 Gregory J. Kaczorowski1 Maria L. Garcia1
Gregory J. Kaczorowski1 Maria L. Garcia1 Jacob Bode2
Jacob Bode2- 1 Kanalis Consulting, Limited Liability Company, Edison, NJ, USA
- 2 Millipore Corporation, Billerica, MA, USA
Profiling of putative lead compounds against a representative panel of relevant enzymes, receptors, ion channels, and transporters is a pragmatic approach to establish a preliminary view of potential issues that might later hamper development. An early idea of which off-target activities must be minimized can save valuable time and money during the preclinical lead optimization phase if pivotal questions are asked beyond the usual profiling at hERG. The best data for critical evaluation of activity at ion channels is obtained using functional assays, since binding assays cannot detect all interactions and do not provide information on whether the interaction is that of an agonist, antagonist, or allosteric modulator. For ion channels present in human cardiac muscle, depending on the required throughput, manual-, or automated-patch-clamp methodologies can be easily used to evaluate compounds individually to accurately reveal any potential liabilities. The issue of expanding screening capacity against a cardiac panel has recently been addressed by developing a series of robust, high-throughput, cell-based counter-screening assays employing fluorescence-based readouts. Similar assay development approaches can be used to configure panels of efficacy assays that can be used to assess selectivity within a family of related ion channels, such as Nav1.X channels. This overview discusses the benefits of in vitro assays, specific decision points where profiling can be of immediate benefit, and highlights the development and validation of patch-clamp and fluorescence-based profiling assays for ion channels (for examples of fluorescence-based assays, see Bhave et al., 2010; and for high-throughput patch-clamp assays see Mathes, 2006; Schrøder et al., 2008).
Why Use in vitro Profiling Assays to Understand Lead Selectivity and Specificity?
The drug discovery and development process is long, and resource-intensive, often fraught with unforeseeable pitfalls, and historically challenged by low probability of success (Tollman et al., 2011). However, vast unmet medical needs justify the efforts expended in this area of research, and new methodologies are always being explored to streamline the research process and improve the success rate of drug registration. Perhaps the most important factor at the beginning of any drug discovery project, regardless of the target, is the identification of the best lead structure amongst the collection of hits discovered either using in silico or through high-throughput screening techniques to target the process of interest (Rudolph and Knoflach, 2011).
Many factors must be taken into account during lead selection, such as chemical tractability, chemical stability, physical–chemical properties, metabolic stability, PK characteristics, protein binding, and suitability for formulation, in addition to potency and mechanism of action. However, none are more important than the specificity and selectivity of the initial compound. Subjecting putative leads to thorough scrutiny with respect to potential off-target pharmacological activities can identify liabilities, some of them potentially fatal, early on in a program, and thereby allow medicinal chemists to explore the feasibility of correcting these problems during exploratory analoging studies. Thus, broad profiling of early hits to determine selectivity, and then use of this information to prioritize compounds for follow-up, rather than simply rank-ordering hits based solely on chemical tractability, can prevent wasting time and effort due to making a poor initial decision based on a limited and incomplete data set. As an example, Millipore offers a wide variety of profiling services that utilize biochemical assays for over 300 kinases and phosphatases and cell-based functional assays for over 150 G-protein coupled receptors (GPCRs) and over 50 ion channels that can be utilized to query compounds at any development stage for specificity and selectivity for primary targets, related family members, and for cardiac safety.
Selectivity and Specificity Screening
The most straightforward means of generating selectivity data on a group of hits is to test for activity against a panel of similar targets from the same and closely related super families of proteins (e.g., classes of related enzymes, GPCRs, nuclear receptors, ion channels, etc.). Such studies provide important data for medicinal chemists and help focus their structure–activity determinations as part of a feasibility assessment of a compound’s potential as a lead candidate. In addition, these studies also immediately identify molecules that are non-selective in their mechanism of action because such agents often display pleiotropic activities across an entire family of related proteins. Functional assays are the preferred methodology to use for these analyses (see below).
In general, two other means are commonly employed initially to gather specificity data (i.e., to identify activities on proteins/processes unrelated to the initial target, some of which could cause serious adverse drug side effects) on a series of hits (c.f. Coburger et al., 2010). One is to profile test compounds in vivo in an animal efficacy model and determine therapeutic index based on comparing exposure for efficacy versus observed side effects. The other is to profile hits employing a panel of enzyme and receptor binding assays, gather the respective profile of off-target activities predicted for each compound, and assess their profile in vivo, in an animal model chosen to reveal ancillary pharmacological activities. Usually, it is assumed that if no untoward activities are observed in vivo, the risk of any predictable ancillary pharmacology will be minimal.
Limitations of in vivo Specificity and Selectivity Assessment
There are a number of confounding aspects related to using such in vivo assessment approaches, and the data generated can be highly misleading without a through understanding gained from in vitro assays (e.g., Raehal et al., 2011). First, animal experiments are usually done in rodents, either mice or rats, and while these species are adequate for tolerability studies to reveal frank toxicity, they may not be useful for detecting subtle changes in physiology. In addition, limited exposure and high clearance rates (typical in rodents) can result in a misleading sense of ancillary pharmacology. Taken together with the facts that some physiological response pathways are species-dependent, high protein binding can mask ancillary pharmacology, and that poly pharmacy of compounds can cause compensation by altering a number of inter-related pathways whose individual modulation might otherwise produce serious consequences, an unremarkable initial in vivo profile could produce a false sense of security. But most importantly, the rodent cardiovascular (CV) system is a poor predictor of CV side effects in humans. This is due to very high heart rates in the rodent and distinct complements/contributions of ion channels in the two species (e.g., the cardiac action potential wave forms are much different when comparing rodent and human species). Therefore, rodent data, by itself, can yield false-negative misinformation with respect to adverse CV effects. We do recognize the utility of rat models for understanding cardiotoxicity (Herman et al., 2011); however in general we advise caution when using this species to assess potential pro-arrhythmic liabilities (i.e., prolongation of QTc intervals, see below). Indeed, this is one of the reasons that CV liabilities of late stage development candidates are traditionally assessed in a dog model, which is more predictive than rodent of CV safety liabilities in humans. However, it is not routinely feasible to profile putative lead structures in the dog due to the need for large quantities of test compound to initiate both tolerability studies and CV measurements. Employment of cardiomyocytes derived from embryonic or induced pluripotent stem cells that display the electrophysiological and biochemical properties of normal human heart cells is currently being explored as a more predictive in vitro model system for testing a compound’s potential cardiotoxicity, but this approach is still under development (Braam et al., 2010; Peng et al., 2010. Therefore, a more informative approach is required to carry out a formal risk analysis of a lead compound’s potential liabilities than to simply use cursory rodent in vivo data. It is critical to have some early means to identify potential problems that could arise during development of a drug candidate and to minimize such hazards by selecting the most appropriate lead class for the focus of medicinal chemists. Moreover, if potential liabilities exist in an otherwise favorable lead compound, predictive, high-capacity assays must be employed to support Medicinal Chemistry efforts to dial out these unwanted activities in real time during the course of structure–activity relationship (SAR) studies.
It is important to consider the contribution of multiple mechanisms to a physiological response when developing in vivo models to evaluate specificity and selectivity and obtaining proof of concept for novel compounds targeting new mechanisms of action. As the drug industry searches for new druggable targets based on human genetics, rodent genetic paradigms, and reverse pharmacology approaches, it is important to check new leads for poly pharmacy before initiating Medicinal Chemistry campaigns, just to confirm that pharmacodynamic responses correlate strictly with the desired molecular action (Weinglass et al., 2008a,b). For example, the search for agonists of the high-conductance, calcium-activated potassium channel (maxi-K; KCa1.1) has often revealed compounds in the past which activate the channel, promote smooth muscle relaxation, and lower blood pressure in rodents, i.e., the phenotype expected for a KCa1.1 agonist. However, to date, all publicly disclosed channel agonists have been observed to have ancillary pharmacology (e.g., calcium entry blocker activity, phosphodiesterase inhibitory activity) that promote smooth muscle relaxation and blood pressure lowering as well, which confounds proof-of-concept arguments that a selective KCa1.1 agonist will have therapeutic utility in reversing pathophysiology associated with smooth muscle hyper-excitability (Garcia et al., 2007). The importance of profiling lead compounds directed against new mechanisms for ancillary pharmacological activity cannot be overemphasized.
Cross-Target Profiling: The Advantages of Functional Assays
Profiling a putative lead compound against a panel representing a consensus of important enzymes, receptors, ion channels, and transporters is an excellent way to establish a preliminary view of potential issues that might hamper development, and which off-target activities must be minimized accordingly. However, although enzyme assays are activity-based measurements (N. B., their interpretation is subject to the conditions chosen for the assay, especially levels of substrate), the use of binding assays to assess interaction of test compound with receptors, channels, or transporters is not as informative as are functional measurements. The same issues exist, but to a greater extent, with ion channel proteins. The ability of test compounds to interfere with binding of specific ligand probes developed for individual ion channels is often used as a guide for determining potential drug liabilities. However, it is well known that, unless the sites at which test agents interact with a channel are identical or strongly allosterically coupled to that region where the radiolabel probe binds, potential channel interactions can be missed. Tetrodotoxin (TTX) is a very potent and selective inhibitor of many voltage-gated sodium channels. However, except for similar congeners that compete for binding to this inhibitor’s site in the external pore of the TTX-sensitive sodium channel subtype, the rich pharmacology known to exist with this channel family (i.e., the extensive number of both agonists and antagonists that have previously been well-characterized) is completely undetectable using the TTX binding reaction (Cestele and Catterall, 2009). Similarly, using omega conotoxin as a probe for Cav2.2, charybdotoxin as a probe for Kv1.X or KCa1.1 channels, apamin as a probe for KCa2.X channels, or sulfonylureas as a probe of K-ATP channels, are additional insensitive means to determine potential off-target activities directed against these proteins. Indeed, it is known that conotoxin binding is highly refractory to modulation by most small molecule Cav2.2 inhibitors (Abbadie et al., 2010). In the case of sulfonylureas, binding is to a regulatory subunit associated with Kir6.2, the pore-forming subunit of the K–ATP channel complex, and monitoring effects on that binding reaction is conceptually a poor way to detect direct modulators of the Kir6 channel (see relevant sections in Hibino et al., 2010). The same criticism could be applied to many of the probes used to assess interactions with ligand-gated ion channels or transporters. In the case of transporters, there is no guarantee that using ligands which interact at a carrier’s substrate binding site is a sensitive means to detect all compounds that affect transporter function. Thus, there is no bona fide substitute to definitively identify putative ion channel or transporter modulators other than by direct measurement of protein function. This is especially true when evaluating novel chemical structures without previous pharmacological history, because many examples of false negatives and false positives have been noted when using binding protocols to profile such compound decks.
Examples of Profiling Compounds Across Targets and Across Channel Families
While drug discovery programs typically test the activity of lead compounds against other related family member targets, and may also include some critical cardiac targets, a much wider profiling can yield a better understanding of potential liabilities of the structural class of compounds under consideration. As an example, BIRB0796, which was developed as an allosteric inhibitor of p38 MAP kinase (aka SAPK2A/SAPK2B; Pargellis et al., 2002) was profiled at a single concentration of 10 μM in a functional Safety and Liability Screening Panel consisting of 125 targets (76 GPCRs, 39 Kinases, two Phosphatases, and eight Cardiac Ion Channels). Table 1 summarizes the activity of BIRB0796 in biochemical assays employing the kinases and phosphatases. BIRB0796 inhibits the SAPK2a/SAPK2b enzyme at a novel “DFG-out” allosteric site as determined from studies of the compound bound to non-activated human p38 MAP kinase, and has weak or no inhibition of 15 other kinases (Pargellis et al., 2002). In our studies greater than 50% inhibition of four kinases, including SAPK2B, was observed. In addition, 10 μM BIRB0796 inhibited five GPCRs (Table 2) and two of seven Cardiac Ion Channels, hERG, and Kv1.5 (Figure 1), by ≥50%. Inhibition of hERG and Kv1.5 channels could raise a flag of potential cardiac safety concern for this compound. All these data on BIRB0796, derived from a combination of biochemical and cell-based functional assays, provide a better insight into the putative compound’s liabilities than would have been obtained from the use of binding assays alone. This example illustrates the need to broadly characterize any potential drug discovery compound in functional assays to better understand its potential off-target activities.
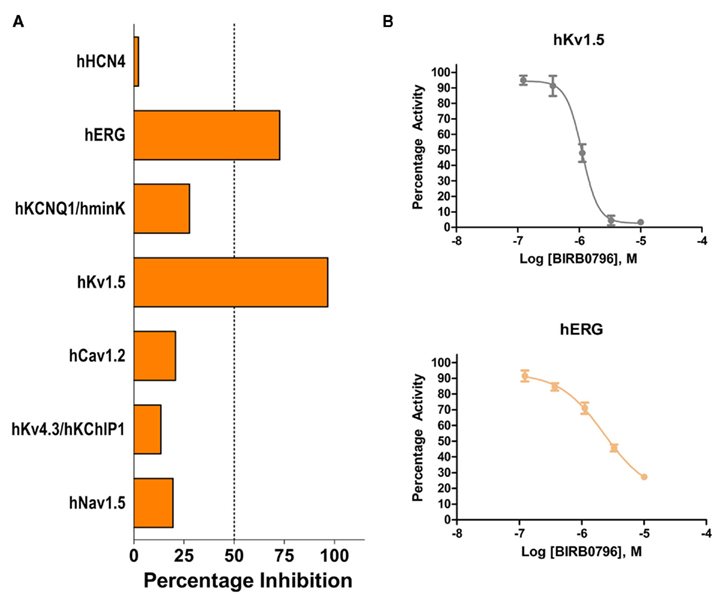
Figure 1. (A) Activity of 10 μM BIRB0796 on cardiac ion channels within Millipore’s Safety and Liability Panel. (B) Because of the significant inhibition observed at 10 μM, full concentration response curves for hERG and Kv1.5 were carried out with BIRB0796. Calculated IC50 values were 2.3 and 1.1 μM for hERG and Kv1.5, respectively.
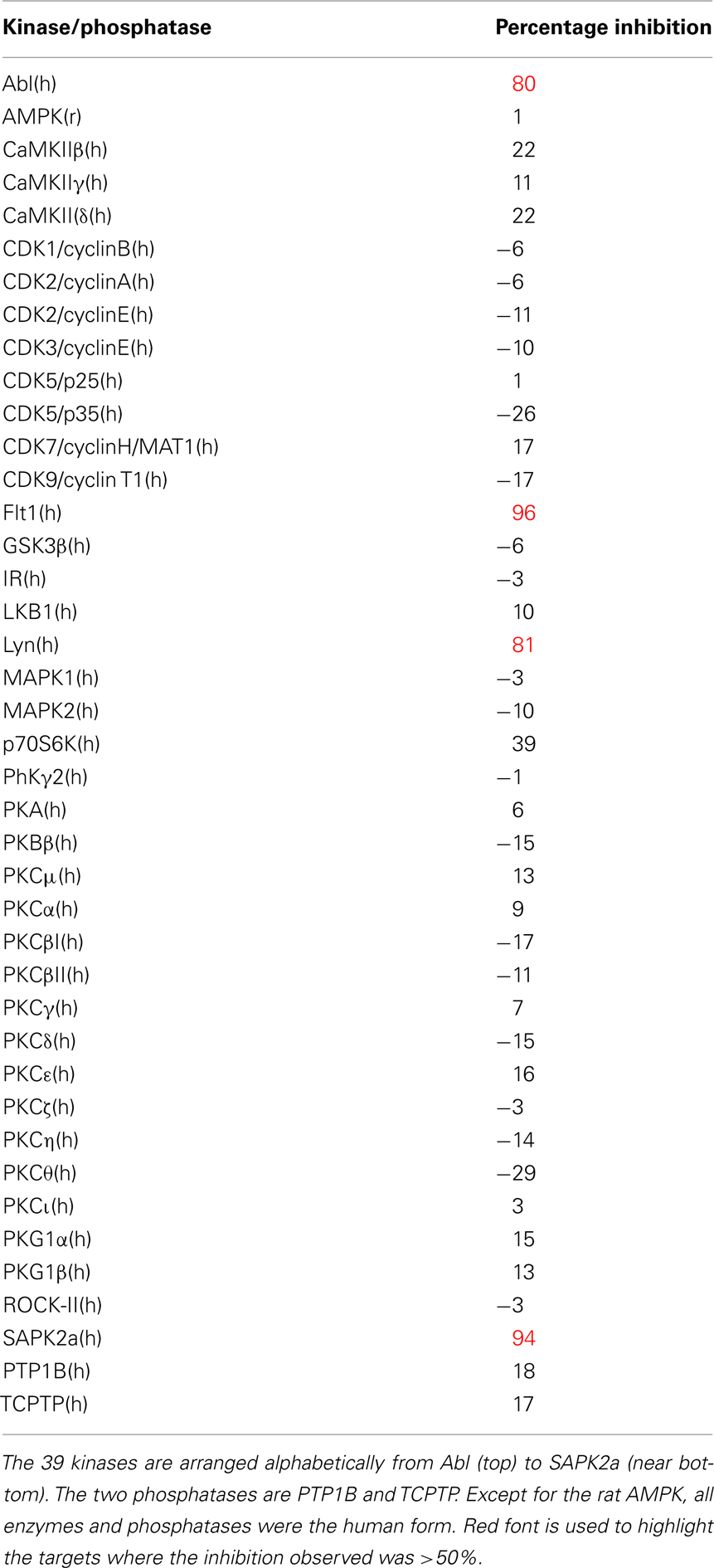
Table 1. Activity expressed as percentage inhibition of 10 μM BIRB0796 on 39 kinases and 2 phosphatases within Millipore’s Safety and Liability Panel.
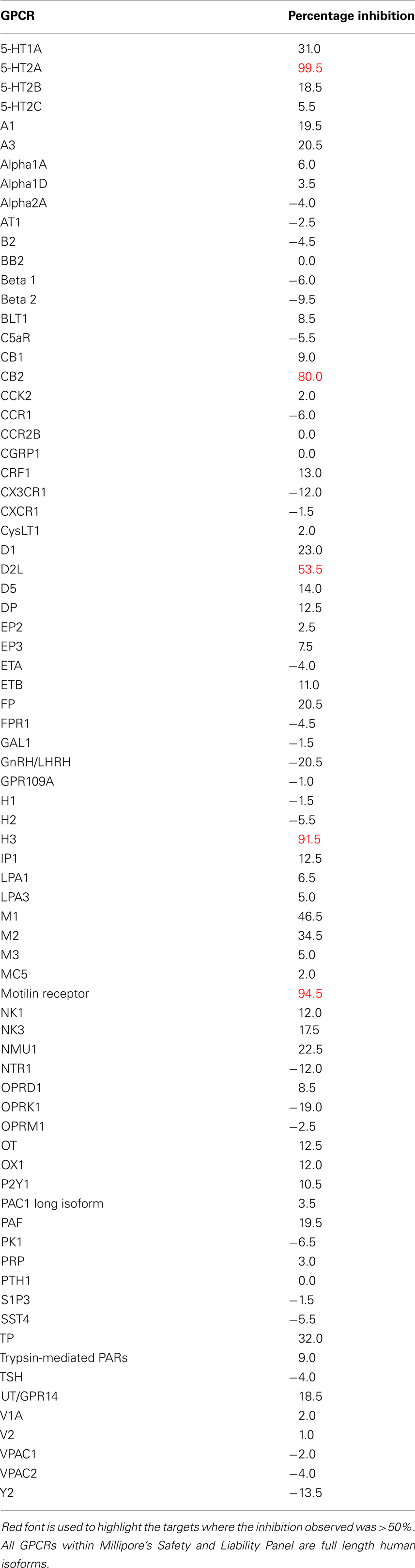
Table 2. Activity expressed as percentage inhibition of 10 μM BIRB0796 on 76 GPCRs within Millipore’s Safety and Liability Panel.
For functional assays to be able to provide accurate and decision-enabling information, it is important to understand how these assays perform over an extended period of time. This is best illustrated in the case of Nav channels, where some specific channel members, such as Nav1.7 and Nav1.8 are of interest for development of novel analgesics, but where such compounds must display restricted activity at the brain Nav1.1 and Nav1.2, and cardiac Nav1.5 isoforms. To identify novel chemical entities with the desired properties, i.e., state- and use-dependence to effectively block peripheral Nav1.7 or Nav1.8 channels involved in pain signaling, while sparing block at the cardiac Nav1.5 channel, drug discovery programs use both biochemical-based assays as well as manual patch-clamp and automated EPhys assays (Felix et al., 2004; Kaczorowski et al., 2008). Using IonWorks and PatchXpress platforms, Millipore can provide rapid turnaround times (within 7 business days from the date of compound receipt) for routine profiling against Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.5, Nav1.6, and Nav1.7 channels since these assays are sufficiently validated and quality control is followed rigorously. In addition, when development of assays using automated platforms is problematic because the amount of current remaining in cells held at V1/2 inactivation is not sufficient to enable robust and reliable IonWorks assays, additional assays can be provided using manual patch-clamp (e.g., Nav1.8).
These assays utilize stable cell lines (e.g., Burbidge et al., 2002; Clare et al., 2009) and voltage-clamp protocols designed to assess multiple features of compounds’ interaction with the Nav channels (Figure 2). A parameter often desired is use-dependent block, a condition where block accumulates due to high frequency firing that can be assayed using multiple-pulse protocols. Block of the inactivated-state of the channel, also known as tonic block, which is thought to be relevant in the disease state, can also be determined by selecting a holding potential that inactivates a certain fraction of the channels, so that one can estimate the affinity of the test compound for the inactivated state (Ki) and compare it with resting state block (Kr; see Hondeghem and Katzung, 1977; Bean et al., 1983). The protocol illustrated in Figure 2 was designed to efficiently profile both modes of interaction of test compounds with Nav1.x channels. Quality-control data for this protocol are shown in Figure 3, where the activity at pulse 25 for two concentrations of the reference compound, and the solvent control are shown. Robust-performing stable cell lines are crucial for these assays; the criteria for initial validation of these cell lines include current amplitude (we set a typical threshold of greater than 250 pA), seal resistance on automated patch-clamp platforms (i.e., is high enough relative to the current amplitude obtained with the assay protocol to give acceptable current resolution; this ranges from hundreds of MΩ to a few GΩ depending on the seal substrate) percent of cells expressing the current (the higher, the better) and continued performance at these levels over at least 25 passages in culture. Data collected over a 6-month time period show that the assays are robust and reproducible (Figure 3). Rigorous quality control checks ensure week-to-week consistencies which are necessary to support Drug Discovery programs.
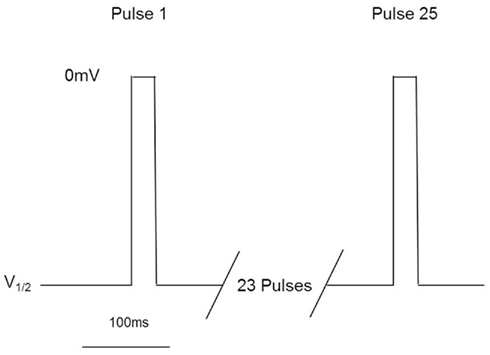
Figure 2. Millipore’s voltage-gated sodium channel IonWorks Quattro™ protocol. Cells are held at a voltage corresponding to ∼V1/2 inactivation, and subjected to 25 depolarization pulses to 0 mV for 20 ms. Pulses are applied every 100 ms. Data from Pulse 1 and Pulse 25 are reported to assess both tonic and use-dependent inhibition.
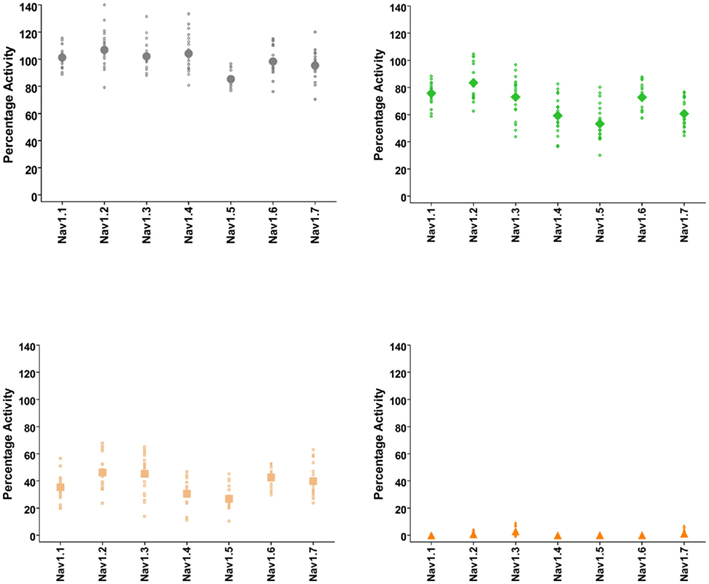
Figure 3. Quality-control data for Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.5, Nav1.6, and Nav1.7 assays collected over a 6 month period of time using IonWorks Quattro. Vehicle control was external buffer with 0.3% DMSO final concentration and reference inhibitors consisted of tetracaine (Nav1.1), lidocaine (Nav1.2, Nav1.3, Nav1.4, Nav1.5, Nav1.6), and lamotrigine (Nav1.7). Data shown were calculated from Pulse 1 and Pulse 25 (as in Figure 2) and displayed as percentage activation relative to the ratio of Post-Compound/Pre-Compound pulses. Where 0.3% DMSO had no effect between the Pre-Compound and the Post-Compound pulses, this was defined as 100% activation. (Top left panel) vehicle control data. (Top right) data obtained at P1 with the IC50 concentration of inhibitor. (Bottom left) data obtained at P25 with the IC50 concentration of inhibitor. (Bottom right) data from P25 using an IC100 concentration of inhibitor. All data points are from cells with a minimum of 250 pA of current, and the mean is shown as the large filled symbol in each panel. The IC50 dose is chosen to estimate the block achieved at P25; since the compounds are state- and use-dependent blockers less than 50% block is frequently achieved at P1.
Additional specific custom automated patch-clamp protocols (see example in Figure 4) can be rapidly developed to identify inhibitors of Nav1.X channels that preferentially bind to the inactivated-state of the channel (Hondeghem and Katzung, 1977; Bean et al., 1983). We have developed custom protocols, with all validation data required, in as little as 3 weeks. Example data obtained using this protocol for seven Nav standards on three Nav1.X channels, Nav1.2, Nav1.5, and Nav1.6, are summarized in Table 3 and example data for Nav1.5 are shown in Figure 5. Lidocaine, tetracaine, lamotrigine, carbamazepine, and riluzole displayed a large leftward shift in their concentration–response curve at pulse 2, consistent with the use-dependent mechanism of block by these agents. Flecainide, on the other hand, displayed a more modest shift in affinity when comparing pulse 1 and 2, whereas for phenytoin there is essentially no block at pulse 1 and pronounced block at pulse 2. These data illustrate that reliable and efficient automated Ephys assays exist which can support Medicinal Chemistry programs targeting Nav channels. Similar protocols are in development for other voltage-gated channels, including Kv channels.
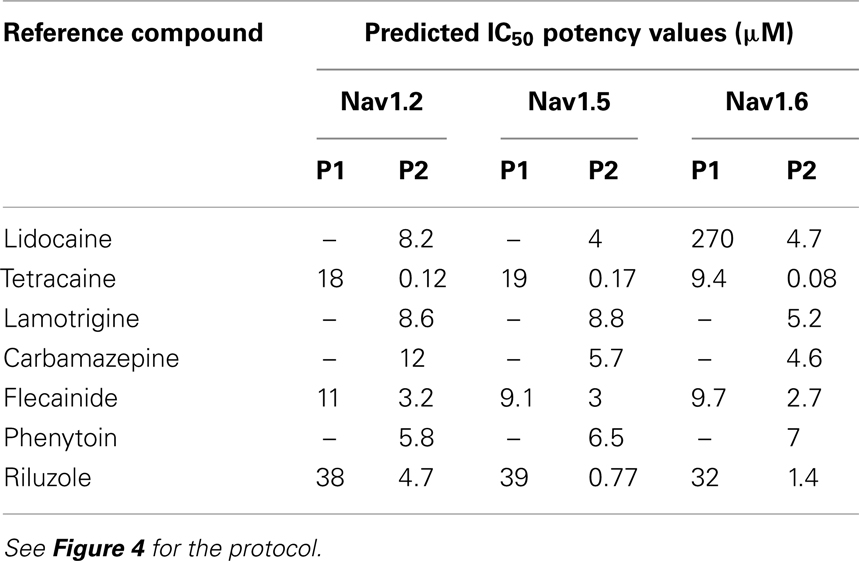
Table 3. Potency values for reference Nav inhibitors as determined with the 2-pulse protocol.
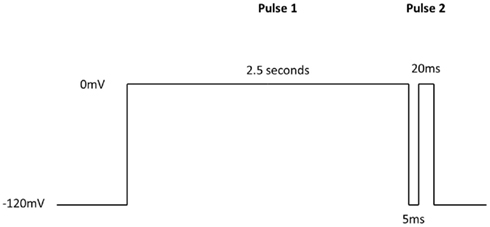
Figure 4. IonWorks Quattro Nav pulse protocol designed specifically to characterize interaction with the inactivated-state of the channel. Pulse 1 drives Nav channels primarily into the inactivated state, the 5 ms step back to −120 mV relieves the fast inactivation, and the second depolarizing pulse (Pulse 2) defines a compound’s ability to bind to the inactivated-state of the channel.
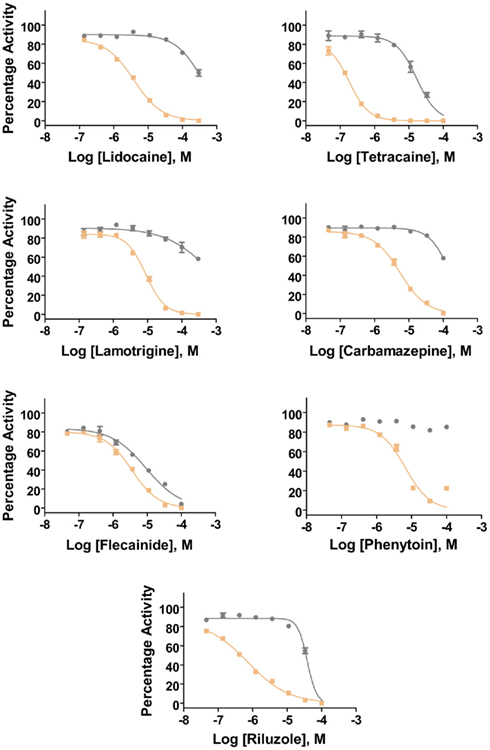
Figure 5. Inhibition of Nav1.5 channels by Nav1.X channel standards using the inactivated-state IonWorks Quattro protocol (Figure 4). Pulse 1 (P1) and Pulse 2 (P2) data were calculated from the maximum inward Nav current amplitudes from each depolarizing pulse and are presented as percentage activation relative to the ratio of Post-Compound/Pre-Compound pulses. Gray symbols and lines are data from P1, and tan symbols and lines are from P2.
Importance of Measuring Activity of Drug Candidates on Ion Channels for Cardiac Safety
Off-target activities of drug candidates are particularly relevant to ion channels expressed in the heart. Untoward modulation of many of these proteins can result in major safety issues, including lethality. Ion channels control conduction of electrical activity over the heart. They are responsible for efficiently orchestrating both cardiac rate and synchronous contraction of cardiac muscles comprising the atria and ventricle to allow efficient pumping of blood thorough the body’s vascular system. Identification of compounds found to alter normal cardiac electrocardiogram (ECG) waveforms, thereby signaling propensity for chronotropic, ionotropic, or arrhythmogenic activities, typically stops development. Therefore, early testing of all drug target lead candidates against a panel of critical cardiac ion channel targets (e.g., Kv11.1, Kv7.1, Kir2.1, Nav1.5, and Cav1.2) can add substantial value in prioritizing initial screening hits, assessing lead specificity, supporting real-time Medicinal Chemistry SAR studies on selected leads in the process of identifying development candidates, rescuing programs with potential major cardiac safety liabilities that are usually only revealed at the end of preclinical development, and supporting final stage drug registration efforts. Medicinal chemists find real value in having access to such selectivity data from a cardiac ion channel panel during lead optimization efforts.
The human ventricular cardiac action potential waveform (Figure 6, top) is shaped primarily by the activity of six different ion channels; Nav1.5, Cav1.2, Kv4.2/4.3, Kv7.1 (KCNQ1/KCNE1 channel complex), Kv11.1 (hERG; the Kv11.1 protein is thought to underlie the IKr current), and Kir2.1 (Roden et al., 2002). Additional channels (Kv1.5, Kir3.1/3.4) also contribute to the action potential waveform in atria. The summation of action potential waveforms over the entire structure of the heart gives rise to the measurements made in a surface ECG (Figure 6, bottom). Human genetics demonstrate that mutations in most of the ventricular ion channels listed above can have profound effects on ECG parameters, such as QTc, QRS, and PR intervals, which thereby signal potential cardiac safety issues (Nattel and Carlsson, 2006; Lu et al., 2008). Some of these waveform alterations may have fatal consequences (e.g., QTc prolongation can lead to torsades de pointes, which can lead to ventricular fibrillation, which can result in sudden cardiac death). Importantly, reduction of the cardiac rapid delayed rectifier potassium channel caused by loss-of-function mutations of the hERG channel will prolong the ventricular action potential, elongate the QTc interval, and cause a lethal arrhythmia. Gain-of-function mutations in hERG produce the opposite functional effects on action potentials and QTc intervals, but could result in either atrial fibrillation (Hong et al., 2005) or ventricular fibrillation; each condition is potentially lethal. Loss-of-function mutations in the cardiac sodium channel, Nav1.5 (Brugada Syndrome), increases QRS interval, slows conduction and may result in initiation of lethal arrhythmias, while gain-of-function mutations prolong QTc, thereby increasing probability of the same fatal end point. Inhibition of the cardiac slow delayed rectifier potassium channel due to loss-of-function of either the pore-forming subunit, KCNQ1, or of the beta regulatory subunit, KCNE1, prolongs QTc, while activation of this channel through gain-of-function mutations in KCNQ1 shortens QTc. Both phenotypes increase the probability of sudden cardiac death. The same spectrum, with respect to effects on QTc interval and arrhythmogenic potential, is also produced by loss- or gain-of-function mutations in the cardiac inwardly rectifying potassium channel, Kir2.1. Finally, gain-of-function mutations (Timothy Syndrome) in the L-type calcium channel, Cav1.2, leads to QTc prolongation, ventricular, and atrial arrhythmias, and can be fatal, while loss of Cav1.2 function shortens QTc intervals and increases the possibility of sudden cardiac death, as well. Taken together, data from studies of human genetics demonstrate that the consequences of altering function of the major ion conduction pathways in the heart often result in an increase in mortality. Thus, drugs which affect these targets can similarly elicit fatal events (Roden, 2004; Knollmann and Roden, 2008) and it is critical to identify such liabilities as early as possible in development and take corrective actions. Indeed as little as 20% block of hERG current by some drugs that do not affect other cardiac currents can produce dangerous prolongation of the ventricular action potential (for a comprehensive analysis of drugs that prolong QT interval, see Redfern et al., 2003). As an example for another cardiac ion channel, we recently profiled a compound from a non-cardiac ion channel program and detected antagonist activity at KCNQ1/minK channels, which could cause delayed ventricular repolarization in vivo. At this stage, profiling of different chemical series and multiple representatives of these chemical series can be performed to provide structure activity relationship data that provided a path forward in the project. CardiacProfiler™ functional patch-clamp assays offered by Millipore provide a convenient path to understand potential cardiac liability of potential lead compounds at the most important eight cardiac ion channels and can serve as an important component of integrated cardiac risk management.

Figure 6. Human ventricular action potential and surface electrocardiogram (ECG). The ventricular action potential (indicated in red) is determined by the activity of several ion channels. When the membrane potential reaches a threshold value, voltage-gated sodium channels (INa) rapidly activate and sodium ions enter the cell down their concentration gradient to cause cell depolarization. INa (Nav1.5), therefore, contributes to the upstroke of the action potential. Following depolarization, calcium channels (ICa; Cav1.2) open to allow calcium ions to enter the cell and initiate the excitation–contraction coupling mechanism. In order to bring the cell back to its resting state, different potassium channels, such as Ito (Kv4.2/Kv4.3), IKr (Kv11.1; HERG), IKs (Kv7.1/mink), and IK1 (Kir2.1) become engaged at different phases of the action potential. The action potential duration represents the time interval between the action potential upstroke and return to the resting level. The surface ECG (purple) is the sum of action potentials over all compartments of the heart. The P wave corresponds to atrial depolarization, whereas the QRS complex is caused by depolarization of the ventricle. The T wave corresponds to repolarization of the ventricle.
Determining Activity on hERG, Nav1.5, and Cav1.2 Channels: Limitations of Ligand-Binding Assays
Several methodologies have been adopted to routinely probe for interactions of test compounds with three of the major cardiac ion channel liability targets; hERG, Nav1.5, and Cav1.2. Historically, the most commonly used technique involves monitoring the effects of test compounds on binding of selective ligands for each of these channels to identify potential safety risks. Of these targets, hERG is the most important to be evaluated, given that: (1) hERG modulation can produce mortality; (2) as little as 20% hERG block can have an effect on QTc interval; (3) some drugs have been withdrawn from the market or received a cautionary warning because of safety issues (lethal arrhythmias) found to be associated with their activity against hERG (Table 4); (4) regulatory agencies will not approve registration of new drugs without a data package demonstrating a minimal hERG liability; (5) the Kv11.1 channel is the most promiscuous ion channel ever characterized in terms of its molecular pharmacology – it is blocked by a wide variety of molecules, perhaps because the channel’s inner cavity located below the selectivity filter can accommodate such a wide range of structures – so there is high probability that some interaction with novel test compounds might be observed (Fermini and Fossa, 2003). Although hERG binding assays have been used in the field for many years, this approach is conceptually flawed when compared with profiling actives directly on hERG function. Many instances of hERG binding by drug candidates have yielded both false positive and false-negative data during testing of novel structural series. False positives can be sorted out by subsequent evaluation in a functional assay. However, false negatives can have disastrous program consequences if the hERG liability is not detected until a potential preclinical candidate is profiled in vivo in a CV dog. The time wasted and lack of SAR information on the newly discovered hERG activity usually results in termination of development efforts. The risk of using hERG binding assays as the only support for Medicinal Chemistry is simply not warranted when alternatives exist for employing medium- and high-capacity functional hERG assay formats.

Table 4. Drugs withdrawn due to QT-prolongation.
Similarly, it may be misleading to monitor the effect of test compounds on batrachotoxin (BTX) binding to Nav1.2 channels in brain tissue as a surrogate for identifying potential interactions with the cardiac sodium channel. Because (1) the BTX site is not coupled to all other drug binding sites on sodium channels (Cestele and Catterall, 2009), (2) the channels under investigation are distinct (Nav1.5 versus Nav1.2), (3) sodium channel subtype-selective inhibitors exist (Williams et al., 2007; Schmalhofer et al., 2008), and (4) electrical environment/biophysical properties can specifically influence Nav1.5 block, testing compounds directly on Nav1.5 function is a much more meaningful approach.
Lastly, monitoring binding of representative ligands for the L-type calcium channel from the dihydropyridine, phenylalkylamine, and benzothiazepine classes to probe for potential Cav1.2 interactions is likewise flawed. Out of the three ligand classes, the diltiazem binding assay is probably the most sensitive means to detect possible channel modulators; however, this assay still yields false positives and negatives. Such findings support use of a functional assay to replace ligand-binding for evaluating potential Cav1.2 interactions.
Development of High-Throughput Functional Assays for Ion Channel Modulation
Depending on the required throughput, manual, or automated patch-clamp methods can be used to evaluate compounds individually against each of the six to eight critical ion channels present in human cardiac muscle to accurately reveal any potential liabilities. However, using such techniques to assess large numbers of compounds can be tedious, time-consuming, and costly. The issue of expanding screening capacity against a cardiac panel has recently been addressed by developing a series of robust, high-throughput, cell-based counter-screening assays employing fluorescence-based readouts. For example, assay conditions were developed and optimized to establish a high-capacity 384- and 1536-well functional thallium flux assay for the hERG channel that recapitulates the molecular pharmacology of a wide range of standard, well-characterized, channel blockers based on their activities measured in electrophysiological protocols (Schmalhofer et al., 2010). These studies were further extended to the analysis of approximately 1000 compounds randomly chosen to represent diverse structural classes from ongoing Medicinal Chemistry programs, where the activities in thallium flux or in a hERG ligand-binding assay were compared with data obtained from electrophysiology determinations made on the same compound deck. Subsequent correlations reveal that the thallium flux inhibitory values correlate more closely with electrophysiology data than do the ligand-binding results for this series of compounds (Figure 7). Thus, use of a well-controlled thallium flux assay is an excellent high-capacity predictive assay of in vitro hERG blocking activity. Such an assay has supported Medicinal Chemistry efforts to significantly dial out problematic off-target interactions with Kv11.1, while potency was simultaneously enhanced on the primary target (Garcia and Kaczorowski, unpublished observations).
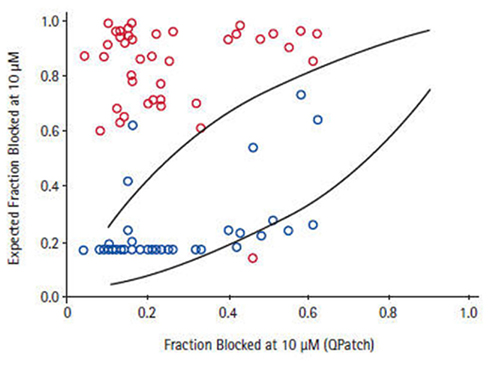
Figure 7. Correlation between QPatch and high-throughput hERG assays. For a subset of compounds for which two high-throughput hERG assays, thallium flux (blue symbol), and 35S-MK499 binding (red symbol) showed >7-fold difference in potency, the estimated fraction of block in QPatch hERG recordings correlates to a greater degree with the thallium flux data Schmalhofer et al. (2010).
Robust HTS assays have been developed for Kir2.1, Nav1.5, and Cav1.2 channels, as well. For example, a HEK hKir2.1 thallium flux assay operant in 384- or 1536-well format has been established, validated, and routinely used as a counter-screen in efforts to develop the molecular pharmacology of other inwardly rectifying potassium channel targets (Garcia and Kaczorowski, unpublished observations). It is expected that a similar HTS assay could be configured with the KCNQ1/KCNE1 channel complex as a probe for modulators of the cardiac slow delayed rectifier potassium current. Using either FRET dye pairs or single fluorescence dyes to monitor membrane potential, a routine, robust, and sensitive hNav1.5 HTS assay has been configured that is operant in formats that can support high-density screening with 1536-well plates (Felix et al., 2004). Use of an agonist initiation protocol allows configuration of assay conditions which yield pharmacologically faithful data when compared to Kinactivation values of literature standards. Finally, a calcium influx assay was developed to detect state-dependent Cav1.2 blockers by stably expressing the Cav1.2 channel subunits (α1C, α2δ1, β2a) along with an inward rectifier potassium channel, Kir2.3, in HEK cells (Dai et al., 2008; Abbadie et al., 2010). Cell membrane potential is controlled by bath external potassium concentrations due to the presence of Kir2.3 and the equilibrium between different Cav1.2 states is shifted by controlling cell membrane potential. By incubating cells with test compounds at different potassium concentrations, triggering channel opening by depolarizing cells with high potassium, and measuring increases in internal calcium with a calcium-sensitive dye, state-dependent Cav1.2 blockers can be detected in an accurate, facile, and reproducible fashion. Importantly, pharmacological sensitivity data from all these protocols display good correlation with patch-clamp results.
This functional HTS approach to assay cardiac ion channels can reduce the risk of unwanted side effects and eliminate such liability early in lead development. The implementation of high-capacity, robust, facile, accurate, and cost effective, cellular-fluorescence-based ion channel counter-screening methodologies can support data delivery on a weekly basis to support Medicinal Chemistry SAR efforts. Moreover, this strategy can be extended to developing a CNS liability panel including functional assays for Kv1.X, Kv2.X, Kv3.X, Kv4.X, Cav2.X, Cav3.X, and KCNQ2-3 using the same general cell-based approach.
Conclusion
Attention to identifying serious off-target activities and supporting Medicinal Chemistry efforts to minimize these activities are as important as the other efforts that go into identifying a potential drug development candidate. Eliminating ancillary pharmacology helps to improve therapeutic index which will be beneficial during clinical trials supporting new drug registration. Often, drug failures in the clinic result from non-mechanism-based side effects which could have been identified and eliminated by proper profiling of lead candidates. Utilizing comprehensive, sensitive, and well-validated profiling assays provides drug developers the ability to ask the crucial questions early and often during the preclinical development process, which should save time, money, and avoid nasty surprises.
Conflict of Interest Statement
Gregory J. Kaczorowski and Maria L. Garcia are President and CEO and Vice President, respectively, of Kanalis Consulting, L. L. C. Jacob Bode, Stephen D. Hess, and Umesh A. Patel are employees of the Millipore Corporation.
References
Abbadie, C., McManus, O. B., Sun, S.-Y., Bugianesi, R. M., Dai, G., Haedo, R. J., Herrington, J. B., Kaczorowski, Smith, M. M., Swensen, A. M., Warren, V. A., Williams, B., Arneric, S. P., Eduljee, C., Snutch, T. P., Tringha, E. W., Jochnowitz, N., Liang, A., MacIntyre, D. E., McGowan, E., Mistry, S., White, V. V., Hoyt, S. B., London, C., Lyons, K. A., Bunting, P. B., Volksdorf, S., and Duffy, J. L. (2010). Analgesic effects of substituted N-triazole oxindole (TROX-1), a state-dependent, voltage-gated calcium channel 2 blocker. J. Pharmcol. Exp. Ther. 334, 545–555.
Bean, B. P., Cohen, C. J., and Tsien, R. W. (1983). Lidocaine block of sodium channels. J. Gen. Physiol. 81, 613–642.
Bhave, G., Lonergan, D., Chauder, B. A. II., and Denton, J. S. (2010). Small-molecule modulators of inward rectifier K+ channels: recent advances and future possibilities. Future Med. Chem. 2, 757–774.
Braam, S. R., Tertoolen, L., van de Stolpe, A., Meyer, T., Passier, R., and Mummery, C. L. (2010). Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 4, 107–116.
Burbidge, S. A., Dale, T. J., Powell, A. J., Whitaker, W. R., Xie, X. M., Romanos, M. A., and Clare, J. J. (2002). Molecular cloning, distribution and functional analysis of the Na(V)1.6. voltage-gated sodium channel from human brain. Brain Res. Mol. Brain Res. 103, 80–90.
Cestele, S., and Catterall, W. A. (2009) “Toxins targeting mammalian sodium channels,” in Animal Toxins: State of the Art; Perspectives in Health and Biotechnology, ed. M. E de Lima (Belo Horizonte: UFMV), 99–121.
Clare, J. J., Chen, M. X., Downie, D. L., Trezise, D. J., and Powell, A. J. (2009). Use of planar array electrophysiology for the development of robust ion channel cell lines. Comb. Chem. High Throughput Screen. 12, 96–106.
Coburger, C., Wollmann, J., Krug, M., Baumert, C., Seifert, M., Molnár, J., Lage, H., and Hilgeroth, A. (2010). Novel structure-activity relationships and selectivity profiling of cage dimeric 1,4-dihydropyridines as multidrug resistance (MDR) modulators. Bioorg. Med. Chem. 18, 4983–4990.
Dai, G., Haedo, R. J., Warren, V. A., Ratliff, K. S., Bugianesi, R. M., Rush, A., Williams, M., Herrington, JJ., Smith, M. M., McMann, O. B., and Swensen, A. M. (2008). A high-throughput assay for evaluating state dependence and subtype selectivity of Cav2 calcium channel inhibitors. Assay Drug Dev. Technol. 6, 195–212.
Felix, J. P., Williams, B. S., Priest, B. T., Brochu, R. M., Dick, I. E., Warren, V. A., Yan, L., Slaughter, R. S., Kaczorowski, G. J., Smith, M. M., and Garcia, M. L. (2004). Functional assay of voltage-gated sodium channels using membrane potential-sensitive dyes. Assay Drug Dev. Technol. 2, 260–268.
Fermini, B., and Fossa, A. A. (2003). The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discov. 2, 439–447.
Garcia, M. L., Shen, D.-M., and Kaczorowski, G. J. (2007). High-conductance calcium-activated potassium channels: validated targets for smooth muscle relaxants? Expert Opin. Ther. Pat. 17, 1–12.
Herman, E. H., Knapton, A., Rosen, E., Thompson, K., Rosenzweig, B., Estis, J., Agee, S., Lu, Q. A., Todd, J. A., Lipshultz, S., Hasinoff, B., and Zhang, J. (2011). A multifaceted evaluation of imatinib-induced cardiotoxicity in the rat. Toxicol. Pathol. 39, 1091–1106.
Hibino, H., Inanobe, A., Furutani, K., Murakami, S., Findlay, I., and Kurachi, Y. (2010). Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366.
Hondeghem, L. M., and Katzung, B. G. (1977). Time- and voltage-dependent interactions of antiarrythmic drugs with cardiac sodium channels. Biochem. Biophys. Acta 472, 373–398.
Hong, K., Bjerregaard, P., Gussak, I., and Brugada, R. (2005). Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J. Cardiovasc. Electrophysiol. 16, 394–396.
Kaczorowski, G. J., McManus, O. B., Priest, B. T., and Garcia, M. L. (2008). Ion channels as drug targets: the next GPCRs. J. Gen. Physiol. 131, 399–405.
Knollmann, B. C., and Roden, D. M. (2008). A genetic framework for improving arrhythmia therapy. Nature 451, 929–936.
Lin, Y. L., Hsiao, C. L., Wu, Y. C., and Kung, M. F. (2011). Electrophysiologic, pharmacokinetic, and pharmacodynamic values indicating a higher risk of torsades de pointes. J. Clin. Pharmacol. 51, 819–829.
Lu, H. R., Vlaminckx, E., Hermans, A. N., Rohrbacher, J., Van Ammel, K., Towart, R., Pugsley, M., and Gallacher, D. J. (2008). Predicting drug-induced changes in QT interval and arrhythmias: QT-shortening drugs point to gaps in the ICHS7B Guidelines. Br. J. Pharmacol. 154, 1427–1438.
Mathes, C. (2006). QPatch: the past, present and future of automated patch clamp. Expert Opin. Ther. Targets 10, 319–327.
Nattel, S., and Carlsson, L. (2006). Innovative approaches to anti-arrhythmic drug therapy. Nat. Rev. 5, 1034–1049.
Olsson, G. (2010). To the editor–market withdrawal of quinidine bisulfate (Kinidin Durules) in 2006. Heart Rhythm 7, 864.
Oshiro, C., Thorn, C. F., Roden, D. M., Klein, T. E., and Altman, R. B. (2010). KCNH2 pharmacogenomics summary. Pharmacogenet. Genomics 20, 775–777.
Pargellis, C., Tong, L., Churchill, L., Cirrilo, P. F., Gilmore, T., Graham, A. G., Grob, P. M., Hickey, E. R., Moss, N., Pav, S., and Regan, J. (2002). Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 9, 268–272.
Peng, S., Lacerda, A. E., Kirsch, G. E., Brown, A. M., and Bruening-Wright, A. (2010). The action potential and comparative pharmacology of stem cell-derived human cardiomyocytes. J. Pharmacol. Toxicol. Methods 61, 277–286.
Raehal, K. M., Schmid, C. L., Groer, C. E., and Bohn, L. M. (2011). Functional selectivity at the μ-opioid receptor: implications for understanding opioid analgesia and tolerance. Pharmacol. Rev. 63, 1001–1019.
Redfern, W., Carlsson, L., Davis, A. S., Lynch, W. G., MacKenzie, I., Palethorpe, S., Siegl, P. K. S., Strang, I., Sullivan, A. T., Wallis, R., Camm, A. J., and Hammond, T. G. (2003). Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 58, 32–45.
Roden, D. M., Balser, J. R., George, A. L., and Anderson, M. E. (2002). Cardiac ion channels. Annu. Rev. Physiol. 64, 431.
Rudolph, U., and Knoflach, F. (2011). Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat. Rev. Drug Discov. 10, 685–697.
Sanofi Aventis. (2011). Available at: http://online.wsj.com/article/BT-CO-20110215-704422.html
Schmalhofer, W., Calhoun, J., Burrows, R., Bailey, T., Kohler, M. G., Weinglass, A. B., Kaczorowski, G. J., Garcia, M. L., Koltzenburg, M., and Priest, B. T. (2008). ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol. Pharmacol. 74, 1476–1484.
Schmalhofer, W. A., Swensen, A., Williams, B. S., Felix, J. P., Haedo, R., Solly, K., Kiss, L., Kaczorowski, G. J., and Garcia, M. L. (2010). A pharmacologically validated, high capacity, functional thallium flux assay for the herg channel. Assay Drug Dev. Technol. 8, 714–726.
Schrøder, R. L., Friis, S., Sunesen, M., Mathes, C., and Willumsen, N. J. (2008). Automated patch-clamp technique: increased throughput in functional characterization and in pharmacological screening of small-conductance Ca2+ release-activated Ca2+ channels. J. Biomol. Screen 13, 638–647.
Shah, R. R. (2006). Can pharmacogenetics help rescue drugs withdrawn from the market? Pharmacogenomics 7, 889–908.
Tollman, P., Morieux, Y., Murphy, J. K., and Schulze, U. (2011). Identifying R&D outliers. Nat. Rev. Drug Discov. 10, 653–654.
Weinglass, A. B., Köhler, M. G., Nketiah, E. O., Liu, J., Schmalhofer, W., Thomas, A., Williams, B., Beers, L., Smith, L., Hafey, M., Bleasby, K., Leone, J., Tang, Y. S., Braun, M., Ujjainwalla, F., McCann, M. E., Kaczorowski, G. J., and Garcia, M. L. (2008a). Madin-Darby canine kidney II cells: a pharmacologically validated system for NPC1L1-mediated cholesterol uptake. Mol. Pharmacol. 73, 1072–8473.
Weinglass, A. B., Kohler, M., Schulte, U., Liu, J., Nketiah, E. O., Thomas, A., Schmalhofer, W., Williams, B., Bildl, W., McMasters, D. R., Dai, K., Beers, L., McCann, M. E., Kaczorowski, G. J., and Garcia, M. L. (2008b). Extracellular loop C of NPC1L1 is important for binding to ezetimibe. Proc. Natl. Acad. Sci. U.S.A. 105, 11140–11145.
Williams, B. S., Felix, J. P., Priest, B. T., Brochu, R. M., Dai, K., Hoyt, S. B., London, C., Tang, Y. S., Duffy, J. L., Parsons, W. H., Kaczorowski, G. J., and Garcia, M. L. (2007). Characterization of a new class of potent inhibitors of the voltage-gated sodium channel NaV1.7. Biochemistry 46, 14693–14703.
Keywords: ion channel
Citation: Kaczorowski GJ, Garcia ML, Bode J Hess SD and Patel UA (2011) The importance of being profiled: improving drug candidate safety and efficacy using ion channel profiling. Front. Pharmacol. 2:78. doi: 10.3389/fphar.2011.00078
Received: 16 August 2011;
Accepted: 19 November 2011;
Published online: 13 December 2011.
Edited by:
Ralf Franz Kettenhofen, Axiogenesis AG, GermanyReviewed by:
Gary Gintant, Abbott, USACopyright: © 2011 Kaczorowski, Garcia, Bode, Hess and Patel. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Umesh A. Patel, Millipore Corporation, 15 Research Park Drive, St. Charles, MO 63304, USA. e-mail:dW1lc2gucGF0ZWxAbWVyY2tncm91cC5jb20=