 Dao W. Wang1,2
Dao W. Wang1,2 Akshitkumar M. Mistry1 Kristopher M. Kahlig3 Jennifer A. Kearney1 Jizhou Xiang2
Akshitkumar M. Mistry1 Kristopher M. Kahlig3 Jennifer A. Kearney1 Jizhou Xiang2 Alfred L. George Jr.1,3*
Alfred L. George Jr.1,3*
- 1 Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, USA
- 2 Department of Pharmacology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3 Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, USA
Propranolol is a widely used, non-selective β-adrenergic receptor antagonist with proven efficacy in treating cardiovascular disorders and in the prevention of migraine headaches. At plasma concentrations exceeding those required for β-adrenergic receptor inhibition, propranolol also exhibits anti-arrhythmic (“membrane stabilizing”) effects that are not fully explained by β-blockade. Previous in vitro studies suggested that propranolol may have local anesthetic effects. We directly tested the effects of propranolol on heterologously expressed recombinant human cardiac (NaV1.5) and brain (NaV1.1, NaV1.2, NaV1.3) sodium channels using whole-cell patch-clamp recording. We found that block was not stereospecific as we observed approximately equal IC50 values for tonic and use-dependent block by R-(+) and S-(−) propranolol (tonic block: R: 21.4 μM vs S: 23.6 μM; use-dependent block: R: 2.7 μM vs S: 2.6 μM). Metoprolol and nadolol did not block NaV1.5 indicating that sodium channel block is not a class effect of β-blockers. The biophysical effects of R-(+)-propranolol on NaV1.5 and NaV1.1 resembled that of the prototypical local anesthetic lidocaine including the requirement for a critical phenylalanine residue (F1760 in NaV1.5) in the domain 4 S6 segment. Finally, we observed that brain sodium channels exhibited less sensitivity to R-(+)-propranolol than NaV1.5 channels. Our findings establish sodium channels as targets for propranolol and may help explain some beneficial effects of the drug in treating cardiac arrhythmias, and may explain certain adverse central nervous system effects.
Introduction
Propranolol is a widely used, non-selective β-adrenergic receptor antagonist with proven efficacy in the treatment of hypertension, in reducing mortality after myocardial infarction, in treatment of other cardiovascular disorders, and in migraine prophylaxis. Clinically, propranolol is administered as a racemic mixture with equal concentrations of R-(+)- and S-(−)-enantiomers. Although the two stereoisomers possess similar physical properties, the R-(+)-enantiomer (previously designated d-propranolol) is practically devoid of β-blocking activity (Mehvar and Brocks, 2001). The pharmacological effects of the drug in treating cardiovascular diseases can be largely explained by β-adrenergic receptor block, but other recognized effects such as anti-arrhythmic “membrane stabilizing” cardiac effects may be due to other molecular targets. By contrast, the effects of propranolol in brain that are responsible for preventing migraine attacks are not clear.
Voltage-gated sodium channels are established pharmacological targets for local anesthetics and many other drugs with shared mechanisms of action including certain anti-arrhythmic and anti-epilepsy agents. Earlier in vitro electrophysiological studies of the effects of propranolol on heart rate and conduction performed in frog atria, rat and canine ventricular myocytes suggested that the drug might be interacting with sodium channels. Further studies demonstrated that racemic propranolol and d-propranolol can suppress ventricular arrhythmias in dogs and humans (Woosley et al., 1979; Dawson et al., 1984; Murray et al., 1990) and exert significant effects on cardiac conduction at doses exceeding that needed for β-adrenergic blockade (Duff et al., 1983). We recently correlated control of life-threatening perinatal ventricular arrhythmias caused by de novo mutation of the cardiac sodium channel gene (SCN5A encoding NaV1.5) with combination propranolol and mexiletine therapy, and demonstrated that propranolol blocks heterologously expressed wild-type and mutant NaV1.5 channels (Wang et al., 2008). A more recent report confirmed that propranolol can suppress the increased persistent current evoked by NaV1.5 mutations associated with congenital long-QT syndrome (LQTS) (Bankston and Kass, 2010). Previously, propranolol was also demonstrated to block heterologously expressed human skeletal muscle NaV1.4 channels (Desaphy et al., 2003). These various experimental findings provided direct evidence that propranolol could block cardiac and skeletal muscle sodium channels. By contrast, there have been no investigations into effects of propranolol on recombinant brain sodium channels.
We examined the effects of propranolol on heterologously expressed cardiac and brain sodium channels with goals of elucidating the biophysical consequences of block, and testing the hypothesis that amino acid residues required for local anesthetic effects are also critical for propranolol effects on sodium channels. We established that cardiac sodium channels have greater affinity for propranolol than brain sodium channels, that block is not stereoselective and that the drug evokes biophysical effects that resemble but are not identical to those of the prototypic local anesthetic agent lidocaine.
Materials and Methods
Heterologous Expression of Sodium Channels
A recombinant human NaV1.5 sodium channel cDNA in a mammalian expression plasmid (pRc/CMV-hH1) (Gellens et al., 1992) was co-expressed with the human β1 subunit (hβ1) in tsA201 cells by transient transfection using FuGene 6 (Roche Diagnostics, Indianapolis, Inc.) and 0.5 μg of each plasmid. A specific NaV1.5 mutation resulting in substitution of phenylalanine-1760 (based on amino acid numbering in Swiss-Prot: Q14524) with alanine was engineered by polymerase chain reaction (PCR) mediated site-directed mutagenesis and the final construct was sequenced in its entirety to confirm the nucleotide change and exclude inadvertent polymerase errors. Transiently transfected cells were transferred to a chamber (Warner Instrument Corp, Hamden, CT, USA) 48–72 h after transfection for electrophysiological measurements. Expression of hβ1 was coupled to expression of enhanced green fluorescent protein in the plasmid pEGFP-IRES-hβ1, and only cells exhibiting green fluorescence were selected for patch-clamp recording.
Three human neuronal sodium channels (NaV1.1, NaV1.2, NaV1.3) were stably co-expressed with human β1 and β2 subunits in HEK-293 cells using a recently described transposon strategy (Kahlig et al., 2010). Plasmids encoding human NaV1.1 and NaV1.2 were previously described (Lossin et al., 2002; Misra et al., 2008; Kahlig et al., 2010) except that two missense variants in NaV1.2 (V1325A and V1768L) were corrected to match the reference sequence (GenBank accession number NP_066287.2). Both NaV1.1 and NaV1.2 were cloned into a transposon vector (Kahlig et al., 2010). A full-length human NaV1.3 cDNA was obtained from Origene Technologies Inc. (product code SC316037, Rockville, MD, USA). The open reading frame (5856 bp) corresponding to the major splice isoform (GenBank accession number NM_001081676.1) was introduced into a mammalian expression vector with the CMV promoter and an HSV-TK polyadenylation signal followed by a neomycin resistance gene. Inverted repeat elements from the piggyBac transposon were introduced flanking the CMV promoter and neomycin selection cassette. For all neuronal sodium channel constructs, full-length cDNAs were propagated in Stbl2 cells (Invitrogen, Carlsbad, CA, USA) at 30°C. The open reading frame of each sodium channel plasmid was fully sequenced before use.
Conventional Patch-Clamp Recording
Sodium currents were recorded at room temperature (22–23°C) using the whole-cell patch-clamp technique as described previously (Lossin et al., 2002; Wang et al., 2002, 2007; Benson et al., 2003). Specific voltage-clamp protocols are depicted in each figure. The bath solution contained the following (in mmol/l): 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.35 (adjusted with NaOH). The pipette solution (intracellular solution) contained the following (in mmol/l): 10 NaF, 110 CsF, 20 CsCl, 2 EGTA, and 10 HEPES, pH 7.35 (adjusted with CsOH). Osmolarity was adjusted to 310 mOsm with sucrose. Data acquisition was carried out with an Axopatch 200B patch-clamp amplifier and pCLAMP 8.0 software. Electrode resistance ranged from 0.8 to 1.5 MΩ. Pipette capacitance was corrected, and whole-cell capacitance and series resistance were compensated 85–95% (voltage error <3 mV). Cells were allowed to stabilize for precisely 10 min after establishment of the whole-cell configuration before current was measured. Whole-cell currents were acquired at 20–50 kHz and filtered at 5 kHz. Recordings from cells exhibiting peak current amplitudes smaller than 0.8 nA were excluded from analysis to avoid potential endogenous channel contamination. Unless otherwise stated, holding potential was −120 mV. Cells exhibiting very large whole-cell currents (>6 nA) were excluded if voltage control was compromised. Leak current was subtracted using a P/4 procedure. Whole-cell capacitance was assessed by integrating the capacitive transient elicited by a 10-mV voltage step from −120 to −110 mV with 10-kHz filtering.
Automated Planar Patch-Clamp Recording
Whole-cell planar patch-clamp recording was used to examine drug block of stably expressed neuronal sodium channels using an automated electrophysiology platform (NPC-16 Patchliner system, Nanion Technologies GmbH, Germany) as described previously (Kahlig et al., 2010). Cells were detached with 0.25% trypsin–EDTA (Invitrogen, Carlsbad, CA, USA) for 1 min at room temperature, then triturated in growth media, centrifuged (100×g, 2 min) and resuspended at 1 × 106 cells/ml in fresh growth media. Following a recovery period (30 min, 37°C) the cells were resuspended and placed in the Patchliner “cell hotel.” Patchcontrol HT software (HEKA Elektronik, Germany) was used to control application of solutions and pressures necessary to establish the whole-cell configuration. Sodium currents were recorded at room temperature (22–23°C) using external and internal solutions identical to those used for conventional patch-clamp experiments. Following cell contact with the 2 MΩ planar electrode, seal enhancer solution (in mM: 80 NaCl, 3 KCl, 35 CaCl2, 10 HEPES/NaOH pH7.4) was added to the external solution to promote giga-ohm seal formation. After establishing the whole-cell configuration, the seal enhancer solution was replaced with two washes of fresh external solution and the series resistance was compensated (50%, 10 μs). Cells were allowed to stabilize for precisely 10 min after establishment of the whole-cell configuration before current was measured. Patchmaster software (HEKA) was used to automatically compensate for whole-cell capacitance and series resistance, and perform voltage-clamp protocols. Leak currents were subtracted by using an online P/4 procedure. Cells exhibiting peak whole-cell current amplitudes <0.6 nA or leak currents >0.5 nA were excluded from analysis. Whole-cell currents were low-pass filtered at 5 kHz and digitized at 50 kHz.
Pharmacology
All drugs (propranolol, R-(+)-propranolol, S-(−)-propranolol, metoprolol, and nadolol; obtained from Sigma Chemical Co, St. Louis, MO, USA) were freshly prepared before experiments from 10 or 100 mM stock solutions stored at −20°C then applied by rapid exchange of extracellular solution. For NaV1.5, tonic block was measured using conventional whole-cell recording at 0.2 Hz after steady-state was achieved in the presence of drug (6–8 min) and then normalized to the current amplitude recorded in the absence of drug. Steady-state use-dependent block was achieved in response to trains of 150 pulses (to –10 mV, 100 ms) at frequencies indicated in the figure legends. Use-dependent block was calculated as the degree of block during the last pulse in a pulse train normalized to the current amplitude recorded without drug.
Pharmacology of brain sodium channels was assessed using the Patchliner system. Tonic block was assessed with a sequence of 20 ms depolarizing steps from −80 to 60 mV in increments of 10 mV. The peak current generated at 0 mV under varying concentrations of R-(+)-propranolol was normalized to the peak current at 0 mV under control conditions without R-(+)-propranolol. Use-dependent block was assessed with a train of 100 depolarizing pulses (to 0 mV, 5 ms, 5 Hz) from a holding potential of −120 mV. Peak current of the last pulse was normalized to the peak current of the first pulse in the same condition.
Data Analysis
All data were analyzed with pCLAMP 8.0 (Axon Instruments, Inc., Sunnyvale, CA, USA) and plotted using SigmaPlot 2002 (SPSS, Inc., Chicago, IL, USA). Steady-state availability was fit with the Boltzmann equation (1),
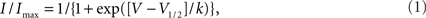
to determine the membrane potential for half-maximal inactivation (V1/2) and slope factor (k). Recovery from inactivation was analyzed by fitting data with either a single (Eq. 2) or double (Eq. 3) exponential function as appropriate:
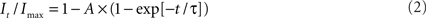
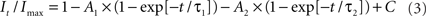
where A, τ, and C refer to amplitudes, time constant, and residual current, respectively. Voltage-dependence of channel activation was estimated by measuring peak sodium current during a variable test potential from a holding potential of −120 mV. Current at each membrane potential was divided by the electrochemical driving force for sodium ions and normalized to the maximum sodium conductance. Data were fit by a Boltzmann function using a non-linear least-squares minimization method (Marquardt–Levenberg algorithm).
To estimate drug blocking effect on sodium channels, the Hill equation (4) was used:

where F is fraction of drug block, L is drug concentration, n is Hill coefficient, and IC50 is the drug concentration producing half-maximal inhibition. Results are presented as mean ± SEM, and statistical comparisons were made between the drug-free (control) condition and during drug application using an unpaired Student’s t-test or one-way ANOVA followed by a Tukey post-test. Statistical significance was assumed for P < 0.05. In some figures, error bars are smaller than data symbols.
Results
Stereoselectivity and Specificity of Propranolol Block of NaV1.5
Propranolol is marketed as a racemic mixture of two enantiomers denoted R-(+) and S-(−), but only the S-(−) form mediates clinically relevant β-blocker effects (Mehvar and Brocks, 2001). We previously demonstrated that racemic propranolol and R-(+)-propranolol block NaV1.5 channels (Wang et al., 2008) but did not investigate the stereoselectivity of this effect. Therefore, we tested tonic and use-dependent block of heterologously expressed human NaV1.5 channels co-expressed with the human sodium channel β1 subunit to compare effects of the two propranolol enantiomers. Figure 1 illustrates representative levels of block and concentration–response relationships for tonic and use-dependent block by R-(+) and S-(−) propranolol. Similar IC50 values were observed for both enantiomers under conditions promoting tonic (R: 21.4 μM vs S: 23.6 μM) and use-dependent (R: 2.7 μM vs S: 2.6 μM) block. These data indicate that both enantiomers exhibit equivalent blocking potency for NaV1.5 channels. Further experiments were performed using only R-(+)-propranolol.
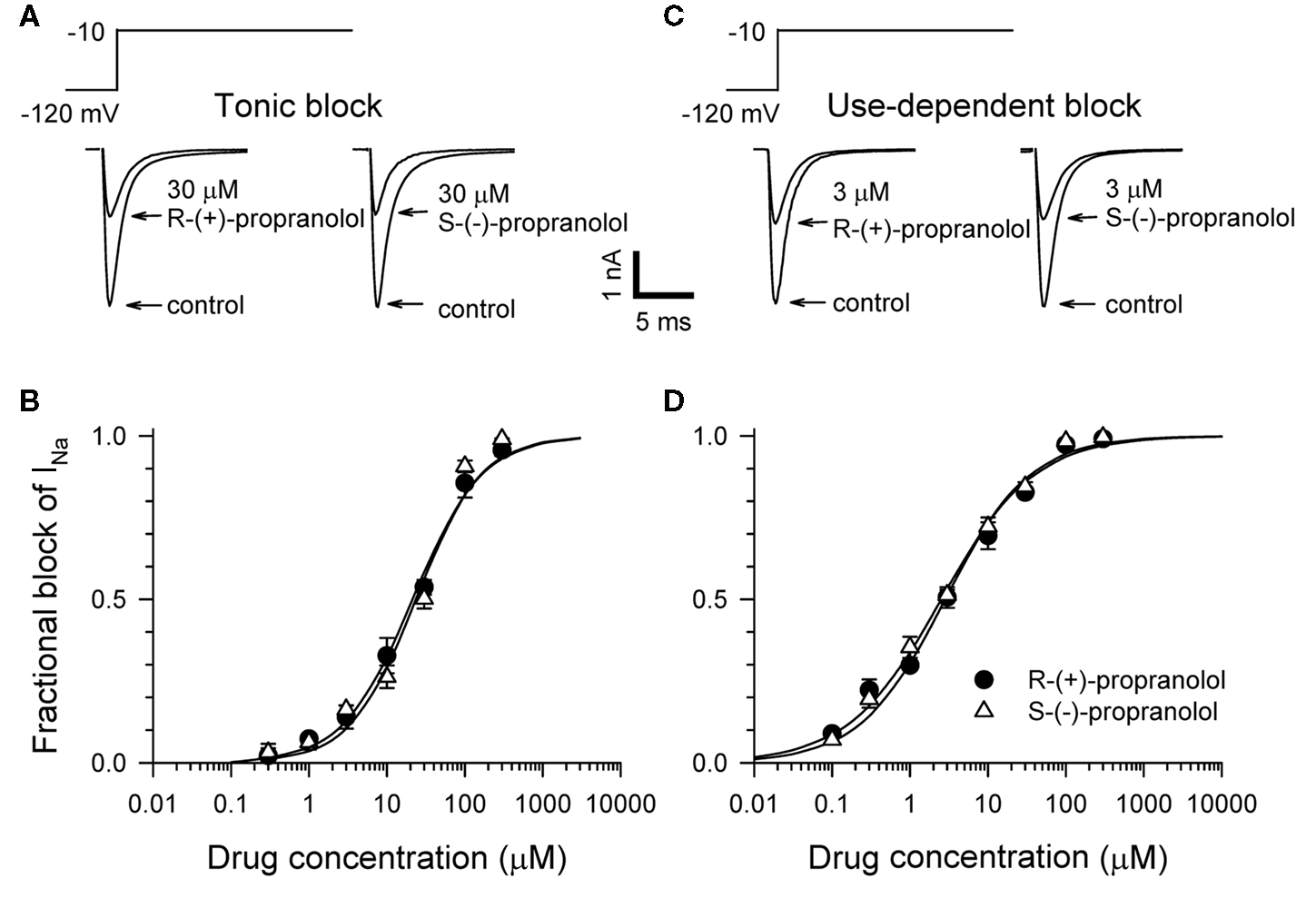
Figure 1. Stereoselectivity of propranolol block of NaV1. 5 sodium channels. (A) Representative whole-cell voltage-clamp recordings made from tsA201 cells expressing NaV1.5 with the human β1 subunit before and after application of 30 μM R-(+)-propranolol or S-(−)-propranolol. Tonic block was elicited by 100 ms depolarizations to −10 mV from a holding potential of −120 mV delivered at a frequency of 0.2 Hz. Recordings made in the presence of drug illustrate the steady-state level of block achieved after 5–7 min. (B) Concentration–response relationships for tonic block (n = 7–14 cells per data point). (C) Representative traces illustrating use-dependent block by 3 μM R-(+)-propranolol or S-(−)-propranolol. Use-dependent block was elicited at a pulsing frequency of 1 Hz. (D) Concentration–response relationships for use-dependent block (n = 7–14 cells per data point).
To test whether propranolol is unique among β-adrenergic receptor blockers in having sodium channel blocking activity, we tested two other commonly prescribed drugs of this class, nadolol (non-selective β-blocker) and metoprolol (selective β1-blocker). Figure 2 illustrates that neither 10 μM nadolol nor 10 μM metoprolol exhibits any appreciable tonic or use-dependent blocking effects on NaV1.5. In experiments testing use-dependent block (Figure 2B), the fraction of residual peak sodium current after 100 successive depolarizing pulses delivered at 1 Hz was 0.93 ± 0.02 and 0.97 ± 0.01 for metoprolol and nadolol, respectively, compared with 0.49 ± 0.03 for 3 μM R-(+)-propranolol. The chemical basis for this selectivity is not entirely obvious but differences in the aromatic ring structures are the most distinctive features of these compounds (Figure 2C). Our findings indicate that propranolol block of NaV1.5 is not stereoselective and not a universal property of all β-blockers.
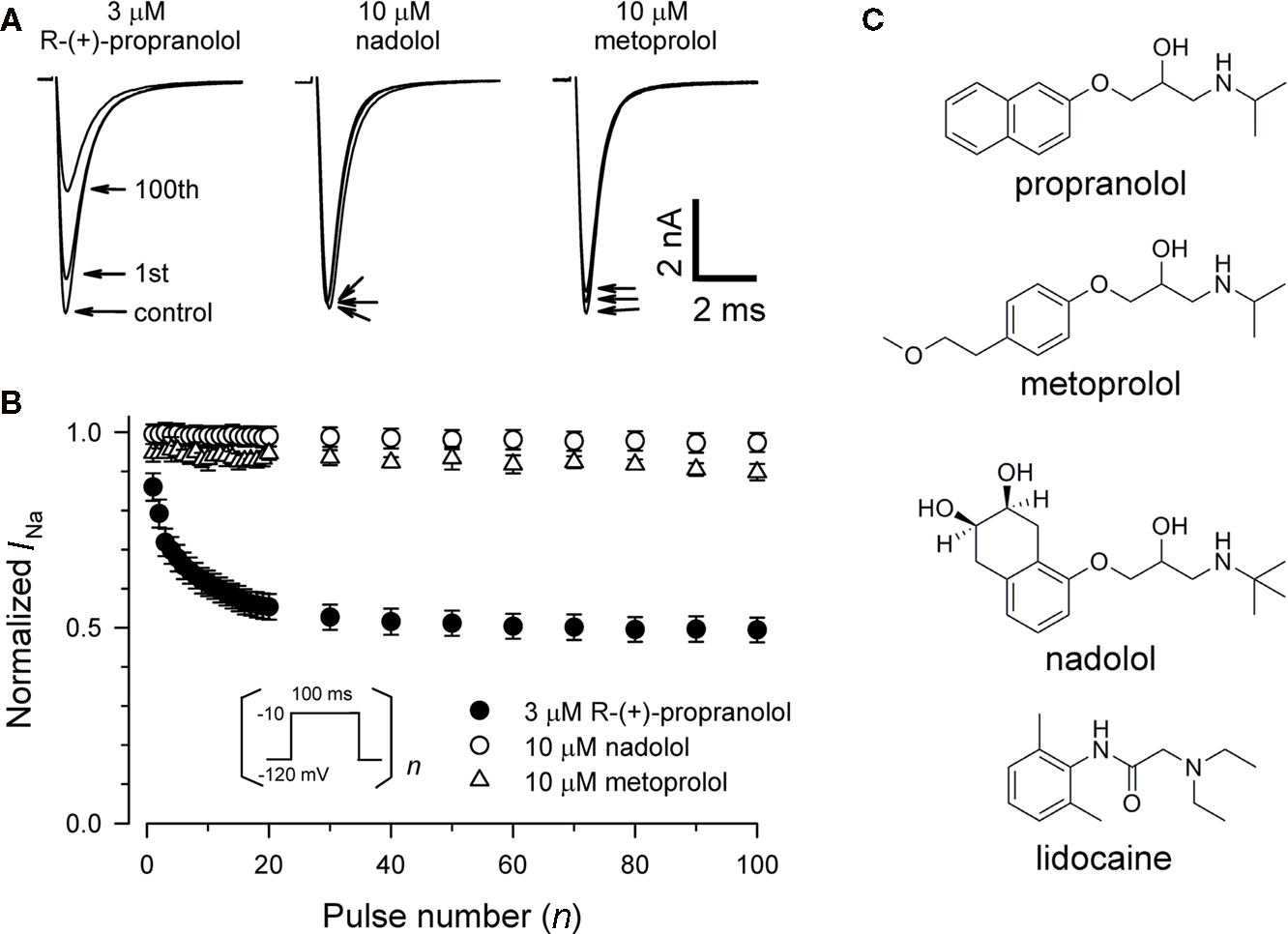
Figure 2. Chemical selectivity of propranolol block of NaV1. 5 sodium channels. (A) Representative whole-cell recordings made from NaV1.5 expressing cells before and after application of 3 μM R-(+)-propranolol, 10 μM nadolol, or 10 μM metoprolol during depolarizing pulse trains to −10 mV at 1 Hz. Pulse protocol was the same as depicted in Figure 1A. Current traces were obtained in drug-free control solution, after steady-state tonic block (first trace) and use-dependent block at 1 Hz (100th trace) measured 4–5 min after drugs were applied to the bath solution. (B) Comparison of use-dependent inhibition of NaV1.5 by 3 μM R-(+)-propranolol, 10 μM nadolol, or 10 μM metoprolol during 1 Hz pulse trains. Peak currents of each subsequent trace were normalized to the peak of the first current trace, and plotted as a function of pulse number (n = 7–12). For clarity, not all data points are plotted. There was no use-dependent reduction of peak sodium current at 1 Hz in the control condition. (C) Chemical structures of propranolol, metoprolol, nadolol, and lidocaine.
Biophysical Effects of Propranolol on NaV1.5
Additional experiments were performed to determine effects of R-(+)-propranolol on the biophysical properties of NaV1.5 channels and to make comparisons with effects exerted by the prototypic local anesthetic agent lidocaine. Figure 3 illustrates the effects of 3 μM R-(+)-propranolol under conditions favoring tonic block on current–voltage relationship, voltage-dependence of activation and inactivation, and on the time course of recovery from inactivation. The drug caused significant hyperpolarizing shifts in the peak of the I–V relationship (Figure 3A), in midpoint of the conductance–voltage relationship (Figure 3B) and in the voltage-dependence of channel inactivation (Figure 3C; Table 1). The effects on steady-state inactivation were similar for 3 μM R-(+)-propranolol and 100 μM lidocaine, but the magnitude of shift in the activation curve with R-(+)-propranolol was significantly greater than with lidocaine (Table 1). Recovery from inactivation was also slowed by R-(+)-propranolol, but this effect was much less dramatic than that observed for lidocaine (Figure 3D; Table 1). Both drugs induced a slow component of recovery from inactivation and increased the magnitude of the time constant corresponding to the dominant, faster component (Table 1). These results demonstrate that R-(+)-propranolol exhibits biophysical effects on NaV1.5 that are similar but not identical to lidocaine.
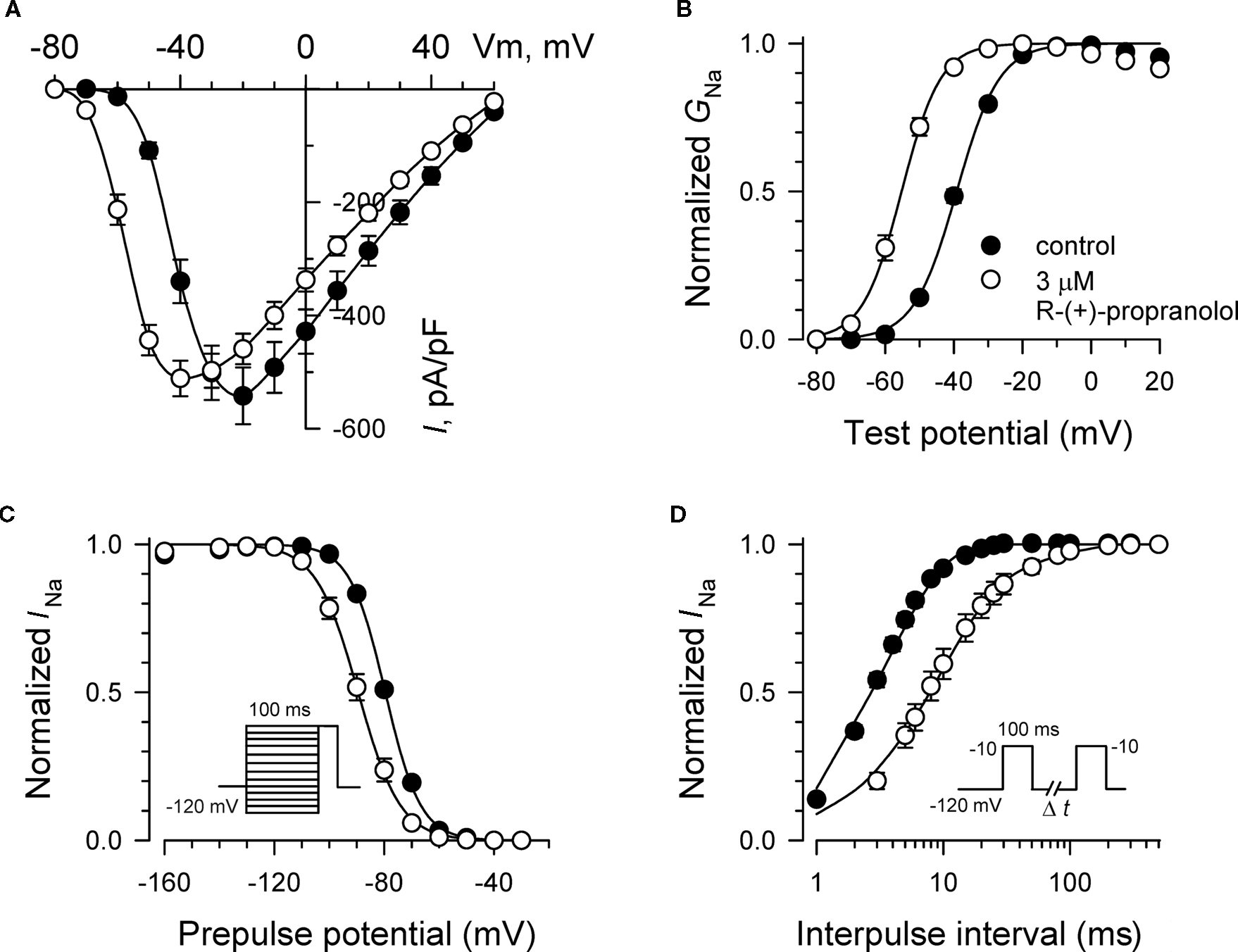
Figure 3. Biophysical effects of R-(+)-propranolol on NaV1. 5. (A) Current–voltage relationships in the absence and presence of 3 μM R-(+)-propranolol. Peak current amplitudes were elicited from a holding potential of −120 mV to various test potentials then normalized to cell capacitance. (B) Conductance–voltage relationship in the absence and presence of 3 μM R-(+)-propranolol. (C) Voltage-dependence of sodium channel availability (“steady-state inactivation”) in the absence and presence of drug measured using a two-pulse protocol illustrated in the inset. (D) Effect of (R)-(+)-propranolol on recovery from inactivation (holding potential −120 mV). Fit parameters for data in (B–D) are provided in Table 1, and lines in (B,C) represent Boltzmann fits. Each point represents mean ± SEM from 12 cells.
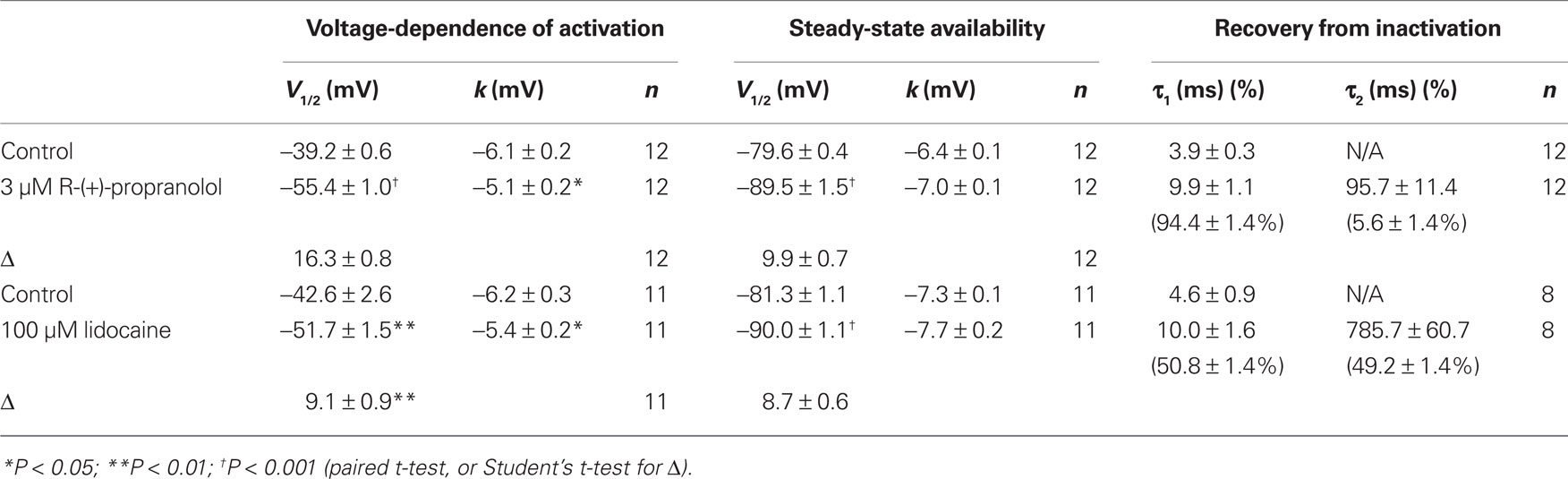
Table 1. Effects of R-(+)-propranolol and lidocaine on NaV1.5 biophysical properties.
We compared the use-dependent effects of lidocaine and R-(+)-propranolol on channels subjected to repetitive depolarizations at different frequencies. The data in Figure 4 illustrate similar levels of use-dependent inhibition of peak current for 100 μM lidocaine and 3 μM R-(+)-propranolol across a range of pulsing frequencies, whereas 40 μM R-(+)-propranolol produced near complete block at 5–10 Hz. These observations indicate that despite some biophysical differences, the two drugs exhibited similar use-dependent blocking effects on NaV1.5 channels.
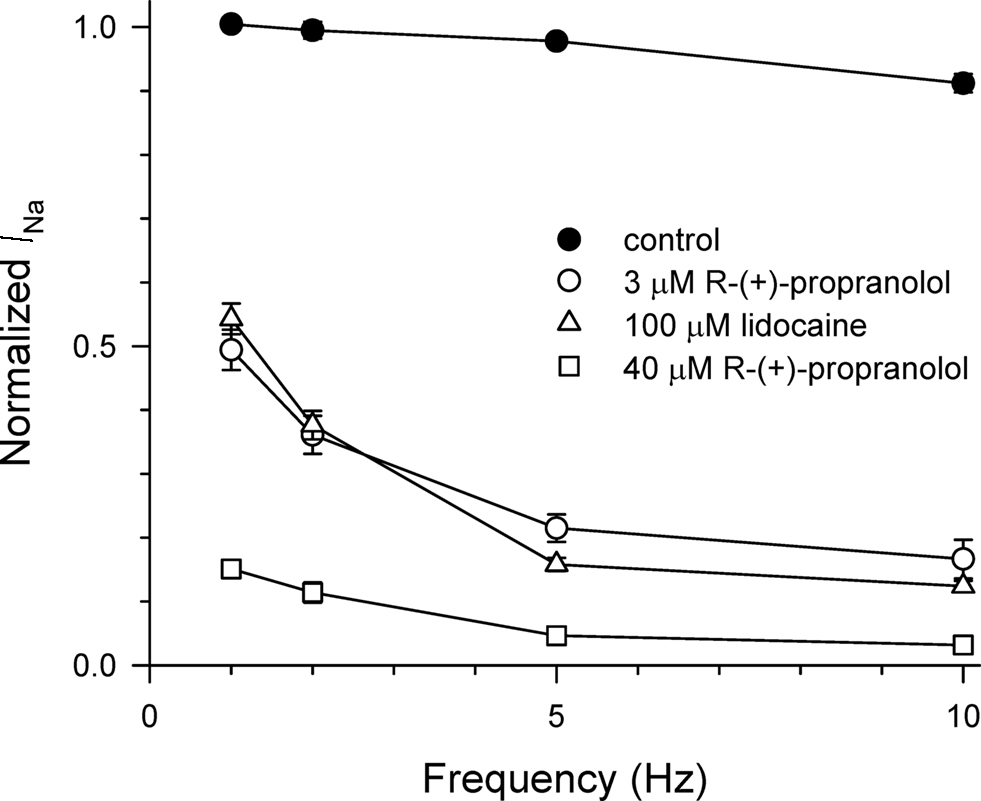
Figure 4. Use-dependent block of NaV1.5 by propranolol and lidocaine. Steady-state use-dependent inhibition of NaV1.5 by 100 μM lidocaine, 3 and 40 μM R-(+)-propranolol at 1, 2, 5, and 10 Hz pulsing frequencies. The data are averaged peak currents following the 100th pulse in the presence of drug normalized to peak current measured in the absence of drug. Each point represents mean ± SEM from 7 to 14 cells.
Propranolol and Lidocaine Block Share Dependence Upon a Critical D4/S6 Residue
Specific residues in the D4/S6 segment including phenylalanine-1760 in human NaV1.5 have been identified as critical for sodium channel block by local anesthetic drugs (Ragsdale et al., 1994). We examined whether mutation of this residue alters block by R-(+)-propranolol. Figure 5 illustrates results from experiments that tested use-dependent block of WT or NaV1.5-F1760A mutant channels. Whereas the WT channel was blocked ∼50% by 100 μM lidocaine or 3 μM R-(+)-propranolol, NaV1.5-F1760A exhibited near complete insensitivity to both compounds. The effects of both drugs on recovery from inactivation were also greatly attenuated (Figure 6), but the other biophysical effects previously noted for WT channels were still evident for both drugs (Table 2; Figures 7 and 8). Our results suggest that use-dependent block by R-(+)-propranolol requires a structural motif in NaV1.5 that is also required by lidocaine.
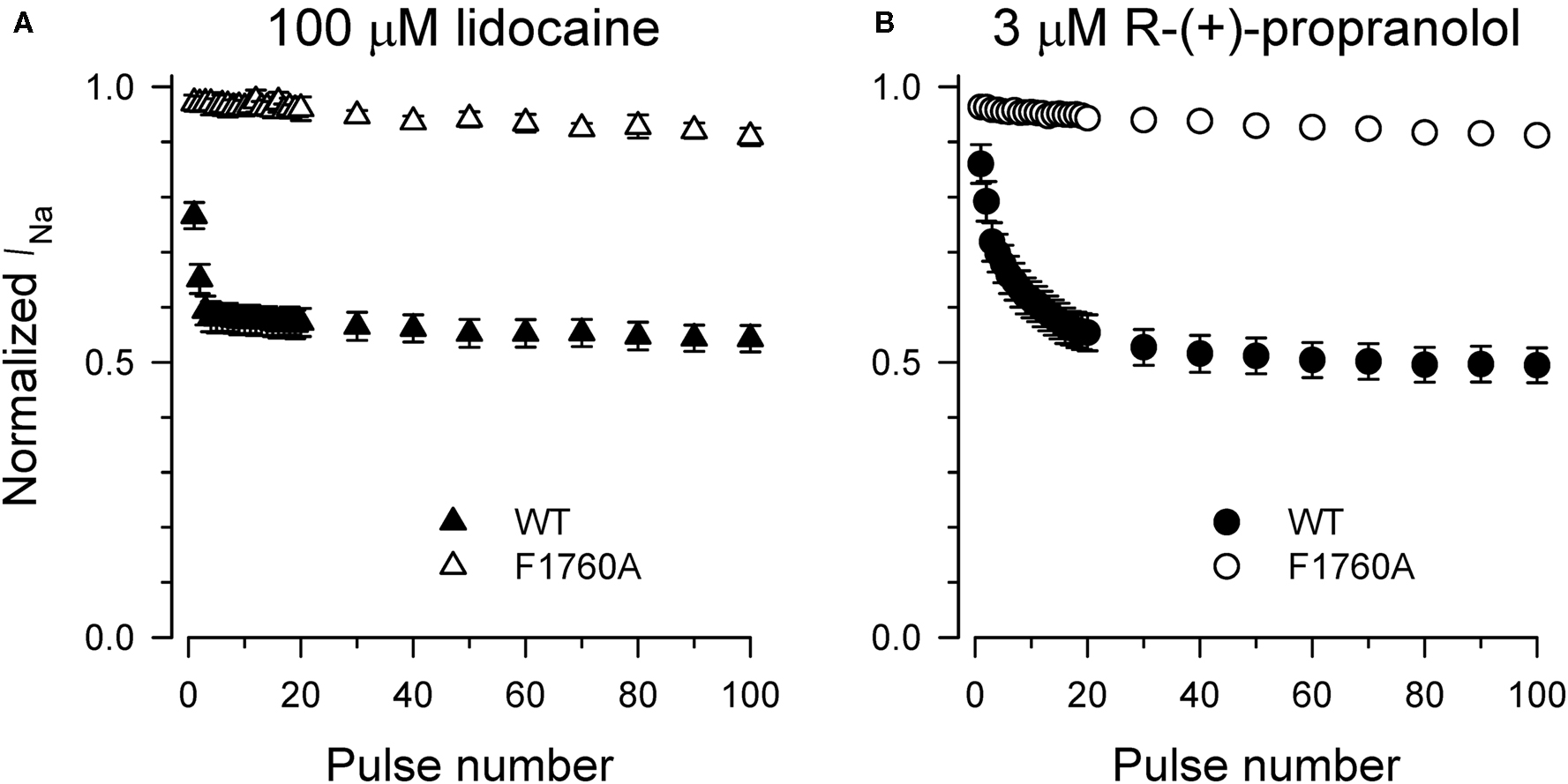
Figure 5. Phenylalanine-1760 is required for NaV1. 5 block by propranolol. Steady-state use-dependent inhibition of WT-NaV1.5 and F1760A channels by 100 μM lidocaine (A) and 3 μM R-(+)-propranolol (B) during 1 Hz pulsing to −10 mV from a holding potential of −120 mV. Each point represents mean ± SEM from 7 to 14 cells.
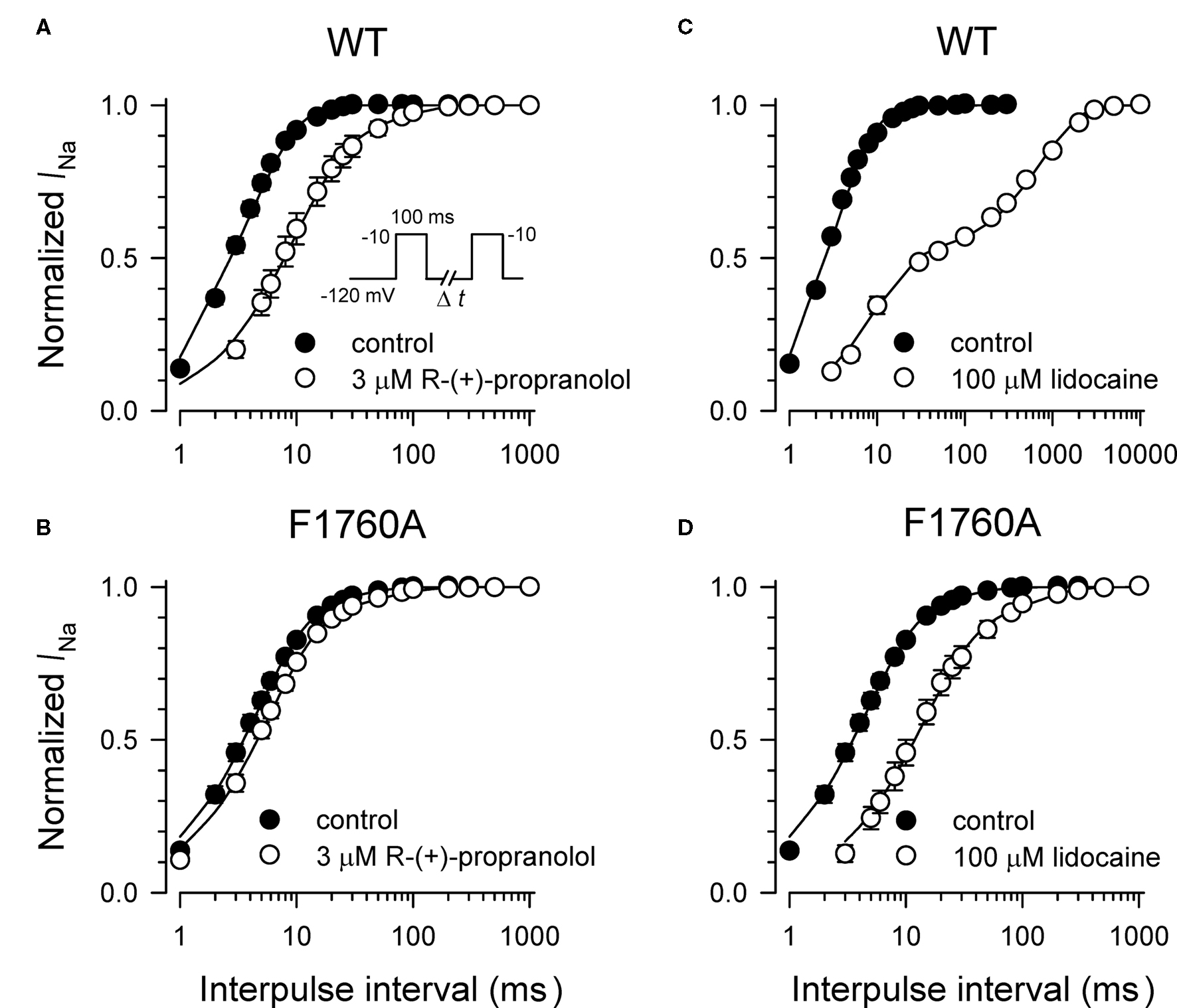
Figure 6. Effects of propranolol and lidocaine on recovery from inactivation for WT-NaV1. 5 and F1760A channels. Recovery from inactivation was determined using a two-pulse protocol illustrated by the inset in (A). Current amplitude recorded after each time interval was normalized to peak current measured during the first pulse. The effects of 3 μM R-(+)-propranolol and 100 μM lidocaine on recovery from inactivation for WT-NaV1.5 channels are illustrated in (A,B), respectively. The effects of 3 μM R-(+)-propranolol and 100 μM lidocaine on recovery from inactivation for F1760A channels are illustrated in (C,D), respectively. Each point represents mean ± SEM from 7 to 14 cells.
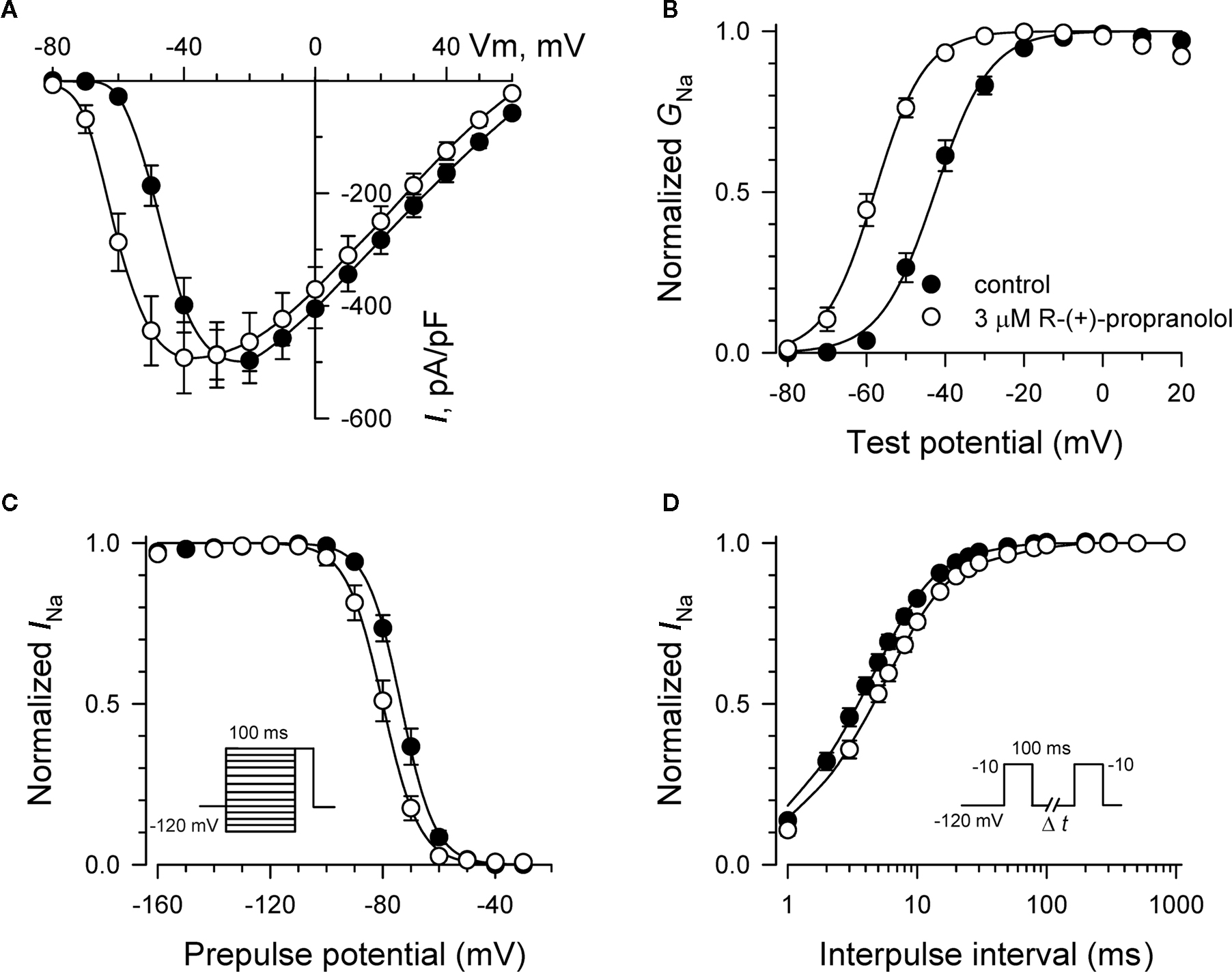
Figure 7. Biophysical effects of propranolol on NaV1. 5-F1760A channels. (A) Current–voltage relationships in the absence and presence of 3 μM R-(+)-propranolol. Peak current amplitudes were elicited from a holding potential of −120 mV to various test potentials then normalized to cell capacitance. (B) Conductance–voltage relationship in the absence and presence of 3 μM R-(+)-propranolol. (C) Voltage-dependence of sodium channel availability (“steady-state inactivation”) in the absence and presence of drug measured using a two-pulse protocol illustrated in the inset. (D) Effect of (R)-(+)-propranolol on recovery from inactivation (holding potential −120 mV). Fit parameters for data in (B–D) are provided in Table 2, and lines in (B,C) represent Boltzmann fits. Each point represents mean ± SEM from 7 to 14 cells.
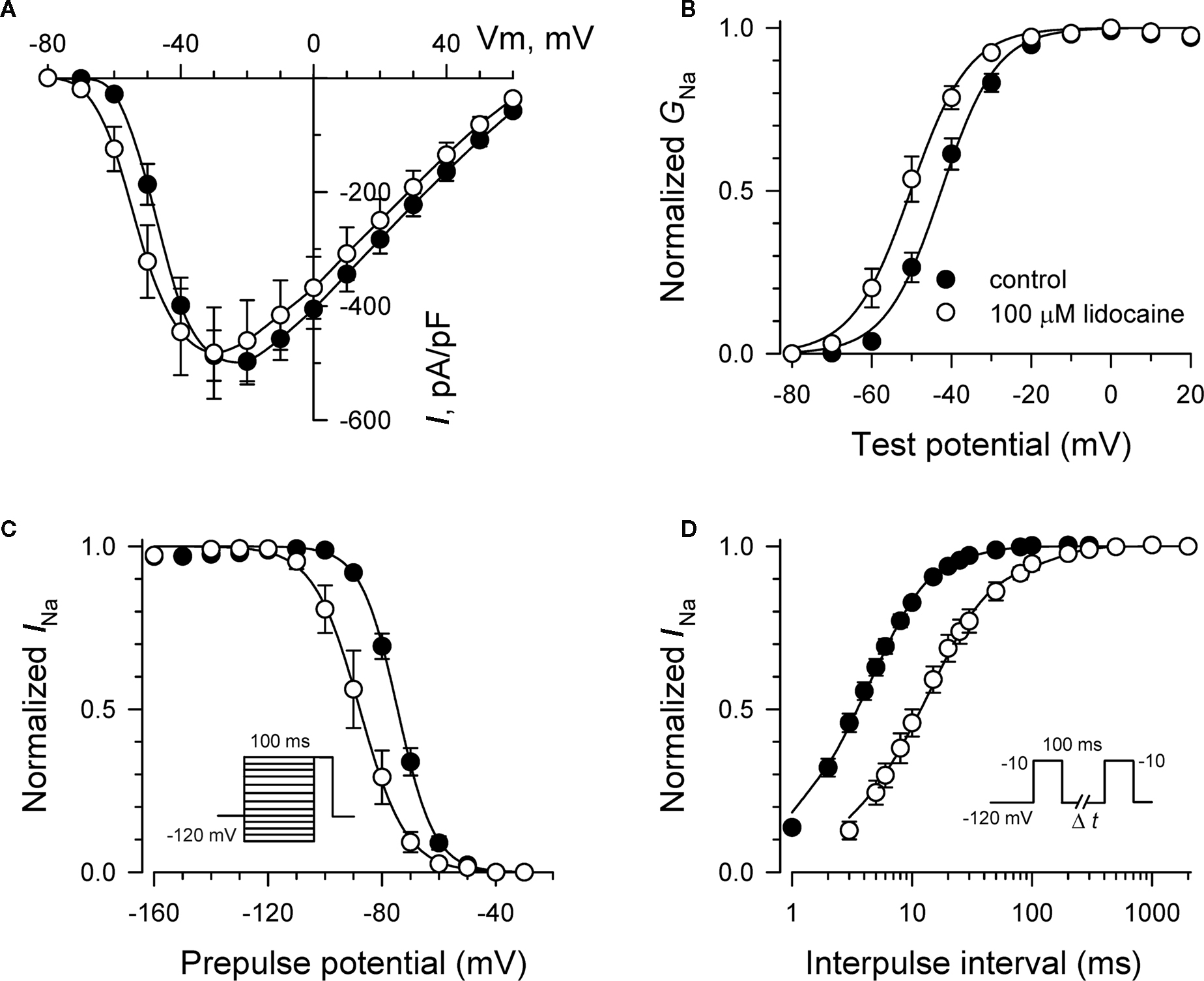
Figure 8. Biophysical effects of lidocaine on NaV1. 5-F1760A channels. (A) Current–voltage relationships in the absence and presence of 100 μM lidocaine. Peak current amplitudes were elicited from a holding potential of −120 mV to various test potentials then normalized to cell capacitance. (B) Conductance–voltage relationship in the absence and presence of 100 μM lidocaine. (C) Voltage-dependence of sodium channel availability (“steady-state inactivation”) in the absence and presence of drug measured using a two-pulse protocol illustrated in the inset. (D) Effect of lidocaine on recovery from inactivation (holding potential −120 mV). Fit parameters for data in (B–D) are provided in Table 2, and lines in panels (B,C) represent Boltzmann fits. Each point represents mean ± SEM from 7 to 14 cells.
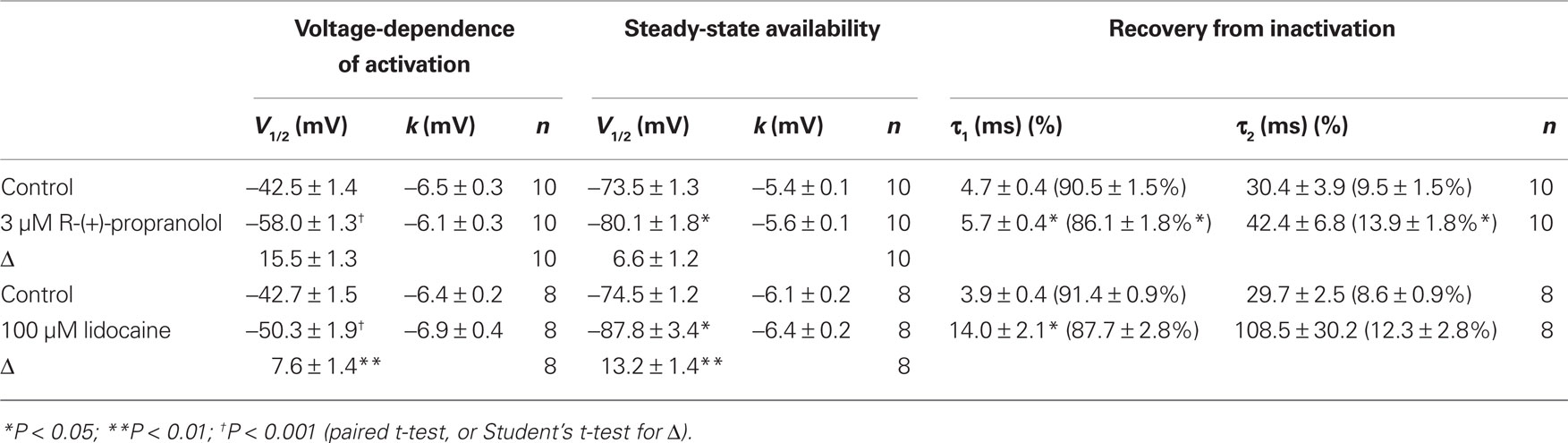
Table 2. Effects of R-(+)-propranolol and lidocaine on NaV1.5-F1760A biophysical properties.
Propranolol Block of Neuronal Sodium Channels
We investigated the effects of R-(+)-propranolol on three human brain sodium channels (NaV1.1, NaV1.2, NaV1.3) stably expressed with human β1 and β2 accessory subunits in HEK-293 cells to test the hypothesis that sodium channels other than NaV1.5 are sensitive to this drug. We focused initially on NaV1.1 as mutations in this channel have been associated with a spectrum of human epilepsies and one familial migraine disorder. Initial studies demonstrated that R-(+)-propranolol exerts tonic block of NaV1.1 with an approximate IC50 of 50 μM (Figure 9A), slightly higher than that observed for NaV1.5 channels. At this concentration, R-(+)-propranolol caused hyperpolarizing shifts in the conductance–voltage relationship (Figure 9B) and steady-state inactivation curve (Figure 9C). The drug also induced a profound slowing of recovery from inactivation (Figure 9D) explained best by an approximate 50-fold increase in magnitude of the time constant representing the slower component of recovery (Table 3). The effects of R-(+)-propranolol on NaV1.1 were qualitatively similar to those observed for NaV1.5. The same concentration of metoprolol (50 μM) exerted only 8 ± 0.01% (n = 4) tonic block on NaV1.1 consistent with our previous conclusion that sodium channel block by propranolol is not a class effect of β-blockers.
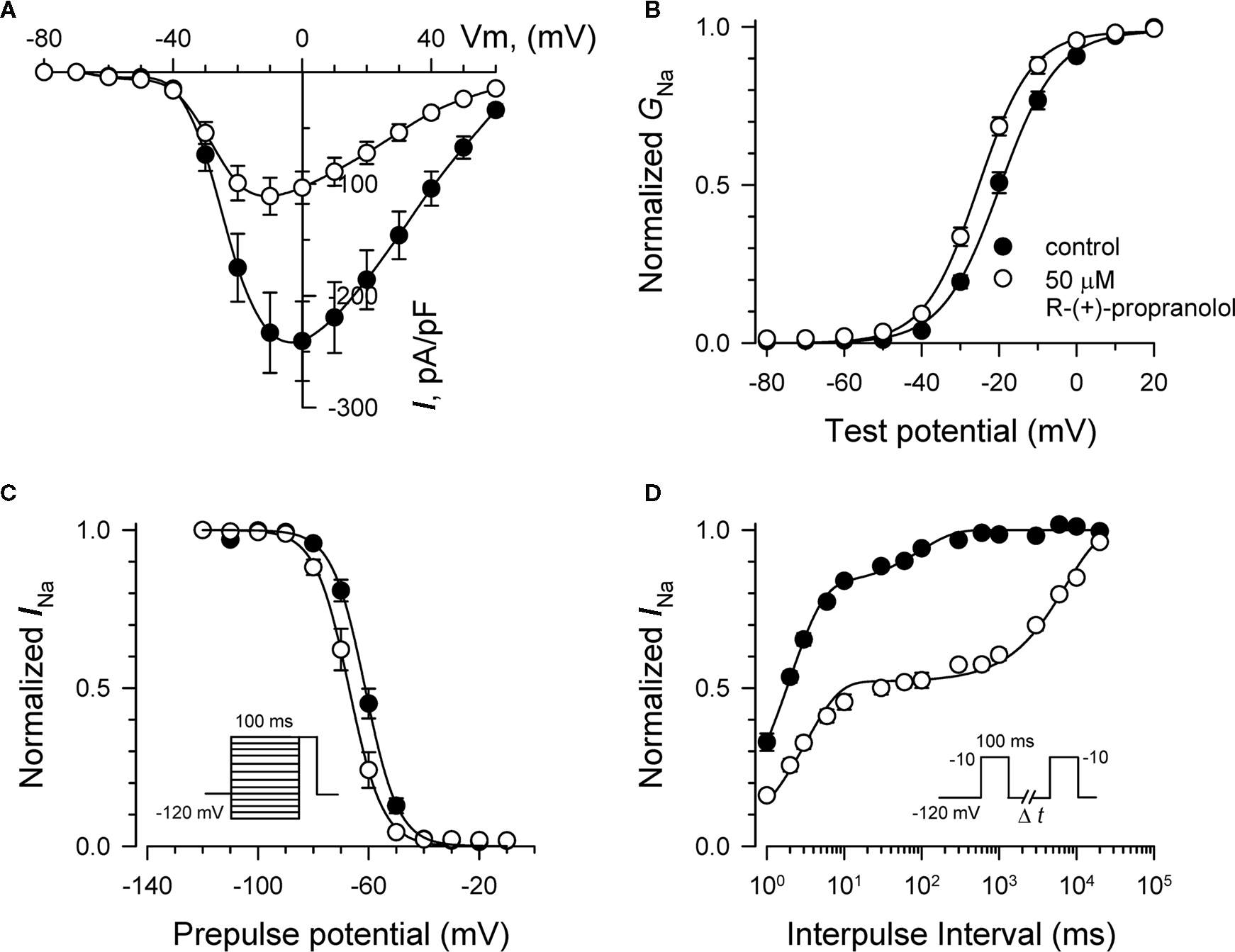
Figure 9. Biophysical effects of R-(+)-propranolol on NaV1. 1. (A) Current–voltage relationships in the absence and presence of 50 μM R-(+)-propranolol. Peak current amplitudes were elicited from a holding potential of −120 mV to various test potentials then normalized to cell capacitance. (B) Conductance–voltage relationship in the absence and presence of 50 μM R-(+)-propranolol. (C) Voltage-dependence of sodium channel availability (“steady-state inactivation”) in the absence and presence of drug measured using a two-pulse protocol illustrated in the inset. (D) Effect of R-(+)-propranolol on recovery from inactivation (holding potential −120 mV). Fit parameters for data in (B–D) are provided in Table 3, and lines in (B,C) represent Boltzmann fits. Each point represents mean ± SEM from 6 to 8 cells.
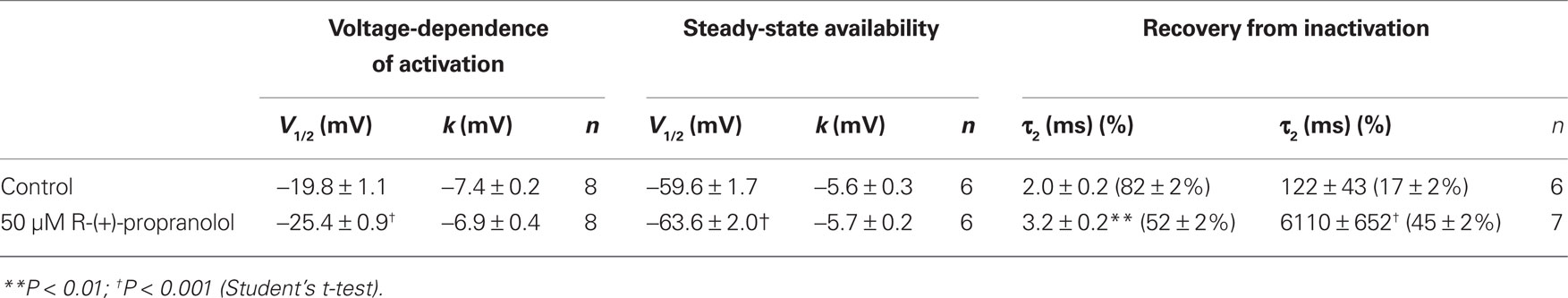
Table 3. Effects of R-(+)-propranolol on NaV1.1 biophysical properties.
To determine the relative sensitivity of brain sodium channels to tonic and use-dependent block by R-(+)-propranolol, we employed an automated planar patch-clamp recording system (Nanion Patchliner) to construct concentration–response curves for NaV1.1, NaV1.2, and NaV1.3. Use of the automated electrophysiology platform enabled the performance of these experiments with greater efficiency than with manual patch-clamp recording. Table 4 presents the IC50 values calculated from these experiments. Using data obtained from cells expressing NaV1.3, we could not reliably fit the concentration–response relationship for use-dependent block, but empirically we observed 53.5% block at 100 μM suggesting that the IC50 was similar for tonic (103 μM) and use-dependent block. These data indicate that brain sodium channels exhibit less sensitivity to R-(+)-propranolol than NaV1.5 channels. Based upon our study and previously published work (Desaphy et al., 2003), the ranking of IC50 values representing sodium channel tonic block by propranolol is (from most to least sensitive): NaV1.5 < NaV1.1 < NaV1.4 < NaV1.2 < NaV1.3.

Table 4. Tonic and use-dependent block of brain voltage-gated sodium channels by R-(+)-propranolol.
Discussion
Propranolol has many clinical uses especially in the treatment of cardiovascular diseases as well as in migraine prophylaxis. Most clinical benefits are readily explainable by the intended drug action of β-adrenergic receptor antagonism. However, off-target effects may contribute to clinical efficacy in some circumstances and could also explain certain adverse effects involving the central nervous system (Gleiter and Deckert, 1996). In this study, we determined the biophysical effects of propranolol on recombinant heart and brain sodium channels. We found that the ability of propranolol to block voltage-gated sodium channels is not stereospecific, nor a class effect of β-blockers. We also demonstrated that block depends upon a critical D4/S6 residue involved with local anesthetic effects, and that propranolol exhibits greater potency against cardiac than brain sodium channels. These findings firmly establish sodium channels as targets for propranolol and may help explain some beneficial effects of the drug.
The mechanism of propranolol block of sodium channels resembles that of local anesthetics. In particular, propranolol exhibits both tonic and use-dependent block probably by stabilizing the inactivated state of the channel as evidenced by a hyperpolarizing shift in the steady-state availability curve and slowing of recovery from inactivation. In NaV1.5 channels, mutation of F1760 greatly attenuates tonic and use-dependent block by both propranolol and lidocaine suggesting that there are some shared structural requirements for the action of both drugs. Mutation of F1760 also partially suppresses the effects of both drugs on recovery from inactivation but curiously has little impact on changes in conductance–voltage relationship and steady-state availability suggesting that other residues or other structural domains contribute to these biophysical effects.
The clinical relevance of propranolol block of sodium channels may be significant in some circumstances. In congenital LQTS caused by SCN5A mutations (LQT3 subtype), an increase in non-inactivating persistent sodium current underlies susceptibility to ventricular arrhythmias (Bennett et al., 1995). Propranolol has been demonstrated to suppress increased persistent current evoked by various SCN5A mutations when expressed heterologously (Bankston and Kass, 2010), and there is anecdotal evidence that propranolol in combination with mexiletine may be effective therapy for severe neonatal presentations of LQT3 (Schulze-Bahr et al., 2004; Wang et al., 2008). However, a recent study demonstrated that propranolol alone did not ameliorate arrhythmia risk in a LQT3 mouse model (Fabritz et al., 2010). Further, clinical studies of LQTS have suggested that β-blockers are less efficacious in LQT3 than in other genetic subtypes (Moss et al., 2000; Schwartz et al., 2001; Priori et al., 2004), but the specific efficacy of propranolol alone or in combination with mexiletine (or other sodium channel blockers) has not been systematically evaluated. The non-stereospecificity of propranolol NaV1.5 block suggests that R-(+)-propranolol might offer utility as a sodium channel blocker in clinical circumstances where β-adrenergic block is undesirable such as LQT3 in which bradycardia could predispose to cardiac arrhythmias. The plasma concentration of propranolol may be a critical factor in achieving clinical benefits in LQT3 as suggested by a recent computational study (Ahrens-Nicklas et al., 2009). Sodium channel block occurs at higher drug concentrations than are required for β-adrenergic antagonism. In clinical practice, propranolol plasma levels are seldom monitored and there is wide inter-individual variation in peak plasma concentrations (Woosley et al., 1979). However, we previously demonstrated that measured propranolol plasma levels in an infant with severe neonatal LQTS caused by a profoundly dysfunctional NaV1.5 mutation were sufficient to cause significant use-dependent block of the mutant channel in vitro (Wang et al., 2008).
In addition to the efficacy of propranolol in cardiovascular diseases, the drug is effective in preventing attacks of common migraine. Migraine is a significant public health problem affecting about 18% of the women and 6% of the men in the United States (Bigal and Lipton, 2009). Migraine attacks can be preceded by transient neurological symptoms (aura) such as visual disturbances, which have been traced to the occurrence of cortical spreading depression, an initial increase in neuronal activity immediately followed by a refractory and long-lasting depression of activity that slowly propagates across the cortex (Lauritzen, 1994). Migraine attacks are typically managed by avoiding known triggers, and with abortive or prophylactic medications. Propranolol has become one of the most regularly used drugs for migraine prophylaxis with many clinical trials consistently proving its efficacy in reducing migraine attacks (Silberstein, 2000; Linde and Rossnagel, 2004). However, despite the well studied and clinically observed effects of propranolol on the central nervous system (McAinsh and Cruickshank, 1990; Gleiter and Deckert, 1996), the molecular basis for these effects remains largely elusive.
Two general mechanisms have been proposed for the prophylactic effect of propranolol in migraine: (1) modulation of the central catecholaminergic system (Schoenen et al., 1986) probably through central β-adrenergic receptors (Casucci et al., 2008); and (2) cross-interaction with serotonin receptors (Casucci et al., 2008). Recently, it has been demonstrated that propranolol suppresses cortical spreading depression frequency in rats (Ayata et al., 2006). Anti-convulsant drugs such as topiramate and sodium valproate have also been shown to be effective in reducing migraine attacks in clinical trials (Chronicle and Mulleners, 2004), and both drugs can block voltage-gated sodium channels. Moreover, propranolol has been shown to have anti-convulsive properties (Jaeger et al., 1979; Fischer, 2002). Based upon these many observations and our current data, we propose that brain sodium channels could be targets for propranolol in preventing migraine. On the other hand, the clinical efficacy of metoprolol (Kangasniemi and Hedman, 1984; Olsson et al., 1984) and the lack of efficacy of d-propranolol (Ayata et al., 2006) in migraine prophylaxis are not consistent with this idea. Further, the rather high IC50 we observed in vitro for propranolol block of brain sodium channels would seem difficult to achieve in clinical settings. Nonetheless, further consideration should be given to sodium channels as molecular targets explaining certain clinical benefits and adverse effects of this widely used drug.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Robert Lefkowitz for suggesting experiments with R-(+)-propranolol, and Katherine Murray for helpful discussions. This work was supported by grants HL083374 and HL065962 from the NIH. Akshitkumar M. Mistry was supported by NIH Summer Research Training Program in Heart, Lung and Vascular Biology (T35 HL090555).
References
Ahrens-Nicklas, R. C., Clancy, C. E., and Christini, D. J. (2009). Re-evaluating the efficacy of β-adrenergic agonists and antagonists in long QT-3 syndrome through computational modeling. Cardiovasc. Res. 82, 439–447.
Ayata, C., Jin, H., Kudo, C., Dalkara, T., and Moskowitz, M. A. (2006). Suppression of cortical spreading depression in migraine prophylaxis. Ann. Neurol. 59, 652–661.
Bankston, J. R., and Kass, R. S. (2010). Molecular determinants of local anesthetic action of beta-blocking drugs: implications for therapeutic management of long QT syndrome variant 3. J. Mol. Cell. Cardiol. 48, 246–253.
Bennett, P. B., Yazawa, K., Makita, N., and George, A. L. Jr. (1995). Molecular mechanism for an inherited cardiac arrhythmia. Nature 376, 683–685.
Benson, D. W., Wang, D. W., Dyment, M., Knilans, T. K., Fish, F. A., Strieper, M. J., Rhodes, T. H., and George, A. L. Jr. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028.
Bigal, M. E., and Lipton, R. B. (2009). The epidemiology, burden, and comorbidities of migraine. Neurol. Clin. 27, 321–334.
Casucci, G., Villani, V., and Frediani, F. (2008). Central mechanism of action of antimigraine prophylactic drugs. Neurol. Sci. 29(Suppl. 1), S123–S126.
Chronicle, E., and Mulleners, W. (2004). Anticonvulsant drugs for migraine prophylaxis. Cochrane Database Syst. Rev. 3, CD003226.
Dawson, A. K., Reele, S. B., Wood, A. J., Duff, H. J., Woosley, R. L., and Smith, R. F. (1984). Electrophysiological effects of high-dose propranolol in dogs: evidence in vivo for effects not mediated by the beta adrenoceptor. J. Pharmacol. Exp. Ther. 229, 91–97.
Desaphy, J. F., Pierno, S., De Luca, A., Didonna, P., and Camerino, D. C. (2003). Different ability of clenbuterol and salbutamol to block sodium channels predicts their therapeutic use in muscle excitability disorders. Mol. Pharmacol. 63, 659–670.
Duff, H. J., Roden, D. M., Brorson, L., Wood, A. J., Dawson, A. K., Primm, R. K., Oates, J. A., Smith, R. F., and Woosley, R. L. (1983). Electrophysiologic actions of high plasma concentrations of propranolol in human subjects. J. Am. Coll. Cardiol. 2, 1134–1140.
Fabritz, L., Damke, D., Emmerich, M., Kaufmann, S., Theis, K., Blana, A., Fortmuller, L., Laakmann, S., Hermann, S., Aleynichenko, E., Steinfurt, J., Volkery, D., Riemann, B., Kirchhefer, U., Franz, M. R., Breithardt, G., Carmeliet, E., Schafers, M., Maier, S. K., Carmeliet, P., and Kirchhof, P. (2010). Autonomic modulation and antiarrhythmic therapy in a model of long QT syndrome type 3. Cardiovasc. Res. 87, 60–72.
Fischer, W. (2002). Anticonvulsant profile and mechanism of action of propranolol and its two enantiomers. Seizure 11, 285–302.
Gellens, M. E., George, A. L., Chen, L., Chahine, M., Horn, R., Barchi, R. L., and Kallen, R. G. (1992). Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. U.S.A. 89, 554–558.
Gleiter, C. H., and Deckert, J. (1996). Adverse CNS-effects of beta- adrenoceptor blockers. Pharmacopsychiatry 29, 201–211.
Jaeger, V., Esplin, B., and Capek, R. (1979). The anticonvulsant effects of propranolol and beta-adrenergic blockade. Experientia 35, 80–81.
Kahlig, K. M., Saridey, S. K., Kaja, A., Daniels, M. A., George, A. L. Jr., and Wilson, M. H. (2010). Multiplexed transposon-mediated stable gene transfer in human cells. Proc. Natl. Acad. Sci. U.S.A. 107, 1343–1348.
Kangasniemi, P., and Hedman, C. (1984). Metoprolol and propranolol in the prophylactic treatment of classical and common migraine. A double-blind study. Cephalalgia 4, 91–96.
Lauritzen, M. (1994). Pathophysiology of the migraine aura. The spreading depression theory. Brain 117, 199–210.
Linde, K., and Rossnagel, K. (2004). Propranolol for migraine prophylaxis. Cochrane Database Syst. Rev. 2, CD003225.
Lossin, C., Wang, D. W., Rhodes, T. H., Vanoye, C. G., and George, A. L. Jr. (2002). Molecular basis of an inherited epilepsy. Neuron 34, 877–884.
McAinsh, J., and Cruickshank, J. M. (1990). Beta-blockers and central nervous system side effects. Pharmacol. Ther. 46, 163–197.
Mehvar, R., and Brocks, D. R. (2001). Stereospecific pharmacokinetics and pharmacodynamics of beta- adrenergic blockers in humans. J. Pharm. Pharm. Sci. 4, 185–200.
Misra, S. N., Kahlig, K. M., and George, A. L. Jr. (2008). Impaired NaV1.2 function and reduced cell surface expression in benign familial neonatal-infantile seizures. Epilepsia 49, 1535–1545.
Moss, A. J., Zareba, W., Hall, W. J., Schwartz, P. J., Crampton, R. S., Benhorin, J., Vincent, G. M., Locati, E. H., Priori, S. G., Napolitano, C., Medina, A., Zhang, L., Robinson, J. L., Timothy, K., Towbin, J. A., and Andrews, M. L. (2000). Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 101, 616–623.
Murray, K. T., Reilly, C., Koshakji, R. P., Roden, D. M., Lineberry, M. D., Wood, A. J., Siddoway, L. A., Barbey, J. T., and Woosley, R. L. (1990). Suppression of ventricular arrhythmias in man by d-propranolol independent of beta-adrenergic receptor blockade. J. Clin. Invest. 85, 836–842.
Olsson, J. E., Behring, H. C., Forssman, B., Hedman, C., Hedman, G., Johansson, F., Kinnman, J., Palhagen, S. E., Samuelsson, M., and Strandman, E. (1984). Metoprolol and propranolol in migraine prophylaxis: a double-blind multicentre study. Acta Neurol. Scand. 70, 160–168.
Priori, S. G., Napolitano, C., Schwartz, P. J., Grillo, M., Bloise, R., Ronchetti, E., Moncalvo, C., Tulipani, C., Veia, A., Bottelli, G., and Nastoli, J. (2004). Association of long QT syndrome loci and cardiac events among patients treated with beta-blockers. JAMA 292, 1341–1344.
Ragsdale, D. S., McPhee, J. C., Scheuer, T., and Catterall, W. A. (1994). Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 265, 1724–1728.
Schoenen, J., Maertens de Noordhout, A., Timsit-Berthier, M., and Timsit, M. (1986). Contingent negative variation and efficacy of beta-blocking agents in migraine. Cephalalgia 6, 229–233.
Schulze-Bahr, E., Fenge, H., Etzrodt, D., Haverkamp, W., Monnig, G., Wedekind, H., Breithardt, G., and Kehl, H. G. (2004). Long QT syndrome and life threatening arrhythmia in a newborn: molecular diagnosis and treatment response. Heart 90, 13–16.
Schwartz, P. J., Priori, S. G., Spazzolini, C., Moss, A. J., Vincent, G. M., Napolitano, C., Denjoy, I., Guicheney, P., Breithardt, G., Keating, M. T., Towbin, J. A., Beggs, A. H., Brink, P., Wilde, A. A., Toivonen, L., Zareba, W., Robinson, J. L., Timothy, K. W., Corfield, V., Wattanasirichaigoon, D., Corbett, C., Haverkamp, W., Schulze-Bahr, E., Lehmann, M. H., Schwartz, K., Coumel, P., and Bloise, R. (2001). Genotype–phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103, 89–95.
Silberstein, S. D. (2000). Practice parameter: evidence-based guidelines for migraine headache (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 55, 754–762.
Wang, D. W., Crotti, L., Shimizu, W., Pedrazzini, M., Cantu, F., De Filippo, P., Kishiki, K., Miyazaki, A., Ikeda, T., Schwartz, P. J., and George, A. L. Jr. (2008). Malignant perinatal variant of long-QT syndrome caused by a profoundly dysfunctional cardiac sodium channel. Circ. Arrhythm. Electrophysiol. 1, 370–378.
Wang, D. W., Desai, R. R., Crotti, L., Arnestad, M., Insolia, R., Pedrazzini, M., Ferrandi, C., Vege, A., Rognum, T., Schwartz, P. J., and George, A. L. Jr. (2007). Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation 115, 368–376.
Wang, D. W., Viswanathan, P. C., Balser, J. R., George, A. L. Jr., and Benson, D. W. (2002). Clinical, genetic, and biophysical characterization of SCN5A mutations associated with atrioventricular conduction block. Circulation 105, 341–346.
Keywords: sodium channel, propranolol, SCN1A, SCN2A, SCN3A, SCN5A
Citation: Wang DW, Mistry AM, Kahlig KM, Kearney JA, Xiang J and George AL Jr. (2010) Propranolol blocks cardiac and neuronal voltage-gated sodium channels. Front. Pharmacol. 1:144. doi: 10.3389/fphar.2010.00144
Received: 22 October 2010;
Paper pending published: 02 November 2010;
Accepted: 13 December 2010;
Published online: 31 December 2010.
Edited by:
Jean-François Desaphy, University of Bari Aldo Moro, ItalyReviewed by:
Mohamed Chahine, Laval University, CanadaJean-François Desaphy, University of Bari Aldo Moro, Italy
Copyright: © 2010 Wang, Mistry, Kahlig, Kearney, Xiang and George Jr. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Alfred L. George Jr., Division of Genetic Medicine, Vanderbilt University, 529 Light Hall, 2215 Garland Avenue, Nashville, TN 37232-0275, USA. e-mail:YWwuZ2VvcmdlQHZhbmRlcmJpbHQuZWR1