

- Department of Pharmacobiology, Faculty of Pharmacy, University of Bari, Italy
The periodic paralysis (PP) are rare autosomal-dominant disorders associated to mutations in the skeletal muscle sodium, calcium, and potassium channel genes characterized by muscle fiber depolarization with un-excitability, episodes of weakness with variations in serum potassium concentrations. Recent advances in thyrotoxic PP and hypokalemic PP (hypoPP) confirm the involvement of the muscle potassium channels in the pathogenesis of the diseases and their role as target of action for drugs of therapeutic interest. The novelty in the gating pore currents theory help to explain the disease symptoms, and open the possibility to more specifically target the disease. It is now known that the fiber depolarization in the hypoPP is due to an unbalance between the novel identified depolarizing gating pore currents (Igp) carried by protons or Na+ ions flowing through aberrant alternative pathways of the mutant subunits and repolarizing inwardly rectifying potassium channel (Kir) currents which also includes the ATP-sensitive subtype. Abnormal activation of the Igp or deficiency in the Kir channels predispose to fiber depolarization. One pharmacological strategy is based on blocking the Igp without affecting normal channel gating. It remains safe and effective the proposal of targeting the KATP, Kir channels, or BK channels by drugs capable to specifically open at nanomolar concentrations the skeletal muscle subtypes with less side effects.
Introduction
The primary periodic paralysis (PP) are rare autosomal-dominant disorders associated to mutations of the skeletal muscle sodium, potassium, and calcium channel genes. They are characterized by episodes of muscle weakness associated with variations in serum potassium concentration. The primary PP are classified as hyperkalemic PP (hyperPP), hypokalemic PP (hypoPP), and Andersen’s syndrome (AS; Platt and Griggs, 2009). Other related disorders are the thyrotoxic PP (TPP) associated with thyrotoxicosis.
HyperPP is characterized by attacks of flaccid limb paralysis with weakness associated with abnormally elevated serum K+ ions levels up to 6 mEq/L. Triggers for these attacks are ingestion of potassium-rich food, rest after strenuous exercise, and cold exposure. Episodes of weakness may last for up to an hour and disappear as the blood potassium concentration decreases. Attacks typically begin in the first decade of life, increase in frequency and severity during puberty, and then decrease in frequency after 40 years of age. Myotonia of the extremities supports the diagnosis of hyperPP.
Hypokalemic PP is generally characterized by reversible attacks of muscle weakness associated with decreased blood potassium concentrations below 3.2 mEq/L. In the most severe cases the blood potassium concentration decrease below 1 mEq/L during the attacks. The attacks are triggered by rest after strenuous exercise, by a meal rich in carbohydrates, or by exposure to cold. The glucose-dependent insulin release from pancreatic beta cells or insulin injection is one of the most effective provocative factors in the human disease. Patients typically wake up paralyzed, and attacks usually last several hours to days in the severe cases. In the elderly, the patients develop progressive, persistent weakness that takes the form of a proximal myopathy.
Andersen’s syndrome is characterized by a unique phenotype consisting of PP, cardiac arrhythmias, facial and skeletal malformations. The disease was firstly discovered by Ellen Andersen in the 1971 but it has been clinically characterized latter on by Al Rabi Tawil (Andersen et al., 1971; Tawil et al., 1994). Paralysis may occurs with either hyperkalemia or hypokalemia that exacerbate the cardiac arrhythmia. Heart manifestations include ventricular arrhythmia (84% of patients), long QT syndrome (50% of patients), abnormal TU wave patterns (73%), and sudden cardiac arrest (10% of patients). A bidirectional ventricular tachycardia (BVT) is a characteristic rhythm disturbance present only in AS, digitalis intoxication, and ryanodine receptor mutations (Morita et al., 2007).
Thyrotoxic PP is a sporadic muscle disorder characterized by episodic attacks of weakness and hypokalemia in some, but not all, thyrotoxic individuals. Patients are normal between attacks, which resolve with treatment of thyrotoxicosis. The adrenergic symptoms potentiated by the abnormal thyroide hormone release play a mayor role in the disease. Episodic weakness in TPP is clinically similar to the hypoPP and AS. It is the most common form of PP being most prevalent in young Asian and Latin American men, about 10% of thyrotoxic males develop episodic weakness. In Caucasians, is less than 0.1. Although thyrotoxicosis is more common in women, most individuals with TPP are men.
Secondary forms of PP are also known and are normally associated with hypokalemia caused by depletion of serum K+ ions associated with a K+ free diet or malnutrition, licorice intoxication, lassative and diuretic abuse, digitalis intoxication, barium intoxication, beta-2 agonists abuse in asthmatic patients or insulin overdose in diabetic patients. Clinical signs such as weakness, flaccid paralysis, and cardiac dysrhythmia are normally transient and their severity is dependent on the serum K+ ions levels reached during the attacks. The clinical signs may be permanent in those conditions associated with K+ free diet as observed in the K-depleted rats or malnutrition. The K-depleted rats, in the absence of transgenic animals, is considered a valid animal model of the familial hypoPP resembling the human phenotype. This is obtained by feeding the animals for 14–18 days with a K+ free diet (Tricarico et al., 1998). These animals show the flaccid paralysis induced by glucose/insulin injection, the vacuolar myopathy and tubular aggregates in the muscle biopsies, the serum K+ ions levels between 1 and 2 mEq/L during the attacks. However, in contrast to patients affected by the familial hypoPP in which the serum K+ ions levels between the attacks is normal, the K-depleted rats have a general weakness associated with serum K+ ions levels below 3.2 mEq/L even between the attacks. Secondary hyperkalemia is observed following glucocorticoids infusions or K+ sparing diuretic abuse. KCl (3–5%) solution infusion overdose is the most frequent and dangerous form of induced hyperkalemia in humans even leading to fatal arrhythmias. This have led to alerts managing this type of drugs.
Pathogenesis of Periodic Paralysis
Pathogenesis of HyperPP
In hyperPP the pathomechanism is well established and it is explained by the gain-of-function mutations in the α-subunit of the skeletal muscle voltage-gated sodium channel, Nav1.4. The T704M and M1592V mutations in the SCN4A gene account for the majority of cases. The mutant channels show failure of the inactivation process which results in continual sodium influx and persistent cell depolarization; stronger and persistent depolarization in hyperPP leads to general inactivation of sodium channels and depolarization block, which manifests as paralysis (Jurkat-Rott and Lehmann-Horn, 2007). The sustained membrane depolarization in hyperPP amplifies K+ ions efflux from the muscle through voltage-dependent K+ channels. The enhanced activity of the voltage-dependent K+ channels elevate the serum K+ ions concentration.
Sporadic cases of patients have however normal serum potassium levels during attacks of weakness. Some of the families carry hyperPP mutations resulting as a phenotypic variant of hyperPP, some others carry unique mutations of the R3 gating charge in the voltage sensor of domain 2 of the Nav1.4 channel. Patients from these families have phenotypes similar to hypoPP, others have phenotypes similar to hyperPP with weakness induced by K+ ions supplementation (Vicart et al., 2005).
Pathogenesis of HypoPP
The pathomechanism of hypoPP has been debated from many years but only recently has been clearly elucidated. HypoPP has been associated with mutations in both the α1-subunit of the skeletal muscle calcium channel, Cav1.1 (hypoPP type-1) and the α-subunit of the Nav1.4 channel (hypoPP type-2). Missense mutations were first identified in the calcium channel gene, CACNA1S, which accounts for approximately 60% of cases (Jurkat-Rott et al., 1994; Ptacek et al., 1994). In hypoPP type-2 10% of the cases are due to mutations in the sodium channel gene, SCN4A (Jurkat-Rott et al., 2000; Sternberg et al., 2001). Phenotypic differences between hypoPP-1 and hypoPP-2 include:
(1) earlier onset of disease in HypoPP-1;
(2) myalgias in hypoPP-2;
(3) predominance of tubular aggregates in hypoPP-2 and vacuoles in hypoPP-1 in muscle biopsy;
(4) aggravation of hypoPP-2 by acetazolamide.
In hypoPP-1 the mutations of the Cav1.1-DHP receptor genes replace arginine residues in the S4 voltage sensor in domains II, III, or IV with neutral amino acids with loss-of-function mutations (Jurkat-Rott et al., 1994; Ptacek et al., 1994; Lapie et al., 1996). Similarly, in hypoPP-2 previous works showed that sodium channel mutations cause a loss-of-function by enhancing channel inactivation, mainly by stabilizing the inactivated state (Jurkat-Rott et al., 2000; Sternberg et al., 2001; Kuzmenkin et al., 2002). Mutations specifically target positively charged arginine residues in the S4 voltage sensor in domains II or III, which triggers voltage sensor dysfunction. These findings suggested that the pathological effects of these mutations in hypoPP-1 and hypoPP-2 are caused by voltage sensor dysfunction and impaired channel gating with impairment of the excitation–contraction coupling. We can say that these mutations are essentially loss-of-function of Cav1.1 and Nav1.4 channels.
However, the general idea is that this theory does not account for the sarcolemmal depolarization or the hypokalemia and paralysis induced by insulin and glucose during the attacks that characterize hypoPP phenotype.
Involvement of K+ channels in the hypoPP pathogenesis
Kir channels have a key role in the pathogenesis of hypoPP. Kir currents carried by the inwardly rectifier K+ channels which also include the ATP-sensitive type set the resting potentials close to the equilibrium potentials for K+ ions which is around −90 mV as calculated by Nernst equation in normal fibers. The inwardly rectifier K+ channels show a non-linear I–V curves with high permeability to K+ ions at negative membrane potentials which generates an elevated inward currents, and lower permeability at depolarized potentials generating much lower outward currents. This graphically generates an hump of the I–V curves which is an intrinsic properties of the Kir channel currents (Figure 1A). This is caused by the internal Mg2+ ions, polyamines and proton-block of the pore at millimolar concentrations which compete with K+ ions for the internal pore binding sites (Figures 1A,B). The blocking actions of these positively charged molecules and ions is voltage-dependent being more effective at depolarizing voltages. A potential gradient across the cell membrane removes this blockage during hyperpolarization but allow these cations to occlude the ion-conducting pore during depolarizations (Hibino et al., 2009). Rectification is important for setting the resting potential and aiding in repolarization of cells while shunting K+ currents during depolarizations allowing it. In normal fibers the lowering of external K+ ions from 4 to 2.5 mEq/L shift the I–V relationships of Kir currents and of total membrane currents to the left toward more negative values as predicted by the Nernst equation (Figure 1A; Cannon, 2010). However, the lowering of ext. K+ ions below 1.5 mEq/L reduces Kir outward currents shifting the I–V relationships of Kir currents and of total membrane currents to the right toward more positive values setting the resting potential at a new depolarized values. This is also called paradoxical membrane depolarization in low ext. K+ ions concentrations which is explained by the enhanced affinity of the Mg2+ ions and polyamines or protons for their inhibitory binding sites unmasked by the extremely low ext. K+ ions concentration (Figures 1 and 2; Hibino et al., 2009; Cannon, 2010). This is an intrinsic property of the Kir channel that play a key role in the pathogenesis of hypoPP and related diseases. One mechanism by which low intracellular pH inhibit channel opening is related with the reductions of the binding affinity of the channel to PtdIns(4,5)P2 interaction. This is observed at pH values of about 6.5 (Qu et al., 2000; Hibino et al., 2009). Pharmacological investigations support the involvement of the Kir channels in hypoPP. Barium toxicity produces a secondary form of hypoPP, and the Kir channel is blocked by Ba2+.
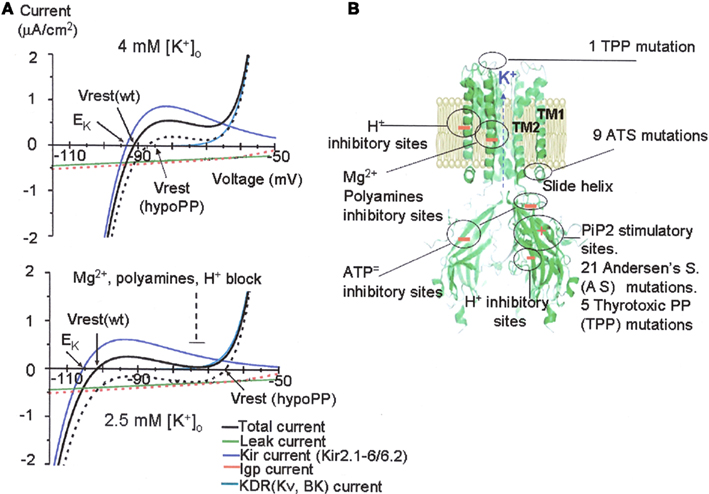
Figure 1. Steady-state I–V relationships simulation in normal and hypoPP conditions, and molecular architecture of a Kir channel [modified from Kurachi and colleagues (Hibino et al., 2009) and Cannon (2010)]. The I–V relation for mammalian skeletal muscle was simulated by the combination of an inward rectifier K+ current, IKir, a delayed rectifier K+ current, IKDR, and a leakage current with a reversal potential of 0 mV, ILeak (A). In 4 mM [K+]o the resting potential of −91.3 mV is determined primarily from the balance of an inward ILeak and outward IKir. Addition of a gating pore current, Igp, to simulate hypoPP (dashed red line), shifts the I–V relation downward (dashed black line) but results in only a small depolarization of Vrest to −87.3 mV. The reduction of [K+]o to 2.5 mM shifts EK and IKir to more negative potentials, with a predicted hyperpolarization of Vrest to −101.6 mV in WT fibers. For hypoPP fibers, however, Vrest depolarizes to −65.3 mV (arrow) because the combination of inward currents (ILeak + Igp) exceeds the outward current from IKir. Modified from the original version of Cannon (2010) to evidence that in low external K+ ions the affinity of Mg2+ions, protons and other endogenous blockers of Kir channels for their inhibitory sites increase leading to the characteristic hump in the I–V relationship of the Kir current component. (B) A schematic representation of the structure of a generic Kir channel. The Kir channel is divided into transmembrane and cytoplasmic domains. The NH2 and COOH termini are cytosolic. Tetrameric assembly of Kir channels. The molecular architecture of a tetrameric Kir channel (protein database ID 2QKS: Kir3.1–KirBac3.1 chimera) is represented as a cartoon model reproduced and modified from Kurachi and colleagues (Hibino et al., 2009). The front subunit has been omitted for clarity. The organization of the tetramer of NH2 and COOH termini leads to an extended pore for ion permeation. The transmembrane domain comprises three helices: TM1, H5, and TM2. At the membrane–cytoplasm interface, there is also an amphiphilic slide helix which is a site for some AS mutations. The residue that is largely responsible for the interaction with polyamines and Mg2+ and thus inward rectification are located on TM2. In Kir6.2, the inhibitory binding sites for ATP are located in the cytoplasmic domains. TM1 contains the inhibitory binding sites for protons in the Kir1.1. Inhibitory sites for protons in other Kir channels as Kir2.3 are located also in the cytoplasmic regions affecting PiP2 binding. The opening of Kir channels requires PtdIns(4,5)P2 (PiP2). Those amino acid residues associated with the interaction with PiP2 are distributed on the surface of the cytoplasmic domain toward the plasma membrane. These are also the sites for most of the AS mutations in the Kir2.1. The cytoplasmic domain also contains most of the TPP mutations recently found in the Kir2.6.
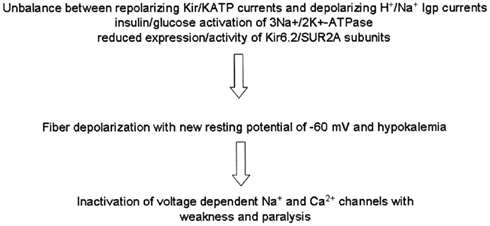
Figure 2. Proposed cascade of the pathogenic events in the hypokalemic periodic paralysis. The unbalance between the depolarizing and repolarizing current components is due to an abnormal activation of depolarizing gating pore currents (Igp) with increased influx of protons or Na+ ions through mutants and to the abnormal reduction of the repolarizing Kir currents carried by various subtypes of Kir channels. The insulin/glucose administration induces activation of the 3Na+/2K+-ATPase but failed to activate KATP channels causing a marked hypokalemia. The hypokalemia reduces the gene expression/activity of the Kir6.2 and SUR2A subunits. All these factors contribute to the fiber depolarization setting the resting potentials to a new value of −60 mV (Vm). The depolarization of the fibers inactivates the voltage-dependent Na+ and Ca2+ channels with paralysis.
However other than Kir block by protons also a down-regulation of the Kir subunits due to low-external K+ ions levels appear to be responsible for hypoPP. Abnormal reduction of KATP channel activity has been reported in excised patches from human hypoPP fibers with the CaV1.1-R528H mutation and in muscle fibers from K-depleted rats (Tricarico et al., 1998, 1999b). These channels show several subconductance states with lower open probability as compared with that recorded in human biopsies from healthy subjects and control rats. The “in vitro” administration of insulin to muscle fibers from K-depleted rats and human hypoPP patients reduced the Kir currents and the KATP currents making them more susceptible to aberrant depolarization (Ruff, 1999; Tricarico et al., 1999a). Reduced channel open probability and single channel conductance has been reproduced “in vitro” following short term exposure (80 min) of the rat fibers to low external K+ ions (0.5 mEq/L) suggesting that an extremely low levels of K+ ions block the Kir6.2 channel of normal rats (Tricarico et al., 1999b). More recently, we demonstrated that in K-depleted rats the impaired KATP activity is associated with a reduced expression of SUR2A and Kir6.2 subunits composing the main KATP channel complex in skeletal muscle (Tricarico et al., 2008a). Case reports showed that patients suffering from hypoPP have reduced expression of the Kir6.2 subunit and/or abnormal membrane localization of SUR2A suggesting defect in membrane trafficking of this subunit (Jovanovic et al., 2008; Kim et al., 2010).
Gating pore current hypothesis in the hypoPP pathogenesis
Recently, several researchers have suggested a new pathomechanism to explain hypoPP-2 and possibly hypoPP-1. Catterall and colleagues (Sokolov et al., 2007) firstly showed that three common hypoPP-2 mutations induced a hyperpolarization-activated cationic leak current through the voltage sensor of the sodium channel, which they called “gating pore current” (Sokolov et al., 2007). According to their model, hypoPP-2 mutations are essentially gain-of-function because the cation leak increases resting membrane conductance and sodium influx, leading to excessive depolarization. Struyk and Cannon (2007) initially found that the gating pore permitted transmembrane permeation of protons. The predicted magnitude of this leak was not large enough to produce aberrant depolarization by itself, but the authors theorized that sustained proton leak could indirectly contribute to instability of the resting membrane potential by interfering with pH balance inside the cell. More recently these and others shown that all five hypoPP mutations in IIS4 screened by expression studies in oocytes produced gating pore currents activated at hyperpolarized potentials (Sokolov et al., 2007; Cannon, 2010). The gating pore currents from IIS4-R1H and IIS4-R2H hypoPP mutations were carried exclusively by protons, whereas the gating pore currents produced by non-histidine mutations at IIS4-R2G/C/S/Q were permeable to alkali metal cations (K+ ≈ Cs+ > Na+ ≈ Li+) and guanidinium were active at hyperpolarized potentials resulting in a poor selectivity among monovalent cations with important pathophysiological implications (Cannon, 2010). In normokalemic PP (normoPP) some mutations in the R3 gating charge producing gating pore current were activated by depolarization leading to abnormal persistent sodium influx and depolarization of the muscle fiber (Sokolov et al., 2008). Jurkat-Rott et al. (2009) in their works demonstrated that a sodium overload is occurring in muscle biopsies from hypoPP and normoPP patients either by direct entry of sodium through the gating pore channels or by increased sodium influx through hyperactivation of the sodium–proton exchanger that transform the proton leak into a Na+ leak.
In hypoPP-1 Kuzmenkin et al. (2007) showed that Cav1.1 mutants stabilize the second open state of the calcium channel and proposed that the low selectivity of the channel in this state would allow monovalent ions to pass into the cell and promote depolarization. As in hypoPP-2 the mutations replace arginine residues in the S4 voltage sensor in domains II, III, or IV with neutral amino acids leading to the muscle paralysis syndrome with progressive myopathy. This hypothesis could account for the common downstream phenotype of the periodic paralyses. However, currently there is no experimental evidence to support it for technical limitations in recordings these types of Igp currents even in cell lines over-expressing Cav1.1 mutants (Cannon, 2010).
In hypoPP mutants the abnormal activation of the inward cationic Igp currents increases the susceptibility of the fibers to the depolarization. Indeed, the membrane currents is the sum of at least 3 components: the inwardly rectifier currents (IKir), the delayed rectifier (IKDR), the passive leak that reverse at 0 mV and in hypoPP the gating pore (Igp).
Imemb = IKir + IKDR + ILeak + Igp
As a result, the resting potential is determined by a balance of two relatively small currents, the outward component of the IKir and the inward ILeak. While IKDR is a gating/voltage-dependent currents involved at depolarized potentials which is carried by Kv and BK channels. The cationic inward Igp is summed to the ILeak exceeding the outward IKir. This is observed at 3–3.5 mEq/L concentrations of K+ ions much closer to the physiological range (4–6 mEq/L) (Cannon, 2010). Because the Kir currents counterbalance the depolarizing Igp and leak currents, the Mg2+ and polyamine or proton dependent depolarization-block of the Kir channels in addition to the Igp currents leads to depolarization of few millivolts setting the resting potentials to a new values of about −60 mV (Vm; Figure 1A). The action potential generation from this new level result in weakness and paralysis. Jurkat-Rott et al. (2009) showed that muscle fibers from healthy subjects and hypoPP patients have bimodal distribution of membrane potentials with two values of >−75 mV (P1) and −60 mV (P2; Vm). However, hypoPP muscle fibers show an higher frequency of finding fibers at P2 membrane potentials than controls (Jurkat-Rott et al., 2009). Taken together, these observations suggest that attacks of hypoPP triggered by carbohydrate ingestion is linked to insulin secretion with the attendant reduction of Kir currents specifically from an increased intracellular ratio of [ATP]/[ADP] which would inhibit KATP channels and of the reduced/expression activity of the Kir6.2/SUR2A subunits (Figure 2). A secondary insulin effect would be a shift of extracellular K+ ions into the myoplasm from increased activity of the 3Na+/2K+-ATPase with uncompensated hypokalemia. Both effects, decreased KATP activity and reduced extracellular [K+], contribute to the additional development of paradoxical depolarization and weakness which is a consequence of the unbalance between the repolarizing Kir/KATP and depolarizing Igp (Figure 2).
Further explanations to the pathogenesis of hypoPP take into account other transports systems. Some authors indeed proposed that the activity of the sodium–hydrogen exchanger transform the influx of proton into a Na+ influx with increased intracellular osmolarity, edema, and sodium cytotoxicity (Jurkat-Rott et al., 2009; Matthews and Hanna, 2010). While the proton influx activates the monocarboxylate transporter that catalyze the efflux of proton and lactate with accumulation of these molecules into the t-tubule. Vacuoles are indeed a common morphological finding in primary and secondary hypokalemic PP. They are localized swelling and vacuolation of the t-tubules secondary to increased osmolarity caused by the local accumulation of ions or metabolites including lactate. The K-depleted rats model demonstrated a vacuolar myopathy associated with an increased efflux of lactate from muscle fibers suggesting that this cannot be related to the genotypes (Tricarico et al., 2008b; Matthews and Hanna, 2010).
Carbonic anhydrase (CA) enzymes that buffer the intra-/extracellular pH changes have been proposed as an additional factor involved in the pathogenesis of PP (Matthews and Hanna, 2010). This is supported by the findings that the deleterious effects of the arginine–histidine substitutions observed in some expressed hypoPP mutants could be ameliorated by a more acidic pH whereas the arginine–glycine substitution was insensitive to alterations in pH (Kuzmenkin et al., 2002). However, against the CA involvement in the PP pathogenesis is the notion that the intracellular acidification would block several K+ channels as Kir or even BK channels and would activate carrier transports as above reported with deleterious effects.
Pathogenesis of Andersen’s Syndrome
In AS, the majority of the patients have loss-of-function mutations in the KCNJ2 gene, which encodes the inward rectifier potassium channel, Kir2.1 (Plaster et al., 2001; Platt and Griggs, 2009). This channel is abundantly expressed in excitable tissues throughout the body, and channel dysfunction explain most of the disease symptoms including facial and skeletal malformations. The mutations in the KCNJ2 gene cause the disease by suppression of outward potassium currents and/or enhancement of inward currents, which can occur by dominant negative effects or haplo-insufficiency (Plaster et al., 2001). In normal heart and skeletal muscle, the inwardly rectifying potassium channel, Kir2.1, stabilizes the resting membrane potential and regulates action potential duration, especially in myocytes. Human patients suffer for longer action potentials and prolonged Q-T interval, and arrhythmia. Many cellular modulators affect potassium channel activity and are necessary for proper channel function; the loss-of-function mutations in AS have been linked to decreased PIP2 sensitivity and an exaggerated inhibitory effects of intracellular Mg2+ or protons (Figure 1B). Another mechanism involves dysfunction of a portion of the channel known as the slide helix, which represents a cluster point for AS mutations. It is thought that interaction of the slide helix with the C-terminus is required for normal gating of the Kir2 family of channels. Skeletal muscle dysmorphisms can be explained by mutations of Kir2.1. This channel is active in differentiating cells inducing hyperpolarization and setting the Em in a range where Ca2+can enter the myoblasts through Ca2+-permeable channels, which promotes the differentiation and fusion of myoblasts. Differentiation is at least in part mediated by expression and activity of myogenic transcription factors, such as myogenin and myocyte enhancer factor-2. Their expression is triggered by Kir2.1-induced hyperpolarization. In addition, abnormal bone structure in the disease would be caused by provoking dysfunction of osteoclasts. Low extracellular pH in the extracellular matrix maintained by H+ secretion via an ATP-driven proton pump is critical for proper degradation of bone by osteoclasts. Since this H+ secretion is achieved in exchange for K+ transport through Kir channels, disruption of these channels would cause osteoclast dysfunction that could lead to severe bone deformity. As demonstrated in Kir2.1 knockout mice that exhibit a narrow maxilla and complete cleft of the secondary palate that may mimic the facial dysmorphology observed in humans (Hibino et al., 2009).
Pathogenesis of Thyrotoxic Periodic Paralysis
In TPP, the muscle PP has been linked to mutations of the inwardly rectifying potassium channel called Kir2.6 encoded by a new KCNJ18 gene (Figure 1B; Ryan et al., 2010). Ryan et al. (2010) demonstrated that some TPP mutants show decreased current density at −60 mV (Vm), some others lack the modulation by PKC which is normally activated during thyrotoxicosis leading to inhibitory action of WT channels but not mutants. Another effects of some mutations is to reduce the binding affinity for PiP2 (Figure 1B). This leads to abnormally prolonged channel openings uncoupled to PiP2. Protein regulation by thyroid hormones occur by transcriptional and post-translational events. Transcriptional regulation is particularly important in skeletal muscle, where both decreased (hypothyroidism) and increased (thyrotoxicosis) T3 has a profound effect on muscle performance. Several ion channels as Kv1.5 and Kv4.2 and now also Kir2.6 channels are known to be transcriptionally regulated by T3 through the thyroid response elements on the genes. Increased or decreased ion channel transcription during thyrotoxicosis may cause drastic changes in resting membrane potential and potassium accumulation and can lead to weakness in otherwise normal patients. The Kir2.6 levels are increased during thyrotoxicosis to aid in proper membrane potential maintenance, warding off more severe weakness. For instance increased Kir currents of some mutants during thyrotoxicosis may lead to hyperpolarization and difficulty to reach the threshold potentials with altered excitability. As opposite the reduced current density in some other TPP mutants lead to depolarization and inactivation of voltage-dependent Na+ and Ca2+ channels with weakness and paralysis (Ryan et al., 2010).
Mechanisms of Drug Action in Periodic Paralysis and Drug Therapy
Drug therapies can be separated into two categories, treatments used for acute attacks and prophylactic treatments.
In hyperPP, acute attacks may respond to inhaled salbutamol or glucose/insulin therapy that exert their repolarizing actions by activating the 3Na+/2K+-ATPase and restoring the serum K+ levels. Whereas in hypoPP, acute attacks are usually treated with oral potassium chloride glucose-free.
The treatment of the hyperkalemia and hypokalemia in hyperPP or hypoPP is achieved with some success by the administration of the benzothiazide diuretics as hydrochlorothiazide and of K+ sparing diuretics, respectively. But the most effective medications for the prevention of attacks in both disorders remain the CA inhibitors, particularly acetazolamide and dichlorphenamide. These drugs ameliorate paralysis reducing the frequency of the attacks. One of the main mechanism responsible for the therapeutic effects of these drugs is related to their capability to open the muscular calcium-activated potassium channels (Tricarico et al., 2000, 2004). These drugs activate BK channels in excised patch experiments from normal and depleted rat fibers at micromolar concentrations indicating that the observed actions are due to a direct interaction of the molecules with the channel subunits in the isolated membrane (Figure 3). The actions of acetazolamide and dichlorphenamide are structure-related; indeed, these molecules share the same pharmacophoric characteristics of benzimidazolone class of compounds which are well known small synthetic BK channel openers (Tricarico et al., 2004). Acetazolamide and dichlorphenamide repolarize skeletal muscle fibers and prevent muscle paralysis and weakness in K-depleted rats induced by insulin/glucose injection by openings the BK channels (Tricarico et al., 2006). Acetazolamide at 100 × 10−4 M is also capable to repolarize muscle fibers from human hypoPP patients even in the presence of low ext. K+ ions (Jurkat-Rott et al., 2009). The effects of acetazolamide and dichlorphenamide on human BK channel have been also recently investigated, and we found that these two drugs are effective against the hslo channel subunit expressed in HEK293 cell at submicromolar concentrations being 10 times more potent against human channel vs. rat channel. These effects were observed at −60 mV (Vm) justifying their use in the attack prevention rather than during the attack of PP (unpublished observations). Currently no direct interaction of these drugs with Kir including KATP channels have been observed in native fibers or recombinant clones expressed in cell lines indicating that acetazolamide by targeting BK channel increase the KDR current component thereby shifting the control of the resting potential under a voltage-dependent current component (Figure 1A).
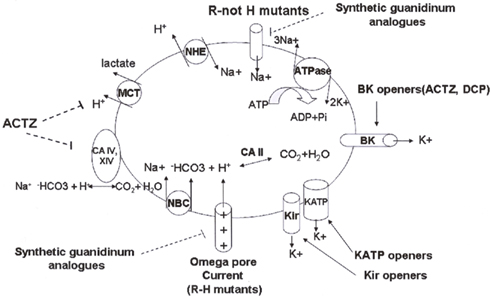
Figure 3. Targets of drug actions of potential interest in the hypokalemic periodic paralysis (modified from Matthews and Hanna, 2010). Acetazolamide or dichlorphenamide are capable to activate the calcium-activated K+ channels (BK) at submicromolar concentrations with fiber repolarization. Acetazolamide is also capable to inhibit the monocarboxylate transporter (MCT) reducing the efflux of lactate thereby preventing vacuolar myopathy. This drug also inhibits the membrane bound carbonic anhydrase (CA) enzymes with effects on the extra-/intracellular proton exchange mechanisms. KATP openers activate the ATP-sensitive K+ channels (KATP) at submicromolar concentrations. Other pumps and co-transports such as the natrium-bicarbonate co-transport (NBC), natrium hydrogen exchanger (NHE), or sodium–potassium ATPase may have a role in hypoPP pathogenesis are however not directly affected by drug actions. Drugs targeting inwardly rectifying K+ channels (Kir) can be effective in preventing fiber depolarization but are not currently available. Even the novel small synthetic guanidinium analogs appear to block at millimolar concentrations the R-H or the R-not H mutant subunits of the voltage-dependent Na+ channels but are however far from the clinical development.
Acetazolamide has been shown to prevent vacuolar myopathy by inhibiting the proton–lactate co-transport in the skeletal muscle of K-depleted rats which implies that it may averts the progressive myopathy seen in hypoPP as well as the inter-attack of weakness in humans (Figure 3; Tricarico et al., 2008b; Matthews and Hanna, 2010).
However, in some hypoPP patients acetazolamide failed to prevent paralysis and in some cases show detrimental effects. It is reported that none of the patients with glycine substitutions, i.e., R528G, R1239G, or R672G had benefit by acetazolamide treatment and some have detrimental responses (Bendahhou et al., 2001; Sternberg et al., 2001; Kim et al., 2007; Kil and Kim, 2009). One explanation is that the gating defects of mutants with a glycine substitution expressed in cell lines are not alleviated by lowering intracellular pH and therefore the patients affected by these mutations would not benefit from acetazolamide, while histidine substitutions are alleviated by low pH and patients carrying those mutations should benefit from this drug (Kuzmenkin et al., 2002).
Currently no Igp blockers are available for clinical use. Catterall and colleagues (Sokolov et al., 2010) recently showed that gating pore current generated by Nav1.4 mutants R666H or R666G can be blocked by trivalent cations, but in a nearly voltage-independent manner, with an apparent K(d) for Gd(3+) of 238 μM at −80 mV. They found that an the 1-(2,4-xylyl)guanidinium, an aliphatic guanidine derivative, can block gating pore current in the millimolar concentration range without affecting normal Na(V)1.4 channel function. This demonstrates voltage-dependent and voltage-independent block by divalent and trivalent cations, respectively, and provide initial support for the concept that guanidine-based molecules can be the first synthetic gating pore blockers (Sokolov et al., 2010).
The treatment of AS is complicated by the necessity of treating two distinct excitable tissues, skeletal muscle and cardiac muscle. For the PP, therapy involves minimizing known triggers and prophylaxis against recurrent attacks with the CA inhibitors. Potassium supplementation may also prevent paralytic attacks while simultaneously shortening the QT interval and lessening arrhythmias. Treatment for the cardiac symptoms of AS is based on anecdotal evidence supporting the use of β-blockers or calcium channel blockers. Recent evidence has shown flecainide to be particularly successful in the treatment of BVT, ventricular arrhythmias, and tachycardia-induced cardiomyopathy in AS.
Treatment of TPP is based on the surgical and iodine 131 treatments, while adrenergic symptoms are cured by beta blockers. No specific treatments for the weakness are available in TPP. Acetazolamide should not be used in these patients for the expected aggravation of the adrenergic symptoms. This drug indeed potentiates the adrenergic cascade of reactions in several tissues.
Acetazolamide and dichlorphenamide are used as off label treatment of PP. Three clinical studies were found in the Clinicaltrial.gov database:
Phase III Randomized, Double-Blind, Placebo-Controlled Study of Dichlorphenamide for Periodic paralysis and Associated Sodium Channel Disorders (NCT00004802) evaluated the efficacy of dichlorphenamide in PP and paramyotonia congenita with PP sponsored by National Center for Research Resources (NCRR) and prof. Jerry R. Mendell as principal investigator.
Hyper- and Hypokalemic Periodic Paralysis Study (HYP-HOP) (NCT00494507) sponsored by National Institute of Neurological Disorders and Stroke (NINDS) and University of Rochester and as principal investigator prof. Robert C. Griggs, is a Phase III multi-center, parallel, randomized trial comparing the effects of dichlorphenamide vs. placebo in patients with hyperkalemic (HYP) and hypokalemic (HOP) periodic paralysis.
Characteristics of Andersen-Tawil Syndrome (NCT00521794) is an observational study to better define the genetic basis, clinical features, and disease progression of AS. The study will also establish clinically relevant endpoints for use in future clinical studies sponsored by Office of Rare Diseases (ORD) and principal investigator prof. Rabi Tawil (Platt and Griggs, 2009).
Conclusions
In conclusion, the unbalance of the depolarizing gating pore currents and of the repolarizing Kir/KATP currents explain the paradoxical depolarization characterizing the hypoPP fibers and predispose the fibers to the paralysis induced by glucose/insulin. Currently, the gain-of-function of the gating pore proton or Na+ currents flowing through alternative pathways of the mutant subunits has been found in the type-2 hypoPP, while a reduced/expression activity of KATP channels and inwardly rectifying potassium channels has been found mostly in the type-1 hypoPP.
The three possible targets that have been proposed for the treatment of hypoPP are: skeletal muscle selective Kir/KATP or BK openers, gating pore blockers and isoform specific CA inhibitors (Figure 3; Matthews and Hanna, 2010).
The recent advance in Igp theory help to explain the PP opening the possibility to more specifically target the hypoPP disease. The ideal drugs would be the Igp blockers but this appears to be very difficult to achieve without affecting normal channel gating of the WT channels. Na+ channel blockers or Ca2+ channel antagonists failed in the treatment of hypoPP and even hyperPP often precipitating the attack of weakness indicating that unselective Igp pore blockers would precipitate the attacks. It remains safe and effective the proposal of targeting the KATP, Kir channels or BK channels by drugs capable to specifically open at nanomolar concentrations the skeletal muscle subtypes with less side effects (Tricarico et al., 2008c).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The work was supported by Telethon Grants.
References
Andersen, E. D., Krasilnikoff, P. A., and Overvad, H. (1971). Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. A new syndrome? Acta Paediatr. Scand. 60, 559–564.
Bendahhou, S., Cummins, T. R., Griggs, R. C., Fu, Y. H., and Ptácek, L. J. (2001). Na channel mutation in DIIS4 (Type2 HypoPP), whose patients exhibit worsening of the phenotype with acetazolamide intake: sodium channel inactivation defects are associated with acetazolamide-exacerbated hypokalemic periodic paralysis. Ann. Neurol. 50, 417–420.
Cannon, S. C. (2010). Voltage-sensor mutations in channelopathies of skeletal muscle. J. Physiol. 588, 1887–1895.
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2009). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366.
Jovanovic, S., Du, Q., Mukhopadhyay, S., Swingler, R., Buckley, R., McEachen, J., Jovanovic, A. (2008). A patient suffering from hypokalemic periodic paralysis is deficient in skeletal muscle ATP-sensitive K+ channels. Clin. Transl. Sci. 1, 71–74.
Jurkat-Rott, K., and Lehmann-Horn, F. (2007). Genotype–phenotype correlation and therapeutic rationale in hyperkalemic periodic paralysis. Neurotherapeutics 4, 216–224.
Jurkat-Rott, K., Lehmann-Horn, F., Elbaz, A., Heine, R., Gregg, R. G., Hogan, K., Powers, P. A., Laple, P., Vale-Santos, J. E., Weissenbach, J., and Fontaine, B. (1994). A calcium channel mutation causing hypokalemic periodic paralysis. Hum. Mol. Genet. 3, 1415–1419.
Jurkat-Rott, K., Marc-André, W., Faulera, M., Guoa, X., Holzherra, B. D., Paczulla, A., Nordsborga, N., Joechle, W., and Lehmann-Horn, F. (2009). K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc. Natl. Acad. Sci. U.S.A. 106, 4036–4041.
Jurkat-Rott, K., Mitrovic, N., Hang, C., Kouzmekine, A., Iaizzo, P., Herzog, J., Lerche, H., Nicole, S., Vale-Santos, J., Chauveau, D., Fontaine, B., and Lehmann-Horn, F. (2000). Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc. Natl. Acad. Sci. U.S.A. 97, 9549–9554.
Kil, T. H., and Kim, J. B. (2009). Severe respiratory phenotype caused by a de novo Arg528Gly mutation in the CACNA1S gene in a patient with hypokalemic periodic paralysis. Eur. J. Paediatr. Neurol. 14, 278–281.
Kim, J. B., Kim, M. H., Lee, S. J., Kim, D. J., and Lee, B.C. (2007). The genotype and clinical phenotype of Korean patients with familial hypokalemic periodic paralysis. J. Korean Med. Sci. 22, 946–951.
Kim, S. J., Lee, Y. J., and Kim, J. B. (2010). Reduced expression and abnormal localization of the K(ATP) channel subunit SUR2A in patients with familial hypokalemic periodic paralysis. Biochem. Biophys. Res. Commun. 391, 974–978.
Kuzmenkin, A., Hang, C., Kuzmenkin, E., and Jurkat-Rott, K. (2007). Gating of the HypoPP-1 mutations: I. Mutant specific effects and cooperativity. Pflugers Arch. 454, 495–505.
Kuzmenkin, A., Muncan, V., Jurkat-Rott, K., Hang, C., Lerche, H., Lehmann-Horn, F., and Mitrovic, M. (2002). Enhanced inactivation and pH sensitivity of Na(+) channel mutations causing hypokalaemic periodic paralysis type II. Brain 125(Pt 4), 835–843.
Lapie, P., Goudet, C., Nargeot, J., Fontaine, B., and Lory, P. (1996). Electrophysiological properties of the hypokalaemic periodic paralysis mutation (R528H) of the skeletal muscle alpha 1s subunit as expressed in mouse L cells. FEBS Lett. 382, 244–248.
Matthews, E., and Hanna, M. G. (2010). Muscle channelopathies: does the predicted channel gating pore offer new treatment insights for hypokalaemic periodic paralysis? J. Physiol. 588, 1879–1886.
Morita, H., Zipes, D. P., Morita, S. T., and Wu, J. (2007). Mechanism of U wave and polymorphic ventricular tachycardia in a canine tissue model of Andersen–Tawil syndrome. Cardiovasc. Res. 75, 510–518.
Plaster, N. M., Tawil, R., Tristani-Firouzi, M., Canun, S., Bendahhou, S., Tsunoda A., Donaldson, M. R., Iannaccone, S. T., Brunt, E, Barohn, R., Clark, J., Deymeer, F., George, A. L., Jr., Fish, F. A., Hahn, A., Nitu, A., Ozdemir, C., Serdaroglu, P., Subramony, S. H., Wolfe, G., Fu, Y. H., and Ptácek, L. J. (2001). Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen’s syndrome. Cell 105, 511–519.
Platt, D., and Griggs, R. (2009). Skeletal muscle channelopathies: new insights into the periodic paralyses and nondystrophic myotonias. Curr. Opin. Neurol. 22, 524–531.
Ptacek, L. J., Tawil, R., Griggs, R. C., Engel, A. G., Layzer, R. B., and Kwiecinski, H. (1994). Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell 77, 863–868.
Qu, Z, Yang, Z., Cui, N., Zhu, G., Liu, C., Xu, H., Chanchevalap, S., Shen, W., Wu, J., Li, Y., and Jiang, C. (2000). Gating of inward rectifier K+ channels by proton-mediated interactions of N- and C-terminal domains. J. Biol. Chem. 275, 31573–31580.
Ruff, R. L. (1999). Insulin acts in hypokalemic periodic paralysis by reducing inward rectifier K+ current. Neurology 53, 1556–1563.
Ryan, D. P., Dias da Silva, M. R., Soong, T. W., Fontaine, B., Donaldson, M. R., Kung, A. W. C., Jongjaroenprasert, W., Liang, M. C., Khoo, D. H. C., Cheah, J. S., Ho, S. C., Bernstein, H. S., Maciel, R. M. B., Brown, R. H. Jr., and Ptácek, L. J. (2010). Mutations in potassium channel Kir2.6 cause susceptibility to thyrotoxic hypokalemic periodic paralysis. Cell 140, 88–98.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2007). Gating pore current in an inherited ion channelopathy. Nature 446, 76–78.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2008). Depolarization-activated gating pore current conducted by mutant sodium channels in potassium-sensitive normokalemic periodic paralysis. Proc. Natl. Acad. Sci. U.S.A. 105, 19980–19985.
Sokolov, S., Scheuer, T., and Catterall, W. A. (2010). Ion permeation and block of the gating pore in the voltage sensor of NaV1.4 channels with hypokalemic periodic paralysis mutations. J. Gen. Physiol. 136, 225–236.
Sternberg, D., Maisonobe, T., Jurkat-Rott, K., Nicole, S., Launay, E., and Chauveau, D., Tabti, N., Lehmann-Horn, F., Hainque, B., and Fontaine, B. (2001). Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain 124(Pt 6), 1091–1099.
Struyk, A. F., and Cannon, S. C. (2007). A Na+ channel mutation linked to hypokalemic periodic paralysis exposes a proton-selective gating pore. J. Gen. Physiol. 130, 11–20.
Tawil, R., Ptacek, L. J., Pavlakis, S. G., DeVivo, D. C., Penn, A. S, Ozdemir, C., and Griggs, R. C. (1994). Andersen’s syndrome: potassium-sensitive periodic paralysis, ventricular ectopy, and dysmorphic features. Ann. Neurol. 35, 326–330.
Tricarico, D., Barbieri, M., and Camerino, D. C. (2000). Acetazolamide opens the muscular KCa2+ channel: a novel mechanism of action that may explain the therapeutic effect of the drug in hypokalemic periodic paralysis. Ann. Neurol. 48, 304–312.
Tricarico, D., Barbieri, M., Mele, A., Carbonara, G., and Camerino, D. C. (2004). Carbonic anhydrase inhibitors are specific openers of skeletal muscle BK channel of K+-deficient rats. FASEB J. 18, 760–761.
Tricarico, D., Capriulo, R., and Conte Camerino, D. (1999a). Insulin modulation of ATP-sensitive K+ channel of rat skeletal muscle is impaired in the hypokalaemic state. Pflugers Arch. 437, 235–240.
Tricarico, D., Servidei, S., Tonali, P., Jurkat-Rott, K., and Camerino, D. C. (1999b). Impairment of skeletal muscle adenosine triphosphate-sensitive K+ channels in patients with hypokalemic periodic paralysis. J. Clin. Invest. 103, 675–682.
Tricarico, D., Mele, A., and Conte Camerino, D. (2006). Carbonic anhydrase inhibitors ameliorate the symptoms of hypokalaemic periodic paralysis in rats by opening the muscular Ca2+-activated-K+ channels. Neuromuscul. Disord. 16, 39–45.
Tricarico, D., Mele, A., Liss, B., Ashcroft, F. M., Lundquist, A. L., Desai, R. R., George, A. L. Jr., and Conte Camerino, D. (2008a). Reduced expression of Kir6.2/SUR2A subunits explains KATP deficiency in K+-depleted rats. Neuromuscul. Disord. 18, 74–80.
Tricarico, D., Lovaglio, S., Mele, A., Rotondo, G., Mancinelli, E., Meola, G., and Camerino, D. C. (2008b). Acetazolamide prevents vacuolar myopathy in skeletal muscle of K(+)-depleted rats. Br. J. Pharmacol. 154, 183–190.
Tricarico, D., Mele, A., Camerino, G. M., Laghezza, A., Carbonara, G., Fracchiolla, G., Tortorella, P., Loiodice, F., and Conte Camerino, D. (2008c). Molecular determinants for the activating/blocking actions of the 2H-1,4-benzoxazine derivatives, a class of potassium channel modulators targeting the skeletal muscle KATP channels. Mol. Pharmacol. 74, 1–9.
Tricarico, D., Pierno, S., Mallamaci, R., Brigiani, G. S., Capriulo, R., Santoro, G., and Camerino, D. C. (1998). The biophysical and pharmacological characteristics of skeletal muscle ATP-sensitive K+ channels are modified in K+-depleted rat, an animal model of hypokalemic periodic paralysis. Mol. Pharmacol. 54, 197–206.
Keywords: periodic paralysis, potassium channels, potassium channel openers, gating pore currents, pharmacology, skeletal muscle, thyrotoxicosis, channelopathies
Citation: Tricarico D and Camerino DC (2011) Recent advances in the pathogenesis and drug action in periodic paralyses and related channelopathies. Front. Pharmacol. 2:8. doi: 10.3389/fphar.2011.00008
Received: 03 January 2011;
Paper pending published: 18 January 2011;
Accepted: 08 February 2011;
Published online: 28 February 2011.
Edited by:
Mohamed Chahine, Laval University, CanadaReviewed by:
Saïd Bendahhou, Centre National de la Recherche Scientifique, FranceAlfred L. George, Vanderbilt University, USA
Copyright: © 2011 Tricarico and Camerino. This is an open-access article subject to an exclusive license agreement between the authors and Frontiers Media SA, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Domenico Tricarico, Department of Pharmacobiology, Faculty of Pharmacy, University of Bari, Via E. Orabona 4, 70125 Bari, Italy. e-mail:ZHRyaWNhcmljb0BmYXJtYmlvbC51bmliYS5pdA==