 Nobuaki Takahashi1,2
Nobuaki Takahashi1,2 Yasuo Mori1,3,4*
Yasuo Mori1,3,4*- 1 Laboratory of Molecular Biology, Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Kyoto, Japan
- 2 Advanced Biomedical Engineering Research Unit, Kyoto University, Kyoto, Japan
- 3 Core Research for Evolutional Science and Technology, Japan Science and Technology Agency, Chiyoda-ku, Tokyo, Japan
- 4 Laboratory of Environmental Systems Biology, Department of Technology and Ecology, Hall of Global Environmental Studies, Kyoto University, Kyoto, Japan
Proteins are capable of sensing the redox status of cells. Cysteine residues, which react with oxidants, reductants, and electrophiles, have been increasingly recognized as the mediators of this redox sensitivity. Cation channels encoded by the transient receptor potential (trp) gene superfamily are characterized by a wide variety of activation triggers that act from outside and inside the cell. Recent studies have revealed that a class of TRP channels is sensitive to changes in redox status and is notably susceptible to modifications of cysteine residues, such as oxidation, electrophilic reaction, and S-nitrosylation of sulfhydryls. In this review, we focus on TRP channels, which directly sense redox status, and discuss the biological significance of cysteine modifications and the consequences of this chemical reaction for physiological responses.
Introduction
The cellular signals initiated by reactive electrophilic species, such as reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive carbonyl species (RCS) are delivered through redox chemistry. This primarily involves the post-translational modification of specific amino acid residues, especially cysteine sulfhydryls, on signaling proteins (Satoh and Lipton, 2006). According to the classical view, the reaction of cysteine sulfhydryl groups maintains protein structure through disulfide bond formation and regulates catalytic centers through metal binding (Lipton et al., 2002). However, the paradigms for function of cysteine sulfhydryl reactions have been extended by the discovery of S-nitrosylation in biological systems. The transfer of a nitric oxide (NO) group to a key cysteine sulfhydryl represents a new model of cysteine sulfhydryls acting as sensors of redox status to regulate protein functions.
The physiological significance of NO in signal transduction was established by the demonstration that NO generated by endothelial cells relaxes vascular smooth muscle through the activation of guanylate cyclase and cyclic GMP-dependent kinase (Murad, 1986). The extensive range of NO-based signaling was illustrated by the discovery of NO synthases (NOSs) with well-conserved phylogenetics and pervasive tissue distributions (Murad, 2006). In addition to the well-characterized binding of NO to the heme iron of guanylate cyclase, S-nitrosylation was suggested to be an alternative physiological NO-based protein modification (Stamler et al., 1992a,b) and this theory has been proven by the identification of more than a hundred substrates for S-nitrosylation (Stamler et al., 2001). Important examples include S-nitrosylation of the NR2A subunit of N-methyl-D-aspartate (NMDA) receptors (Lipton et al., 2002), ryanodine receptors (RyR; (Eu et al., 2000), matrix metalloproteinases (MMPs; Gu et al., 2002), GAPDH (Hara et al., 2005), and protein-disulfide isomerase (PDI; Uehara et al., 2006). There is a growing awareness of S-nitrosylation as a post-translational protein modification regulated with precise temporal and spatial characteristics (Stamler et al., 2001; Barouch et al., 2002; Gow et al., 2002; Boehning and Snyder, 2003). These characteristics confer specificity to NO-derived effects, allowing S-nitrosylation to function as a prototype of mechanisms that convey redox-based cellular signals (Stamler et al., 2001).
Analogous to NO, other electrophiles can activate or inhibit specific signal transduction pathways by reacting with cysteine sulfhydryls and thus causing modification of protein structure. ROS can directly oxidize cysteine sulfhydryls in vitro and in vivo (Berlett and Stadtman, 1997). For example, H2O2 oxidizes sulfhydryl groups to sulfenic acid, which is rather unstable and reactive and rapidly forms stable intra- or intermolecular disulfide bonds if other adjacent sulfhydryl groups are present (Claiborne et al., 1999). Post-translational modifications include the addition of RCS on proteins, generically termed as “protein carbonylation,” with the most reactive and common form of these carbonyl groups being aldehydes. α,β-Unsaturated aldehydes, including 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), 4-hydroxy-(2E)-non-enal (4-HNE), and acrolein are reactive aldehydes generated from polyunsaturated fatty acid oxidation. Because of the presence of electron-withdrawing functional groups, the double bond of these compounds serves as a site for Michael addition with the sulfur atom of cysteine. Through the modification of key cysteine sulfhydryls, these electrophiles can mediate various biological actions including functional regulation of the IκB kinase (IKK) β subunit, the nuclear factor (NF)-κB p65 subunit (Rossi et al., 2000; Straus et al., 2000), kelch-like ECH-associated protein (Keap1; Eggler et al., 2005), thioredoxin (Shibata et al., 2003), and peroxisome proliferator activated receptor (PPAR)Γ (Shiraki et al., 2005). Thus, highly conserved redox reactions of cysteine sulfhydryls can be elementary molecular processes in the same way as phosphorylation reactions of threonine, serine, and tyrosine residues are in cell signaling.
Studies of transient receptor potential (trp) proteins (TRP), which form a variety of Ca2+-permeable cation channels, have significantly extended our knowledge of the molecular basis of sensory biology. TRP homologs are grouped into six subfamilies [canonical (C), vanilloid (V), melastatin (M), polycystic kidney disease (P), mucolipin (ML), and ankyrin (A)] by the homology of their protein sequences (Clapham, 2003; Voets et al., 2005). Because of their distinct activation mechanisms and biophysical properties, TRP channels are highly suited to function in sensory receptor cells, either as molecular sensors for environmental or endogenous stimuli or as modulators of signal transduction cascades downstream of metabotropic receptors. In fact, TRP channels play crucial roles in many types of senses, including touch, taste, and smell in mammals (Clapham, 2003; Voets et al., 2005). Recently, it has been demonstrated that a group of TRP channels are cell sensors for changes in redox status (Hara et al., 2002; Aarts et al., 2003; Yoshida et al., 2006; Xu et al., 2008). The TRPM2 channel, the first identified ROS-sensitive TRP channel, is activated indirectly by H2O2 through the production of nicotinamide adenine dinucleotide and its metabolites, ADP-ribose (ADPR) and cyclic ADPR (Hara et al., 2002; Perraud et al., 2005). Accumulated evidence indicates that TRPM2 mediates several cellular responses, including the H2O2-activated Ca2+ influx that mediates cell death (Hara et al., 2002) and, in pancreatic β-cells, the Ca2+ or cation influx that drives insulin secretion (Togashi et al., 2006; Uchida et al., 2011). Recently, through studies using Trpm2 knockout (KO) mice, we have demonstrated that H2O2-activated Ca2+ influx through TRPM2 induces chemokine production in monocytes, which aggravates inflammatory neutrophil infiltration (Yamamoto et al., 2008). In addition to TRPM2, certain members of the TRPC and TRPV subfamily, including TRPC5 and TRPV1, are activated directly by NO, oxidants, and other chemical agents through modification of cysteine free sulfhydryl groups (Yoshida et al., 2006). TRPC5 is also activated by reducing substances such as thioredoxin (Xu et al., 2008). More recently, TRPA1 channel activation has been shown to occur following oxidative cysteine modification by pungent compounds and inflammatory mediators (Hinman et al., 2006; Macpherson et al., 2007; Takahashi et al., 2008). Thus, TRP channels are targets of cysteine modification.
In this review, we focus on the three types of TRPs: TRPC5, TRPV1, and TRPA1 to extend our understanding of the biological significance of cysteine modifications by oxidants and electrophiles, and the physiological consequences of these chemical reactions in signal transduction pathway and in sensory neuronal responses.
TRPC5
TRPC5 was cloned from the mouse brain and functionally identified as a receptor-activated Ca2+-permeable cation channel linked to phospholipase C (PLC; Okada et al., 1998; Philipp et al., 1998). Although it is still controversial whether depletion of Ca2+ stores can activate TRPC5, a number of proteins and factors have been shown to act as direct triggers and modulators of TRPC5 channel activation. For example, binding of intracellular Ca2+ and calmodulin (CaM) have been implicated in TRPC5 activation and modulation (Ordaz et al., 2005; Shimizu et al., 2006; Blair et al., 2009; Gross et al., 2009), while membrane polyphosphoinositides, such as phosphatidylinositol 4,5-bisphosphate (PIP2), exert both stimulatory and inhibitory effects in regulating TRPC5 channel activity (Trebak et al., 2009). TRPC4 and TRPC1, the closest structural homologs of TRPC5, interact with the TRPC5 protein (Lockwich et al., 2000; Tang et al., 2000; Yuan et al., 2003; Obukhov and Nowycky, 2004; Goel et al., 2005; Schindl et al., 2008; Miehe et al., 2010). As TRPC1 interacts with both TRPC5 and caveolin-1 (Lockwich et al., 2000; Strübing et al., 2001), it is likely that TRPC5 forms protein complexes with caveolin-1 (the importance of which will discussed below). An array of these proteins and factors can cooperatively control the function of TRPC5 channelsomes.
TRPC5 is potently regulated by cysteine modifications and is activated by NO via cysteine S-nitrosylation (Yoshida et al., 2006). By performing labeling and functional assays with cysteine mutants, we showed that cysteine residues accessible from the cytoplasm, namely Cys553 and nearby Cys558 on the N-terminal side of the putative pore-forming region between the fifth and sixth transmembrane domains, are essential for mouse TRPC5 activation in response to NO (Figure 1). The corresponding cysteine sites of TRPC1, TRPC4, TRPV1, TRPV3, and TRPV4 are potential targets of nitrosylation that leads to channel activation (see also below for nitrosylation of TRPV1; Figure 2). NO-activated TRPC5 channels were significantly but not entirely suppressed by ascorbate, which reduces S-nitrosothiols to thiols but not disulfides. However, DTT, which reduces both S-nitrosothiols and disulfides to thiols, fully suppressed NO-activated TRPC5 channel activity. Thus, both nitrosylation and disulfide bond formation are very likely to be involved in NO-induced TRPC5 activation. In an S-nitrosylation assay (Jaffrey et al., 2001), S-nitrosylation was abolished by the mutation of Cys553 of TRPC5, whereas it was unaffected by mutation of Cys558. As proposed for the acid–base catalysis of hemoglobin nitrosylation in proteins with high NO sensitivity, basic and acidic amino acids surrounding S-nitrosylated cysteines enhance the nucleophilicity of the sulfhydryl and therefore the S-nitrosylation of this group (Hess et al., 2005). Indeed, in TRPC5, charged residues flanking Cys553 and Cys558 may confer modification susceptibility to NO. Our data may suggest that the TRPC5 channel is opened via the S-nitrosylation of Cys553 and a subsequent nucleophilic attack of nitrosylated Cys553 by the free sulfhydryl group of Cys558 to form a disulfide bond that stabilizes the open state (Figure 1). However, the NO sensitivity of TRPC5 channels has been disputed by several groups (Xu et al., 2008; Wong et al., 2010). It is possible that NO sensitivity of TRPC5 is dependent on culturing conditions, application systems of drugs, cell density during measurements, or other experimental conditions, which may affect the modification state of TRPC5 proteins, levels of antioxidants, or other molecular and cellular states.
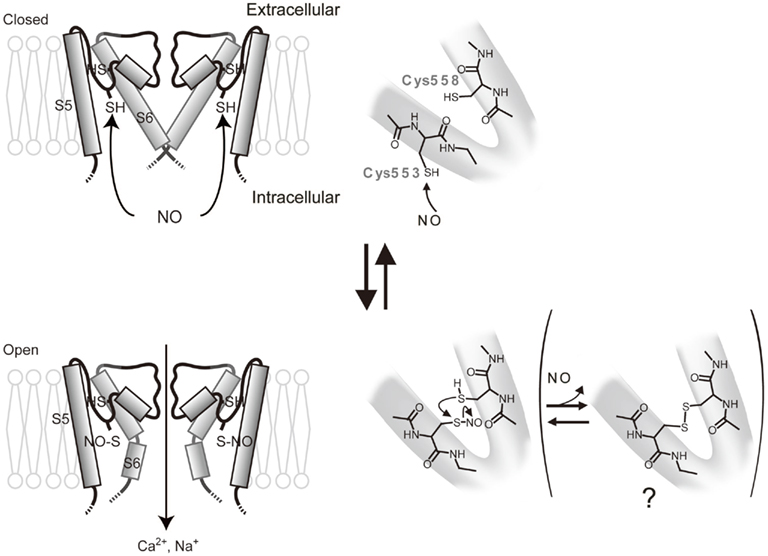
Figure 1. Model for activation of TRPC5 by NO. Possible protein conformation changes and chemical reactions during activation of TRPC5 by NO. NO modify the free sulfhydryl group of Cys553 accessible from the cytoplasmic side to open the activation gate. The modified Cys553 can be further attacked nucleophilically by the free sulfhydryl group of Cys558 to form an intramolecular disulfide bond, which may stabilize the channel in activation states.

Figure 2. Conserved cysteine residues on the N-terminal side of putative pore-forming regions in TRPs. Alignment of various TRPs with the Cys553/Cys558-containing TRPC5 sequence.
High extracellular concentrations of thioredoxin are apparent in rheumatoid arthritis, an inflammatory joint disease that disables millions of people worldwide (Burke-Gaffney et al., 2005; Smolen et al., 2007). TRPC5 is expressed in secretory fibroblast-like synoviocytes from patients with rheumatoid arthritis. TRPC5 is also activated by the reducing agent, DTT, and by extracellular reduced thioredoxin, which both cleave a disulfide bridge in the predicted extracellular loop adjacent to the ion-selectivity filter of TRPC5 (Xu et al., 2008). Blockade of thioredoxin-activated TRPC5 enhances secretory activity and prevents the suppression of secretion, suggesting that TRPC5 has a protective role against the progression of rheumatoid arthritis. Thus, the cysteine residues of TRPC5 that are important for the actions of NO and oxidants are also functional targets for reducing agents.
Previous reports have provided important information with respect to the TRPC5 “channelsome,” a molecular assembly centered upon a channel, in endothelial cells. Firstly, as mentioned above, TRPC1 has been described to form heterotetrameric channels with TRPC5 (Strübing et al., 2001) and a protein complex with caveolin-1 in caveolae/lipid raft domains (Lockwich et al., 2000; Bergdahl et al., 2003), which regulate plasma membrane trafficking of TRPC1 (Brazer et al., 2003). It is therefore possible that TRPC5 forms indirect protein complexes with caveolin-1 via TRPC1. In fact, we have found an interaction of TRPC5 with caveolin-1 and eNOS by co-immunoprecipitation experiments as well as by co-localization of TRPC5 with caveolin-1 (Mori et al., unpublished data). Secondly, among numerous caveolin-associated proteins linked to signaling cascades (Couet et al., 1997; García-Cardeña et al., 1997; Sato et al., 2004; Quest et al., 2008), three isoforms of NOS, such as eNOS, have been identified (Kone et al., 2003). The inhibitory association of caveolin is disrupted by the binding of Ca2+–CaM to eNOS, leading to eNOS activation (Ju et al., 1997; Michel et al., 1997a,b; Rizzo et al., 1998; Bernatchez et al., 2005). Thirdly, eNOS is known to be activated by different kinases, including Akt, protein kinase A, and protein kinase C (García-Cardeña et al., 1996; Fulton et al., 1999; Michell et al., 2001; Boo and Jo, 2003; Heijnen et al., 2004). Based on these papers and our own data, we can propose a plausible model to describe the role of the TRPC5 channelsome in regulating receptor-activated NO production in vascular endothelial cells (Figure 3). In this model, TRPC5 proteins form complexes with vasodilator receptors, G-proteins, PLCβs, and eNOS, and these complexes are anchored in caveolae by the scaffolding protein, caveolin-1. Upon vasodilator receptor stimulation, TRPC5 is activated by the PLCβ cascade to induce Ca2+ influx, which elevates the intracellular Ca2+ concentration ([Ca2+]i) and forms Ca2+–CaM. This releases eNOS from the inhibitory control of caveolin-1 and leads to an initial NO production, which activates uncomplexed TRPC5 channels. Ca2+ influx via NO-activated TRPC5 channels then induces secondary activation of eNOS to amplify the production of NO, resulting in a positive feedback cycle of receptor-activated Ca2+ and NO signaling. This model has been neatly summarized by Stamler and colleagues in a short review (Foster et al., 2006) based on our data (Yoshida et al., 2006).
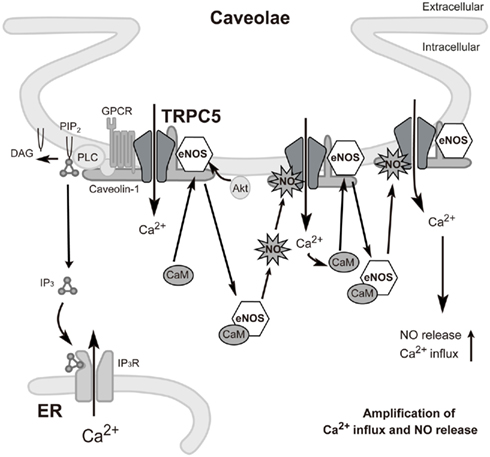
Figure 3. Proposed model for TRPC5-mediated feedback cycle of receptor-activated Ca2+ and NO signaling in caveolae of endothelial cells. Stimulation of GPCRs (such as the ATP-activated P2Y receptor) induces Ca2+ influx and activation of eNOS as a consequence of binding of Ca2+–CaM and release of eNOS from caveolin-1. TRPC5 undergoes eNOS-dependent S-nitrosylation after GPCR stimulation, resulting in amplified Ca2+ entry and secondary activation of eNOS to amplify production of NO. GPCR: G protein-coupled receptor.
Our immunolocalization studies have revealed that TRPC5 is distributed on both the apical and basal membrane in the endothelial cell layer of vascular tissue (Mori, unpublished data). Given that vasodilator receptors are distributed at the apical luminal surface of the endothelial cell layer, an initial burst of NO produced there may diffuse across the cytoplasm and activate TRPC5 located at the basolateral membrane. This could lead to an efficient propagation of Ca2+ signals directed toward the basal membrane. This feedback mechanism might further contribute to global [Ca2+]i elevation/oscillation and full activation of eNOS at the Golgi (Fulton et al., 2002) of endothelial cells, leading to synchronization of neighboring smooth muscle cells during relaxation of vascular tissues. This idea is substantiated by the fact that genetic disruption of TRPC4 (the closest relative of TRPC5), which we demonstrated to be colocalized with TRPC5 in the endothelial cell membrane (Yoshida et al., 2006), impairs agonist-dependent vasorelaxation (Freichel et al., 2001). Interestingly, the importance of TRPC5 has been demonstrated in neurite extension (Greka et al., 2003). As NO signals are reported to regulate neurite extension (Zhang et al., 2005), the feedback mechanism might also be important in growth cone morphology. TRPC5 channelsomes might also be involved in the activation of eNOS by shear stress (Fulton et al., 1999; Boo et al., 2002), which is purported to proceed through a Ca2+-independent mechanism because membrane stretch has been reported to activate TRPC5 independently of PLC function (Gomis et al., 2008). Even where initiation of NO production is evoked independently of Ca2+, the secondary amplification phase might be mediated by Ca2+ influx via NO-activated TRPC5 channels. Thus, the positive feedback regulation of Ca2+ signals by NO-activated TRP channels can be involved in diverse biological systems.
TRPV1
TRPV1 is the most extensively studied and best characterized among TRP family members. TRPV1 has been implicated in a wide variety of cellular and physiological processes, including detection of noxious physical and chemical stimuli, making it a promising target for the development of analgesic drugs with ultra-specificity for the origin of the pain.
TRPV1 is a non-selective cation channel that is activated by noxious stimuli such as heat (>43°C; Caterina et al., 1997), acidic pH (Tominaga et al., 1998), and environmental irritants and endogenous algesic substances including capsaicin (Caterina et al., 1997), camphor (Xu et al., 2005), allyl isothiocyanate (AITC) from mustard oil and wasabi (Everaerts et al., 2011), 12-hydroperoxyeicosatetraenoic acid (12-HPETE; Hwang et al., 2000; Shin et al., 2002), bradykinin (Premkumar and Ahern, 2000), and anandamide (Zygmunt et al., 1999). Although expression of TRPV1 was originally reported to be restricted to primary afferent nociceptors of the dorsal root ganglia (DRG), trigeminal ganglia, and nodose ganglia (Szallasi et al., 1995; Caterina et al., 1997; Helliwell et al., 1998; Tominaga et al., 1998; Ward et al., 2003; Brierley et al., 2005; Christianson et al., 2006), recent studies have argued for a much wider distribution, both in the central nervous system and in non-neuronal tissues (Mezey et al., 2000; Roberts et al., 2004; Tóth et al., 2005; Cristino et al., 2006; Steenland et al., 2006; Cavanaugh et al., 2011). Analysis of TRPV1-deficient mice confirmed the specificity and activation kinetics of various receptor-specific stimuli (Caterina et al., 2000; Davis et al., 2000). TRPV1 is a useful model for studying the biochemical regulation of the multimodal sensitivity of cells to stimuli. For example, the sensitivity of TRPV1 to capsaicin and noxious heat can be greatly enhanced by mild acidosis (Chuang et al., 2001; Ji et al., 2002; Amadesi et al., 2004, 2006; Sugiuar et al., 2004) and inflammatory agents that activate PLC signaling pathways (Premkumar and Ahern, 2000; Tominaga et al., 2001). Such integration of different signaling pathways allows TRPV1 to detect subthreshold stimuli, and provides a mechanism through which tissue injury produces thermal hypersensitivity (Julius and Basbaum, 2001).
Cysteine modifications also mediate the TRPV1-activating actions of several regulatory factors (Yoshida et al., 2006). The alignment of amino acid sequences surrounding Cys553 and Cys558 of TRPC5 with counterpart sequences shows cysteines conserved on the N-terminal side of the putative pore-forming region, which is located between the fifth and sixth transmembrane domains in TRPV1. Indeed, TRPV1 channels are activated by NO by itself, while a TRPV1 mutant with substitutions at these conserved cysteines gives significantly suppressed responses to NO. NO also enhances the sensitivity of TRPV1 to H+ and heat, suggesting that nitrosylation-induced Ca2+ entry through TRPV1 is involved in heat or pain sensation. Sensitizing effects of oxidizing agents such as diamide and chloramine-T support the existence of these counterpart cysteines in TRPV1 (Susankova et al., 2006).
TRPV1 also shows sensitivity to pungent compounds from onion and garlic, such as allicin (Macpherson et al., 2005), through covalent modification of a single cysteine residue located in the N-terminal region (Salazar et al., 2008). TRPV1 activation by allicin is of physiological significance, because TRPV1 mediates part of the response to this compound in isolated DRG neurons and in an in vivo model. Recently, C-terminal cytoplasmic cysteine residues that sensitize TRPV1 activation upon oxidative challenge have been identified (Chuang and Lin, 2009). Robust oxidative modulation recovers the agonist sensitivity of receptors desensitized by prolonged exposure to capsaicin. Moreover, oxidative modulation operates synergistically with kinases and proton modulation. Considering that tissue damage and inflammation produce ROS, sensitization of TRPV1 under oxidative challenge is likely to play a role in nociceptor pain sensation during inflammation, infection, and tissue injury.
TRPA1
TRPA1 is the only member of the TRPA sub-branch of the TRP gene superfamily in mammals, characterized by a large number (17) of amino-terminal ankyrin repeats (Gaudet, 2008). Although TRPA1 was first cloned from a fibroblast cell line (Jaquemar et al., 1999), expression of TRPA1 is largely restricted to a subset of nociceptive C-fiber nerves, including somatosensory and vagal nerves (Story et al., 2003; Bandell et al., 2004; Jordt et al., 2004). In sensory neurons, TRPA1 was initially identified as a cold-sensitive ion channel in a small subset of cells (Story et al., 2003). Pharmacological experiments have revealed that TRPA1 is the sensory neuronal receptor for pungent compounds such as AITC (Bandell et al., 2004; Jordt et al., 2004), cinnamaldehyde from cinnamon (Bandell et al., 2004), and allicin from onion and garlic (Macpherson et al., 2005). These compounds are potentially susceptible to nucleophilic attack by the sulfhydryl groups of cysteine residues (Bautista et al., 2005; Macpherson et al., 2005). Similar to capsaicin, mustard oil activates sensory neurons, causing acute pain, thermal and mechanical hyperalgesia, and neurogenic inflammation (Jancsó et al., 1967; Bautista et al., 2006). TRPA1 is also activated by receptor stimulation (Bandell et al., 2004; Dai et al., 2007; Wang et al., 2008; Schmidt et al., 2009), cannabinoids (Jordt et al., 2004), caffeine (Nagatomo and Kubo, 2008), nicotine (Talavera et al., 2009), and heavy metals including zinc, cadmium, and copper (Hu et al., 2009; Gu and Lin, 2010). Recent reports have demonstrated that the biophysical and pharmacological properties of TRPA1 expressed in trigeminal and vagal neurons have intriguing parallels with those of the proposed reactive airway irritant receptor (Bessac and Jordt, 2008; Taylor-Clark and Undem, 2011).
Different approaches have provided strong support for the proposal that the activation mechanism of TRPA1 by AITC and cinnamaldehyde is dependent on covalent cysteine modifications (Figure 4; Table 1). Systematic mutation of candidate acceptor sites identified three neighboring cysteines within the cytoplasmic N-terminus on human TRPA1 (Cys621, Cys641, and Cys665) whose simultaneous mutation negates the channel-activating effects of several cysteine-modifying reagents (Hinman et al., 2006). Mass spectrometry independently implicated three cysteines in mouse TRPA1 (Cys415, Cys422, and Cys622), which are conserved in the human homolog (as Cys414, Csy421, and Cys621), as the target site for electrophilic agonists (Macpherson et al., 2007). Recently, we have shown that a variety of inflammatory mediators [15d-PGJ2, NO, H2O2, and protons (H+)] activate human TRPA1 in heterologous systems and mouse TRPA1 in dissociated sensory neurons (Takahashi et al., 2008). This finding suggests that TRPA1 channels are targeted by an array of inflammatory mediators to elicit inflammatory pain in the nervous system. Functional characterization of site-directed cysteine mutants of TRPA1 in combination with labeling experiments using biotinylated 15d-PGJ2 demonstrated that modifications of cytoplasmic N-terminal Cys421 and Cys621 are responsible for the activation of TRPA1 by 15d-PGJ2. In TRPA1 responses to other cysteine-reactive inflammatory mediators, such as NO and H2O2, the extent of impairment by respective cysteine mutations differed from those in TRPA1 responses to 15d-PGJ2. This characteristic can be attributed to a difference in the mechanisms employed by 15d-PGJ2 and NO/H2O2 to achieve the modification of free sulfhydryl groups of cysteine residues in TRPA1 proteins: 15d-PGJ2 modifies sulfhydryl groups through the Michael addition reaction, while NO and H2O2 modify sulfhydryl groups through the redox reaction. Interestingly, the Cys421 mutation critically impaired the TRPA1 response to H+ as well. Cysteine residues such as Cys421 can be deprotonated by nearby basic residues to form a thiolate anion in resting states and are protonated by acidic pH in activated states. This is a likely physiological scenario, considering that thiolate anions exert nucleophilic attack on 15d-PGJ2, NO, and H2O2. According to the structural model, Cys421 is spatially located close to the basic residue His418 on the same side of an α-helix (Gaudet, 2008). Notably, TRPA1 is activated by external 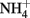 -induced intracellular alkalization (Fujita et al., 2008) as well as acidic pH (Takahashi et al., 2008; Wang et al., 2010), suggesting that the pH dependency of TRPA1 activity has an inverted bell-shape with the minimum around physiological pH of 7.4. Interestingly, different sets of cysteine residues have been identified by independent groups (Table 1). This can be attributed to differences among mechanisms underlying modification of cysteine residues as described above for 15d-PGJ2 and NO/H2O2. In RyR1, which is a redox-sensitive Ca2+ channel, only 12 of the 100 cysteine residues are redox-modified (“hyper-reactive” cysteines) and two of the 12 hyper-reactive cysteines are S-nitrosylated but not S-glutathionylated, whereas for a further two, the reverse applies (Aracena-Parks et al., 2006). In Keap1, which is a molecular sensor for intracellular redox changes, different cysteine residues have been reported to display different preferences for alkylating reagents (Dinkova-Kostova et al., 2002; Eggler et al., 2005; Hong et al., 2005). The discrepancy may be due to the differences among the mutants employed by the groups: Hinman et al. (2006) showed data only for triple TRPA1 mutant, which carries mutations of Cys621, Cys641, and Cys665, while others used single mutants. To fully understand cysteine residues responsible for TRPA1 activation, analyses of its three dimensional structure is essential.
-induced intracellular alkalization (Fujita et al., 2008) as well as acidic pH (Takahashi et al., 2008; Wang et al., 2010), suggesting that the pH dependency of TRPA1 activity has an inverted bell-shape with the minimum around physiological pH of 7.4. Interestingly, different sets of cysteine residues have been identified by independent groups (Table 1). This can be attributed to differences among mechanisms underlying modification of cysteine residues as described above for 15d-PGJ2 and NO/H2O2. In RyR1, which is a redox-sensitive Ca2+ channel, only 12 of the 100 cysteine residues are redox-modified (“hyper-reactive” cysteines) and two of the 12 hyper-reactive cysteines are S-nitrosylated but not S-glutathionylated, whereas for a further two, the reverse applies (Aracena-Parks et al., 2006). In Keap1, which is a molecular sensor for intracellular redox changes, different cysteine residues have been reported to display different preferences for alkylating reagents (Dinkova-Kostova et al., 2002; Eggler et al., 2005; Hong et al., 2005). The discrepancy may be due to the differences among the mutants employed by the groups: Hinman et al. (2006) showed data only for triple TRPA1 mutant, which carries mutations of Cys621, Cys641, and Cys665, while others used single mutants. To fully understand cysteine residues responsible for TRPA1 activation, analyses of its three dimensional structure is essential.
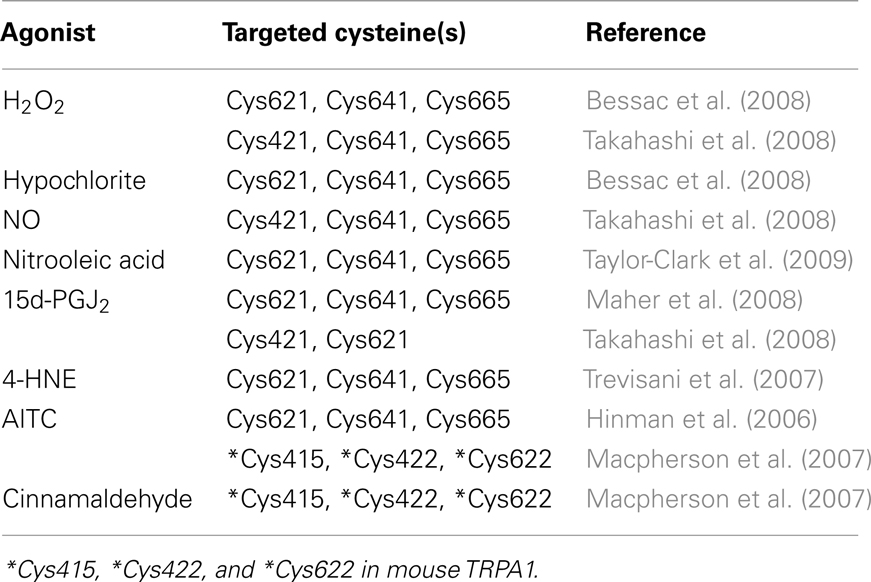
Table 1. Targeted cysteine residues in human TRPA1.
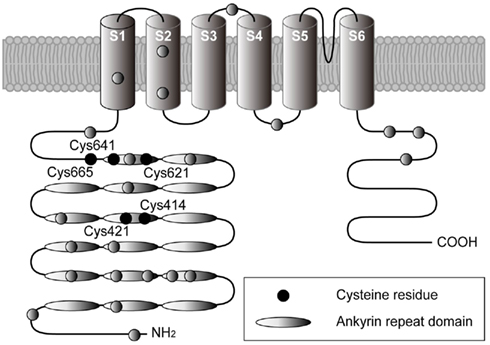
Figure 4. Structural model for TRPA1 protein. Four identical TRPA1 subunits are believed to be combined in the formation a functional channel. Each subunit spanning the plasma membrane six times (transmembrane domains S1–S6) has a long cytoplasmic N-terminal domain. Ovals indicate ankyrin repeats, while filled circles indicate cysteine residues identified as crucial sites for covalent modification of TRPA1 (Hinman et al., 2006; Macpherson et al., 2007; Trevisani et al., 2007; Bessac et al., 2008; Maher et al., 2008; Takahashi et al., 2008; Taylor-Clark et al., 2009).
An array of cysteine-reactive electrophiles has been proposed as TRPA1 activators. These include a variety of RCS such as acrolein (2-propenal; Bautista et al., 2006), 4-HNE (Trevisani et al., 2007), 4-oxonon-enal (Taylor-Clark et al., 2008a), and 15d-PGJ2 (Andersson et al., 2008; Maher et al., 2008; Takahashi et al., 2008; Taylor-Clark et al., 2008b); ROS such as hypochlorite (Bessac et al., 2008), H2O2 (Andersson et al., 2008; Bessac et al., 2008; Sawada et al., 2008; Takahashi et al., 2008), and O3 (Taylor-Clark and Undem, 2010); and RNS such as NO (Sawada et al., 2008; Takahashi et al., 2008), peroxynitrite (Sawada et al., 2008), and nitrooleic acid (Taylor-Clark et al., 2009).
Many TRPA1 stimulants activate bronchopulmonary C-fibers of vagal nerves, which innervate the airways and play a critical role in the detection of the airway microenvironment (Kubin et al., 2006). Oxidative stress and associated compounds have been shown to activate unmyelinated bronchopulmonary C-fibers, initiating action potentials in these nerves that conduct centrally to evoke unpleasant sensations such as urge to cough, dyspnea, and chest-tightness and to stimulate/modulate reflexes such as cough, bronchoconstriction, respiratory rate, and inspiratory drive (Bessac and Jordt, 2008). In vivo inhalation of H2O2 and hypochlorite given as an aerosol evokes a decrease in respiratory rate and an increase in end expiratory pause, which was abolished in Trpa1 KO mice (Bessac et al., 2008). In addition, O3 evokes robust action potential discharges from cinnamaldehyde-sensitive mouse bronchopulmonary C-fibers, which were reduced by approximately 80% by the generic TRP channel blocker, ruthenium red (Taylor-Clark and Undem, 2010). O3 also causes airway nociceptor hyperexcitability (Ho and Lee, 1998; Taylor-Clark and Undem, 2010), although it is unclear if this is due to TRPA1 function. Recently, our systematic evaluation of the oxidation sensitivity of TRP cation channels using reactive disulfides with different electrophilicity reveals the capability of TRPA1 to sense O2 in bronchopulmonary C-fibers (Takahashi et al., 2011).
The role of TRPA1 in airway afferent signaling has been studied in association with cigaret smoke and inflammation. It is well established that cigaret smoke contains nicotine and inflammation induces bradykinin, lipoxygenase products, and cyclooxygenase products, all of which can modulate airway nociceptor function independent of oxidative stress (Carr et al., 2003; Kwong and Lee, 2005; Lee et al., 2007; Taylor-Clark et al., 2008c; Talavera et al., 2009). However, both cigaret smoke (Andrè et al., 2008, 2009; Lin et al., 2010) and inflammation (Joseph et al., 2008; Kuhad and Chopra, 2009; Kamboj et al., 2010) also have the potential to modulate afferent excitability downstream of ROS production. Another important role for TRPA1 has been suggested from a mouse model of allergic asthma (Caceres et al., 2009). Allergen-induced eosinophilia, mucin production, and airway hyper-reactivity were reduced by a TRPA1 inhibitor, HC-030031 in wild-type mice by 35, 90, and 90%, respectively. These markers of allergen-induced airway inflammation were reduced in Trpa1 KO mice. Because TRPA1 is exquisitely sensitive to oxidants, further analyses are necessary to determine whether TRPA1 activation induced by smoke and inflammation is independent of ROS.
Conclusions
Transient receptor potential channels can respond to multiple activation triggers and therefore serve as polymodal signal detectors. An important aspect of this multimodal activation of TRP channels is its role in signal integration and amplification. When a TRP channel participates in a specific signaling cascade and is activated by downstream or upstream constituents (molecules/proteins/enzymes), in addition to the primary activation trigger immediately upstream, the TRP channel equips the cascade with positive feedback or feed-forward loops. This mechanism, which is capable of ensuring the fidelity of cellular responses and minimizing variation in their magnitude, may synchronize the responses of neighboring cells that comprise functional domains within tissues. For example, in vascular endothelial cells upon vasodilator receptor stimulation, Ca2+ influx via NO-activated TRPC5 channels can amplify production of NO by eNOS, resulting in the enhancement of NO production in nearby endothelial cells and NO-dependent relaxation of smooth muscle cells. The intercellular amplification of NO production eventually leads to vasodilation synchronized at the vascular tissue level.
The studies summarized above clearly indicate that multiple inflammatory signals conveyed by oxidants, lipid products, and protons converge at TRPV1 and TRPA1 to increase the excitability of sensory and vagal neurons during inflammation. Thus, TRPV1 and TRPA1 are sensors that translate oxidant and electrophilic stimuli into electrical signals in sensory and vagal neurons. However, the role(s) of TRPV1 and TRPA1 Ca2+ permeability in controlling Ca2+ signaling pathways is still elusive in neurons. How cellular signals amplified by activation of redox-sensitive TRP channels are terminated is also unclear. Our study of TRP channels is now extending from functional description of single molecules to analysis and integration of molecular systems controlled by TRP channels.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aarts, M., Iihara, K., Wei, W. L., Xiong, Z. G., Arundine, M., Cerwinski, W., MacDonald, J. F., and Tymianski, M. (2003). A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877.
Amadesi, S., Cottrell, G. S., Divino, L., Chapman, K., Grady, E. F., Bautista, F., Karanjia, R., Barajas-Lopez, C., Vanner, S., Vergnolle, N., and Bunnett, N. W. (2006). Protease-activated receptor 2 sensitizes TRPV1 by protein kinase C epsilon- and A-dependent mechanisms in rats and mice. J. Physiol. 575, 555–571.
Amadesi, S., Nie, J., Vergnolle, N., Cottrell, G. S., Grady, E. F., Trevisani, M., Manni, C., Geppetti, P., McRoberts, J. A., Ennes, H., Davis, J. B., Mayer, E. A., and Bunnett, N. W. (2004). Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J. Neurosci. 24, 4300–4312.
Andersson, D. A., Gentry, C., Moss, S., and Bevan, S. (2008). Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J. Neurosci. 28, 2485–2494.
Andrè, E., Campi, B., Materazzi, S., Trevisani, M., Amadesi, S., Massi, D., Creminon, C., Vaksman, N., Nassini, R., Civelli, M., Baraldi, P. G., Poole, D. P., Bunnett, N. W., Geppetti, P., and Patacchini, R. (2008). Cigarette smoke-induced neurogenic inflammation is mediated by α,β-unsaturated aldehydes and the TRPA1 receptor in rodents. J. Clin. Invest. 118, 2574–2582.
Andrè, E., Gatti, R., Trevisani, M., Preti, D., Baraldi, P. G., Patacchini, R., and Geppetti, P. (2009). Transient receptor potential ankyrin receptor 1 is a novel target for pro-tussive agents. Br. J. Pharmacol. 158, 1621–1628.
Aracena-Parks, P., Goonasekera, S. A., Gilman, C. P., Dirksen, R. T., Hidalgo, C., and Hamilton, S. L. (2006). Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J. Biol. Chem. 281, 40354–40368.
Bandell, M., Story, G. M., Hwang, S. W., Viswanath, V., Eid, S. R., Petrus, M. J., Earley, T. J., and Patapoutian, A. (2004). Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 25, 849–857.
Barouch, L. A., Harrison, R. W., Skaf, M. W., Rosas, G. O., Cappola, T. P., Kobeissi, Z. A., Hobai, I. A., Lemmon, C. A., Burnett, A. L., O’Rourke, B., Rodriguez, E. R., Huang, P. L., Lima, J. A., Berkowitz, D. E., and Hare, J. M. (2002). Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 416, 337–339.
Bautista, D. M., Jordt, S. E., Nikai, T., Tsuruda, P. R., Read, A. J., Poblete, J., Yamoah, E. N., Basbaum, A. I., and Julius, D. (2006). TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124, 1269–1282.
Bautista, D. M., Movahed, P., Hinman, A., Axelsson, H. E., Sterner, O., Högestätt, E. D., Julius, D., Jordt, S. E., and Zygmunt, P. M. (2005). Pungent products from garlic activate the sensory ion channel TRPA1. Proc. Natl. Acad. Sci. U.S.A. 102, 12248–12252.
Bergdahl, A., Gomez, M. F., Dreja, K., Xu, S. Z., Adner, M., Beech, D. J., Broman, J., Hellstrand, P., and Swärd, K. (2003). Cholesterol depletion impairs vascular reactivity to endothelin-1 by reducing store-operated Ca2+ entry dependent on TRPC1. Circ. Res. 93, 839–847.
Berlett, B. S., and Stadtman, E. R. (1997). Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 272, 20313–20316.
Bernatchez, P. N., Bauer, P. M., and Yu, J. (2005). Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc. Natl. Acad. Sci. U.S.A. 102, 761–766.
Bessac, B. F., and Jordt, S. E. (2008). Breathtaking TRP channels: TRPA1 and TRPV1 in airway chemosensation and reflex control. Physiology (Bethesda) 23, 360–370.
Bessac, B. F., Sivula, M., von Hehn, C. A., Escalera, J., Cohn, L., and Jordt, S. E. (2008). TRPA1 is a major oxidant sensor in murine airway sensory neurons. J. Clin. Invest. 118, 1899–1910.
Blair, N. T., Kaczmarek, J. S., and Clapham, D. E. (2009). Intracellular calcium strongly potentiates agonist-activated TRPC5 channels. J. Gen. Physiol. 133, 525–546.
Boo, Y. C., Hwang, J., Sykes, M., Michell, B. J., Kemp, B. E., Lum, H., and Jo, H. (2002). Shear stress stimulates phosphorylation of eNOS at Ser635 by a protein kinase A-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 283, H1819–H1828.
Boo, Y. C., and Jo, H. (2003). Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am. J. Physiol. Cell Physiol. 285, C499–C508.
Brazer, S. C., Singh, B. B., Liu, X., Swaim, W., and Ambudkar, I. S. (2003). Caveolin-1 contributes to assembly of store-operated Ca2+ influx channels by regulating plasma membrane localization of TRPC1. J. Biol. Chem. 278, 27208–27215.
Brierley, S. M., Carter, R., Jones, W., Xu, L., Robinson, D. R., Hicks, G. A., Gebhart, G. F., and Blackshaw, L. A. (2005). Differential chemosensory function and receptor expression of splanchnic and pelvic colonic afferents in mice. J. Physiol. 567, 267–281.
Burke-Gaffney, A., Callister, M. E., and Nakamura, H. (2005). Thioredoxin: friend or foe in human disease? Trends Pharmacol. Sci. 26, 398–404.
Caceres, A. I., Brackmann, M., Elia, M. D., Bessac, B. F., del Camino, D., D’Amours, M., Witek, J. S., Fanger, C. M., Chong, J. A., Hayward, N. J., Homer, R. J., Cohn, L., Huang, X., Moran, M. M., and Jordt, S. E. (2009). A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc. Natl. Acad. Sci. U.S.A. 106, 9099–9104.
Carr, M. J., Kollarik, M., Meeker, S. N., and Undem, B. J. (2003). A role for TRPV1 in bradykinin-induced excitation of vagal airway afferent nerve terminals. J. Pharmacol. Exp. Ther. 304, 1275–1279.
Caterina, M. J., Leffler, A., Malmberg, A. B., Martin, W. J., Trafton, J., Petersen-Zeitz, K. R., Koltzenburg, M., Basbaum, A. I., and Julius, D. (2000). Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288, 306–313.
Caterina, M. J., Schumacher, M. A., Tominaga, M., Rosen, T. A., Levine, J. D., and Julius, D. (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824.
Cavanaugh, D. J., Chesler, A. T., Jackson, A. C., Sigal, Y. M., Yamanaka, H., Grant, R., O’Donnell, D., Nicoll, R. A., Shah, N. M., Julius, D., and Basbaum, A. I. (2011). Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J. Neurosci. 31, 5067–5077.
Christianson, J. A., Traub, R. J., and Davis, B. M. (2006). Differences in spinal distribution and neurochemical phenotype of colonic afferents in mouse and rat. J. Comp. Neurol. 494, 246–259.
Chuang, H. H., and Lin, S. (2009). Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc. Natl. Acad. Sci. U.S.A. 106, 20097–20102.
Chuang, H. H., Prescott, E. D., Kong, H., Shields, S., Jordt, S. E., Basbaum, A. I., Chao, M. V., and Julius, D. (2001). Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411, 957–962.
Claiborne, A., Yeh, J. I., Mallett, T. C., Luba, J., Crane, E. J., Charrier, V., and Parsonage, D. (1999). Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry 38, 15407–15416.
Couet, J., Li, S., Okamoto, T., Ikezu, T., and Lisanti, M. P. (1997). Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 272, 6525–6533.
Cristino, L., de Petrocellis, L., Pryce, G., Baker, D., Guglielmotti, V., and Di Marzo, V. (2006). Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 139, 1405–1415.
Dai, Y., Wang, S., Tominaga, M., Yamamoto, S., Fukuoka, T., Higashi, T., Kobayashi, K., Obata, K., Yamanaka, H., and Noguchi, K. (2007). Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J. Clin. Invest. 117, 1979–1987.
Davis, J. B., Gray, J., Gunthorpe, M. J., Hatcher, J. P., Davey, P. T., Overend, P., Harries, M. H., Latcham, J., Clapham, C., Atkinson, K., Hughes, S. A., Rance, K., Grau, E., Harper, A. J., Pugh, P. L., Rogers, D. C., Bingham, S., Randall, A., and Sheardown, S. A. (2000). Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187.
Dinkova-Kostova, A. T., Holtzclaw, W. D., Cole, R. N., Itoh, K., Wakabayashi, N., Katoh, Y., Yamamoto, M., and Talalay, P. (2002). Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. U.S.A. 99, 11908–11913.
Eggler, A. L., Liu, G., Pezzuto, J. M., van Breemen, R. B., and Mesecar, A. D. (2005). Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. U.S.A. 102, 10070–10075.
Eu, J. P., Sun, J., Xu, L., Stamler, J. S., and Meissner, G. (2000). The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell 102, 499–509.
Everaerts, W., Gees, M., Alpizar, Y. A., Farre, R., Leten, C., Apetrei, A., Dewachter, I., van Leuven, F., Vennekens, R., De Ridder, D., Nilius, B., Voets, T., and Talavera, K. (2011). The capsaicin receptor TRPV1 is a crucial mediator of the noxious effects of mustard oil. Curr. Biol. 21, 316–321.
Foster, M. W., Hess, D. T., and Stamler, J. S. (2006). S-nitrosylation TRiPs a calcium Switch. Nat. Chem. Biol. 2, 570–571.
Freichel, M., Suh, S. H., Pfeifer, A., Schweig, U., Trost, C., Weissgerber, P., Biel, M., Philipp, S., Freise, D., Droogmans, G., Hofmann, F., Flockerzi, V., and Nilius, B. (2001). Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat. Cell Biol. 3, 121–127.
Fujita, F., Uchida, K., Moriyama, T., Shima, A., Shibasaki, K., Inada, H., Sokabe, T., and Tominaga, M. (2008). Intracellular alkalization causes pain sensation through activation of TRPA1 in mice. J. Clin. Invest. 118, 4049–4057.
Fulton, D., Fontana, J., Sowa, G., Gratton, J. P., Lin, M., Li, K. X., Michell, B., Kemp, B. E., Rodman, D., and Sessa, W. C. (2002). Localization of endothelial nitric-oxide synthase phosphorylated on serine 1179 and nitric oxide in golgi and plasma membrane defines the existence of two pools of active enzyme. J. Biol. Chem. 277, 4277–4284.
Fulton, D., Gratton, J. P., McCabe, T. J., Fontana, J., Fujio, Y., Walsh, K., Franke, T. F., Papapetropoulos, A., and Sessa, W. C. (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597–601.
García-Cardeña, G., Fan, R., Stern, D. F., Liu, J., and Sessa, W. C. (1996). Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J. Biol. Chem. 271, 27237–27240.
García-Cardeña, G., Martasek, P., Masters, B. S., Skidd, P. M., Couet, J., Li, S., Lisanti, M. P., and Sessa, W. C. (1997). Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 272, 25437–25440.
Gaudet, R. (2008). A primer on ankyrin repeat function in TRP channels and beyond. Mol. Biosyst. 4, 372–379.
Goel, M., Sinkins, W., Keightley, A., Kinter, M., and Schilling, W. P. (2005). Proteomic analysis of TRPC5- and TRPC6-binding partners reveals interaction with the plasmalemmal Na+/K+-ATPase. Pflügers Arch. 451, 87–98.
Gomis, A., Soriano, S., Belmonte, C., and Viana, F. (2008). Hypoosmotic- and pressure-induced membrane stretch activate TRPC5 channels. J. Physiol. 586, 5633–5649.
Gow, A. J., Chen, Q., Hess, D. T., Day, B. J., Ischiropoulos, H., and Stamler, J. S. (2002). Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J. Biol. Chem. 277, 9637–9640.
Greka, A., Navarro, B., Oancea, E., Duggan, A., and Clapham, D. E. (2003). TRPC5 is a regulator of hippocampal neurite length and growth cone morphology. Nat. Neurosci. 6, 837–845.
Gross, S. A., Guzmán, G. A., Wissenbach, U., Philipp, S. E., Zhu, M. X., Bruns, D., and Cavalié, A. (2009). TRPC5 is a Ca2+-activated channel functionally coupled to Ca2+-selective ion channels. J. Biol. Chem. 284, 34423–34432.
Gu, Q., and Lin, R. L. (2010). Heavy metals zinc, cadmium, and copper stimulate pulmonary sensory neurons via direct activation of TRPA1. J. Appl. Physiol. 108, 891–897.
Gu, Z., Kaul, M., Yan, B., Kridel, S. J., Cui, J., Strongin, A., Smith, J. W., Liddington, R. C., and Lipton, S. A. (2002). S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science 297, 1186–1190.
Hara, M. R., Agrawal, N., Kim, S. F., Cascio, M. B., Fujimuro, M., Ozeki, Y., Takahashi, M., Cheah, J. H., Tankou, S. K., Hester, L. D., Ferris, C. D., Hayward, S. D., Snyder, S. H., and Sawa, A. (2005). S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674.
Hara, Y., Wakamori, M., Ishii, M., Maeno, E., Nishida, M., Yoshida, T., Yamada, H., Shimizu, S., Mori, E., Kudoh, J., Shimizu, N., Kurose, H., Okada, Y., Imoto, K., and Mori, Y. (2002). LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 9, 163–173.
Heijnen, H. F., Waaijenborg, S., Crapo, J. D., Bowler, R. P., Akkerman, J. W., and Slot, J. W. (2004). Colocalization of eNOS and the catalytic subunit of PKA in endothelial cell junctions: a clue for regulated NO production. J. Histochem. Cytochem. 52, 1277–1285.
Helliwell, R. J., McLatchie, L. M., Clarke, M., Winter, J., Bevan, S., and McIntyre, P. (1998). Capsaicin sensitivity is associated with the expression of the vanilloid (capsaicin) receptor (VR1) mRNA in adult rat sensory ganglia. Neurosci. Lett. 250, 177–180.
Hess, D. T., Matsumoto, A., Kim, S. O., Marshall, H. E., and Stamler, J. S. (2005). Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166.
Hinman, A., Chuang, H. H., Bautista, D. M., and Julius, D. (2006). TRP channel activation by reversible covalent modification. Proc. Natl. Acad. Sci. U.S.A. 103, 19564–19568.
Ho, C. Y., and Lee, L. Y. (1998). Ozone enhances excitabilities of pulmonary C fibers to chemical and mechanical stimuli in anesthetized rats. J. Appl. Physiol. 85, 1509–1515.
Hong, F., Sekhar, K. R., Freeman, M. L., and Liebler, D. C. (2005). Specific patterns of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. J. Biol. Chem. 280, 31768–31775.
Hu, H., Bandell, M., Petrus, M. J., Zhu, M. X., and Patapoutian, A. (2009). Zinc activates damage-sensing TRPA1 ion channels. Nat. Chem. Biol. 5, 183–190.
Hwang, S. W., Cho, H., Kwak, J., Lee, S. Y., Kang, C. J., Jung, J., Cho, S., Min, K. H., Suh, Y. G., Kim, D., and Oh, U. (2000). Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc. Natl. Acad. Sci. U.S.A. 97, 6155–6160.
Jaffrey, S. R., Erdjument-Bromage, H., Ferris, C. D., Tempst, P., and Snyder, S. H. (2001). Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 3, 193–197.
Jancsó, N., Jancsó-Gábor, A., and Szolcsányi, J. (1967). Direct evidence for neurogenic inflammation and its prevention by denervation and by pretreatment with capsaicin. Br. J. Pharmacol. Chemother. 31, 138–151.
Jaquemar, D., Schenker, T., and Trueb, B. (1999). An ankyrin-like protein with transmembrane domains is specifically lost after oncogenic transformation of human fibroblasts. J. Biol. Chem. 274, 7325–7333.
Ji, R. R., Samad, T. A., Jin, S. X., Schmoll, R., and Woolf, C. J. (2002). p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36, 57–68.
Jordt, S. E., Bautista, D. M., Chuang, H. H., McKemy, D. D., Zygmunt, P. M., Högestätt, E. D., Meng, I. D., and Julius, D. (2004). Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265.
Joseph, E. K., Chen, X., Bogen, O., and Levine, J. D. (2008). Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. J. Pain 9, 463–472.
Ju, H., Zou, R., Venema, V. J., and Venema, R. C. (1997). Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 272, 18522–18525.
Kamboj, S. S., Vasishta, R. K., and Sandhir, R. (2010). N-acetylcysteine inhibits hyperglycemia-induced oxidative stress and apoptosis markers in diabetic neuropathy. J. Neurochem. 112, 77–91.
Kone, B. C., Kuncewicz, T., Zhang, W., and Yu, Z. Y. (2003). Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am. J. Physiol. Renal Physiol. 285, F178–F190.
Kubin, L., Alheid, G. F., Zuperku, E. J., and McCrimmon, D. R. (2006). Central pathways of pulmonary and lower airway vagal afferents. J. Appl. Physiol. 101, 618–627.
Kuhad, A., and Chopra, K. (2009). Tocotrienol attenuates oxidative-nitrosative stress and inflammatory cascade in experimental model of diabetic neuropathy. Neuropharmacology 57, 456–462.
Kwong, K., and Lee, L. Y. (2005). Prostaglandin E2 potentiates a TTX-resistant sodium current in rat capsaicin-sensitive vagal pulmonary sensory neurones. J. Physiol. 564, 437–450.
Lee, L. Y., Burki, N. K., Gerhardstein, D. C., Gu, Q., Kou, Y. R., and Xu, J. (2007). Airway irritation and cough evoked by inhaled cigarette smoke: role of neuronal nicotinic acetylcholine receptors. Pulm. Pharmacol. Ther. 20, 355–364.
Lin, Y. S., Hsu, C. C., Bien, M. Y., Hsu, H. C., Weng, H. T., and Kou, Y. R. (2010). Activations of TRPA1 and P2X receptors are important in ROS-mediated stimulation of capsaicin-sensitive lung vagal afferents by cigarette smoke in rats. J. Appl. Physiol. 108, 1293–1303.
Lipton, S. A., Choi, Y. B., Takahashi, H., Zhang, D., Li, W., Godzik, A., and Bankston, L. A. (2002). Cysteine regulation of protein function–as exemplified by NMDA-receptor modulation. Trends Neurosci. 25, 474–480.
Lockwich, T. P., Liu, X., Singh, B. B., Jadlowiec, J., Weiland, S., and Ambudkar, I. S. (2000). Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J. Biol. Chem. 275, 11934–11942.
Macpherson, L. J., Dubin, A. E., Evans, M. J., Marr, F., Schultz, P. G., Cravatt, B. F., and Patapoutian, A. (2007). Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445, 541–545.
Macpherson, L. J., Geierstanger, B. H., Viswanath, V., Bandell, M., Eid, S. R., Hwang, S., and Patapoutian, A. (2005). The pungency of garlic: activation of TRPA1 and TRPV1 in response to allicin. Curr. Biol. 15, 929–934.
Maher, M., Ao, H., Banke, T., Nasser, N., Wu, N. T., Breitenbucher, J. G., Chaplan, S. R., and Wickenden, A. D. (2008). Activation of TRPA1 by farnesyl thiosalicylic acid. Mol. Pharmacol. 73, 1225–1234.
Mezey, E., Tóth, Z. E., Cortright, D. N., Arzubi, M. K., Krause, J. E., Elde, R., Guo, A., Blumberg, P. M., and Szallasi, A. (2000). Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc. Natl. Acad. Sci. U.S.A. 97, 3655–3660.
Michel, J. B., Feron, O., Sacks, D., and Michel, T. (1997a). Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J. Biol. Chem. 272, 15583–15586.
Michel, J. B., Feron, O., Sase, K., Prabhakar, P., and Michel, T. (1997b). Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J. Biol. Chem. 272, 25907–25912.
Michell, B. J., Chen, Z. P., Tiganis, T., Stapleton, D., Katsis, F., Power, D. A., Sim, A. T., and Kemp, B. E. (2001). Coordinated Control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J. Biol. Chem. 276, 17625–17628.
Miehe, S., Bieberstein, A., Arnould, I., Ihdene, O., Rütten, H., and Strübing, C. (2010). The phospholipid-binding protein SESTD1 is a novel regulator of the transient receptor potential channels TRPC4 and TRPC5. J. Biol. Chem. 285, 12426–12434.
Murad, F. (1986). Cyclic guanosine monophosphate as a mediator of vasodilation. J. Clin. Invest. 78, 1–5.
Murad, F. (2006). Shattuck lecture. nitric oxide and cyclic GMP in cell signaling and drug development. N. Engl. J. Med. 355, 2003–2011.
Nagatomo, K., and Kubo, Y. (2008). Caffeine activates mouse TRPA1 channels but suppresses human TRPA1 channels. Proc. Natl. Acad. Sci. U.S.A. 105, 17373–17378.
Obukhov, A. G., and Nowycky, M. C. (2004). TRPC5 activation kinetics are modulated by the scaffolding protein ezrin/radixin/moesin-binding phosphoprotein-50 (EBP50). J. Cell. Physiol. 201, 227–235.
Okada, T., Shimizu, S., Wakamori, M., Maeda, A., Kurosaki, T., Takada, N., Imoto, K., and Mori, Y. (1998). Molecular cloning and functional characterization of a novel receptor-activated TRP Ca2+ channel from mouse brain. J. Biol. Chem. 273, 10279–10287.
Ordaz, B., Tang, J., Xiao, R., Salgado, A., Sampieri, A., Zhu, M. X., and Vaca, L. (2005). Calmodulin and calcium interplay in the modulation of TRPC5 channel activity. Identification of a novel C-terminal domain for calcium/calmodulin-mediated facilitation. J. Biol. Chem. 280, 30788–30796.
Perraud, A. L., Takanishi, C. L., Shen, B., Kang, S., Smith, M. K., Schmitz, C., Knowles, H. M., Ferraris, D., Li, W., Zhang, J., Stoddard, B. L., and Scharenberg, A. M. (2005). Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 280, 6138–6148.
Philipp, S., Hambrecht, J., Braslavski, L., Schroth, G., Freichel, M., Murakami, M., Cavalié, A., and Flockerzi, V. (1998). A novel capacitative calcium entry channel expressed in excitable cells. EMBO J. 17, 4274–4282.
Premkumar, L. S., and Ahern, G. P. (2000). Induction of vanilloid receptor channel activity by protein kinase C. Nature 408, 985–990.
Quest, A. F., Gutierrez-Pajares, J. L., and Torres, V. A. (2008). Caveolin-1: an ambiguous partner in cell signalling and cancer. J. Cell. Mol. Med. 12, 1130–1150.
Rizzo, V., McIntosh, D. P., Oh, P., and Schnitzer, J. E. (1998). In situ flow activates endothelial nitric oxide synthase in luminal caveolae of endothelium with rapid caveolin dissociation and calmodulin association. J. Biol. Chem. 273, 34724–34729.
Roberts, J. C., Davis, J. B., and Benham, C. D. (2004). [3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res. 995, 176–183.
Rossi, A., Kapahi, P., Natoli, G., Takahashi, T., Chen, Y., Karin, M., and Santoro, M. G. (2000). Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature 403, 103–108.
Salazar, H., Llorente, I., Jara-Oseguera, A., García-Villegas, R., Munari, M., Gordon, S. E., Islas, L. D., and Rosenbaum, T. (2008). A single N-terminal cysteine in TRPV1 determines activation by pungent compounds from onion and garlic. Nat. Neurosci. 11, 255–261.
Sato, Y., Sagami, I., and Shimizu, T. (2004). Identification of caveolin-1-interacting sites in neuronal nitric-oxide synthase. Molecular mechanism for inhibition of NO formation. J. Biol. Chem. 279, 8827–8836.
Satoh, T., and Lipton, S. A. (2006). Redox regulation of neuronal survival mediated by electrophilic compounds. Trends Neurosci. 30, 37–45.
Sawada, Y., Hosokawa, H., Matsumura, K., and Kobayashi, S. (2008). Activation of transient receptor potential ankyrin 1 by hydrogen peroxide. Eur. J. Neurosci. 27, 1131–1142.
Schindl, R., Frischauf, I., Kahr, H., Fritsch, R., Krenn, M., Derndl, A., Vales, E., Muik, M., Derler, I., Groschner, K., and Romanin, C. (2008). The first ankyrin-like repeat is the minimum indispensable key structure for functional assembly of homo- and heteromeric TRPC4/TRPC5 channels. Cell Calcium 43, 260–269.
Schmidt, M., Dubin, A. E., Petrus, M. J., Earley, T. J., and Patapoutian, A. (2009). Nociceptive signals induce trafficking of TRPA1 to the plasma membrane. Neuron 64, 498–509.
Shibata, T., Yamada, T., Ishii, T., Kumazawa, S., Nakamura, H., Masutani, H., Yodoi, J., and Uchida, K. (2003). Thioredoxin as a molecular target of cyclopentenone prostaglandins. J. Biol. Chem. 278, 26046–26054.
Shimizu, S., Yoshida, T., Wakamori, M., Ishii, M., Okada, T., Takahashi, M., Seto, M., Sakurada, K., Kiuchi, Y., and Mori, Y. (2006). Ca2+-calmodulin-dependent myosin light chain kinase is essential for activation of TRPC5 channels expressed in HEK 293 cells. J. Physiol. 570, 219–235.
Shin, J., Cho, H., Hwang, S. W., Jung, J., Shin, C. Y., Lee, S. Y., Kim, S. H., Lee, M. G., Choi, Y. H., Kim, J., Haber, N. A., Reichling, D. B., Khasar, S., Levine, J. D., and Oh, U. (2002). Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc. Natl. Acad. Sci. U.S.A. 99, 10150–10155.
Shiraki, T., Kamiya, N., Shiki, S., Kodama, T. S., Kakizuka, A., and Jingami, H. (2005). α,β-unsaturated ketone is a core moiety of natural ligands for covalent binding to peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 280, 14145–14153.
Smolen, J. S., Aletaha, D., Koeller, M., Weisman, M. H., and Emery, P. (2007). New therapies for treatment of rheumatoid arthritis. Lancet 370, 1861–1874.
Stamler, J. S., Lamas, S., and Fang, F. C. (2001). Nitrosylation. the prototypic redox-based signaling mechanism. Cell 106, 675–683.
Stamler, J. S., Simon, D. I., Jaraki, O., Osborne, J. A., Francis, S., Mullins, M., Singel, D., and Loscalzo, J. (1992a). S-nitrosylation of tissue-type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc. Natl. Acad. Sci. U.S.A. 89, 8087–8091.
Stamler, J. S., Simon, D. I., Osborne, J. A., Mullins, M. E., Jaraki, O., Michel, T., Singel, D. J., and Loscalzo, J. (1992b). S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proc. Natl. Acad. Sci. U.S.A. 89, 444–448.
Steenland, H. W., Ko, S. W., Wu, L. J., and Zhuo, M. (2006). Hot receptors in the brain. Mol. Pain 2, 34.
Story, G. M., Peier, A. M., Reeve, A. J., Eid, S. R., Mosbacher, J., Hricik, T. R., Earley, T. J., Hergarden, A. C., Andersson, D. A., Hwang, S. W., McIntyre, P., Jegla, T., Bevan, S., and Patapoutian, A. (2003). ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829.
Straus, D. S., Pascual, G., Li, M., Welch, J. S., Ricote, M., Hsiang, C. H., Sengchanthalangsy, L. L., Ghosh, G., and Glass, C. K. (2000). 15-deoxy-Δ12,14-prostaglandin J2 inhibits multiple steps in the NF-κB signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 97, 4844–4849.
Strübing, C., Krapivinsky, G., Krapivinsky, L., and Clapham, D. E. (2001). TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron 29, 645–655.
Sugiuar, T., Bielefeldt, K., and Gebhart, G. F. (2004). TRPV1 function in mouse colon sensory neurons is enhanced by metabotropic 5-hydroxytryptamine receptor activation. J. Neurosci. 24, 9521–9530.
Susankova, K., Tousova, K., Vyklicky, L., Teisinger, J., and Vlachova, V. (2006). Reducing and oxidizing agents sensitize heat-activated vanilloid receptor (TRPV1) current. Mol. Pharmacol. 70, 383–394.
Szallasi, A., Nilsson, S., Farkas-Szallasi, T., Blumberg, P. M., Hökfelt, T., and Lundberg, J. M. (1995). Vanilloid (capsaicin) receptors in the rat: distribution in the brain, regional differences in the spinal cord, axonal transport to the periphery, and depletion by systemic vanilloid treatment. Brain Res. 703, 175–183.
Takahashi, N., Kuwaki, T., Kiyonaka, S., Numata, T., Kozai, D., Mizuno, Y., Yamamoto, S., Naito, S., Knevels, E., Carmeliet, P., Oga, T., Kaneko, S., Suga, S., Nokami, T., Yoshida, J., and Mori, Y. (2011). TRPA1 underlies a sensing mechanism for O2. Nat. Chem. Biol. 7, 701–711.
Takahashi, N., Mizuno, Y., Kozai, D., Yamamoto, S., Kiyonaka, S., Shibata, T., Uchida, K., and Mori, Y. (2008). Molecular characterization of TRPA1 channel activation by cysteine-reactive inflammatory mediators. Channels (Austin) 2, 287–298.
Talavera, K., Gees, M., Karashima, Y., Meseguer, V. M., Vanoirbeek, J. A., Damann, N., Everaerts, W., Benoit, M., Janssens, A., Vennekens, R., Viana, F., Nemery, B., Nilius, B., and Voets, T. (2009). Nicotine activates the chemosensory cation channel TRPA1. Nat. Neurosci. 12, 1293–1299.
Tang, Y., Tang, J., Chen, Z., Trost, C., Flockerzi, V., Li, M., Ramesh, V., and Zhu, M. X. (2000). Association of mammalian Trp4 and phospholipase C isozymes with a PDZ domain-containing protein, NHERF. J. Biol. Chem. 275, 37559–37564.
Taylor-Clark, T. E., Ghatta, S., Bettner, W., and Undem, B. J. (2009). Nitrooleic acid, an endogenous product of nitrative stress, activates nociceptive sensory nerves via the direct activation of TRPA1. Mol. Pharmacol. 75, 820–829.
Taylor-Clark, T. E., McAlexander, M. A., Nassenstein, C., Sheardown, S. A., Wilson, S., Thornton, J., Carr, M. J., and Undem, B. J. (2008a). Relative contributions of TRPA1 and TRPV1 channels in the activation of vagal bronchopulmonary C-fibres by the endogenous autacoid 4-oxononenal. J. Physiol. 586, 3447–3459.
Taylor-Clark, T. E., Undem, B. J., Macglashan, D. W., Ghatta, S., Carr, M. J., and McAlexander, M. A. (2008b). Prostaglandin-induced activation of nociceptive neurons via direct interaction with transient receptor potential A1 (TRPA1). Mol. Pharmacol. 73, 274–281.
Taylor-Clark, T. E., Nassenstein, C., and Undem, B. J. (2008c). Leukotriene D4 increases the excitability of capsaicin-sensitive nasal sensory nerves to electrical and chemical stimuli. Br. J. Pharmacol. 154, 1359–1368.
Taylor-Clark, T. E., and Undem, B. J. (2010). Ozone activates airway nerves via the selective stimulation of TRPA1 ion channels. J. Physiol. 588, 423–433.
Taylor-Clark, T. E., and Undem, B. J. (2011). Sensing pulmonary oxidative stress by lung vagal afferents. Respir. Physiol. Neurobiol. 178, 406–413.
Togashi, K., Hara, Y., Tominaga, T., Higashi, T., Konishi, Y., Mori, Y., and Tominaga, M. (2006). TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J. 25, 1804–1815.
Tominaga, M., Caterina, M. J., Malmberg, A. B., Rosen, T. A., Gilbert, H., Skinner, K., Raumann, B. E., Basbaum, A. I., and Julius, D. (1998). The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 21, 531–543.
Tominaga, M., Wada, M., and Masu, M. (2001). Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc. Natl. Acad. Sci. U.S.A. 98, 6951–6956.
Tóth, A., Boczán, J., Kedei, N., Lizanecz, E., Bagi, Z., Papp, Z., Edes, I., Csiba, L., and Blumberg, P. M. (2005). Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res. Mol. Brain Res. 135, 162–168.
Trebak, M., Lemonnier, L., DeHaven, W. I., Wedel, B. J., Bird, G. S., and Putney, J. W. (2009). Complex functions of phosphatidylinositol 4,5-bisphosphate in regulation of TRPC5 cation channels. Pflügers Arch. 457, 757–769.
Trevisani, M., Siemens, J., Materazzi, S., Bautista, D. M., Nassini, R., Campi, B., Imamachi, N., Andrè, E., Patacchini, R., Cottrell, G. S., Gatti, R., Basbaum, A. I., Bunnett, N. W., Julius, D., and Geppetti, P. (2007). 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc. Natl. Acad. Sci. U.S.A. 104, 13519–13524.
Uchida, K., Dezaki, K., Damdindorj, B., Inada, H., Shiuchi, T., Mori, Y., Yada, T., Minokoshi, Y., and Tominaga, M. (2011). Lack of TRPM2 impaired insulin secretion and glucose metabolisms in mice. Diabetes 60, 119–126.
Uehara, T., Nakamura, T., Yao, D., Shi, Z. Q., Gu, Z., Ma, Y., Masliah, E., Nomura, Y., and Lipton, S. A. (2006). S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441, 513–517.
Voets, T., Talavera, K., Owsianik, G., and Nilius, B. (2005). Sensing with TRP channels. Nat. Chem. Biol. 1, 85–92.
Wang, S., Dai, Y., Fukuoka, T., Yamanaka, H., Kobayashi, K., Obata, K., Cui, X., Tominaga, M., and Noguchi, K. (2008). Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain 131, 1241–1251.
Wang, Y. Y., Chang, R. B., and Liman, E. R. (2010). TRPA1 is a component of the nociceptive response to CO2. J. Neurosci. 30, 12958–12963.
Ward, S. M., Bayguinov, J., Won, K. J., Grundy, D., and Berthoud, H. R. (2003). Distribution of the vanilloid receptor (VR1) in the gastrointestinal tract. J. Comp. Neurol. 465, 121–135.
Wong, C. O., Sukumar, P., Beech, D. J., and Yao, X. (2010). Nitric oxide lacks direct effect on TRPC5 channels but suppresses endogenous TRPC5-containing channels in endothelial cells. Pflugers Arch. 460, 121–130.
Xu, H., Blair, N. T., and Clapham, D. E. (2005). Camphor activates and strongly desensitizes the transient receptor potential vanilloid subtype 1 channel in a vanilloid-independent mechanism. J. Neurosci. 25, 8924–8937.
Xu, S. Z., Sukumar, P., Zeng, F., Li, J., Jairaman, A., English, A., Naylor, J., Ciurtin, C., Majeed, Y., Milligan, C. J., Bahnasi, Y. M., Al-Shawaf, E., Porter, K. E., Jiang, L. H., Emery, P., Sivaprasadarao, A., and Beech, D. J. (2008). TRPC channel activation by extracellular thioredoxin. Nature 451, 69–72.
Yamamoto, S., Shimizu, S., Kiyonaka, S., Takahashi, N., Wajima, T., Hara, Y., Negoro, T., Hiroi, T., Kiuchi, Y., Okada, T., Kaneko, S., Lange, I., Fleig, A., Penner, R., Nishi, M., Takeshima, H., and Mori, Y. (2008). TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat. Med. 14, 738–747.
Yoshida, T., Inoue, R., Morii, T., Takahashi, N., Yamamoto, S., Hara, Y., Tominaga, M., Shimizu, S., Sato, Y., and Mori, Y. (2006). Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2, 596–607.
Yuan, J. P., Kiselyov, K., Shin, D. M., Chen, J., Shcheynikov, N., Kang, S. H., Dehoff, M. H., Schwarz, M. K., Seeburg, P. H., Muallem, S., and Worley, P. F. (2003). Homer binds TRPC family channels and is required for gating of TRPC1 by IP3 receptors. Cell 114, 777–789.
Zhang, N., Beuve, A., and Townes-Anderson, E. (2005). The nitric oxide-cGMP signaling pathway differentially regulates presynaptic structural plasticity in cone and rod cells. J. Neurosci. 25, 2761–2770.
Keywords: TRP channels, oxidative stress, Ca2+ signaling, cysteine modification, S-nitrosylation, TRPC5, TRPV1, TRPA1
Citation: Takahashi N and Mori Y (2011) TRP channels as sensors and signal integrators of redox status changes. Front. Pharmacol. 2:58. doi: 10.3389/fphar.2011.00058
Received: 09 June 2011; Paper pending published: 30 June 2011;
Accepted: 20 September 2011; Published online: 13 October 2011.
Edited by:
Tuck Wah Soong, National University of Singapore, SingaporeReviewed by:
Rudi Vennekens, Katholieke Universiteit Leuven, BelgiumUhtaek Oh, Seoul National University, South Korea
Copyright: © 2011 Takahashi and Mori. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Yasuo Mori, Laboratory of Molecular Biology, Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, and Laboratory of Environmental Systems Biology, Department of Technology and Ecology, Hall of Global Environmental Studies, Kyoto University, Kyoto 615-8510, Japan. e-mail:bW9yaUBzYmNoZW0ua3lvdG8tdS5hYy5qcA==