 David E. Stec*
David E. Stec* Peter A. Hosick and Joey P. Granger
Peter A. Hosick and Joey P. Granger
- Department of Physiology and Biophysics, Center for Excellence in Cardiovascular – Renal Research, University of Mississippi Medical Center, Jackson, MS, USA
Bilirubin is generated from the breakdown of heme by heme oxygenase and the reduction of biliverdin by the enzyme biliverdin reductase. Several large population studies have reported a significant inverse correlation between plasma bilirubin levels and the incidence of cardiovascular disease. Protection from cardiovascular disease is also observed in patients with Gilbert’s syndrome which is a disease characterized by mutations in hepatic UGT1A1, the enzyme responsible for the conjugation of bilirubin into the bile. Despite the strong correlation between plasma bilirubin levels and the protection from cardiovascular disease, the mechanism by which increases in plasma bilirubin acts to protect against cardiovascular disease is unknown. Since the chronic antihypertensive actions of bilirubin are likely due to its renal actions, the effects of moderate increases in plasma bilirubin on renal hemodynamics as well as bilirubin’s potential effects on renal tubule function will be discussed in this review. Mechanisms of action as well as the potential for antihypertensive therapies targeting moderate increases in plasma bilirubin levels will also be highlighted.
Introduction
Bilirubin is derived in the plasma from the breakdown of red blood cells in the spleen. In the spleen, heme oxygenase (HO) enzymes catabolize heme released from the breakdown of red blood cells to carbon monoxide (CO) and biliverdin. Biliverdin is then reduced to bilirubin by the enzyme biliverdin reductase (BVR) and released into the blood stream. In the blood, most of the bilirubin is bound to albumin (δ bilirubin) where it travels to the liver and is conjugated by hepatic UDP-glucuronosyltransferase (UGT1A1) enzymes. Most of the conjugated bilirubin then exits the liver through the biliary ducts into the bile where it is eliminated through the digestive system. Gilbert’s syndrome is a disease characterized by elevations in the levels of unconjugated bilirubin in the plasma due to mutations in hepatic UGT1A1 which decrease conjugation of bilirubin in the liver. Levels of unconjugated bilirubin in the plasma can also be increased by drugs which compete with bilirubin for conjugation by UGT1A1. Analysis of patients in the Framingham heart study concluded that mutations in UGT1A1 which resulted in moderate increases in plasma bilirubin (twofold) were associated with a decrease risk for the development of cardiovascular disease (Lin et al., 2006). This is in agreement with previous human population studies which found that moderated increases in plasma bilirubin were protective against atherosclerosis, coronary heart disease, metabolic syndrome, diabetic nephropathy, and end stage renal disease (Hopkins et al., 1996; Novotny and Vitek, 2003; Chin et al., 2009a; Han et al., 2010; Wu et al., 2011).
It is well established that the kidney’s play a central role the regulation of arterial pressure. Thus, the chronic antihypertensive actions of bilirubin are likely due to its renal actions. As mentioned above, bilirubin is derived from the breakdown of heme by HO enzymes. Our knowledge of the effects of bilirubin in the kidney is mainly derived from studies in which the levels of HO enzymes have been altered. However, the results obtained from these studies are complicated by the fact that CO and bilirubin are generated in equimolar amounts by HO enzymes. This makes it impossible to separate out the contribution of each metabolite to any observed response to alterations in HO activity. Given this fact, we will mainly focus this review on the effects of increases in plasma bilirubin levels on the regulation of renal vascular and tubular function as well as the effects of plasma bilirubin on blood pressure. Studies utilizing alterations of HO enzymes will be discussed in areas in which data from models of hyperbilirubinemia are not available and which point to a potential role for bilirubin.
Bilirubin and the Regulation of Renal Hemodynamics
Alterations in renal hemodynamics underlie the development and maintenance of hypertension in several forms including angiotensin II (Ang II) dependent hypertension (Granger and Schnackenberg, 2000). Marked elevations in Ang II levels as observed with models of chronic Ang II infusion decrease glomerular filtration rate (GFR) due to effects on the afferent arteriole. The Gunn rat, a rodent model of severe (<20 fold) hyperbilirubinemia due to loss of hepatic UGT1A1, exhibits a significant 50% decrease in the blood pressure response to Ang II infusion. The Gunn rat also does not exhibit a significant decline in GFR in response to chronic, 4 week infusion Ang II infusion as compared to control rats (Pflueger et al., 2005). In a mouse model of moderate hyperbilirubinemia due to blockade of hepatic UGT1A1 with either indinavir, an antiretroviral protease inhibitor, or specific antisense morpholino oligonucleotides, chronic Ang II infusion did not result in a decrease in GFR (Figure 1A; Vera and Stec, 2010). Moderate hyperbilirubinemia also preserves renal blood flow and normalizes renal vascular resistance in Ang II treated mice (Figures 1B,C; Vera and Stec, 2010).
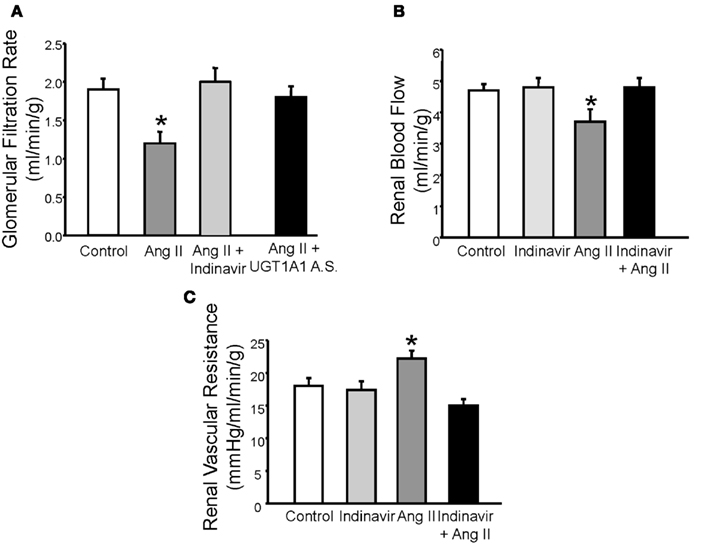
Figure 1. (A) Effects of moderate hyperbilirubinemia with indinavir or UGT1A1 antisense morpholino (UGT1A1 A.S.) on glomerular filtration rate (GFR) in Ang II infused mice. Ang II infusion resulted in a significant decrease in GFR which was normalized by moderate hyperbilirubinemia. (B) Effect of moderate hyperbilirubinemia with indinavir on renal blood flow. Ang II infusion resulted in a significant decline in renal blood flow which was normalized by moderate hyperbilirubinemia. (C) Effect of moderate hyperbilirubinemia on renal vascular resistance. Renal vascular resistance was significantly increased by Ang II infusion and was normalized by moderate hyperbilirubinemia. * = P < 0.05 as compared to control. Figure modified from Vera and Stec (2010).
Renal afferent arteriole regulation of GFR is accomplished by two major mechanisms; the myogenic response and tubuloglomerular feedback (TGF). The myogenic response is an endogenous response of blood vessels to constrict when stretched by increases in translumenal pressure. The myogenic constriction of the afferent arteriole is believed to help protect the glomerulus from increases in perfusion pressure (Bidani et al., 2009). The role of bilirubin in modifying myogenic vasoconstriction of the afferent arteriole has not been investigated. TGF is the second mechanism by which vascular tone of the afferent arteriole is regulated. TGF response utilizes sodium delivery to macula densa cells of the kidney in order to regulate the tone of the afferent arteriole. The macula densa cells convey a signal to the afferent arteriole to constrict in response to increases in sodium delivery to the macula densa (signal of increase in GFR) and to dilate in response to decreases in sodium delivery to the macula densa (signal of decreased GFR). Recent studies have demonstrated an important role for bilirubin (via conversion from biliverdin) in protecting against excessive TGF mediated constriction of the afferent arteriole (Ren et al., 2008; Wang et al., 2011). The mechanism by which bilirubin can protect against TGF mediated vasoconstriction is not known. It is not clear if moderate increases in plasma bilirubin can have similar effects on TGF mediated vasoconstriction or whether this response is dependent on HO generated bilirubin in the macula densa cells or in the vascular smooth muscle cells of the afferent arteriole.
Current evidence suggests that increases in bilirubin (via conversion from biliverdin) protect against excessive TGF mediated vasoconstriction and can attenuate constriction to high levels of vasoconstrictors such as Ang II; however, the mechanism by which bilirubin can maintain vascular tone is not fully understood. Bilirubin is one of the most potent antioxidants in the body (Stocker et al., 1987; Stocker and Peterhans, 1989; Bulmer et al., 2008). Given that vasoconstrictors such as Ang II can stimulate superoxide anion production in the vasculature, one potential mechanism for the protective role of moderate hyperbilirubinemia in the renal vasculature is quenching of reactive oxygen species (ROS) such as superoxide (Griendling et al., 1994; Rajagopalan et al., 1996; Hanna et al., 2002). This hypothesis is supported by the observation that Ang II mediated superoxide production is significantly attenuated in aortic ring segments from moderately hyperbilirubinemic mice (Figure 2A; Vera et al., 2009). Bilirubin can directly scavenge superoxide but also reduces superoxide production via direct inhibition of NAD(P)H oxidases in the vasculature (Lanone et al., 2005). The reduction of superoxide production in the vasculature by bilirubin is associated with the increase in the bioavailability of nitric oxide as reflected in the nitrate/nitrite levels in the plasma of chronically infused Ang II hypertensive mice made moderately hyperbilirubinemic (Figures 2B and 3; Vera et al., 2009). Nitric oxide (NO) can react with superoxide to produce peroxynitrite which is a potent oxidant (Figure 3). It is possible that moderate increases in plasma bilirubin are able to preserve renal blood flow and maintain GFR in Ang II-dependent hypertension through increases in NO; however, the importance of increased NO to this response remains to be specifically tested.
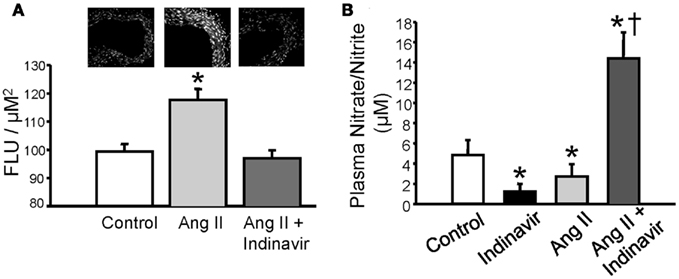
Figure 2. (A) Moderate hyperbilirubinemia decrease Ang II-dependent superoxide production in aortic tissue. Superoxide levels were determined using dihydroethidium (DHE) staining of aortic tissue segments (upper panel). Ang II treatment resulted in a significant increase in aortic superoxide production which was normalized by moderate hyperbilirubinemia. (B) Moderate hyperbilirubinemia increases plasma NO levels in Ang II infused mice. * = P < 0.05 vs. Control, † = P < 0.05 vs. Ang II. Figure modified from Vera et al. (2009).
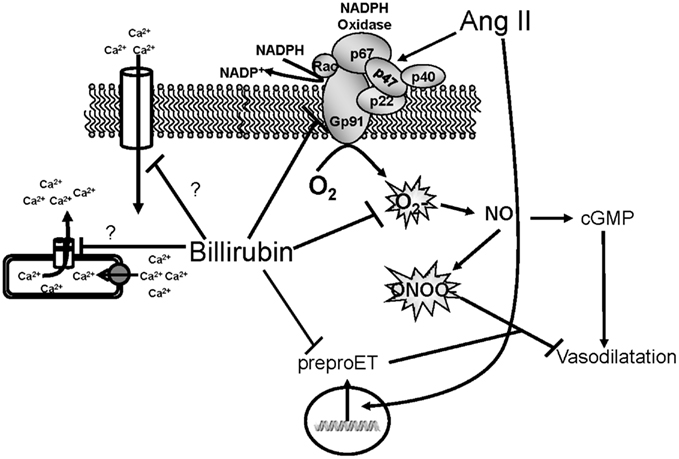
Figure 3. Schematic of vascular actions of bilirubin. Ang II acts on the NADPH oxidase to increase superoxide production. Superoxide can then react with nitric oxide (NO) to form peroxynitrite which inhibits vasodilatation. Ang II can also stimulate the expression of preproendothelin (preproET) which is a vasoconstrictor. Bilirubin can directly scavenge superoxide as well as block NADPH oxidase to increase the bioavailability of NO and promote vasodilatation. Bilirubin can also inhibit Ang II mediated increases in preproET and may also block intracellular calcium through membrane and intracellular calcium channels.
Bilirubin may also preserve renal blood flow and GFR through mechanisms independent of ROS production. For example, bilirubin can attenuate Ang II mediated increase in preproendothelin gene transcription in endothelial cells (unpublished observation). Several studies have demonstrated an important role for endothelin signaling through the endothelin A (ETA) receptor in Ang II-dependent hypertension (d’Uscio et al., 1997; Rajagopalan et al., 1997; Ballew and Fink, 2001). Thus, attenuation of Ang II mediated endothelin signaling could contribute to the preservation of renal blood flow and GFR by moderate hyperbilirubinemia. Bilirubin may also have effects on calcium handling in vascular smooth muscle cells through interactions with calcium channels or the intracellular storing and release of calcium (Figure 3). The effects of bilirubin on endothelin production and calcium handling need to be further explored to determine the importance of these potential mechanisms which protect the renal vasculature from excessive constriction in Ang II-dependent hypertension.
Bilirubin and the Regulation of Renal Tubule Function
Very little is currently known about the role of bilirubin in the regulation of renal tubule function. Sodium and water homeostasis in models of elevated plasma bilirubin such as the Gunn rat or the moderately hyperbilirubinemic mouse has not been evaluated. The effect that increased renal bilirubin via direct infusion into the kidney has on sodium excretion is not known. Likewise, the effect of blockade of bilirubin production on sodium excretion is also unknown. Several studies in rodents have demonstrated that induction of renal HO increases sodium and water excretion while inhibition of renal HO promotes sodium reabsorption (Rodriguez et al., 2003; Li et al., 2007; Jackson et al., 2011). However, the specific role of bilirubin in this response is not known. Studies in cultured renal thick ascending loop of Henle (TALH) cells have demonstrated that blockade of bilirubin production by targeting BVR results in increased Ang II mediated superoxide production and sodium reabsorption (Figure 4; Young et al., 2009). This would suggest a potential role for endogenous cellular bilirubin production in the regulation oxidative stress and sodium reabsorption in the TALH.
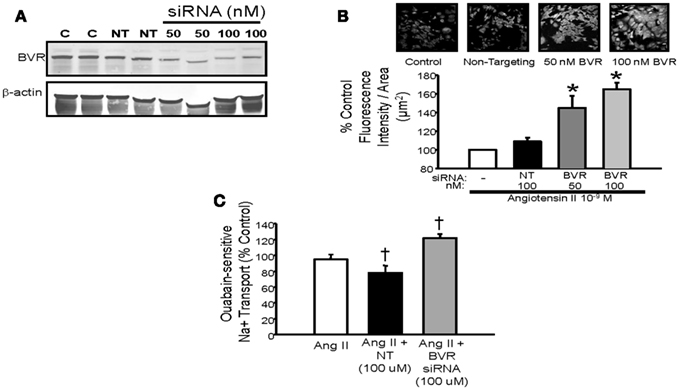
Figure 4. (A) Representative Western blot of protein lysates from mouse thick ascending loop of Henle (TALH) cells treated with biliverdin reductase (BVR) or non-targeting (NT) siRNAs. Treatment with 50 or 100 nM BVR siRNA resulted in significant decrease in BVR protein. (B) Top panel – representative images of dihydroethidium (DHE) staining of Ang II treated TALH cells receiving BVR siRNA. (B) Bottom panel – treatment with 50 or 100 nM BVR siRNA resulted in significant increase in DHE staining. (C) Effect of BVR siRNA treatment on ouabain-sensitive sodium transport in cultured mouse TALH cells. BVR siRNA significantly increase Ang II-mediated sodium transport. *P < 0.05 as compared control. † P < 0.05 vs. Ang II. Figure modified from Young et al. (2009).
Bilirubin may affect tubular function through similar mechanism as proposed in the renal vasculature (Figure 3). Several studies have reported an important role for superoxide in stimulating sodium reabsorption in the TALH (Ortiz and Garvin, 2002a; Juncos and Garvin, 2005). Superoxide can also quench the levels of nitric oxide to further increase sodium reabsorption in this nephron segment (Ortiz and Garvin, 2002b). Thus, decreases in bilirubin production via targeting of biliverdin reductase could increase sodium reabsorption in the TALH via increased superoxide production. Whether decreases in BVR activity could result in similar increases in sodium reabsorption in the TALH in vitro is not known. The importance of cellular bilirubin generation to the natriuresis exhibited with induction of HO-1 in the kidney is also an area which is unclear. The role of cellular bilirubin generation will require the development of new experimental models in which the activity of BVR is reduced genetically either through chronic knockdown with siRNAs in vivo or the creation of specific knockout mice or rats.
Bilirubin and the Regulation of Blood Pressure
The blood pressure lowering actions of systemic HO-1 induction have been demonstrated in several rodent models of experimental hypertension (Sacerdoti et al., 1989; Sabaawy et al., 2001; Botros et al., 2005; Vera et al., 2007; George et al., 2011). Additional studies have also revealed that alterations in renal heme oxygenase activity alone can have profound effects on the development of hypertension (Li et al., 2007; Vera et al., 2008). While the effects of alterations of HO activity on blood pressure have been established, the specific role for bilirubin generation in this response has remained elusive. As mentioned above, it is impossible to separate the generation of bilirubin from that of CO by HO enzymes so the relative importance of one vs. the other metabolite in the actions of HO on blood pressure remains unresolved. Several large scale human population studies have demonstrated an inverse relationship between plasma bilirubin levels and the incidence of hypertension (Chin et al., 2009b). As stated above, alterations in plasma bilirubin levels are also associated with other cardiovascular risk factors such as atherosclerosis, diabetic nephropathy, and renal disease all of which could contribute to the development of hypertension.
Although it is difficult to determine the specific role of bilirubin in the blood pressure response to alterations in HO activity, several studies have investigated the specific role of plasma bilirubin levels in the development of hypertension using experimental animal models in which confounding factors such as atherosclerosis, diabetes, and renal injury can be carefully controlled. As mentioned above, the Gunn rat is a model of severe hyperbilirubinemia characterized by very high levels of plasma bilirubin resulting from decreased hepatic conjugation due to loss of the UGT1A1 gene (van et al., 1968; van der Wegen et al., 2006). The Gunn rat has been demonstrated to be resistant to the development of both Ang II and deoxycorticosterone acetate (DOCA)-salt dependent hypertension (Pflueger et al., 2005; Nath et al., 2007). The resistance to the development of hypertension in the Gunn rat was associated with preservation of NO dependent vasorelaxation which is altered in both of these forms of hypertension. Both of these forms of hypertension are also characterized by increases in superoxide anion production which is significantly attenuated in the Gunn rat. The results obtained in the Gunn rat clearly demonstrate the difficulty in obtaining a hypertensive response to known pressors; however, the level of bilirubin in the plasma of these rats is significantly higher than those reported to be beneficial in humans (Hopkins et al., 1996; Lin et al., 2006).
In order to determine if physiologic increases in bilirubin (∼twofold) could have similar effects on the development of hypertension, our lab created a mouse model of moderate hyperbilirubinemia achieved through targeting of hepatic UGT1A1 either with a drug which competes with bilirubin for conjugation (indinavir) or antisense oligonucleotides directed against UGT1A1. We were able to find concentrations of these agents which resulted in two to threefold increases in the levels of plasma unconjugated bilirubin (Figure 5A) which are in contrast with the greater than 20 fold elevations in unconjugated observed in the Gunn rat (van der Wegen et al., 2006). Using this mouse model, we demonstrated that moderate hyperbilirubinemia achieved with indinavir prevented the development of Ang II mediated hypertension (Figure 5B; Vera et al., 2009). Similar effects on the development of Ang II hypertension were also observed in mice which received direct intravenous infusion of bilirubin which increased the levels of unconjugated bilirubin to levels observed in indinavir treated mice (Vera et al., 2009). As mentioned above, moderate hyperbilirubinemia in this mouse model was also associated with increases in renal blood flow and GFR as well as decreases in renal vascular resistance and vascular superoxide production. These results indicate that selective targeting of hepatic UGT1A1 to increase plasma unconjugated bilirubin levels by two to threefold may be a novel antihypertensive approach. In another study, intraperitoneal injection of bilirubin was found to significantly decrease Ang II mediated proteinuria by 60% while having no significant effect on Ang II-dependent hypertension (LeBlanc et al., 2010). The results of this study suggest that alterations in bilirubin may have beneficial effects on the kidney independent of changes in blood pressure. However, since no measurements of plasma or renal bilirubin levels were made in the treated animals; it is difficult to determine whether the bilirubin treatment protocol was sufficient to elevate the levels of unconjugated bilirubin in the plasma enough to lower blood pressure in this model. Nonetheless, this study indicates that bilirubin may have renoprotective effects in the absence of any changes in blood pressure.
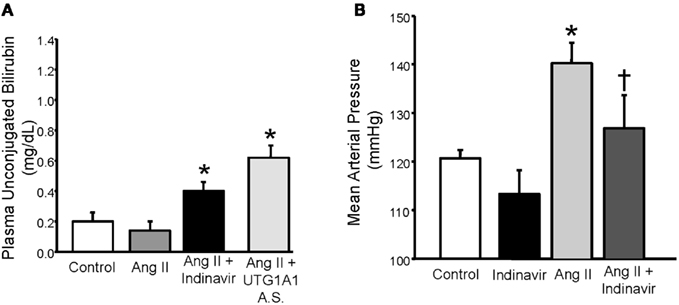
Figure 5. (A) Effect of indinavir or UGT1A1 antisense morpholino oligonucleotides on plasma unconjugated bilirubin levels in Ang II treated mice. (B) Effect of moderate hyperbilirubinemia with indinavir treatment on blood pressure in Ang II infused mice. Mice were made moderately hyperbilirubinemic by treatment with Indinavir (500 mg/kg/day, oral gavage) starting 3 days before the implantation of minipumps that delivered Ang II at 1 μg/kg/min and continuing throughout the 12 day infusion. Blood pressure was measured in conscious freely moving mice by fluid filled catheters. Ang II infusion increased blood pressure and this increase was significantly attenuated by moderate hyperbilirubinemia. *P < 0.05 as compared control. † P < 0.05 vs. Ang II. Figure modified from Vera et al. (2009).
Future Directions and Perspectives
Bilirubin either derived intracellularly by generation through actions of HO or in the plasma regulated by the activity of hepatic UGT1A1 has been demonstrated to play an important role in the regulation of blood pressure as well as renal vascular and tubular function. The challenge in the future is to determine the mechanism by which the levels of bilirubin in both the cell and plasma regulate renal vascular and tubular function in the hopes of creating new targets for therapies. The understanding of cellular bilirubin generation in the regulation of renal function will rely on targeting BVR in the kidney either through siRNA mediated viral approaches or the creation of genetically modified mice or rats. Interestingly, a recent human population study correlated a loss of function mutation in the BVR-A gene with increases in blood pressure suggesting a potential role for this enzyme in the regulation of blood pressure (Lin et al., 2011).
The manipulation of plasma levels of bilirubin is an area which could be further explored as a potential therapeutic approach for the treatment of hypertension. Targeting the conjugation of bilirubin in the liver via UGT1A1 has the potential to be one mechanism to increase plasma bilirubin levels; however, given the important role of UGT1A1 in the elimination of drugs from the body other potential targets in the handling of bilirubin by the liver may need to be developed. For example, modification of proteins responsible for the transport of bilirubin into and out of hepatocytes may be another potential target to increase plasma levels of unconjugated bilirubin for therapeutic purposes. Before targeting of hepatic bilirubin metabolism can be considered for a therapeutic approach to lower blood pressure and improve renal vascular function, the mechanism by which increases in plasma bilirubin levels act to lower blood pressure and improve renal vascular function need to be identified. Determination of these mechanisms may take several years to develop. It is possible that bilirubin may act as more than just an antioxidant to elicit its effects on renal hemodynamics and blood pressure. This possibility also needs to be further tested before targeting of hepatic bilirubin metabolism can move forward as a potential therapeutic option for hypertension. What is clear is that bilirubin has moved beyond a simple component of the plasma responsible for jaundice, to a complex molecule with several beneficial properties that could be exploited for treatment of hypertension and renal disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge grant support from the National Heart, Lung, and Blood Institute 1RO1HL088421 (David E. Stec), 1T32HL105324-01 (Peter A. Hosick), 1R01HL108618-01 (Joey P. Granger), and PO1HL-51971.
References
Ballew, J. R., and Fink, G. D. (2001). Role of ET(A) receptors in experimental ANG II-induced hypertension in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R150–R154.
Bidani, A. K., Griffin, K. A., Williamson, G., Wang, X., and Loutzenhiser, R. (2009). Protective importance of the myogenic response in the renal circulation. Hypertension 54, 393–398.
Botros, F. T., Schwartzman, M. L., Stier, C. T. Jr., Goodman, A. I., and Abraham, N. G. (2005). Increase in heme oxygenase-1 levels ameliorates renovascular hypertension. Kidney Int. 68, 2745–2755.
Bulmer, A. C., Blanchfield, J. T., Toth, I., Fassett, R. G., and Coombes, J. S. (2008). Improved resistance to serum oxidation in Gilbert’s syndrome: a mechanism for cardiovascular protection. Atherosclerosis 199, 390–396.
Chin, H. J., Cho, H. J., Lee, T. W., Na, K. Y., Oh, K. H., Joo, K. W., Yoon, H. J., Kim, Y. S., Ahn, C., Han, J. S., Kim, S., Jeon, E. S., Jin, D. C., Kim, Y. L., Park, S. H., Kim, C. D., Song, Y. R., Kim, S. G., Kim, Y. G., Lee, J. E., Oh, Y. K., Lim, C. S., Lee, S. K., Chae, D. W., Cho, W. Y., Kim, H. K., and Jo, S. K. (2009a). The mildly elevated serum bilirubin level is negatively associated with the incidence of end stage renal disease in patients with IgA nephropathy. J. Korean Med. Sci. 24(Suppl.), S22–S29.
Chin, H. J., Song, Y. R., Kim, H. S., Park, M., Yoon, H. J., Na, K. Y., Kim, Y., Chae, D. W., and Kim, S. (2009b). The bilirubin level is negatively correlated with the incidence of hypertension in normotensive Korean population. J. Korean Med. Sci. 24(Suppl.), S50–S56.
d’Uscio, L. V., Moreau, P., Shaw, S., Takase, H., Barton, M., and Luscher, T. F. (1997). Effects of chronic ETA-receptor blockade in angiotensin II-induced hypertension. Hypertension 29, 435–441.
George, E. M., Cockrell, K., Aranay, M., Csongradi, E., Stec, D. E., and Granger, J. P. (2011). Induction of heme oxygenase 1 attenuates placental ischemia-induced hypertension. Hypertension 57, 941–948.
Granger, J. P., and Schnackenberg, C. G. (2000). Renal mechanisms of angiotensin II-induced hypertension Semin. Nephrology 20, 417–425.
Griendling, K. K., Minieri, C. A., Ollerenshaw, J. D., and Alexander, R. W. (1994). Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 74, 1141–1148.
Han, S. S., Na, K. Y., Chae, D. W., Kim, Y. S., Kim, S., and Chin, H. J. (2010). High serum bilirubin is associated with the reduced risk of diabetes mellitus and diabetic nephropathy Tohoku. J. Exp. Med. 221, 133–140.
Hanna, I. R., Taniyama, Y., Szocs, K., Rocic, P., and Griendling, K. K. (2002). NAD(P)H oxidase-derived reactive oxygen species as mediators of angiotensin II signaling. Antioxid. Redox Signal. 4, 899–914.
Hopkins, P. N., Wu, L. L., Hunt, S. C., James, B. C., Vincent, G. M., and Williams, R. R. (1996). Higher serum bilirubin is associated with decreased risk for early familial coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 16, 250–255.
Jackson, K. E., Jackson, D. W., Quadri, S., Reitzell, M. J., and Navar, L. G. (2011). Inhibition of heme oxygenase augments tubular sodium reabsorption. Am. J. Physiol. Renal Physiol. 300, F941–F946.
Juncos, R., and Garvin, J. L. (2005). Superoxide enhances Na-K-2Cl cotransporter activity in the thick ascending limb. Am. J. Physiol. Renal Physiol. 288, F982–F987.
Lanone, S., Bloc, S., Foresti, R., Almolki, A., Taille, C., Callebert, J., Conti, M., Goven, D., Aubier, M., Dureuil, B., El Benna, J., Motterlini, R., and Boczkowski, J. (2005). Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J. 19, 1890–1892.
LeBlanc, R. M., Navar, L. G., and Botros, F. T. (2010). Bilirubin exerts renoprotective effects in angiotensin II-hypertension. Am. J. Med. Sci. 340, 144–146.
Li, N., Yi, F., Dos Santos, E. A., Donley, D. K., and Li, P. L. (2007). Role of renal medullary heme oxygenase in the regulation of pressure natriuresis and arterial blood pressure. Hypertension 49, 148–154.
Lin, J. P., O’Donnell, C. J., Schwaiger, J. P., Cupples, L. A., Lingenhel, A., Hunt, S. C., Yang, S., and Kronenberg, F. (2006). Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham heart study circulation 114, 1476–1481.
Lin, R., Wang, X., Zhou, W., Fu, W., Wang, Y., Huang, W., and Jin, L. (2011). Association of a BLVRA common polymorphism with essential hypertension and blood pressure in Kazaks. Clin. Exp. Hypertens. 33, 294–298.
Nath, K. A., d’Uscio, L. V., Juncos, J. P., Croatt, A. J., Manriquez, M. C., Pittock, S. T., and Katusic, Z. S. (2007). An analysis of the DOCA-salt model of hypertension in HO-1-/- mice and the Gunn rat. Am. J. Physiol. Heart Circ. Physiol. 293, H333–H342.
Novotny, L., and Vitek, L. (2003). Inverse relationship between serum bilirubin and atherosclerosis in men: a meta-analysis of published studies. Exp. Biol. Med. (Maywood) 228, 568–571.
Ortiz, P. A., and Garvin, J. L. (2002a). Superoxide stimulates NaCl absorption by the thick ascending limb. Am. J. Physiol. Renal Physiol. 283, F957–F962.
Ortiz, P. A., and Garvin, J. L. (2002b). Interaction of O(2)(−) and NO in the thick ascending limb. Hypertension 39, 591–596.
Pflueger, A., Croatt, A. J., Peterson, T. E., Smith, L. A., d’Uscio, L. V., Katusic, Z. S., and Nath, K. A. (2005). The hyperbilirubinemic Gunn rat is resistant to the pressor effects of angiotensin II. Am. J. Physiol. Renal Physiol. 288, F552–F558.
Rajagopalan, S., Kurz, S., Munzel, T., Tarpey, M., Freeman, B. A., Griendling, K. K., and Harrison, D. G. (1996). Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J. Clin. Invest. 97, 1916–1923.
Rajagopalan, S., Laursen, J. B., Borthayre, A., Kurz, S., Keiser, J., Haleen, S., Giaid, A., and Harrison, D. G. (1997). Role for endothelin-1 in angiotensin II-mediated hypertension. Hypertension 30, 29–34.
Ren, Y., D’Ambrosio, M. A., Wang, H., Liu, R., Garvin, J. L., and Carretero, O. A. (2008). Heme oxygenase metabolites inhibit tubuloglomerular feedback (TGF). Am. J. Physiol. Renal Physiol. 295, F1207–F1212.
Rodriguez, F., Kemp, R., Balazy, M., and Nasjletti, A. (2003). Effects of exogenous heme on renal function: role of heme oxygenase and cyclooxygenase. Hypertension 42, 680–684.
Sabaawy, H. E., Zhang, F., Nguyen, X., ElHosseiny, A., Nasjletti, A., Schwartzman, M., Dennery, P., Kappas, A., and Abraham, N. G. (2001). Human heme oxygenase-1 gene transfer lowers blood pressure and promotes growth in spontaneously hypertensive rats. Hypertension 38, 210–215.
Sacerdoti, D., Escalante, B., Abraham, N. G., McGiff, J. C., Levere, R. D., and Schwartzman, M. L. (1989). Treatment with tin prevents the development of hypertension in spontaneously hypertensive rats. Science 243, 388–390.
Stocker, R., Glazer, A. N., and Ames, B. N. (1987). Antioxidant activity of albumin-bound bilirubin. Proc. Natl. Acad. Sci. U.S.A. 84, 5918–5922.
Stocker, R., and Peterhans, E. (1989). Antioxidant properties of conjugated bilirubin and biliverdin: biologically relevant scavenging of hypochlorous acid. Free Radic. Res. Commun. 6, 57–66.
van, L. R., Go, I. H., and Scharm, L. (1968). Metabolism of bilirubin in the Gunn rat. Lancet 2, 973–980.
van der Wegen, P., Louwen, R., Imam, A. M., Buijs-Offerman, R. M., Sinaasappel, M., Grosveld, F., and Scholte, B. J. (2006). Successful treatment of UGT1A1 deficiency in a rat model of Crigler-Najjar disease by intravenous administration of a liver-specific lentiviral vector. Mol. Ther. 13, 374–381.
Vera, T., Granger, J. P., and Stec, D. E. (2009). Inhibition of bilirubin metabolism induces moderate hyperbilirubinemia and attenuates ANG II-dependent hypertension in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R738–R743.
Vera, T., Kelsen, S., and Stec, D. E. (2008). Kidney-specific induction of heme oxygenase-1 prevents angiotensin II hypertension. Hypertension 52, 660–665.
Vera, T., Kelsen, S., Yanes, L. L., Reckelhoff, J. F., and Stec, D. E. (2007). HO-1 induction lowers blood pressure and superoxide production in the renal medulla of angiotensin II hypertensive mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1472–R1478.
Vera, T., and Stec, D. E. (2010). Moderate hyperbilirubinemia improves renal hemodynamics in ANG II-dependent hypertension. Am. J. Physiol Regul. Integr. Comp. Physiol. 299, R1044–R1049.
Wang, H., Garvin, J. L., D’Ambrosio, M. A., Falck, J. R., Leung, P., Liu, R., Ren, Y., and Carretero, O. A. (2011). Heme oxygenase metabolites inhibit tubuloglomerular feedback in vivo. Am. J. Physiol. Heart Circ. Physiol. 300, H1320–H1326.
Wu, Y., Li, M., Xu, M., Bi, Y., Li, X., Chen, Y., Ning, G., and Wang, W. (2011). Low serum total bilirubin concentrations are associated with increased prevalence of metabolic syndrome in Chinese. J. Diabetes 3, 217–224.
Young, S. C., Storm, M. V., Speed, J. S., Kelsen, S., Tiller, C. V., Vera, T., Drummond, H. A., and Stec, D. E. (2009). Inhibition of biliverdin reductase increases ANG II-dependent superoxide levels in cultured renal tubular epithelial cells. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1546–R1553.
Keywords: biliverdin, heme oxygenase, biliverdin reductase, carbon monoxide, UGT1A1, liver, glomerular filtration rate, renal blood flow
Citation: Stec DE, Hosick PA and Granger JP (2012) Bilirubin, renal hemodynamics, and blood pressure. Front. Pharmacol. 3:18. doi: 10.3389/fphar.2012.00018
Received: 22 November 2011;
Accepted: 01 February 2012;
Published online: 14 February 2012.
Edited by:
Jaime Kapitulnik, The Hebrew University of Jerusalem, IsraelReviewed by:
Yoseph Caraco, Hadassah University Hospital, IsraelFranz Worek, Bundeswehr Institute of Pharmacology and Toxicology, Germany
Copyright: © 2012 Stec, Hosick and Granger. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: David E. Stec, Department of Physiology and Biophysics, Center for Excellence in Cardiovascular – Renal, Research, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216, USA. e-mail:ZHN0ZWNAdW1jLmVkdQ==