J., Knoth, K. M., Roberds, S. L., and Tamkun, M. M. (1991). Time-, voltage-, and state-dependent block by quinidine of a cloned human cardiac potassium channel. Mol. Pharmacol. 41, 322–330.
Spector, P. S., Curran, M. E., Keating, M. T., and Sanguinetti, M. C. (1996). Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier K + channel. Open-channel block by methanesulfonanilides. Circ. Res. 78, 499–503.
Stansfeld, P. J., Gedeck, P., Gosling, M., Cox, B., Mitcheson, J. S., and Sutcliffe, M. J. (2007). Drug block of the hERG potassium channel: insight from modeling. Proteins 68, 568–580.
Sticherling, C., Schaer, B. A., Ammann, P., Maeder, M., and Osswald, S. (2005). Methadone-induced torsade de pointes tachycardias. Swiss Med. Wkly 135, 282–285.
Sun, Z., Milos, P. M., Thompson, J. F., Lloyd, D. B., Mank-Seymour, A., Richmond, J., Cordes, J. S., and Zhou, J. (2004). Role of a KCNH2 polymorphism (R1047 L) in dofetilide-induced Torsades de Pointes. J. Mol. Cell Cardiol. 37, 1031–1039.
Thomas, D., Gut, B., Wendt-Nordahl, G., and Kiehn, J. (2002). The antidepressant drug fluoxetine is an inhibitor of human ether-a-go-go-related gene (HERG) potassium channels. J. Pharmacol. Exp. Ther. 300, 543–548.
Thomas, D., Wu, K., Kathöfer, S., Katus, H. A., Schoels, W., Kiehn, J., and Karle, C. A. (2003). The antipsychotic drug chlorpromazine inhibits HERG potassium channels. Brit. J. Pharmacol. 139, 567–574.
Tierney, M. G., Uhthoff, T. L., Kravcik, S., and Wielgosz, A. T. (1997). Potential cisapride-erythromycin interaction. Can. J. Clin. Pharmacol. 4, 82–84.
Triggle, D. J. (1996). Ion channels as pharmacologic receptors: the chirality of drug interactions. Chirality 8, 35–38.
Valenzuela, C., Delpon, E., Tamkun, M. M., Tamargo, J., and Snyders, D. J. (1995a). Stereoselective block of a human cardiac potassium channel (Kv1.5) by bupivacaine enantiomers. Biophys. J. 69, 418–427.
Valenzuela, C., Snyders, D. J., Bennett, P. B., Tamargo, J., and Hondeghem, L. M. (1995b). Stereoselective block of cardiac sodium channels by bupivacaine in guinea pig ventricular myocytes. Circulation 92, 3014–3024.
Valko, K., Du, C. M., Bevan, C., Reynolds, D. P., and Abraham, M. H. (2001). Rapid method for the estimation of octanol/water partition coefficient (log Poct) from gradient RP-HPLC retention and a hydrogen bond acidity term (Σα2H). Curr. Med. Chem. 8, 1137–1146.
Vanhoutte, F., Vereecke, J., Carmeliet, E., and Verbeke, N. (1991). Effects of the enantiomers of disopyramide and its major metabolite on the electrophysiological characteristics of the guinea-pig papillary muscle. Naunyn-Schmiedberg’s Arch. Pharmacol. 344, 662–673.
Waldegger, S., Niemeyer, G., Mörike, K., Wagner, C. A., Suessbrich, H., Busch, A. E., Lang, F., and Eichelbaum, M. (1999). Effect of verapamil enantiomers and metabolites on cardiac K + channels expressed in xenopus oocytes. Cell. Physiol. Biochem. 9, 81–89.
Walker, B. D., Valenzuela, S. M., Singleton, C. B., Tie, H., Bursill, J. A., Wyse, K. R., Qiu, M. R., Breit, S. N., and Campbell, T. J. (1999). Inhibition of HERG channels stably expressed in a mammalian cell line by the antianginal agent perhexiline maleate. Br. J. Pharmacol. 127, 243–251.
Wesche, D. L., Schuster, B. G., Wang, W.-X., and Woosley, R. L. (2000). Mechanism of cardiotoxicity of halofantrine. Clin. Pharmacol. Ther. 521–529.
Wilcock, A., and Beattie, J. M. (2009). Prolonged QT interval and methadone: implications for palliative care. Curr. Opin. Support Palliat. Care 3, 252–257.
Witchel, H. J., Dempsey, C. E., Sessions, R. B., Perry, M., Milnes, J. T., Hancox, J. C., and Mitcheson, J. S. (2004). The low-potency, voltage-dependent HERG blocker propafenone – molecular determinants and drug trapping. Mol. Pharmacol. 66, 1201–1212.
Yang, I. C. H., Scherz, M. W., Bahinski, A., Bennett, P. B., and Murray, K. T. (2000). Stereoselective interactions of the enantiomers of chromanol 293B with human voltage-gated potassium channels1. J. Pharmacol. Exp. Ther. 294, 955–962.
Yang, P., Kanki, H., Drolet, B., Yang, T., Wei, J., Viswanathan, P. C., Hohnloser, S. H., Shimizu, W., Schwartz, P. J., Stanton, M., Murray, K. T., Norris, K., George, A. L. Jr., and Roden, D. M. (2002). Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation 105, 1943–1948.
Yang, T., Snyders, D. J., and Roden, D. M. (1997). Rapid inactivation determines the rectification and [K + ]o dependence of the rapid component of the delayed rectifier K + current in cardiac cells. Circ. Res. 80, 782–789.
Keywords: cardiotoxicity, enantiomer, hERG1 channel, long QT syndrome, methadone, stereoselectivity
Citation: Sintra Grilo L, Carrupt PA and Abriel H (2010) Stereoselective inhibition of the hERG1 potassium channel. Front. Pharmacol. 1:137. doi: 10.3389/fphar.2010.00137
Received: 17 September 2010;
Paper pending published: 05 October 2010;
Accepted: 03 November 2010;
Published online: 22 November 2010.
Edited by:
Mohamed Chahine, Laval University, CanadaReviewed by:
Georges Christé, INSERM, FranceJacques Barhanin, Université de Nice Sophia Antipolis, France
Copyright: © 2010 Sintra Grilo, Carrupt and Abriel. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Hugues Abriel, Department of Clinical Research, University of Bern, Murtenstrasse 35, 3010 Bern, Switzerland. e-mail:aHVndWVzLmFicmllbEBka2YudW5pYmUuY2g=