


- Neurosurgery and Neurobiology, Research and Surgery Services, Durham VA Medical Center, Duke University, Durham, NC, USA
Neuronal activity, astrocytic responses to this activity, and energy homeostasis are linked together during baseline, conscious conditions, and short-term rapid activation (as occurs with sensory or motor function). Nervous system energy homeostasis also varies during long-term physiological conditions (i.e., development and aging) and with adaptation to pathological conditions, such as ischemia or low glucose. Neuronal activation requires increased metabolism (i.e., ATP generation) which leads initially to substrate depletion, induction of a variety of signals for enhanced astrocytic function, and increased local blood flow and substrate delivery. Energy generation (particularly in mitochondria) and use during ATP hydrolysis also lead to considerable heat generation. The local increases in blood flow noted following neuronal activation can both enhance local substrate delivery but also provides a heat sink to help cool the brain and removal of waste by-products. In this review we highlight the interactions between short-term neuronal activity and energy metabolism with an emphasis on signals and factors regulating astrocyte function and substrate supply.
Introduction
The goal of this review is to summarize the linkage between neurons and astrocytes during both short-term perturbations in activity and metabolic function and long-term metabolic adaptation. In addition, we compare where possible observed and predicted differences in metabolic function between in vivo and in vitro preparations, since the varying methods of substrate delivery in these two conditions lead to contrasting tissue measurements in spite of well preserved neuronal function and activity. In the first section we introduce the responses associated with neuronal activation, in the second section we describe in more detail a number of signals critical for communication across the metabolic unit, and in the third section discuss long-term metabolic homeostasis.
Cerebral Metabolic Unit
The three critical cellular elements in determining the response of the brain to physiological events include: (1) neurons, which determine cerebral physiological activity based on electrical processing; (2) astrocytes and other glial cells, which directly cooperate between blood vessels and neurons for regulation of energy needs and maintenance of the extracellular environment; and (3) blood vessels for both delivery of substrate to the brain (i.e., glucose and oxygen) and removal of by-products, such as CO2 and heat. These three elements interact for all aspects of metabolic function, forming a coherent and functional metabolic unit.
Basal Neuronal Metabolism
Neurons are never completely quiescent, with modulation of action potential firing during specific brain states. For example, in the awake or conscious state there is considerable spontaneous activity, which is progressively decreased with deeper levels of anesthesia (Shulman et al., 2009) and increased with focal neuronal activation such as sensory or motor function. Neuronal and glial function is crucially dependent on the maintenance of electrochemical gradients across cellular membranes. Therefore, even during the resting state (i.e., conscious but in the absence of specific sensory, mental physical activity) a significant portion of ATP is utilized to fuel Na+/K+-ATPase activity to maintain sodium and potassium gradients (Pellerin and Magistretti, 1994; Chatton et al., 2000; Voutsinos-Porche et al., 2003) and also needed to maintain neurotransmitter recycling and packaging for synaptic vesicles, cell signaling pathways in both neurons and astrocytes, and repair of structural membrane, protein assemblies, and DNA. In addition, further studies have also revealed the importance of the specific composition of energy substrate pool and its utilization for controlling of neuronal excitability and maintaining resting membrane potential (Rheims et al., 2009; Holmgren et al., 2010; Zilberter et al., 2010; Ivanov and Zilberter, 2011; Mukhtarov et al., 2011). For example, lactate is utilized more effectively in the presence of glucose as an oxidative metabolic substrate, to maintain synaptic function (Ivanov et al., 2011).
Brain primarily utilizes glucose as its main energy source (in adults) to synthesize ATP (Clarke and Sokoloff, 1999) and 90% of cerebral ATP production occurs in the mitochondria though oxidative phosphorylation. Therefore, cerebral metabolic activity can be evaluated by measuring cerebral metabolic rates of glucose (CMR glucose) and oxygen (CMRO2) using either [18F]-fluoro-2-deoxy-D-glucose positron emission tomography (FDG PET) together with oxygen-15 PET or calibrated blood oxygen level dependent functional magnetic resonance imaging (BOLD fMRI) methods, respectively (Davis et al., 1998; Hoge et al., 1999). Although rates of energy turnover in the alert resting state are difficult to estimate, metabolic brain activity remains significant during this awake, non-stimulated state (Shulman et al., 2004). Several PET and fMRI studies indicate that during the transition from the “awake state” to deeper levels of general anesthesia (Smith et al., 2002; Rothman et al., 2003; Shulman et al., 2004) there is a widespread, substantial decrease in energy demand; this decrease is larger in magnitude than the increments in energy consumption noted during activation (Hyder et al., 2001; Shulman et al., 2009).
For example, ∼15% increase in metabolic activity over baseline is observed in conscious humans during tasks to study cognitive and other neurological functions (Phelps and Mazziotta, 1985; Grafton et al., 1992; Elman et al., 1999). In contrast, after the induction of anesthesia brain oxygen and glucose consumption are reduced by ∼40–50% as revealed by PET measurements; these values decline further to ∼15–20% of baseline upon reaching deeper levels of anesthesia with pentobarbital (Shulman et al., 2009). Similarly, results from 31P magnetic resonance spectroscopy (MRS) studies, combined with magnetization transfer method to measure the rate of Pinorganic conversion to ATP, determined that the progressive decline of ATP metabolism with increasing levels of anesthesia was parallel to the decline in brain EEG activity; at an isoelectric state the oxidative production rate of ATP was reduced to 50% compared to light isoflurane anesthesia condition (Du et al., 2008). Thus, the conscious state provides a baseline for activation but also represents a high level of spontaneous neural activity and accompanying metabolic demand, which can be substantially reduced with progressively deeper general anesthesia.
Short-Term Metabolic Responses
Metabolic Events Occurring during Neuronal Activation
Neuronal activation commonly is evoked during sensory or cognitive stimulation or motor tasks. The stimulation activates neurons in brain regions which respond to the intense incoming synaptic input, resulting typically in firing a sequence of action potentials. Moreover, intermittent periods of increased neuronal activity also can occur during the rapid-eye-movement (REM) stage of sleep, which is associated with dream formation and consolidation of learning and memory. Neuronal activation can also be evoked in in vitro preparations, by a short train of electrical stimulation pulses to excite axons and presynaptic terminals, resulting in postsynaptic action potentials and often a compound evoked potential response representing a combination of synaptic potentials and action potentials (Galeffi et al., 2007, 2011). The timing and density of such stimulation can be varied but commonly the duration is 5–30 s, to temporarily “activate” neurons with an intense incoming synaptic input. The energy demands associated with the neuronal activity result in a cascade of metabolic events which can be measured in a number of different ways depending on the preparation and the technique, shown on a timeline in Figure 1 (in vitro) and Figure 2 (in vivo). Common measurements shown include extracellular K+, Ca2+, oxygen, as well as metabolic imaging, such as NADH or FAD.
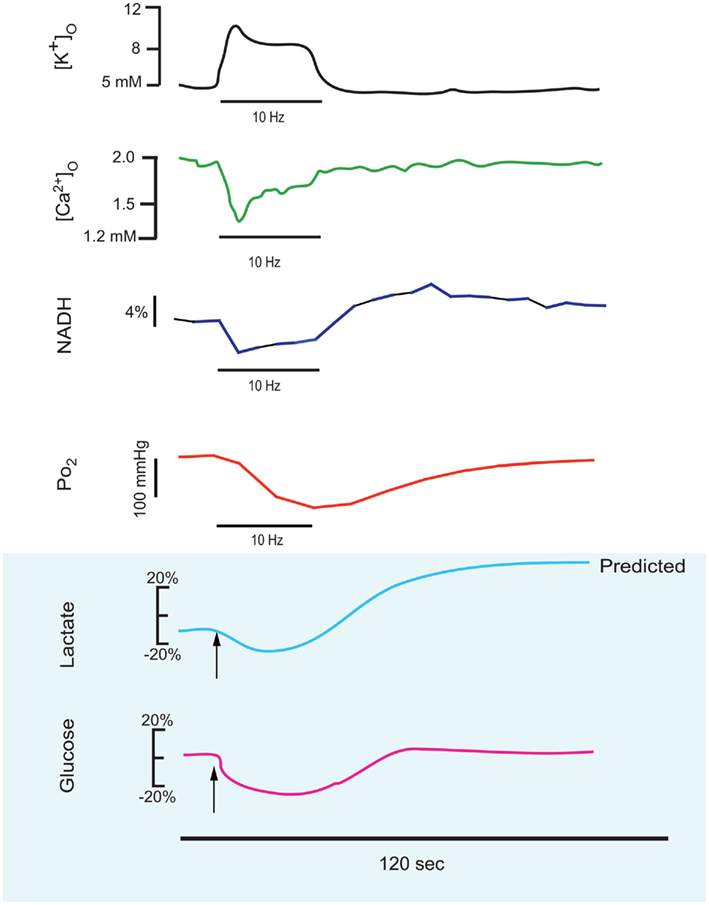
Figure 1. This series of traces illustrates the response to a 10–25 short stimulus train applied to the stratum radiatum of the CA1 region of an in vitro hippocampal tissue slice, and the physiological responses which can be measured in real-time during this stimulus train. These reconfigured traces have been redrawn from primary sources to be on the same time scale (shown as 120 s below). Measured responses include extracellular K+ and Ca2+ (Benninger et al., 1980) and NADH imaging and tissue PO2 (Galeffi et al., 2007). Predicted in vitro tissue slice responses are shown below with blue shading, with lactate and glucose extrapolated from in vivo measurements performed by Hu and Wilson(1997a,b). Note that extracellular K+ rises during the train and Ca2+ decreases, due to K+ release from neurons and Ca2+ uptake into neurons. NADH shows both an oxidative phase (i.e., a decrease) and a reduction phase (i.e., an increase), whereas in tissue slices the tissue oxygen can only decrease from the baseline level. Extracellular glucose can likewise only decrease, whereas lactate synthesis (stimulated by the train) results in an initial dip (due to demand) but a subsequent elevation, as lactate is further extruded from astrocytes.
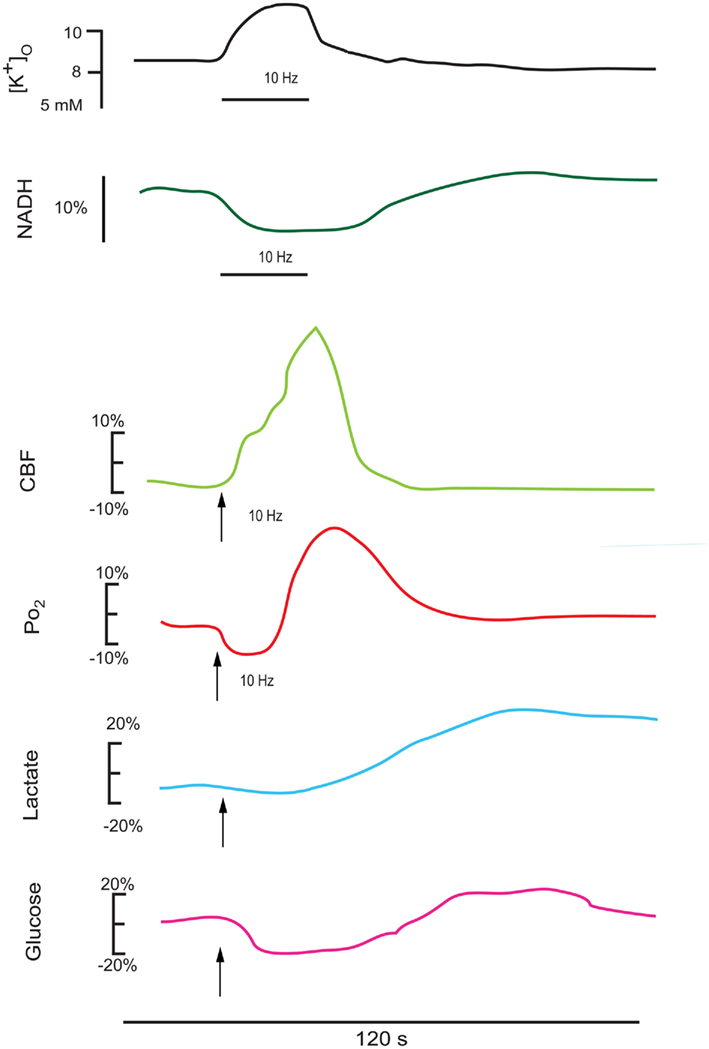
Figure 2. This series of redrawn and rescaled traces show measured responses in vivo from intact brain (though different structures), where changes in cerebral blood flow can alter the basic responses to a stimulation train. The upper K+ and NADH traces are in response to a 25-s stimulus train at 10 Hz in the cortex (Lothman et al., 1975), showing a consistent K+ elevation and an NADH oxidation (Turner et al., 2007). The CBF and PO2 responses follow a 15-s stimulation train at 10 Hz in the cerebellum (Offenhauser et al., 2005). The glucose and lactate measurements are redrawn from Hu and Wilson(1997a,b). Note that the oxygen response shows an initial dip in the activated region then an elevation as the enhanced cerebral blood flow response occurs. There is an initial decrease and a delayed enhancement in both cerebral lactate (due to astrocytic extrusion) and glucose (due to enhanced transport into the brain) as measured extracellularly.
For example, trigeminal nerve or whisker stimulation increases glucose metabolism in the thalamus and brain stem (Ciricillo et al., 1994) based on the 2-deoxy-glucose imaging method (the animal equivalent to human FDG PET). Similarly, classical functional imaging studies have reported an increase in blood flow and in the metabolic rate for oxygen and glucose during a sensory, motor, or cognitive simulation after subtraction of baseline activity. However, during sensory stimulation, measurements of glucose and oxygen uptake using PET in human subjects have revealed greater glucose consumption compared to oxygen (Fox et al., 1988). Human detection methods (i.e., fMRI and PET) are sometimes insensitive to early events following neuronal activation due to the small areas involved and rapid time course. During the first few seconds (i.e., up to 10 s) intracellular Na+ and Ca2+ starts to build up and K+ is released from neurons whereas mitochondrial intermediates are rapidly oxidized (together with oxygen) for ATP generation, as noted in Figure 1. Real-time imaging methods are more sensitive to these early changes but typically are more invasive in nature.
For example, activity-dependent changes in NADH/NAD+ redox state can be monitored with real-time fluorescence imaging of NADH in vivo as well as in vitro. Physiological stimulation leads to an NADH fluorescence decrease in well-oxygenated tissue (in vitro and in vivo), followed by either recovery to baseline or an increase (i.e., overshoot) above baseline (Rosenthal and Jobsis, 1971; Lothman et al., 1975; Schuchmann et al., 2001; Shuttleworth et al., 2003). Influx of glucose and lactate to neurons results in a transient decrease in lactate and glucose concentration in the extracellular space (ECS; Hu and Wilson, 1997a,b). Perfusion of the brain also depends on the tight coupling between O2 supply and metabolic demand; stimulation of neurons results in a transient drop in O2 concentration due to increased oxidative phosphorylation (Foster et al., 2005), which stimulates a series of reactions to increase cerebral blood flow and match demand. Increases in intracellular and mitochondrial Ca2+ also can enhance enzymatic activity within the tricarboxylic acid (TCA) cycle along with release of oxygen free radicals and reactive oxygen species (ROS; Duchen, 1992; Keelan et al., 1999; Kovacs et al., 2001; Schuchmann et al., 2001). Tissue oxygen measurements with NADH imaging during prolonged stimulation in vitro also revealed persistent NADH oxidation and O2 utilization with continued metabolic demand (Galeffi et al., 2011).
Recovery after Neuronal Activation
During recovery neuronal activity is decreasing to baseline but metabolic needs can persist as well as delayed enhancement of blood flow. For example, NADH remains elevated temporarily (Foster et al., 2005; Galeffi et al., 2011) due to persistent TCA cycle dehydrogenase activity (Denton, 2009) and lactate uptake, extracellular lactate rises as astrocytic metabolism continues, and local blood flow stays elevated, while short-term intermediate molecules within the mitochondria are repleted, as noted in Figure 1. Eventually, neuronal activation winds down, leading to O2 consumption trending down to basal levels as metabolism wanes, restoration of membrane potentials by pumps, stabilization of NADH and other mitochondrial intermediates once replenished, and finally decreases in local blood flow to control levels. Eventually NADH levels recover back to baseline levels (Rosenthal and Jobsis, 1971; Lothman et al., 1975; Schuchmann et al., 2001; Shuttleworth et al., 2003).
Cellular Localization of Energy Utilization after Neuronal Activation – Neurons vs Astrocytes?
Synaptic activity stimulates glucose uptake and glucose utilization by both neurons and astrocytes (Sokoloff, 1999; Chih and Roberts, 2003). Cellular and tissue studies show that neurons likely use a combination of extracellular lactate (derived from astrocytic glycolysis) and glucose (Figure 3) as their energy substrates (Galeffi et al., 2007). In vivo data reveals that there is a prevalence of glycolysis over oxidative phosphorylation during neuronal activation which results in increased lactate production, likely primarily within astrocytes (Dienel and Hertz, 2001).
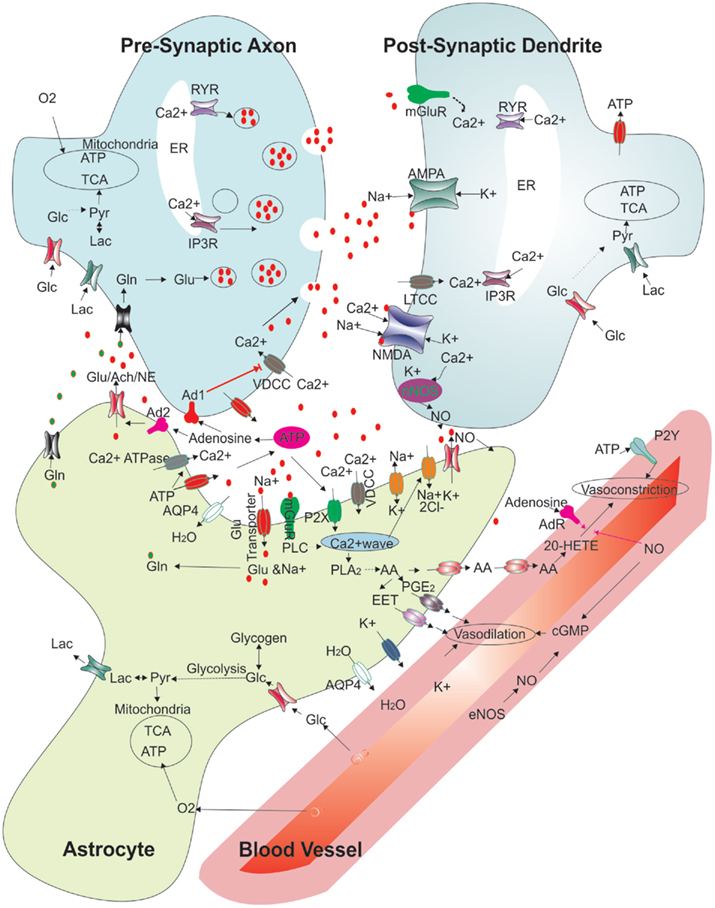
Figure 3. Regulation of synaptic function and cerebral blood flow: modified schematic diagram (Attwell et al., 2010; Kapogiannis and Mattson, 2011) showing the multiple interactions between an astrocyte, neurons, and a blood vessel. The various functions depicted show presynaptic release, glutamate receptor binding and handling, various energy transport functions between blood vessels, the astrocyte and neurons, vasoconstriction and vasodilation pathways, and extracellular molecules. Abbreviations: RYR, ryanodine receptor; ER, endoplasmic reticulum; Glu, glutamate; Gln, glutamine; IP3R, Inositol 3 phosphate receptor; Lac, lactate; Glc, glucose; Pyr, pyruvate; TCA, Tricaboxylic acid cycle; ATP, adenosine 5’- tri phosphate; Ach, acetyl choline; NE, norepinephrine; No, nitric oxide; Ad1, adenosine receptor 1; Ad2, adenosine receptor 2; mGluR, metabotropic glutamate receptor; NMDAR, N-methyl-D-aspartic acid receptor; AMPAR, 2-amino-3-(5-methyl-3-oxo-1;2- oxazol-4-yl)propanoic acid receptor; nNOS, neuronal nitric oxide synthase; eNOS, endothelial nitric oxide synthase EET, epoxyeicosatrienoic acids; AA, arachidonic acid; PGE2, prostaglandin 2; 20-HETE, 20-hydroxy arachidonic acid; PLA2, phospholipase A2; cGMP, cyclic guanosine monophosphate; P2X; purinergicreceptor P2X; P2Y, purinergic receptor P2Y; PLC, phospholipase C; AQP4, aquaporin-4; VDCC, voltage dependent calcium channel; LTCC, L-type calcium channel.
However, evidence supporting direct lactate utilization by neurons continues to have skeptics (Korf, 2006), including data linking glucose (but not lactate) utilization with NMDA-induced neurotransmission (Bak et al., 2006, 2009). In contrast, there is activity-dependent oxidation of lactate in vivo (Wyss et al., 2011), whereas astrocytic-derived lactate is necessary to maintain long-term potentiation (Suzuki et al., 2011).
In astrocytes, the presence of extracellular glutamate following synaptic release induces rapid glutamate uptake [through glutamate transporters (GLTs)] associated with Na+ influx (Figure 3). Uptake of glutamate and its conversion to glutamine and subsequent recycling back to neurons requires energy provided by glycolysis within astrocytes (Pellerin and Magistretti, 1996, 1997). In addition, de novo synthesis of glutamate from glucose in astrocytes also requires ATP, through the TCA cycle intermediate α-keto-glutarate. The summary view at this time is that glucose can be metabolized by both neurons and astrocytes, but neurons utilize a large proportion of extracellular lactate for oxidative metabolism (perhaps greater than 50%, drawn from a large extracellular pool of lactate generated by astrocytes) and the balance from glucose (Zielke et al., 2007).
Differences in Substrate Delivery in vivo vs in vitro Slice Tissue Preparations
The large difference between the in vivo condition and in vitro slice preparations is oxygen and glucose delivery, which occurs via diffusion from tissue borders in vitro and through an extensive vessel and capillary network in vivo (Turner et al., 2007). Since there is no secondary blood flow response in vitro, then the tissue levels of oxygen (Galeffi et al., 2007, 2011) and glucose can only dip to lowered levels and recover in response to the enhanced metabolic demand. In contrast to in vivo conditions where hemoglobin can possess high oxygen saturation and content at low PO2 tensions, in in vitro preparations a high oxygen tension is needed to maintain tissue oxygen levels, since oxygen content and flux through the tissue are very low with diffusion alone, particularly at the long distances required to maintain slices (i.e., up to 400–500 μm depth). As a comparison, the oxygen dip following significant in vivo cortical activation may be ∼5–10 mm Hg O2 (Thompson et al., 2003) whereas similar activation in a tissue slice can lead to ∼180 mm Hg O2 change (Galeffi et al., 2011). This large difference in oxygen carrying capacity or content can lead to considerable confusion about what may be a “sufficient” level of oxygen in tissue slices (i.e., Gordon et al., 2008); clearly, the nature of the tissue slice preparation and the degree of neuronal activation and metabolic demand may help clarify the levels of oxygen required in the tissue bath, which should give rise to values greater than 10–15 mm Hg in the center of a slice even with significant metabolic demand. Our experience is that relatively normal neuronal function requires at least 50% ambient oxygen levels (i.e., >350 mm Hg) to maintain even low levels in the middle of slices during metabolic demand (Galeffi et al., 2011).
Cerebral Blood Flow
Stimulation of local neuronal activity increases local blood flow through a variety of neuronal and astrocytic mechanisms (Koehler et al., 2009; Paulson et al., 2009). Metabolic signals derived from both neurons and astrocytes can modulate both blood flow and substrate availability (Furchgott and Zawadzki, 1980; Gordon et al., 2008). For example, NO generated by neurons leads to vessel dilation and acetylcholine action on muscarinic receptors of the endothelial cells can cause relaxation of vascular smooth muscle. Bradykinin is also an important vasodilator, known to induce KCa channel activation in endothelial cells through the generation of ROS (Nelson and Quayle, 1995; Sobey et al., 1997). Glutamate also has a major role in regulating blood flow by increased NO generation from neurons and arachidonic acid derived from astrocytes (Gordon et al., 2008). In astrocytes Ca2+ also activates phospholipase A2 and increases the production of arachidonic acid metabolites (Figure 3), such as prostaglandins and epoxyeicosatrienoic acids, which dilate nearby arterioles (Iadecola and Nedergaard, 2007; Koehler et al., 2009). Prostaglandins binding to the EP4 PG receptors activate protein kinase A by cAMP and thus decrease phosphorylation of myosin chains; EETs inhibit the receptors for thromboxane thereby inhibiting vasoconstriction. Adenosine, a metabolite of ATP, also modulates blood flow, through A2A receptors localized on arterioles (Gordon et al., 2008). Rapid glycolysis during neuronal activation increases the conversion of pyruvate to lactate and an increased NADH/NAD+ ratio, which in turn increases cerebral blood flow (Ido et al., 2004; Pellerin et al., 2007; Gordon et al., 2008). A number of these mechanisms are directly in response to neuronal and astrocytic activation, linking neuronal activity to metabolic demand and supply in a number of ways (Figure 2). Measurements in vivo in intact preparations include K+, NADH, cerebral blood flow, oxygen, as well as lactate and glucose, as shown in Figure 2.
Heat Production
Direct heat emission can reveal the source(s) of ongoing metabolism (both through ATP hydrolysis as well as mitochondrial heat release) at a cellular and circuit level, as well as the efficiency of metabolism [i.e., the coupling of mitochondrial energy with ATP production or heat release (LaManna et al., 1980, 1989; Yablonskiy et al., 2000; Trubel et al., 2005)]. Maintenance of mitochondrial membrane potential for ATP generation releases ∼33% of total energy as heat, but ATP hydrolysis also leads to heat production, whereas there is minimal heat production by anaerobic glycolysis (LaManna et al., 1980; Yablonskiy et al., 2000). The considerable resulting heat production is rapidly diffused into surrounding tissue, but since the brain resides in a closed space the heat must be dissipated by a large cerebrovascular response (LaManna et al., 1989; Trubel et al., 2005). In most instances, the cerebrovascular (dilation) response to neuronal metabolism is far out of proportion to the substrate supply need, suggesting that a secondary function of the vascular response is heat shunting (Sukstanskii and Yablonskiy, 2006). In spite of the considerable heat dissipation through the vascular system neuronal activation can still lead to up to 0.5°C temperature elevation in a local region, whereas spreading depression, seizures and recovery from ischemia can generate up to 1.0°C in intact cortex (LaManna et al., 1980, 1989). Direct heat emission imaging with specialized infrared cameras may resolve the efficiency of metabolism since uncoupling of ATP generation from mitochondrial membrane potential releases nearly all of metabolism as heat.
Changes in Extracellular Space
Both astrocytes and neurons swell with activation due to Na+ and water entry, leading to shrinkage of the ECS (Bourke et al., 1980; Fayuk et al., 2002). For example, the ECS of the mature optic nerve and also hippocampus rapidly and reversibly decreases by as much as 20% (Ransom et al., 1985; Fayuk et al., 2002). Intrinsic optical signal (IOS) imaging and measurements also have revealed spontaneous neuronal hyperactivity associated with ECS shrinkage in vitro (Buchheim et al., 1999). This swelling is rapid and reversible and is related to duration of the physiological stimuli (Dietzel et al., 1982) but also is associated with anoxia and inflammation (Hochwald et al., 1981). The shrinkage of ECS in central nervous system is also observed developmentally due to activity-dependent changes in postnatal development (Bondareff and Pysh, 1968; Van Harreveld et al., 1969; Connors et al., 1982). Thus, ECS or IOS imaging may augment other methods of following neuronal activation and metabolism.
Summary
Following neuronal activation a complex sequence of responses in neurons, astrocytes and blood vessels orchestrates enhanced substrate supply and blood flow to be able to maintain the energy requirements for the activation regardless of duration. We now summarize some of these critical intermediate signals and how they function to preserve metabolic homeostasis within the cerebral metabolic unit.
Factors Involved in Short-Term Energy Homeostasis
Critical Intermediate Signals Underlying Neuronal–Glial Communication
There are a number of factors and signals responsible for homeostatic maintenance of neuronal function and energy metabolism. As soon as neuronal activity is stimulated neurons initially show Na+ and Ca2+ inward currents and then subsequently release K+, glutamate, NO, CO2, and H2O2 into the ECS; this increased neuronal activity requires increased oxygen, lactate and glucose uptake to maintain metabolic function. The neuronal uptake of metabolic substrates and the release of secondary signals (i.e., K+, NO, and glutamate in particular) can function as intermediate cues needed for enhancement of metabolic substrate utilization. These cues then are used as signals by astrocytes and blood vessels to enhance blood flow and substrate delivery, which are amplified into secondary signals, such as ATP release, to spread within astrocytic networks. We will discuss these critical intermediate signals and the astrocytic responses which occur for short-term maintenance of metabolism.
Na+/K+ Homeostasis
Na+ entry via voltage-dependent channels and action potential generation underlay the basic function of neurons for rapid communication and information processing, with a large number of K+-voltage channels needed to repolarize neurons at various time intervals; thus, tight regulation of extracellular K+ is important for maintaining neuronal activation through rapid membrane repolarization. Astrocytes maintain extracellular K+ homeostasis via both passive and active mechanism of uptake through a variety of voltage-dependent channels and transporters, which is then dissipated back to the ECS, other astrocytes or blood vessels (Figure 3). Membrane-bound Na+/K+ ATPase (Skou and Esmann, 1992) is the key enzyme responsible for the maintenance of intracellular ion homeostasis in both neurons and astrocytes (Walz and Hertz, 1982). Na+/K+ ATPase hydrolyzes ATP to ADP in the process of extruding three Na+ against two K+ molecules; inhibition of Na+/K+ ATPase results in a marked decrease in astrocytic metabolism. Thus, astrocytes may exert considerable energy for K+ extracellular stabilization, which helps to maintain neuronal excitability, and in turn K+ provides an immediate extracellular signal to astrocytes demonstrating neuronal action potential firing.
Glutamate Metabolism
Glutamate uptake into astrocytes via GLT following synaptic release is another major signaling factor to astrocytes, reflecting neuronal activation (Parpura et al., 1994; Anderson and Swanson, 2000; Auger and Attwell, 2000; Danbolt, 2001; Malarkey and Parpura, 2008). Glutamate is the major excitatory neurotransmitter released throughout the brain, and glutamate action is primarily terminated by glutamate uptake into astrocytes via GLTs. Neurons express both ionotropic and metabotropic glutamate receptors, including (NMDA N-methyl-D-aspartate), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA), and kainate subtypes (Figure 3). Astrocytes also express metabotropic receptors (mGlur 1–8), which are a family of G protein coupled receptors, mediating effects primarily through second messengers. Astrocytes expresses mainly mGlur receptors subtypes 2, 3, and 5: mGlur 5 is coupled to phosphoinositide (PI) hydrolysis and can increase intracellular calcium, whereas mGlur 2 and 3 are negatively coupled to adenylate cyclase to inhibit cAMP formation.
In addition to glutamate actions on these receptors, astrocytes also uptake glutamate through glutamate transport protein-mediated endocytosis and then astrocytes recycle the glutamate back to neurons for further use by synthesis of glutamine (via glutamine synthetase, exclusively in astrocytes) with secondary release for neuronal uptake and repackaging of glutamate. This recycling creates a glutamate–glutamine cycle (Figure 3) occurring between neurons and astrocytes (Van den Berg et al., 1969; Balazs et al., 1970; Van den Berg and Garfinkel, 1971; Norenberg and Martinez-Hernandez, 1979). The vesicular uptake of glutamate requires glutamate transport protein and vacuolar-type H+-ATPase, which couples ATP hydrolysis and glutamate transport. (Naito and Ueda, 1983; Ozkan and Ueda, 1998) and which also make the interior of the vesicle acidic. Glutamine uptake into neurons increases during hypoglycemia since glutamine can also be oxidized for energy production in low glucose conditions (Amaral et al., 2011).
Most of the glutamate taken up by the astrocytes is eventually converted to glutamine for extrusion and recycling back to neurons, via glutamine synthase. However, oxidation of glutamate can be increased up to 45% when the extracellular glutamate concentration increases beyond 100 μM (McKenna et al., 1996a,b). Exposure of cultured astrocytes to glutamate in glucose can also stimulate oxidative phosphorylation, reducing glucose utilization (Qu et al., 2001; Dienel and Cruz, 2006). Oxidation of glutamate provides more ATP than that generated from lactate, and spares the need for glucose utilization. Oxidation of glutamate in the cell can also generate pyruvate and lactate (Moreadith and Lehninger, 1984). Under steady state basal metabolism the levels of TCA derived intermediates are stable but the rate of degradation increases with neuronal activation (Gruetter et al., 2001; Sibson et al., 2001). During activation oxidation of TCA cycle derived amino acids, such as glutamate, GABA, and aspartate can provide a significant energy source. Glutamate may also be recycled in the form of lactate and alanine or other TCA cycle intermediates (Dringen et al., 1993; Westergaard et al., 1995). These intermediates may be important since neurons lack the capacity to synthesize TCA cycle constituents de novo due to absence of pyruvate carboxylase (Yu et al., 1983; Shank et al., 1985; Cesar and Hamprecht, 1995).
Ca2+ Homeostasis
The extracellular concentration of Ca2+ is typically ∼1.2 mM, compared to the intracellular concentration of 100 nM (Somjen, 2002). This large transmembrane gradient of Ca2+ can create a high inward flux and incoming Ca2+ ions can make a large impact on intracellular Ca2+ levels. The reverse transport of calcium across the membrane back into the ECS is maintained by energy dependent extrusion. For example, inward Ca2+ flux (Figure 3) via voltage sensitive calcium channels (L-type) activated by elevated K+ stimulates both the Na+, K+, Cl− co-transporter (Su et al., 2000) and also oxidative metabolism (Hertz and Dienel, 2002). These transporters are also regulated by intracellular messengers such as cAMP, cGMP, Ca2+/calmodulin and also by cell shrinkage or swelling (Jensen et al., 1993; O’Neill, 1999). The degree of calcium influx is dependent on the frequency of stimulation, particularly above 10 Hz. Increased Ca2+ influx promote a large number of reactions for metabolic function, particularly modulation of the rate of the TCA cycle for NADH/FAD production and ATP generation through direct influence on the dehydrogenase reactions.
Tight buffering of cytoplasmic Ca2+ is maintained by Ca2+ extrusion, sequestration with Ca2+ binding molecules, and uptake into organelles including endoplasmic reticulum (ER) and mitochondria. Each molecule of Ca2+ transported either into endoplasmic reticulum or out of the plasma membrane requires one molecule of ATP, through Ca2+ ATPase activity or the Na+/Ca2+ exchanger. Influx of Ca2+ is important for initiation of long-term neuronal signaling (i.e., long-term potentiation) and also for activation of TCA cycle enzymes and mitochondrial oxidative phosphorylation. Buffering of mitochondrial Ca2+ is important to prevent toxic effects exerted by increased ROS generation and loss of signaling efficacy. However, extrusion of Ca2+ from mitochondria occurs at time points when Ca2+ levels in the cytosol return to baseline and when ATP demand and O2 consumption are low, through either a mitochondrial Ca2+ proton exchanger or the mitochondrial permeability transition pore (MTP). The kinetics of transport for these exchangers is low, which can enhance Ca2+ overload in mitochondria. During neuronal activation astrocytes show elevation of intracellular Ca2+, which may be spontaneous or triggered by ATP released extracellularly (Charles et al., 1991). In neurons Ca2+ influx occurs at synapses via NMDA receptors and L-type calcium channels (LTCCs). CaV1.2 is a widely expressed LTCC in the brain, localized mainly at dendritic shafts and spines (Hell et al., 1993; Clark et al., 2003; Obermair et al., 2004; Schlick et al., 2010); this LTCC can be internalized through tumor suppressor eIF3e mediated calcium and dynamin-dependent endocytosis (Green et al., 2007; Di Biase et al., 2011).
ATP Signaling
Both neurons and astrocytes can release ATP during basal metabolic activity and during neuronal activation. ATP acts as a neurotransmitter by binding to purinergic receptors (Burnstock, 1999; Fields and Stevens, 2000). However, ATP release from astrocytes is likely the main source of extracellular ATP (Butt, 2011) and is mediated by exocytosis (Pangrsic et al., 2007). ATP can also trigger a rise in intracellular calcium within astrocytes, particularly as a delayed Ca2+ wave (Figure 3). This action is mediated by an elevation in nicotinic acid-adenine dinucleotide phosphate by ATP (Barceló-Torns et al., 2011). Purinergic receptors are either ionotropic (P2X) or metabotropic (P2Y). P2X receptors consists of seven transmembrane subunits (P2 × 1–P2 × 7) involved in fast synaptic transmission (Bardoni et al., 1997). Metabotropic receptors (P2Y1, Y2, Y4, Y6, Y11) are abundant throughout the nervous system (Abbracchio and Burnstock, 1994; Cook et al., 1997) and are coupled with Gq/G11 proteins which activate phospholipase c/inositol triphosphate pathways to increase intracellular Ca2+ (Kastritsis et al., 1992). In contrast, P2Y receptors are linked to GI/0 proteins.
Quantal release of ATP from neuronal synaptic terminals has been characterized by the analysis of P2X receptor-mediated evoked and spontaneous miniature excitatory postsynaptic currents (mEPSCs) in hippocampal neurons (Pankratov et al., 2006). P2X mediated mEPSCs are different from glutamate mediated mEPSCs in terms of amplitude and kinetics. It has been speculated that ATP is released either from a separate pool of vesicles (White and MacDonald, 1990) or vesicles associated with acetylcholine, GABA, or glutamate (William, 2003; Pankratov et al., 2006). Astrocytes also release ATP from vesicles containing SNARE complexes; the release of ATP is suppressed in transgenic mice expressing a dominant negative SNARE domain (Pascual et al., 2005). ATP is also a potent vasoconstrictor and astrocytes demonstrate secondary Ca2+ waves through highly enriched purinergic receptors (P2Y2 and 4; Butt, 2011). However, ATP as a vasoconstrictor is opposed by adenosine, a metabolite of ATP, which acts as a potent vasodilator; the relative balance of these two metabolites at the astrocytic-vascular interface plays an important role in neurovascular coupling mechanisms (Butt, 2011).
Adenosine
Adenosine, a metabolite of ATP, exerts potent vasodilation effects and inhibits neuronal transmission via AD1 receptors (Figure 3); this can result in inhibition of glutamate release at presynaptic terminals of Schaffer collaterals in the CA1 hippocampus and reduce the release of glutamate from these fibers (Fowler, 1992; Lupica et al., 1992; Zhu and Krnjevic, 1993; Butt, 2011). Adenosine can also enhance the release of several neurotransmitters, such as acetylcholine, glutamate, and noradrenaline, via AD2 receptors (Sebastiao and Ribeiro, 1996). Extracellular adenosine formation from ATP/ADP/AMP depends upon the activity of ecto-5′-nucleotidase and the overall levels of energy metabolism and ATP levels. Extracellular concentration of adenosine is maintained by specific nucleoside transporters present in both neurons and astrocytes and by its conversion to inosine. For example, insulin-induced hypoglycemia increases extracellular adenosine levels and depresses evoked synaptic potentials (Chapman et al., 1981; Fan et al., 1988).
Addition of alternative substrates, such as lactate, during glucose deprivation can maintain ATP levels and significantly reduce adenosine levels, leading to improved neuronal function (Fowler, 1993). Addition of AD1 receptor antagonists also can extend synaptic activity for a limited period of time in low glucose conditions, suggesting that adenosine levels rise before complete energy failure occurs (Shetty et al., 2012). Adenosine can also block voltage-dependent calcium channels (Phillis and Wu, 1981; Scholz and Miller, 1991; Bennett and Ho, 1992; Mogul et al., 1993) and thereby inhibits presynaptic calcium fluxes. Differential effects of adenosine on calcium channels depends on adenosine receptor subtype activation and has significant implications for neuronal excitability (Mogul et al., 1993). Adenosine also activates presynaptic potassium channels and can attenuate evoked neurotransmitter release by hyperpolarizing the presynaptic membrane during aglycemia (Bachelard et al., 1984; Knopfel et al., 1990).
Glycogen Utilization and Synaptic Activity
Though brain utilizes glucose for its energy substrate there is also the capacity to store glucose in the form of glycogen. Although the enzyme for glycogen formation, glycogen synthase, is present in both astrocytes and neurons glycogen storage capability and glycogen degrading enzymes are localized only in astrocytes (Vilchez et al., 2007). However, innate glycogen levels in the brain appear small compared to liver and muscle glycogen content (3–12 μmol glycosyl units/g; Cataldo and Broadwell, 1986; Cruz and Dienel, 2002; Choi and Gruetter, 2003; Suh et al., 2007). In certain conditions glycogen levels can be augmented; for example, between periods of intermittent hypoglycemia glycogen levels can increase above normal levels (termed supercompensation). The ability of brain tissue to accumulate extra glycogen has been also demonstrated in brain slices following supplementation with extra glucose, pyruvate, or lactate (Shetty et al., 2012). Because glycogen is stored only in astrocytes, its utilization by neurons (likely as extruded lactate) reveals highly coordinated energy metabolism interactions between astrocytes and neurons (Suzuki et al., 2011).
Astrocytic stored glycogen can be rapidly converted directly into pyruvate via glycolysis or perhaps glucose (Ghosh et al., 2005). Glycogen storage is advantageous in some ways since no further ATP is required to initiate metabolism, unlike glucose and fatty acids (Hertz et al., 2007). Glycogen-derived lactate is likely released into the ECS (Figure 3) and taken up by neurons for energy, particularly in low glucose conditions where glycogenolysis is initiated (Dringen et al., 1993; Pellerin and Magistretti, 1994; Magistretti and Pellerin, 1999; Brown et al., 2004). In contrast, the utilization of glycogen during euglycemic conditions is controversial. Inhibition of glycogen breakdown by 1,4-dideaxy-1, 4-imino-D-arabinitol (DAB; glycogen phosphorylase inhibitor) did not suppress ongoing neuronal activity during euglycemic conditions (Shetty et al., 2012). In contrast, blocking glycogen metabolism in vivo with DAB may impair hippocampal memory formation (Suzuki et al., 2011). Glycogen may also be utilized for glutamate and glutamine synthesis (Gibbs et al., 2006, 2007; Sickmann et al., 2009). Glycogen-derived energy is also involved in the uptake of glutamate, since inhibition of glycogen breakdown impairs the uptake of the glutamate analog D-[3H]aspartate irrespective of the presence of glucose in cultured astrocytes (Sickmann et al., 2009). Glycogenolysis can be stimulated directly by low glucose levels, norepinephrine, and vasoactive intestinal peptide (VIP) (Magistretti et al., 1998; Magistretti, 2006).
Summary
Short-term regulation of the metabolic unit occurs through tight regulation of these multiple local factors, acting between neurons, astrocytes and blood vessels, on the time scale of seconds to minutes. Some factors, such as glycogen, can also potentially help prevent substrate starvation over a period of up to an hour. However, during long-term issues such as development and aging there are a number of other cues needed to maintain homeostasis, beyond these factors implicated in short-term regulation of metabolism.
Long-Term Regulation of CNS Energy Metabolism
Complex neuronal networks regulate long-term energy homeostasis, using a variety of peripheral and brain derived hormones to regulate appetite and body weight. For example, brain function responds to changes in insulin, leptins, orexin, neuropeptide Y, propiomelanocortin, epinephrine, norepinephrine, glucagon, glutamate, and a wide variety of other systemic stimuli and regulatory molecules, some of which can act as growth factors for long-term support of the brain. We discuss a few of these factors relevant to energy homeostasis in the brain.
Leptin
Leptin and orexin are involved in long-term weight regulation. Leptin functions to regulate body fat, but leptin receptors are widely expressed throughout the CNS, particularly in hippocampus, cerebellum, brain stem and amygdala (Elmquist et al., 1998; Hakansson et al., 1998; Harvey, 2003), suggesting that leptin may be involved in other roles in CNS function. Leptin can increase memory formation and hippocampal synaptic plasticity through facilitation of Ca2+ influx via NMDA receptors (Shanley et al., 2001), functioning by phosphorylation of MAPK, Src, or PI3 Kinase mediated pathways. However, incubation of leptin with brain slices at resting conditions without glutamate failed to increase Ca2+ concentrations. Leptin can also inhibit mTOR and can function as an antiepileptogenic hormone. Leptin also activates AMP-activated protein kinase (AMPK) and Silent information regulator proteins (Sirtuins) in neurons and is neuroprotective (Greco et al., 2011). Sirtuins are histone deacetylases and are dependent on nicotinamide adenine dinucleotide (NAD+) and upregulation of proteins required for energy metabolism and survival (Zhang et al., 2011).
Insulin
The hormone insulin, which has the ability to activate cell signaling pathways similar to leptin and can function as a neurotrophic factor, facilitates NMDA receptor-mediated responses. Insulin may also decrease Alzheimer pathology by regulating tau phosphorylation and by inhibiting GSK-3 function (Greco et al., 2009, 2011). Neurons can secrete insulin in a calcium-dependent manner when depolarized but this appears to be more of a neurotrophic role rather than associated with energy homeostasis. Insulin can inhibit neuronal activity by activating the Ca2+ activated potassium channel via mitogen activated protein kinase dependent process. There are now trials beginning administering intranasal insulin for possible treatment of Alzheimer’s disease, since this may be a less invasive method of delivery.
Starvation and Ketosis Responses
During severe fasting or with rigorous exercise circulating blood glucose levels fall, leading to initiation of regulatory responses by the liver, particularly increased glucose synthesis and glycogen degradation (Amiel, 1997). Activation of sympathetic neurons, stimulation of adrenalin, cortisol, and enhanced growth hormone secretion all favor increased blood glucose but through different mechanisms, after a more prolonged period of fasting (i.e., 8–12 h); growth hormone in particular can liberate fat from lipid stores as ketone bodies, beta-hydroxybutyrate and acetoacetate. Brain can utilize ketone bodies as a alternative energy source during starvation or hypoglycemia. Because of their permeability through the blood brain barrier via monocarboxylate transporters, these compounds do have ready access to the nervous system and are easily metabolized by the brain (Puchowicz et al., 2007). Beta-hydroxybutyrate, the major ketone body in the circulation, can preserve ATP levels in the brain during low glucose conditions and early development. The rate of utilization of beta-hydroxybutyrate is very high during brain development, particularly during postnatal suckling in mammals, but reduced as animals wean from breast milk after P14 in rats (Izumi et al., 1998). Thus, during starvation a long-term series of changes occur in metabolism to adapt to different energy sources, and particularly to utilize internal fat and eventually protein for energy substrates.
Role of Sirtuins
Sirtuins (Sirt) are a family of nicotinamide adenine nucleotide (NAD) dependent deacetylases, which are implicated in energy metabolism and life span (Michan and Sinclair, 2007; Yu and Auwerx, 2009). Among the known Sirt isoforms (Sirt1–7), Sirt3 has been identified as a stress responsive deacetylase recently shown to have a role in protecting cells under stress conditions (Sundaresan et al., 2008; Kim et al., 2011). Sirt3 was shown to regulate the activity of acetyl-CoA synthetase 2 (AceCS2), an important mitochondrial enzyme involved in generating acetyl-CoA for the TCA cycle. In these studies, Sirt3 knockout resulted in a marked decrease of basal ATP level in vivo (Hirschey et al., 2010, 2011b; Shimazu et al., 2010). Overexpression of Sirt1 and the use of the Sirt1 agonist, resveratrol, were neuroprotective against amyloid beta toxicity by reducing microglial NF-kappaB signaling (Chen et al., 2005). Mitochondrial protein acetylation is markedly elevated in Sirt3(−/−) tissues (Ahn et al., 2008; Sundaresan et al., 2008; Huang et al., 2010). NAD-dependent deacetylation regulates energy homeostasis and the absence of the deacetylase leads to a marked reduction in ATP. Complex I activity can be regulated by acetylation and deacetylation, a posttranslational modification, and Sirt3 also deacetylates GDH and AceCS2 (Hallows et al., 2006; Hirschey et al., 2011a). Sirt has been implicated in caloric restriction as a treatment to enhance life span during aging as well (Someya et al., 2010; Hallows et al., 2011; Shetty et al., 2011; Zhang et al., 2011). This is one of several regulatory molecules spanning the gap between nuclear regulation of metabolism and actual metabolic processing.
Pathological Conditions Leading to Energy Crises
Inadequate supply of oxygen and glucose to the brain can lead to impaired brain function, and if sustained, to permanent cell loss and damage. The clinical conditions where this occurs are usually termed stroke, which commonly arises from changes in blood vessels, such as thrombosis or embolic reasons. Energy metabolism deficits during ischemia can inhibit Na+/K+ ATPase activity and lead to increased ROS generation. Drugs that increase Na+/K+ ATPase activity have a positive effect on neuronal energy metabolism during oxidative stress (Simao et al., 2011). During hypoxia, decreased ATP content leads to rapid depression of the evoked synaptic potentials (Lipton and Whittingham, 1979, 1982). The brain can eventually adapt to these energy crises by increasing collateral blood flow and eventually demarcating regions of the brain into areas with partial or intact neuronal function, separating out regions of permanent damage.
Conclusion
Energy homeostasis is an ongoing process under baseline conscious conditions, during and after brief periods of neuronal activation with accompanying changes in metabolic demand and supply, and also over long-term periods. There are multiple links between neurons, astrocytes, and blood vessels to maintain homeostasis during a wide range of energy demand and supply conditions in the nervous system. Some of these links can now be directly measured by in vivo or in vitro imaging techniques, including PET, fMRI, intrinsic fluorescent imaging (such as NADH or FAD), direct imaging of ion fluxes (i.e., Na+, Ca2+, Cl−), ECS imaging (with IOS), blood flow and oxygen or nitric oxide tissue measurements, and possibly direct heat emission imaging, taking advantage of the large heat signature released by metabolism.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH Grant AG037599 and a VA Merit Review Award.
References
Abbracchio, M. P., and Burnstock, G. (1994). Purinoceptors: are there families of P2X and P2Y purinoceptors? Pharmacol. Ther. 64, 445–475.
Ahn, B. H., Kim, H. S., Song, S., Lee, I. H., Liu, J., Vassilopoulos, A., Deng, C. X., and Finkel, T. (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. U.S.A. 105, 14447–14452.
Amaral, A. I., Teixeira, A. P., Sonnewald, U., and Alves, P. M. (2011). Estimation of intracellular fluxes in cerebellar neurons after hypoglycemia: importance of the pyruvate recycling pathway and glutamine oxidation. J. Neurosci. Res. 89, 700–710.
Amiel, S. A. (1997). Hypoglycaemia in diabetes mellitus – protecting the brain. Diabetologia 40(Suppl. 2), S62–S68.
Anderson, C. M., and Swanson, R. A. (2000). Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32, 1–14.
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., Macvicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243.
Auger, C., and Attwell, D. (2000). Fast removal of synaptic glutamate by postsynaptic transporters. Neuron 28, 547–558.
Bachelard, H. S., Cox, D. W., and Drower, J. (1984). Sensitivity of guinea-pig hippocampal granule cell field potentials to hexoses in vitro: an effect on cell excitability? J. Physiol. 352, 91–102.
Bak, L. K., Schousboe, A., Sonnewald, U., and Waagepetersen, H. S. (2006). Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J. Cereb. Blood Flow Metab. 26, 1285–1297.
Bak, L. K., Walls, A. B., Schousboe, A., Ring, A., Sonnewald, U., and Waagepetersen, H. S. (2009). Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 109(Suppl. 1), 87–93.
Balazs, R., Machiyama, Y., Hammond, B. J., Julian, T., and Richter, D. (1970). The operation of the gamma-aminobutyrate bypath of the tricarboxylic acid cycle in brain tissue in vitro. Biochem. J. 116, 445–461.
Barceló-Torns, M., Lewis, A. M., Gubern, A., Barneda, D., Bloor-Young, D., Picatoste, F., Churchill, G. C., Claro, E., and Masgrau, R. (2011). NAADP mediates ATP-induced Ca2+ signals in astrocytes. FEBS Lett. 585, 2300–2306.
Bardoni, R., Goldstein, P. A., Lee, C. J., Gu, J. G., and Macdermott, A. B. (1997). ATP P2X receptors mediate fast synaptic transmission in the dorsal horn of the rat spinal cord. J. Neurosci. 17, 5297–5304.
Bennett, M. R., and Ho, S. (1992). Adenosine modulation of potassium currents in preganglionic nerve terminals of avian ciliary ganglia. Neurosci. Lett. 137, 41–44.
Benninger, C., Kadis, J., and Prince, D. A. (1980). Extracellular calcium and potassium changes in hippocampal slices. Brain Res. 187, 165–182.
Bondareff, W., and Pysh, J. J. (1968). Distribution of the extracellular space during postnatal maturation of rat cerebral cortex. Anat. Rec. 160, 773–780.
Bourke, R. S., Kimelberg, H. K., Nelson, L. R., Barron, K. D., Auen, E. L., Popp, A. J., and Waldman, J. B. (1980). Biology of glial swelling in experimental brain edema. Adv. Neurol. 28, 99–109.
Brown, A. M., Baltan Tekkok, S., and Ransom, B. R. (2004). Energy transfer from astrocytes to axons: the role of CNS glycogen. Neurochem. Int. 45, 529–536.
Buchheim, K., Schuchmann, S., Siegmund, H., Gabriel, H. J., Heinemann, U., and Meierkord, H. (1999). Intrinsic optical signal measurements reveal characteristic features during different forms of spontaneous neuronal hyperactivity associated with ECS shrinkage in vitro. Eur. J. Neurosci. 11, 1877–1882.
Burnstock, G. (1999). Current status of purinergic signalling in the nervous system. Prog. Brain Res. 120, 3–10.
Butt, A. M. (2011). ATP: a ubiquitous gliotransmitter integrating neuron-glial networks. Semin. Cell Dev. Biol. 22, 205–213.
Cataldo, A. M., and Broadwell, R. D. (1986). Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II. Choroid plexus and ependymal epithelia, endothelia and pericytes. J. Neurocytol. 15, 511–524.
Cesar, M., and Hamprecht, B. (1995). Immunocytochemical examination of neural rat and mouse primary cultures using monoclonal antibodies raised against pyruvate carboxylase. J. Neurochem. 64, 2312–2318.
Chapman, A. G., Westerberg, E., and Siesjo, B. K. (1981). The metabolism of purine and pyrimidine nucleotides in rat cortex during insulin-induced hypoglycemia and recovery. J. Neurochem. 36, 179–189.
Charles, A. C., Merrill, J. E., Dirksen, E. R., and Sanderson, M. J. (1991). Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron 6, 983–992.
Chatton, J.-Y., Marquet, P., and Magistretti, P. J. (2000). A quantitative analysis of l-glutamate-regulated Na+ dynamics in mouse cortical astrocytes: implications for cellular bioenergetics. Eur. J. Neurosci. 12, 3843–3853.
Chen, J., Zhou, Y., Mueller-Steiner, S., Chen, L. F., Kwon, H., Yi, S., Mucke, L., and Gan, L. (2005). SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 280, 40364–40374.
Chih, C.-P., and Roberts, E. L. Jr. (2003). Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J. Cereb. Blood Flow Metab. 23, 1263–1281.
Choi, I. Y., and Gruetter, R. (2003). In vivo 13C NMR assessment of brain glycogen concentration and turnover in the awake rat. Neurochem. Int. 43, 317–322.
Ciricillo, S. P., Hill, M. P., Gonzalez, M. F., Smalley, S., Morton, M. T., and Sharp, F. R. (1994). Whisker stimulation metabolically activates thalamus following cortical transplantation but not following cortical ablation. Neuroscience 59, 975–992.
Clark, N. C., Nagano, N., Kuenzi, F. M., Jarolimek, W., Huber, I., Walter, D., Wietzorrek, G., Boyce, S., Kullmann, D. M., Striessnig, J., and Seabrook, G. R. (2003). Neurological phenotype and synaptic function in mice lacking the CaV1.3 alpha subunit of neuronal L-type voltage-dependent Ca2+ channels. Neuroscience 120, 435–442.
Clarke, D., and Sokoloff, L. (1999). “Circulation and energy metabolism of the brain,” in Basic Neurochemistry: Molecular, Cellular, and Medical Aspects, 5th Edn, eds G. Siegel, B. Agranoff, R. W. Albers, and P. Molinoff (New York: Raven Press), 645–680.
Connors, B. W., Ransom, B. R., Kunis, D. M., and Gutnick, M. J. (1982). Activity-dependent K+ accumulation in the developing rat optic nerve. Science 216, 1341–1343.
Cook, S. P., Vulchanova, L., Hargreaves, K. M., Elde, R., and Mccleskey, E. W. (1997). Distinct ATP receptors on pain-sensing and stretch-sensing neurons. Nature 387, 505–508.
Cruz, N. F., and Dienel, G. A. (2002). High glycogen levels in brains of rats with minimal environmental stimuli: implications for metabolic contributions of working astrocytes. J. Cereb. Blood Flow Metab. 22, 1476–1489.
Davis, K. D., Kwan, C. L., Crawley, A. P., and Mikulis, D. J. (1998). Functional MRI study of thalamic and cortical activations evoked by cutaneous heat, cold, and tactile stimuli. J. Neurophysiol. 80, 1533–1546.
Denton, R. M. (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316.
Di Biase, V., Tuluc, P., Campiglio, M., Obermair, G. J., Heine, M., and Flucher, B. E. (2011). Surface traffic of dendritic CaV1.2 calcium channels in hippocampal neurons. J. Neurosci. 31, 13682–13694.
Dienel, G. A., and Cruz, N. F. (2006). Astrocyte activation in working brain: energy supplied by minor substrates. Neurochem. Int. 48, 586–595.
Dienel, G. A., and Hertz, L. (2001). Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 66, 824–838.
Dietzel, I., Heinemann, U., Hofmeier, G., and Lux, H. D. (1982). Stimulus-induced changes in extracellular Na+ and Cl- concentration in relation to changes in the size of the extracellular space. Exp. Brain Res. 46, 73–84.
Dringen, R., Wiesinger, H., and Hamprecht, B. (1993). Uptake of L-lactate by cultured rat brain neurons. Neurosci. Lett. 163, 5–7.
Du, F., Zhu, X.-H., Zhang, Y., Friedman, M., Zhang, N., Ugurbil, K., and Chen, W. (2008). Tightly coupled brain activity and cerebral ATP metabolic rate. Proc. Natl. Acad. Sci. U.S.A. 105, 6409–6414.
Duchen, M. R. (1992). Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem. J. 283(Pt 1), 41–50.
Elman, I., Sokoloff, L., Adler, C. M., Weisenfeld, N., and Breier, A. (1999). The effects of pharmacological doses of 2-deoxyglucose on cerebral blood flow in healthy volunteers. Brain Res. 815, 243–249.
Elmquist, J. K., Maratos-Flier, E., Saper, C. B., and Flier, J. S. (1998). Unraveling the central nervous system pathways underlying responses to leptin. Nat. Neurosci. 1, 445–450.
Fan, P., O’Regan, P. A., and Szerb, J. C. (1988). Effect of low glucose concentration on synaptic transmission in the rat hippocampal slice. Brain Res. Bull. 21, 741–747.
Fayuk, D., Aitken, P. G., Somjen, G. G., and Turner, D. A. (2002). Two different mechanisms underlie reversible, intrinsic optical signals in rat hippocampal slices. J. Neurophysiol. 87, 1924–1937.
Fields, R. D., and Stevens, B. (2000). ATP: an extracellular signaling molecule between neurons and glia. Trends Neurosci. 23, 625–633.
Foster, K. A., Beaver, C. J., and Turner, D. A. (2005). Interaction between tissue oxygen tension and NADH imaging during synaptic stimulation and hypoxia in rat hippocampal slices. Neuroscience 132, 645–657.
Fowler, J. C. (1992). Escape from inhibition of synaptic transmission during in vitro hypoxia and hypoglycemia in the hippocampus. Brain Res. 573, 169–173.
Fowler, J. C. (1993). Purine release and inhibition of synaptic transmission during hypoxia and hypoglycemia in rat hippocampal slices. Neurosci. Lett. 157, 83–86.
Fox, P. T., Mintun, M. A., Reiman, E. M., and Raichle, M. E. (1988). Enhanced detection of focal brain responses using intersubject averaging and change-distribution analysis of subtracted PET images. J. Cereb. Blood Flow Metab. 8, 642–653.
Furchgott, R. F., and Zawadzki, J. V. (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288, 373–376.
Galeffi, F., Foster, K. A., Sadgrove, M. P., Beaver, C. J., and Turner, D. A. (2007). Lactate uptake contributes to the NAD(P)H biphasic response and tissue oxygen response during synaptic stimulation in area CA1 of rat hippocampal slices. J. Neurochem. 103, 2449–2461.
Galeffi, F., Somjen, G. G., Foster, K. A., and Turner, D. A. (2011). Simultaneous monitoring of tissue PO2 and NADH fluorescence during synaptic stimulation and spreading depression reveals a transient dissociation between oxygen utilization and mitochondrial redox state in rat hippocampal slices. J. Cereb. Blood Flow Metab. 31, 626–639.
Ghosh, A., Cheung, Y. Y., Mansfield, B. C., and Chou, J. Y. (2005). Brain contains a functional glucose-6-phosphatase complex capable of endogenous glucose production. J. Biol. Chem. 280, 11114–11119.
Gibbs, M. E., Anderson, D. G., and Hertz, L. (2006). Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 54, 214–222.
Gibbs, M. E., Lloyd, H. G., Santa, T., and Hertz, L. (2007). Glycogen is a preferred glutamate precursor during learning in 1-day-old chick: biochemical and behavioral evidence. J. Neurosci. Res. 85, 3326–3333.
Gordon, G. R., Choi, H. B., Rungta, R. L., Ellis-Davies, G. C., and Macvicar, B. A. (2008). Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 456, 745–749.
Grafton, S. T., Mazziotta, J. C., Presty, S., Friston, K. J., Frackowiak, R. S., and Phelps, M. E. (1992). Functional anatomy of human procedural learning determined with regional cerebral blood flow and PET. J. Neurosci. 12, 2542–2548.
Greco, S. J., Hamzelou, A., Johnston, J. M., Smith, M. A., Ashford, J. W., and Tezapsidis, N. (2011). Leptin boosts cellular metabolism by activating AMPK and the sirtuins to reduce tau phosphorylation and beta-amyloid in neurons. Biochem. Biophys. Res. Commun. 414, 170–174.
Greco, S. J., Sarkar, S., Casadesus, G., Zhu, X., Smith, M. A., Ashford, J. W., Johnston, J. M., and Tezapsidis, N. (2009). Leptin inhibits glycogen synthase kinase-3beta to prevent tau phosphorylation in neuronal cells. Neurosci. Lett. 455, 191–194.
Green, E. M., Barrett, C. F., Bultynck, G., Shamah, S. M., and Dolmetsch, R. E. (2007). The tumor suppressor eIF3e mediates calcium-dependent internalization of the L-type calcium channel CaV1.2. Neuron 55, 615–632.
Gruetter, R., Seaquist, E. R., and Ugurbil, K. (2001). A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am. J. Physiol. Endocrinol. Metab. 281, E100–E112.
Hakansson, M. L., Brown, H., Ghilardi, N., Skoda, R. C., and Meister, B. (1998). Leptin receptor immunoreactivity in chemically defined target neurons of the hypothalamus. J. Neurosci. 18, 559–572.
Hallows, W. C., Lee, S., and Denu, J. M. (2006). Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. U.S.A. 103, 10230–10235.
Hallows, W. C., Yu, W., Smith, B. C., Devries, M. K., Ellinger, J. J., Someya, S., Shortreed, M. R., Prolla, T., Markley, J. L., Smith, L. M., Zhao, S., Guan, K. L., and Denu, J. M. (2011). Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell 41, 139–149.
Hell, J. W., Westenbroek, R. E., Warner, C., Ahlijanian, M. K., Prystay, W., Gilbert, M. M., Snutch, T. P., and Catterall, W. A. (1993). Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 123, 949–962.
Hertz, L., and Dienel, G. A. (2002). Energy metabolism in the brain. Int. Rev. Neurobiol. 51, 1–102.
Hertz, L., Peng, L., and Dienel, G. A. (2007). Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 27, 219–249.
Hirschey, M. D., Shimazu, T., Capra, J. A., Pollard, K. S., and Verdin, E. (2011a). SIRT1 and SIRT3 deacetylate homologous substrates: AceCS1,2 and HMGCS1,2. Aging (Albany NY) 3, 635–642.
Hirschey, M. D., Shimazu, T., Huang, J. Y., Schwer, B., and Verdin, E. (2011b). SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb. Symp. Quant. Biol. doi: 10.1101/sqb.2011.76.010850. [Epub ahead of print].
Hirschey, M. D., Shimazu, T., Goetzman, E., Jing, E., Schwer, B., Lombard, D. B., Grueter, C. A., Harris, C., Biddinger, S., Ilkayeva, O. R., Stevens, R. D., Li, Y., Saha, A. K., Ruderman, N. B., Bain, J. R., Newgard, C. B., Farese, R. V. Jr., Alt, F. W., Kahn, C. R., and Verdin, E. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125.
Hochwald, G. M., Nakamura, S., and Camins, M. B. (1981). The rat in experimental obstructive hydrocephalus. Z. Kinderchir. 34, 403–410.
Hoge, R. D., Atkinson, J., Gill, B., Crelier, G. R., Marrett, S., and Pike, G. B. (1999). Stimulus-dependent BOLD and perfusion dynamics in human V1. Neuroimage 9, 573–585.
Holmgren, C. D., Mukhtarov, M., Malkov, A. E., Popova, I. Y., Bregestovski, P., and Zilberter, Y. (2010). Energy substrate availability as a determinant of neuronal resting potential, GABA signaling and spontaneous network activity in the neonatal cortex in vitro. J. Neurochem. 112, 900–912.
Hu, Y., and Wilson, G. S. (1997a). Rapid changes in local extracellular rat brain glucose observed with an in vivo glucose sensor. J. Neurochem. 68, 1745–1752.
Hu, Y., and Wilson, G. S. (1997b). A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J. Neurochem. 69, 1484–1490.
Huang, J. Y., Hirschey, M. D., Shimazu, T., Ho, L., and Verdin, E. (2010). Mitochondrial sirtuins. Biochim. Biophys. Acta 1804, 1645–1651.
Hyder, F., Kida, I., Behar, K. L., Kennan, R. P., Maciejewski, P. K., and Rothman, D. L. (2001). Quantitative functional imaging of the brain: towards mapping neuronal activity by BOLD fMRI. NMR. Biomed. 14, 413–431.
Iadecola, C., and Nedergaard, M. (2007). Glial regulation of the cerebral microvasculature. Nat. Neurosci. 10, 1369–1376.
Ido, Y., Chang, K., and Williamson, J. R. (2004). NADH augments blood flow in physiologically activated retina and visual cortex. Proc. Natl. Acad. Sci. U.S.A. 101, 653–658.
Ivanov, A., Mukhtarov, M., Bregestovski, P., and Zilberter, Y. (2011). Lactate effectively covers energy demands during neuronal network activity in neonatal hippocampal slices. Front Neuroenergetics 3:2. doi:10.3389/fnene.2011.00002
Ivanov, A., and Zilberter, Y. (2011). Critical state of energy metabolism in brain slices: the principal role of oxygen delivery and energy substrates in shaping neuronal activity. Front Neuroenergetics 3:9. doi:10.3389/fnene.2011.00009
Izumi, Y., Ishii, K., Katsuki, H., Benz, A. M., and Zorumski, C. F. (1998). beta-Hydroxybutyrate fuels synaptic function during development. Histological and physiological evidence in rat hippocampal slices. J. Clin. Invest. 101, 1121–1132.
Jensen, B. S., Jessen, F., and Hoffmann, E. K. (1993). Na+, K+, Cl- cotransport and its regulation in Ehrlich ascites tumor cells. Ca2+/calmodulin and protein kinase C dependent pathways. J. Membr. Biol. 131, 161–178.
Kapogiannis, D., and Mattson, M. P. (2011). Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 10, 187–198.
Kastritsis, C. H., Salm, A. K., and Mccarthy, K. (1992). Stimulation of the P2Y purinergic receptor on type 1 astroglia results in inositol phosphate formation and calcium mobilization. J. Neurochem. 58, 1277–1284.
Keelan, J., Vergun, O., and Duchen, M. R. (1999). Excitotoxic mitochondrial depolarisation requires both calcium and nitric oxide in rat hippocampal neurons. J. Physiol. (Lond.) 520(Pt 3), 797–813.
Kim, S. H., Lu, H. F., and Alano, C. C. (2011). Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS ONE 6, e14731. doi:10.1371/journal.pone.0014731
Knopfel, T., Spuler, A., Grafe, P., and Gahwiler, B. H. (1990). Cytosolic calcium during glucose deprivation in hippocampal pyramidal cells of rats. Neurosci. Lett. 117, 295–299.
Koehler, R. C., Roman, R. J., and Harder, D. R. (2009). Astrocytes and the regulation of cerebral blood flow. Trends Neurosci. 32, 160–169.
Korf, J. (2006). Is brain lactate metabolized immediately after neuronal activity through the oxidative pathway? J. Cereb. Blood Flow Metab. 26, 1584–1586.
Kovacs, R., Schuchmann, S., Gabriel, S., Kardos, J., and Heinemann, U. (2001). Ca2+ signalling and changes of mitochondrial function during low-Mg2+-induced epileptiform activity in organotypic hippocampal slice cultures. Eur. J. Neurosci. 13, 1311–1319.
LaManna, J. C., Mccracken, K. A., Patil, M., and Prohaska, O. J. (1989). Stimulus-activated changes in brain tissue temperature in the anesthetized rat. Metab. Brain Dis. 4, 225–237.
LaManna, J. C., Rosenthal, M., Novack, R., Moffett, D. F., and Jobsis, F. F. (1980). Temperature coefficients for the oxidative metabolic responses to electrical stimulation in cerebral cortex. J. Neurochem. 34, 203–209.
Lipton, P., and Whittingham, T. S. (1979). The effect of hypoxia on evoked potentials in the in vitro hippocampus. J. Physiol. (Lond.) 287, 427–438.
Lipton, P., and Whittingham, T. S. (1982). Reduced ATP concentration as a basis for synaptic transmission failure during hypoxia in the in vitro guinea-pig hippocampus. J. Physiol. (Lond.) 325, 51–65.
Lothman, E., Lamanna, J., Cordingley, G., Rosenthal, M., and Somjen, G. (1975). Responses of electrical potential, potassium levels, and oxidative metabolic activity of the cerebral neocortex of cats. Brain Res. 88, 15–36.
Lupica, C. R., Proctor, W. R., and Dunwiddie, T. V. (1992). Presynaptic inhibition of excitatory synaptic transmission by adenosine in rat hippocampus: analysis of unitary EPSP variance measured by whole-cell recording. J. Neurosci. 12, 3753–3764.
Magistretti, P. J. (2006). Neuron-glia metabolic coupling and plasticity. J. Exp. Biol. 209, 2304–2311.
Magistretti, P. J., Cardinaux, J. R., and Martin, J. L. (1998). VIP and PACAP in the CNS: regulators of glial energy metabolism and modulators of glutamatergic signaling. Ann. N. Y. Acad. Sci. 865, 213–225.
Magistretti, P. J., and Pellerin, L. (1999). Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354, 1155–1163.
Malarkey, E. B., and Parpura, V. (2008). Mechanisms of glutamate release from astrocytes. Neurochem. Int. 52, 142–154.
McKenna, M. C., Sonnewald, U., Huang, X., Stevenson, J., and Zielke, H. R. (1996a). Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 66, 386–393.
McKenna, M. C., Tildon, J. T., Stevenson, J. H., and Huang, X. (1996b). New insights into the compartmentation of glutamate and glutamine in cultured rat brain astrocytes. Dev. Neurosci. 18, 380–390.
Michan, S., and Sinclair, D. (2007). Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13.
Mogul, D. J., Adams, M. E., and Fox, A. P. (1993). Differential activation of adenosine receptors decreases N-type but potentiates P-type Ca2+ current in hippocampal CA3 neurons. Neuron 10, 327–334.
Moreadith, R. W., and Lehninger, A. L. (1984). The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J. Biol. Chem. 259, 6215–6221.
Mukhtarov, M., Ivanov, A., Zilberter, Y., and Bregestovski, P. (2011). Inhibition of spontaneous network activity in neonatal hippocampal slices by energy substrates is not correlated with intracellular acidification. J. Neurochem. 116, 316–321.
Naito, S., and Ueda, T. (1983). Adenosine triphosphate-dependent uptake of glutamate into protein I-associated synaptic vesicles. J. Biol. Chem. 258, 696–699.
Nelson, M. T., and Quayle, J. M. (1995). Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 268, C799–C822.
Norenberg, M. D., and Martinez-Hernandez, A. (1979). Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 161, 303–310.
Obermair, G. J., Szabo, Z., Bourinet, E., and Flucher, B. E. (2004). Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur. J. Neurosci. 19, 2109–2122.
Offenhauser, N., Thomsen, K., Caesar, K., and Lauritzen, M. (2005). Activity-induced tissue oxygenation changes in rat cerebellar cortex: interplay of postsynaptic activation and blood flow. J. Physiol. 565, 279–294.
O’Neill, W. C. (1999). Physiological significance of volume-regulatory transporters. Am. J. Physiol. 276, C995–C1011.
Ozkan, E. D., and Ueda, T. (1998). Glutamate transport and storage in synaptic vesicles. Jpn. J. Pharmacol. 77, 1–10.
Pangrsic, T., Potokar, M., Stenovec, M., Kreft, M., Fabbretti, E., Nistri, A., Pryazhnikov, E., Khiroug, L., Giniatullin, R., and Zorec, R. (2007). Exocytotic release of ATP from cultured astrocytes. J. Biol. Chem. 282, 28749–28758.
Pankratov, Y., Lalo, U., Verkhratsky, A., and North, R. (2006). Vesicular release of ATP at central synapses. Pflügers Arch. 452, 589–597.
Parpura, V., Basarsky, T. A., Liu, F., Jeftinija, K., Jeftinija, S., and Haydon, P. G. (1994). Glutamate-mediated astrocyte-neuron signalling. Nature 369, 744–747.
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J.-Y., Takano, H., Moss, S. J., Mccarthy, K., and Haydon, P. G. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116.
Paulson, O. B., Hasselbalch, S. G., Rostrup, E., Knudsen, G. M., and Pelligrino, D. (2009). Cerebral blood flow response to functional activation. J. Cereb. Blood Flow Metab. 30, 2–14.
Pellerin, L., Bouzier-Sore, A. K., Aubert, A., Serres, S., Merle, M., Costalat, R., and Magistretti, P. J. (2007). Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262.
Pellerin, L., and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. U.S.A. 91, 10625–10629.
Pellerin, L., and Magistretti, P. J. (1996). Excitatory amino acids stimulate aerobic glycolysis in astrocytes via an activation of the Na+/K+ ATPase. Dev. Neurosci. 18, 336–342.
Pellerin, L., and Magistretti, P. J. (1997). Glutamate uptake stimulates Na+, K+ -ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J. Neurochem. 69, 2132–2137.
Phelps, M. E., and Mazziotta, J. C. (1985). Positron emission tomography: human brain function and biochemistry. Science 228, 799–809.
Phillis, J. W., and Wu, P. H. (1981). The role of adenosine and its nucleotides in central synaptic transmission. Prog. Neurobiol. 16, 187–239.
Puchowicz, M. A., Xu, K., Sun, X., Ivy, A., Emancipator, D., and Lamanna, J. C. (2007). Diet-induced ketosis increases capillary density without altered blood flow in rat brain. Am. J. Physiol. Endocrinol. Metab. 292, E1607–E1615.
Qu, H., Eloqayli, H., Unsgard, G., and Sonnewald, U. (2001). Glutamate decreases pyruvate carboxylase activity and spares glucose as energy substrate in cultured cerebellar astrocytes. J. Neurosci. Res. 66, 1127–1132.
Ransom, B. R., Yamate, C. L., and Connors, B. W. (1985). Activity-dependent shrinkage of extracellular space in rat optic nerve: a developmental study. J. Neurosci. 5, 532–535.
Rheims, S., Holmgren, C. D., Chazal, G., Mulder, J., Harkany, T., Zilberter, T., and Zilberter, Y. (2009). GABA action in immature neocortical neurons directly depends on the availability of ketone bodies. J. Neurochem. 110, 1330–1338.
Rosenthal, M., and Jobsis, F. F. (1971). Intracellular redox changes in functioning cerebral cortex. II. Effects of direct cortical stimulation. J. Neurophysiol. 34, 750–762.
Rothman, D. L., Behar, K. L., Hyder, F., and Shulman, R. G. (2003). In vivo NMR studies of the glutamate neurotransmitter flux and neuroenergetics: implications for brain function. Annu. Rev. Physiol. 65, 401–427.
Schlick, B., Flucher, B. E., and Obermair, G. J. (2010). Voltage-activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience 167, 786–798.
Scholz, K. P., and Miller, R. J. (1991). Analysis of adenosine actions on Ca2+ currents and synaptic transmission in cultured rat hippocampal pyramidal neurones. J. Physiol. 435, 373–393.
Schuchmann, S., Kovacs, R., Kann, O., Heinemann, U., and Buchheim, K. (2001). Monitoring NAD(P)H autofluorescence to assess mitochondrial metabolic functions in rat hippocampal-entorhinal cortex slices. Brain Res. Brain Res. Protoc. 7, 267–276.
Sebastiao, A. M., and Ribeiro, J. A. (1996). Adenosine A2 receptor-mediated excitatory actions on the nervous system. Prog. Neurobiol. 48, 167–189.
Shank, R. P., Bennett, G. S., Freytag, S. O., and Campbell, G. L. (1985). Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 329, 364–367.
Shanley, L. J., Irving, A. J., and Harvey, J. (2001). Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 21, RC186.
Shetty, P. K., Galeffi, F., and Turner, D. A. (2011). Age-induced alterations in hippocampal function and metabolism. Aging Dis. 2, 196–218.
Shetty, P. K., Sadgrove, M. P., Galeffi, F., and Turner, D. A. (2012). Pyruvate incubation enhances glycogen stores and sustains neuronal function during subsequent glucose deprivation. Neurobiol. Dis. 45, 177–187.
Shimazu, T., Hirschey, M. D., Hua, L., Dittenhafer-Reed, K. E., Schwer, B., Lombard, D. B., Li, Y., Bunkenborg, J., Alt, F. W., Denu, J. M., Jacobson, M. P., and Verdin, E. (2010). SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 12, 654–661.
Shulman, R. G., Hyder, F., and Rothman, D. L. (2009). Baseline brain energy supports the state of consciousness. Proc. Natl. Acad. Sci. U.S.A. 106, 11096–11101.
Shulman, R. G., Rothman, D. L., Behar, K. L., and Hyder, F. (2004). Energetic basis of brain activity: implications for neuroimaging. Trends Neurosci. 27, 489–495.
Shuttleworth, C. W., Brennan, A. M., and Connor, J. A. (2003). NAD(P)H fluorescence imaging of postsynaptic neuronal activation in murine hippocampal slices. J. Neurosci. 23, 3196–3208.
Sibson, N. R., Mason, G. F., Shen, J., Cline, G. W., Herskovits, A. Z., Wall, J. E., Behar, K. L., Rothman, D. L., and Shulman, R. G. (2001). In vivo (13)C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during. J. Neurochem. 76, 975–989.
Sickmann, H. M., Walls, A. B., Schousboe, A., Bouman, S. D., and Waagepetersen, H. S. (2009). Functional significance of brain glycogen in sustaining glutamatergic neurotransmission. J. Neurochem. 109(Suppl. 1), 80–86.
Simao, F., Matte, A., Matte, C., Soares, F. M., Wyse, A. T., Netto, C. A., and Salbego, C. G. (2011). Resveratrol prevents oxidative stress and inhibition of Na(+)K(+)-ATPase activity induced by transient global cerebral ischemia in rats. J. Nutr. Biochem. 22, 921–928.
Smith, A. J., Blumenfeld, H., Behar, K. L., Rothman, D. L., Shulman, R. G., and Hyder, F. (2002). Cerebral energetics and spiking frequency: the neurophysiological basis of fMRI. Proc. Natl. Acad. Sci. U.S.A. 99, 10765–10770.
Sobey, C. G., Heistad, D. D., and Faraci, F. M. (1997). Mechanisms of bradykinin-induced cerebral vasodilatation in rats. Evidence that reactive oxygen species activate K+ channels. Stroke 28, 2290–2294; discussion 2295.
Sokoloff, L. (1999). Energetics of functional activation in neural tissues. Neurochem. Res. 24, 321–329.
Someya, S., Yu, W., Hallows, W. C., Xu, J., Vann, J. M., Leeuwenburgh, C., Tanokura, M., Denu, J. M., and Prolla, T. A. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812.
Somjen, G. G. (2002). Ion regulation in the brain: implications for pathophysiology. Neuroscientist 8, 254–267.
Su, G., Haworth, R. A., Dempsey, R. J., and Sun, D. (2000). Regulation of Na(+)-K(+)-Cl(-) cotransporter in primary astrocytes by dibutyryl cAMP and high [K(+)](o). Am. J. Physiol. Cell Physiol. 279, C1710–C1721.
Suh, S. W., Hamby, A. M., and Swanson, R. A. (2007). Hypoglycemia, brain energetics, and hypoglycemic neuronal death. Glia 55, 1280–1286.
Sukstanskii, A. L., and Yablonskiy, D. A. (2006). Theoretical model of temperature regulation in the brain during changes in functional activity. Proc. Natl. Acad. Sci. U.S.A. 103, 12144–12149.
Sundaresan, N. R., Samant, S. A., Pillai, V. B., Rajamohan, S. B., and Gupta, M. P. (2008). SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 28, 6384–6401.
Suzuki, A., Stern, S. A., Bozdagi, O., Huntley, G. W., Walker, R. H., Magistretti, P. J., and Alberini, C. M. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823.
Thompson, J. K., Peterson, M. R., and Freeman, R. D. (2003). Single-neuron activity and tissue oxygenation in the cerebral cortex. Science 299, 1070–1072.
Trubel, H. K. F., Sacolick, L. I., and Hyder, F. (2005). Regional temperature changes in the brain during somatosensory stimulation. J. Cereb. Blood Flow Metab. 26, 68–78.
Turner, D. A., Foster, K. A., Galeffi, F., and Somjen, G. G. (2007). Differences in O2 availability resolve the apparent discrepancies in metabolic intrinsic optical signals in vivo and in vitro. Trends Neurosci. 30, 390–398.
Van den Berg, C. J., and Garfinkel, D. (1971). A stimulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem. J. 123, 211–218.
Van den Berg, C. J., Krzalic, L., Mela, P., and Waelsch, H. (1969). Compartmentation of glutamate metabolism in brain. Evidence for the existence of two different tricarboxylic acid cycles in brain. Biochem. J. 113, 281–290.
Van Harreveld, A., Khattab, F. I., and Steiner, J. (1969). Extracellular space in the central nervous system of the leech, Mooreobdella fervida. J. Neurobiol. 1, 23–40.
Vilchez, D., Ros, S., Cifuentes, D., Pujadas, L., Valles, J., Garcia-Fojeda, B., Criado-Garcia, O., Fernandez-Sanchez, E., Medrano-Fernandez, I., Dominguez, J., Garcia-Rocha, M., Soriano, E., Rodriguez de Cordoba, S., and Guinovart, J. J. (2007). Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 10, 1407–1413.
Voutsinos-Porche, B., Bonvento, G., Tanaka, K., Steiner, P., Welker, E., Chatton, J. Y., Magistretti, P. J., and Pellerin, L. (2003). Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron 37, 275–286.
Walz, W., and Hertz, L. (1982). Ouabain-sensitive and ouabain-resistant net uptake of potassium into astrocytes and neurons in primary cultures. J. Neurochem. 39, 70–77.
Westergaard, N., Sonnewald, U., and Schousboe, A. (1995). Metabolic trafficking between neurons and astrocytes: the glutamate/glutamine cycle revisited. Dev. Neurosci. 17, 203–211.
White, T. D., and MacDonald, W. F. (1990). Neural release of ATP and adenosinea. Ann. N. Y. Acad. Sci. 603, 287–298.
William, V. D. K. (2003). Loading and recycling of synaptic vesicles in the Torpedo electric organ and the vertebrate neuromuscular junction. Prog. Neurobiol. 71, 269–303.
Wyss, M. T., Jolivet, R., Buck, A., Magistretti, P. J., and Weber, B. (2011). In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 31, 7477–7485.
Yablonskiy, D. A., Ackerman, J. J., and Raichle, M. E. (2000). Coupling between changes in human brain temperature and oxidative metabolism during prolonged visual stimulation. Proc. Natl. Acad. Sci. U.S.A. 97, 7603–7608.
Yu, A. C., Drejer, J., Hertz, L., and Schousboe, A. (1983). Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J. Neurochem. 41, 1484–1487.
Yu, J., and Auwerx, J. (2009). The role of sirtuins in the control of metabolic homeostasis. Ann. N. Y. Acad. Sci. 1173(Suppl. 1), E10–E19.
Zhang, D., Liu, Y., and Chen, D. (2011). SIRT-ain relief from age-inducing stress. Aging (Albany NY) 3, 158–161.
Zhu, P. J., and Krnjevic, K. (1993). Adenosine release is a major cause of failure of synaptic transmission during hypoglycemia in rat hippocampal slices. Neurosci. Lett. 155, 128–131.
Zielke, H. R., Zielke, C. L., Baab, P. J., and Tildon, J. T. (2007). Effect of fluorocitrate on cerebral oxidation of lactate and glucose in freely moving rats. J. Neurochem. 101, 9–16.
Keywords: neuronal metabolism, hippocampus, ATP, NADH, oxygen, lactate, pyruvate, cerebral metabolism
Citation: Shetty PK, Galeffi F and Turner DA (2012) Cellular links between neuronal activity and energy homeostasis. Front. Pharmacol. 3:43. doi: 10.3389/fphar.2012.00043
Received: 21 December 2011; Accepted: 24 February 2012;
Published online: 20 March 2012.
Edited by:
Yuri Zilberter, INSERM U751, FranceReviewed by:
Yuri Zilberter, INSERM U751, FranceMuzamil Ahmad, Indian Institute of Integrative Medicine, India
Copyright: © 2012 Shetty, Galeffi and Turner. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Dennis A. Turner, Neurosurgery and Neurobiology, Research and Surgery Services, Duke University, Box 3807, NSU, DUMC, Durham, NC 27710, USA. e-mail:ZGVubmlzLnR1cm5lckBkdWtlLmVkdQ==