Abstract
We previously showed that the β-adrenoceptor modulators, clenbuterol and propranolol, directly blocked voltage-gated sodium channels, whereas salbutamol and nadolol did not (Desaphy et al., 2003), suggesting the presence of two hydroxyl groups on the aromatic moiety of the drugs as a molecular requisite for impeding sodium channel block. To verify such an hypothesis, we synthesized five new mexiletine analogs by adding one or two hydroxyl groups to the aryloxy moiety of the sodium channel blocker and tested these compounds on hNav1.4 channels expressed in HEK293 cells. Concentration–response relationships were constructed using 25-ms-long depolarizing pulses at −30 mV applied from an holding potential of −120 mV at 0.1 Hz (tonic block) and 10 Hz (use-dependent block) stimulation frequencies. The half-maximum inhibitory concentrations (IC50) were linearly correlated to drug lipophilicity: the less lipophilic the drug, minor was the block. The same compounds were also tested on F1586C and Y1593C hNav1.4 channel mutants, to gain further information on the molecular interactions of mexiletine with its receptor within the sodium channel pore. In particular, replacement of Phe1586 and Tyr1593 by non-aromatic cysteine residues may help in the understanding of the role of π–π or π–cation interactions in mexiletine binding. Alteration of tonic block suggests that the aryloxy moiety of mexiletine may interact either directly or indirectly with Phe1586 in the closed sodium channel to produce low-affinity binding block, and that this interaction depends on the electrostatic potential of the drug aromatic tail. Alteration of use-dependent block suggests that addition of hydroxyl groups to the aryloxy moiety may modify high-affinity binding of the drug amine terminal to Phe1586 through cooperativity between the two pharmacophores, this effect being mainly related to drug lipophilicity. Mutation of Tyr1593 further impaired such cooperativity. In conclusion, these results confirm our former hypothesis by showing that the presence of hydroxyl groups to the aryloxy moiety of mexiletine greatly reduced sodium channel block, and provide molecular insights into the intimate interaction of local anesthetics with their receptor.
Introduction
Mexiletine is a class Ib antiarrhythmic drug, also considered as the first choice drug for treating myotonic syndromes (Conte Camerino et al., 2007). Moreover, recent clinical trials demonstrate its therapeutic value to relieve neuropathic pain (Challapalli et al., 2005). Mexiletine exerts its pharmacological action through blockade of voltage-gated sodium channels, thus reducing cell excitability. Preferential binding to inactivated channels and use-dependent block are the basis of the selective action of mexiletine on pathologic hyperactive tissues. The structure of mexiletine is related to that of local anesthetic (LA) drugs, presenting a protonable amine group connected to a hydrophobic aromatic ring through an intermediate ether link (Table 1). We have shown that little modification of these three components can substantially affect sodium channel blockade in vitro and antimyotonic effects in vivo (Desaphy et al., 1999, 2001; De Luca et al., 2000, 2003, 2004; De Bellis et al., 2006). The use of mexiletine analogs has allowed to get new important information on the molecular dynamic interaction between the drug and its receptor within the sodium channel pore (De Luca et al., 2000, 2003). Besides, sodium channel mutagenesis experiments have defined the amino acids most probably involved in the interaction with LAs (Ragsdale et al., 1994, 1996; Wright et al., 1998; Nau et al., 1999; Wang et al., 2000; Sunami et al., 2001; Yarov-Yarovoy et al., 2001, 2002; O’Leary and Chahine, 2002; McNulty et al., 2007; Ahern et al., 2008).
Table 1
| Compound | Structure | Log P | pKa | Log D (pH 7.4) | Ionization (mol%, pH 7.4) | Electrostatic potential (kcal/mol) |
|---|---|---|---|---|---|---|
| Mexiletine | 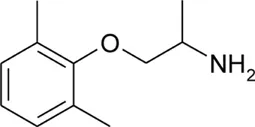 | 2.21 ± 0.01 | 9.28 ± 0.01 | 0.53 | 98.7 | −50.3 |
| mHM | 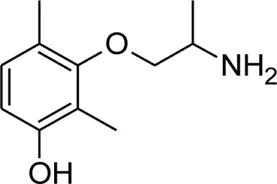 | 1.67 ± 0.01 | 9.04 ± 0.01 | 0.02 | 97.7 | −48.0 |
| pHM | 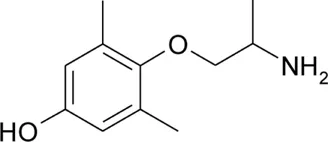 | 1.53 ± 0.01 | 8.97 ± 0.01 | −0.05 | 97.3 | −50.4 |
| HMM | 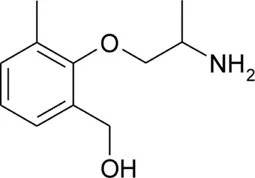 | 1.15 ± 0.01 | 9.13 ± 0.01 | −0.59 | 98.1 | −64.9 |
| bHMM | 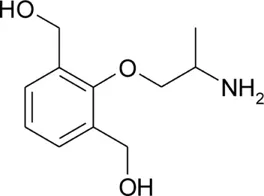 | 0.25 ± 0.05 | 9.21 ± 0.03 | −0.85 | 98.5 | −62.6 |
| pHHMM | 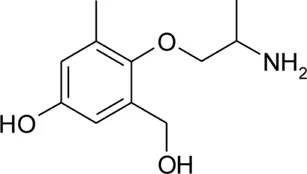 | 0.23 ± 0.02 | 8.89 ± 0.02 | −1.27 | 96.8 | −65.0 |
Chemical structures and physicochemical properties.
The Log P, pKa, Log D, and electrostatic potential values were determined experimentally, as described in the Section “Materials and Methods.” Values of Log P and pKa are given as mean ± SEM. Ionization of the amine group at pH 7.4 was calculated from Henderson–Hasselbalch equation,
In a previous study, we showed that drugs known as modulators of β-adrenergic receptors are able to block sodium channels in a manner reminiscent of LA drugs (Desaphy et al., 2003). The β-agonist clenbuterol and the β-blocker propranolol showed efficacies comparable to mexiletine in producing use-dependent inhibition of sodium currents in rat skeletal muscle fibers as well as in tsA201 cells transiently expressing the human muscle sodium channel isoform, hNav1.4. In contrast, the β-agonist salbutamol and the β-blocker nadolol had no effect on sodium currents at 1 mM concentration. Examination of chemical structures and physicochemical properties of these drugs revealed the presence of the two pharmacophores important for sodium channel blocking activity, the amine group and the aromatic ring, which respectively confer a high pKa and high lipophilicity. However, the two inactive compounds were characterized by the presence of two hydroxyl groups on the aromatic moiety, which we proposed to be determinant for impeding sodium channel blockade.
To verify this hypothesis, we have synthesized new analogs of mexiletine by introducing one or two hydroxyl groups on the aryloxy moiety of the drug and tested them on sodium currents in mammalian cells permanently transfected with hNav1.4. Moreover site-directed mutagenesis was performed to introduce non-aromatic cysteine residues in place of the Phe1586 and Tyr1593 residues of hNav1.4, which are thought to be part of the binding site for LAs within the domain IV segment 6 of sodium channels (Ragsdale et al., 1994).
The results demonstrate that the presence of hydroxyl groups on the aromatic moiety drastically reduces the ability of compounds to block sodium channels and provide further insight in the molecular interaction between LA drugs and their binding site within the sodium channel pore.
Materials and Methods
Site-directed mutagenesis and expression of recombinant sodium channels
Full-length cDNA encoding the main α-subunit of wild-type (WT) skeletal muscle isoform of voltage-gated sodium channel (hNav1.4) was subcloned in the mammalian expression vector pRc–CMV. The two hNav1.4 mutants, F1586C and Y1593C, were engineered by standard two-step PCR-based site-directed mutagenesis. All PCR reactions were performed using Pfu DNA polymerase (Stratagene, La Jolla, CA, USA) for high-fidelity amplification (Desaphy et al., 2010). The complete coding region of channel mutant cDNAs was sequenced to exclude any polymerase errors. Permanent sodium channel expression was achieved by transfecting 1 mg ml−1 of plasmid in human embryonic kidney HEK293 cells using the calcium phosphate precipitation method and clone selection using geneticin (Gibco-Invitrogen, Milan, Italy).
Sodium current measurement in HEK293 cells
Whole-cell sodium currents (INa) were recorded at room temperature (20–22°C) using an Axopatch 1D amplifier (Axon Instruments, Union City, CA, USA). Voltage clamp protocols and data acquisition were performed with pCLAMP 9.2 software (Axon Instruments) through a 12-bit A–D/D–A interface (Digidata 1340, Axon Instruments). Pipettes made with Corning 7052 glass (Garner Glass, Claremont, CA, USA) had resistance that ranged from 1 to 3 MΩ. Currents were low-pass filtered at 2 kHz (–3 dB) by the four-pole Bessel filter of the amplifier and digitized at 10–20 kHz. After the patch membrane had been ruptured, a 25-ms-long test pulse to −30 mV from a holding potential (HP) of −120 mV was applied to the cell at a low frequency until stabilization of INa amplitude and kinetics was achieved (typically 5 min). Only those data obtained from cells exhibiting series resistance errors <5 mV were considered for analysis. Little (<5%) or no rundown was observed within the experiments. The concentration–response relationships were produced by obtaining the peak current amplitude measured in the presence of drug (IDRUG), normalized to the peak current amplitude measured in the same cell before drug application (ICTRL), as a function of drug concentration [DRUG]. Each data point is the mean ± SEM from at least three cells. The relationships were fitted with a first-order binding function:
where IC50 is the half-maximum inhibitory concentration, and nH is the slope factor. The variable Imax was introduced only for dose–response relationships of compounds effects on Y1593C channel mutant at 10 Hz, otherwise Imax was fixed to 1.
Mutant cycle analysis
Mutant cycle analysis was performed to evaluate the cross influence between amino acid mutations in the hNav1.4 channel and hydroxyl substitutions in mexiletine, in order to characterize the molecular interactions. The coupling energy ΔG is thus calculated as RT·lnΩ, where R is the perfect gas constant (8.314 J K mol−1), T is the temperature expressed in Kelvin (295.15 K = 22°C), and Ω is the coupling constant (Hidalgo and MacKinnon, 1995).
Drugs and solutions
Patch clamp pipette solution contained in mM: 120 CsF, 10 CsCl, 10 NaCl, 5 EGTA, and 5 HEPES, and the pH was set to 7.2 with CsOH. Bath solution for patch clamp recordings contained (in mM): 150 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, and 5 glucose. The pH was set to 7.4 with NaOH. Mexiletine hydrochloride [Mex, 1-(2,6-dimethylphenoxy)propan-2-amine] was purchased from Sigma (Milan, Italy). The hydroxylated analogs of mexiletine {metahydroxymexiletine, mHM, 3-(2-aminopropoxy)-2,4-dimethylphenol; parahydroxymexiletine, pHM, 4-(2-aminopropoxy)-3,5-dimethylphenol; hydroxymethylmexiletine, HMM, [2-(2-aminopropoxy)-3-methylphenyl]methanol; bis(hydroxymethyl)mexiletine, bHMM, [2-(2-aminopropoxy)-1,3-phenylene]dimethanol; parahydroxyhydroxymethyl mexiletine, pHHMM, 4-(2-aminopropoxy)-3-(hydroxymethyl)-5-methylphenol} were synthesized in our laboratories (Catalano et al., 2004, 2010; Cavalluzzi et al., 2007). All drugs were dissolved directly in external patch solution at the desired final concentration. The patched cell was continuously exposed to a stream of control or drug-supplemented bath solution flowing out from a plastic capillary.
Experimental determination of drug physicochemical properties
The pKa and Log P values of mexiletine and its analogs were determined with a potentiometric method using Sirius GlpKa (Sirius Analytical Instrument Ltd., Forest Row, East Sussex, UK) as described previously (Catalano et al., 2010). Because the compounds showed a low water solubility, methanol was used as co-solvent (methanol–water 10–30% w/w) for the determination of pKa.
The electrostatic potential of each analog was determined as the molecular charge distribution of the corresponding methyl ether moiety [2,6-dimethylphenyl methyl ether, for Mex; 3-methoxy-2,4-dimethylphenol, for mHM; 4-methoxy-3,5-dimethylphenol, for pHM; (2-methoxy-3-methylphenyl)methanol, for HMM; (2-methoxybenzene-1,3-diyl)dimethanol, for bHMM; 3-(hydroxymethyl)-4-methoxy-5-methylphenol, for pHHMM] was investigated. Molecular models were constructed by fragments offered by SPARTAN PRO (Wavefunction, Inc., 18401 Von Karman Avenue, Suite 370, Irvine, CA 92715, USA) inner fragment library and assuming the suggested default starting geometries. The generated geometries were optimized by the molecular mechanics MMFF routine proposed by the software and then submitted to a systematic conformer distribution analysis. Conformers were classified according to the ab initio gas phase energy content calculated at the HF/3-21G* level. All conformers falling within a window of 5 kcal/mol above global minimum were retained. After removal of conformers differing for dihedral values lower than 10°, the retained conformers were submitted to HF/3-21G* geometry optimization. The most negative electrostatic potential (i.e., the highest energy of interaction with a positive charge used as a probe) value for the most stable conformer of each analog was used for structure–activity relationship considerations.
Results
Effects of mexiletine and analogs on wild-type hNav1.4 channels
To compare the effects of mexiletine and its hydroxylated analogs, block of sodium channels was evaluated by measuring the drug-induced reduction of INa elicited at −30 mV for 25 ms every 10 (0.1 Hz) or 0.1 (10 Hz) s from the HP of −120 mV. Applying this protocol in the absence of drug, there was no significant change in current amplitude for hNav1.4 channels (not shown). Typical time course of peak INa amplitude during application of 100 μM mexiletine is illustrated in Figure 1A. At the stimulation of 0.1 Hz, application of mexiletine reduced gradually INa down to a plateau. This block will be called tonic block hereafter. It is due mainly to the block of closed channels with low affinity, but depends also in part on the block of closed-state inactivated channels with high affinity (Desaphy et al., 2001, 2004). Then increasing the stimulation to 10 Hz further induced the reduction of INa to a lower level, determining the so-called use-dependent block, which depends on the high-affinity binding to inactivated channels. These effects were fully reversible. Examples of current traces recorded at steady state before (control) and in the presence of 100 μM mexiletine at 0.1 Hz then 10 Hz are illustrated in Figure 1B. The reduction of INa induced by 100 μM mexiletine was 32 and 74% at 0.1 and 10 Hz, respectively.
Figure 1
The same protocol was used to test all the exploratory compounds at various concentrations. Figure 2 shows representative current traces recorded at 0.1 and 10 Hz stimulation during application of 1 mM of each compound. Effects of mexiletine and mHM were quite similar, blocking INa by 77% at 0.1 Hz and 95% at 10 Hz. All the other compounds were less potent than mexiletine, following the rank order of potency: pHM > HMM > bHMM > pHHMM. The less potent pHHMM at 1 mM had no effect at 0.1 Hz and very little effect at 10 Hz. All the effects were fully reversible (not shown). The peak INa amplitude measured in presence of drug at 0.1 or 10 Hz was normalized with respect to control peak INa and reported as a function of drug concentration to draw concentration–response curves (Figure 3). All the curves were satisfactorily fitted with a first-order binding function, given the half-maximum inhibitory concentrations (IC50) reported in Table 2. Addition of one hydroxyl group to mexiletine had different effects on efficacy depending on the position of substituent on the aryloxy moiety, ranging from merely no effect (mHM) to an eightfold increase of IC50 value (HMM) at 0.1 Hz. The addition of two hydroxyl groups induced a dramatic reduction of efficacy by more than 45 times for bHMM and 122 times for pHHMM. The effects of chemical maneuvers were more pronounced at 10 Hz stimulation, with an increase of IC50 values ranging from 1.7-fold for mHM to 567-fold for pHHMM, compared to mexiletine. Thus the 0.1- to 10-Hz IC50 ratio decreased from 9.8 for mexiletine to 6.3 for mHM and about 2.5 for the other compounds.
Figure 2
Figure 3
Table 2
| Drug | Stimulation frequency | WT hNav1.4 | F1586C | Y1593C | ||
|---|---|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | IC50 (μM) | Imax | |||
| Mexiletine | 0.1 Hz | 246 ± 15 | 1340 ± 72 | 476 ± 36 | 1 | |
| 10 Hz | 24.4 ± 1.8 | 1089 ± 181 | 169 ± 94 | 0.84 ± 0.17 | ||
| 0.1-to-10 Hz ratio | 10.1 | 1.2 | 2.8 | |||
| mHM | 0.1 Hz | 259 ± 28 | n.d. | n.d. | n.d. | |
| 10 Hz | 40.9 ± 4.7 | n.d. | n.d. | n.d. | ||
| 0.1-to-10 Hz ratio | 6.3 | n.d. | n.d. | |||
| pHM | 0.1 Hz | 1086 ± 183 | 1132 ± 36 | 908 ± 30 | 1 | |
| 10 Hz | 394 ± 82 | 847 ± 81 | 626 ± 45 | 0.74 ± 0.02 | ||
| 0.1-to-10 Hz ratio | 2.8 | 1.3 | 1.5 | HMM | ||
| 0.1 Hz | 2028 ± 255 | 2994 ± 74 | 1638 ± 14 | 1 | ||
| 10 Hz | 790 ± 91 | 2362 ± 187 | 1452 ± 111 | 0.76 ± 0.02 | ||
| 0.1-to-10 Hz ratio | 2.6 | 1.3 | 1.1 | bHMM | ||
| 0.1 Hz | 11883 ± 2992 | 28055 ± 10520 | 12740 ± 3141 | 1 | ||
| 10 Hz | 5190 ± 1021 | 17296 ± 2571 | 10353 ± 4102 | 0.78 ± 0.11 | ||
| 0.1-to-10 Hz ratio | 2.3 | 1.6 | 1.2 | pHHMM | ||
| 0.1 Hz | 30073 ± 7059 | 67730 ± 9125 | 24782 ± 2161 | 1 | ||
| 10 Hz | 13845 ± 2516 | 21056 ± 7650 | 13482 ± 1304 | 0.81 ± 0.03 | ||
| 0.1-to-10 Hz ratio | 2.2 | 3.2 | 1.8 | |||
Half-maximum inhibitory concentration values (IC50).
These values were determined at the holding potential of −120 mV from the fit of concentration–response curves with the first-order bindingEq. 1described in Section “Materials and Methods.” The IC50 values are given together with SE of the fit. Regarding Y1593C, the Imax value was fixed to 1 at 0.1 Hz, but was variable at 10 Hz. n.d., Not determined.
The most prominent effect of hydroxylation on the physicochemical properties of compounds is a reduction of the lipophilicity at the experimental pH, expressed as the Log D determined experimentally (Table 1). The relationships of IC50 values as a function of Log P or Log D, plotted on semilogarithmic axes, were linearly correlated: The higher the lipophilicity, the more efficient was the compound (Figure 4). Notwithstanding, if the lipophilicity was the unique factor influencing compound efficacy, mHM should have appeared less efficient than it was.
Figure 4
Effects of mexiletine and derivatives on F1586C hNav1.4 mutant
Mexiletine is thought to bind the putative high-affinity receptor for LAs within the pore of sodium channels. Indeed, the non-conservative mutation of phenylalanine to cysteine in position 1586 (hNav1.4) within the 6th segment of domain IV drastically reduced mexiletine block of inactivated channels (Desaphy et al., 2009). The hydroxylated compounds, except mHM, were tested on the same sodium channel mutant F1586C. Dose–response relationships of compound effects on F1586C INa are shown in Figure 5, and the IC50 values are reported in Table 2. On FC channels, the compound pHM appeared as much potent as mexiletine, whereas HMM was 2.2 times less efficient, bHMM 15–20 times less, and pHHMM 20–50 times less. Compared to WT channels, the mutation reduced sodium channel block for all the compounds, especially at 10 Hz stimulation. Indeed use-dependence was almost abolished for mexiletine, pHM, HMM, and bHMM, the 0.1- to 10-Hz IC50 ratio being reduced to 1.2–1.6. In contrast, use-dependent block of F1586C mutant by pHHMM was maintained.
Figure 5
Effects of mexiletine and derivatives on Y1593C hNav1.4 mutant
Another amino acid potentially involved in the binding of LA-like drugs is the tyrosine in position 1593 of hNav1.4 channel (Desaphy et al., 2010). This residue was proposed to interact with the hydrophobic moiety of sodium channel blockers through a π–π interaction (Ragsdale et al., 1994), although this hypothesis has been recently challenged (Ahern et al., 2008). We tested our compounds, except mHM, on the Y1593C mutant. Representative INa current traces recorded before and after application of 1 mM mexiletine are shown in Figure 6. In contrast to WT and F1586C channels, the Y1593C-mutant INa developed significant use-dependent reduction at 10 Hz in absence of drug, ranging from 15 to 30% of control current depending on the cell. The dose–response relationships are shown in Figure 6. To take into account the use-dependent reduction of Y1593C-mutant INa in absence of drugs, an additional variable, namely Imax, was introduced in the fitting equation. The IC50 and Imax values are reported in Table 2. Regarding the IC50 values at 0.1 Hz, the Y1593C mutation was less disturbing than F1586C. Compared to WT, the IC50 of mexiletine on Y1593C INa at 0.1 Hz was twofold higher, whereas no significant change was observed for the other compounds. In contrast, the Y1593C mutation reduced dramatically use-dependent block at 10 Hz, increasing IC50 sevenfold for mexiletine, and 1.5- to 2-fold for the other compounds, except for pHHMM. Compared to WT, the 0.1- to 10-Hz IC50 ratio decreased to 2.8 for mexiletine and 1–1.5 for the other compounds. No significant effect of Y1593C mutation was observed for pHHMM.
Figure 6
Mutant cycle analysis
Being modified solely on the aryloxy moiety, the derivatives of mexiletine can be considered as simple mexiletine mutants. Thus the combined evaluation of effects of mexiletine mutants on hNav1.4 channel mutants allows the calculation of mutant cycles (Hidalgo and MacKinnon, 1995; Lipkind and Fozzard, 2005). By this way, the cross influence of one mutation on the effect of the other can be quantified using a thermodynamic cycle in order to characterize the intimate molecular interactions. Such a mutant cycle analysis is illustrated for pHM in Figure 7, and coupling energies are reported in Table 3. Since the tonic block is determined mainly by interaction of drugs with the closed channel, mutant cycle calculation using the IC50 values for tonic block likely reveals information about interaction of drugs with the low-affinity binding site (Figure 7A). In these conditions, the coupling energy (ΔG) for interaction with F1586 is 1 kcal/mol, whereas ΔG is 0.5 kcal/mol for interaction with Y1593, thereby suggesting that mexiletine is closer to F1586 than to Y1593 in the closed channel. Since OH substitutions reduce ΔG for F1586 but leave unchanged ΔG for Y1593, we may hypothesize that the former amino acid may interact directly or indirectly with the aryloxy mexiletine moiety whereas the latter may not be involved in low-affinity binding. On the other hand, mutant cycle calculation using the IC50 values for use-dependent block (10 Hz stimulation) may allow to get information about the interaction of drugs with the high-affinity binding site (Figure 7B). In this case, the coupling energy between the drug and Y1593 is 0.9 kcal/mol, whereas ΔG is ∼1.8 kcal/mol for F1586, which suggests that mexiletine may interact with both amino acids, and that interaction with F1586 is more critical to high-affinity binding.
Figure 7
Table 3
| Drug | Tonic block | Use-dependent block | ||
|---|---|---|---|---|
| F1586 | Y1593 | F1586 | Y1593 | |
| pHM | 0.97 | 0.49 | 1.79 | 0.87 |
| HMM | 0.75 | 0.50 | 1.60 | 0.78 |
| bHMM | 0.49 | 0.35 | 1.53 | 0.73 |
| pHHMM | 0.52 | 0.50 | 2.00 | 1.15 |
Coupling energy (ΔG, kcal/mol) calculated from mutant cycles, as shown in Figure 7.
Discussion
In this study, we used chemical maneuvers on mexiletine and site-directed mutagenesis on hNav1.4 voltage-gated sodium channel to get further insight in the intimate drug-channel interaction at the LA receptor. The binding affinity of LA drugs depends on the state of the channel, being higher for the open/inactivated with respect to the closed channel (Hille, 1977; Fozzard et al., 2011). Please note here that tonic block measured at the stimulation of 0.1 Hz results from the combination of low-affinity drug binding to closed channels and, to a lesser extent, high-affinity binding to closed-state inactivated channels, while use-dependent block measured at 10 Hz stimulation depends mainly on the high-affinity binding to inactivated channels (Desaphy et al., 2001, 2004).
Lipophilicity of drugs is a key determinant of sodium channel blockade
The results first confirm our former hypothesis (Desaphy et al., 2003), that the presence of two hydroxyl groups on the aryloxy moiety of LA-like drugs impedes high-affinity sodium channel blockade. We also show that the presence of a single hydroxyl group is less deleterious, depending however on the position of the OH group. The good correlation between the log D of compounds and the IC50 values, either at 0.1 or 10 Hz, suggests that compound lipophilicity is a major parameter. Because the putative LA receptor lies within the channel pore, lipophilicity might be determinant allowing the compound to reach/escape its receptor through the closed or inactivated channel. For instance, we previously showed that the drug pilsicainide, which has a negative log D, blocks channels inactivated from the closed state with very low efficiency compared to mexiletine, although affinities for inactivated channels of both drugs are comparable (Desaphy et al., 2010). Thus altering hydrophobicity of compounds can modify drug access to or escape from the binding site. A reduced hydrophobicity is expected to reduce drug access to closed channels, which will result in a reduced apparent affinity for the closed and closed-inactivated states. Such a mechanism likely contributes significantly to the effects observed in the present study. Nevertheless, such an explanation is not completely sufficient because inhibition of sodium current at 10 Hz stimulation is greatly reduced as well as use-dependence expressed as 0.1- to-10 Hz IC50 ratio, arguing against a trapping mechanism of the more hydrophilic compounds within the closed/inactivated channel (O’Leary et al., 2003). This result indicates that lipophilicity is also a critical determinant of the intimate interaction between the compound and its receptor. We thus tried to get more information by using contemporaneously mexiletine analogs and sodium channel mutants.
Effects of F1586C mutation on sodium channel blockade by mexiletine and hydroxylated derivatives
The phenylalanine in position 1586 is thought to be a key component of LA drug binding to the LA receptor (Ragsdale et al., 1994; Sheets et al., 2010). The aromaticity of this residue is required for use-dependent inhibition, while its hydrophobicity may be relevant for resting block (Li et al., 1999). By incorporating unnatural fluorinated phenylalanine analogs in place of the natural phenylalanine, it was recently demonstrated that disruption of the electrostatic potential at this position abolishes use-dependent inhibition without affecting resting block by lidocaine and mexiletine (Ahern et al., 2008; Pless et al., 2011). Since the fluorination did not affect blockade by the neutral benzocaine, the authors concluded that a π–cation interaction between phenylalanine side chain and the charged amine of drugs is a key player in high-affinity binding, at least for class Ib antiarrhythmics such as mexiletine (Pless et al., 2011). Accordingly, we observed that the mutation of F1586 by a non-aromatic cysteine nearly zeroed use-dependent inhibition by mexiletine and its derivatives, except pHHMM. In addition, we found that low-affinity block by mexiletine was also lightly affected by F1586C, because the IC50 value calculated at 0.1 Hz stimulation was 50% greater than mexiletine affinity for WT channels in the closed state (∼800 μM, Desaphy et al., 2001).
Interestingly, the inhibition of F1586C by pHM and mexiletine was comparable, indicating that the presence of one hydroxyl group in pHM mainly reduced high-affinity, use-dependent block of WT channels (as in mHM). Considering the π–cation interaction with F1586 as the main determinant of high-affinity binding, this suggests that alteration of the aryloxy moiety by introduction of the OH in mHM and pHM is able to weaken the critical interaction of the charged amine with F1586, through a mechanism resembling cooperativity between two pharmacophores. Our results suggest that this may be related to the reduction of drug lipophilicity (see Log D value in Table 1). Nevertheless, it has been recently shown that addition of a methyl group in para position of lidocaine is also able to weaken the π–cation interaction with F1586 (see supplemental data in Pless et al., 2011), suggesting that changes other than lipophilicity may be also involved. As hypothesized from previous experiments using aryloxy-substituted mexiletine analogs (Franchini et al., 2003), a possibility is that the presence of substituents on the aromatic ring may slightly modify the orientation of the molecule, thereby weakening the π–cation interaction. It is also interesting that while the overall hydrophobicity does not change between mHM and pHM, the IC50 for use-dependent block of WT channels decreases 10-fold with the OH in para position, thereby confirming that not only lipophilicity is involved. Thus, in function of its position, the substituent may modify more or less the orientation of the entire molecule within the binding site. It is possible to appreciate from Figure 8 how OH addition in meta have limited effects on aryloxy hindrance and on electrostatic distribution within the aryloxy (compared to mexiletine), whereas the OH in para concentrates more electronegativity close to the molecule extremity and induces a major steric hindrance. We may speculate that, in case of π–π stacking between the aryloxy and Y1593 side chain (see below), the OH in para position as in pHM may interact very negatively with the OH in Y1593, whereas the OH in meta position may be less unfavorable. The former negative interaction may result from steric hindrance or local electrostatic repulsion between the two OH.
Figure 8
When the strong interaction between the charged amine group and F1586 is disrupted by cysteine substitution, pHM has nearly the same activity than mexiletine even if it displays a reduced Log D. This latter observation suggests that an additional OH, as in mHM and pHM, or substitution of F1586 have no additive effect on low-affinity binding. Such an observation would be in accord with the proposed horizontal orientation of LA drugs in the closed channel, where the aromatic moiety of drugs may be exposed toward F1586 (Bruhova et al., 2008; Hanck et al., 2009; Sheets et al., 2010). Thus the alteration of one or the other may determine similar effect on low-affinity binding.
Interestingly, we noted that mHM and pHM show an electrostatic potential similar to that of mexiletine (Figure 8). In addition we calculated the value of energy coupling for tonic block calculated from mexiletine/pHM and F1586C mutants cycle, which resulted close to 1 kcal/mol (Table 3), being in the range expected for a through-space electrostatic coupling (Hidalgo and MacKinnon, 1995). Altogether, these results suggest that the aromatic moiety of the drug may interact with F1586 in the closed channel through π–π stacking thereby accounting for low-affinity binding. It should be noted however that, in contrast with this hypothesis, the variation of F1586 electrostatic potential by fluorination was shown to have no effect on low-affinity binding of lidocaine (Ahern et al., 2008). Thus an alternative hypothesis to be considered may be an indirect effect of F1586C mutation, through an allosteric mechanism, on the electrostatic coupling between the aryloxy drug moiety and another aromatic residue in the closed pore.
Regarding HMM and bHMM, a similar impairment of positive cooperativity between the aromatic and protonable drug extremities may account for the reduction of high-affinity blockade of WT channels, and use-dependent block was also nearly zeroed by the F1586C mutation. Moreover, tonic block of WT channels by HMM and bHMM was greatly reduced compared to that exerted by mexiletine. Thus the IC50 values for tonic block by HMM and bHMM were more than 2- and 10-fold higher than the specific affinity of mexiletine for closed channels, respectively (∼800 μM, Desaphy et al., 2001), indicating that these compounds significantly affect the low-affinity binding. Interestingly, we observed that the electrostatic potentials of the aryloxy moiety of HMM and bHMM were 10-kcal/mol more negative than that of mexiletine, mHM, and pHM. We may thus hypothesized that alteration of electrostatic coupling may contribute to weaken the interaction of the aromatic tail of HMM/bHMM with the low-affinity receptor. Such a hypothesis is supported by the low energy coupling value (<1 kcal/mol) for tonic block by HMM and bHMM. Most probably, other parameters, including hydrophobicity, should be however involved to explain the large difference in tonic block exerted by HMM and bHMM. In addition, we observed that both HMM and bHMM exert a lower tonic block of F1586C channel with respect to mexiletine and pHM, indicating that substituents on HMM and bHMM have additional effects to those resulting from the suppression of aromatic side chain at position 1586.
The compound pHHMM shows an electrostatic potential similar to HMM/bHMM, and tonic block was similarly reduced twofold by F1586C. Quite surprisingly, the mutation had no significant effect on pHHMM use-dependence, indicating that the drug does not bind to F1586 at all. This latter observation suggests that F1586 is no more involved in the higher-affinity binding of pHHMM.
Effects of Y1593C mutation on sodium channel blockade by mexiletine and hydroxylated derivatives
The tyrosine in position 1593 was originally individuated as an important component of the intimate drug-receptor interaction, possibility through hydrophobic interaction with the aromatic moiety of LA drugs (Ragsdale et al., 1994). This hypothesis has been somewhat challenged because Y1593 mutations can modify sodium channel gating, which rends difficult the interpretation of pharmacological results. In addition to various effects on steady-state activation and inactivation, a slowing of recovery from inactivation has been reported (O’Reilly et al., 2000; Xiao et al., 2001), which may explain the use-dependent reduction of Y1593C currents we observed in the absence of drug. Recently, the contribution of Y1593 to π–cation or π–π interactions with lidocaine has been ruled out by experiments using fluorinated amino acids (Ahern et al., 2008).
As for pilsicainide in a previous study (Desaphy et al., 2010), we observed that Y1593C reduced use-dependent sodium current blockade by mexiletine but to a lesser extent than F1586C.
The mutation may impede a direct interaction of Y1593 with the drug, or reduce the lipophilicity in proximity of the binding site. Thus considering Y1593 being involved either directly or indirectly on mexiletine binding, its mutation to C1593 may cooperatively reduce the strong interaction between the charged amine and F1586. The Y1593C mutation had a similar effect on mexiletine derivatives (except pHHMM), reducing use-dependence without altering tonic block. This observation indicates that Y1593 is not involved in the low-affinity binding, still in accord with the proposed position of LA drugs in the closed channel (Sheets et al., 2010). Tonic block and use-dependence of pHHMM was not altered by Y1593C, most probably because hydrophobicity is no more critical for a drug having a very negative Log D.
Synopsis
We made an attempt to summarize the main findings in a graphical sketch (Figure 9). Regarding the low-affinity binding to closed channels (Figure 9A), our results suggest that the aryloxy moiety of drugs may interact either directly or indirectly with F1586 and suggest that electrostatic coupling is critical in drug binding. Regarding the high-affinity binding to inactivated channels (Figure 9B), addition of OH groups on the aryloxy moiety cooperatively reduced the interaction of drug charged amine group with F1586. The amplitude of this effect depends mainly on lipophilicity and also on the position of the OH substituent.
Figure 9
Statements
Acknowledgments
The financial support from Telethon, Italy (non-for-profit, grants GGP04140 and GGP10101) and Association Française contre les Myopathies (non-for-profit, grant #15020) to Diana Conte Camerino is gratefully acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AhernC. A.EastwoodA. L.DoughertyD. A.HornR. (2008). Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ. Res.102, 86–94.10.1161/CIRCRESAHA.107.160663
2
BruhovaI.TikhonovD. B.ZhorovB. S. (2008). Access and binding of local anesthetics in the closed sodium channel. Mol. Pharmacol.74, 1033–1045.10.1124/mol.108.049759
3
CatalanoA.CarocciA.CavalluzziM. M.Di MolaA.LentiniG.LoveceA.DipalmaA.CostanzaT.DesaphyJ.-F.Conte CamerinoD.FranchiniC. (2010). Hydroxylated analogs of mexiletine as tools for structural-requirements investigation of the sodium channel blocking activity. Arch. Pharm. (Weinheim)343, 325–332.10.1002/ardp.200900218
4
CatalanoA.CarocciA.FracchiollaG.FranchiniC.LentiniG.TortorellaV.De LucaA.De BellisM.DesaphyJ.-F.Conte CamerinoD. (2004). Stereospecific synthesis of “para-hydroxymexiletine” and sodium channel blocking activity evaluation. Chirality16, 72–78.10.1002/chir.10307
5
CavalluzziM. M.CatalanoA.BrunoC.LoveceA.CarocciA.CorboF.FranchiniC.LentiniG.TortorellaV. (2007). Synthesis of (R)-, (S)-, and (RS)-hydroxymethylmexiletine, one of the major metabolites of mexiletine. Tetrahedron Asymmetry18, 2409–2417.10.1016/j.tetasy.2007.10.002
6
ChallapalliV.Tremont-LukatsI. W.McNicolE. D.LauJ.CarrD. B. (2005). Systemic administration of local anesthetic agents to relieve neuropathic pain. Cochrane Database Syst. Rev.4, CD003345.
7
Conte CamerinoD.TricaricoD.DesaphyJ.-F. (2007). Ion channel pharmacology. Neurotherapeutics4, 184–198.10.1016/j.nurt.2007.01.013
8
De BellisM.De LucaA.RanaF.CavalluzziM. M.CatalanoA.LentiniG.FranchiniC.TortorellaV.Conte CamerinoD. (2006). Evaluation of the pharmacological activity of the major mexiletine metabolites on skeletal muscle sodium currents. Br. J. Pharmacol.149, 300–310.10.1038/sj.bjp.0706867
9
De LucaA.NatuzziF.DesaphyJ.-F.LoniG.LentiniG.FranchiniC.TortorellaV.Conte CamerinoD. (2000). Molecular determinants of mexiletine structure for potent and use-dependent block of skeletal muscle sodium channels. Mol. Pharmacol.57, 268–277.
10
De LucaA.PiernoS.LiantonioA.DesaphyJ.-F.NatuzziF.DidonnaM. P.FerranniniE.JockuschH.FranchiniC.LentiniG.CorboF.TortorellaV.Conte CamerinoD. (2004). New potent mexiletine and tocainide analogues evaluated in vivo and in vitro as antimyotonic agents on myotonic ADR mouse. Neuromuscul. Disord.14, 405–416.10.1016/j.nmd.2004.04.006
11
De LucaA.TalonS.De BellisM.DesaphyJ.-F.FranchiniC.LentiniG.CatalanoA.CorboF.TortorellaV.Conte CamerinoD. (2003). Inhibition of skeletal muscle sodium currents by mexiletine analogues: specific hydrophobic interactions rather than lipophilia per se account for drug therapeutic profile. Naunyn Schmiedebergs Arch. Pharmacol.367, 318–327.10.1007/s00210-002-0669-0
12
DesaphyJ.-F.Conte CamerinoD.FranchiniC.LentiniG.TortorellaV.De LucaA. (1999). Increased hindrance on the chiral carbon atom of mexiletine enhances the block of rat skeletal muscle Na+ channels in a model of myotonia induced by ATX. Br. J. Pharmacol.128, 1165–1174.10.1038/sj.bjp.0702901
13
DesaphyJ.-F.De LucaA.DidonnaM. P.GeorgeA. L.Jr.Conte CamerinoD. (2004). Different flecainide sensitivity of hNav1.4 channels and myotonic mutants explained by state-dependent block. J. Physiol.554, 321–334.10.1113/jphysiol.2003.046995
14
DesaphyJ.-F.De LucaA.TortorellaP.De VitoD.GeorgeA. L.Jr.Conte CamerinoD. (2001). Gating of myotonic Na channel mutants defines the response to mexiletine and a potent derivative. Neurology57, 1849–1857.
15
DesaphyJ.-F.DipalmaA.CostanzaT.BrunoC.LentiniG.FranchiniC.GeorgeA. L.Jr.Conte CamerinoD. (2010). Molecular determinants of state-dependent block of voltage-gated sodium channels by pilsicainide. Br. J. Pharmacol.160, 1521–1533.
16
DesaphyJ.-F.DipalmaA.De BellisM.CostanzaT.GaudiosoC.DelmasP.GeorgeA. L.Jr.Conte CamerinoD. (2009). Involvement of voltage-gated sodium channels blockade in the analgesic effects of orphenadrine. Pain142, 225–235.10.1016/j.pain.2009.01.010
17
DesaphyJ.-F.PiernoS.De LucaA.DidonnaP.Conte CamerinoD. (2003). Different ability of clenbuterol and salbutamol to block sodium channels predicts their therapeutic use in muscle excitability disorders. Mol. Pharmacol.63, 659–670.10.1124/mol.63.3.659
18
FozzardH. A.SheetsM. F.HanckD. A. (2011). The sodium channel as a target of local anesthetic drugs. Front. Pharmacol.2:68.10.3389/fphar.2011.00068
19
FranchiniC.CarocciA.CatalanoA.CavalluzziM. M.CorboF.LentiniG.ScilimatiA.TortorellaP.Conte CamerinoD.De LucaA. (2003). Optically active mexiletine analogues as stereoselective blockers of voltage-gated Na+ channels. J. Med. Chem.46, 5238–5248.10.1021/jm030865y
20
HanckD. A.NikitinaE.McNultyM. M.FozzardH. A.LipkindG. M.SheetsM. F. (2009). Using lidocaine and benzocaine to link sodium channel molecular conformations to state-dependent antiarrhythmic drug affinity. Circ. Res.105, 492–499.10.1161/CIRCRESAHA.109.198572
21
HidalgoP.MacKinnonR. (1995). Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science268, 307–310.10.1126/science.7716527
22
HilleB. (1977). Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol.69, 497–515.10.1085/jgp.69.4.497
23
LiH. L.GalueA.MeadowsL.RagsdaleD. S. (1999). A molecular basis for the different local anesthetic affinities of resting versus open and inactivated states of the sodium channel. Mol. Pharmacol.55, 134–141.
24
LipkindG. M.FozzardH. A. (2005). Molecular modelling of local anesthetic drug binding by voltage-gated sodium channels. Mol. Pharmacol.68, 1611–1622.
25
McNultyM. M.EdgertonG. B.ShahR. D.HanckD. A.FozzardH. A.LipkindG. M. (2007). Charge at the lidocaine binding site residues Phe-1759 affects permeation in human cardiac voltage-gated sodium channels. J. Physiol.581, 741–755.10.1113/jphysiol.2007.130161
26
NauC.WangS.-H.StrichartzG. R.WangG. K. (1999). Point mutations at N434 in D1-S6 of m1 Na+ channels modulate binding affinity and stereoselectivity of local anesthetic enantiomers. Mol. Pharmacol.56, 404–413.
27
O’LearyM. E.ChahineM. (2002). Cocaine binds to a common site on open and inactivated human heart (Nav1.5) sodium channels. J. Physiol.541, 701–716.10.1113/jphysiol.2001.016139
28
O’LearyM. E.DigregorioM.ChahineM. (2003). Closing and inactivation potentiate the cocaethylene inhibition of cardiac sodium channels by distinct mechanisms. Mol. Pharmacol.64, 1575–1585.10.1124/mol.64.6.1575
29
O’ReillyJ. P.WangS.-Y.WangG. K. (2000). A point mutation in domain 4-segment 6 of the skeletal muscle sodium channel produces an atypical inactivation state. Biophys. J.78, 773–784.10.1016/S0006-3495(00)76635-5
30
PlessS. A.GalpinJ. D.FrankelA.AhernC. A. (2011). Molecular basis for class Ib anti-arrhythmic inhibition of cardiac sodium channels. Nat. Commun.2, 351.10.1038/ncomms1351
31
RagsdaleD. S.McPheeJ. C.ScheuerT.CatterallW. A. (1994). Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science265, 1724–1728.10.1126/science.8085162
32
RagsdaleD. S.McPheeJ. C.ScheuerT.CatterallW. A. (1996). Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated sodium channels. Proc. Natl. Acad. Sci. U.S.A.93, 9270–9275.10.1073/pnas.93.17.9270
33
SheetsM. F.FozzardH. A.LipkindG. M.HanckD. A. (2010). Sodium channel molecular conformations and antiarrhythmic drug affinity. Trends Cardiovasc. Med.20, 16–21.10.1016/j.tcm.2010.03.002
34
SunamiA.GlaaseJ. W.FozzardH. A. (2001). Structural and gating changes of the sodium channel induced by mutation of a residue in the upper third of IVS6, creating an external access path for local anesthetics. Mol. Pharmacol.59, 684–691.
35
WangS. Y.NauC.WangG. K. (2000). Residues in Na+ channel D3-S6 segment modulate both batrachotoxin and local anesthetic affinities. Biophys. J.79, 1379–1387.10.1016/S0006-3495(00)76390-9
36
WrightS. N.WangS. Y.WangG. K. (1998). Lysine point mutations in Na+ channel D4-S6 reduce inactivated channel block by local anesthetics. Mol. Pharmacol.54, 733–739.
37
XiaoY.-F.KeQ.WangS.-Y.YangY.WangG. K.MorganJ. P.LeafA. (2001). Point mutations in α-subunit of human cardiac Na+ channels alter Na+ current kinetics. Biochem. Biophys. Res. Commun.281, 45–52.10.1006/bbrc.2001.4309
38
Yarov-YarovoyV.BrownJ.SharpE. M.ClareJ. J.ScheuerT.CatterallW. A. (2001). Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na+ channel α subunit. J. Biol. Chem.276, 20–27.10.1074/jbc.M006992200
39
Yarov-YarovoyV.McPheeJ. C.IdsvoogD.PateC.ScheuerT.CatterallW. A. (2002). Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel α subunit in voltage-dependent gating and drug block. J. Biol. Chem.277, 35393–35401.10.1074/jbc.M206126200
Summary
Keywords
sodium channel, hNav1.4, mexiletine analogs, local anesthetic receptor
Citation
Desaphy J-F, Dipalma A, Costanza T, Carbonara R, Dinardo MM, Catalano A, Carocci A, Lentini G, Franchini C and Camerino DC (2012) Molecular Insights into the Local Anesthetic Receptor within Voltage-Gated Sodium Channels Using Hydroxylated Analogs of Mexiletine. Front. Pharmacol. 3:17. doi: 10.3389/fphar.2012.00017
Received
26 October 2011
Accepted
30 January 2012
Published
15 February 2012
Volume
3 - 2012
Edited by
Mohamed Chahine, Laval University, Canada
Reviewed by
Georges Christé, INSERM, France; Michael E. O’Leary, Jefferson University, USA
Copyright
© 2012 Desaphy, Dipalma, Costanza, Carbonara, Dinardo, Catalano, Carocci, Lentini, Franchini and Camerino.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jean-François Desaphy, Sezione di Farmacologia, Dipartimento Farmaco-Biologico, Facoltà di Farmacia, Università degli Studi di Bari “Aldo Moro,” via Orabona 4 – Campus, I-70125 Bari, Italy. e-mail: jfdesaphy@farmbiol.uniba.it
This article was submitted to Frontiers in Pharmacology of Ion Channels and Channelopathies, a specialty of Frontiers in Pharmacology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.