 Martin K. Childers1,2*
Martin K. Childers1,2* Janet R. Bogan3,4
Janet R. Bogan3,4 Daniel J. Bogan3,4 Hansel Greiner1†
Daniel J. Bogan3,4 Hansel Greiner1† Melanie Holder2
Melanie Holder2 Robert W. Grange5
Robert W. Grange5 Joe N. Kornegay3,4
Joe N. Kornegay3,4
- 1 Department of Neurology, Wake Forest University Health Sciences, Winston-Salem, NC, USA
- 2 Wake Forest Institute for Regenerative Medicine, Wake Forest University Health Sciences, Winston-Salem, NC, USA
- 3 Department of Pathology and Laboratory Medicine, School of Medicine, University of North Carolina, Chapel Hill, NC, USA
- 4 Department of Neurology, School of Medicine, University of North Carolina, Chapel Hill, NC, USA
- 5 Department of Human Nutrition, Foods and Exercise, College of Agriculture and Life Sciences, Virginia Tech, Blacksburg, VA, USA
Calpains likely play a role in the pathogenesis of Duchenne muscular dystrophy (DMD). Accordingly, calpain inhibition may provide therapeutic benefit to DMD patients. In the present study, we sought to measure benefit from administration of a novel calpain inhibitor, C101, in a canine muscular dystrophy model. Specifically, we tested the hypothesis that treatment with C101 mitigates progressive weakness and severe muscle pathology observed in young dogs with golden retriever muscular dystrophy (GRMD). Young (6-week-old) GRMD dogs were treated daily with either C101 (17 mg/kg twice daily oral dose, n = 9) or placebo (vehicle only, n = 7) for 8 weeks. A battery of functional tests, including tibiotarsal joint angle, muscle/fat composition, and pelvic limb muscle strength were performed at baseline and every 2 weeks during the 8-week study. Results indicate that C101-treated GRMD dogs maintained strength in their cranial pelvic limb muscles (tibiotarsal flexors) while placebo-treated dogs progressively lost strength. However, concomitant improvement was not observed in posterior pelvic limb muscles (tibiotarsal extensors). C101 treatment did not mitigate force drop following repeated eccentric contractions and no improvement was seen in the development of joint contractures, lean muscle mass, or muscle histopathology. Taken together, these data do not support the hypothesis that treatment with C101 mitigates progressive weakness or ameliorates severe muscle pathology observed in young dogs with GRMD.
Introduction
Several lines of evidence (Figarella-Branger et al., 2002; Spencer and Mellgren, 2002; Gissel, 2005; Takamure et al., 2005; Courdier-Fruh and Briguet, 2006; Whitehead et al., 2006; Gailly et al., 2007; Hopf et al., 2007) support a rationale to explore the potential therapeutic value of calpain inhibitors in Duchenne muscular dystrophy (DMD) because calpains likely contribute directly to DMD pathophysiology (Saez et al., 2006; Gailly et al., 2007; Hopf et al., 2007; Briguet et al., 2008; Urasawa et al., 2008). Transient membrane ruptures in the muscles of DMD patients allow an influx of calcium that subsequently triggers calcium-activated proteases (m- and μ-calpain) that, in turn, modify calcium leak channels to cause further calcium ingress. Thus, a vicious cycle occurs whereby calcium homeostasis becomes dysregulated. Calpain may initiate the early steps in protein turnover by creating nicks in myofibrillar and cytoskeletal proteins for subsequent degradation by other enzymes (Hopf et al., 2007). In normal skeletal muscle, the sarcolemmal protein, dystrophin, forms a glycoprotein complex (Hoffman et al., 1987) that protects the muscle from mechanical damage. In dystrophin-deficient DMD muscle, the sarcolemma is highly susceptible to mechanical damage and subsequent calcium leak (Turner et al., 1991, 1993; McCarter and Steinhardt, 2000). Calcium leaks activate calpain that in turn degrades additional membrane proteins leading to even further calcium leakage.
Dystrophin-deficient dogs with golden retriever muscular dystrophy (GRMD) carry a mutation in the dystrophin gene resulting from an RNA processing error and termination of the dystrophin reading frame (Sharp et al., 1992; Schatzberg et al., 1999). The GRMD dog remains the best-studied large animal model of DMD as it recapitulates the severity and progression of the disease as it appears in humans (Cooper et al., 1988; Cooper, 1989; Dubowitz, 1992; Childers et al., 2001; Kornegay et al., 2011). The present preclinical study tested the idea that calpain inhibition with C101, a novel calpain inhibitor derived from leupeptin (Selsby et al., 2010), can mitigate the progressive decline in strength and ameliorate the severe muscle pathology observed in young dogs with GRMD. The compound, C101, was chosen for this study based on several unique features. First, C101 is covalently linked to leupeptin, to provide efficient uptake into muscle cells via the carnitine organic cation transporter found on the sarcolemma (Selsby et al., 2010), this unique design has the potential to improve both muscle tissue penetration and blood clearance. Second, C101 inhibits calpain (as shown previously by Selsby et al., 2010) due to its derivation from leupeptin. Finally, the dose of C101 chosen for the present study was based on our earlier (unpublished) pilot of leupeptin in GRMD dogs. GRMD dogs were previously given oral leupeptin (15 mg/kg twice daily). Analysis of blood plasma leupeptin levels verified that twice daily oral dosing was appropriate to maintain a stable amount of leupeptin in the blood.
Materials and Methods
Study Design and Use of Animals
Golden retriever cross-bred dogs (n = 16) from a GRMD colony were studied (Kornegay et al., 1988). Dogs were used and cared for according to principles outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The study was conducted as a randomized blinded placebo-controlled intervention trial to test the efficacy and safety of oral C101 in a canine model of DMD. Experimental treatments were concealed from investigators responsible for functional muscle evaluations. Dogs were randomized into two groups: an interventional (treatment) group (n = 9) and a placebo group (n = 7). Newborn GRMD dogs were identified based on elevation of serum creatine kinase and subsequent development of characteristic clinical signs (Kornegay et al., 2011). Genotype was confirmed by polymerase chain reaction (PCR) as described (Bartlett et al., 1996). The experimental treatment, C101, is a proprietary compound consisting of a leupeptin derivative bound to L-aminocarnityl through a linker sequence (Selsby et al., 2010). C101 (17 mg/kg) was given orally twice daily. A placebo (vehicle only) was given as a control. Biopsies were obtained from the cranial sartorius, vastus lateralis, cranial tibialis, and lateral head of the gastrocnemius muscles from C101- and placebo-treated GRMD dogs at the beginning of the study. Samples from the same muscles were obtained at the end of the 8-week study.
Primary Muscle Cell Cultures
For fluorogenic calpain assays and calpain-specific degradation of talin immunoblots, normal, and GRMD myoblasts were grown in culture as previously described (McClorey et al., 2006). Briefly, cranial sartorius muscles from an untreated normal and a GRMD dog 5 months of age, were minced and placed in culture media with collagenase, incubated at 37°C for 1 h, and then plated onto gelatin-coated culture plates in rich serum media for 3–4 days until cells reached 50% confluence.
Assay for μ- and M-Calpain Activity
This assay is based on the unique ability of calpain to catalyze the release of the fluorogenic compound 7-amino-4-methyl-coumarin (AMC) from the substrate Suc-LLVY AMC. We followed the manufacturer’s instructions for a commercial kit (Calpain Assay Kit, Calbiochem, #QIA120). Calpain activity was determined by subtracting the activity obtained using the inhibition buffer from the activity detected with the activation buffer. Standards and controls were run in duplicate on a fluorometer (Synergy HT, Bio-Tek). Two independent experiments from four separate muscle cell cultures were assayed.
Muscle Tissue Preparation
Biopsy samples were minced and homogenized in buffer containing protease inhibitors (Sigma, P2714). After homogenization, lysates were centrifuged for 10 min at 10,000 rpm to separate the insoluble aggregates from the supernatant. The Bradford assay was used to determine protein concentration in each GRMD sample (Zor and Selinger, 1996). Each sample was prepared in duplicate and assayed in triplicate.
SDS Gel Electrophoresis and Immunoblots
Muscle lysates were mixed with Laemmli sample buffer (Bio-Rad) boiled and electrophoresed on 3–8% Tris-Acetate gels (Nupage, Invitrogen) for 75 min at 150 V. Proteins (20 μg/lane) were transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Millipore, MA,USA) over-night at 4°C. The success of protein transfer to the membrane was confirmed by Ponceau stain and by staining gels with Coomassie blue to confirm complete and equal transfer of all protein. Following transfer, the membrane was blocked over-night in 5% milk.
Antibodies
Calpain biomarker activity was accomplished by measuring the relative amount of calpain-specific cleavage products of talin, vinculin, or αII-spectrin on immunoblots using the following antibodies: anti-talin (1:400; Abcam 9290), anti-vinculin (1:2000; Sigma V4505), and anti-αII-spectrin (1:5000, Enzo Life Sciences; Nath et al., 1996; McGinn et al., 2009; Zhang et al., 2009). Following over-night blocking, membranes were rinsed, probed with the primary antibodies followed by Horseradish Peroxidase (HRP)-linked anti-Biotin and goat anti-mouse IgG (Cell Signaling Technology, Beverly, MA, USA) secondary antibodies. Immunoblots were imaged by chemiluminescence (Super Signal West Femto Maximum Sensitivity Substrate, Thermo Scientific) using digital photography. All gel electrophoresis, transfer, and blotting procedures were repeated to produce three independent immunoblots for each sample.
Image Acquisition and Quantification
ImageJ densitometry software (Version 1.4, National Institute of Health, Bethesda, MD, USA) was used to measure density of immunoblot bands (Kobeissy et al., 2006; McGinn et al., 2009; Zhang et al., 2009). Optical density for each band was measured three times and values were averaged.
In vivo Strength Assessment of the Hind Limb
To assess effects of C101 on muscle strength in GRMD dogs, we used a non-invasive method (Kornegay et al., 1999) to measure tarsal joint flexor and extensor muscle strength in anesthetized dogs. Briefly, the dog was positioned in a stereotactic frame that aligned the tibia at a right angle to the femur. Hind limb isometric force was measured by wrapping the foot to a pedal affixed to a force transducer. Percutaneous stimulation (26 gage needle electrodes) of the peroneal nerve at the stifle (knee) activated hind limb muscles to pull the foot up toward the body to generate isometric flexion force. Percutaneous stimulation of the tibial nerve activated muscles to push the foot away from the body to generate isometric extension of the tarsal joint. Hind limb strength (isometric flexion and extension of the tarsal joint) was measured at baseline and at the end of the 8-week study.
To determine if C101 could mitigate effects of muscle damage in GRMD dogs, we applied repeated eccentric (lengthening) contractions to the cranial tibial compartment muscles using established methods (Childers et al., 2002). Briefly, stimulating the sciatic nerve via percutaneous needle electrodes induced eccentric contractions in tibiotarsal flexor muscles. The sciatic nerve innervates both the cranial (anterior) and caudal (posterior) tibial compartment muscles. Stimulation of the muscles of the caudal compartment, larger and stronger than muscles of the cranial compartment, resulted in lengthening (eccentric) contractions of the flexor muscles.
Joint Angle Measurements
To determine if C101 could mitigate joint contractures in GRMD dogs, anesthetized dogs were placed in dorsal recumbency and the angle between the tarsus and the tibia (with the stifle at 90°) was measured as described (Kornegay et al., 1994). The average of two measurements was recorded.
Lean Body Muscle Mass Composition by Dual-Energy X-Ray Absorptiometry
Dual-energy X-ray absorptiometry (DEXA) was used to assess whether C101 can maintain or improve total lean body muscle mass in GRMD. Whole-body DEXA scans were performed at baseline and after 8 weeks of treatment to quantify lean body and fat mass. The Hologic Whole-Body Composition Analysis (version 5.47P) software package was used as previously described for dogs (Lauten et al., 2001).
Histology
To determine effects of C101 on muscle histopathology, biopsy frozen sections (10 μm) were cut on a cryostat (Leica Jung CM1800, Bremen, Germany) and stored at −80°C. Muscle samples were processed, stained (see below), and evaluated with a light microscope. Sections were stained with hematoxylin and eosin (H and E) for general morphology, alizarin red (pH 4.2) for the detection of calcium precipitates, and modified Gomori trichrome for assessment of fibrosis (Childers et al., 2001). One investigator (Joe N. Kornegay) examined images after randomization and coding using an Olympus light microscope. Each slide was previewed under low magnification to identify areas devoid of staining artifacts. Lesions considered included the degree of myofiber necrosis with particular reference to numbers of hyaline and mineralized fibers, fibrosis, and regeneration (Badalamente and Stracher, 2000). Depending on the severity of lesions, scores of 0 (normal), 1 (mild), 2 (moderate), and 3 (severe) were assigned to each slide.
Statistical Analyses
The student’s t-test (unpaired) was used to test the hypothesis that muscle damage, as measured by the force deficit, resulted as a consequence of experimental contractions and was also used to test for differences in treatment groups with respect to percent change in force. Student’s t-test was further used in image analyses, histologic, and calpain assays to test for differences between the C101 and placebo-treated samples as well as between GRMD and normal samples. Statistical significance was set at the 5% level (p < 0.05).
Results
Endogenous Calpain Activity is Increased in vitro to a Greater Extent in GRMD Compared to Normal Primary Cultured Muscle Cells
To measure in vitro changes in endogenous calpain activity in GRMD myoblasts, in vitro calpain activity was compared between normal and GRMD cultured muscle cells. A fluorogenic calpain assay in two independent experiments from four separate primary muscle cell cultures demonstrated in vitro calpain activity was about 60% greater in GRMD versus normal canine muscle cell lysates, respectively (Table 1). Calpain activity (assessed using the biomarker, talin, and its 190 kDa cleavage product) suggested greater activity in GRMD versus normal. As shown in Figure 1, immunoblot densitometry analysis of intact to cleaved talin was 2.61 ± 0.1 and 2.92 ± 0.3, for normal and GRMD cultured muscle cells, respectively (p = 0.11). These findings indicate that in vitro calpain activity is increased in dystrophin-deficient GRMD compared to normal canine muscle cells.
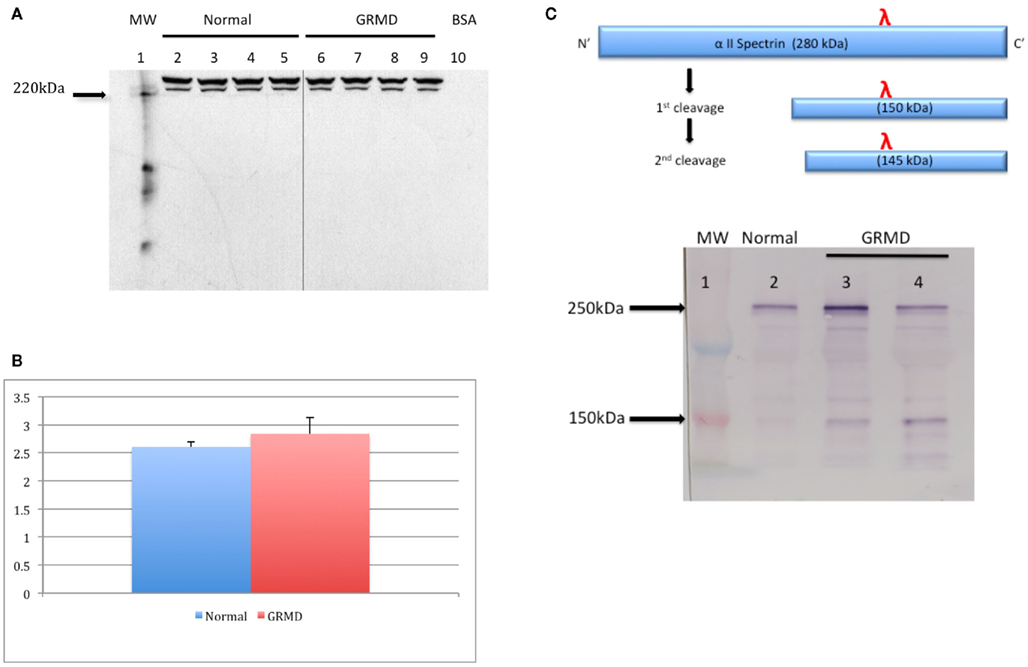
Figure 1. Calpain-specific degradation of talin in primary muscle cell culture lysates from GRMD and normal dogs. A representative immunoblot of cell lysates probed with an anti-talin antibody (1:400; Abcam cat. #9290) (A). Densitometry ratios of the intact talin to cleaved 190 kDa breakdown product (B) between normal (n = 4) and GRMD (n = 4) cell lysates. α-2 spectrin, a major cytoskeletal substrate of calpains, is cleaved in succession to yield two fragments of 150 and 145 kDa [(C), upper panel]. A representative immunoblot of muscle lysates probed with an anti- α-2 spectrin antibody (1:5000; Biomol cat. #fg6090) [(C), lower panel]. In vivo calpain-specific degradation of α-2 spectrin in muscle biopsy sample lysates, 20 μg each, is shown for a normal adult dog (lane 2) and age-matched GRMD littermates (lanes 3 and 4). Note that the 150-kDa spectrin breakdown product appears visibly darker in lanes 3 and 4.

Table 1. Calpain activity in primary cultured muscle cell lysates.
Calpain-Specific Spectrin Degradation in vivo is Increased to a Greater Extent in GRMD Compared To Normal Skeletal Pelvic Limb Muscle
To investigate whether endogenous calpain activity is increased in adult GRMD skeletal muscle tissue in vivo, a biomarker for calpain activity was assessed in pelvic limb muscle biopsy samples from age-matched GRMD and normal littermates. Muscle lysates prepared from adult GRMD dogs demonstrated higher than normal levels of calpain-specific breakdown of α-2 spectrin (Figure 1C). This result indicates that in vivo calpain activity is greater-than-normal in GRMD skeletal pelvic limb muscle.
Effects of C101 on Muscle Function in GRMD Dogs
To investigate potential functional benefit of C101 in GRMD muscle function, a series of mechanical muscle tests were performed in the pelvic limb. Isometric flexion and extension force measures (Kornegay et al., 1999), resting tibiotarsal joint angles (Kornegay et al., 1994), and mechanical response to repeated lengthening (eccentric) contractions (Childers et al., 2002) were measured in GRMD dogs at baseline (6 weeks-of-age) and after 8 weeks of daily administration of C101 (n = 9) or placebo (n = 7). Forces (N) were measured relative to body mass (N/kg).
Hind Limb Strength
Compared to untreated controls, isometric tibiotarsal flexion force (i.e., the force generated by the cranial compartment muscles, the cranial tibialis, and extensor digitorum longus, EDL) was generally maintained in C101-treated dogs (Figure 2A). GRMD dogs treated with C101 generated 0.380 ± 0.11 N/kg at baseline and 0.363 ± 0.07 N/kg at the end of the 8-week study. In contrast, flexion force in placebo-treated GRMD dogs declined from 0.494 ± 0.15 at baseline to 0.325 ± 0.11 N/kg at the study completion (p < 0.05). These results indicate that the cranial compartment muscles were relatively stronger in the C101-treated group compared to the placebo group.
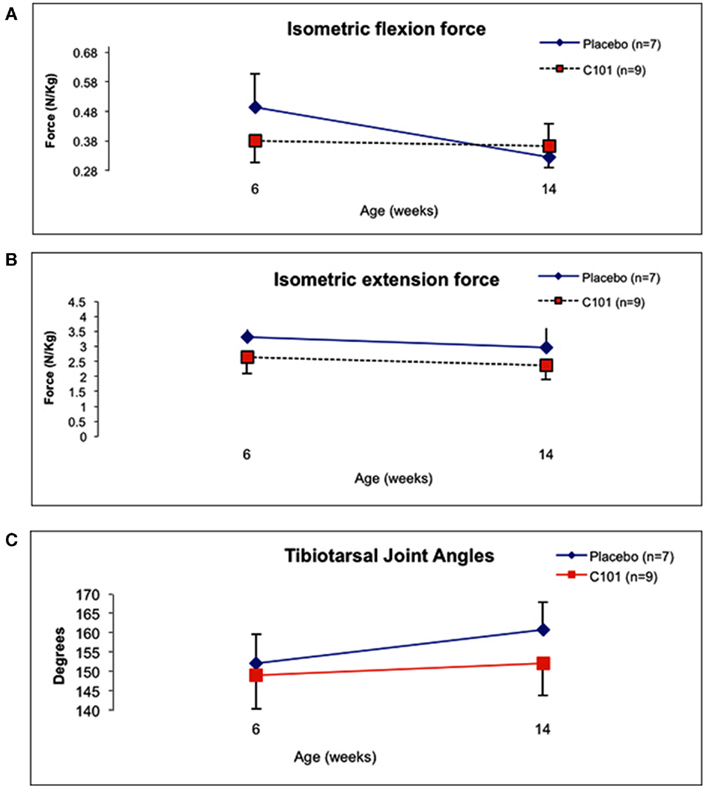
Figure 2. Comparison of in vivo pelvic limb isometric force normalized to body mass between placebo and C101 – treated GRMD dogs. Flexion (A) and extension (B) isometric force responses for both treatment groups at baseline (6 weeks-of-age) and at the end of the treatment (14 weeks-of-age). Resting tibiotarsal joint angles at ages 6 and 14 weeks (C) Data are presented as mean ± SD. Blue-filled diamonds = placebo group; red-filled boxes = C101 group.
A general decline in isometric extension force (i.e., the force generated primarily by the gastrocnemius and superficial digital flexor muscles) occurred in both the C101 and placebo-treated GRMD dogs (Figure 2B). Extension force in C101-treated dogs measured 2.64 ± 0.5 N/kg at baseline and 2.37 ± 0.5 N/kg at 8 weeks. Extension force in placebo-treated dogs also declined from 3.30 ± 0.2 N/kg at baseline to 2.96 ± 0.7 N/kg at the study end. These results indicate that the caudal compartment muscles were weaker at the end of the study in both treatment groups, and that there was no detectable benefit from C101 treatment in these muscles.
Joint Contractures
Resting tibiotarsal joint angles were comparable (149° ± 9° versus 152° ± 8°) between treated and untreated 6-week-old GRMD dogs at the beginning of the study. After chronic administration of C101 or placebo, no significant differences were detected between treatment groups (152° ± 8° versus 161° ± 7°, respectively, Figure 2C). This finding indicates that C101 treatment had no measureable effect on the progression of tibiotarsal joint contractures in GRMD dogs.
Response to Repeated Eccentric Contractions
To assess the possibility that C101 might mitigate contraction-induced muscle damage in dystrophin-deficient skeletal muscle, GRMD pelvic limbs were subjected to repeated in vivo eccentric contractions. Muscles of the cranial compartment (i.e., the cranial tibialis and EDL) were subjected to repeated stretch-activations that typically result in muscle damage and loss of force (Childers et al., 2002). Thus, to measure the extent of stretch-induced muscle damage, force drop was measured before and 3 days after repeated eccentric contractions. At the end of the study, significant differences were not detected between treatment groups: the force drop was 21.7 ± 9 and 30.7 ± 16%, for C101- and placebo-treated dogs, respectively. Interestingly, analysis of force drop results over the course of the study indicated that very young GRMD dogs (6 weeks-of-age) respond differently than older dogs. Flexion force for 6-week-old GRMD dogs was 0.408 ± 0.10 N/kg and dropped to 0.382 ± N/kg after eccentric contractions. In contrast, at the end of the study, flexion force in 16-week-old GRMD dogs was 0.346 ± 0.09 N/kg and dropped to 0.266 ± 0.08 N/kg after eccentric contractions. Thus, in GRMD dogs, 6-week-old pups manifested only a small force drop of 11.6 ± 20% compared to 26.6 ± 7% in 15-week-old animals, p < 0.05 (Figure 3). Together, these results suggest that age has an effect on the extent of stretch-induced muscle damage in dystrophin-deficient muscle.
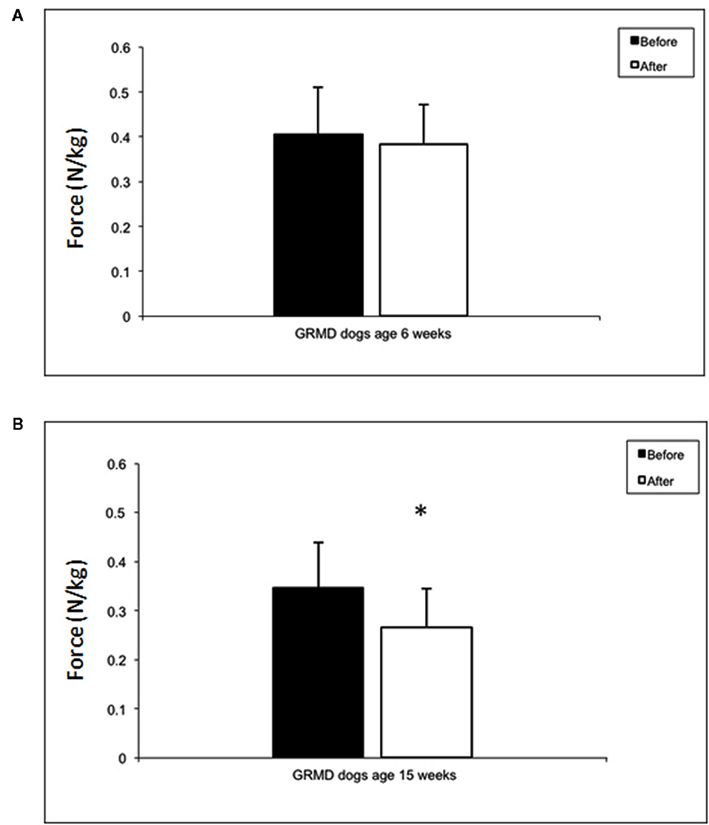
Figure 3. Maximum isometric tibiotarsal flexion force normalized to body mass before (white bars) and after (black bars) a series of eccentric (lengthening) contractions. At 6 weeks-of-age, GRMD dogs do not demonstrate a loss of isometric force (A), whereas at 14 weeks, normalized isometric force drops significantly (p < 0.05) (B). There were no differences in the extent of isometric force drop after eccentric contractions between placebo and C101-treated GRMD dogs (data not shown).
Lean Body Mass
To assess whether or not C101 might have an effect in maintaining or improving total lean body muscle mass in dogs with GRMD, whole-body DEXA scans were performed at baseline and at 15 weeks-of-age to quantify fat and lean body mass. At the beginning of the study, lean body mass was comparable between treatment groups (not shown). At the study completion, differences were not detected among the groups. The percent fat and lean body mass were 16.2 ± 2 and 81.3 ± 2 compared to 15.4 ± 5 and 82.3 ± 5 for C101- and placebo- treated dogs, respectively. This result indicates that C101 treatment did not lead to an improvement in total lean body mass in young GRMD dogs.
Muscle Histology
To investigate effects of C101 treatment on muscle histopathology in GRMD dogs, pelvic limb muscle biopsy samples taken at baseline and at the end of the study were sectioned, stained, and scored for evidence of myofiber necrosis and calcification (Childers et al., 2001). Briefly, assessment of myofiber necrosis and calcification was performed by examining serial transverse cryosections stained with H and E and alizarin red (pH 4.2) using light microscopy. Evidence of myofiber necrosis consisted of identifying “hyaline” or hypercontracted fibers, fibers undergoing myophagocytosis (fibers surrounded by phagocytic cells), or empty sarcolemmal tubes. Evidence of myofiber calcification consisted of identifying bright red myofibers stained by calcium precipitates. The combined mean necrosis scores for all of the muscles examined were 2.85 ± 3.4 and 3.20 ± 3.5 for the C101 and placebo groups, respectively. The combined means scores of necrosis and calcification for each muscle evaluated over time and among treatment groups did not reveal significant differences between any of the variables examined (Table 2). These results indicate that C101 treatment did not lead to a detectable improvement in muscle histopathology in young GRMD dogs.
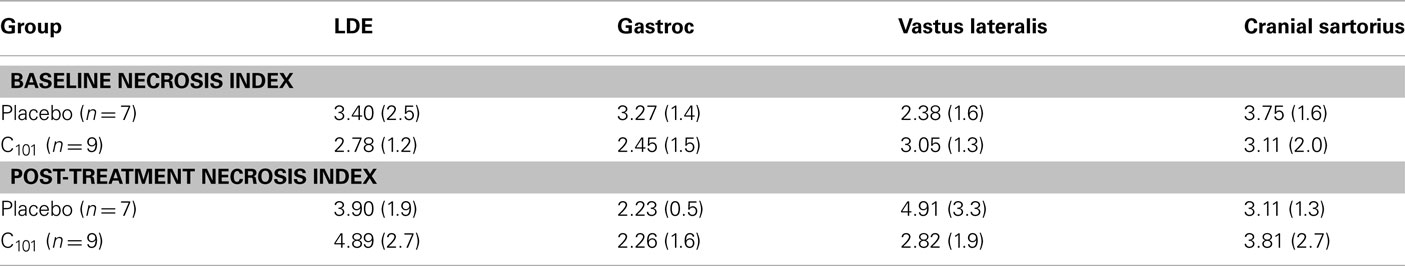
Table 2. Comparison of pelvic muscle necrosis scores in GRMD dogs treated with C101 or placebo.
Calpain-Specific Spectrin Degradation in vivo
To determine if chronic daily administration of C101 inhibited calpain-specific spectrin degradation in vivo to a greater extent than placebo, the 150-kDa breakdown product of α-2 spectrin in muscle lysates was measured and compared between the treatment groups. As shown in Figure 4, GRMD muscle lysates demonstrate significantly greater-than-normal degradation of the 250-kDa intact α-2 spectrin (p < 0.05). A change in the 150-kDa product was noted in the placebo group over time (the baseline relative intensity increased from 0.45 to 0.67 by the end of the study, p = 0.14). In contrast, the C101 group remained stable (0.54–0.56, p = 0.44). However, significant differences were not detected between treatment groups for inhibition of the 150-kDa spectrin breakdown product.
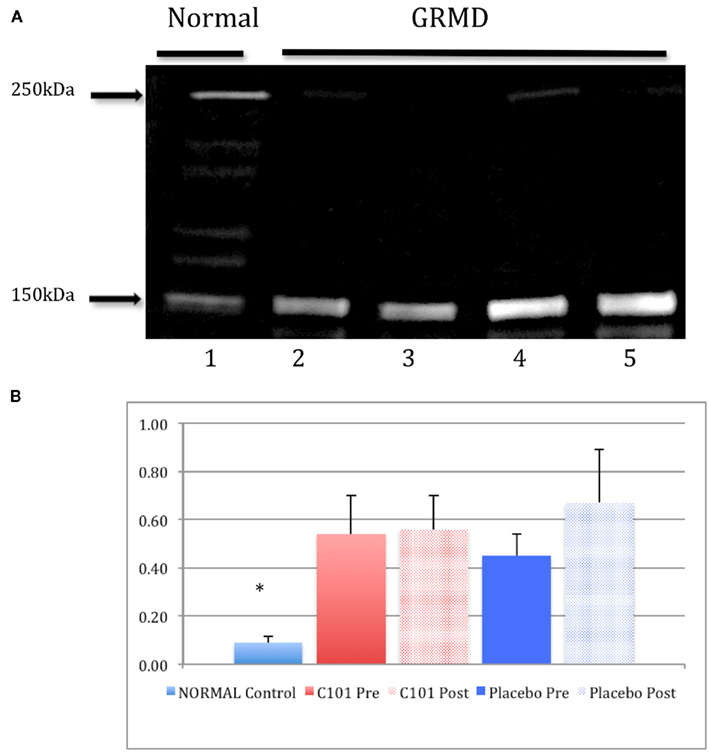
Figure 4. Calpain-specific degradation of α-2 spectrin in muscle biopsy lysates from normal and GRMD dogs. In vivo calpain-specific degradation of α-2 spectrin in muscle biopsy sample lysates, 20 μg each, is shown for a normal dog (lane 1) and age-matched GRMD dogs (lanes 2–5, (A). Densitometry ratios (B) of the intact α-2 spectrin to the cleaved 150 kDa fragment among treatment groups. Significant differences were detected between normal and GRMD dogs (p < 0.05) but no differences were detected between placebo or C101 treatment groups. Legend: red-filled bars = C101-treated groups (pre- and post-treatment); blue-filled bars = placebo groups. Data are presented as mean ± SD.
Clinical Safety
C101 (17 mg/kg twice daily oral administration) was generally well tolerated without notable adverse clinical effects. Blood serum chemistries and clotting profiles remained within normal limits throughout the treatment timeline in all dogs (not shown).
Discussion
We tested the hypothesis that treatment with the novel calpain inhibitor, C101, can mitigate the progressive weakness and severe muscle pathology observed in young GRMD dogs. Our findings do not support this hypothesis and are in agreement with a recent report of leupeptin-based calpain inhibitors in mdx mice. In their report, Selsby et al. (2010) demonstrated that C101 given intra-peritoneal (i.p.) over 4 weeks failed to improve the phenotype of mdx mice. In our study, we administered oral C101 at a dose based on oral leupeptin biodistribution in GRMD dogs (data not shown). We found that C101 given over 8 weeks failed to improve the phenotype of GRMD dogs. The only functional difference between C101 and placebo-treated dogs was preservation of flexor force; however, results from a prednisone treatment study in GRMD indicated that flexion was paradoxically decreased while extension force improved (Liu et al., 2004). We believe this resulted from reduced necrosis early in the disease process, thus preventing functional hypertrophy in flexor muscles. In this context, preserved flexor force actually is more compatible with an overall decline in function.
Study Limitations
C101 was designed as a small molecule leupeptin derivative covalently linked to carnitine (Selsby et al., 2010). The sarcolemmal membrane contains an organic carnitine transporter that has the potential to rapidly clear C101 from the circulation and actively transport the compound into muscle cells (Grigat et al., 2009). Indeed, peak serum C101 levels (∼29 μM) occur within 30 min but fall to 3 μM 2 h later (Selsby et al., 2010). Our results suggest that C101 failed to inhibit calpain activity in vivo as determined by quantitative assessment of the biomarker α-spectrin. However, we did not measure serum or muscle lysate C101 content, and it is possible that the dose given was not sufficient to reach inhibitory activity. This possibility is unlikely, as Selsby et al. previously demonstrated the ability of C101 to inhibit calpain both in vitro and in vivo.
In general, there is a shift to a MHC type I phenotype in dystrophic muscle over time. We previously observed this in GRMD dogs, that a gradual shift in fiber type occurs in selected hind limb muscles between 3 and 6 months of age with a general trend toward type I (slow) fiber predominance in by 6 months of age. In the flexors, this appears to be particularly evident. Thus, it is conceivable that some beneficial effects of C101 might have escaped detection in the present study because the eccentric contraction assays were only performed in the flexor muscles. However, it is generally accepted that dystrophin-deficient myofibers are more highly susceptible to mechanical damage due to the lack of dystrophin (rather than due to a fiber type shift). The disease also affects the heart and diaphragm muscles, and it is conceivable that some beneficial effect from the experimental intervention may have been present, and not tested. Further experiments specifically targeted at these critical muscles are required to address this possibility.
New Findings
Unexpectedly, we found that pelvic limb muscles of very young (6-week-old) GRMD dogs did not demonstrate a drop in isometric force 3 days after repeated eccentric contractions, whereas older (15-week-old) dogs demonstrate a significant force drop (Figure 3), a feature shown to represent the functional effects of muscle damage (Childers et al., 2002, 2005; Markert et al., 2006). The absence of a force drop in young GRMD dogs may be related to age. In a related study of EDL muscles from young mdx mice (9–12 days of age), there was no force drop between the first and last of a series of five acute eccentric contractions (Grange et al., 2002). However, in EDL muscles of older (∼90–125 days) mdx mice subjected to the same injury protocol, force drop markedly increased. Thus, the age of both GRMD dogs and mdx mice may influence the extent of force drop in muscles subjected to repeated eccentric contractions.
Clinical Implications
Failure of C101 to improve the phenotype in dystrophin-deficient mice and dogs might be explained by the observation that long-term exposure to leupeptin in mdx mice results in an increase in the autolysis of m-calpain (Cong et al., 1989; Goll et al., 1992, 2003). Chronic administration of a calpain inhibitor (e.g., C101) might not change the calpain activity set point because of a compensatory increase in calpain autolysis in dystrophin-deficient muscle. Together, both murine and canine studies generally indicate that pharmacological inhibition of calpain alone may not benefit patients with DMD probably due to several protease mechanisms functioning in dystrophin-deficient skeletal muscle.
Conflict of Interest Statement
Ceptor, Inc., the manufacturer of C101, provided financial support for a portion of this work.
Acknowledgments
We thank J. Staley, MD for performing some of the calpain assays. This work was supported by a grant (1R21NS050135) from the National Institutes of Neurological Diseases and Stroke (NINDS).
References
Badalamente, M. A., and Stracher, A. (2000). Delay of muscle degeneration and necrosis in mdx mice by calpain inhibition. Muscle Nerve 23, 106–111.
Bartlett, R. J., Winand, N. J., Secore, S. L., Singer, J. T., Fletcher, S., Wilton, S., Bogan, D. J., Metcalf-Bogan, J. R., Bartlett, W. T., Howell, J. M., Cooper, B. J., and Kornegay, J. N. (1996). Mutation segregation and rapid carrier detection of X-linked muscular dystrophy in dogs. Am. J. Vet. Res. 57, 650–654.
Briguet, A., Erb, M., Courdier-Fruh, I., Barzaghi, P., Santos, G., Herzner, H., Lescop, C., Siendt, H., Henneboehle, M., Weyermann, P., Magyar, J. P., Dubach-Powell, J., Metz, G., and Meier, T. (2008). Effect of calpain and proteasome inhibition on Ca2+-dependent proteolysis and muscle histopathology in the mdx mouse. FASEB J. 22, 4190–4200.
Childers, M. K., Okamura, C. S., Bogan, D. J., Bogan, J. R., Petroski, G. F., Mcdonald, K., and Kornegay, J. N. (2002). Eccentric contraction injury in dystrophic canine muscle. Arch. Phys. Med. Rehabil. 83, 1572–1578.
Childers, M. K., Okamura, C. S., Bogan, D. J., Bogan, J. R., Sullivan, M. J., and Kornegay, J. N. (2001). Myofiber injury and regeneration in a canine homologue of Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil. 80, 175–181.
Childers, M. K., Staley, J. T., Kornegay, J. N., and Mcdonald, K. S. (2005). Skinned single fibers from normal and dystrophin-deficient dogs incur comparable stretch-induced force deficits. Muscle Nerve 31, 768–771.
Cong, J., Goll, D. E., Peterson, A. M., and Kapprell, H. P. (1989). The role of autolysis in activity of the Ca2+-dependent proteinases (mu-calpain and m-calpain). J. Biol. Chem. 264, 10096–10103.
Cooper, B. J. (1989). Animal models of Duchenne and Becker muscular dystrophy. Br. Med. Bull. 45, 703–718.
Cooper, B. J., Winand, N. J., Stedman, H., Valentine, B. A., Hoffman, E. P., Kunkel, L. M., Scott, M. O., Fischbeck, K. H., Kornegay, J. N., Avery, R. J., Williams, J. R., Schmickel, R. D., and Sylvester, J. E. (1988). The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature 334, 154–156.
Courdier-Fruh, I., and Briguet, A. (2006). Utrophin is a calpain substrate in muscle cells. Muscle Nerve 33, 753–759.
Figarella-Branger, D., El-Dassouki, M., Saenz, A., Cobo, A. M., Malzac, P., Tong, S., Cassotte, E., Azulay, J. P., Pouget, J., and Pellissier, J. F. (2002). Myopathy with lobulated muscle fibers: evidence for heterogeneous etiology and clinical presentation. Neuromuscul. Disord. 12, 4–12.
Gailly, P., De Backer, F., Van Schoor, M., and Gillis, J. M. (2007). In situ measurements of calpain activity in isolated muscle fibres from normal and dystrophin-lacking mdx mice. J. Physiol. (Lond.) 582, 1261–1275.
Goll, D. E., Thompson, V. F., Li, H., Wei, W., and Cong, J. (2003). The calpain system. Physiol. Rev. 83, 731–801.
Goll, D. E., Thompson, V. F., Taylor, R. G., and Christiansen, J. A. (1992). Role of the calpain system in muscle growth. Biochimie 74, 225–237.
Grange, R. W., Gainer, T. G., Marschner, K. M., Talmadge, R. J., and Stull, J. T. (2002). Fast-twitch skeletal muscles of dystrophic mouse pups are resistant to injury from acute mechanical stress. Am. J. Physiol. Cell Physiol. 283, C1090–C1101.
Grigat, S., Fork, C., Bach, M., Golz, S., Geerts, A., Schomig, E., and Grundemann, D. (2009). The carnitine transporter SLC22A5 is not a general drug transporter, but it efficiently translocates mildronate. Drug Metab. Dispos. 37, 330–337.
Hoffman, E. P., Brown, R. H. Jr., and Kunkel, L. M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928.
Hopf, F. W., Turner, P. R., and Steinhardt, R. A. (2007). Calcium misregulation and the pathogenesis of muscular dystrophy. Subcell. Biochem. 45, 429–464.
Kobeissy, F. H., Ottens, A. K., Zhang, Z., Liu, M. C., Denslow, N. D., Dave, J. R., Tortella, F. C., Hayes, R. L., and Wang, K. K. (2006). Novel differential neuroproteomics analysis of traumatic brain injury in rats. Mol. Cell Proteomics 5, 1887–1898.
Kornegay, J. N., Bogan, D. J., Bogan, J. R., Childers, M. K., Cundiff, D. D., Petroski, G. F., and Schueler, R. O. (1999). Contraction force generated by tarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J. Neurol. Sci. 166, 115–121.
Kornegay, J. N., Bogan, J. R., Bogan, D. J., Childers, M. K., and Grange, R. W. (2011). Golden retriever muscular dystrophy (GRMD): developing and maintaining a colony and physiological functional measurements. Methods Mol. Boil. 709, 105–123.
Kornegay, J. N., Sharp, N. J., Schueler, R. O., and Betts, C. W. (1994). Tarsal joint contracture in dogs with golden retriever muscular dystrophy. Lab. Anim. Sci. 44, 331–333.
Kornegay, J. N., Tuler, S. M., Miller, D. M., and Levesque, D. C. (1988). Muscular dystrophy in a litter of golden retriever dogs. Muscle Nerve 11, 1056–1064.
Lauten, S. D., Cox, N. R., Brawner, W. R. Jr., and Baker, H. J. (2001). Use of dual energy x-ray absorptiometry for noninvasive body composition measurements in clinically normal dogs. Am. J. Vet. Res. 62, 1295–1301.
Liu, J. M., Okamura, C. S., Bogan, D. J., Bogan, J. R., Childers, M. K., and Kornegay, J. N. (2004). Effects of prednisone in canine muscular dystrophy. Muscle Nerve 30, 767–773.
Markert, C., Petroski, G. F., Childers, C. K., Mcdonald, K. S., and Childers, M. K. (2006). Stretch-induced force deficits in murine extensor digitorum longus muscles after cardiotoxin injection. Muscle Nerve 34, 485–488.
McCarter, G. C., and Steinhardt, R. A. (2000). Increased activity of calcium leak channels caused by proteolysis near sarcolemmal ruptures. J. Membr. Biol. 176, 169–174.
McClorey, G., Moulton, H. M., Iversen, P. L., Fletcher, S., and Wilton, S. D. (2006). Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 13, 1373–1381.
McGinn, M. J., Kelley, B. J., Akinyi, L., Oli, M. W., Liu, M. C., Hayes, R. L., Wang, K. K., and Povlishock, J. T. (2009). Biochemical, structural, and biomarker evidence for calpain-mediated cytoskeletal change after diffuse brain injury uncomplicated by contusion. J. Neuropathol. Exp. Neurol. 68, 241–249.
Nath, R., Raser, K. J., Stafford, D., Hajimohammadreza, I., Posner, A., Allen, H., Talanian, R. V., Yuen, P., Gilbertsen, R. B., and Wang, K. K. (1996). Non-erythroid alpha-spectrin breakdown by calpain and interleukin 1 beta-converting-enzyme-like protease(s) in apoptotic cells: contributory roles of both protease families in neuronal apoptosis. Biochem. J. 319(Pt 3), 683–690.
Saez, M. E., Ramirez-Lorca, R., Moron, F. J., and Ruiz, A. (2006). The therapeutic potential of the calpain family: new aspects. Drug Discov. Today 11, 917–923.
Schatzberg, S. J., Olby, N. J., Breen, M., Anderson, L. V., Langford, C. F., Dickens, H. F., Wilton, S. D., Zeiss, C. J., Binns, M. M., Kornegay, J. N., Morris, G. E., and Sharp, N. J. (1999). Molecular analysis of a spontaneous dystrophin ‘knockout’ dog. Neuromuscul. Disord. 9, 289–295.
Selsby, J., Pendrak, K., Zadel, M., Tian, Z., Pham, J., Carver, T., Acosta, P., Barton, E., and Sweeney, H. L. (2010). Leupeptin-based inhibitors do not improve the mdx phenotype. Am. J. Physiol. Regul. Integr. Comp. Physiol. 299, R1192–R1201.
Sharp, N. J., Kornegay, J. N., Van Camp, S. D., Herbstreith, M. H., Secore, S. L., Kettle, S., Hung, W. Y., Constantinou, C. D., Dykstra, M. J., Roses, A. D., and Bartlett, R. J. (1992). An error in dystrophin mRNA processing in golden retriever muscular dystrophy, an animal homologue of Duchenne muscular dystrophy. Genomics 13, 115–121.
Spencer, M. J., and Mellgren, R. L. (2002). Overexpression of a calpastatin transgene in mdx muscle reduces dystrophic pathology. Hum. Mol. Genet. 11, 2645–2655.
Takamure, M., Murata, K. Y., Tamada, Y., Azuma, M., and Ueno, S. (2005). Calpain-dependent alpha-fodrin cleavage at the sarcolemma in muscle diseases. Muscle Nerve 32, 303–309.
Turner, P. R., Fong, P. Y., Denetclaw, W. F., and Steinhardt, R. A. (1991). Increased calcium influx in dystrophic muscle. J. Cell Biol. 115, 1701–1712.
Turner, P. R., Schultz, R., Ganguly, B., and Steinhardt, R. A. (1993). Proteolysis results in altered leak channel kinetics and elevated free calcium in mdx muscle. J. Membr. Biol. 133, 243–251.
Urasawa, N., Wada, M. R., Machida, N., Yuasa, K., Shimatsu, Y., Wakao, Y., Yuasa, S., Sano, T., Nonaka, I., Nakamura, A., and Takeda, S. (2008). Selective vacuolar degeneration in dystrophin-deficient canine Purkinje fibers despite preservation of dystrophin-associated proteins with overexpression of Dp71. Circulation 117, 2437–2448.
Whitehead, N. P., Yeung, E. W., and Allen, D. G. (2006). Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin. Exp. Pharmacol. Physiol. 33, 657–662.
Zhang, Z., Larner, S. F., Liu, M. C., Zheng, W., Hayes, R. L., and Wang, K. K. (2009). Multiple alphaII-spectrin breakdown products distinguish calpain and caspase dominated necrotic and apoptotic cell death pathways. Apoptosis 14, 1289–1298.
Keywords: muscular dystrophy, animal, calpain, skeletal muscle, protease, canine
Citation: Childers MK, Bogan JR, Bogan DJ, Greiner H, Holder M, Grange RW and Kornegay JN (2012) Chronic administration of a leupeptin-derived calpain inhibitor fails to ameliorate severe muscle pathology in a canine model of Duchenne muscular dystrophy. Front. Pharmacol. 2:89. doi: 10.3389/fphar.2011.00089
Received: 14 August 2011; Paper pending published: 19 September 2011;
Accepted: 18 December 2011; Published online: 09 January 2012.
Edited by:
Eileen M. Hasser, University of Missouri, USAReviewed by:
Tom Cunningham, University of North Texas Health Science Center, USAMarybeth Brown, University of Missouri, USA
Copyright: © 2012 Childers, Bogan, Bogan, Greiner, Holder, Grange and Kornegay. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Martin K. Childers, Wake Forest Institute for Regenerative Medicine, Room 258 Dean Biomedical Research Building, 391 Technology Way, Winston-Salem, NC 27101, USA. e-mail:bWNoaWxkZXJAd2Z1Ym1jLmVkdQ==
†Present address: Hansel Greiner , Departments of Pediatrics and Neurology, Cincinnati Children's Hospital Medical Center, 3333 Burnet Avenue, Cincinnati, OH 45229, USA