 Maija Pesonen1,2*
Maija Pesonen1,2* Kirsi Vähäkangas1
Kirsi Vähäkangas1 Mia Halme3
Mia Halme3 Paula Vanninen3
Paula Vanninen3 Heikki Seulanto4
Heikki Seulanto4 Matti Hemmilä4
Matti Hemmilä4 Markku Pasanen1
Markku Pasanen1 Tapio Kuitunen2
Tapio Kuitunen2
- 1 Faculty of Health Sciences, School of Pharmacy, University of Eastern Finland, Kuopio, Finland
- 2 Research and Development Department, Centre for Military Medicine, Helsinki, Finland
- 3 Finnish Institute for Verification of the Chemical Weapons Convention, University of Helsinki, Finland
- 4 Defence Forces, Technical Research Centre, Lakiala, Finland
Incapacitating and irritating agents produce temporary disability persisting for hours to days after the exposure. One can be exposed to these agents occupationally in industrial or other working environments. Also general public can be exposed in special circumstances, like industrial accidents or riots. Incapacitating and irritating agents discussed in this review are chloropicrin and capsaicinoids. In addition, we include sulfur mustard, which is an old chemical warfare agent and known to cause severe long-lasting injuries or even death. Chloropicrin that was used as a warfare agent in the World War I is currently used mainly as a pesticide. Capsaicinoids, components of hot pepper plants, are used by police and other law enforcement personnel as riot control agents. Toxicity of these chemicals is associated particularly with the respiratory tract, eyes, and skin. Their acute effects are relatively well known but the knowledge of putative long-term effects is almost non-existent. Also, mechanisms of effects at cellular level are not fully understood. There is a need for further research to get better idea of health risks, particularly of long-term and low-level exposures to these chemicals. For this, exposure biomarkers are essential. Validated exposure biomarkers for capsaicinoids, chloropicrin, and sulfur mustard do not exist so far. Metabolites and macromolecular adducts have been suggested biomarkers for sulfur mustard and these can already be measured qualitatively, but quantitative biomarkers await further development and validation. The purpose of this review is, based on the existing mechanistic and toxicokinetic information, to shed light on the possibilities for developing biomarkers for exposure biomonitoring of these compounds. It is also of interest to find ideas for early effect biomarkers considering the need for studies on subchronic and chronic toxicity.
Introduction
Incapacitating and irritating agents produce temporary disability persisting for hours to days after the exposure. Occupational exposure to incapacitating and irritating chemicals may occur in industry or in other work environment (e.g., transportation, stockpiling). Also general public may be exposed to these compounds in special circumstances, like during riots or industrial accidents. A terrorist attack with chemical agents is a more likely threat than exposure in military open battlefields. Exposure in buildings or other confined spaces with limited air conditioning may result in serious injury or death as concentrations may rise substantially high. Personnel working in crisis managements may repeatedly be exposed to low concentrations of different hazardous chemicals of unknown origin.
Sulfur mustard, chloropicrin, and capsaicinoids have been selected into this review because they represent chemically diverse irritating and incapacitating agents, with various modes of action. Sulfur mustard is an old chemical warfare agent (CWA) and the exposure to this vesicant has caused human casualties. Chloropicrin was used as a CWA during the World War I and is nowadays mainly used as a pesticide. Capsaicinoids are used by police and soldiers as riot control agents. Although acute effects of these compounds are relatively well known, knowledge of their putative long-term effects is almost non-existent. Also mechanisms of action at cellular level are not fully understood. The recent follow-up of Iranian victims has revealed long-term debilitating diseases of the lungs, eyes, and skin after expose to sulfur mustard (Balali-Mood and Hefazi, 2006). Similar long-term health effects have been described for some other warfare agents (Volans and Karalliedde, 2002) and the possibility for toxic effects by any chemical after chronic exposure needs to be taken on account.
There is an urgent need for the development and use of biomarkers in order to assess human exposures to these chemicals at occupational or other occasions. Exposure biomarkers that measure internal exposure or dose provide more reliable data to establish the link between possible adverse health effects and exposures than traditional measurements from environmental, e.g., from air or food (Watson and Mutti, 2004). The application of specific exposure biomarkers is of critical importance in the detection of high acute exposure at accidental or military situations in order to ensure rapid medical aid. In addition, sensitive and accurate biomarkers of exposure would be even more critical when monitoring low-level exposures in connection with chronic effects in follow-up studies.
Our purpose is, based on the existing mechanistic and toxicokinetic information, to review data about existing ideas of biomarkers and possibilities for developing new biomarkers to capsaicinoids, chloropicrin, and sulfur mustard for exposure biomonitoring. Also, any ideas for early effect biomarkers considering the need for studies on subchronic and chronic toxicity would be very useful. Special emphasis is given on any existing human data.
Capsaicinoids
Capsaicinoids are components in hot pepper plants, genus Capsicum sp. Six naturally occurring components, i.e., capsaicin, dihydrocapsaicin, nordihydrocapsaicin, homocapsaicin, homodihydrocapsaicin, and nonivamide have been identified in pepper products. Capsaicinoids belong to a group of vanillyl fatty acid amides. The characteristic structure of a capsaicinoid analog has a vanillamide moiety (4-hydroxy-3-methoxybenzylamide) and an acyl chain containing 10–11 carbon atoms (Table 1; Cordell and Araujo, 1993; Surh and Lee, 1995). Capsaicin and dihydrocapsaicin are the principal and most abundant analogs constituting approximately 60–90% of the total capsaicinoid concentration in the natural pepper products and the remainder (10–40%) is combination of other analogs (Cordell and Araujo, 1993; Reilly et al., 2001).
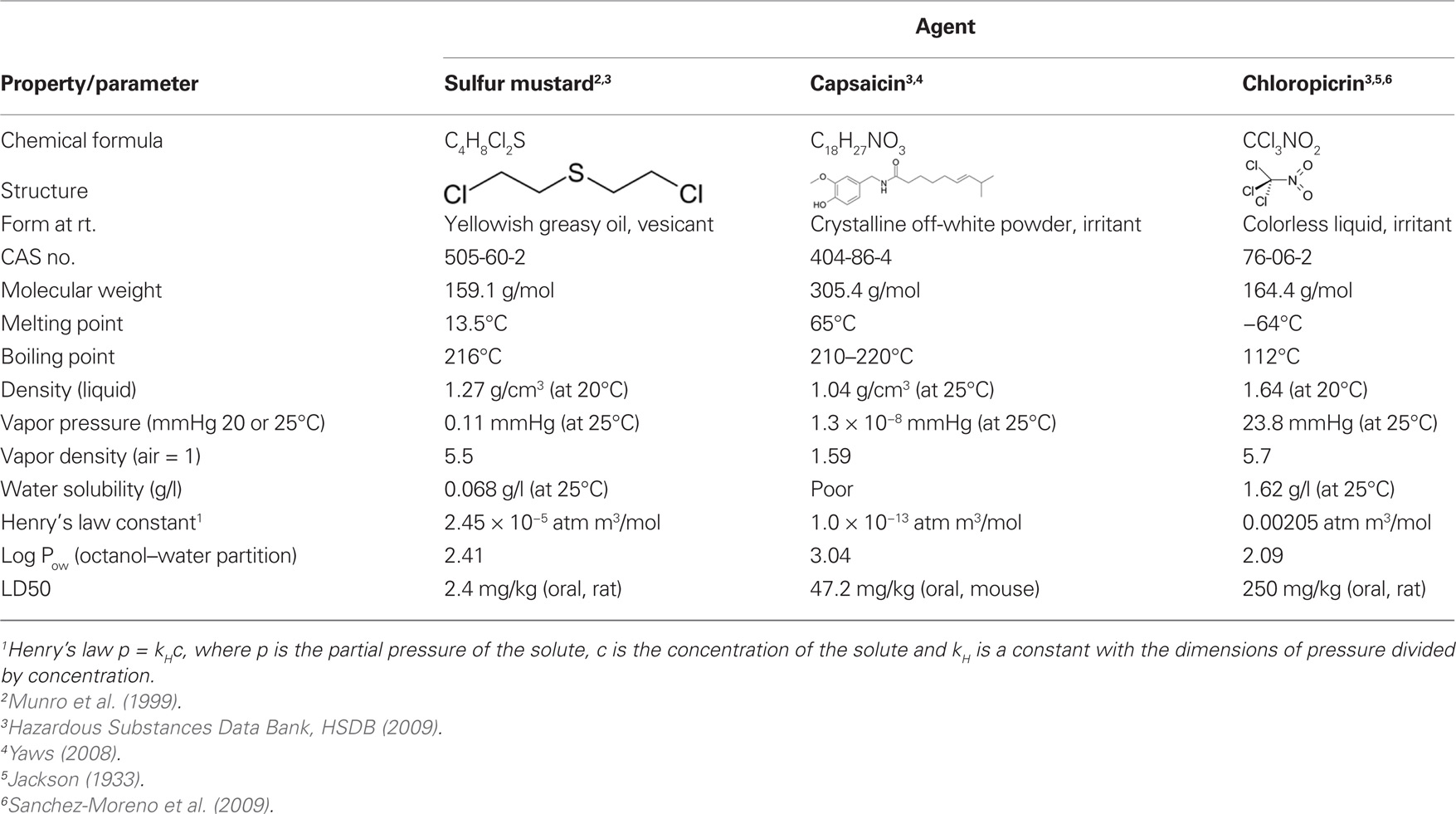
Table 1. Chemical and physical properties of sulfur mustard, capsaicin, and chloropicrin.
Capsaicinoids are used for many purposes, particularly as spices (Cordell and Araujo, 1993). Interesting from toxicological point of view is their use as irritating ingredients in “pepper gas” sprays or riot control weapons. Capsaicinoids containing sprays are utilized by the police or other low enforcement personnel to temporally incapacitate violent and rebellious individuals (Busker and van Helden, 1998). Capsaicinoids are known to act particularly on the small afferent sensory neurons but also on non-neuronal tissues. Irritation and burning pain are especially perceived in the skin, eyes, nose, tongue, and the respiratory tract or generally in the tissues where a number of sensory nerve fibers terminate. Due to causing intense pain, uncontrolled coughing, and temporary loss of vision, exposed individuals are easily captured (Watson et al., 1996; Busker and van Helden, 1998).
Capsaicinoids are pharmacologically active compounds, which have been shown to elicit multiple effects in humans and experimental animals (Szallasi and Blumberg, 1999). Most studies concerning pharmacological properties and toxicity of capsaicinoids have been conducted using capsaicin. There are only a few studies done using other capsaicinoid analogs or mixture of analogs. According to the available literature, the adverse effects and toxicity are not generally high, but the use of these compounds is not entirely without risk. The risk for toxicity increases with higher exposure levels and prolonged exposure. Several cases of poisoning and fatalities have been reported particularly following the use of the pepper gas spray even though it has been regarded as “a relative safe weapon” (Miller, 1996; Watson et al., 1996; Busker and van Helden, 1998; Snyman et al., 2001; Smith and Greaves, 2002). In such cases the patients have suffered from airway obstructions and respiratory arrest after inhaling pepper products (Busker and van Helden, 1998).
Toxicokinetics
A few studies on absorption and elimination of capsaicinoids in humans are available. Recently, Babbar et al. (2009) exposed human subjects for 60–90 min using high-concentration capsaicin skin patches (640 μg/cm2). Skin absorption of capsaicin was low and elimination rate from the plasma rapid. The maximal plasma concentration reached 17.8 ng/ml and the estimated mean elimination half-life in the plasma was 1.64 h. Human stratum corneum may restrict permeation of capsaicin (Pershing et al., 2004; Babbar et al., 2009). In vitro permeation studies of topical capsaicin indicate that human and pig skin have rather similar rates of absorption whereas the skin of rat, mouse, and rabbit are more permeable (Fang et al., 1995).
In animal models, capsaicinoids (capsaicin and dihydrocapsaicin) have been shown to absorb readily from the gastrointestinal tract (85% absorbed in 3 h). They are further extensively metabolized in the liver before reaching the general circulation. Excretion in free and glucuronide conjugates in urine and feces has been demonstrated (Saria et al., 1982; Kawada et al., 1984; Donnerer et al., 1990). After intravenous (2 mg/kg) and subcutaneous (50 mg/kg) administrations, unchanged capsaicin is distributed to the brain, spinal cord, blood, kidney, and liver within 3–10 min. Later on, the level quickly decreases in blood and liver while being still high in the brain and spinal cord (Saria et al., 1982; Donnerer et al., 1990).
Studies, using microsomal, and S9-fractions from humans and other species, or recombinant cytochrome P450 enzymes (CYP-enzymes) have shown efficient metabolism of capsaicin by hepatic enzymes but less efficient by enzymes from the extrahepatic tissues like skin and respiratory tract (Reilly et al., 2003a; Reilly and Yost, 2006; Chanda et al., 2008). Early studies demonstrated that capsaicin is converted to hydroxylated metabolites through aromatic and alkyl side chain hydroxylation and modifications of these structures were suggested to reduce its biological activity (Surh et al., 1995; Surh and Lee, 1995). Later on, Reilly et al. (2003a), using cell fractions from human liver and respiratory tract and combination of LC/MS, LC/MS/MS and LC/NMR techniques identified nine capsaicin metabolites. These were generated by aromatic and alkyl hydroxylation, O-demethylation, N-alkyldehydrogenation and ring oxygenation of capsaicin. Several CYP-enzymes (CYP1A1, 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4) were demonstrated to catalyze these reactions. The metabolite patterns formed by human liver and lung microsomal fractions were similar but the overall rate of capsaicin metabolism was markedly less by lung than liver microsomal fraction (Reilly et al., 2003a; Reilly and Yost, 2006).
Capsaicin was metabolized by CYP-enzymes also to electrophilic, reactive metabolites that produce adducts with GSH and inhibited CYP2E1 and possibly some other CYP-enzymes (Reilly et al., 2003a). The studies by Reilly et al. (2003a) also implicated CYP-mediated formation of quinone methide metabolite at the 4-OH-group of the vanilloid ring. The mechanism by which quinones could produce deleterious effects is by alkylation of proteins and DNA, depletion of GSH or by the formation of reactive oxygen species (ROS; Bolton et al., 2000). On the other hand, metabolism of capsaicinoids may also diminish their toxicity as shown in cell culture studies. Inhibition of CYP-dependent metabolism of capsaicin by the common CYP inhibitor (1-aminobenzotriazole) increases toxicity in liver and lung cells (Reilly et al., 2003a; Reilly and Yost, 2006). Thus, reactive metabolites formed by CYP-mediated metabolism of capsaicin may increase toxicity particularly in tissues, where protective systems against reactive metabolites are weak. However, the reactive metabolites formed from CYP-enzymes are probably not the only reason for toxicity by capsaicinoids. The studies on capsaicinoid metabolism have focused mainly on CYP-metabolism. It would be important to gain more information on potential damage to DNA or other macromolecules in the commonly exposed tissues.
General Toxicity
Capsaicinoids have multiple effects extending from mild irritation to severe toxic effects in various tissues. The main source of adverse human exposures to capsaicinoids is pepper spray aerosols. Exposure can be highly toxic if the pepper spray is used in closed places, e.g., inside a house. Pepper spray induces inflammation in the skin, eyes, and epithelia of the airways leading to swelling of capillaries in the exposed tissues. The effects on eyes result in copious lacrimation, pain, immediate closure of the eyes (blepharospasm), and temporary blindness. In the skin, capsaicinoids cause an intense burning pain sensation, vasodilatation, erythema, and subsequent desensitization of the exposed skin area. The symptoms are generally brief and disappear within 2 h (Busker and van Helden, 1998).
The major responses to an acute exposure of pepper spray can be found in pulmonary system. Capsaicin-sensitive sensory nerves innervate all levels of the respiratory tract and are widely distributed also in the cardiovascular system. The capsaicin activated sensory nerves are associated with initiation of many reflexes in pulmonary system, i.e., mucus hypersecretion, cough, bronchoconstriction, and sensation of dyspnea (Lee and Pisarri, 2001). Low concentrations of capsaicinoids induce coughing and sneezing, while higher doses can lead to bronchoconstriction, mucosal edema, and dyspnea (Collier and Fuller, 1984; Fuller, 1990). The mechanism of bronchoconstriction and airway mucosal edema seen in laboratory animals and humans has been explained by capsaicin-induced release of neuropeptides, particularly substance P and promotion of proinflammatory cytokine production (e.g., IL6, IL8, tumor necrosis factor-α), which lead to inflammation in respiratory tissue (Lundberg et al., 1983; Veronesi et al., 1999; Reilly et al., 2003b). High doses of capsaicin containing sprays or other capsaicin-products may be risk particularly for children, asthmatics, and for the patients suffering from cardiovascular diseases (Doherty et al., 2000; Geppetti et al., 2006). The airway resistance and other respiratory problems have been the most serious symptoms in children and adults after inhaling high concentrations of pepper products (Watson et al., 1996; Miller, 1996). Respiratory symptoms and changes in pulmonary functions have also been reported in hot pepper workers (Blanc et al., 1991). However, chronic effects of low-dose inhalation of capsaicinoids are not well known.
In animal studies, capsaicinoids (by nose-only inhalation, approximately 1.0–1.2 mg/kg for 30 min) produce marked acute inflammation, epithelial cell dysplasia, and necrosis in the upper and lower respiratory tract. The lesions can be particularly severe in the terminal bronchioles and alveoli where the capsaicinoids cause epithelial cell injury, and mild and marked vascular congestion with alveolar hemorrhage (Reilly et al., 2003b). The doses used by Reilly et al. (2003b) were estimated to be about the same as humans could receive during 5–10 s exposures to pepper spray.
Capsaicinoids have been known to act selectively on the small diameter primary sensory neurons (C- and Aδ). Irritation of these capsaicin-sensitive neurons, which originate from the dorsal root, trigeminal, and nodose ganglia (Holzer, 1991; Szallasi and Blumberg, 1999) results in an initial burning sensation and a period of enhanced sensitivity to painful thermal and mechanical stimuli. This is followed by inactivation or desensitization (defunctionalization) during which an individual is relatively resistant to capsaicin or other painful stimuli. Defunctionalization can be reversible but can also be followed by morphological changes or neurotoxicity and loss of a subpopulation of small sensory neurons (see Holzer, 1991; Caterina and Julius, 2001).
Neurotoxicity of capsaicin is dependent of the developmental stage. When capsaicin is administrated (50 mg/kg s.c. in rat) at neonatal stage, it acts as a neurotoxin and destroys terminal parts of the sensory neurons supplied by vanilloid receptors and induces long-lasting mitochondrial swelling in the soma (see Holzer, 1991; Hiura et al., 2002). As adults these animals are unresponsive to capsaicinoids (see Hiura 2000; Holzer, 1991; Caterina and Julius, 2001). The toxicity of capsaicin on developing neurons has been explained by the inhibition of axonal transport of nerve growth factor (NGF). NGF can prevent the neuronal loss, but it does not, however, repair damaged mitochondria (Szöke et al., 2002). In adult rats, degeneration of primary sensory neurons of dorsal root and nodose ganglia by capsaicin has also been demonstrated (Jancsó et al., 1985; Czaja et al., 2008). However, adult rats show neuronal recovery that can last for several weeks or months (Holzer, 1991; Czaja et al., 2008).
In addition to itching and burning pain sensation, in humans, repeated topical application of capsaicin (0.075% four times per day for 3 weeks) produces decreased sensitivity to mechanically and heat induced pain. This is followed by degeneration of epidermal nerve fibers, as revealed by histological studies (Nolano et al., 1999). The degeneration of epidermal nerve fibers is, however, local and presumably only in those nerve fibers that came in contact with capsaicin. As in animal studies, limited re-innervations of epidermis and partial return of sensation was seen 6 weeks after the treatment (Nolano et al., 1999).
Mechanisms of Toxicity
The molecular mechanism(s) involved in the action of capsaicinoids are complex cascades of reactions, which can end up in necrosis or apoptosis. The mechanisms are associated with the vanilloid or capsaicin receptor (transient receptor potential vanilloid type 1 receptor, TRPV1) that is predominantly expressed in the primary sensory neurons and known to be involved in various physiological functions, e.g., inflammation and painful stimuli (for review see Holzer, 2008). The TRPV1-receptor is a non-selective cation channel that is activated by acidosis (protons), heat (>42°C) and various chemicals, the best-known activator being capsaicin (Caterina et al., 1997; Szallasi et al., 2007). TRPV1-receptor has been found also in many other tissues, e.g., in the central nervous system, epithelial cells in urinary bladder, keratinocytes, cells of the gastric mucosa, and mast cells of the skin (Szallasi and Blumberg, 1999; Hayes et al., 2000; Mezey et al., 2000; Ständer et al., 2004). However, it is not clear whether the function of this receptor in non-neuronal tissues or in the central nervous system is similar as in sensory neurons or whether it is involved in the effects of capsaicinoids. In any case sensory neurons are about an order of magnitude more sensitive to capsaicinoids than non-neuronal cells (Holzer, 1991).
Stimulation of sensory neurons by capsaicin causes vanilloid receptor mediated influx of extracellular calcium and release of intracellular calcium from the endoplasmic reticulum (Caterina et al., 1997; Karai et al., 2004). This releases neuropeptides such as substance P, calsitonin-gene-activated peptide (CGRP), and neurokinin A from peripheral nerve terminals. These bioactive compounds are important in the communication of primary sensory neurons with other cells. Various immune, smooth muscle, and endothelial cells are known to respond to these neuropeptides and mediate neurogenic inflammation (Richardson and Vasko, 2002). Change of calcium homeostasis by prolonged or excessive activation of TRPV1-receptor is probably the main cause for neurotoxicity indicated by swelling of mitochondria and degeneration of the neurons (Szallasi and Blumberg, 1999; Hiura et al., 2002). Cell culture studies have shown that capsaicin elevated intracellular calcium causes mitochondrial membrane permeability transition, ROS-formation, and DNA fragmentation. Mitochondrial dysfunction and mitochondrial-derived caspases have been suggested to be the mechanism of apoptotic cell death in the sensory neurons (Shin et al., 2003). In addition to mitochondrial dysfunction, endoplasmic reticulum stress (ER-stress) or reduction of endoplasmic reticulum functions have been suggested to increase apoptosis in bronchial epithelial and lung epithelial fibroblast cells following treatment with capsaicin (Thomas et al., 2007; Oh and Lim, 2009). Under ER-stress, the efficiency of protein-processing decreases and unfolded proteins accumulate in the ER lumen leading ultimately to apoptotic cascades (Kaufman, 2002). Mitochondria- and ER-stress-mediated apoptosis has also been proposed as a mechanism for cell death in human prostate and breast cancer cell lines following capsaicin treatment (Sanchez et al., 2008; Lee et al., 2009).
Some effects of capsaicinoids are probably not mediated via TRPV1 receptor. The effects in non-neuronal tissues do not generally show desensitization like the responses in sensory neurons (Holzer, 1991). Capsaicin has been shown, e.g., to decrease mitochondrial membrane potential and release cytochrome c (Athanasiou et al., 2007), to change plasma membrane fluidity and structure (Aranda et al., 1995) to promote lipid peroxidation (Richeux et al., 2000), to inhibit the plasma membrane electron–transport system (NADH-oxidoreductase) and to promote the generation of ROS in various cell lines (Wolvetang et al., 1996; Macho et al., 1999). However, it is not known whether metabolic intermediates or the parent compound are responsible for these effects.
Chloropicrin
Chloropicrin (trichloronitromethane) is a lung irritant and strong lacrimator (Table 1). It is a volatile liquid at room temperature and standard pressure, and decomposes at high temperatures forming toxic gases such as phosgene, nitrogen oxides, and nitrosylchloride. Because of its strong irritating property it was used in combination with other war gases as a harassing and casualty agent in the World War I (Sutherland, 2008). Today chloropicrin is used particularly as a soil fumigant against insects, fungi, and nematodes (Ruzo, 2006). Currently the main source for human exposure is occupational via inhalation or dermal contact. Another but minor source may be chlorination of drinking water (Castro et al., 1983).
The knowledge of human health effects of chloropicrin exposure is limited and only a few accidental cases have been reported. The main findings of post-mortem examination of exposed victims have been severe pulmonary edema and spotty discoloration of the skin (Gonmori et al., 1987). Persons living close to the area where chloropicrin is used as a fumigant have been reported to suffer from symptoms of irritation such as cough, dyspnea, lacrimation, pain, burning, and chest pain (óMalley et al., 2004).
Toxicokinetics
Metabolism and elimination of chloropicrin is poorly characterized in mammals. Sparks et al. (1997, 2000) studied metabolism and interaction of chloropicrin with biological thiols in mice using radiocarbon labeled chloropicrin. This study showed that chloropicrin was readily absorbed and distributed, primarily to the liver, kidney, and blood after oral or i.p. administration. Over 50% of administered radiocarbon was found in urine and around 9% in feces 48 h after the administration. The metabolites identified were thiophosgene and dechlorination products; dichloronitromethane (CHCl2NO2) chloronitromethane (CH2ClNO2), nitromethane (CH3NO2), and numerous unidentified polar products. Similarly mouse liver microsomal and cytosolic fractions metabolized chloropicrin rapidly to dechlorinated metabolites and addition of NADPH to the microsomal reaction mixture accelerated the formation of these products (Sparks et al., 1997). The enzymes responsible for the metabolism are not known but CYP, glutathione-S-transferase, and β-lyase reactions have been suggested as the contributing pathways (Castro et al., 1988; Sparks et al., 1997, 2000).
Toxicity and Mechanism of Action
General effects after chloropicrin exposure are severe irritation of the eyes, skin, and mucosal membranes of the respiratory and gastrointestinal tracts. Because of its high volatility, the main route for human exposure to chloropicrin is inhalation. In addition to irritation, acute exposure may cause nausea, vomiting, breathing difficulties, and inflammation of the respiratory tract. At severe cases, damage in respiratory tract can lead to pulmonary edema and death (AEGL, 2008). In vitro testing implicates genetic toxicity in the presence of an exogenous metabolic system or glutathione (Schneider et al., 1999; Kundu et al., 2004; Plewa et al., 2004). Mammalian cells in vitro have shown some evidence of a clastogenic effect (Garry et al., 1990). There are no relevant studies on potential health effects of low and long-term exposure to chloropicrin.
Very little is known of the molecular mechanisms of chloropicrin. It has been shown to form protein adducts with thiol groups (e.g., Hb-SH; Sparks et al., 2000). The early work by Mackworth (1948) suggests that lachrymators like chloropicrin act by inhibiting thiol-containing enzymes such as succinate dehydrogenase and pyruvic oxidase, enzymes, which play an important role in energy metabolism. Later on, Sparks et al. (1997, 2000) studied chloropicrin reaction with thiols and its glutathione-mediated metabolism to dechlorinated products using various cells lines in vitro and mice in vivo. According to their results, toxicity of chloropicrin was due to the parent compound or metabolites other than the dechlorinated ones (CHCl2NO2, CH2ClNO2) and may be associated with inhibition of pyruvate dehydrogenase complex and elevated liver oxyhemoglobin. Chloropicrin was also shown to react with sulfhydryl groups of hemoglobin and decreasing thereby oxygen transport but this is not, according to Sparks et al. (2000), the main reason for toxicity.
Sulfur Mustard
Sulfur mustard [bis-2-(chloroethyl)disulfide] is a toxic vesicant type of warfare agent (Table 1). It is a colorless oily liquid at room temperature but its volatility results in rapid generation of vapors that can pass readily through clothing and skin. The hazard of human contact is not only with droplets of liquid, but also with vapor. In water, sulfur mustard hydrolyzes slowly forming hydrochloric acid and thiodiglycol (Watson and Griffin, 1992).
The principal use of sulfur mustard has been as a warfare chemical and it was first used in the World War I, near Ypres in Belgium. After that it has been utilized in several military conflicts, and used also against Kurdish civilians in the 1980s, (Watson and Griffin, 1992; Geraci, 2008). Although sulfur mustard can be lethal (about 5% of the exposed died during World War I), it is more likely to cause severe acute and long-term incapacitating injuries to the exposed persons (Watson and Griffin, 1992; Balali-Mood and Hefazi, 2006; Kehe et al., 2009b). There is also strong evidence that occupational exposure to sulfur mustard can cause cancer particularly in the respiratory tract and larynx area (Easton et al., 1988).
Although sulfur mustard is an old chemical, it is still a threat to military personnel and civilian population as well as an environmental hazard. There are no good antidotes other than decontamination and supportive care to treat the exposed persons. Sulfur mustard has been manufactured in large quantities and stored and dumped in soil and seas in many areas. In addition to the Iranian victims exposed in 1980s, there are several more recent cases of human exposure after accidental leakage from old reservoirs and stockpiles during the last 10 years (Geraci, 2008).
Toxicokinetics
Sulfur mustard is known to be absorbed through the skin or lungs. It is a lipophilic and very reactive chemical and when delivered as liquid or vapor, skin is the main route of exposure (Somani and Babu, 1989; Geraci, 2008). Applied to the skin, about 80% is evaporated and the remaining 20% penetrates the skin. Around 10% of the penetrated sulfur mustard is estimated to remain locally bound to skin and the rest transported with the circulation (Somani and Babu, 1989). Adipose tissue and particularly the subcutaneous fat form probably a depot from which sulfur mustard can be further delivered to circulation (Paromov et al., 2007). Chemical analyses of tissues from an Iranian soldier who died 7 days after exposure to sulfur mustard showed main accumulation in subcutaneous fat, brain, kidney, liver, spleen, and lungs (Drasch et al., 1987). Rather similar distribution has been seen in rodents after intravenous, inhalation, or percutaneous administration (Langenberg et al., 1998; Maisonneuve et al., 1994).
Davison et al. (1961) administered radiolabeled sulfur mustard (i.v.) to two terminal cancer patients. 80–90% of the radioactivity was cleared from human blood within some minutes after administration. The residual level in plasma remained constant at least for 2 days, suggesting binding to some blood constituents. The metabolites excreted were present mainly as conjugates. The majority of radioactivity, excreted in urine was suggested to represent compounds formed from alkylation by sulfur mustard, rather than metabolites from enzyme action (Davison et al., 1961). However, it is unclear how this reflects events in healthy people, e.g., liver metabolism. Chemical analysis of urinary metabolites and adducts with macromolecules from rodents (Black et al., 1992) and humans (Davison et al., 1961; Black and Read, 1995; Barr et al., 2008) suggest that sulfur mustard is metabolized by hydrolysis, oxidation, conjugation with glutathione, and β-lyase reactions. As shown by Davison et al. (1961) with the cancer patients, the main metabolites, thiodiglycol sulfoxide, and two β-lyase metabolites in urine have also been found from casualties of sulfur mustard (Black and Read, 1995; Read and Black, 2004; Barr et al., 2008).
General Toxicity
The data from both animal and human exposures show that sulfur mustard is extremely toxic through all routes of exposure causing severe lesions in multiple organs at very low doses (Dacre and Goldman, 1996). The immediate tissues affected are the eyes, skin, and respiratory tract. Extensive exposure leads also to systemic effects; these include the gastrointestinal tract, hematopoietic and lymphoid tissues and nervous system. The effects generally increase with increasing temperature and humidity (Balali-Mood and Hafazi, 2006; Watson and Griffin, 1992; ). In addition to acute toxicity, delayed chronic responses occur several months or years (even 15–20 years) after a single exposure. At present, about 45,000 Iranians, exposed in the 1980s, are reported to suffer from delayed adverse effects of sulfur mustard (Kehe et al., 2009a; Ghanei and Harandi, 2007; Rowell et al., 2009). However, there is no data about the putative effects of a long low-dose exposure.
Eyes are the most sensitive organs to the acute effects of sulfur mustard. The moist mucous surface of the cornea and conjunctiva, as well as high metabolic activity of corneal epithelial cells contribute to the sensitivity of eyes to sulfur mustard. Mild and moderate exposure to vapor or liquid causes irritation, lacrimation, burning, swollen eyelids, blepharospasm, and temporary blindness. Severely injured victims have corneal lesions with destruction of blood vessels, hemorrhages, opacity, and ulceration. Severely injured eyes may develop delayed keratopathy that leads to blindness (Baladi-Mood and Hefazi, 2006; Solberg et al., 1997; ).
Skin exposure results in erythema, edema, and blistering. Moist parts of the body (underarms, neck, and back) are generally most affected (Kehe et al., 2009a). Histological studies of lesions have shown degenerative changes particularly in the basal keratinocytes of epidermis and separation of epidermis from dermis (Sabourin et al., 2000; Smith et al., 1998). The lesions heal slowly and blisters may ulcerate promoting secondary infections (Paromov et al., 2007). In the skin as in the eyes, late toxic effects include hyper- or hypopigmentation, atrophy, keratitis, and chronic neuropathic symptoms (Balali-Mood and Hefazi, 2006).
All parts of the respiratory tract are vulnerable to sulfur mustard vapor (see Balali-Mood and Hefazi, 2006). Inhalation causes inflammation in the upper and lower respiratory tract that can progresses over several days. High doses cause primarily bronchospasm and pseudomembranes, which can lead to edema and secondary infections. Delayed effects include chronic obstructive pulmonary diseases, bronchiectasis, pulmonary fibrosis, and lung and pleural cancer (Ghanei and Harandi, 2007; Rowell et al., 2009).
High doses of sulfur mustard absorbed systemically can cause permanent damage particularly in proliferating tissues like bone marrow, lymphoid tissue, and the intestinal tract (see Balali-Mood and Hefazi, 2006; Rowell et al., 2009). Examination of sulfur mustard-victims have shown a decrease in the number of leucocytes, depression of cell mediated immunity and anemia (Balali-Mood and Hefazi, 2006). Sulfur mustard is classified as carcinogenic to humans (class 1) by the International Agency for Research on Cancer (IARC, 1975).
Proposed Mechanisms of Toxicity
Sulfur mustard is a reactive chemical causing disruption and complex impairment of a variety of cellular functions (see reviews by Paromov et al., 2007; Kehe et al., 2009b; Shakarjian et al., 2010). Proliferating epithelial cells are the cell type with the highest susceptibility to sulfur mustard-induced toxicity in primary target tissues eyes, skin, and respiratory tract. Many studies provide evidence that acute toxicity of sulfur mustard is related to its alkylating capacity, and ability to induce oxidative stress and inflammation in affected tissues. These events in turn cause detrimental changes in metabolic, genetic, and other cellular functions leading to cell death by apoptosis or necrosis (Papirmeister et al., 1985; Paromov et al., 2007; Kehe et al., 2009b).
The strong reactivity of sulfur mustard is due to ionization of its chlorine atoms that can lead to the formation of an ethylene sulfonium ion intermediate and carbonium ion. These are strong alkylating agents, which react with cellular nucleophilic centers (-SH, -NH2- groups) in DNA, RNA, proteins, and lipids (Somani and Babu, 1989; Dacre and Goldman, 1996). Several studies have shown that sulfur mustard can modify DNA through the formation of DNA mono-adducts and DNA cross-links and thus inhibit cell division (Fidder et al., 1994; Lundlum et al., 1994).
DNA alkylation is considered as the primary alteration of sulfur mustard toxicity, which then leads to further changes in cellular functions. DNA-damage activates DNA-repair mechanisms, particularly poly(ADP-ribose) polymerase (PARP; Papirmeister et al., 1985). The repair reactions, which use β-nicotinamide adenine dinucleotide (NAD+) as a substrate, deplete cellular NAD+ stores. Since NAD+ is an important cofactor in the glycolysis and tricarboxylic acid cycle, this leads to impairment of energy production. Increase of ROS, nitric oxide, and peroxynitrite concentrations, i.e., oxidative stress has also been reported to be an important initial effect. The disturbance of proper oxidation–reduction potential leads to lipid peroxidation that further contributes damage in the exposed cells (Gross et al., 1993; Paromov et al., 2007). In addition, activation of several inflammatory mediators (proinflammatory cytokines, eicosanoids), release of tissue proteases (matrix metalloproteases), and change of calcium balance in cells have been shown to be important in blister formation, and epidermal–dermal separation in skin toxicity (Arroyo et al., 2000; Mol et al., 2009; Ruff and Dillman, 2010).
Although the exact mechanisms underlying chronic toxicity of sulfur mustard are not clear, it is apparent that the events in acute toxicity are related to long-term toxicity. Some studies suggest autoimmune basis for the cause of chronic toxicity. Sulfur mustard alters proteins resulting in protein-sulfur mustard products that may stimulate autoimmune reactions in exposed tissues (Solberg et al., 1997; Javadi et al., 2005). Also, high level of inflammatory cytokines after the initial injury can hinder apoptosis of polymorphonuclear leukocytes, thus increasing their active lifespan and contributing to the chronic tissue damage (Asensi et al., 2004). Furthermore, toxicity of free radicals and other degenerative changes after the initial injury have been proposed as causes of chronic effects (Aasted et al., 1987; Hosseini-Khalili et al., 2009; Ghanei and Harandi, 2010). Recently, Hosseini-Khalili et al. (2009) showed mutations in the tumor suppressor gene, TP53, when characterizing lung cancer in mustard gas victims from the Iran–Iraq war. These mutations included similar types of point mutations as found in factory workers chronically exposed to sulfur mustard (Takeshima et al., 1994) implicating TP53 mutations as a biomarker (Vähäkangas, 2003).
Experimental in vivo rabbit studies (Kadar et al., 2009) suggest that the chronic lesions in the eyes, leading later to epithelial defect, and visual impairment, are due to epithelial stem cell degeneration. This may be based on prolonged impaired corneal innervation and chronic inflammation, associated with increased activity of matrix metalloproteinases. In rabbit eyes, protease inhibitors attenuate acute injury, and postpone the delayed toxicity (Kadar et al., 2009).
Possibilities for Biomarkers and Biomarker Developments
A biomarker is a distinctive biological or biologically derived indicator (e.g., a metabolite) of a process, event, or condition (see Hecht, 2008). Various types of biomarkers for toxic agents have been developed, including biomarkers of exposure, e.g., lead in blood (Barbosa et al., 2005), and biomarkers for early effects, e.g., micronuclei in peripheral blood lymphocytes indicating damage by genotoxic agents (Decordier et al., 2009). The objective to use biomarkers of exposure is to increase the accuracy of exposure evaluation (Phillips, 2008). To verify exposure and to link the exposure with putative health effects of harmful agents, good validated biomarkers of exposure are essential. For the development of exposure biomarkers for specific compounds, mechanistic, and toxicokinetic information of the compound is the basis.
Biomarkers of exposure can be developed based on the compound itself or its metabolites in body fluids, or tissue bound products, e.g., protein and DNA-adducts (Vineis and Perera, 2000, 2007). Tissue responses to the chemical like an increase in serum enzyme-activities (alanine aminotransferase) in connection with drug-induced liver damage may also serve as an exposure biomarker (Ozer et al., 2008). In the development of biomarkers, as important as the relevance of the putative biomarker is the analytical technique for its detection and finally validation of the biomarker for its purpose. This step-wise development and validation of both the analytical method (reproducibility, accuracy, sensitivity, and specificity) and the biomarker (e.g., inter- and intraindividual variability and confounding factors) in molecular epidemiology studies separately is the prerequisite for a routinely usable biomarker (see e.g., Vineis and Garte, 2008; Vähäkangas, 2008). Furthermore, it has to be shown that the analytical method works in available biological matrix, like tissue, urine, or blood. These different matrixes have different implications as to the timing of exposure. Urinary metabolites are usually short-lived and only reflect acute exposure (Watson and Mutti, 2004), whereas adducts in tissue or blood cells may be retained as long as the cells live. DNA-adducts in white blood cells implicate longer-term exposure, and hemoglobin-adducts in red blood cells reflect more chronic exposure considering that the life span of red blood cells is over 100 days. Advantage of the protein adducts is that they have a longer life span than DNA-adducts. They are generally more stable and have the same lifetime as the native proteins. For instance the half-life of human albumin is 20–25 days and hemoglobin 120 days, respectively (Törnqvist et al., 2002).
In pepper products, capsaicin and dihydrocapsaicin together constitute 60–90% of capsaicinoids. Both of them can be detected by LC–tandem MS in blood of treated rats (Reilly et al., 2002). Unfortunately, only anecdotal studies reporting human blood or urinary levels of capsaicinoids exist (Babbar et al., 2009). Thus, no biomarkers or validated methods for such markers as to capsaicinoid exposure exist. Stratum corneum of the human skin is known to serve as an efficient barrier to the absorption of chemicals. According to Pershing et al. (2004) this applies also to capsaicinoids, which are lipid soluble. They found the half-life of capsaicinoids to be about 24 h in stratum corneum, which probably explains the re-emergence of symptoms with heat or water-exposure after the acute phase. Skin cells may be a useful tissue for exposure monitoring of capsaicinoids.
Quantifying nitromethane, a metabolite of chloropicrin (trichloronitromethane) has been proposed as an indicator of chloropicrin exposure (Alwis et al., 2008). Halonitromethanes are spontaneously dehalogenated in blood and nitromethane is the endproduct. However, nitromethane may also be formed endogenously from peroxynitrate, and thus this metabolite is not specific for chloropicrin. The only known metabolic pathway in mammals is conversion of chloropicrin to raphanusamic acid via thiophosgene intermediate. Raphanusamic acid (Figure 1), which is a cyclic cysteine adduct, was found in urine of chloropicrin-exposed mice by GC–MS (Sparks et al., 2000). Whether this metabolic route exists in humans remains to be seen. Thus, it will require further work before useful exposure biomarkers for chloropicrin have been identified and validated for its meant use.
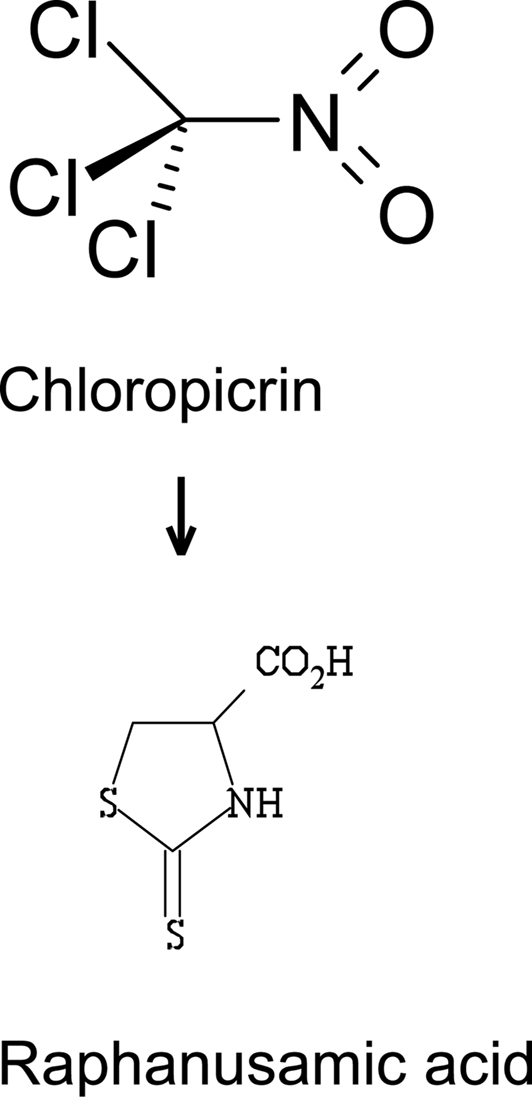
Figure 1. Raphanusamic acid, a metabolite found in mice urine after exposure to chloropicrin.
Several metabolites and adducts with proteins and DNA have been suggested as biomarkers of exposure to sulfur mustard (for a recent review see Black, 2008). The major urinary metabolites of sulfur mustard are derived from hydrolysis, oxidation or conjugation with glutathione. Gas chromatography–tandem mass spectrometry (GC–MS/MS) and liquid chromatography–tandem mass spectrometry (LC–MS–MS) methods have been developed and used for the detection of two closely related urinary β-lyase metabolites and their common reduction products (Table 2) in exposed rats and human casualties (Black and Reed, 1995; Read and Black, 2004). These metabolites in urine are regarded as potentially good and specific markers for exposure since they can be present at high concentrations in exposed persons but are not found in control persons.
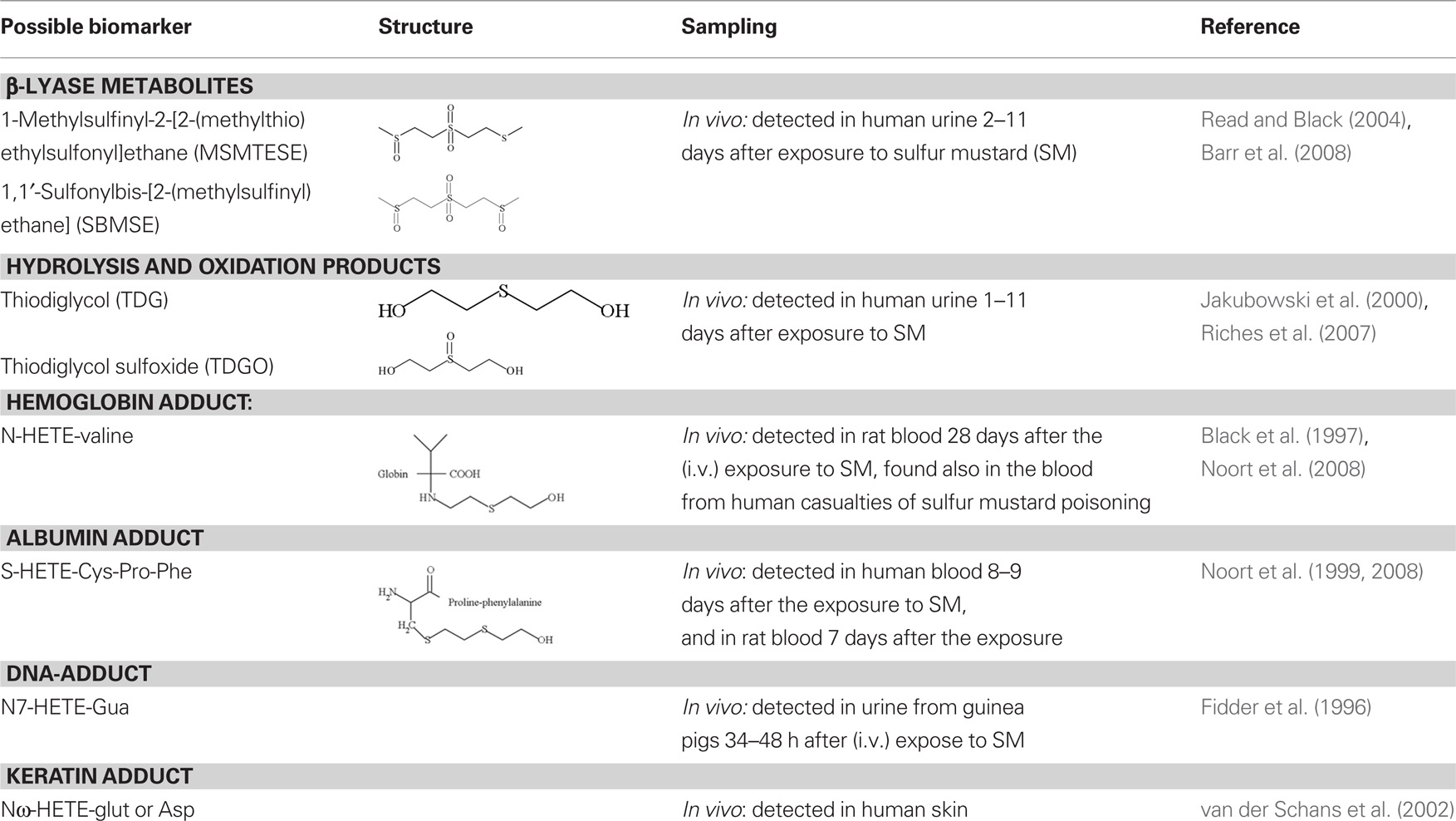
Table 2. Metabolites and adducts suggested as exposure biomarkers for sulfur mustard.
In addition to beta-lyase metabolites, two other, the major hydrolysis products, thiodiglycol, and its subsequent oxidation product, thiodiglycol sulfoxide (Table 2) are abundant in human urine exposed to sulfur mustard (Jakubowski et al., 2000; Riches et al., 2007). Although high levels in urine provide evidence of exposure to sulfur mustard, disadvantage is that low level (<2 ng/ml), of thiodiglycol sulfoxide is present in non-exposed human urine (Riches et al., 2007). It is also not known, if ethnic background, food supply or other life style factors could contribute to thiodiglycol sulfoxide levels in the urine. All these aspects need to be taken into consideration.
The primary alkylation site of DNA by sulfur mustard is the N7-position of deoxyguanosine leading to the formation of N7-HETE-Guanine adduct (Fidder et al., 1994). Experimental studies with guinea pigs have shown that excretion of N7-HETE-Guanine in urine increases within short time (2–3 h) after the exposure but decreases rapidly thereafter. On basis of this Fidder et al. (1996) proposed the detection of N7-HETE-Guanine in urine as suitable to assess acute exposure to sulfur mustard (Table 2). An antibody has been raised against this adduct and it has been used in competitive ELISA assay to detect the adduct in human white blood cells after in vitro treatment (van der Schans et al., 1994). However, no human biomonitoring studies exist to verify the usefulness of this adduct.
Binding of sulfur mustard to valine or histidine residues of hemoglobin (N7-HETE-valine) and a cysteine residue in a digested small peptide fragment of albumin (S-HETE-Cys-Pro-Phe) have been explored as possible biomarkers for exposure and effect (Table 2; Noort et al., 1999, 2008). Gas chromatographic–mass spectrometric methods exist and have also been used to qualitatively analyze these adducts in blood samples from humans and experimental animals exposed to sulfur mustard (Noort et al., 1999, 2008; Smith et al., 2008). The adducts provide information of exposure for several weeks or months, because the life span of the read blood cells is about 4 months and that of albumin around 21 days.
The skin is one of the major targets for sulfur mustard toxicity. Monoclonal antibodies that recognize sulfur mustard keratin adducts in stratum corneum of human skin have been raised (van der Schans et al., 2002). These antibodies bind directly, without preconditioning of the samples, to the skin cross-sections of sulfur mustard-treated human callus. This approach does not involve laborious procedures before the detection and may be used to develop a detection method applicable for showing acute exposure. Whether such a method is sensitive enough for low-dose exposure remains to be studied.
Conclusions
Biomarkers for low-level and long-term exposures of chloropicrin, sulfur mustard, and capsaicinoids do not exist so far. However, possible biomarkers have been suggested especially in the case of sulfur mustard, but no routinely usable quantitative biomarkers exist for exposure monitoring. The suggested biomarkers of sulfur mustard, metabolites, and macromolecular adducts can already be analyzed qualitatively. However, quantitative measurements await further development and validation of the analytical methods especially for low exposure level. Studies on the mechanisms of long-term toxicity may provide further ideas in addition to the potential metabolism-based biomarkers. It is essential that potential biomarkers for routine use are validated in molecular epidemiology studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by Scientific Advisory Board for Finnish Defence and Finnish Defence Forces.
References
Aasted, A., Darre, E., and Wulf, H. C. (1987). Mustard gas: clinical, toxicological, and mutagenic aspects based on modern experience. Ann. Plast. Surg. 19, 330–333.
AEGL. (2008). Acute Exposure Guideline Levels, Chloropicrin. Available at: http://www.epa.gov/oppt/aegl/pubs/chemlist.htm
Alwis, K. U., Blount, B. C., Silva, L. K., Smith, M. M., and Loose, K.-H. (2008). Method for quantifying nitromethane in blood as a potential biomarker of halonitromethane exposure. Environ. Sci. Technol. 42, 2522–2527.
Aranda, F., Villalain, J., and Gomez-Fernandes, J. C. (1995). Capsaicin affects the structure and phase organization of phospholipid membranes. Biochim. Bhiophys. Acta 1234, 225–234.
Arroyo, C. M., Schafer, R. J., Kurt, E. M., Broomfield, C. A., and Carmichael, A. J. (2000). Response of normal human keratinocytes to sulfur mustard: cytokine release. J. Appl. Toxicol. 20, 63–72.
Asensi, V., Valle, E., Meana, A., Fierer, J., Celada, A., Alvarez, V., Paz, J., Coto, E., Carton, J. A., Maradona, J. A., Dieguez, A., Sarasua, J., Ocana, M. G., and Arribas, J. M. (2004). In vivo interleukin-6 protects neutrophils from apoptosis in osteomyelitis. Infect. Immun. 72, 3823–3828.
Athanasiou, A., Smith, P. A., Vakilpour, S., Kumuran, N. M., Turner, A. E., Bagiokou, D., Layfield, R., Ray, D. E., Westwell, A. D., Alexander, S. P., Kendall, D. A., Lobo, D. N., Watson, S. A., Lophatanon, A., Muir, K. A., Guo, D., and Bates, T. E. (2007). Vanilloid receptor agonists and antagonists are mitochondrial inhibitors: How vanilloids cause non-vanilloid receptor mediated cell death. Biochem. Biophys. Res. Commun. 354, 50–55.
Babbar, S., Marier, J.-F., Mouksassi, M.-S., Beliveau, M., Vanhove, G. V., Chanda, S., and Bley, K. (2009). Pharmacokinetic analysis of capsaicin after topical administration of high-concentration capsaicin patch to patients with peripheral neuropathic pain. Drug Monit. 31, 502–510.
Balali-Mood, M., and Hefazi, M. (2006). Comparison of early and late effects of sulfur mustard in Iranian veterans. Basic Clin. Pharmacol. Toxicol. 99, 273–282.
Barbosa, F., Tanus-Santos, J. E., Gerlach, R. F., and Parsons, P. J. (2005). A critical review of biomarkers used for monitoring human exposure to lead: advantages, limitations, and future needs. Environ. Health Perspect. 113, 1669–1674.
Barr, J. R., Pierce, C. L., Smith, J. R., Capacio, B. R., Woolfitt, A. R., Solano, M. I., Wooten, J. V., Lemire, S. W., Thomas, J. D., Ash, D. H., and Ashley, D. L. (2008). Analysis of urinary metabolites of sulfur mustard in two individuals after accidental exposure. J. Anal. Toxicol. 32, 10–16.
Black, R. M. (2008). An overview of biological markers of exposure to chemical warfare agents. J. Anal. Toxicol. 32, 2–9.
Black, R. M., Brewster, K., Clarke, R. J., Hambrook, J. L., Harrison, J. M., and Howells, D. J. (1992). Biological fate of sulfur mustard, 1,1-thiobis(2-chloroethane): isolation and identification of urinary metabolites following intraperitoneal administration to rat. Xenobiotica 22, 405–418.
Black, R. M., Clarke, R. J., Harrison, J. M., and Read, R. W. (1997). Biological fate of sulfur mustard: identification of valine and histidine adducts in haemoglobin from casualties of sulfur mustard poisoning. Xenobiotica 27, 499–512.
Black, R. M., and Read, R. W. (1995). Biological fate of sulfur mustard, 1,1’-thiobis(2-chloroethane): identification of β-lyase metabolites and hydrolysis products in human urine. Xenobiotica 25, 167–173.
Blanc, P., Liu, D., Juarez, C., and Boushey, H. A. (1991). Cough in hot pepper workers. Chest 99, 27–32.
Bolton, J. L., Trush, M. A., Penning, T. M., Dryhurst, G., and Monks, T. J. (2000). Role of quinones in toxicology. Chem. Res. Toxicol. 13, 136–160.
Busker, R., and van Helden, H. P. (1998). Toxicologic evaluation of pepper spray as a possible weapon for the Dutch police force: risk assessment and efficacy. Am. J. Forensic Med. Pathol. 19, 309–316.
Castro, C. E., Wade, R. S., and Belser, N. O. (1983). Biodehalogenation. The metabolism of chloropicrin by Pseudomonas sp. J. Agric. Food Chem. 31, 1184–1187.
Castro, C. E., Yokoyama, W. H., and Belser, N. O. (1988). Biohalogenation. Reactivities of microbial and mammalian cytochrome P450 compared with heme and whole-cell models. J. Agric. Food Chem. 36, 915–919.
Caterina, M. J., and Julius, D. (2001). The vanilloid receptor: a molecular gateway to the pain pathway. Annu. Rev. Neurosci. 24, 487–517.
Caterina, M. J., Schumacher, M. A., Tominaga, M., Rosen, T., Levine, J. D., and Julius, D. (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824.
Chanda, S., Bashir, M., Babbar, S., Koganti, A., and Bley, K. (2008). In vitro hepatic and skin metabolism of capsaicin. Drug Metab. Dispos. 36, 670–675.
Collier, J. G., and Fuller, R. W. (1984). Capsaicin inhalation in man and the effects of sodium cromoglycate. Br. J. Pharmacol. 81, 113–117.
Cordell, G. A., and Araujo, O. E. (1993). Capsaicin: identification, nomenclature, and pharmacotherapy. Ann. Pharmacother. 27, 330–336.
Czaja, K., Burns, G. A., and Ritter, R. C. (2008). Capsaicin-induced neuronal death and proliferation of primary sensory neurons located in the nodose ganglia of adult rats. Neuroscience 154, 621–630.
Dacre, J. C., and Goldman, M. (1996). Toxicology and pharmacology of the chemical warfare agent sulfur mustard. Pharmacol. Rev. 48, 289–326.
Davison, C., Rozman, R. S., and Smith, P. K. (1961). Metabolism of bis β chloroethyl sulfide (sulfur mustard gas). Biochem. Pharmacol. 7, 65–74.
Decordier, I., Papine, A., Plas, G., Roesems, S., Vande Loock, K., Moreno-Palomo, J., Cemeli, E., Anderson, D., Fucic, A., Marcos, R., Soussaline, F., and Kirsch-Volders, M. (2009). Automated image analysis of cytokinesis-blocked micronuclei: an adapted protocol and a validated scoring procedure for biomonitoring. Mutagenesis 24, 85–93.
Doherty, M. J., Mister, R., Pearson, M. G., and Calverley, P. M. (2000). Capsaicin responsiveness and cough in asthma and chronic obstructive pulmonary disease. Thorax 55, 643–649.
Donnerer, J., Amann, R., Schuligoi, R., and Lembeck, F. (1990). Absorption and metabolism of capsaicinoids following intragastric administration in rats. Naunyn Schmiedebergs Arch. Pharmacol. 342, 357–361.
Drasch, G., Kretschmer, E., Pharm, M., Kauert, G., and von Meyer, L. (1987). Concentrations of mustard gas [bis(2 chloroethyl)sulfide] in the tissues of a victim of a vesicant exposure. Case report. J. Forensic Sci. 32, 1788–1793.
Easton, D. F., Peto, J., and Doll, R. (1988). Cancers of the respiratory tract in mustard gas workers. Br. J. Ind. Med. 45, 625–659.
Fang, J.-Y., Wu, P.-C., Huang, Y.-B., and Tsai, Y.-H. (1995). In vitro permeation study of capsaicin and its synthetic derivatives from ointment bases using various skin types. Int. J. Pharm. 126, 119–128.
Fidder, A., Moes, G. W., Scheffe, A. G., van der Schans, G. P., Baan, R. A., de Jong, L. P., and Benchop, H. P. (1994). Synthesis, characterization, and quantitation of the major adducts formed between sulfur mustard and DNA of calf thymus and human blood. Chem. Res. Toxicol. 7, 199–204.
Fidder, A., Noort, D., deJong, L. P., and Benschop, H. P. (1996). N7-(2-hydroxyethylthioethy)-guanine: a novel urinary metabolite following exposure to sulfur mustard. Arch. Toxicol. 70, 854–855.
Garry, V. F., Nelson, R. L., Griffith, J., and Harkins, M. (1990). Preparation for human study of pesticide applicators: sister chromatid exchanges and chromosome aberrations in cultured human lymphocytes exposed to selected fumigants. Teratog. Carcinog. Mutagen 10, 21–29.
Geppetti, P., Materazzi, S., and Nicoletti, P. (2006). The transient receptor potential vanilloid 1: role in airway inflammation and disease. Eur. J. Pharmacol. 533, 207–214.
Ghanei, M., and Harandi, A. A. (2007). Long term consequences from exposure to sulfur mustard: a review. Inhal. Toxicol. 19, 451–456.
Ghanei, M., and Harandi, A. A. (2010). Lung carcinogenicity of sulfur mustard. Clin. Lung Cancer 11, 13–17.
Gonmori, K., Muto, H., Yamamoto, T., and Takahashi, K. (1987). A case of homicidal intoxication by chloropicrin. Am. J. Forensic Med. Pathol. 8, 135–138.
Gross, C. L., Innace, J. K., Hovatter, R. C., Meier, H. I., and Smith, W. J. (1993). Biochemical manipulation of intracellular glutathione levels influences cytotoxicity to isolated human lymphocytes by sulfur mustard. Cell Biol. Toxicol. 9, 259–267.
Hayes, P., Meadows, H. J., Gunthorpe, M. J., Harries, M. H., Duckworth, D. M., Cairns, W., Harrison, D. C., Clarke, C. E., Ellington, K., Prinjha, R. K., Barton, A. J., Medhurst, A. D., Smith, G. D., Topp, S., Murdock, P., Sanger, G. J., Terrett, J., Jenkins O., Benham, C. D., Randall, A. D., Gloger, I. S., and Davis J. B. (2000). Cloning and functional expression of a human orthologue of rat vanilloid receptor. Pain 88, 205–215.
Hazardous Substances Data Bank (HSDB). (2009). Available at: http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen
Hecht, S. S. (2008). “Carcinogen metabolites as biomarkers,” in Molecular Epidemiology of Chronic Diseases, eds C. Wild, P. Vineis, and S. Garte (England: John Wiley and Sons, Ltd), 97–110.
Hiura, A. (2000). Neuroanatomical effects of capsaicin on the primary afferent neurons. Arch. Histol. Cytol. 63, 199–215.
Hiura, A., Nakae, Y., and Nakagawa, H. (2002). Cell death of primary afferent nerve cells in neonatal mice treated with capsaicin. Anat. Sci. Int. 77, 47–50.
Holzer, P. (1991). Capsaicin: cellular targets, mechanism of action, and selectivity for thin sensory neurons. Pharmacol. Rev. 43, 143–201.
Holzer, P. (2008). The pharmacological challenge to tame the transient receptor potential vanilloid-1 (TRPV1) nocisensor. Br. J. Pharmacol. 115, 1145–1162.
Hosseini-Khalili, A., Haines, D. D., Modirian, E., Soroush, M., Khateri, S., Joshi, R., Zendehdel, K., Ghanei, M., and Giardina, C. (2009). Mustard gas exposure and carcinogesis of lung. Mutat. Res. 678, 1–6.
Jakubowski, E. M., Sidell, F. R., Evans, R. A., Carter, M. A., Keeler, J. R., McMonagle, J. D., Swift, A., Smith, J. R., and Dolzine, T. W. (2000). Quantification of thiodiglycol in human urine after an accidental sulfur mustard exposure. Toxicol. Methods 10, 143–150.
Jancsó, G., Kiraly, E., Joo, F., Such, G., and Nagy, A. (1985). Selective degeneration by capsaicin of a subpopulation of primary sensory neurons in adult rat. Neurosci. Lett. 30, 209–2014.
Javadi, M.-A., Yazdani, S., Sajjadi, H., Jadidi, K., Karimian, F., Einollahi, B., Jáfarinasab, M.-R., and Zare, M. (2005). Chronic and delayed-onset mustard gas keratitis. Ophthalmology 112, 617–625.
Kadar, T., Dachir, S., Cohen, L., Sahar, R., Fishbine, E., Cohen, M., Turetz, J., Gutman, H., Buch, H., Brandeis, R., Horwitz, V., Solomon, A., and Amir, A. (2009). Ocular injuries following sulfur mustard exposure – pathological mechanism and potential therapy. Toxicology 263, 59–69.
Karai, L. J., Russell, J. T., Iandarola, M. J., and Olah, Z. (2004). Vanilloid receptor 1 regulates calcium compartments and contributes to Ca2+ -induced Ca2+ release in sensory neurons. J. Biol. Chem. 279, 16377–16387.
Kaufman, R. J. (2002). Orchestrating the unfolded protein response in health and disease. J. Clin. Invest. 110, 1389–1398.
Kawada, T., Suzuki, T., Takahashi, M., and Iwai, K. (1984). Gastrointestinal absorption and metabolism of capsaicin and dihydrocapsaicin in rats. Toxicol. Appl. Pharmacol. 72, 449–456.
Kehe, K., Thiermann, H., Balszuweit, F., Eyer, F., Steinritz, D., and Zilker, T. (2009a). Acute effects of sulfur mustard injury – Munich experiences. Toxicology 263, 3–8.
Kehe, K., Thiermann, H., Balszuweit, F., Eyer, F., Steinritz, D., and Zilker, T. (2009b). Molecular toxicology of sulfur mustard-induced cutaneous inflammation and blistering. Toxicology 263, 12–19.
Kundu, B., Richardson, S. D., Swartz, P. D., Matthews, P. P., Richard, A. M., and DeMarini, D. M. (2004). Mutagenicity in Salmonella of halonitromethanes: a recently recognized class of disinfection by-products in drinking water. Mutat. Res. 562, 39–65.
Langenberg, J. P., van der Schans, G. P., Spruit, H. E., Kuijpers, W. C., Mars-Groenendijk, R. H., van Dijk-Knijnenburg, H. C., Trap, H. T., van Helden, H. P., and Benschop, H. P. (1998). Toxicokinetics of sulfur mustard and its DNA-adducts in the hairless guinea pig. Drug Chem. Toxicol. 21, 131–147.
Lee, L. Y., and Pisarri, T. E. (2001). Afferent properties and reflex functions of bronchopulmonary C-fibers. Respir. Physiol. 125, 47–65.
Lee, M. J., Kee, K. H., Suh, C. H., Lim, S. C., and Oh, S. H. (2009). Capsaicin-induced apoptosis is regulated by endoplasmic reticulum stress- and calpain-mediated mitochondrial cell death pathways. Toxicology 264, 205–214.
Lundberg, J. M., Martling, C. R., and Saria, A. (1983). Substance P. and capsaicin-induced contraction of human bronchi. Acta Physiol. Scand. 119, 49–53.
Lundlum, D. B., Austin-Ritchie, P., Hagopian, M., Niu, T.-Q., and Yu, D. (1994). Detection of sulfur mustard-induced DNA modifications. Chem. Biol. Interact. 91, 39–49.
Macho, A., Calzado, M. A., Munoz-Blanco, J., Gomez-Diaz, C., Gajate, C., Mollinedo, F., Navas, P., and Munoz, E. (1999). Selective induction of apoptosis by capsaicin in transformed cells: the role of reactive oxygen species and calcium. Cell Death Differ. 6, 155–165.
Maisonneuve, A., Callebat, I., Debourdes, L., and Coppet, L. (1994). Distribution of [14C]sulfur mustard in rats after intravenous exposure. Toxicol. Appl. Pharmacol. 125, 281–287.
Mezey, E., Toth, Z. E., Cortright, D. N., Arzubi, M. K., Krause, J. E., Elde, R., Blumber, M., and Szallasi, A. (2000). Distribution of mRNA for vanilloid receptor subtype 11 (VR1), and VR1-like immunoreactivity, in the central nervous system of rat and human. Proc. Natl. Acad. Sci. U.S.A. 97, 3655–3660.
Mol, M. A., van der Berg, R. M., and Benschop, H. P. (2009). Involvement of caspases and transmembrane metalloproteases in sulfur mustard-induced microvesication in adult human skin in organ culture: directions for therapy. Toxicology 258, 39–46.
Munro, N. B., Talmage, S. S., Griffin, G. D., Waters, L. C., Watson, A. P., King, J. F., and Hauschild, V. (1999). The source, fate and toxicity of chemical warfare agent degradation products. Environ. Health Perspect. 107, 933–974.
Nolano, M., Simone, D. A., Wendelschafer-Crabb, G., Johnson, T., Hazen, E., and Kennedy, T. (1999).Topical capsaicin in humans: parallel loss of epidermal nerve fibers and pain sensation. Pain 81, 135–145.
Noort, D., Fidder, A., Degenhardt-Langelaan, C. E., and Hulst, A. G. (2008). Retrospective detection of sulfur mustard exposure by mass spectrometric analysis of adducts to albumin and hemoglobin: an in vivo study. J. Anal. Toxicol. 32, 25–30.
Noort, D., Hulst, A. G., de Jong, L. A., and Benschop, H. P. (1999). Alkylation of human serum albumin by sulfur mustard in vitro and in vivo: Mass spectrometric analysis of a cysteine adduct as a sensitive biomarker of exposure. Chem. Res. Toxicol. 12, 7115–7721.
Oh, S.-H., and Lim, S.-C. (2009). Endoplasmic reticulum stress-mediated autophagy/apoptosis induced by capsaicin (8-methyl-N-vanillyl-6-nonenamide) and dihydrocapsaicin is regulated by extent of c-Jun NH2-terminal kinase/extracellular signal-regulated kinase activation in WI38 lung epithelial fibroblast cells. J. Pharmacol. Exp. Ther. 329,112–122.
óMalley, M. A., Richmond, D., Ibarra, M., Barry, T., Smith, M., and Calvert, G. M. (2004). Illness associated with drift of chloropicrin soil fumigant onto a residential area-Kern County, California 2003. MMWR Morb. Mortal. Wkly. Rep. 53, 740–742.
Ozer, J., Ratner, M., Shaw, M., Bailey, W., and Schomaker, S. (2008). The current state of serum biomarkers of hepatotoxicity. Toxicology 245, 194–205.
Papirmeister, B., Gross. C. L., Meier, H. L., Petrali, J. P., and Johnson. J. B. (1985). Molecular basis for mustard-induced vesication. Fundam. Appl. Toxicol. 5,134–149.
Paromov, V., Suntres, Z., Smith, M., and Stone, W. L. (2007). Sulfur mustard following dermal exposure; role of oxidative stress, and antioxidant therapy. J. Burns Wounds 7, 60–85.
Pershing, L. K., Reilly, C. A., Corlett, J. L., and Crouch, D. J. (2004). Effects of vehicle on the uptake and elimination kinetics of capsaicinoids in human skin in vivo. Toxicol. Appl. Pharmacol. 200, 73–81.
Phillips, D. H. (2008). “Biomarkers of exposure: adducts,” in Molecular Epidemiology of Chronic Diseases, eds C. Wild, P. Vineis, and S. Garte (England: Jonh Wiley and Sons, Ltd), 111–125.
Plewa, M. J., Wagner, E. D., and Jazwierska, P. (2004). Halomethane drinking water disinfection by-products; chemical characterization and mammalian cell cytotoxicity and genotoxicity. Environ. Sci. Technol. 38, 62–68.
Read, R. W., and Black, R. M. (2004). Analysis of β-lyase metabolites of sulfur mustard in urine by electrospray liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 28, 346–351.
Reilly, C. A., Crouch, D. J., and Yost, G. S. (2001). Quantitative analysis of capsaicinoids in fresh peppers, oleoresin capsicum and pepper spray products. J. Forensic Sci. 46, 502–509.
Reilly, C. A., Crouch, D. J., Yost, G., and Fatah, A. A. (2002). Determination of capsaicin, nonivamide, and dihydrocapsaicin in blood and tissue by liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 26, 313–319.
Reilly, C. A., Elhlhardt, W. J., Jackson, D. A., Kulanthaivel, P., Mutlib, A. E., Espina, R. J., Moody, D. E., Crouch, D. J., and Yost, G. S. (2003a). Metabolism of capsaicin by cytochrome P450 produces novel dehydrogenated metabolites and decreases cytotoxicity to lung and liver cells. Chem. Res. Toxicol. 16, 336–349.
Reilly, C. A., Taylor, J. L., Lanza, D. L., Carr, B. A., Crouch, D. J., and Yost, G. S. (2003b). Capsaicinoids cause inflammation and epithelial cell death through activation of vanilloid receptors. Toxicol. Sci. 73, 170–181.
Reilly, C. A., and Yost, G. S. (2006). Metabolism of capsainoids by P450 enzymes: A review of recent findings on reaction mechanisms, bioactivation, and detoxification processes. Drug Metab. Rev. 38, 685–706.
Richardson, J. D., and Vasko, M. R. (2002). Cellular mechanisms of neurogenic inflammation. J. Pharmacol. Exp. Ther. 302, 839–845.
Riches, J., Read, R. W., and Black, R. M. (2007). Analysis of the sulfur mustard metabolites thiodiglycol and thiodiglycol sulphoxide in urine using isotope-dilution gas chromatography-ion trap tandem mass spectrometry. J. Chromatogr. 845, 114–120.
Richeux, F., Cascante, M., Ennamany, R., Sachez, D., Sanni, A., Saboureau, D., and Creppy, E. E. (2000). Implications of oxidative stress and inflammatory process in the toxicity of capsaicin in human endothelial cells: lack of DNA strand breakage. Toxicology 147, 41–49.
Rowell, M., Kehe, K., Balszuweit, F., and Thiermann, H. (2009). The chronic effects of sulfur mustard exposure. Toxicology 263, 9–11.
Ruff, A. L., and Dillman, J. F. (2010). Sulfur mustard induced cytokine production and cell death: Investigating the potential roles of the p38, p53 and NF-κB signalling pathways with RNA interference. J. Biochem. Mol. Toxicol. 0, 1–10.
Ruzo, L. O. (2006). Physical, chemical and environmental properties of selected chemical alternatives for the pre-plant use of methyl bromide as soil fumigant. Pest Manag. Sci. 62, 99–113.
Sabourin, C. L., Petrali, J. P., and Casillas, R. P. (2000). Alterations in inflammatory cytokine gene expression in sulfur mustard-exposed mouse skin. J. Biochem. Mol. Toxicol. 14, 291–302.
Sanchez, A. M., Martinez-Botas, J., Malagarie-Cazenave, M., Olea, N., Vara, D., Lasuncion, M. A., and Diaz-Laviada, I. (2008). Induction of the endoplasmic reticulum stress protein GADD153/CHOP by capsaicin in prostate PC-3 cells: a microarray study. Biochem. Biophys. Res. Commun. 372, 785–791.
Sanchez-Moreno, S., Alonso-Prados, E., Alonso-Prados, J. L., and Garcia-Baudin, J. M. (2009). Multivariate analysis of toxicological and environmental properties of soil nematicides. Pest Manag. Sci. 65, 82–92.
Saria, A., Skofitsch, G., and Lembeck, F. (1982). Distribution of capsaicin in rat tissues after systemic administration. J. Pharm. Pharmacol. 34, 273–275.
Schneider, M., Quistad, G. B., and Casida, J. E. (1999). Glutathione activation of chloropicrin in the Salmonella mutagenicity test. Mutat. Res. 439, 233–238.
Shakarjian, M. P., Heck, D. E., Gray, J. P., Sinko, P. J., Gordon, M. K., Casillas, R. P., Heindel, N., Gerecke, D. R., Laskin, D. L., and Laskin, J. D. (2010). Mechanisms mediating the vesicant actions of sulfur mustard after cutaneous exposure. Toxicol. Sci. 114, 5–19.
Shin, C. Y., Shin, J., Kim, B.-M., Wang, M.-H., Jang, J.-H., Surh, Y.-J., and Oh, U. (2003). Essential role of mitochondrial permeability transition in vanilloid receptor 1-dependent cell death of sensory neurons. Mol. Cell. Neurosci. 24, 57–68.
Smith, J., and Greaves, I. (2002). The use of chemical incapacitant sprays: a review. J. Trauma 52, 595–600.
Smith, J. R., Capacio, B. R., Korte, W. D., Woolfitt, A. R., and Barr, J. R. (2008). Analysis for plasma protein biomarkers following an accidental human exposure to sulfur mustard. J. Anal. Toxicol. 32, 17–24.
Smith, K. J., Smith, W. J., Hamilton, T., Skelton, H. G., Graham, J. S., Okerberg, C. D., Moeller, R., and Hackley, B. E. (1998). Histopathologic an immunohistochemical features in human skin after exposure to nitrogen and sulfur mustard. Am. J. Dermatopathol. 20, 22–28.
Snyman, T., Stewart, M. J., and Steenkamp, V. (2001). A fatal case of pepper poisoning. Forensic Sci. Int. 124, 43–46.
Solberg, Y., Alcalay, M., and Belkin, M. (1997). Ocular injury by mustard. Surv. Ophthalmol. 41, 461–466.
Somani, S. M., and Babu, S. R. (1989). Toxicodynamics of sulfur mustard. Therapeutic review. Int. J. Clin. Pharmacol. Ther. Toxicol. 27, 419–435.
Sparks, S. E., Quistad, G. B., and Casida, J. E. (1997). Chloropicrin: reactions with biological thiols and metabolism in mice. Chem. Res. Toxicol. 10, 1001–1007.
Sparks, S. E., Quiatad, G. B., Li W., and Casida, J. E. (2000). Chloropicrin dechlorination in relation to toxic action. J. Biochem. Mol. Toxicol. 14, 26–32.
Ständer, S., Moormann, C., Schumacher, M., Buddenkotte, J., Artuc, M., Shpacovitch, V., Brzoska, T., Lippert, U., Henz, B. M., Luger, T. A., Metze, D., and Steinhoff, M. (2004). Expression of vanilloid receptor subtype 1 in cutaneous sensory nerve fibers, mast cells, and epithelial cells of appendage structures. Exp. Dermatol. 13, 129–139.
Surh, Y.-J., Ahn, S. A., Kim, K.-C., Park, J.-B., Sohn, Y. W., and Lee, S. S. (1995). Metabolism of capsaicinoids: evidence for aliphatic hydroxylation and its pharmacological implications. Life Sci. 56, 305–311.
Surh, Y.-J., and Lee, S. S. (1995). Capsaicin, a double sword: toxicity, metabolism, and chemopreventive potential. Life Sci. 56, 1845–1855.
Szallasi, A., and Blumberg, P. M. (1999). Vanilloid (capsaicin) receptor and mechanisms. Pharmacol. Rev. 51, 159–211.
Szallasi, A., Cortright, D. N., Blum, C. A., and Eid, S. R. (2007). The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat. Rev. Drug Discov. 6, 357–372.
Szöke, E., Seress, L., and Szolcsanyi, J. (2002). Neonatal capsaicin treatment results in prolonged mitochondrial damage and delayed cell death of B cells in the rat trigeminal ganglia. Neuroscience 113, 925–937.
Takeshima, Y., Inai, K., Bennett, W. P., Metcalf, R. A., Welsh, J. A., Yonehara, S., Hayashi, Y., Fujihara, M., Yamakido, M., Akiyama, M., Tokouka, S., Land, C. E., and Harris, C. C. (1994). p54 mutations in lung cancers from Japanese mustard gas workers. Carcinogenesis 15, 2075–2079.
Thomas, K. C., Sabnis, A. S., Johansen, M. E., Lanza, D. L., Moos, P. J., Yost, G. S., and Reilly, C. A. (2007). Transient receptor potential vanilloid 1 agonists cause endoplasmic reticulum stress and cell death in human lung cells. J. Pharmacol. Exp. Ther. 321, 830–838.
Törnqvist, M., Fred C., Haglung,J., Helleberg, H., Paulsson, B., and Rydberg, P. (2002). Protein adducts: quantitative and qualitative aspects of their formation, analysis and application. J. Chromatogr. B 778, 279–308.
van der Schans, G. P., Noort, D., Mars-Groenendijk, R. H., Fidder, A., Chau, L. F., de Jong, P. A., and Benschop, H. P. (2002). Immunochemical detection of sulfur mustard adducts with keratins in the stratum corneum of human skin. Chem. Res. Toxicol. 15, 21–25.
van der Schans, G. P., Scheffer, A. G., Mars-Groenendijk, R. H., Fidder, A., Benschop, H. P., and Baan, R. A. (1994). Immunochemical detection of adducts of sulfur mustard to DNA of calf thymus and white blood cells. Chem. Res. Toxicol. 7, 408–413.
Veronesi, B., Carter, J. D., Devlin, R. B., Simon, S. A., and Oortgiesen, M. (1999). Neuropeptides and capsaicin stimulate the release of inflammatory cytokines in a human bronchial cell line. Neuropeptides 33, 447–456.
Vineis, P., and Garte, S. (2008). “Biomarker validation,” in Molecular Epidemiology of Chronic Diseases, eds C. Wild, P. Vineis, and S. Garte (England: Jonh Wiley and Sons, Ltd.), 71–81.
Vineis, P., and Perera, F. (2000). DNA adducts as markers of exposure to carcinogens and risk of cancer. Int. J. Cancer 88, 325–328.
Vineis, P., and Perera, F. (2007). Molecular epidemiology and biomarkers in etiologic cancer research: the new in light of the old. Cancer Epidemiol. Biomarkers Prev. 6, 1954–1965.
Volans, G. N., and Karalliedde, L. (2002). Long-term effects of chemical weapons. Lancet 360, 35–36.
Vähäkangas, K. (2003). TP53 mutations in workers exposed to occupational carcinogens. Hum. Mutat. 21, 240–251.
Vähäkangas, K. (2008). “Molecular epidemiology and ethics: biomarkers for disease susceptibility,” in Molecular Epidemiology of Chronic Diseases, eds C. Wild, P. Vineis, and S. Garte (England: John Wiley and Sons, Ltd.), 281–297.
Watson, A. P., and Griffin, G. D. (1992). Toxicity of vesicant agent scheduled for destruction by the chemical stockpile disposal program. Environ. Health Perspect. 98, 259–280.
Watson, W. A., Stremel, K., and Westdorp, E. J. (1996). Oleoresin capsicum (cap-stun) toxicity from aerosol exposure. Ann. Pharmacother. 30, 733–735.
Watson, W. P., and Mutti, A. (2004). Role of biomarkers in monitoring exposures to chemicals: present position, future prospects. Biomarkers 9, 211–242.
Wolvetang, E. J., Larm, J. A., Moutsoulas, P., and Lawen, A. (1996). Apoptosis induced by inhibitors of the plasma membrane NADH-oxidase involves Bcl-2 calcineurin. Cell Growth Differ. 7, 1315–1325.
Yaws, C. L. (2008). Yaws’ Handbook of Physical Properties for Hydrocarbons and Chemicals. Available at: http://www.knovel.com/web/portal/basic_search/display
Keywords: biomarkers, toxicity, molecular mechanisms of action, irritating and incapacitating agents
Citation: Pesonen M, Vähäkangas K, Halme M, Vanninen P, Seulanto H, Hemmilä M, Pasanen M and Kuitunen T (2010) Capsaicinoids, chloropicrin and sulfur mustard: possibilities for exposure biomarkers. Front. Pharmacol. 1:140. doi: 10.3389/fphar.2010.00140
Received: 11 June 2010;
Accepted: 26 November 2010;
Published online: 20 December 2010.
Edited by:
Ursula Gundert-Remy, Drug Commission of the German Medical Association, GermanyReviewed by:
Hans Mielke, Federal Institute for Risk Assessment, GermanyUrsula Gundert-Remy, Drug Commission of the German Medical Association, Germany
Copyright: © 2010 Pesonen, Vähäkangas, Halme, Vanninen, Seulanto, Hemmilä, Pasanen and Kuitunen. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Maija Pesonen, Faculty of Health Sciences, School of Pharmacy/Toxicology, University of Eastern Finland, P.O. Box 1627, 70211 Kuopio, Finland. e-mail:bWFpamEucGVzb25lbkB1ZWYuZmk=